

Abstract
The development of C(sp3)−H functionalizations of free carboxylic acids has provided a wide range of C−C and C−Y (Y = heteroatom) bond-forming reactions that both complement and expand upon what can be achieved through conventional methods. Additionally, C−H functionalizations have lent themselves to the one-step preparation of a number of valuable, synthetic motifs, that are often difficult to prepare through conventional methods. Herein, we report a β- or γ-C(sp3)−H carbonylation of free carboxylic acids using Mo(CO)6 as a convenient solid CO source and enabled by a bidentate ligand, leading to convenient syntheses of cyclic anhydrides. Among these, the succinic anhydride products are versatile stepping stones for the mono-selective introduction of alkyl, alkenyl, aryl, alkynyl, boryl, silyl, fluoro, iodo, and thiophenyl groups at the β position of the parent acids by decarboxylative functionalizations, thus providing a divergent strategy to synthesize a myriad of carboxylic acids inaccessible by previous β-C−H activation reactions. The enantioselective carbonylation of free cyclopropanecarboxylic acids has also been achieved using a chiral bidentate thioether ligand.
Keywords: C−H activation, Palladium, Ligand design, Carboxylic acids, Carbonylation
Graphical Abstract
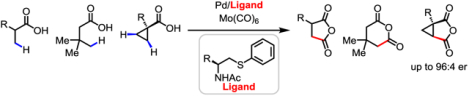
Pd(II)-catalyzed β- or γ-C(sp3)−H carbonylation of free carboxylic acids is reported. This protocol affords a new method for the syntheses of a wide range of cyclic anhydrides. Decarboxylative functionalization of the installed carboxyl group provides access to diverse β-functionalized aliphatic acids. The enantioselective C−H carbonylation of cyclopropanecarboxylic acids is also achieved.
Aliphatic carboxylic acids are ubiquitous and highly versatile motifs and are often inexpensive reagents in organic chemistry; as such, they are privileged starting materials in synthesis campaigns. While conjugate addition to α,β-unsaturated carbonyl compounds remains a strategic disconnection to install β-substituents, the past two decades have witnessed rapid developments in the β-C(sp3)−H functionalizations of carboxylic acids, enabling the construction of C−C and C−Y (Y = heteroatom) bonds far beyond what can be achieved through established methods.[1–3] Among these endeavors, the use of native carboxylic acids as directing groups (DGs) is highly desirable, since it obviates the need to install and remove bespoke DGs.[3] Moreover, C(sp3)−H functionalization reactions directed by native functional groups enable the one-step synthesis of synthetically important structural motifs from the parent compounds, which is otherwise impossible if using exogenous DGs. For example, we recently reported a β-C(sp3)−H activation reaction to synthesize β-lactones from inexpensive and abundant free aliphatic acids.[3i] This appealing system, which uses inexpensive and practical peroxide oxidants, was further extended to allow the preparation of γ-, δ-, and ε-lactones as well as tetralin, chromane, and indane motifs from free carboxylic acids (Scheme 1A).[3j,l] Despite the great value these protocols might have for synthetic chemistry, the development of new C(sp3)−H activation reactions to prepare common structural motifs from native functional groups remains a significant challenge.
Scheme 1.

Construction of common structural motifs using a C(sp3)−H functionalization strategy.
Herein, we report a β- or γ-C(sp3)−H carbonylation of free carboxylic acids using Mo(CO)6 as a convenient, solid CO source and a single Pd(II) catalyst bearing a bidentate ligand for the syntheses of a wide range of substituted cyclic anhydrides (Scheme 1B). The formation of cyclic anhydride-based products ensures exclusive mono-selectivity in the presence of multiple C−H bonds. The newly installed carboxyl groups are versatile linchpins for the introduction of diverse alkyl, alkenyl, aryl, alkynyl, boryl, silyl, fluoro, iodo, and thiophenyl groups at the β position of the parent acids via decarboxylative functionalizations, and so provide a divergent strategy to synthesize a myriad of carboxylic acids inaccessible through previous β-C−H activation reactions (Scheme 1C). The enantioselective carbonylation of free cyclopropanecarboxylic acids has also been achieved using a chiral bidentate thioether ligand.
The first example of Pd-catalyzed β-C(sp3)−H carbonylation of carboxylic acid derivatives using acidic aryl amides as the DGs was disclosed previously.[4] Further efforts on β-C(sp3)−H carbonylations from other groups have focused on the use of different DGs, transition metal catalysts, and CO sources for various substrates.[5–7] However, the requisite exogenous DGs on the final succinimide products were difficult to remove and so hampered the synthetic utility of these protocols. Bearing this in mind, we initiated our investigation of β-C(sp3)−H carbonylation reactions by selecting free aliphatic carboxylic acid 1c as a model substrate. Following our recent disclosure of the β-C(sp3)−H olefination of free carboxylic acids,[3f] we were delighted to observe a 5% 1H NMR yield of the desired succinic anhydride product 2c and trace amounts of product 2cʹ, derived from opening of the anhydride (2cʹ could be quantitatively cyclized to 2c by refluxing with 1 equiv. Ac2O in toluene), using the optimal catalyst of the olefination under 1 atm. CO. Further investigation of the CO source and terminal oxidant revealed that the combination of Mo(CO)6 as a convenient and air-stable solid source of CO[6d,g] and AgNO3 could further improve the yield to 57% (see Table S2). The use of AgNO3 as an effective oxidant has been reported in other C−H activation reactions such as fluorination and acetoxylation.[8] In light of recent advances in ligand-accelerated Pd(II)-catalyzed C−H activation,[9] we next searched for ligands that could substantially improve the reactivity of the catalyst (Table 1). A range of representative monodentate ligands developed in our laboratory, such as pyridine L1, quinoline L2, and 2-pyridone L3, were first tested but yields were unsatisfactory (10–15%).[10] Guided by previous MPAA ligand-enabled C(sp3)−H activation reactions of free carboxylic acids, we tested a series of previously optimal MPAA ligands (L4–L7):[3c,d,g,i,j,l,9b] these ligands promoted the carbonylation reaction slightly, delivering the carbonylation product in 20–31% yields. Bidentate ligands (L8–L11), previously found to promote enantioselective C(sp3)−H activation, gave poor yields (4–22%).[3e,11] To our delight, the optimal ligand L12, developed for the olefination of free carboxylic acids, significantly improved the yield to 74% (72% isolated yield of the benzyl (Bn)-protected diacid 2c’).[3f,12] Notably, the thioether ligand L12 could be easily prepared in one step with a single recrystallization in nearly quantitative yield (95%) by refluxing 2-methyl-2-oxazoline and thiophenol in toluene (see the preparation of ligand L12 in the Supporting Information). Efforts to introduce substitution on the ligand backbone (L13 and L14) did not further enhance the reactivity. Control experiments showed that the yields were low in the absence of the ligand or in the presence of the bidentate sulfoxide ligand L15 (2% or 1%, respectively), indicating the importance of the soft σ donor thioether for reactivity. We reasoned that, in addition to promote C(sp3)−H activation, thioether ligand might stabilize Pd(0) species and promote oxidation of Pd(0) by Ag, which might contribute to its superior reactivity over other ligands.
Table 1.

|
Conditions: 1c (0.1 mmol), Pd(OAc)2 (10 mol%), ligand (L) (10 mol%), Mo(CO)6 (0.5 equiv), AgNO3 (2.0 equiv), HFIP (1.0 mL), 80 °C, 24 h.
Sum of 2c and 2c’. The yields were determined by 1H NMR analysis of the crude product using CH2Br2 as the internal standard.
Isolated yield of Bn-protected diacid 2c’.
With the optimal ligand and reaction conditions in hand, we evaluated the scope of the C(sp3)−H carbonylation reaction (Table 2). The cyclic anhydride products were hydrolyzed and subsequently protected by Bn for ease of purification and analysis. A wide range of tertiary aliphatic acids bearing an α-gem-dimethyl group (1a–m) or a single α-methyl group (1n and 1o) were all compatible, affording the succinic anhydrides containing α-quaternary centers in moderate to good yields (36–75%), which are inconvenient to access from maleic anhydride. Less reactive free carboxylic acids containing α-hydrogens (1p–u) also reacted in synthetically useful yields (38–51%). Cyclopropanecarboxylic acids (1v–ad) were all amenable substrates, delivering cis-cyclopropanedicarboxylic anhydrides in yields ranging from modest to good (47–70%). Other free cyclic carboxylic acids (e.g., cyclobutane, cyclopentane, and cyclohexane) failed to afford succinic anhydride products. Among these, cyclic rings, including six- (2f), five- (2o and 2t), four- (2e), and three-membered (2s and 2v–ad) rings, were viable in the carbonylation reaction; a variety of functionalities such as fluoro (2i), trifluoromethyl (2j), tetrahydropyran (2f), methoxy (2k), Bn-protected hydroxyl (2l), methoxymethyl (MOM)-protected hydroxyl (2m), and phthalimide (2ad) were all well-tolerated, with the protected hydroxyls (2l and 2m) serving as useful synthetic handles for subsequent derivatization. While carboxylic acids containing α-phenyl groups (e.g., 2-phenylpropionic acid) afforded C(sp2)−H carbonylation product,[7d] free carboxylic acids bearing β- or γ-phenyl groups (2g, 2h, and 2y–ab) were compatible with the current conditions and remained intact despite the potentially reactive aryl C(sp2)−H bonds.[7a,b,e] A range of substituents on the aryl ring from electron-withdrawing (fluoro and chloro) groups (2h, 2u, 2ab, and 2ac) to electron-donating (benzyloxy and methyl) groups (2z and 2aa) were also well-tolerated. This protocol could be extended to form synthetically valuable glutaric anhydride 2ae via γ-C(sp3)−H carbonylation, albeit in moderate yield (38%) under the current conditions. Current conditions failed to synthesize adipic anhydrides probably due to sluggish δ-C(sp3)−H activation via seven-membered palladacycle.
Table 2.

|
Conditions: 1 (0.1 mmol), Pd(OAc)2 (10 mol%), L12 (10 mol%), Mo(CO)6 (0.5 equiv), AgNO3 (2.0 equiv), HFIP (1.0 mL), 80 °C, 24 h.
Isolated yields of Bn-protected diacid 2’ are shown.
The yield was determined by 1H NMR analysis of the crude product (4% yield of 2v and 64% yield of 2v’).
Conditions: 1ae (0.1 mmol), Pd(OAc)2 (10 mol%), Ac-Val-OH (15 mol%), Mo(CO)6 (0.3 equiv), AgNO3 (2.0 equiv), HFIP (1.0 mL), 90 °C, 24 h.
This protocol could also be extended to the enantioselective β-C(sp3)−H carbonylation of cyclopropanecarboxylic acids (Table 3).[3e,g] Through systematic modifications to the backbone of the bidentate thioether ligand, we found that introduction of a methyl group (L13) gave optimal reactivity and enantioselectivity; a range of cyclopropanecarboxylic acids (1y–ac) were compatible with the current protocol, affording cyclopropane-fused succinic anhydrides with good enantioselectivities (up to 96:4 er) and moderate yields (36–60%).
Table 3.
Substrate Scope of the Enantioselective β-C(sp3)−H Carbonylation of Cylcopropanecarboxylic Acids[a,b]

|
Conditions: 1 (0.1 mmol), Pd(OAc)2 (10 mol%), L13 (10 mol%), Mo(CO)6 (0.5 equiv), AgNO3 (2.0 equiv), HFIP (1.0 mL), 70 °C, 24 h.
Isolated yields of Bn-protected diacid 2’ are shown. The er values were determined on the SFC system using commercially available chiral columns.
The succinic anhydride products of this reaction are versatile linchpins for the mono-selective introduction of C−C and C−Y (Y = heteroatom) bonds at the β position of the parent acids. For example, the Ni-catalyzed decarbonylative cross-coupling of cyclic anhydrides (e.g., 2a) with alkyl bromides delivers free acid products featuring formal β-alkylation, with decarbonylation occurring regioselectively at the sterically less hindered side of the parent anhydride.[13] This protocol features the use of readily available alkyl bromides as well as compatibility with secondary alkyls, which are usually unreactive in Pd-catalyzed C−H alkylation reactions.[2d,f,h] In addition, the regioselective opening of anhydride products to unmask just one of the carboxyl groups has been demonstrated.[14] The resultant carboxyl groups are themselves versatile synthetic handles: in addition to their conversion to amide, ketone, ester or hydroxyl moieties, via addition of appropriate nucleophiles or reducing agents, they can also engage in a wide range of decarboxylative functionalizations.[15,16] To demonstrate the utility of our succinic anhydride products, 2a was converted to the β-carboxyl ester 3b , a stepping stone for the mono-selective installation of a range of aryl, alkenyl, alkynyl, boryl, silyl, fluoro, iodo, and thiophenyl groups (Table 4). Aza-heterocycles could be efficiently installed by either decarboxylative cross-coupling with a pyridinylboronic acid (4a, 55%)[16b] or a decarboxylative Minisci-type reaction with isoquinoline (4b, 48%).[16c,d] Notably, aza-heterocyclic halides are usually incompatible with Pd-catalyzed C−H arylation reactions due to catalyst poisoning from coordination of these aza-heterocycles.[2c,d,f,3a–e,g,l] The analogous decarboxylative cross-coupling with a vinyl boronic acid delivered β-vinyl ester 4c in moderate yield (53%),[16b] which provided a complementary strategy to Pd-catalyzed β-C−H olefination of acids and their derivatives, where only electron-deficient olefins are effective.[2e,3f] β-Alkynyl ester 4d was directly accessible in a synthetically useful yield (39%) through a mild decarboxylative alkynylation with more general and diverse alkynyl Grignard reagents.[16e] For C−Y (Y = heteroatom) bond-forming reactions, boryl (B(pin), 4e),[16f] silyl (4f),[16g] and iodo (4g)[16h] groups could be efficiently installed at the β position of the parent acid in modest yields (39–43%), producing versatile linchpins for further elaboration. The CH2F fragment, a highly sought-after bioisostere in medicinal chemistry, could be successfully introduced by decarboxylative fluorination, albeit in low yield (4h, 11%).[16i] Finally, the formal β-chalcogenation product 4i was obtained in moderate yield (56%).[16j]
Table 4.

|
Conditions for regioselective opening of 2a: 2a, DMAP (30 mol%), tBuOH, 80 °C, 12 h; K2CO3 (2.0 equiv), BnBr (2.0 equiv), CH3CN, 80 °C, 6 h; TFA (2.0 equiv), DCM, rt, 3 h. Conditions for decarboxylative functionalization: 4a–g and 4i from redox-active ester (RAE) of 3b; 4h from 3b. See Supporting Information for details.
Isolated yields are shown.
In summary, we have realized an effective C−H carbonylation of ubiquitous aliphatic acids enabled by a single Pd(II) catalyst bearing a bidentate ligand. This reaction affords a new method for the syntheses of a wide range of cyclic anhydrides using Mo(CO)6 as a convenient solid CO source. Exploitation of the versatile reactivity of the newly installed carboxyl group provides access to diverse β-functionalized aliphatic acids in exclusive mono-selectivity and unparalleled scope. The enantioselective C−H carbonylation of cyclopropanecarboxylic acids has also been achieved using a chiral bidentate thioether ligand.
Supplementary Material
Acknowledgements
We gratefully acknowledge the NIH (NIGMS, R01GM084019) and The Scripps Research Institute for financial support. We thank Kessil Lighting for a kind loan of PR160L-370nm and PR160L-390nm lamps.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Conflict of interest
The authors declare no conflict of interest.
References
- [1].For reviews, see:; a) Chen X, Engle KM, Wang D-H, Yu J-Q, Angew. Chem. Int. Ed 2009, 48, 5094–5115; Angew. Chem. 2009, 121, 5196–5217; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Daugulis O, Roane J, Tran LD, Acc. Chem. Res 2015, 48, 1053–1064; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lyons TW, Sanford MS, Chem. Rev 2010, 110, 1147–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].For early examples of β-C(sp3)−H functionalization reactions of carboxylic acids using DGs, see:; a) Giri R, Chen X, Yu J-Q, Angew. Chem. Int. Ed 2005, 44, 2112–2115; Angew. Chem. 2005, 117, 2150–2153; [DOI] [PubMed] [Google Scholar]; b) Giri R, Liang J, Lei J-G, Li J-J, Wang D-H, Chen X, Naggar IC, Guo C, Foxman BM, Yu J-Q, Angew. Chem. Int. Ed 2005, 44, 7420–7424; Angew. Chem. 2005, 117, 7586–7590; [DOI] [PubMed] [Google Scholar]; c) Zaitsev VG, Shabashov D, Daugulis O, J. Am. Chem. Soc 2005, 127, 13154–13155; [DOI] [PubMed] [Google Scholar]; d) Wang D-H, Wasa M, Giri R, Yu J-Q, J. Am. Chem. Soc 2008, 130, 7190–7191; [DOI] [PubMed] [Google Scholar]; e) Wasa M, Engle KM, Yu J-Q, J. Am. Chem. Soc 2010, 132, 3680–3681; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Shabashov D, Daugulis O, J. Am. Chem. Soc 2010, 132, 3965–3972; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Ano Y, Tobisu M, Chatani N, J. Am. Chem. Soc 2011, 133, 12984–12986; [DOI] [PubMed] [Google Scholar]; h) Zhang S-Y, Li Q, He G, Nack WA, Chen G, J. Am. Chem. Soc 2013, 135, 12135–12141. [DOI] [PubMed] [Google Scholar]
- [3].For β-C(sp3)−H functionalization reactions of free carboxylic acids, see:; a) Giri R, Maugel N, Li J-J, Wang D-H, Breazzano SP, Saunder LB, Yu J-Q, J. Am. Chem. Soc 2007, 129, 3510–3511; [DOI] [PubMed] [Google Scholar]; b) Chen G, Zhuang Z, Li G-C, Saint-Denis TG, Hsiao Y, Joe CL, Yu J-Q, Angew. Chem. Int. Ed 2017, 56, 1506–1509; Angew. Chem. 2017, 129, 1528–1531; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhu Y, Chen X, Yuan C, Li G, Zhang J, Zhao Y, Nat. Commun 2017, 8, 14904; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ghosh KK, van Gemmeren M, Chem. Eur. J 2017, 23, 17697–17700; [DOI] [PubMed] [Google Scholar]; e) Shen P-X, Hu L, Shao Q, Hong K, Yu J-Q, J. Am. Chem. Soc 2018, 140, 6545–6549; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Zhuang Z, Yu C-B, Chen G, Wu Q-F, Hsiao Y, Joe CL, Qiao JX, Poss MA, Yu J-Q, J. Am. Chem. Soc 2018, 140, 10363–10367; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Hu L, Shen P-X, Shao Q, Hong K, Qiao JX, Yu J-Q, Angew. Chem. Int. Ed 2019, 58, 2134–2138; Angew. Chem. 2019, 131, 2156–2160; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Ghosh KK, Uttry A, Koldemir A, Ong M, van Gemmeren M, Org. Lett 2019, 21, 7154–7157; [DOI] [PubMed] [Google Scholar]; i) Zhuang Z, Yu J-Q, Nature 2020, 577, 656–659; [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Zhuang Z, Herron AN, Fan Z, Yu J-Q, J. Am. Chem. Soc 2020, 142, 6769–6776; [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Ghiringhelli F, Uttry A, Ghosh KK, van Gemmeren M, Angew. Chem. Int. Ed 2020, 59, 23127–23131; Angew. Chem. 2020, 132, 23327–23331; [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Zhuang Z, Herron AN, Liu S, Yu J-Q, J. Am. Chem. Soc 2021, 143, 687–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].For the first example of Pd-catalyzed β-C(sp3)−H carbonylation reaction of carboxylic acids using DGs, see:; Yoo EJ, Wasa M, Yu J-Q, J. Am. Chem. Soc 2010, 132, 17378–17380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].For other metals-catalyzed or mediated β-C(sp3)−H carbonylation reactions of carboxylic acids using DGs, see:; a) Hasegawa N, Charra V, Inoue S, Fukumoto Y, Chatani N, J. Am. Chem. Soc 2011, 133, 8070–8073; [DOI] [PubMed] [Google Scholar]; b) Wu X, Zhao Y, Ge H, J. Am. Chem. Soc 2015, 137, 4924–4927; [DOI] [PubMed] [Google Scholar]; c) Wu X, Miao J, Li Y, Li G, Ge H, Chem. Sci 2016, 7, 5260–5264; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Barsu N, Bolli SK, Sundararaju B, Chem. Sci 2017, 8, 2431–2435; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Williamson P, Galvan A, Gaunt MJ, Chem. Sci 2017, 8, 2588–2591; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Liu Y-H, Xia Y-N, Shi B-F, Chin. J. Chem 2020, 38, 635–662. [Google Scholar]
- [6].For Pd-catalyzed C(sp3)−H carbonylation reactions of other classes of substrates, see:; a) Fujiwara Y, Jintoku T, Uchida Y, New J. Chem 1989, 13, 649–650; [Google Scholar]; b) McNally A, Haffemayer B, Collins BSL, Gaunt MJ, Nature 2014, 510, 129–133; [DOI] [PubMed] [Google Scholar]; c) Wang C, Zhang L, Chen C, Han J, Yao Y, Zhao Y, Chem. Sci 2015, 6, 4610–4614; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hernando E, Villalva J, Martínez ÁM, Alonso I, Rodríguez N, Arrayás RG, Carretero JC, ACS Catal 2016, 6, 6868–6882; [Google Scholar]; e) Willcox D, Chappell BGN, Hogg KF, Calleja J, Smalley AP, Gaunt MJ, Science 2016, 354, 851–857; [DOI] [PubMed] [Google Scholar]; f) Tanaka K, Ewing WR, Yu J-Q, J. Am. Chem. Soc 2019, 141, 15494–15497; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Zhuang Z, Yu J-Q, J. Am. Chem. Soc 2020, 142, 12015–12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].For Pd-catalyzed C(sp2)−H carbonylation reactions, see:; a) Fujiwara Y, Kawauchi T, Taniguchi H, J. Chem. Soc., Chem. Commun 1980, 220–221; [Google Scholar]; b) Fujiwara Y, Kawata I, Sugimoto H, Taniguchi H, J. Organomet. Chem 1983, 256, C35–C36; [Google Scholar]; c) Orito K, Horibata A, Nakamura T, Ushito H, Nagasaki H, Yuguchi M, Yamashita S, Tokuda M, J. Am. Chem. Soc 2004, 126, 14342–14343; [DOI] [PubMed] [Google Scholar]; d) Giri R, Yu J-Q, J. Am. Chem. Soc 2008, 130, 14082–14083; [DOI] [PubMed] [Google Scholar]; e) Wang P, Verma P, Xia G, Shi J, Qiao JX, Tao S, Cheng PTW, Poss MA, Farmer ME, Yeung K-S, Yu J-Q, Nature 2017, 551, 489–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Lou S-J, Xu D-Q, Xu Z-Y, Angew. Chem. Int. Ed 2014, 53, 10330–10335; Angew. Chem. 2014, 126, 10498–10503; [DOI] [PubMed] [Google Scholar]; b) Stowers KJ, Kubota A, Sanford MS, Chem. Sci 2012, 3, 3192–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].For reviews, see:; a) He J, Wasa M, Chan KSL, Shao Q, Yu J-Q, Chem. Rev 2017, 117, 8754–8786; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shao Q, Wu K, Zhuang Z, Qian S, Yu J-Q, Acc. Chem. Res 2020, 53, 833–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Wasa M, Chan KSL, Zhang X-G, He J, Miura M, Yu J-Q, J. Am. Chem. Soc 2012, 134, 18570–18572; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) He J, Li S, Deng Y, Fu H, Laforteza BN, Spangler JE, Homs A, Yu J-Q, Science 2014, 343, 1216–1220; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chen Y-Q, Wang Z, Wu Y, Wisniewski SR, Qiao JX, Ewing WR, Eastgate MD, Yu J-Q, J. Am. Chem. Soc 2018, 140, 17884–17894; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Xia G, Zhuang Z, Liu L-Y, Schreiber SL, Melillo B, Yu J-Q, Angew. Chem. Int. Ed 2020, 59, 7783–7787; Angew. Chem. 2020, 132, 7857–7861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Chen G, Gong W, Zhuang Z, Andrä MS, Chen Y-Q, Hong X, Yang Y-F, Liu T, Houk KN, Yu J-Q, Science 2016, 353, 1023–1027; [DOI] [PMC free article] [PubMed] [Google Scholar]; b ) Wu Q-F, Shen P-X, He J, Wang X-B, Zhang F, Shao Q, Zhu R-Y, Mapelli C, Qiao JX, Poss MA, Yu J-Q, Science 2017, 355, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].For reviews on thioether ligands, see:; a) Bayón JC, Claver C, Masdeu-Bultó AM, Coord. Chem. Rev 1999, 193–195, 73–145; [Google Scholar]; b) Mellah M, Voituriez A, Schulz E, Chem. Rev 2007, 107, 5133–5209. [DOI] [PubMed] [Google Scholar]
- [13].a) Lin T, Gu Y, Qian P, Guan H, Walsh PJ, Mao J, Nat. Commun 2020, 11, 5638; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yamamoto T, Sano K, Yamamoto A, J. Am. Chem. Soc 1987, 109, 1092–1100; [Google Scholar]; c) O’Brien EM, Bercot EA, Rovis T, J. Am. Chem. Soc 2003, 125, 10498–10499. [DOI] [PubMed] [Google Scholar]
- [14].Atodiresei I, Schiffers I, Bolm C, Chem. Rev 2007, 107, 5683–5712. [DOI] [PubMed] [Google Scholar]
- [15].a) Parida SK, Mandal T, Das S, Hota SK, De Sarkar S, Murarka S, ACS Catal 2021, 11, 1640–1683; [Google Scholar]; b) Crespi S, Fagnoni M, Chem. Rev 2020, 120, 9790–9833; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Smith JM, Harwood SJ, Baran PS, Acc. Chem. Res 2018, 51, 1807–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Chen T-G, Barton LM, Lin Y, Tsien J, Kossler D, Bastida I, Asai S, Bi C, Chen JS, Shan M, Fang H, Fang FG, Choi H, Hawkins L, Qin T, Baran PS, Nature 2018, 560, 350–354; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang J, Qin T, Chen T-G, Wimmer L, Edwards JT, Cornella J, Vokits B, Shaw SA, Baran PS, Angew. Chem. Int. Ed 2016, 55, 9676–9679; Angew. Chem. 2016, 128, 9828–9831; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cheng W, Shang R, Fu M, Fu Y, Chem. Eur. J 2017, 23, 2537–2541; [DOI] [PubMed] [Google Scholar]; d) Sherwood TC, Li N, Yazdani AN, Dhar TGM, J. Org. Chem 2018, 83, 3000–3012; [DOI] [PubMed] [Google Scholar]; e) Smith JM, Qin T, Merchant RR, Edwards JT, Malins LR, Liu Z, Che G, Shen Z, Shaw SA, Eastgate MD, Baran PS, Angew. Chem. Int. Ed 2017, 56, 11906–11910; Angew. Chem. 2017, 129, 12068–12072; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Wang J, Shang M, Lundberg H, Feu KS, Hecker SJ, Qin T, Blackmond DG, Baran PS, ACS Catal 2018, 8, 9537–9542; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Xue W, Oestreich M, Angew. Chem. Int. Ed 2017, 56, 11649–11652; Angew. Chem. 2017, 129, 11808–11811; [DOI] [PubMed] [Google Scholar]; h) Fu M-C, Wang J-X, Shang R, Org. Lett 2020, 22, 8572–8577; [DOI] [PubMed] [Google Scholar]; i) Ventre S, Petronijevic FR, MacMillan DWC, J. Am. Chem. Soc 2015, 137, 5654–5657; [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Jin Y, Yang H, Fu H, Chem. Commun 2016, 52, 12909–12912. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.