

Abstract
Hyperactivated EGFR signaling is a driver of various human cancers, including glioblastoma (GBM). Effective EGFR-targeted therapies rely on knowledge of key signaling hubs that transfer and amplify EGFR signaling. Here we focus on the transcription factor TAZ, a potential signaling hub in the EGFR signaling network. TAZ expression was positively associated with EGFR expression in clinical GBM specimens. In patient-derived GBM neurospheres, EGF induced TAZ through EGFR-ERK and EGFR-STAT3 signaling, and the constitutively active EGFRvIII mutation caused EGF-independent hyperactivation of TAZ. Genome-wide analysis showed that the EGFR-TAZ axis activates multiple oncogenic signaling mechanisms, including an EGFR-TAZ-RTK positive feedback loop, as well as upregulating HIF1α and other oncogenic genes. TAZ hyperactivation in GBM stem-like cells induced exogenous mitogen-independent growth and promoted GBM invasion, radioresistance, and tumorigenicity. Screening a panel of brain-penetrating EGFR inhibitors identified osimertinib as the most potent inhibitor of the EGFR-TAZ signaling axis. Systemic osimertinib treatment inhibited the EGFR-TAZ axis and in vivo growth of GBM stem-like cell xenografts. Overall these results show that the therapeutic efficacy of osimertinib relies on effective TAZ inhibition, thus identifying TAZ as a potential biomarker of osimertinib sensitivity.
Keywords: receptor tyrosine kinase, oncogene, osimertinib, transcription factor, brain tumor
Introduction
Gliomas are the most common neoplasms of the central nervous system (CNS), accounting for 80% of primary malignant brain tumors (1). Patients with glioblastoma (GBM), the grade IV glioma, have a median survival of 14–15 months and less than 6% 5-year survival. The Cancer Genome Atlas (TCGA) project identified a group of GBM-associated mutations that mainly reside in three core signaling pathways involving the receptor tyrosine kinases (RTKs), p53, and RB (2). EGFR is a GBM-associated RTK that is hyperactivated through gene amplification or mutation in about 45% of GBM cases and drives GBM malignancy (2). EGFRVIII, the most common mutation in GBM, has a deletion of exons 2–7 of EGFR resulting in ligand-independent constitutive EGFR activation. Hyperactivation of the EGFR signaling pathway promotes GBM cell proliferation, invasion, therapeutic resistance, stemness and other malignant phenotypes (3). The EGFR signaling axis activates a transcription network relying on transcription factors (TFs) as signaling hubs to transfer and amplify oncogenic signals (3). Better understanding of key EGFR-activated TFs will facilitate the development of therapies to target EGFR signaling more effectively in GBM and other EGFR-driven malignancies.
We studied the transcription factor TAZ (also known as WW domain containing transcription regulator 1, WWTR1) in the context of the EGFR signaling axis. TAZ and its paralog YAP (the Yes-associated protein) are transcriptional co-activators initially known as the downstream mediators of the Hippo signaling pathway (4). TAZ/YAP form complex with their transcriptional partners (e.g. TEAD1–4, SMAD and RUNX) to activate gene targets commonly hyperactive in human cancers (4). TAZ and YAP function as oncogenes through promoting aberrant cell proliferation and survival, enhancing cancer stemness and maintaining a pro-tumor microenvironment (4, 5). TAZ/YAP are contextually regulated by various upstream signaling pathways in tumors, e.g. the Hippo signaling pathway in liver cancer (6), the WNT signaling pathway in mammary (7) and colon cancers (8), and the GPCR signaling pathway in melanoma (9). Bhat et al. were the first to show that TAZ is hyperactivated in GBM specimens and TAZ silencing inhibited GBM neurosphere growth in vitro and xenograft formation in vivo, supporting TAZ as a potential therapeutic target for GBM (10). Findings from Bhat et al. (10) are supported by recent single-cell analyses defining TAZ/YAP as key regulators of GBM stemness (5), and notably their results also indicate that TAZ hyperactivation in GBM cells may not be regulated by the Hippo signaling pathway, thus warranting more studies to define upstream driver of TAZ hyperactivation and develop potential TAZ-targeted therapies for GBM.
Here, we identified EGFR signaling as a TAZ inducer in GBM. We established a genome-wide map of the EGFR-activated TAZ-dependent transcription program and further identified a brain-penetrating cancer drug for inhibiting the EGFR-TAZ axis. Our discoveries reveal a novel EGFR-TAZ signaling axis and provide a clinically translatable strategy for treating GBM and other TAZ-hyperactivated tumors.
Materials and Methods
Cell culture and reagents
GBM neurosphere (GBM1A and GBM1B) were established by Dr. Vescovi, and referred to as GBM0913 and GBM0627, respectively, previously in (11). The M1123 (1123) GBM neurosphere line was established by Dr. Nakano (12). Cells were cultured in N2/B27 medium with EGF) and FGF. A172 and U87MG cells were purchased from ATCC and grown in DMEM with 10% FBS under the condition of 5% CO2/95% air at 37 Celsius. The EGFR status in each cell model is EGFR amplified (GBM1A and GBM1B) (11), EGFRvIII+ (M1123) (Fig. 3A), EGFR wild-type (U87MG) (13), or mutant EGFR with an in-frame tandem duplication of tyrosine kinase and calcium internalization domains (A172) (14). Primary GBM neurospheres (JH273 and JH551) were established from GBM tumors at Johns Hopkins University and used within 5 passages. All cell lines are tested every two weeks for Mycoplasma and routinely authenticated with short tandem repeat profiling. All cell lines are used in our experiments within 10 passages from isolation or thawing.
Figure 3: The EGFRvIII mutation causes EGF-independent TAZ hyperactivation in GBM cells.

A: EGFR western blotting using total protein lysates from GBM cells. 293FT cells with lentiviral EGFRvIII expression were used as the positive control.
B and C: GBM1B and U87MG cells with +/− EGFRvIII expression were subjected to TAZ western blotting (B). GBM1B cells with +/− EGFRvIII expression were subjected to qRT-PCR for two TAZ gene targets (C).
D: GBM cells as marked were depleted of growth factors for 16 hours and treated with EGF for the indicated times (Con: EGF-untreated cells). Total protein lysates were subjected to western blotting of TAZ and EGFR-activated kinases.
Protein fold expression normalized to β-Actin, total ERK1/2 or STAT3 is shown below each lane. Data are represented as mean ± SEM (*: p<0.01).
All chemicals (Table S8) were from Sigma Aldrich unless otherwise mentioned.
KEY RESOURCES
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-TAZ | Cell Signaling Technology | # 4883 |
| mouse anti-Actin | Sigma-Aldrich | # A1978 |
| rabbit anti-TBP | Cell Signaling Technology | # 40059 |
| rabbit anti-YAP | Cell Signaling Technology | # 14074 |
| rabbit anti-phospho-LATS1 | Cell Signaling Technology | # 9157 |
| rabbit anti-LATS1 | Cell Signaling Technology | # 9153 |
| rabbit anti-phospho-STAT3 | Cell Signaling Technology | # 9145 |
| mouse anti-STAT3 | Cell Signaling Technology | # 9139 |
| rabbit anti-phospho-ERK1/2 | Cell Signaling Technology | # 4370 |
| mouse anti-ERK1/2 | Cell Signaling Technology | # 4696 |
| rabbit anti-phospho-AKT | Cell Signaling Technology | # 4060 |
| mouse anti-AKT | Cell Signaling Technology | # 2920 |
| rabbit anti-EGFR | Cell Signaling Technology | # 4267 |
| rabbit anti-phospho-EGFR | Cell Signaling Technology | # 2231 |
| rabbit anti-PDGFRβ | Cell Signaling Technology | # 3169 |
| rabbit anti-phospho-PDGFRβ | Cell Signaling Technology | # 3166 |
| Rabbit anti-phospho-FGFR1–4 | R&D | # AF3285-SP |
| Rabbit anti-FGFR1 | Cell Signaling Technology | # 9740 |
| rabbit anti-phospho-MET | Cell Signaling Technology | # 3077 |
| rabbit anti-MET | Cell Signaling Technology | # 8198 |
| rabbit anti-JUN | Cell Signaling Technology | # 9165 |
| rabbit anti-CD274 | Cell Signaling Technology | # 13684 |
| rabbit anti-CXCR4 | Thermo Fisher | # 35–8800 |
| rabbit anti-HIF1A | Cell Signaling Technology | # 36169 |
| rabbit anti-JAK2 | Cell Signaling Technology | # 3230 |
| rabbit anti-PDGFRA | Cell Signaling Technology | # 3174 |
| rabbit anti-TEAD1 | Cell Signaling Technology | #12292 |
| mouse anti-FLAG HRP | Sigma-Aldrich | # A8592 |
| Rabbit anti-Ki67 | Cell Signaling Technology | #9129 |
| mouse anti-human Nuclei | Millipore | #MAB1281 |
| rabbit anti-STAT3 (ChIP) | Cell Signaling Technology | # 12640 |
| rabbit IgG (ChIP) | Cell Signaling Technology | # 2729 |
| Bacterial and Virus Strains | ||
| STBL3 Competent E. coli | Thermo Fisher | C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Osimertinib (0.5μM for screening) | Medkoo | # 206042 |
| AEE788 (0.05μM for screening) | Cayman | # 18416 |
| PF299804 (0.1μM for screening) | Cayman | # 9001879 |
| Lapatinib (0.2μM for screening) | Medkoo | # 100490A |
| BPI-2009 (10μM for screening) | Medkoo | # 205897 |
| Lazertinib (0.1μM for screening) | Medkoo | # 206860 |
| Lidocaine hydrochloride (1mM for screening) | Selleckchem | # S4667 |
| Erlotinib (10μM for screening) | Cell Signaling Technology | # 5083 |
| Gefitinib (10μM for screening) | Cell Signaling Technology | # 4765 |
| JNJ-42756493 (0.05μM for screening) | Cayman | # 21813 |
| Sunitinib (1μM for screening) | Cayman | # 13159 |
| Sorafenib (1μM for screening) | Cayman | # 10009644 |
| 5,15-DPP | Santa Cruz | # sc-204305 |
| Stattic | Santa Cruz | # sc-202818 |
| PD98059 | Cell Signaling Technology | # 9900 |
| FR180204 | Sigma-Aldrich | # SML0320 |
| Akti-1/2 | Tocris | # 5773 |
| Critical Commercial Assays | ||
| Trans-Lentiviral Packaging System | Thermo Fisher | TLP4614 |
| PDGFβ ELISA Kit | R&D Systems | DBB00 |
| EGF ELISA Kit | R&D Systems | DEG00 |
| alamarBlue Cell Viability Reagent | Thermo Fisher | DAL1025 |
| Illumina TrueSeq RNA Sample Preparation kit v2 | Illumina | RS-122–2001 |
| ChIP-Seq DNA Sample Prep kit | Illumina | IP-202–1012 |
| MAGnify ChIP system | Thermo Fisher | 492024 |
| RNeasy kit | Qiagen | 74104 |
| Deposited Data | ||
| ChIP-seq datasets | This paper | GEO GSE151994 |
| RNA-seq datasets | This paper | GEO GSE151995 |
| Experimental Models: Cell Lines | ||
| GBM1B, GBM1A | Dr. Vescovi (Galli et al., 2004) | |
| M1123 (1123) | Dr. Nakano (Mao et al., 2013) | |
| U87MG | ATCC | HTB-14 |
| A172 | ATCC | CRL 1620 |
| 293FT | Thermo Fisher | R70007 |
| Experimental Models: Organisms/Strains | ||
| NOD/SCID mice (female, 6–8 weeks) | Jackson Laboratory | 001303 |
| Oligonucleotides | ||
| Primers for qRT-PCR, see below | This paper | |
| Primers for ChIP-PCR, see below | This paper | |
| Recombinant DNA | ||
| pLEX-MCS lentiviral vector | Thermo Scientific | OHS4735 |
| GCGATGAATCAGCCTCTGAAT (shTAZ1 lentiviral shRNA vector) | Sigma-Aldrich | TRCN0000019469 |
| CCAGGAACAAACGTTGACTTA (shTAZ2 lentiviral shRNA vector) | Sigma-Aldrich | TRCN0000019470 |
| pLKO.1-puro TurboGFP™ Control shRNA Lentiviral Vector | Sigma-Aldrich | SHC004 |
| EGFRvIII ORF | Dr. Alonzo Ross | Addgene #20737 |
| TAZ ORF | Dr. Kunliang Guan | Addgene #32839 |
| Software and Algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 | |
| MACS2 | Zhang et al., 2008 | |
| BEDTools | Lawrence et al., 2013 | |
| CisGenome | Ji et al., 2008 | |
| HOMER | Heinz et al., 2010 | |
| Primers | ||
| qRT-PCR | ||
| Gene | Primer 5’ | Primer 3’ |
| TAZ | GATCCTGCCGGAGTCTTTCTT | CACGTCGTAGGACTGCTGG |
| CTGF | CCAATGACAACGCCTCCTG | TGGTGCAGCCAGAAAGCTC |
| MYC | CCTGGTGCTCCATGAGGAGAC | CAGACTCTGACCTTTTGCCAGG |
| 18s | ACAGGATTGACAGATTGATAGCTC | CAAATCGCTCCACCAACTAAGAA |
| EGFR | AACACCCTGGTCTGGAAGTACG | TCGTTGGACAGCCTTCAAGACC |
| PDGFB | GAGATGCTGAGTGACCACTCGA | GTCATGTTCAGGTCCAACTCGG |
| EGF | TGCGATGCCAAGCAGTCTGTGA | GCATAGCCCAATCTGAGAACCAC |
| PDGFRA | GACTTTCGCCAAAGTGGAGGAG | AGCCACCGTGAGTTCAGAACGC |
| PDGFA | CAGCGACTCCTGGAGATAGACT | CGATGCTTCTCTTCCTCCGAATG |
| VEGFA | TTGCCTTGCTGCTCTACCTCCA | GATGGCAGTAGCTGCGCTGATA |
| FGF2 | AGCGGCTGTACTGCAAAAACGG | CCTTTGATAGACACAACTCCTCTC |
| FGF1 | ATGGCACAGTGGATGGGACAAG | TAAAAGCCCGTCGGTGTCCATG |
| FGFR4 | AACACCGTCAAGTTCCGCTGTC | CATCACGAGACTCCAGTGCTGA |
| ERBB3 | CTATGAGGCGATACTTGGAACGG | GCACAGTTCCAAAGACACCCGA |
| JUN | CCTTGAAAGCTCAGAACTCGGAG | TGCTGCGTTAGCATGAGTTGGC |
| HIF1A | TATGAGCCAGAAGAACTTTTAGGC | CACCTCTTTTGGCAAGCATCCTG |
| JAK2 | CCAGATGGAAACTGTTCGCTCAG | GAGGTTGGTACATCAGAAACACC |
| CXCR4 | CTCCTCTTTGTCATCACGCTTCC | GGATGAGGACACTGCTGTAGAG |
| CD274 | TGCCGACTACAAGCGAATTACTG | CTGCTTGTCCAGATGACTTCGG |
| TEAD1 | CCTGGCTATCTATCCACCATGTG | TTCTGGTCCTCGTCTTGCCTGT |
| ChIP-PCR | ||
| Gene | Primer 5’ | Primer 3’ |
| EGFR | CGTCCTTTCCTGTTTCCTTG | GGGGAAAGTGAGGGAAGAAA |
| PDGFB | ACTGAATGGGCAAAAACTGG | TGCAAACAGGGACAATTTGA |
| EGF | TATTGGCTGTGGGTTTGTCA | ACCATGATCAAGTGGGCTTC |
| PDGFRA | CAGGTGGATTGCTTGAGGTC | CTTGAGTGCAATGGCATGAT |
| PDGFA | CTGAGCCCAGAGTCAGGTTC | GGAGTAGCGACTGCATGTGA |
| VEGFA | CCAACTTCTGGGCTGTTCTC | CCCCTCTCCTCTTCCTTCTC |
| FGF2 | ATGATGACAATGGCGGTTTT | GGATCCCAAGGCAGAAGAAT |
| FGF1 | TGCCTTTTGCCTTTGACTCT | ATGGAGGAGCTGAGCTTTGA |
| FGFR4 | CCCAGAGCCAGTTCACAAAC | CGAGTAAGGAGCTTGGCAGT |
| ERBB3 | GTTGGGGGTAAGGTCACAGA | GAATAGAAAGGCGGGAAAGG |
| HIF1A | CATCACTGGCCATCAGAGAA | TGGCTGGGTCAAATGGTATT |
| JAK2 | AAATGGTGCTGGGAAAACTG | GCCATTGCTTTTGGTGTTTT |
| CXCR4 | TTCTGGGCTTCAAGCAACTT | AAACGCGCCAAGTGATAAAC |
| CD274 | CAGCACCTGTTGTTTCCTGA | GGGCGAAGGATATGAACAGA |
| TEAD1 | GTCCTGGCGTTCATTTCATT | AGTCTGCAAAGTGCTTGGTG |
| Negative Control NC1 | GTCTGTACTCCCAGCTACTC | GACAGAGCAAGACTCCATC |
| Negative Control NC2 | GACTGGTTCATTCACTGCTC | CACTTCCCTACCCGAAAC |
| STAT3 ChIP-PCR | TCCTCTTCCTCCTCCTCCTC | AAAGGAATGCAGATGCAGGT |
Analysis of GBM specimens
TCGA, CGGA and Rembrandt datasets were analyzed in the GlioVis data portal (15).
Western blotting
Total cellular proteins extraction and western blotting follow our publication (16).
Immunostaining
Cells were collected by cytospin onto glass slides and fixed with 4% paraformaldehyde. Immunostaining using antibodies listed in Table S8, and images were analyzed using the ApoTome System (Zeiss) or ImageJ software.
ELISA
PDGFβ and EGF levels were measured using kits from R&D.
Neurosphere formation, clonogenic assay and radiation
Neurosphere formation analysis follows our publication (16). Soft agar clonogenic assay follows our publication using 5 × 103 cells (17). Cells in soft agar were irradiated using the Gammacell 40 Exactor irradiator.
Transwell and scratch wound healing assay
These assays follow our publication (18).
Lentiviral transduction
shRNAs are listed in Table S8. TAZ (from Kunliang Guan, Addgene # 32839) and EGFRvIII (from Alonzo Ross, Addgene # 20737) ORF were PCR-amplified and cloned into pLEX-MCS (Thermo Scientific). Lentivirus packaging, infection (MOI = 5) and puromycin selection (1μg/ml) follow our publication (16).
RNA isolation and RNA-sequencing
RNA was extracted using the RNeasy kit (Qiagen). RNA-seq follows our publication (19) using the HiSeq 2500 system (Illumina). Raw reads were aligned to human genome build hg19 using HISAT2 (20) with default parameters. Read numbers aligned to each exon were summarized into gene level counts by StringTie (21). Sample normalization was performed by edgeR (22).
Chromatin immunoprecipitation (ChIP) and sequencing
TAZ ChIP was performed following our publication (19), using the TAZ antibody and normal rabbit IgG (Table S8). Library preparation follows our previous publication (19) for sequencing using Illumina HiSeq 2500. Raw reads were aligned to the human genome build hg19 using Bowtie2 (23) allowing at most 2 mismatches in the first 28-bp bases. TAZ binding sites were called using MACS2 (24) with default settings. Read numbers aligned to peak regions was counted by BEDTools (25) and normalized for the library size. Normalized counts for each peak region were further subtracted by its mean normalized counts across samples and then divided by its standard deviation, which gave the scaled binding intensity. The 150-bp-long sequences for the top ranked peaks were extracted for the de novo motif discovery algorithm of CisGenome (26). Control regions were randomly chosen to match the GC content and distributional properties of peak regions. Motifs similar to the TAZ binding motif are obtained by applying HOMER de novo motif analysis to the top 2000 TAZ peaks.
Peak data from histone modifications H3K4me3, H3K27ac, and H3K27me3 were obtained from ENCODE and downsampled to 300,000 peaks.
Quantitative real-time PCR (qRT-PCR) and ChIP-PCR
qRT-PCR follows our publication (16) with normalization to 18S rRNA (primers listed in Table S8).
Tumor xenografts and drug treatment
All animal protocols were approved by the Johns Hopkins School of Medicine Animal Care and Use Committee. Animal numbers per group are calculated by power analysis (power: 80%; significance level: 5%). NOD/SCID mice (female, 6–8 weeks old) were transplanted with 104 viable cells in 2 μL DMEM medium into the right caudate/putamen. Animals received osimertinib in 200 μL Oral-Plus solution (Paddock) by oral gavage. Drug toxicity was monitored daily during treatment (maximal weight loss <10%). Mice were perfused with 4% paraformaldehyde. Tumor sizes were quantified by observers blinded to the experimental groups to measure maximum tumor volume on H&E stained coronal sections using computer-assisted morphometry as reported by us (16).
Data Availability
Sequencing datasets are available from the Gene Expression Omnibus (GEO) Repository (GSE151994 and GSE151995).
Statistical analysis
All results represent at least three replicates. Statistical analysis was performed using the Prizm software (GraphPad) or R/Bioconductor package. All data are represented as mean value ± standard error of mean (SEM). Unpaired, two-tailed Student’s t tests were performed and p < 0.05 was accepted as significant. If more than two groups existed, one-way ANOVA tests with Tukey corrections were used (minimal requirement: p < 0.05).
Results
EGF induces TAZ expression and TAZ gene targets in GBM cells.
In three glioma databases (TCGA, CGGA and Rembrandt), TAZ (WWTR1) mRNA levels are elevated in GBM specimens compared to low-grade glioma and normal brain specimens (Fig. S1A) and are positively correlated with tumor grade (Fig. S1A). TAZ hyperexpression is also associated with poor prognosis in GBM specimens from the TCGA and CGGA datasets (Fig. S1B). To identify upstream drivers of TAZ hyperactivation, we performed gene correlation assays using the TCGA and CGGA GBM database. The mRNA levels of RTKs (e.g. EGFR, FGFR1, MET and PDGFRB) and their ligands (e.g. EGF, FGF1, FGF2, HGF, and PDGFA) showed positive correlation with TAZ mRNA levels in the TCGA (Fig. 1A and S1C) and CGGA datasets (Fig. S1D), suggesting a possible link between TAZ and RTKs. To test this hypothesis, we studied the response of TAZ expression to four pro-GBM RTK ligands (EGF, FGF2, HGF and PDGF) (27). GBM neurospheres were cultured in growth factor-depleted medium overnight followed by RTK ligand stimulation that induced phosphorylation of individual RTK receptors (Fig. 1B). EGF is the most potent TAZ inducer in both protein and mRNA levels (Fig. 1B and 1C). We assessed the kinetics of TAZ induction by EGF. In GBM cell models, EGF rapidly induced ERK phosphorylation within 30 minutes followed by peak TAZ induction at about 4 hours (Fig. 1D). Low-passage primary GBM-derived neurospheres also showed consistent TAZ induction by EGF (Fig. 1D).
Figure 1: EGF induces TAZ expression in GBM cells.

A: TAZ (WWTR1) mRNA levels positively correlate with EGF and FGF2 levels in GBM specimens from the TCGA database (n=454).
B: GBM1B cells were depleted of growth factors for 16 hours and treated with RTK ligands as indicated for 1h or 5min for testing TAZ induction or phosphorylation of their receptors, respectively (Con: RTK ligand-untreated cells).
C: GBM1B cells were deprived from growth factors for 16 hours and treated with EGF for 2 hours. TAZ mRNA was measured by qRT-PCR.
D: After growth factor depletion for 16 hours, GBM cells as marked received EGF treatment for the indicated times (Con: untreated cells). TAZ protein was quantified by western blotting.
E and F: After growth factor depletion for 16 hours, GBM1B cells were treated with EGF for the indicated times (Con: untreated cells). TAZ western blotting was performed using proteins from nuclear and cytosol fractions (E). Cells with +/− EGF treatment (4 hours) were subjected to TAZ immunostaining and quantification of TAZ signal intensity in DAPI+ nuclei (F; DAPI: nuclear counterstaining; Bar = 10 μM; n=50)
G and H: The same cells as used in E were subjected to TAZ ChIP-PCR with IgG as the control (G, NC: negative control regions randomly selected from the genome). CTGF and MYC transcription was also quantified (H).
Protein fold expression normalized to β-Actin, total levels of receptors or TBP (E) is shown below each lane. Data are represented as mean ± SEM (*: p < 0.01).
TAZ functions as a transcriptional co-activator. Here, we showed that EGF treatment upregulated nuclear TAZ levels (2.3-fold at 4 hours and 14.6-fold at 8 hours, Fig. 1E). TAZ immunostaining also showed nuclear translocation of TAZ protein after EGF treatment (Fig. 1F, left panel), consistent with higher signal intensity of TAZ nuclear staining (Fig. 1F, right panel). As the functional outcome, EGF treatment enhanced TAZ binding to the promoters/enhancers of CTGF and MYC (Fig. 1G), two well-defined TAZ targets (10, 28), and induced their upregulation (Fig. 1H). These results support that EGF activates TAZ expression and its transcriptional activity in GBM cells.
TAZ and its paralog YAP are both regulated by the Hippo signaling pathway (4, 29, 30). We found that EGF did not significantly induce YAP expression in GBM neurosphere and adherent cell lines (Fig. S2A), contrasting with its capacity to induce TAZ (Fig. 1D). EGF also did not alter levels of phosphorylated and total LATS1, a key Hippo-signaling kinase mediating TAZ/YAP phosphorylation and degradation (4, 29) (Fig. S2B). Thus, EGF may have the potential to activate TAZ but not YAP in GBM cells via Hippo-independent mechanisms.
EGF induces TAZ through EGFR and downstream kinases STAT3 and ERK1/2.
Next, we asked if EGF-induced TAZ activation is inhibited by well-validated inhibitors of EGFR and its downstream kinases (ERK1/2, STAT3 and AKT). GBM neurospheres were pre-treated with these inhibitors followed by EGF stimulation. Two EGFR inhibitors (Erlotinib and Gefitinib) significantly attenuated TAZ induction by EGF, as examined using whole cell lysates and nuclear/cytosolic fractions of EGF-treated cells (Fig. 2A and 2B). Two STAT3 inhibitors (5,15-DPP and Stattic) potently inhibited STAT3 phosphorylation and EGF-induced TAZ induction (Fig. 2C and S3A). Two ERK1/2 inhibitors (PD98059 and FR180204) also effectively suppressed ERK1/2 phosphorylation and TAZ induction by EGF (Fig. 2D and S3B). In contrast, the AKT inhibitor Akti-1/2 failed to inhibit TAZ induction in EGF-stimulated cells, under conditions showing effective inhibition of AKT phosphorylation (Fig. 2E). These results identified STAT3 and ERK1/2 as potential mediators of EGFR-induced TAZ activation.
Figure 2: EGF induces TAZ expression through EGFR and its downstream signaling molecules.

A and B: GBM1B cells were treated with +/− Erlotinib or Gefitinib (10 μM, 16 hours) in growth factor-depleted medium followed by EGF treatment for the indicated times. TAZ western blotting was performed using total protein lysates (A) or proteins from the nuclear and cytosol fractions (B).
C - E: GBM1B cells were treated with or without the inhibitors as marked for 16 hours in growth factor-depleted medium followed by 4-hour EGF stimulation (5,15-DPP: 100 μM; PD98059: 20 μM; Akti-1/2: 20 μM). Total protein lysates were subjected to western blotting for TAZ and other proteins.
F: ChIP-PCR using STAT3 antibody and control IgG showed the interaction between STAT3 and a putative STAT3-binding site on the TAZ promoter (−500bp to the TSS).
Protein fold expression normalized to β-Actin is shown below each lane. Data are represented as mean ± SEM (*: p < 0.01).
Certain EGFR-activated kinases (e.g. STAT3) also function as TFs to activate the promoters/enhancers of downstream gene targets (31). In the TAZ promoter, we identified a putative STAT3 binding site at −500 bp to the transcriptional start site (TSS) of TAZ. In GBM neurospheres, STAT3 ChIP followed by qPCR showed the enrichment of STAT3 on this putative binding site over the IgG-ChIP control (Fig. 2F), indicating that EGFR acts through STAT3 to transactivate TAZ.
The constitutively active EGFRvIII mutation leads to EGF-independent TAZ hyperactivation in GBM cells.
We established GBM cell lines with lentiviral EGFRvIII expression from two parental EGFRvIII− cell lines (GBM1B and U87MB), and also used the EGFRvIII+ GBM neurosphere M1123 (Fig. 3A). TAZ levels were 100–150% higher in EGFRvIII+ cells compared to their EGFRvIII− isogenic controls (Fig. 3B). TAZ gene targets (CTGF and MYC) were also significantly upregulated in EGFRvIII+ cells (Fig. 3C). Regarding TAZ induction kinetics in EGF-stimulated EGFRvIII+ cells, all three EGFRvIII+ models showed weak or no detectable TAZ induction after EGF treatment (Fig. 3D), compared to significant TAZ induction by EGF in EGFRvIII− cells (Fig. 1D). Phosphorylation of ERK1/2 and STAT3 was also not induced by EGF in EGFRvIII+ cells (Fig. 3D) compared to their strong induction in the parental EGFRvIII− cells (Fig. 2C and 2D). These results suggest that the EGFRvIII mutation leads to EGF-independent TAZ hyperactivation in GBM cells.
Genome-wide analysis of gene targets regulated by the EGFR-TAZ signaling axis.
We applied a high-throughput sequencing strategy (Fig. 4A) to establish a genome-wide map of EGFR-TAZ-regulated genes. GBM neurospheres were depleted of growth factors for 16 hours followed by EGF stimulation for 4 and 24 hours. RNA-seq was used to identify differentially-expressed (DE) genes after EGF treatment. Over 25 million cDNA reads were generated for each of three conditions (Control, EGF-4h and EGF-24h, n=2 for each condition) and showed a >87% alignment rate to the human genome. Consistency between two replicates was demonstrated by heatmap clustering (Fig. 4B). We first defined DE genes for each time point of EGF treatment (Fig. 4C, FDR ≤ 0.05, log2(Fold-change) Cut-off: ± 0.8, listed in Table S1 and S2). DE genes from two time points were further merged to generate lists of 1,624 up-regulated and 1,238 down-regulated genes after EGF treatment (Table S3).
Figure 4: Genome-wide profiling of EGFR-regulated TAZ gene targets in GBM neurospheres.
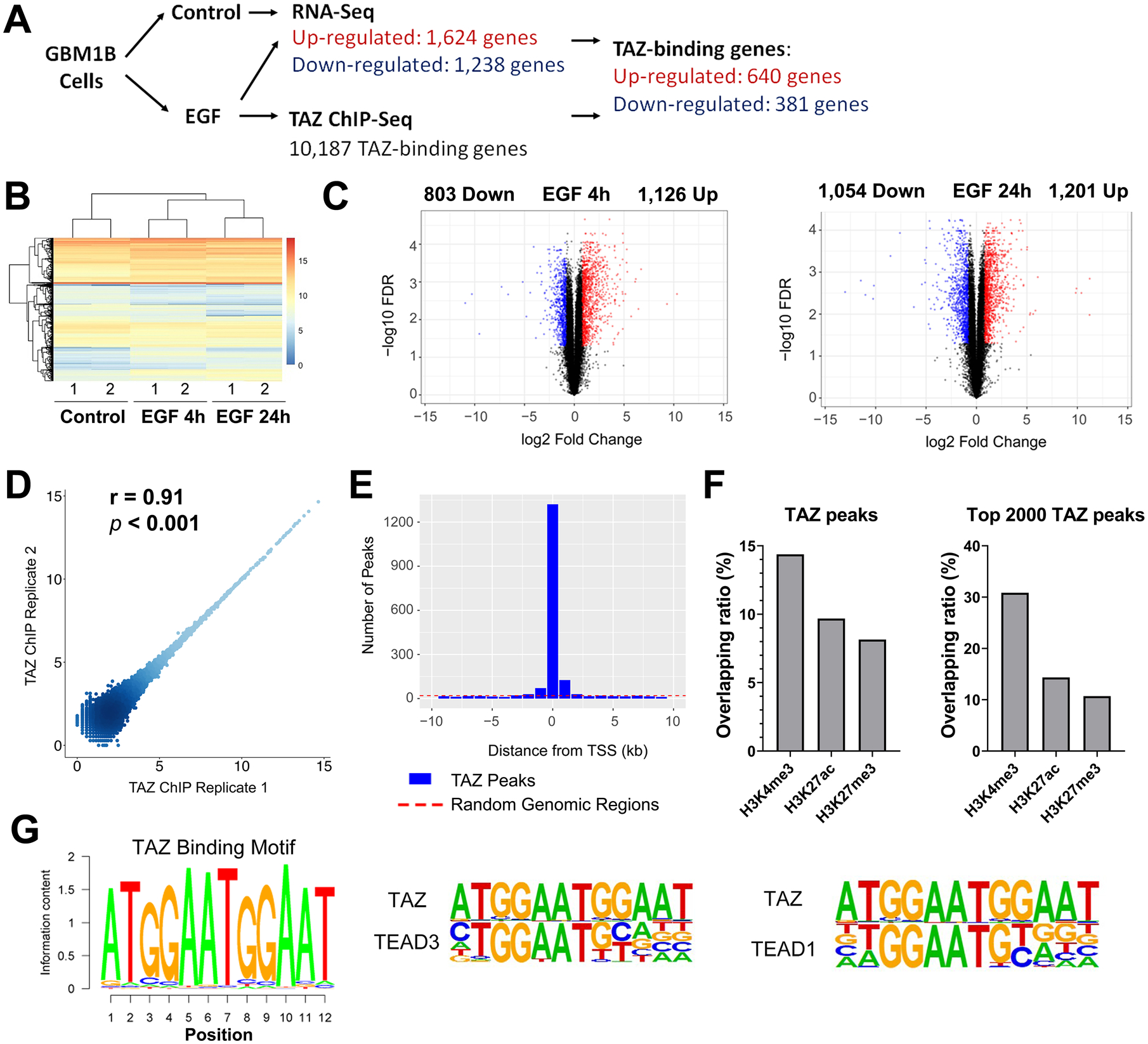
A: Outline of the genome-wide analysis.
B: Heatmap clustering of 6 RNA-Seq samples from 3 conditions. Gene expression was calculated by RPKM.
C: Volcano plot showing DE genes (red and blue dots: genes with FDR ≤ 0.05 and log2(fold change) ≥ 0.8 or ≤ −0.8, respectively).
D: ChIP-Seq reproducibility was analyzed by scatter plot comparing peak intensities for two ChIP-Seq replicates.
E: TAZ binding peaks show enrichment around TSSs, compared to randomly selected genomic regions.
F: Overlapping between TAZ peaks and histone marks as marked.
G: TAZ binding motif was identified by de novo motif discovery and compared to TEAD3 and TEAD1 motifs.
TAZ ChIP-Seq was performed to define genome-wide TAZ binding sites in GBM neurospheres after EGF treatment for 4 hours, the time point showing peak TAZ induction after EGF stimulation (Fig. 1D). Greater than 90% of the ChIP-seq reads aligned to the human genome. 40,225 TAZ binding peaks were called at an FDR of 5% and mapped to 10,187 genes (Table S4). The ChIP intensities within TAZ binding peaks were consistent between two replicates (Fig. 4D, Pearson’s r = 0.91, p <0.001). TAZ binding sites, but not randomly selected genomic regions, were highly enriched around the ±1kb region of TSS (Fig. 4E). We further calculated the overlapping ratio between TAZ binding peaks and peaks of histone modification marks for promoters, enhancers and suppressors sourced from the ENCODE database (32) (Fig. 4F). 14.4% of all TAZ binding peaks overlapped with the H3K4me3 activation mark enriched in promoters, higher than that with the H3K27ac enhancer mark (9.7%) or the H3K27me3 suppressor mark (8.2%). The top 2000 TAZ binding peaks also overlapped more with H3K4me3 (30.8%) than either H3K27ac (14.4%) or H3K27me3 (10.7%). These results suggest that TAZ functions predominantly to activate gene transcription in EGF-stimulated GBM cells. De novo motif discovery analysis defined a consensus sequence (5’-ATGGAATGGAAT-3’) as the most enriched TAZ-binding motif (Fig. 4G). In the HOMER motif database, this TAZ-binding motif closely resembles the motifs of TEAD1 and TEAD3, two known TAZ-interacting transcription activators (Fig. 4G and all motifs ranked in Table S5).
TAZ mediates the activation of multiple oncogenic signaling pathways by EGF.
By combining RNA-Seq and TAZ ChIP-Seq datasets, we generated lists of TAZ target genes that are differentially expressed after EGF treatment and have at least one TAZ binding site. These EGF-regulated TAZ gene targets were divided into EGF-activated and EGF-repressed groups, referred to as TAZ-Up and TAZ-Down genes (Table S6), respectively. In the KEGG pathway analysis (33), top-ranked signaling pathways enriched in TAZ-Up genes contained multiple oncogenic signaling pathways, e.g. focal adhesion, MAPK, HIF-1, RAS and PI3K-AKT signaling pathways (Fig. 5A and Table S7). In comparison, these oncogenic pathways were not enriched in TAZ-Down genes (Table S7). The clustergram analysis was used to rank all TAZ-Up genes based on their enrichment in the top 20 TAZ-Up KEGG terms (Fig. 5B and S4). 8 of the top 10 enriched genes code for oncogenic RTK ligands and receptors (EGFR, EGF, PDGFB, PDGFRA, PDGFA, VEGFA, FGF2 and FGF1), suggesting an EGFR-TAZ-RTK signaling axis with positive feedback mechanisms. In the TAZ-Up gene list, there are additional genes with other well-defined oncogenic functions (e.g. HIF1A, JUN, JAK2, CXCR4 and CD274 (PD-L1)) and known TAZ-binding partners (e.g. TEAD1) essential for its transcriptional activity.
Figure 5: TAZ acts downstream of EGFR to activate key oncogenic molecules.

A: Top 20 KEGG pathway terms enriched in TAZ-Up genes. Pathways are ranked based on -log(p value) as calculated by the Fisher’s exact test.
B: TAZ-Up genes were ranked in the clustergram graph by their enrichment in the top 20 KEGG pathways. The top-ranked 20 genes are shown.
C - E: GBM1B cells were deprived from growth factors for 16 hours and treated with +/− EGF for 4 hours (qRT-PCR and ELISA) and 24 hours (western blotting and ELISA). The expression of TAZ-Up genes was quantified by qRT-PCR (C), western blotting (D) and PDGFβ ELISA (E). Control: EGF-untreated cells.
F: Representative tracks from ChIP-Seq results show TAZ binding peaks validated by ChIP-PCR. The orientation of each transcript is marked by the arrow. Arrowheads mark peaks for validation (T: TSS).
G: GBM1B cells after 16-hour growth factor depletion were treated with EGF for 4 hours and were subjected to ChIP using TAZ antibody and control IgG. qPCR was used to measure TAZ binding to putative TAZ binding sites (NC1 and NC2: negative control genomic regions without TAZ binding).
H: GBM1B cells were infected by lentiviral TAZ shRNAs (shTAZ1 and shTAZ2) or control shRNA (Con) for 72 hours in EGF-containing medium. The expression of TAZ and TAZ-Up genes was measured by western blotting or ELISA (PDGFβ).
I and J. GBM1B cells were infected by lentiviral TAZ shRNAs (shTAZ1 and shTAZ2) or control shRNA for 24 hours. Equal numbers of viable cells (105) after infection were cultured for 6 days to form neurospheres. Representative microscopic fields of control and shTAZ1 samples are shown (I, left; Bar = 100 μm). Neurospheres (>100 μm diameter) were counted (I, right, n=6, N.D.: not detected). Equal numbers (10,000) of viable cells after infection were transplanted into mouse brains (n=4). Coronal brain sections (20 μm, with H&E staining) were shown (day 80 post implantation, Bar = 0.5 mm) with tumor size quantification (J).
Data are represented as mean ± SEM (*: p < 0.01 compared to control).
We focused on validating 16 TAZ targets, including 10 RTK genes (EGFR, EGF, PDGFB, PDGFRA, PDGFA, VEGFA, FGF1, FGF2, FGFR4 and ERBB3) and another 7 oncogenic regulators (JUN, HIF1A, JAK2, CXCR4, CD274 and TEAD1). EGF induction of these genes in GBM neurospheres was validated by qRT-PCR (Fig. 5C). Protein induction of nine genes by EGF was also validated by western blotting or ELISA (Fig. 5D and 5E). Next, we used ChIP-PCR to validate TAZ-binding peaks proximal to 12 TAZ-Up genes as identified by ChIP-Seq (Fig. 5F, 5G and S5). To support their TAZ-dependent expression, we showed that TAZ silencing by two shRNAs led to significant downregulation of TAZ and TAZ-Up genes, as measured by western blotting or ELISA (Fig. 5H). This TAZ silencing also led to loss of GBM neurosphere formation in vitro (Fig. 5I) and inhibited their tumorigenicity in vivo (Fig. 5J). Overall, this genome-wide study and following validation reveal the direct activation of multiple GBM-promoting genes by the EGFR-TAZ signaling axis.
Next, we asked if these findings are applicable to clinical GBM specimens in the TAGA database. Among the 16 TAZ-Up genes we focused on, 13 genes are positively correlated with TAZ expression in the TCGA GBM specimens, including RTK genes shown in Fig. 1A and S1C (EGF/EGFR, FGF1/2 and PDGFA/B) and other TAZ-Up genes (Fig. S6; VEGFA, JUN, HIF1A, JAK2, CXCR4, CD274 and TEAD1). More broadly, we analyzed all TAZ-Up and -Down genes in the TCGA GBM database. After excluding genes without expression data, 78% of TAZ-Up genes have positive correlation with TAZ expression (Pearson’s r > 0.2, p<0.001), as compared to only 35% of TAZ-Down genes showing this positive correlation.
TAZ hyperactivation promotes various phenotypes associated with GBM malignancy.
We discovered an EGFR-TAZ signaling axis that leads to the hyperactivation of TAZ and its downstream oncogenic program in GBM cells. To better define the effects of TAZ hyperactivation on GBM malignancy, we established GBM cells with enforced TAZ expression to recapitulate TAZ hyperactivation found in EGF-stimulated and EGFRvIII+ cells. We established GBM neurospheres and adherent cells with lentiviral enforced expression of FLAG-tagged TAZ (Fig. 6A). All 16 validated TAZ-Up genes showed strong mRNA induction in response to enforced TAZ expression, compared to control cells harboring an empty lentiviral vector (Fig. 6B). Protein induction of eight TAZ-Up genes (EGFR, PDGFRA, HIF1A, JAK2, CXCR4, JUN, TEAD1 and CD274) was detected in cells with enforced TAZ expression (Fig. 6C). Next, we provided multiple lines of evidence to demonstrate that enforced TAZ expression potently promotes various GBM-associated malignant phenotypes. First, enforced TAZ expression significantly accelerated GBM cell growth (Fig. 6D). Second, TAZ induction increased clonogenicity of GBM neurospheres by 78% in EGF-containing medium (Fig. 6E), and, strikingly, rendered these cells colony-forming in growth-factor-depleted medium, hereby referred to as a mitogen independent phenotype that is absent in control cells (Fig. 6E). Notably, GBM neurospheres with enforced TAZ expression, compared to control cells, released >20-fold more EGF, a TAZ-Up gene and key mitogen supporting neurosphere growth (Fig. 6F). Conditioned medium from GBM neurospheres with enforced TAZ expression sustained colony formation of control neurospheres (Fig. 6G). This effect was not detected when using conditioned medium from control neurospheres. This result is consistent with the EGF-releasing capability of neurospheres with enforced TAZ expression (Fig. 6F). Third, TAZ hyperactivation increased the invasiveness of two GBM cell models in the Transwell cell migration assay and scratch wound healing assay (Fig. 6H, 6I, and S7). Finally, GBM neurospheres with enforced TAZ expression showed more radioresistance than the isogenic control, as evidenced by a 2-fold increase of cell survival after one or three daily radiation treatment (Fig. 6J).
Figure 6: Enforced TAZ expression promotes GBM-associated malignant phenotypes.

A: GBM1B and A172 cells were infected with lentiviruses harboring TAZ (FLAG-tagged) cDNA or no cDNA insert as the control to established stable cell lines. Transgene expression was measured by western blotting.
B and C: Validated TAZ-Up genes were analyzed by qRT-PCR (B) in GBM1B and GBM1B-TAZ cells, and by western blotting (C) using whole cell lysates from GBM1B and A172 cells with +/− enforced TAZ expression.
D: Cell growth curves were drawn from the number of trypan blue stained GBM1B and A172 cells with +/− enforced TAZ expression.
E: Colony formation was quantified in GBM1B and GBM1B-TAZ cells in medium with +/− EGF for 10 days.
F: GBM1B with +/− enforced TAZ expression were grown in medium without growth factors for 48 hours, and EGF release in medium was measured by ELISA.
G: GBM1B cells were grown in soft agar with EGF-containing medium or EGF-free conditioned medium (24h) from GBM1B and GBM1B-TAZ cells. Colony formation after 10 days was quantified.
H: GBM1B and GBM1B-TAZ cells were plated onto laminin-coated Transwell membranes. Cell migration was compared after 24 h by quantifying DAPI+ cells per field.
I: A172 and A172-TAZ cells were subjected to scratch wound healing assay to quantify migrating cells in the scratch area as shown in Figure S7.
J: GBM1B and GBM1B-TAZ cells received +/− irradiation (1 or 3 daily doses of 3 Gy). Clonogenic survival was quantified and normalized to untreated control cells.
K: 10,000 viable A172 and A172-TAZ cells were transplanted into mouse brains (n=5). Coronal brain sections (20 μm, with H&E staining) were shown (post-implantation day 76, Bar = 0.5 mm (left) and 50 μm (right); arrowhead: necrotic area).
L and M: 10,000 viable GBM1B and GBM1B-TAZ cells were transplanted into mouse brains (n=5). Coronal brain sections (20 μm, with H&E staining) were shown (day 60 post implantation, Bar = 0.5 mm) with tumor size quantification (L). hNu immunostaining (M) was used to detect and quantify tumor cells invading the contralateral corpus callosum area (marked by black rectangles in L). hNu+ cell numbers were quantified in 5 fields with equal sizes as marked underneath the picture (Bar = 100 mm).
Data are represented as mean ± SEM (*: p < 0.01).
The effects of TAZ hyperactivation on GBM tumorigenicity were further studied in vivo. The parental A172 GBM cell line has been defined as non-tumorigenic when transplanted into the brains of immunodeficient mice (34, 35). This phenotype was confirmed by our results (Fig. 6K). In contrast, A172 cells with enforced TAZ expression formed orthotopic tumor xenografts with necrosis, a histopathological hallmark of GBM (Fig. 6K, n=5). Mice bearing xenografts from GBM1B-TAZ neurospheres also showed more aggressive and invasive tumors than control GBM1B xenografts (Fig. 6L), as supported by >2.8-fold increase in tumor size (19.5 ± 3.5 vs 75.3 ± 5.1 mm3, n=5), and >3.7-fold more abundant tumor cells invading the contralateral hemisphere (Fig. 6M). Taken together, all these results support that TAZ hyperactivation aggravates GBM malignancy.
The EGFR inhibitor Osimertinib inhibits the TAZ-driven oncogenic program and GBM tumorigenicity.
We explored pharmacological strategies for TAZ targeting. Based on the EGFR-TAZ signaling model, we screened a set of brain-penetrating compounds for TAZ inhibition, including nine EGFR inhibitors, two PDGFR inhibitors and one FGFR inhibitor (see Table S8 for compound information). Three GBM cell lines expressing wild-type EGFR (GBM1B) or EGFRvIII (GBM1B-EGFRvIII and M1123) were pre-treated with individual compounds followed by 4-hour EGF stimulation. TAZ inhibition potency determined by western blotting (Fig. 7A) showed that the third-generation EGFR inhibitor osimertinib (OS, 0.5 μM) most potently downregulated TAZ in all three cell lines, as compared to the first-generation EGFR inhibitor Erlotinib (Er, 10 μM) and other compounds. OS also blunted the activation of ERK1/2, STAT3 and AKT in response to EGF stimulation (Fig. S8A). OS (0.5 μM) more effectively downregulated TAZ-Up genes, as compared to Er at higher concentration (10 μM) (Fig. 7B). In addition, OS showed higher anti-proliferation activity in vitro than Er (Fig. 7C). These results justified our focus on in-vivo efficacy of OS for TAZ inhibition and GBM treatment.
Figure 7: Pharmacological TAZ inhibition by the EGFR inhibitor osimertinib (OS).
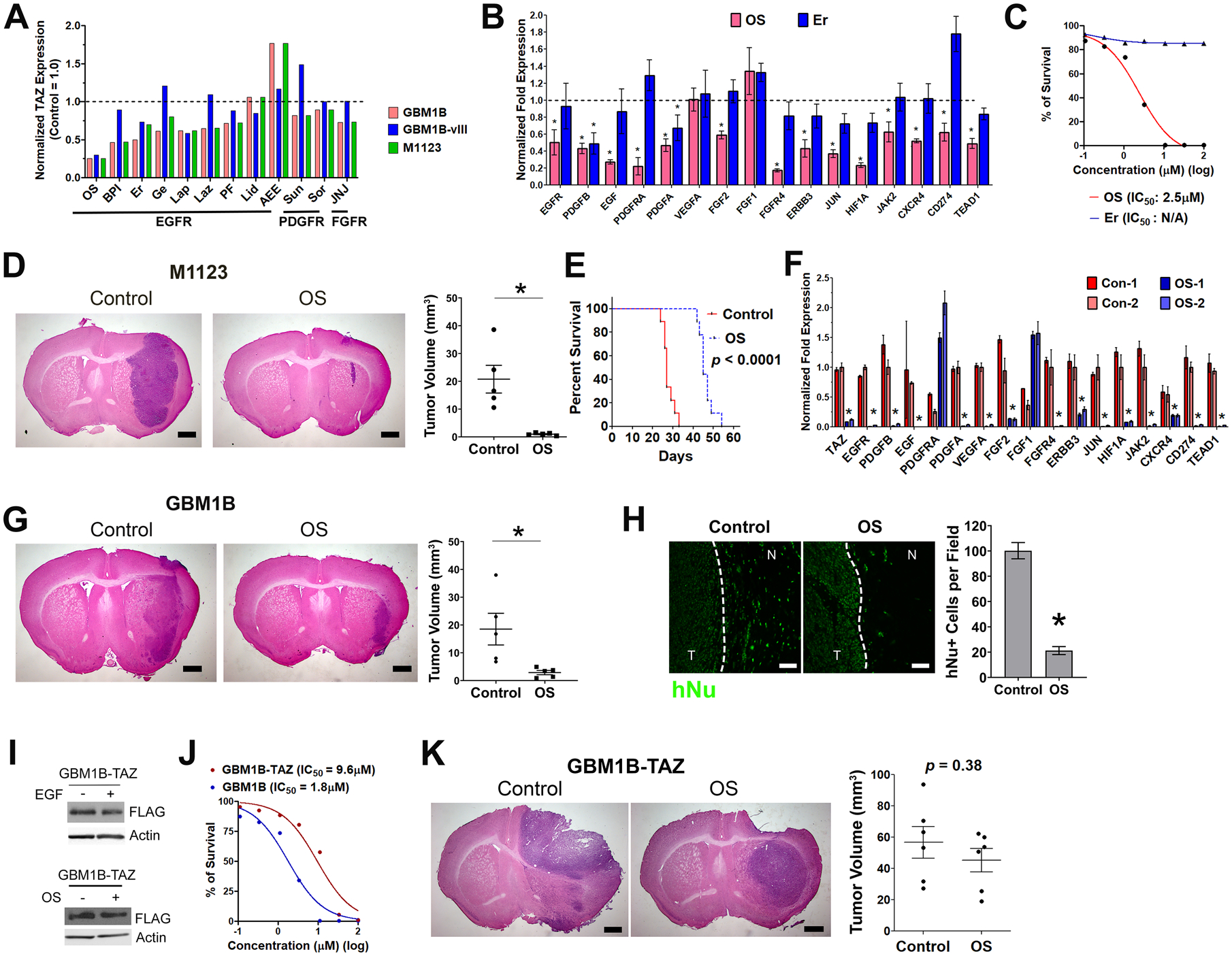
A: Three GBM cell lines were pre-treated with 12 RTK inhibitors as marked for 16 hours in growth factor-depleted medium. After 4-hour EGF stimulation, total protein lysates were subjected to TAZ western blotting. Protein fold expressions normalized to β-Actin were quantified and normalized to control cells without compound treatment (dash line: TAZ level in compound-untreated cells).
B: GBM1B cells with +/− OS (0.5 μM) and Er (10 μM) treatment as prepared in A were subjected to qRT-PCR for TAZ-Up genes (compound-untreated control = 1.0 (dash line); *: p < 0.01 compared to the untreated control).
C: Alamar blue cell proliferation assay in GBM1B cells with OS and Er treatment.
D-F: Coronal brain sections (20 μm, with H&E staining) were used for tumor size quantification of M1123 xenografts with +/− OS treatment (D, n=5, post-implantation day 15, Bar = 0.5 mm). This OS treatment also extended the survival of mice bearing M1123 xenografts (n = 9, p < 0.0001, log-rank test). Tumor specimens (n=2 for each group) were microdissected from regions close to injection needle tracts to measure TAZ and TAZ-Up gene expression by qRT-PCR (F, n=2).
G and H: Coronal brain sections (20 μm, with H&E staining) were prepared for tumor size quantification of GBM1B xenografts with +/− OS treatment (F, n=5, post-implantation day 82, Bar = 0.5 mm). hNu immunostaining detected tumor cells in the invading tumor edge (dash line) for quantification (G, Bar = 50 μm)
I: GBM1B-TAZ cells were depleted of growth factors for 16 hours and treated with +/− EGF for 4 hours (Top panel). GBM1B-TAZ cells have +/− OS treatment for 16 hours in growth factor-depleted medium followed by 4-hour EGF stimulation (Bottom panel). Total protein lysates were subjected to TAZ western blotting.
J: Alamar blue cell proliferation assay in GBM1B and GBM1B-TAZ cells with OS treatment.
K: The size of GBM1B-TAZ xenografts with +/− OS treatment was quantified using coronal brain sections (20 μm, with H&E staining, n=6, post-implantation day 50, Bar = 0.5 mm, p = 0.38).
Data are represented as mean ± SEM (*: p < 0.01).
We performed systemic OS treatment in mice bearing pre-established orthotopic xenografts from two GBM neurospheres (EGFR wild-type GBM1B and EGFRvIII+ M1123). Mice bearing M1123 xenografts received OS treatment (100 mg/kg oral, daily) starting from post-implantation day 5 and were subjected to tumor size measurement after 10 days. OS treatment inhibited tumor growth by 95% (Fig. 7D, 0.98 ± 0.2 vs 20.76 ± 5.0 mm3, p < 0.01, n=5), decreased the percentage of Ki67+ proliferative tumor cells and TAZ expression (Fig. S8B and S8C). OS treatment also extended the survival of mice bearing M1123 xenografts (Fig. 7E, median survival: 27 vs 45 days, p < 0.0001, n=9). From a parallel set of similarly treated animals, we microdissected tumor specimens for qRT-PCR. OS treatment downregulated TAZ (>90%) and 14 out of the 16 validated TAZ-Up genes (Fig. 7F) in tumor xenografts. Using GBM1B xenografts as the biological replicate, we showed that OS treatment (50 mg/kg oral, every other day, post-implantation day 20 to 82) inhibited tumor growth by 85% (Fig. 7G, 18.5 ± 5.7 vs 2.9 ± 0.8 mm3, p < 0.01, n=5) and showed inhibitory effects on Ki67 and TAZ (Fig. S8B and S8C). In this highly invasive xenograft model, OS-treated tumors showed >80% reduction of tumor cells in the invading tumor edge as compared to control tumors (Fig. 7H).
Considering the capability of OS to induce extensive molecular changes in GBM tumors, we asked if the efficacy of OS relies on effective TAZ inhibition. We used GBM1B-TAZ neurospheres harboring a lentiviral FLAG-tagged TAZ transgene under the control of the CMV promotor. This TAZ transgene was neither induced by EGFR activation nor downregulated after EGFR inhibition by OS (Fig. 7I), in contrast to the positive association between EGFR activity and endogenous TAZ expression as presented earlier (Fig. 1D and 7A). GBM1B-TAZ cells were found to be less sensitive to OS treatment in vitro compared to isogenic GBM1B cells (Fig. 7J; IC50: 9.6 vs 1.8 μM). Systemic OS treatment also did not significantly inhibit GBM1B-TAZ xenografts (Fig. 7K, 56.7 ± 10.2 vs 45.2 ± 7.5 mm3, p = 0.38, n=6), contrasting the sensitivity of isogenic GBM1B xenografts to OS treatment (Fig. 7G). Taken together, these results support that TAZ inhibition is required for potent GBM inhibition by OS.
Discussion
The RTK signaling network consists of various TFs as essential signaling hubs to transfer and amplify oncogenic signals. Here, we identified TAZ as a novel signaling hub activated by EGFR and downstream kinases STAT3 and ERK1/2. Since TAZ is most potently induced by EGF in GBM neurospheres, we focused on studying this EGFR-TAZ signaling axis in GBM models with ligand-stimulated EGFR or the ligand-independent hyperactivated EGFRvIII mutation. TAZ was also found to be induced in GBM neurospheres by other GBM-associated RTK ligands (PDGFβ, HGF and bFGF), suggesting a more general RTK-TAZ signaling axis. Thus, it will be of value to study how TAZ responds to single- and multi-RTK activation. Our result support the direct activation of TAZ transcription by STAT3. It is possible that STAT3 and other RTK-activated TFs (e.g. ELK1 and AP-1) may cooperate the TAZ promoter/enhancer to initiate and sustain TAZ expression, as indicated by the STAT3/ELK1 cooperation on the c-Fos promoter (36). TAZ and its paralog YAP were first known to be regulated by the canonical Hippo pathway and later known to be contextually regulated by other signaling pathways. Evidence reported here and previously by others (10) supports a non-Hippo mechanism for TAZ hyperactivation in GBM. We showed that phosphorylation of the Hippo-regulated LATS kinase is unchanged during the induction of TAZ by EGF, indicating that the EGFR-TAZ axis may not signal through the canonical Hippo signaling. Similarly, Bhat et. al. reported that clinical GBM specimens have higher TAZ expression than low-grade gliomas, but GBM and low-grade gliomas show no differences in LATS kinase activation (10), also suggesting a Hippo-independent mechanism of TAZ regulation in GBM. In addition, we showed that YAP levels in GBM cells are not significantly altered by EGF, consistent with no LATS response after EGF stimulation, as TAZ/YAP proteins are commonly co-regulated by the LATS kinase. Growing lines of evidence support the contextual and differential regulation of TAZ/YAP in various cancer contexts (30), thus warranting more context-specific studies and cross-context comparisons to dissect underlying mechanisms.
We identified potential TAZ-binding genes by ChIP-seq in a specific GBM context that was synchronized by growth factor depletion followed by EGF stimulation to induce peak TAZ induction. This strategy better fits our goal to dissect the EGFR-TAZ oncogenic network in a physiological relevant context, compared to previous TAZ ChIP-Seq using unsynchronized breast cancer cells in serum-containing medium with various RTK ligands and other exogenous factors (28). Transcriptomic analysis identified EGF-regulated genes to overlap with TAZ-binding genes for building the EGFR-induced TAZ transcriptome. TAZ-binding genomic sites are enriched in promoter regions near TSS, consistent with their higher colocalization with the active promoter mark (H3K4me3) rather than the enhancer (H3K27ac) and repressor (H3K27me3) marks. The predicted TAZ-binding motif also shows high similarity with the binding motifs of TEAD1 and TEAD3, in line with the well-established function of TAZ as a transcription co-activator in the TEAD complex.
In the context of GBM, we found that 39% (640/1,624) of the EGF-induced genes have TAZ-binding sites, supporting TAZ as an essential hub for EGFR signaling. TAZ silencing led to downregulation of TAZ-Up genes, further validating the reproducibility of our transcriptomic results. KEGG terms of TAZ-Up genes contain signaling pathways essential for GBM malignancy (e.g. focal adhesion, HIF1 and MAPK signaling). Consistently, molecular and phenotypic analysis of TAZ-hyperactivated GBM cells showed upregulation of TAZ-Up genes and TAZ-induced malignancy, including hyperproliferation, enhanced invasion, radioresistance and tumor propagation. An especially notable finding is the high enrichment of multiple RTK signaling components in the TAZ-Up genes. Those components include RTKs and their ligands (e.g. EGF/EGFR, PDGF/PDGFR, FGF/FGFR) with their TAZ-dependent expression validated by TAZ gain- and loss-of-function assays. EGF and EGFR induction by TAZ supports an EGFR-TAZ positive feedback loop predicted to amplify the EGFR-TAZ signal in GBM cells, consistent with other publications also supporting the EGFR-TAZ signaling model (37–39). Furthermore, TAZ activation by EGFR was found to induce other RTK pathway genes including PDGF/PDGFR and FGF/FGFR. Promiscuous RTK pathway activation by TAZ may involve additional post-translational mechanisms, as suggested by the fact that TAZ-Up genes also contain known regulators of RTK phosphorylation and downstream kinase cascades (e.g. CDC42 and SHC3). Activating a network of RTKs would provide a tumor growth advantage and contribute to resistance to single-RTK-targeted therapies. As an example, one malignant phenotype driven by this network is mitogen independence, as found in TAZ-hyperactivated GBM neurospheres that are capable to release high levels of mitogens (e.g. EGF and PDGFβ) and provide adequate autocrine and paracrine support of GBM neurosphere growth. Predicted by these findings, we further showed that TAZ hyperactivation promotes xenograft formation from GBM neurospheres and induces tumor-propagating capacity in non-tumorigenic A172 cells.
Additional TAZ-Up genes enriched in top KEGG terms (e.g. HIF1A, CXCR4, CD274, JUN, JAK2, and TEAD1) are highly associated with oncogenesis in GBM and other malignancies. Their expression was found to positively correlate with TAZ expression in clinical GBM specimens, providing additional support for the clinical relevance of our discoveries. We showed that TAZ-hyperactivated GBM cells are more invasive in vitro and in vivo, reflecting at least in part the induction of GBM-invasion regulators by TAZ (e.g. CXCR4 and ITGA2/6 and ITGB8) (40–42). CXCR4 and CD274 (PD-L1) are both associated with immune suppression and evasion of anti-tumor immunity in GBM and other cancers (43, 44). Co-targeting CXCR4 and PD-L1 was found to be therapeutically synergistic in syngeneic murine models of colon cancer and melanoma (45). Co-targeting them for GBM immunotherapy may be feasible through inhibiting the EGFR-TAZ axis by the brain-penetrating EGFR inhibitor osimertinib as shown by us to suppress CXCR4 and PD-L1 expression in GBM xenografts. HIF1α is a well-known oncogenic TF that orchestrates the oncogenic adaption of tumor cells to hypoxia (46). Our results identify TAZ as a novel mediator of EGFR-induced HIF1α activation. HIF1α is induced by TAZ in breast cancer, and complex formation between TAZ and HIF1α augment their transactivation functions in breast cancer cells (47). These results suggest potential mechanisms of TAZ/HIF1α feedback and cooperation, and HIF1α targeting may be rationally designed by inhibiting the EGFR-TAZ axis. JUN and TEAD1 are both TAZ-interacting proteins also induced by the EGFR-TAZ axis. A TF complex containing YAP/TAZ/TEAD and AP-1 (dimer of JUN and FOS) has been shown to synergistically activate target genes essential for S-phase entry and mitosis in breast cancer cells (28). Together, these findings suggest that the EGFR-TAZ transcriptional network may drive oncogenesis by augmenting cooperative TF hubs to activate their shared gene targets, such as PD-L1.
TAZ and YAP are promising targets for cancer therapy based on their frequent hyperactivation in human cancers and validated oncogenic functions (30). Current development of TAZ/YAP inhibitors mainly focuses on compounds that bind to and inhibit TAZ/YAP and/or their partners (e.g. TEADs), such as Verteporfin (48). Considering that TAZ/YAP also play essential roles in normal tissue homeostasis, targeting TAZ/YAP upstream regulators hyperactivated in cancer (e.g. EGFR) may provide effective inhibition as well as better tumor selectivity and safety profiles. Osimertinib (OS) is a third-generation EGFR inhibitor approved for the first-line treatment of metastatic NSCLC with T790M and other EGFR mutations. We identified OS as the most potent inhibitor of the EGFR-TAZ oncogenic network. Systemic OS treatment effectively inhibited the EGFR-TAZ signal and xenograft growth in two orthotopic GBM xenograft models, including an aggressive EGFRvIII+ model, justifying the clinical evaluation of OS in GBM. This result is also consistent with a recent publication supporting the pre-clinical efficacy of this drug on EGFRvIII+ GBM models (49). It is noteworthy to mention that OS downregulates validated TAZ-Up genes (e.g. key RTK components, HIF1α, CD274 and CXCR4), indicating the feasibility to simultaneously inhibit multiple oncogenic drivers, the targeting of which is still challenging. The efficacy of OS in GBM is also supported by a recent case report of OS treatment in a recurrent GBM patient (50). Moreover, we showed that EGFR-independent TAZ hyperactivation attenuated the therapeutic efficacy of OS both in vitro and in vivo, demonstrating the importance of TAZ downregulation for achieving therapeutically effective EGFR targeting. TAZ activity might serve as a biomarker of OS sensitivity, and it is feasible to measure TAZ responses to OS treatment using personalized GBM models (e.g. patient-derived neurospheres and xenografts). The TAZ-Up gene signature we defined here can also provide a functional readout of TAZ activity.
In conclusion, our found that TAZ functions as a major transcriptional hub in EGFR signaling that activates positive feed-back loops, cooperative TFs, and multifunctional oncogenic drivers. We identified the FDA-approved drug osimertinib as a potent inhibitor of this EGFR-TAZ axis, providing a clinically translatable strategy for TAZ targeting, which may be applicable across EGFR-positive human cancers with TAZ hyperactivation.
Supplementary Material
Significance:
This study establishes a genome-wide map of EGFR-TAZ signaling in glioblastoma and finds osimertinib effectively inhibits this signaling, justifying its future clinical evaluation to treat glioblastoma and other cancers with EGFR/TAZ hyperactivation.
Acknowledgement
This work was supported by grants from the NIH/NINDS R01NS099460 R21NS106407 R21NS101400 (M.Y.), 5R01NS110087 5R01NS096754 (J.L.), the American Brain Tumor Association Discovery Grant (M.Y.), and the Career Development Award from the United States Department of Defense (W81XWH-14-1-0176, M.Y.). W.Z. and H.J. were partially supported by the NIH/NHGRI grant R01HG009518. We thank Dr. Angelo Vescovi and Dr. Ichiro Nakano for providing human GBM cell models.
Footnotes
Conflict of interest statement
The authors declare no potential conflicts of interest.
References
- 1.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. [DOI] [PubMed] [Google Scholar]
- 2.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.An Z, Aksoy O, Zheng T, Fan QW, Weiss WA. Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene. 2018;37:1561–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiological reviews. 2014;94:1287–312. [DOI] [PubMed] [Google Scholar]
- 5.Castellan M, Guarnieri A, Fujimura A, Zanconato F, Battilana G, Panciera T, et al. Single-cell analyses reveal YAP/TAZ as regulators of stemness and cell plasticity in Glioblastoma. Nature cancer. 2021;2:174–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci U S A. 2010;107:1437–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park J, Jeong S. Wnt activated beta-catenin and YAP proteins enhance the expression of non-coding RNA component of RNase MRP in colon cancer cells. Oncotarget. 2015;6:34658–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai J, Maitra A, Anders RA, Taketo MM, Pan D. beta-Catenin destruction complex-independent regulation of Hippo-YAP signaling by APC in intestinal tumorigenesis. Genes Dev. 2015;29:1493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012;150:780–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhat KP, Salazar KL, Balasubramaniyan V, Wani K, Heathcock L, Hollingsworth F, et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 2011;25:2594–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–21. [DOI] [PubMed] [Google Scholar]
- 12.Mao P, Joshi K, Li J, Kim SH, Li P, Santana-Santos L, et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc Natl Acad Sci U S A. 2013;110:8644–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Learn CA, Hartzell TL, Wikstrand CJ, Archer GE, Rich JN, Friedman AH, et al. Resistance to tyrosine kinase inhibition by mutant epidermal growth factor receptor variant III contributes to the neoplastic phenotype of glioblastoma multiforme. Clin Cancer Res. 2004;10:3216–24. [DOI] [PubMed] [Google Scholar]
- 14.Fenstermaker RA, Ciesielski MJ, Castiglia GJ. Tandem duplication of the epidermal growth factor receptor tyrosine kinase and calcium internalization domains in A-172 glioma cells. Oncogene. 1998;16:3435–43. [DOI] [PubMed] [Google Scholar]
- 15.Bowman RL, Wang Q, Carro A, Verhaak RG, Squatrito M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 2017;19:139–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tilghman J, Wu H, Sang Y, Shi X, Guerrero-Cazares H, Quinones-Hinojosa A, et al. HMMR maintains the stemness and tumorigenicity of glioblastoma stem-like cells. Cancer Res. 2014;74:3168–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ying M, Sang Y, Li Y, Guerrero-Cazares H, Quinones-Hinojosa A, Vescovi AL, et al. Kruppel-like family of transcription factor 9, a differentiation-associated transcription factor, suppresses Notch1 signaling and inhibits glioblastoma-initiating stem cells. Stem Cells. 2011;29:20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wan J, Su Y, Song Q, Tung B, Oyinlade O, Liu S, et al. Methylated cis-regulatory elements mediate KLF4-dependent gene transactivation and cell migration. eLife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ying M, Wang S, Sang Y, Sun P, Lal B, Goodwin CR, et al. Regulation of glioblastoma stem cells by retinoic acid: role for Notch pathway inhibition. Oncogene. 2011;30:3454–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome biology. 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, et al. Software for computing and annotating genomic ranges. PLoS Comput Biol. 2013;9:e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ji H, Jiang H, Ma W, Johnson DS, Myers RM, Wong WH. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat Biotechnol. 2008;26:1293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pearson JRD, Regad T. Targeting cellular pathways in glioblastoma multiforme. Signal transduction and targeted therapy. 2017;2:17040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol. 2015;17:1218–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421–34. [DOI] [PubMed] [Google Scholar]
- 30.Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer cell. 2016;29:783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tripathi SK, Chen Z, Larjo A, Kanduri K, Nousiainen K, Aijo T, et al. Genome-wide Analysis of STAT3-Mediated Transcription during Early Human Th17 Cell Differentiation. Cell reports. 2017;19:1888–901. [DOI] [PubMed] [Google Scholar]
- 32.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kiseleva LN, Kartashev AV, Vartanyan NL, Pinevich AA, Samoilovich MP. Characteristics of A172 and T98g Cell Lines. Tsitologiia. 2016;58:349–55. [PubMed] [Google Scholar]
- 35.Lopez-Bertoni H, Lal B, Li A, Caplan M, Guerrero-Cazares H, Eberhart CG, et al. DNMT-dependent suppression of microRNA regulates the induction of GBM tumor-propagating phenotype by Oct4 and Sox2. Oncogene. 2015;34:3994–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang E, Lerner L, Besser D, Darnell JE, Jr. Independent and cooperative activation of chromosomal c-fos promoter by STAT3. J Biol Chem. 2003;278:15794–9. [DOI] [PubMed] [Google Scholar]
- 37.Gargini R, Segura-Collar B, Herranz B, Garcia-Escudero V, Romero-Bravo A, Nunez FJ, et al. The IDH-TAU-EGFR triad defines the neovascular landscape of diffuse gliomas. Sci Transl Med. 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang R, Wu Y, Zou J, Zhou J, Wang M, Hao X, et al. The Hippo transducer TAZ promotes cell proliferation and tumor formation of glioblastoma cells through EGFR pathway. Oncotarget. 2016;7:36255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang R, Wu Y, Wang M, Sun Z, Zou J, Zhang Y, et al. HDAC9 promotes glioblastoma growth via TAZ-mediated EGFR pathway activation. Oncotarget. 2015;6:7644–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reyes SB, Narayanan AS, Lee HS, Tchaicha JH, Aldape KD, Lang FF, et al. alphavbeta8 integrin interacts with RhoGDI1 to regulate Rac1 and Cdc42 activation and drive glioblastoma cell invasion. Molecular biology of the cell. 2013;24:474–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo P, Moses-Gardner A, Huang J, Smith ER, Moses MA. ITGA2 as a potential nanotherapeutic target for glioblastoma. Scientific reports. 2019;9:6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yadav VN, Zamler D, Baker GJ, Kadiyala P, Erdreich-Epstein A, DeCarvalho AC, et al. CXCR4 increases in-vivo glioma perivascular invasion, and reduces radiation induced apoptosis: A genetic knockdown study. Oncotarget. 2016;7:83701–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caccese M, Indraccolo S, Zagonel V, Lombardi G. PD-1/PD-L1 immune-checkpoint inhibitors in glioblastoma: A concise review. Critical reviews in oncology/hematology. 2019;135:128–34. [DOI] [PubMed] [Google Scholar]
- 44.Eckert F, Schilbach K, Klumpp L, Bardoscia L, Sezgin EC, Schwab M, et al. Potential Role of CXCR4 Targeting in the Context of Radiotherapy and Immunotherapy of Cancer. Frontiers in immunology. 2018;9:3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.D’Alterio C, Buoncervello M, Ierano C, Napolitano M, Portella L, Rea G, et al. Targeting CXCR4 potentiates anti-PD-1 efficacy modifying the tumor microenvironment and inhibiting neoplastic PD-1. Journal of experimental & clinical cancer research : CR. 2019;38:432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Semenza GL. HIF-1 and human disease: one highly involved factor. Genes Dev. 2000;14:1983–91. [PubMed] [Google Scholar]
- 47.Xiang L, Gilkes DM, Hu H, Luo W, Bullen JW, Liang H, et al. HIF-1alpha and TAZ serve as reciprocal co-activators in human breast cancer cells. Oncotarget. 2015;6:11768–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chagoya G, Kwatra SG, Nanni CW, Roberts CM, Phillips SM, Nullmeyergh S, et al. Efficacy of osimertinib against EGFRvIII+ glioblastoma. Oncotarget. 2020;11:2074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Makhlin I, Salinas RD, Zhang D, Jacob F, Ming GL, Song H, et al. Clinical activity of the EGFR tyrosine kinase inhibitor osimertinib in EGFR-mutant glioblastoma. CNS oncology. 2019;8:CNS43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing datasets are available from the Gene Expression Omnibus (GEO) Repository (GSE151994 and GSE151995).