

Abstract
Synthetic glucocorticoids (sGCs) such as dexamethasone (DEX), while used to mitigate inflammation and disease progression in premature infants with severe bronchopulmonary dysplasia (BPD), are also associated with significant adverse neurologic effects such as reductions in myelination and abnormalities in neuroanatomical development. Ciclesonide (CIC) is a sGC prodrug approved for asthma treatment that exhibits limited systemic side effects. Carboxylesterases enriched in the lower airways convert CIC to the glucocorticoid receptor (GR) agonist des-CIC. We therefore examined whether CIC would likewise activate GR in neonatal lung but have limited adverse extra-pulmonary effects, particularly in the developing brain. Neonatal rats were administered subcutaneous injections of CIC, DEX or vehicle from postnatal days 1-5 (PND1-PND5). Systemic effects linked to DEX exposure, including reduced body and brain weight, were not observed in CIC treated neonates. Furthermore, CIC did not trigger the long-lasting reduction in myelin basic protein expression in the cerebral cortex nor cerebellar size caused by neonatal DEX exposure. Conversely, DEX and CIC were both effective at inducing the expression of select GR target genes in neonatal lung, including those implicated in lung-protective and anti-inflammatory effects. Thus, CIC is a promising, novel candidate drug to treat or prevent BPD in neonates given its activation of GR in neonatal lung and limited adverse neurodevelopmental effects. Furthermore, since sGCs such as DEX administered to pregnant women in pre-term labor can adversely affect fetal brain development, the neurological-sparing properties of CIC, make it an attractive alternative for DEX to treat pregnant women severely ill with respiratory illness, such as with asthma exacerbations or COVID-19 infections.
Keywords: ciclesonide, dexamethasone, bronchopulmonary dysplasia, glucocorticoids
Introduction
Preterm infants, especially those born earlier than 28 weeks of gestation, have a significant risk of developing bronchopulmonary dysplasia (BPD) (Davidson and Berkelhamer, 2017), with an incidence estimated to be ~45% (Rutkowska et al., 2019). It remains the most expensive complication of prematurity in terms of average cost per hospitalization and is a primary factor contributing to length of hospital stay. In addition, there are increased costs and potential morbidities associated with therapies related to the treatment of BPD, such as nitric oxide, oxygen supplementation, potential for tracheostomy, and other associated care (Kalikkot Thekkeveedu et al., 2017). Current clinical strategies to reduce the incidence and severity of BPD include gentle ventilation, avoiding extremes of oxygenation, and pharmacotherapy including diuretics, and bronchodilators, as well as inhaled or intratracheal budesonide or systemic dexamethasone (DEX), a long-acting synthetic glucocorticoid (sGC). While helpful in many cases for reducing inflammation and disease progression in BPD, systemic sGC administration unfortunately can be associated with adverse side effects, and it has been implicated in detrimental and persistent effects on infant neurodevelopment, including alterations in brain architecture and behavior (Cheong et al., 2014; Kraft et al., 2019).
Recent outcome studies after postnatal DEX administration demonstrate increased risk for cerebral palsy, abnormal neurologic examinations, and higher Major Depression Inventory scores depending upon both the dose and the age of the infant at the time of sGC initiation (Kraft et al., 2019; LeFlore and Engle, 2011; Malaeb and Stonestreet, 2014). Despite efforts to minimize adverse neurodevelopmental outcomes by limiting use of sGC to older (7-14 days postnatal) and more severely affected infants, neonates in these groups continue to remain at risk for other significant systemic side effects such as hyperglycemia, hypertension, gastrointestinal bleeding, and infection (Malaeb and Stonestreet, 2014). Hydrocortisone, a less potent and short acting GC often used in place of DEX, also results in side effects such as adrenal insufficiency (Ng et al., 2004) and increased risk of spontaneous intestinal perforation (Peltoniemi et al., 2005). In summary, there remains a need for a treatment that will have the desired anti-inflammatory and tissue maturation effects on neonatal lungs, while limiting negative systemic side effects, including those in the developing brain.
Ciclesonide (CIC) is a new generation inhaled sGC currently approved for the treatment of asthma and allergic rhinitis. It is a prodrug that is converted by carboxylesterases (CES) enriched in the lower airway into the active compound des-ciclesonide (des-CIC) (Mutch et al., 2007; Sato et al., 2007), which has a 100-fold higher binding affinity to the GC receptor (GR) than the parent (inactive) compound (Daley-Yates, 2015). In addition, des-CIC has optimal pharmacokinetics, with 99% of drug being protein-bound in the serum (bioavailability of <1%), and >99% first-pass metabolism in the liver – which, combined with targeted activation by airway CESs, makes it an ideal candidate for avoidance of systemic, and in particular, neurologic effects (Daley-Yates, 2015; Nave et al., 2004; Nonaka et al., 2007). The pharmacokinetic (PK) properties of CIC and des-CIC evaluated in adult humans and rodents further enhance its potential benefit to treat neonatal lung injury. Specifically, fatty acid esterification of des-CIC increases its tissue residency time in lung and is the basis for the current once daily dosing regimen of CIC recommended in adults and children (Nave and McCracken, 2008). Furthermore, des-CIC accumulation in rodent brain is highly limited and can be an order of magnitude less than those in lung even after multiple doses (Mars et al., 2013). Clinical studies, including with pediatric subjects, have shown non-inferiority of CIC in terms of managing respiratory symptoms compared to other standard sGCs used in asthma management, and a safety profile comparable to current first-line therapies (Maglione et al., 2018; Postma et al., 2017). Additionally, CIC does not appear to cause unwanted systemic adverse effects often observed with other systemic or inhaled sGCs, such as adrenal suppression, reduced long bone growth, or neurologic impairment. In fact, CIC has been used successfully in children as young as 6 years old as an alternative asthma therapy to current first-line agents to reverse adrenal suppression while still controlling respiratory symptoms (Liddell et al., 2017; Pedersen et al., 2006; Pedersen et al., 2010; Skoner, 2016).
Given the potential for serious systemic side effects following neonatal sGC administration, alternative GR-targeting drugs for BPD treatment need to be evaluated, particularly those drugs with potential to limit adverse effects in the brain. Many of the persistent detrimental effects of postnatal sGCs in humans have also been observed in rodent models (Bhatt et al., 2013; de Kloet et al., 2014; Feng et al., 2009; Lee et al., 2012), allowing us to evaluate CIC mechanism and targets in comparison to DEX using a model organism. For example, because the saccular stage of newborn rat lung development (i.e., E21-PND4) matches the stage of human lung development typically attained in premature infants (i.e., saccular stage between 24-38 gestational weeks of age) (Schittny, 2017) they provide an ideal initial paradigm to evaluate localization-specific CIC responses. Herein, we demonstrate that DEX and CIC can regulate various GR targets in the newborn rodent lung, including both ubiquitously responsive genes and those implicated in lung protective effects. However, CIC does not suppress somatic growth nor induce the neuroanatomical changes in the cerebral cortex and cerebellum that are established consequences of DEX exposure in neonates. Therefore, CIC, a drug that has been evaluated in clinical trials for children as young as 2 years old (ClinicalTrials.gov Identifier: NCT00261287 (Brand et al., 2011)) is an attractive candidate for future evaluation as an alternative sGC to treat or prevent BPD.
Material and methods
Animal care
Animal experiments were performed in accordance with the NIH Guide for the Care and Use of Animals and approved by the University of Pittsburgh Animal Care and Use Committee as were images used in this publication. In addition, the use of C57Bl/6 mice in this study was approved by the Monash University Animal Research Ethics Committee.
sGC injections
Timed-pregnant Sprague Dawley rats (Charles River) were housed individually in standard microisolator cages with food (Prolab Isopro RMH 3000, LabDiet) and water ad libitum under a 12-hour light cycle from 7a-7p and allowed to give birth. Within 24 hours of birth (PND1), pups were weighed individually and divided among three treatment groups, vehicle (VEH), 0.5mg/kg DEX, or 0.5mg/kg CIC, a dose which has been used in a similar study (Kim et al., 2013). Stock solutions (10−2 M) of DEX and CIC were prepared in 100% ethanol and stored at −20°C. Prior to injections, the stocks were diluted to a concentration of 50 mg/ml in sterile saline. VEH consisted of 100% ethanol diluted in sterile saline using identical dilution parameters to DEX. Compounds were administered subcutaneously (SC) at the nape of the neck via a 31G needle not to exceed 10 μl/g once a day for up to 5 consecutive days on postnatal days (PND1-5) . Pups were weighed daily prior to injection. DEX treated neonates were given additional injections of sterile saline as needed if showing signs of dehydration. Both male and female rats were included in this study.
Tissue Fixation, Immunohistochemistry (IHC) and Anatomical Evaluation
For analysis of neonatal C57Bl/6 mouse lung, PND1 and PND5 tissue was collected and fixed in 4% paraformaldehyde, embedded in paraffin blocks, and 5μM sections cut and mounted on slides. Sections were stained using a rabbit polyclonal antibody to detect CES1d (Invitrogen, PA5-19740, 1:200 dilution) with visualization by diaminobenzidine staining and counterstaining with eosin, as described previously (Bird et al., 2014).
For anatomical and myelination evaluations, PND15 rat brains were fixed in 10 % neutral buffered formalin (Formal-Fixx, Shandon, Thermo Scientific) overnight at 4°C then processed through increasing concentrations of sucrose (10, 20, 30% in phosphate buffered saline (PBS)). Photographs were taken of fixed PND15 brains with an iPhone XS (Apple, Inc.) mounted (Universal Cell Phone Adapter Mount, Gosky Optics) under magnification on a stereomicroscope at 7X (MZ6 modular stereomicroscope, Leica Microsystems).
Fixed rat brain tissue was embedded in Tissue Tek (Scigen Scientific) compound, frozen and consecutive sagittal sections (30 μm) were collected of the cerebellum or coronally from the forebrain. Cerebellar sections were incubated in PBS with 0.05% Tween 20 followed with fluorescent Nissl stain for 2 hours in a solution containing 5% BSA, 5% goat serum, 0.3% triton X100, 5% glycine in PBS (NeuroTrace 500/525, Thermo Fisher Scientific, N21480, 1:5000). For myelin staining in the cerebral cortex, consecutive 30μM coronal forebrain sections were collected from every 4th section, incubated with an anti-MBP antibody (Invitrogen PA578397) overnight, and subsequently incubated with a biotinylated secondary antibody, avidin/streptavidin amplification with the Vector ABC kit (Thermo Fisher Sci, P132054) and subsequent visualization with diaminobenzidine staining and counterstained with eosin. Images were visualized at 10X and 20X on an EVOS cell imaging system (Fisher Scientific), photographed, imported and figures assembled in Adobe Photoshop.
Protein Immunoassays
The Jess System (Protein Simple) was used for capillary-based protein immunoassays of CES1d (Abeam, ab68190, 1:10,000) and β-actin (Proteintech, HRP-60004, 1:100) in tissue collected from untreated rat neonates including lung, cerebellum, and cortex. At PND1 and PND5 tissues were collected and protein lysates prepared in RIPA buffer processed according to Protein Simple using the supplied reagents after BCA assay (Pierce) to determine total protein. Separation modules were loaded into the Jess System, and proteins underwent separation by size within capillaries prior to automated immunodetection. The secondary HRP conjugated antibodies were from Protein Simple.
Similarly, total and phosphorylated glucocorticoid receptor (GR) from PND1 and PND2 rat lung was examined in treated animals. At 4 and 24 hours after SC injection with 0.5 mg/kg DEX, 0.5 mg/kg CIC or VEH on PND1, lung tissue was collected, and instructions utilizing the supplied reagent kits. Antibodies used to detect total GR (Cell Signaling Technology, 47411, 1:50) or GR phosphorylated at serine 232 (orthologous to serine 211 in human) (Cell Signaling Technology, 4161, 1:100 ) and serine 246 (226 in human) (Cell Signaling Technology, 97285, 1:100) were from Cell Signaling. The secondary HRP conjugated antibodies against mouse or rabbit were from Protein Simple. Relative protein expression was quantified with the Compass software (Protein Simple) using the total protein normalization module for the Jess System (Protein Simple).
MBP expression was assessed at PND15 following daily SC injections from PND1-PND5 with 0.5 mg/kg DEX, 0.5 mg/kg CIC or VEH. Specifically, forebrain lysates were prepared from tissue isolated using a brain block to obtain 3mm coronal sections of the rat forebrain containing the body of the corpus callosum anterior to the hippocampi. Tissue was homogenized by sonication on ice in RIPA buffer containing 50mM Tris HCI pH 8.0, 1% Triton x-100, 0.5% Na-deoxycholate, 0.1% SDS with 1x HALT Protease and Phosphatase inhibitor cocktail (Thermo Scientific). 10μg of total protein as determined by BCA (Pierce) for each sample was separated on a 10% TruPage premade gel with MOPS running buffer (Sigma). Proteins were then transferred to a PVDL-FL membrane (Millipore). The membrane was blocked in Odyssey Blocking Buffer (LiCor) then incubated overnight with an antibody against MBP (Cell Signaling Technology, 78896, 1:2000) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Santa Cruz Biotech, sc-32233, 1:2500) followed by incubation with secondary antibodies (goat anti-rabbit 800, Licor 92603221, 1:1000) and goat anti mouse 680 LiCor 926-32220, 1:1000). Processed membranes were imaged on an Odyssey CLx infrared scanner (LiCor Bioscience) and analyzed with the Image Studio Lite Ver 5.2 (LiCor Bioscience).
qRT-PCR
RNA was isolated from frozen rat tissues collected at multiple postnatal time points (untreated for Ces expression analysis at PND1, PND2, PND15; and 4 and/or 24 hours post treatment administered on PND1 for expression of genes driven by GR activation) using a Trizol (Invitrogen) extraction method and cDNA prepared using the iScript cDNA kit (Bio-Rad). qRT-PCR analysis was performed using a Bio-Rad SYBR Master Mix and run on a CFX96 Real-Time System/C1000 Touch thermal cycler using Bio-Rad CFX Manager 3.1 software. A table of primers utilized in this study is included in the supplemental material (sTable 1). Fold change was calculated using the 2−ΔΔCt method as described previously (Livak and Schmittgen, 2001). ΔCt values for vehicle-treated animals were averaged within litters for lung inflammation gene assessment, and ΔΔCt values for DEX- and CIC-treated animals were calculated from the average ΔCt values from their respective vehicle-treated littermates. Fold change values were used to compare gene expression between treatment groups.
Statistical analysis
Data analysis was conducted with SAS software (IBM SPSS Statistics 25) or GraphPad statistical software (Prism, version 9). A one-way ANOVA for comparison of multiple groups followed by the appropriate post hoc test as indicated (e.g., Dunnett’s test, Tukey, Bonferroni). If the Brown-Forsythe indicated significantly unequal variance between groups, a nonparametric Kruskal-Wallis test was used (indicated by H statistic), followed by a Dunnett’s post hoc test. For the growth curve, GraphPad version 9 (Prism) was used for two-way ANOVA with repeated measures followed by Bonferonni post hoc at the daily time points. Results are presented as the mean ± SEM.
Results
Tissue specific Ces gene expression in neonatal lung across species exceeds brain expression
To support the feasibility of CIC use in neonates, we evaluated expression levels of CIC-activating enzymes, the Ces family genes (CES in humans, Ces in mouse/rat (Holmes et al., 2010)) by interrogating publicly available genome-wide data sets from multiple species. We assessed expression levels in lung, liver, and brain tissue collected at late embryonic through term neonatal ages to represent the developmental spectrum of human infants at risk for BPD, encompassing gestational ages of premature through term infants. Because CIC hydrolysis and activation are not known to be limited to a specific Ces subgroup (Mutch et al., 2007; Nave and McCracken, 2008) and additional Ces isoenzymes have been identified in mice and rats compared to humans (Holmes et al., 2010; Jones et al., 2013; Lian et al., 2018), we broadly considered all identified Ces isoforms reported.
Analysis of scRNA-Seq datasets from PND1 human lung revealed expression of two CES genes, (i.e., CES1 and CES2) in multiple cell types (sFig. 1) (Du et al., 2015; Du et al., 2017; Guo et al., 2019). Although only a few studies have quantified expression of CES genes in human fetal and neonatal brain and a complete regional inventory is unavailable, CES genes appear to have limited expression in this organ (Keil et al., 2018; Miller et al., 2014) (http://www.brainspan.org/rnaseq/search/index.html http://hbatlas.org/). These observations provide initial support for organ-specific activation of CIC based on CES enzyme expression in human neonates.
Given that mice are frequently used as model organisms for perinatal pathology, we also investigated published single cell scRNA-seq data in late fetal and neonatal mouse lung (Du et al., 2015; Du et al., 2017; Guo et al., 2019) to provide more comprehensive assessments of Ces gene expression, particularly in specific lung cell types. Ces1d, the ortholog of human CES1 (Lian et al., 2018) is the most highly expressed isoform in neonatal mouse lung (sFig. 2A,B) with expression in multiple distinct cell types in the lung (sFig. 2A,B) with Ces2g the only other isoform with expression above background (sFig. 2C,D). Cell type-specific expression of CES1d protein in PND1 and PND5 mouse lung was confirmed by IHC analysis (sFig. 3). To broaden the evaluation of Ces isoform gene expression to other tissues, we interrogated genome-wide bulk RNA-seq data from mice (Darbellay and Necsulea, 2020) (GEO:GSE108348). These analyses revealed relatively high levels of mRNA expression in late embryonic and/or newborn liver of a number of the 18 identified Ces genes (i.e., Ces1c, Ces1d, Ces2a, Ces2e, Ces2g, Ces3a). Contrarily, in the late embryonic or neonatal brain, Ces mRNA expression was limited or undetectable (sTable 2).
We expanded our investigation to rats, because the saccular stage of newborn rat lung development (i.e., E21-PND4) matches the stage of human lung development typically attained in premature infants (i.e., saccular stage between 24-38 gestational weeks of age) (Schittny, 2017). Therefore, the lung developmental stage in newborn rats closely models the premature human lung, an ideal initial paradigm to evaluate localization-specific CIC responses. Consistent with results obtained in transcriptome studies of neonatal mouse tissue, genome-wide microarray analysis of rat lung from various ages (Weng et al., 2006) and RNA-Seq datasets from multiple rat neonatal organs (Darbellay and Necsulea, 2020) revealed only a few isoforms with mRNA expression in neonatal lung and no consistent mRNA expression of any isoform in neonatal brain.
In validation experiments using qRT-PCR, Ces1d and Ces1e were the only isoforms examined with mRNA expression above background in neonatal rat lung and showed differential regulation during the neonatal period. qRT-PCR analysis did not detect Ces2g mRNA above background levels in neonatal rat lung tissue (i.e., PND1, PND2, PND15) (not shown), which contrasts with scRNA-Seq data from mouse neonatal lung (sFig. 2C,D). Specifically, Ces1d (Fig. 1A, F(2,8)=7.363, p=0.015, post hoc*p<0.05, Bonferroni, n=3-4 per group) mRNA expression was significantly higher at later ages (i.e., PND15 versus PND1 and PND2). In contrast, Ces1e mRNA expression peaked at PND2 and was significantly higher than at PND1 and PND15 (Fig. 1A, F(2,8)=9.446, p=0.008, post hoc *p<0.05, Bonferroni, n=3-4 per group). CES1d protein, the highest Ces isoform expressed in rodent neonatal lung (sFig. 2A,B), was present in PND1 and PND5 rat lung as revealed by Western blot analysis but not detected in neonatal brain regions, including the cerebellum and cortex (Fig. 1B). In summary, the wealth of publicly available genome-wide data sets, along with our internal validations, confirms the expression of enzymes primarily responsible for activating CIC in neonatal lung with limited brain expression in humans and rodents.
Figure 1. CES expression in rat tissue.
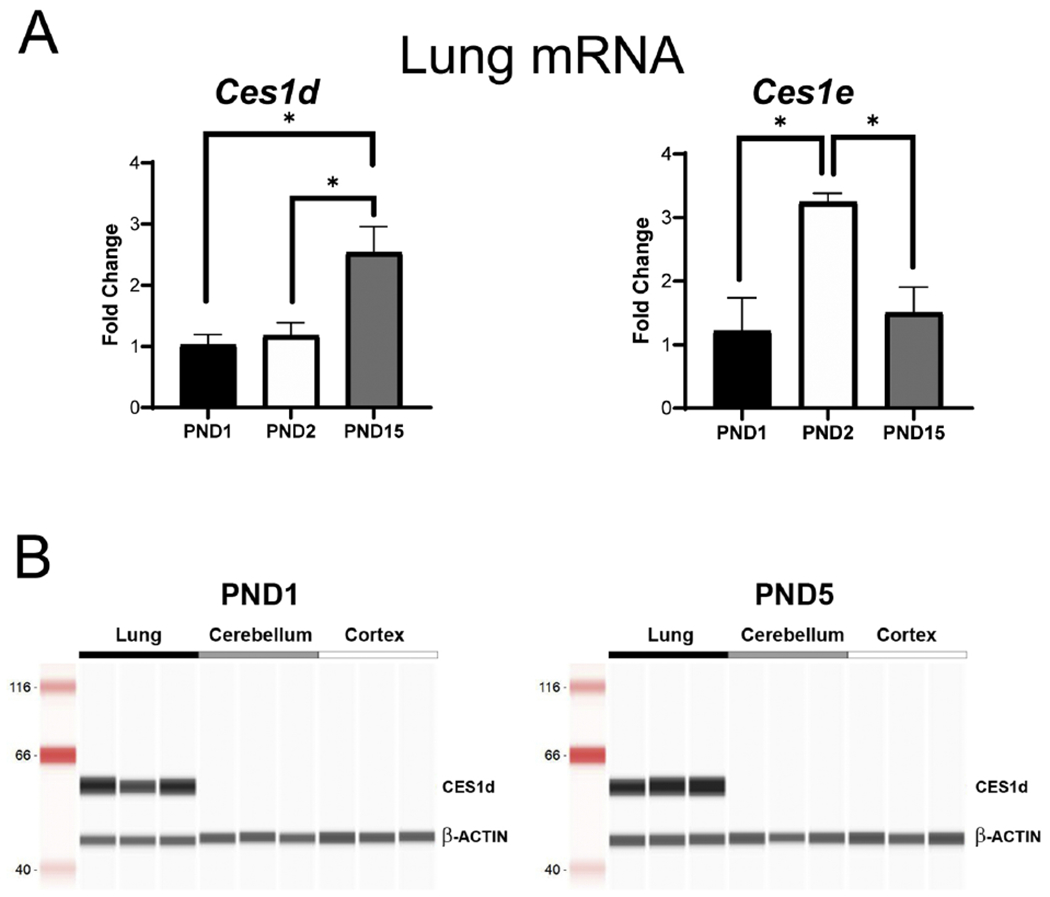
qRT-PCR analysis of (A) Ces1d and Ces1e (A) mRNA expression in lung tissue from neonatal rats collected at PND1, PND2 and PND15. Fold change was calculated compared to expression of ß-actin mRNA levels on PND1 and compared using a one-way ANOVA followed by Bonferroni post hoc analysis (*p<0.05, **p<0.01, n=3-4 per group). (B) Protein expression of CES1d by Simple Western (Protein Simple). Representative immunoblot analysis is shown for CES1d and β-ACTIN in PND1 and PND5 of lung, cerebellum, and cortex. CES1d was highly expressed in lung tissue at all ages with no detectable protein in brain tissue.
DEX but not CIC hinders neonatal growth and reduces brain size
A daily dose of 0.5mg/kg of DEX has previously been shown to generate myelination defects in neonatal rats (Kim et al., 2013). While allometric scaling parameters of therapeutic drug doses to convert between neonatal rats and humans that take into account the unique metabolic parameters of these age groups have not been fully delineated, the Human Equivalent Dose (HED) can be approximated using the FDA-recommended exponent of 0.67 to scale “no-observed-adverse-effect-levels” between species based on surface area and the equation HED = animal dose in mg/kg x (animal weight in kg/human weight in kg)0.33 (Sharma and McNeill, 2009). Assuming an average neonatal rat weight of 10g and human infant weight of 3.5kg, the HED of 0.5mg/kg/d would be approximately 0.07mg/kg/day. This dose is below the AAP recommended maximum “low” DEX dose for BPD (i.e., 0.2mg/kg/day) (Watterberg et al., 2010). However, given that biological effects in rats are observed at 0.5mg/kg and given our goal to seek a minimally effective dose, we evaluated biological effects of this dose for both DEX and CIC. Our studies assume biological equivalence of identical doses of DEX and CIC taking into account the 10-fold higher affinity of des-CIC for GR than DEX (Sato et al., 2007) and a minimum 10% conversion of CIC to des-CIC.
During the course of our treatment paradigm, we observed a distinct phenotype in DEX-treated neonates compared to either vehicle- or CIC-treated animals. Specifically, neonatal rat pups treated with DEX showed significant growth restriction (weight and length), decreased development of fur, as well as thinner and translucent skin as compared to the other groups (Fig. 2A). In addition, DEX-treated pups frequently required supplementation with normal saline during the treatment period to avoid death from dehydration/malnutrition; this complication was not due to inability to nurse, as milk was visualized in the pups’ stomachs regularly throughout the same period, and mothers did not demonstrate neglect of these pups.
Figure 2. Differential effects of DEX and CIC on overall growth and brain size in rat neonates.
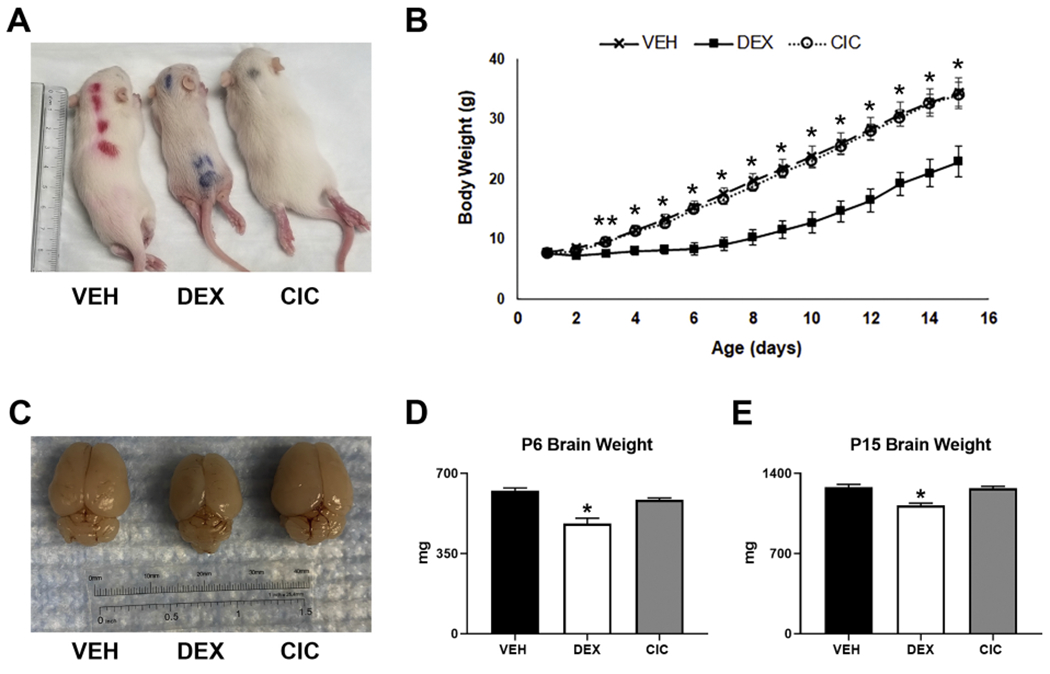
Rats were treated with daily injections with VEH, 0.5mg/kg DEX, or 0.5mg/kg CIC from PND1 - PND5 and allowed to nurse until sacrifice at PND15. Representative images of (A) PND15 pups and (C) whole fixed brains following treatment with VEH (left), DEX (middle) or CIC (right). (B) Growth curves reveal transient neonatal growth restriction following DEX but not CIC treatment. From PND3 to PND15, body weights of CIC and DEX treated animals were significantly different (** p<0.05 two-way repeated measures ANOVA , F(28,252), F=10.75 p<0.0001, Bonferonni post hoc, n=7 per treatment). From PND4 to PND15, the body weights of VEH and DEX treated animals were significantly different (*p<0.02, two-way repeated measures ANOVA , F(28,252), F=10.75, p<0.0001, Bonferonni post hoc, n=7 per treatment). Decreased total brain weight following DEX but not CIC treatment of neonates. DEX treated animals had significantly decreased brain weights than VEH treated at PND6 (panel D, *p=0.005, F(2,27)=23.090, p<0.001, Dunnett’s post hoc, n=3-4 per group) and PND15 (Panel E, *p<0.001, F(2,27)=26.062, p<0.001, Dunnett’s post hoc, n=10 per group). CIC had no significant effect on brain weight.
Weight gain in treated pups was assessed daily from PND1-PND15. As shown in Figure 2B, weight gain in CIC and vehicle treated neonates did not significantly differ throughout the entire period of our study (i.e., PND1-PND15). However, DEX treated neonates exhibited slower weight gain during the 5-day treatment period, a differential which persisted even after completion of treatment up to PND15. From PND10 – PND15, all groups gained approximately 2g daily regardless of treatment. From PND4 to PND15, DEX treated animals were significantly smaller than both VEH and CIC treated animals and were additionally significantly smaller than CIC treated animals on PND3 (Fig. 2B).
Gross examination of brains collected from PND15 demonstrated an overall smaller size and less developed cortex and cerebellum (representative fixed brains shown in Fig. 2C). Total brain weight in DEX treated animals was significantly decreased at PND6 and PND15 (Figs. 2D,E). There was no significant difference between vehicle- and CIC-treated animals in terms of total brain weight, but by PND6, brain weight was decreased by 23% in DEX-treated animals compared to VEH (Fig. 2D); and by 12% at PND15 (Fig. 2E) Therefore, CIC does not generate the systemic adverse effects in somatic and brain growth as triggered by the potent GR agonist, DEX.
Transient neonatal DEX but not CIC reduces MBP accumulation in the cortex
Use of sGCs in human neonates has been linked to a high risk for white matter injury, a serious adverse effect that limits widespread use, despite their potential beneficial anti-inflammatory actions for BPD treatment (Watterberg et al., 2010). In neonatal rodents, myelination initiates after birth in a caudal to rostral manner and continues into adulthood (Davidson and Berkelhamer, 2017; Rutkowska et al., 2019). The young postnatal rodent is consequently sensitive to white matter loss following transient exposure to sGCs (Kim et al., 2013). Therefore, CIC and DEX effects on white matter were evaluated in the forebrain at PND15, when significant myelinated fibers are present in the caudate putamen, globus pallidus, internal capsule, and corpus callosum. IHC analysis demonstrated a notable reduction in MBP expression after DEX exposure in the corpus callosum (Fig. 3B) and the striatum (Fig. 3E), a phenotype which was not triggered after CIC exposure (Figs. 3C, F). Conversely, the anterior commissure appeared less susceptible to DEX driven effects (Figs. 3G–I). The reduction in IHC immunoreactivity was quantified with western blot analysis of MBP in cortex/corpus callosum tissue. Specifically, DEX treatment resulted in a decrease of MBP expression by 62% to that in the VEH animals. CIC did not significantly decrease MBP expression in cortex/corpus callosum (Figs. 3J, K). In summary, IHC and western blot analysis of MBP corroborate the detrimental effects of the GR agonist, DEX, on white matter development during the rodent neonatal period (Kim et al., 2013) but reveal no significant effect in neonates exposed to the GR prodrug CIC.
Figure 3. Differential effects of DEX and CIC on myelination in the forebrain of neonatal rats.
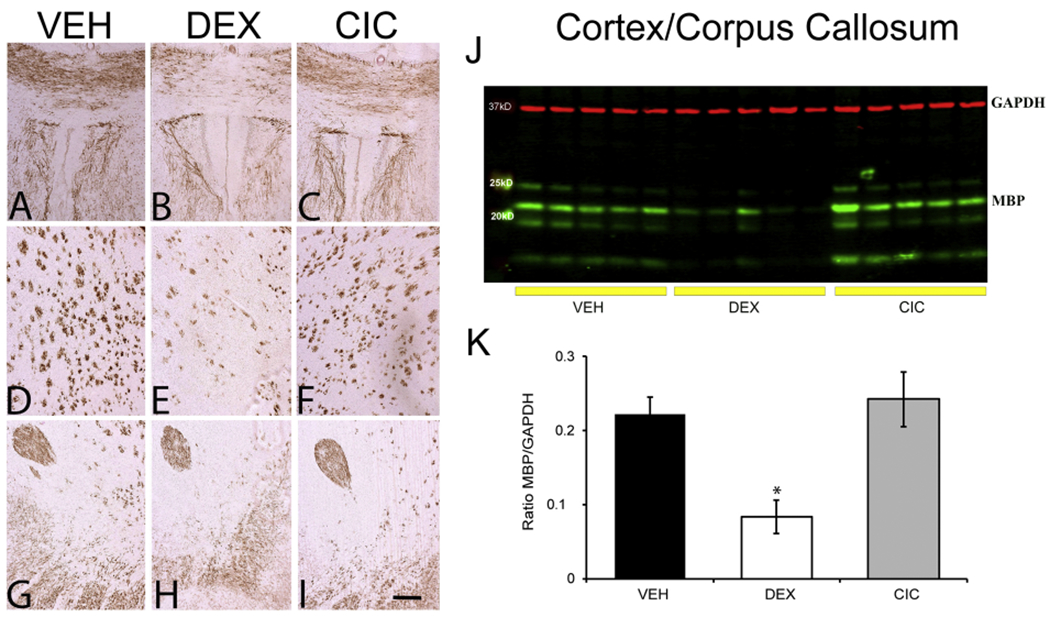
Brains were collected on PND15 from rats that were given daily injections of DEX, CIC or VEH from PND1-PND5. IHC of MBP in PND15 brains from animals treated with VEH (A,D,G), DEX (B, E, H) and CIC (C, F, I). Representative photomicrograph of the corpus callosum (panels A-C), striatum (panels D-F) and anterior commissure (panels G-I) (are shown (Scale bar, 100 μm). (J) Representative Western blot of cortex/corpus callosum tissue lysate of PND15. (K) Relative levels of MBP protein levels to loading control GAPDH. DEX significantly decreased MBP as compared to VEH and CIC (*p <0.05, F(2,12)=9.403, p=0.003, Tukey post hoc, n=5 per group). CIC did not significantly affect MBP expression compared to VEH.
Transient neonatal DEX but not CIC reduces cerebellar size
Previous studies have established that exposure to DEX in the neonatal period can lead to neurodevelopmental consequences, specifically with targeted effects in the cerebellum that present as an overall decrease in cerebellar size (Heine and Rowitch, 2009; Noguchi, 2014). After our treatment paradigm of exposures from PND1-5, we assessed cerebellar anatomy on PND15 and confirmed that our model replicated the established DEX-mediated diminutive phenotype (Fig. 4B) as compared to VEH (Fig. 4A). We also evaluated the PND15 brain after CIC treatment, which did not appear to result in decreased size (Fig. 4C). Examination of midline sections by fluorescent Nissl staining at the level of the cerebellar vermis highlights the overall decrease in size noted after exposure to DEX (Fig. 4E) but not CIC (Fig. 4F) compared to VEH (Fig. 4D). In summary, the adverse effects of neonatal DEX exposure on both the forebrain and cerebellum are not observed with CIC.
Figure 4. Differential effects of DEX and CIC on cerebellar size and foliation in neonatal rats.
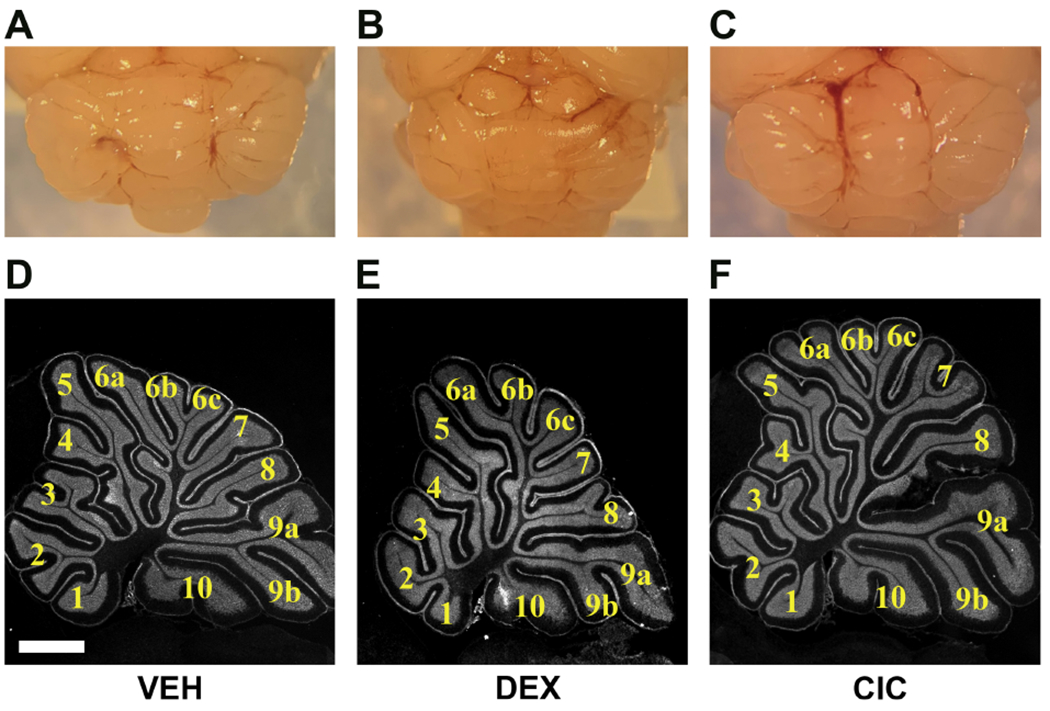
Neonatal rats were treated with daily injections of VEH (A,D),0.5mg/kg DEX (B,E), or 0.5mg/kg CIC (C,F) daily from PND1 - PND5 and brain tissue was collected on PND15. Representative gross images of cerebella from VEH (A), DEX (B), and CIC (C) treated rats showing the smaller size of the cerebellum in the DEX-treated animal compared to VEH and CIC. Representative photomicrographs of cerebellum from VEH (D), DEX (E), and CIC (F) treated rats along midline through the vermis stained with fluorescent Nissl. Three brains were examined per treatment.
CIC and DEX increase site specific GR phosphorylation in neonatal lung
In addition to minimizing systemic and neurologic adverse effects, CIC viability as an alternative to DEX requires activation of GR in neonatal lung. In order to provide a broad assessment of GR activation in neonatal lung, agonist-regulated site-specific phosphorylation of GR was measured. GR can undergo transient phosphorylation at multiple sites (Galliher-Beckley and Cidlowski, 2009), and conserved serine residues at position 232 in rat (S232, S211 in human) and 246 (S246, S226 in human) exhibit increased phosphorylation following agonist binding to the receptor. Phosphorylation status at each of these sites can impact gene-specific transcriptional responses (Chen et al., 2008; Galliher-Beckley and Cidlowski, 2009; Peffer et al., 2014), though the kinetics and magnitude of site-specific GR phosphorylation varies in different cell types (Lynch et al., 2010).
As shown in Figures 5A and 5B, a 4-hour DEX or CIC treatment at PND1 triggered an increase in GR phosphorylation at S232 in lung while DEX also induced S246 phosphorylation, demonstrating that CIC has the capacity to bind to and activate GR in neonatal lung. These hormone-induced phosphorylation events were transient and not observed at 24 post-treatment (Fig. 5C). Increased rates of phosphorylated GR were not due to increases in total steady state levels of intact GR (i.e., 95kDa), for which no significant differences were seen at either 4- or 24-hours post-treatment (Figs. 5B,C). However, 4 hours after CIC treatment, a significant reduction in the 55kDa GR isoform (presumedly representing the previously characterized translational “D” isoform (Lu and Cidlowski, 2005)) was observed (Fig. 5B), but were not significantly different by 24 hours of treatment (Fig. 5C). Thus, CIC has the capacity to activate agonist-specific changes in GR phosphorylation in neonatal lung.
Figure 5. Impact of DEX and CIC on GR protein expression and phosphorylation in neonatal rat lung.
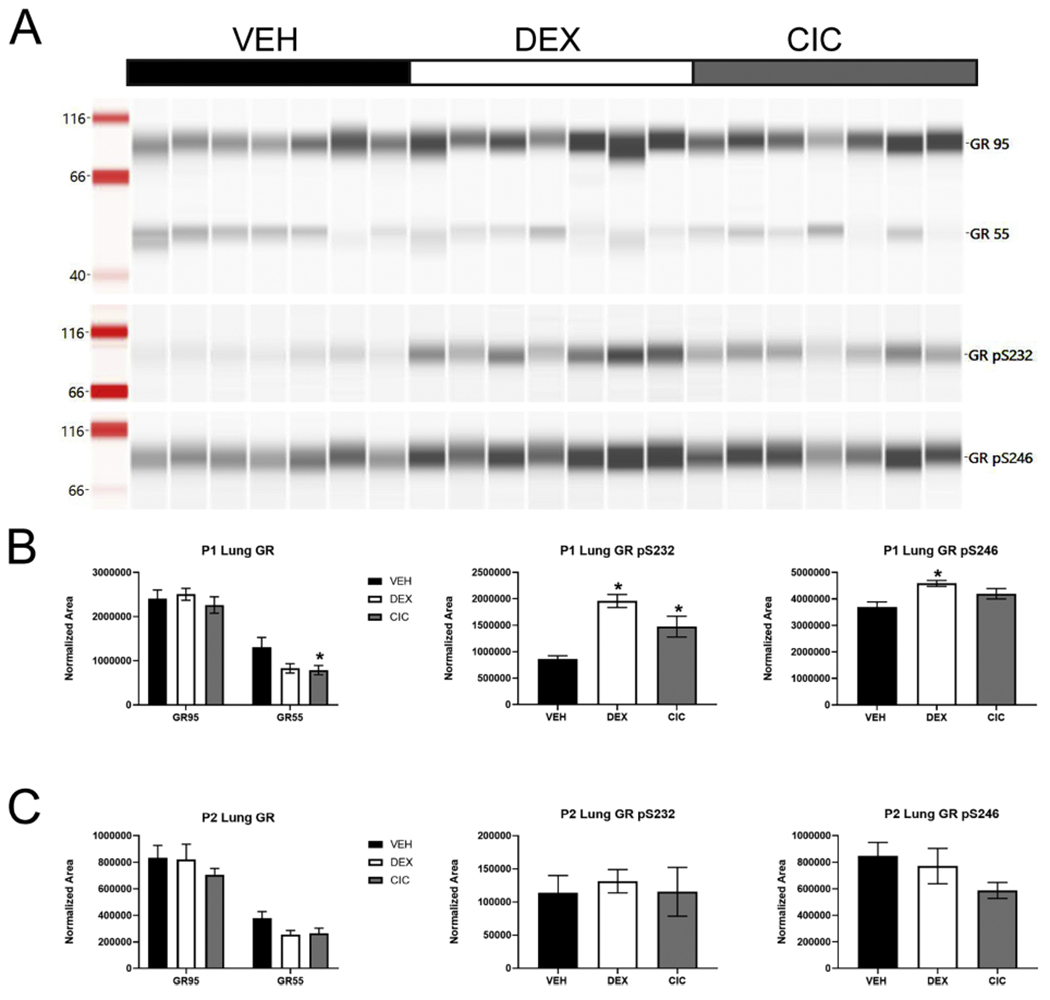
Rats were treated with VEH, 0.5mg/kg DEX, or 0.5mg/kg CIC on PND1 and lung tissue lysates prepared following 4 or 24 hours. (A) Representative Simple Western of PND1 lung tissue following 4-hour treatment to detect total full length 95kDa GR, a 55kDa GR isoform, and GR phosphorylated at Ser 232 (pSer232) or Ser246 (pSer246). (B) Amount of full-length (GR95), 55kDa (GR55), pSer232 or pSer246 isoforms of GR normalized to total protein with Jess System software in lung following 4-hour treatment initiated at PND1. Neither DEX nor CIC affected full length GR. CIC significantly decreased GR55 levels (*p<0.05, F(2,18)=3.567, p=0.05, Dunnett’s post hoc, n=7 per group) while there was a trend for GR55 levels to be decreased following DEX treatment. Conversely DEX increased pS232 ( * p<0.05, F(2,18)=15.79, p=0.0001, Dunnett’s post hoc, n=7 per group) and pS246 (* p<0.05, F(2,18)=6.836, p=0.0062, Dunnett’s post hoc, n=7 per group), while CIC significantly increased pS232 but not pS246. (C) None of the changes in GR55 or phospho-isoforms were observed 24 hours following DEX or CIC treatment.
CIC and DEX mobilize lung-protective transcriptional responses in the neonate
To evaluate if CIC-induced GR activation led to a similar transcriptional induction or suppression of GR-regulated genes, qRT-PCR analysis was performed after VEH, DEX, or CIC treatment in PND1 rats. In order to compare target organ effects in the lung to unintended off-site effects in the brain, we first evaluated a variety of GR target genes that are broadly expressed in multiple organs (e.g., Per1, Gilz, Fkbp5), and which have been previously characterized as widely responsive to sGC exposure by our lab and others (Al-Safadi et al., 2015; Binder, 2009; Peffer et al., 2014; Srinivasan and Lahiri, 2017). Per1, or PERIOD1, is a GC-responsive circadian clock protein that is increases after exogenous or endogenous hormone exposure (Al-Safadi et al., 2015). Gilz, or glucocorticoid induced leucine zipper, is hypothesized to have anti-inflammatory and/or neurogenic functions, and also is induced by GC exposure (Srinivasan and Lahiri, 2017). Fkbp5 (FK506 binding protein 5) is a gene induced by GC exposure and functions in a negative feedback mechanism to desensitize the GR response (Binder, 2009).
At 4 hours post-treatment, both DEX- and CIC- treatment induced the expression of Per1 and Gilz mRNA in lung compared to VEH (Figs. 6A,B), though Fkbp5 was only significantly induced by DEX at 4 hours despite an increasing trend after CIC exposure (Fig. 6C). At 24 hours, Gilz and Fkbp5 continued to be significantly induced by DEX, but not CIC (Figs. 6A–C). These results demonstrate that CIC has the capability to mobilize transcription of GR-regulated genes, although the duration of gene specific responses may differ between CIC and DEX. Furthermore, although DEX significantly induced Gilz at 24 hours and Fkbp5 at 4 hours in the cerebellum, none of these genes were significantly induced by CIC in the cerebellum after 4 or 24 hours (Figs. 6D–F).
Figure 6.
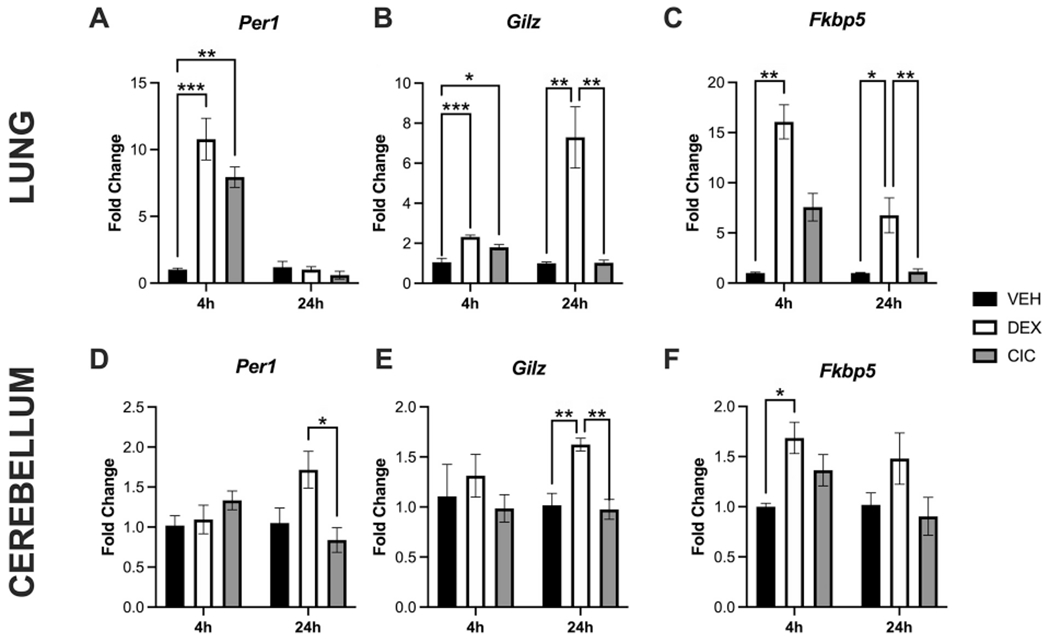
Activation of select GR target genes in neonatal lung and brain following DEX or CIC treatment. PND1 rat pups were treated with 0.5mg/kg CIC, 0.5mg/kg DEX, or VEH, and tissues collected at 4 or 24 hours post injections. Fold change of Per1 (A,D), Gilz (B,E) and Fkbp5 (C,F) mRNA were measured (normalized to ß-actin mRNA levels in VEH-treated rats) by qRT-PCR. Data were analyzed using a one-way ANOVA followed by a post hoc Bonferroni’s multiple comparisons test unless groups were determined to have significantly unequal variance, when a nonparametric Kruskal-Wallis test (H statistic) was used followed by a Dunnett’s post hoc test. Post hoc test significance is annotated as follows: *p<0.05, **p<0.01, ***p<0.001. In lung, 4 hours after treatment on PND1, significant induction of Per1 (F(2,9)=24.92, p=0.0002) and Gilz (F(2,9)=19.56, p=0.0005) mRNA was observed with both DEX and CIC (A,B). In contrast, Fkbp5 was only induced by DEX at 4 (H(2) =9.846, exact p=0.0002) (C) and 24 hours (H(2)=13.07, approximate p=0.0015). At 24 hours in lung, Gilz continued to be significantly induced by DEX (F(2,9)=16.73, p=0.0009) (B). None of these genes were induced by CIC in the cerebellum after 4 or 24 hours (D-F). DEX significantly induced Gilz at 24 hours (F2,9)=14.24, p=0.0016) and Fkbp5 at 4 hours (F(2,9)=7.047, p=0.0144) in the cerebellum (E,F) (n=4 for all groups except lung Fkbp5 expression at 24 hours for which n=7-8, and cerebellar expression of Gilz at 24 hours n=4-5).
Given the ability of CIC to induce transcription of broadly expressed GR target genes in neonatal lung, we then examined effects of CIC treatment on the expression of GR-regulated genes whose response is likely to contribute to the therapeutic effects of sGC in BPD. These genes were assessed 24 hours following treatment to evaluate the viability of CIC to exert enduring effects on putative lung-protective genes. As shown in Fig. 7, basal mRNA expression of a number of pro-inflammatory genes in neonatal lungs was repressed to the same extent 24 hours following DEX or CIC treatment of PND1 pups. The GR target genes repressed by DEX and CIC included Icam1, Il-8 and Tnfα (Figs. 7A–C). DEX and CIC treatment also induced genes that promote angiogenesis (i.e., Vegf, Dll4, Angpt2) (Figs. 7D–F) or alveolar type II and type I epithelial cell function (i.e.,Spc, Aqp5) (Figs. 7G,H) in neonatal lung. Specific differences in the response of these genes were noted for Angpt2 (Fig. 7F), which was significantly induced by CIC but not DEX, whereas Spc achieved a statistically significant induction following DEX but not CIC treatment (Fig. 7G).
Figure 7. CIC and DEX mobilize lung-protective transcriptional responses in the neonate.
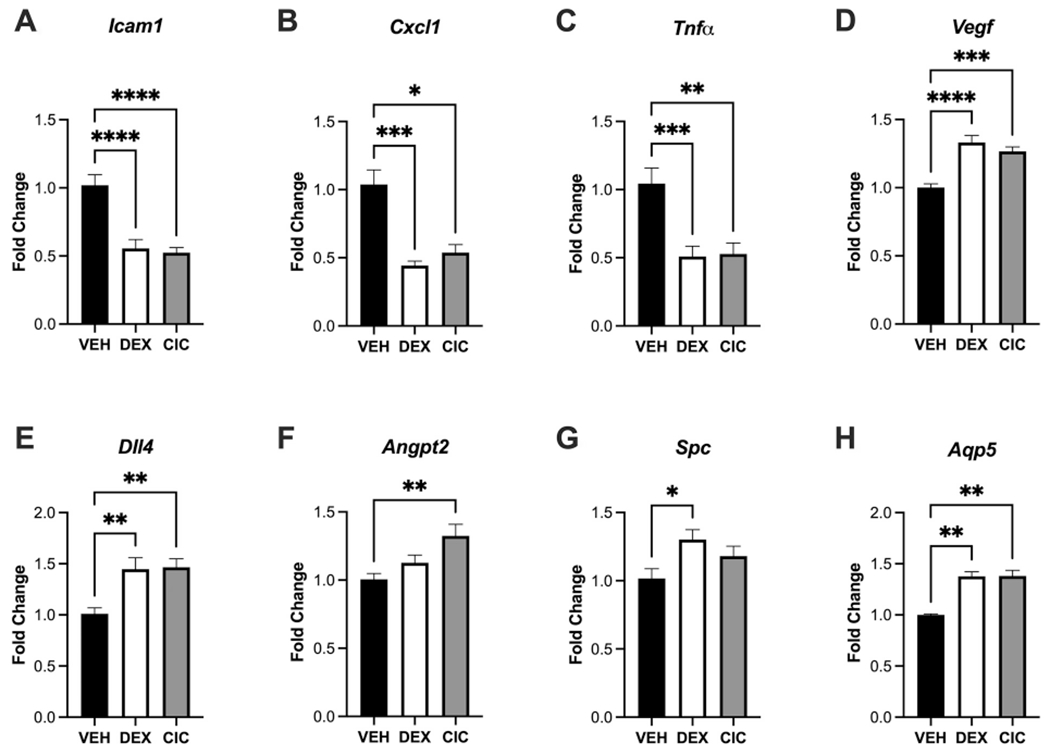
24 hours after treatment of PND1 rat pups with 0.5mg/kg DEX, CIC, or VEH, lung tissue was collected and subjected to qRT-PCR analysis of genes associated with protective and anti-inflammatory responses in the lung. All data were analyzed with a one-way ANOVA followed by a Dunnett’s post-hoc test with *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, n=8/treatment group for all analyses. Both DEX and CIC significantly repressed expression compared to VEH of Icam1 (A), Cxcl1 (B), and Tnfα (C). DEX and CIC treatment both induced pro-angiogenic Vegf (D) and Dll4 (E), although Angpt2 was only significantly induced by CIC (F). Neonatal lung markers of alveolar type II and type I epithelial cell populations were induced by DEX (G,H) although CIC induced Aqp5 (H) but not Spc (G).
In summary, qRT-PCR results show that CIC has the capability to induce changes in expression in both widely expressed and functionally relevant GR-regulated genes, though individual gene responses compared to DEX are not identical. Specifically, while CIC transcription induction of the ubiquitously responsive GR targets in lungs noted at 4 hours did not seem to persist as long as after DEX exposure, significant changes in transcription of lung genes associated with BPD pathology were still observed 24 hours post-exposure. Additionally, CIC treatment does not appear to lead to significant mobilization of GR responses in the brain, reinforcing that the unique pharmacokinetics and pharmacodynamics of CIC may promote organ-specific GC action that spares the developing brain.
Discussion
Significant evidence has accumulated indicating that perinatal sGC exposure leads to adverse consequences on neonatal brain development. Perinatal administration of DEX to infants at risk for BPD has been associated with abnormal brain development in both rodents and humans (Heine and Rowitch, 2009; Kim et al., 2013; Noguchi, 2014). Studies in rodents have shown that DEX leads to a reduction in myelination by targeting oligodendrocyte progenitor cells (Cheong et al., 2014), which may provide some explanation as to why in the neonatal period, the developing brain is particularly vulnerable to white matter injury. Consistent with these findings, in both human and animal models, neonatal DEX exposure is associated with exacerbation of white matter injury with increased risk for cerebral palsy and motor impairments (Kalikkot Thekkeveedu et al., 2017), as well as aberrant synaptic plasticity in the hippocampus and memory deficits (Kalikkot Thekkeveedu et al., 2017). Our findings in neonatal rats after DEX exposure recapitulate the documented reductions in MBP observed in the corpus callosum, caudate, putamen, and globus pallidum (Kim et al., 2013). Similar alterations in neonatal myelination were not observed after exposure to CIC.
Furthermore, in neonatal rats, DEX administration caused a characteristic and robust phenotype of skin and fur abnormalities, somatic growth restriction, and decreased brain weight that was notably absent in the CIC-treated pups. DEX suppresses insulin growth factor 2 (IGF-2) levels to values similar to those found in Igf-2 knockout mice (Pew et al., 2016), which provides a plausible mechanism for reduced neonatal growth we observed in DEX but not CIC treated rats. Unlike in older animals, the neonatal period in mammals including rat (Levine, 2002) is designated as a stress hyporesponsive period, as levels of circulating GCs are relatively low and they do not consistently respond to circadian cues or pituitary-derived adrenocorticotropic hormone. Negative feedback of the hypothalamic/pituitary/adrenal (HPA) axis by natural or synthetic GCs is also not developed in the neonate (Levine, 2002). Therefore, is it unlikely that endogenous hormone dysregulation from HPA feedback is driving the well-established detrimental systemic and neurologic effects of DEX administration.
The SC delivery of CIC employed in our studies, while shown to allow for systemic distribution of des-CIC (Mars et al., 2013) was used for practical reasons given the technical difficulties in administering drugs intravenously (IV) or by inhalation (INH) in a neonatal rat pup. However, nebulization is one option for administering CIC to patients with BPD where local delivery could further enhance its therapeutic targeting to lung and limit adverse sGC side effects that are particularly concerning for the neonate.
Despite utilizing this systemic administration route in our model, we still did not see significant adverse neurologic effects in CIC-treated neonates. A major contributor to this pattern of effects is likely reflective of the site-specific generation of des-CIC in the lung, and thus provides support that CIC is unlikely to be ectopically activated in the brain. The highly protein-bound des-CIC (Nave et al., 2004; Nonaka et al., 2007) may also have a limited ability to cross the blood-brain barrier (BBB) in sufficient quantity prior to metabolic breakdown, although we cannot exclude the possibility that CIC or systemically generated des-CIC could cross the immature neonatal BBB of neonates or induce indirect effects on neonatal brain function. More extensive CIC dosing regimens and comprehensive phenotypic, neuroanatomical and transcriptome analysis in brain and lung could identify an optimal dosing strategy for treating lung injury in neonates but limit adverse neurodevelopmental effects.
When considering the potential for CIC, as a GR prodrug, to treat neonatal human lung disease characterizing its ability to activate GR in newborn lung is crucial for establishing viability of a novel safe treatment approach. GR activation can in turn advance maturation and reduce inflammation, improving respiratory outcomes. Our studies in neonatal rats suggest that CIC may have the potential to fulfill these requirements, as CIC was capable of rapidly activating many GC-responsive genes in neonatal lung to a similar degree as DEX. In addition, CIC can exert a long-lasting effect to reduce the baseline expression of cytokine gene products that contribute to inflammatory lung injury as well as increase the expression of genes that promote angiogenesis and epithelial cell function. The ability of CIC to likewise trigger the analogous site-specific phosphorylation of the GR suggests that phospho-GR specific genome-wide transcriptional responses in lung may demonstrate some overlap between DEX and CIC treatments. However, subtle differences in site-specific GR phosphorylation and translational isoform regulation suggest that there may be some GR targets that exhibit differential responses to DEX versus CIC in neonatal lung that impact downstream effects on respiratory outcome measures (Chen et al., 2008; Galliher-Beckley and Cidlowski, 2009; Oakley et al., 2018; Peffer et al., 2014). Furthermore, Ces gene expression has the potential to vary in response to physiologic state. For example, Ces isoform expression was altered in macrophages of high fat diet-fed mice exposed to lipopolysaccharide (Jones et al., 2013), suggesting that an inflammatory disease state could in turn affect Ces substrate metabolism. While a comprehensive assessment of the individual relative abilities of CES enzymes to hydrolyze CIC has not yet been performed, substrate specificity has been documented with various CES enzymes (Wang et al., 2018). Thus, the therapeutic potential of CIC as a sGC of choice in pediatric patients will need to take into account potential pathologic effects on CES enzyme expression and/or activity.
Given our initial promising data, future studies will move to evaluate CIC within the context of relevant injury and/or prematurity models of BPD in rodents and further characterize its effectiveness as a potential therapeutic (Lesage et al., 2018). As pathologic inflammation and lung remodeling processes are activated in BPD, a comprehensive characterization of CIC-mediated GR activation effects in neonatal lung injury models will provide critical contextual effectiveness data and promote timely translation into clinical studies. Developing a better understanding of the scope of action and PK of CIC may also allow for extrapolation of its use into additional scenarios where established sGC treatments carry associated neurodevelopmental risks (Damsted et al., 2011; Davis et al., 2013; Ilg et al., 2019; McGowan and Matthews, 2018).
In summary, our results provide the impetus to continue to evaluate CES -metabolized GR prodrugs as a novel pharmacotherapy to mitigate lung disease in human neonates, where use of currently available sGCs, is limited due to the potential for severe adverse neurological and systemic side effects. The potential therapeutic use of CIC may not be limited to asthma and BPD. For example, CIC has both anti-inflammatory and anti-viral properties against the SARS CoV-2 virus (Jeon et al., 2020; Matsuyama et al., 2020; Mori et al., 2021). Since sGCs such as DEX administered to pregnant women in pre-term labor can adversely affect fetal brain development, the neurological-sparing properties of CIC, make it an attractive alternative for DEX to treat pregnant women severely ill with respiratory illness, such as with asthma exacerbations or COVID-19 infections (Delahoy et al., 2020; Ellington et al., 2020).
Supplementary Material
Relative expression of human CES-1 (A) and CES-2 (B) mRNA in various cell types of neonatal (PND 1) human lung extracted from publicly available scRNA-seq data set (Keil et al., 2018). Average expression shown in transcripts per kilobase per million mapped reads (TPM).
Relative expression and cellular distribution of mouse Ces1d (A, B) and Ces2g (C, D) mRNA expression in mouse lung extracted from scRNA-seq data set (Du et al., 2015; Du et al., 2017; Guo et al., 2019). Average expression shown as reads per kilobase of transcript per million mapped reads (RPKM).
IHC analysis for CES1d in PND1 (A, B) and PND5 (C,D) mouse lung tissue .(E, F) no primary control staining at PND1. Low power, 40X and high power, 100 X (E, F) images are shown following staining with CES1d specific antibodies.
Carboxylesterase (Ces) gene mRNA expression in perinatal mouse brain and liver extracted from scRNA-Seq data set (Darbellay and Necsulea, 2020).
Primer sequences utilized in qRT-PCR within the study. All sequences represent 5’ → 3’ orientation.
Highlights.
Rodent and human neonatal lung, but not brain, express specific carboxylesterases
Ciclesonide is metabolized to a glucocorticoid receptor agonist by carboxylesterases
Ciclesonide does not generate adverse systemic or brain effects in neonatal rats
Ciclesonide regulates multiple lung protective genes in neonatal rats
ACKNOWLEDGEMENTS
Ross Carson is thanked for technical assistance and Uma Chandran and Andrew Chang for assistance with mining GEO data sets.
Support:
This work was funded by the National Institutes of Health grants R01 HD087288 to DBD and APM, R21 HD097694 to DBD, R01 HL128374 to VS, K12 HD052892 career development award to ALF. EMB was supported by a Magee-Women’s Research Institute Clinical Trainee Research Award and TJC by Australian National Health & Research Council Ideas Research Grant 1185813.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interests: None
References
- Al-Safadi S, et al. , 2015. Glucocorticoids and Stress-Induced Changes in the Expression of PERIOD1 in the Rat Forebrain. PLoS One. 10, e0130085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassler D, van den Anker J, 2017. Inhaled Drugs and Systemic Corticosteroids for Bronchopulmonary Dysplasia. Pediatr Clin North Am. 64, 1355–1367. [DOI] [PubMed] [Google Scholar]
- Bhatt AJ, et al. , 2013. Dexamethasone induces apoptosis of progenitor cells in the subventricular zone and dentate gyrus of developing rat brain. J Neurosci Res. 91, 1191–202. [DOI] [PubMed] [Google Scholar]
- Binder EB, 2009. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. 34 Suppl 1, S186–95. [DOI] [PubMed] [Google Scholar]
- Bird AD, et al. , 2014. Mesenchymal glucocorticoid receptor regulates the development of multiple cell layers of the mouse lung. Am J Respir Cell Mol Biol. 50, 419–28. [DOI] [PubMed] [Google Scholar]
- Brand PL, et al. , 2011. Ciclesonide in wheezy preschool children with a positive asthma predictive index or atopy. Respir Med. 105, 1588–95. [DOI] [PubMed] [Google Scholar]
- Chen W, et al. , 2008. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol. 22, 1754–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong JL, et al. , 2014. Association between postnatal dexamethasone for treatment of bronchopulmonary dysplasia and brain volumes at adolescence in infants born very preterm. J Pediatr. 164, 737–743 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley-Yates PT, 2015. Inhaled corticosteroids: potency, dose equivalence and therapeutic index. Br J Clin Pharmacol. 80, 372–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damsted SK, et al. , 2011. Exogenous glucocorticoids and adverse cerebral effects in children. Eur J Paediatr Neurol. 15, 465–77. [DOI] [PubMed] [Google Scholar]
- Darbellay F, Necsulea A, 2020. Comparative Transcriptomics Analyses across Species, Organs, and Developmental Stages Reveal Functionally Constrained IncRNAs. Mol Biol Evol. 37, 240–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson LM, Berkelhamer SK, 2017. Bronchopulmonary Dysplasia: Chronic Lung Disease of Infancy and Long-Term Pulmonary Outcomes. J Clin Med. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis EP, et al. , 2013. Fetal glucocorticoid exposure is associated with preadolescent brain development. Biol Psychiatry. 74, 647–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kloet ER, et al. , 2014. Context modulates outcome of perinatal glucocorticoid action in the brain. Front Endocrinol (Lausanne). 5, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delahoy MJ, et al. , 2020. Characteristics and Maternal and Birth Outcomes of Hospitalized Pregnant Women with Laboratory-Confirmed COVID-19 - COVID-NET, 13 States, March 1-August 22, 2020. MMWR Morb Mortal Wkly Rep. 69, 1347–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, et al. , 2015. ‘LungGENS’: a web-based tool for mapping single-cell gene expression in the developing lung. Thorax. 70, 1092–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, et al. , 2017. Lung Gene Expression Analysis (LGEA): an integrative web portal for comprehensive gene expression data analysis in lung development. Thorax. 72, 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellington S, et al. , 2020. Characteristics of Women of Reproductive Age with Laboratory-Confirmed SARS-CoV-2 Infection by Pregnancy Status - United States, January 22-June 7, 2020. MMWR Morb Mortal Wkly Rep. 69, 769–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, et al. , 2009. Dexamethasone induces neurodegeneration but also up-regulates vascular endothelial growth factor A in neonatal rat brains. Neuroscience. 158, 823–32. [DOI] [PubMed] [Google Scholar]
- Galliher-Beckley AJ, Cidlowski JA, 2009. Emerging roles of glucocorticoid receptor phosphorylation in modulating glucocorticoid hormone action in health and disease. IUBMB Life. 61, 979–86. [DOI] [PubMed] [Google Scholar]
- Guo M, et al. , 2019. Single cell RNA analysis identifies cellular heterogeneity and adaptive responses of the lung at birth. Nat Commun. 10, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine VM, Rowitch DH, 2009. Hedgehog signaling has a protective effect in glucocorticoid-induced mouse neonatal brain injury through an 11betaHSD2-dependent mechanism. J Clin Invest. 119, 267–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes RS, et al. , 2010. Recommended nomenclature for five mammalian carboxylesterase gene families: human, mouse, and rat genes and proteins. Mamm Genome. 21,427–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilg L, et al. , 2019. Persistent Effects of Antenatal Synthetic Glucocorticoids on Endocrine Stress Reactivity From Childhood to Adolescence. J Clin Endocrinol Metab. 104, 827–834. [DOI] [PubMed] [Google Scholar]
- Jeon S, et al. , 2020. Identification of Antiviral Drug Candidates against SARS-CoV-2 from FDA-Approved Drugs. Antimicrob Agents Chemother. 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RD, et al. , 2013. Carboxylesterases are uniquely expressed among tissues and regulated by nuclear hormone receptors in the mouse. Drug Metab Dispos. 41,40–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalikkot Thekkeveedu R, et al. , 2017. Bronchopulmonary dysplasia: A review of pathogenesis and pathophysiology. Respir Med. 132, 170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman DB, et al. , 1991. Beneficial and detrimental effects of RBC-adsorbed antilymphocyte globulin and prednisone on purified canine islet autograft and allograft function. Transplantation. 51,37–42. [DOI] [PubMed] [Google Scholar]
- Keil JM, et al. , 2018. Brain Transcriptome Databases: A User’s Guide. J Neurosci. 38, 2399–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, et al. , 2013. Administration of dexamethasone to neonatal rats induces hypomyelination and changes in the morphology of oligodendrocyte precursors. Comp Med. 63, 48–54. [PMC free article] [PubMed] [Google Scholar]
- Kraft KE, et al. , 2019. Functional outcome at school age of preterm-born children treated with low-dose dexamethasone in infancy. Early Hum Dev. 129, 16–22. [DOI] [PubMed] [Google Scholar]
- Lee HJ, et al. , 2012. Effects of postnatal dexamethasone or hydrocortisone in a rat model of antenatal lipopolysaccharide and neonatal hyperoxia exposure. J Korean Med Sci. 27, 395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeFlore JL, Engle WD, 2011. Growth and neurodevelopment in extremely low-birth-weight neonates exposed to postnatal steroid therapy. Am J Perinatol. 28, 635–42. [DOI] [PubMed] [Google Scholar]
- Lesage F, et al. , 2018. Preclinical evaluation of cell-based strategies to prevent or treat bronchopulmonary dysplasia in animal models: a systematic review. J Matern Fetal Neonatal Med. 31, 958–966. [DOI] [PubMed] [Google Scholar]
- Levine S, 2002. Regulation of the hypothalamic-pituitary-adrenal axis in the neonatal rat: the role of maternal behavior. Neurotox Res. 4, 557–564. [DOI] [PubMed] [Google Scholar]
- Lian J, et al. , 2018. Carboxylesterases in lipid metabolism: from mouse to human. Protein Cell. 9, 178–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddell BS, et al. , 2017. Inhaled corticosteroid related adrenal suppression detected by poor growth and reversed with ciclesonide. J Asthma. 54, 99–104. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD, 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 25, 402–8. [DOI] [PubMed] [Google Scholar]
- Lu NZ, Cidlowski JA, 2005. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell. 18, 331–42. [DOI] [PubMed] [Google Scholar]
- Lynch JT, et al. , 2010. The role of glucocorticoid receptor phosphorylation in Mcl-1 and NOXA gene expression. Mol Cancer. 9, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglione M, et al. , 2018. New Drugs for Pediatric Asthma. Front Pediatr. 6, 432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaeb SN, Stonestreet BS, 2014. Steroids and injury to the developing brain: net harm or net benefit? Clin Perinatol. 41, 191–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mars U, et al. , 2013. Tissue accumulation kinetics of ciclesonide-active metabolite and budesonide in mice. Basic Clin Pharmacol Toxicol. 112, 401–11. [DOI] [PubMed] [Google Scholar]
- Matsuyama S, et al. , 2020. The Inhaled Steroid Ciclesonide Blocks SARS-CoV-2 RNA Replication by Targeting the Viral Replication-Transcription Complex in Cultured Cells. J Virol. 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan PO, Matthews SG, 2018. Prenatal Stress, Glucocorticoids, and Developmental Programming of the Stress Response. Endocrinology. 159, 69–82. [DOI] [PubMed] [Google Scholar]
- Miller JA, et al. , 2014. Transcriptional landscape of the prenatal human brain. Nature. 508, 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori N, et al. , 2021. Triple therapy with hydroxychloroquine, azithromycin, and ciclesonide for COVID-19 pneumonia. J Microbiol Immunol Infect. 54, 109–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutch E, et al. , 2007. The role of esterases in the metabolism of ciclesonide to desisobutyryl-ciclesonide in human tissue. Biochem Pharmacol. 73, 1657–64. [DOI] [PubMed] [Google Scholar]
- Nave R, et al. , 2004. Pharmacokinetics of [14C]ciclesonide after oral and intravenous administration to healthy subjects. Clin Pharmacokinet. 43, 479–86. [DOI] [PubMed] [Google Scholar]
- Nave R, McCracken N, 2008. Metabolism of ciclesonide in the upper and lower airways: review of available data. J Asthma Allergy. 1, 11–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, et al. , 2004. Transient adrenocortical insufficiency of prematurity and systemic hypotension in very low birthweight infants. Arch Dis Child Fetal Neonatal Ed. 89, F119–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi KK, 2014. Glucocorticoid Induced Cerebellar Toxicity in the Developing Neonate: Implications for Glucocorticoid Therapy during Bronchopulmonary Dysplasia. Cells. 3, 36–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka T, et al. , 2007. Ciclesonide uptake and metabolism in human alveolar type II epithelial cells (A549). BMC Pharmacol. 7, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley RH, et al. , 2018. Glucocorticoid receptor isoform-specific regulation of development, circadian rhythm, and inflammation in mice. FASEB J. 32, 5258–5271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen S, et al. , 2006. A comparative study of inhaled ciclesonide 160 microg/day and fluticasone propionate 176 microg/day in children with asthma. Pediatr Pulmonol. 41,954–61. [DOI] [PubMed] [Google Scholar]
- Pedersen S, et al. , 2010. Efficacy and safety of three ciclesonide doses vs placebo in children with asthma: the RAINBOW study. Respir Med. 104, 1618–28. [DOI] [PubMed] [Google Scholar]
- Peffer ME, et al. , 2014. Caveolin-1 regulates genomic action of the glucocorticoid receptor in neural stem cells. Mol Cell Biol. 34, 2611–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltoniemi O, et al. , 2005. Pretreatment cortisol values may predict responses to hydrocortisone administration for the prevention of bronchopulmonary dysplasia in high-risk infants. J Pediatr. 146, 632–7. [DOI] [PubMed] [Google Scholar]
- Pew BK, et al. , 2016. Structural and transcriptomic response to antenatal corticosteroids in an Erk3-null mouse model of respiratory distress. Am J Obstet Gynecol. 215, 384 e1–384 e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postma DS, et al. , 2017. Asthma-Related Outcomes in Patients Initiating Extrafine Ciclesonide or Fine-Particle Inhaled Corticosteroids. Allergy Asthma Immunol Res. 9, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Principi N, et al. , 2018. Bronchopulmonary dysplasia: clinical aspects and preventive and therapeutic strategies. J Transl Med. 16, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowska M, et al. , 2019. Severe bronchopulmonary dysplasia - incidence and predictive factors in a prospective, multicenter study in very preterm infants with respiratory distress syndrome. J Matern Fetal Neonatal Med. 32, 1958–1964. [DOI] [PubMed] [Google Scholar]
- Sato H, et al. , 2007. In vitro metabolism of ciclesonide in human nasal epithelial cells. Biopharm Drug Dispos. 28, 43–50. [DOI] [PubMed] [Google Scholar]
- Schittny JC, 2017. Development of the lung. Cell Tissue Res. 367, 427–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V, McNeill JH, 2009. To scale or not to scale: the principles of dose extrapolation. Br J Pharmacol. 157, 907–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoner DP, 2016. Inhaled corticosteroids: Effects on growth and bone health. Ann Allergy Asthma Immunol. 117, 595–600. [DOI] [PubMed] [Google Scholar]
- Srinivasan M, Lahiri DK, 2017. Glucocorticoid-Induced Leucine Zipper in Central Nervous System Health and Disease. Mol Neurobiol. 54, 8063–8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, et al. , 2018. Human carboxylesterases: a comprehensive review. Acta Pharm Sin B. 8, 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterberg KL, et al. , 2010. Policy statement--postnatal corticosteroids to prevent or treat bronchopulmonary dysplasia. Pediatrics. 126, 800–8. [DOI] [PubMed] [Google Scholar]
- Weng T, et al. , 2006. Gene expression profiling identifies regulatory pathways involved in the late stage of rat fetal lung development. Am J Physiol Lung Cell Mol Physiol. 291, L1027–37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Relative expression of human CES-1 (A) and CES-2 (B) mRNA in various cell types of neonatal (PND 1) human lung extracted from publicly available scRNA-seq data set (Keil et al., 2018). Average expression shown in transcripts per kilobase per million mapped reads (TPM).
Relative expression and cellular distribution of mouse Ces1d (A, B) and Ces2g (C, D) mRNA expression in mouse lung extracted from scRNA-seq data set (Du et al., 2015; Du et al., 2017; Guo et al., 2019). Average expression shown as reads per kilobase of transcript per million mapped reads (RPKM).
IHC analysis for CES1d in PND1 (A, B) and PND5 (C,D) mouse lung tissue .(E, F) no primary control staining at PND1. Low power, 40X and high power, 100 X (E, F) images are shown following staining with CES1d specific antibodies.
Carboxylesterase (Ces) gene mRNA expression in perinatal mouse brain and liver extracted from scRNA-Seq data set (Darbellay and Necsulea, 2020).
Primer sequences utilized in qRT-PCR within the study. All sequences represent 5’ → 3’ orientation.