

Abstract
The ability to establish, and reactivate from, latent infections is central to the biology and pathogenesis of HSV-1. It also poses a strong challenge to antiviral therapy, as latent HSV-1 genomes do not replicate or express any protein to be targeted. Although the processes regulating the establishment and maintenance of, and reactivation from, latency are not fully elucidated, the current general consensus is that epigenetics play a major role. A unifying model postulates that whereas HSV-1 avoids or counteracts chromatin silencing in lytic infections, it becomes silenced during latency, silencing which is somewhat disrupted during reactivation. Many years of work by different groups using a variety of approaches have also shown that the lytic HSV-1 chromatin is distinct and has unique biophysical properties not shared with most cellular chromatin. Nonetheless, the lytic and latent viral chromatins are typically enriched in post translational modifications or histone variants characteristic of active or repressed transcription, respectively. Moreover, a variety of small molecule epigenetic modulators inhibit viral replication and reactivation from latency. Despite these successes in culture and animal models, it is not obvious how epigenetic modulation would be used in antiviral therapy if the same epigenetic mechanisms governed viral and cellular gene expression. Recent work has highlighted several important differences between the viral and cellular chromatins, which appear to be of consequence to their respective epigenetic regulations. In this review, we will discuss the distinctiveness of the viral chromatin, and explore whether it is regulated by mechanisms unique enough to be exploited in antiviral therapy.
Keywords: Antiviral, Herpes, HSV-1, Epigenetics, Latency, Chromatin dynamics
1. Introduction
HIV, the herpesviruses, and hepatitis B virus are among the important human pathogens that establish life-long infections in which viral double-stranded DNA persists in the nucleus of infected cells. HIV persists as an integrated provirus, whereas the herpes and hepadna viruses persist as extrachromosomal episomes. The maintenance of viral genomes in the absence of genome replication by viral proteins is operationally defined as “latency” whereas the maintenance mediated by continuous low level replication by viral proteins is operationally defined as persistence, although in some cases these differences are difficult to evaluate. Latent genomes periodically “reactivate” producing new infectious virions and resulting in recrudescence of disease. Latency is thus critical in pathogenesis.
Numerous clinical drugs effectively control the replication of herpes simplex virus 1 and 2 (HSV-1, 2) (James and Prichard, 2014), two of the viruses that establish latent infections. However, they have no effect on the latent genomes and cannot prevent the reactivation process, albeit they shorten the recrudescence and minimize shedding. Several innovative approaches have been explored to target latent viral genomes, including ribozymes (Bai et al., 2008; Watson et al., 2018), antisense RNA (Kenney et al., 2001), designer restriction enzymes (Aubert et al., 2016, 2020; Weber et al., 2014) and CRISPR/Cas9 based systems (Aubert et al., 2020; Oh et al., 2019). None has proven to be clinically viable as yet. As an alternative, the persistent viral genomes could be prevented from producing infectious virions by, for example, epigenetic modulation (Kristie, 2015; Whitley and Baines, 2018).
Epigenetic regulation refers to the regulation of gene expression by non-DNA base encoded mechanisms, such as DNA methylation or chromatin modifications. Nuclear double stranded DNA is normally complexed with histones, and non-histone proteins, in chromatin, which regulates DNA accessibility for transcription and other functions. Several mechanisms govern the accessibility to DNA in chromatin, including the specific histones, their post-translational modifications (PTM), remodeling, and the association of several non-histone proteins (Fig. 1). Chromatin epigenetics physiologically regulate the expression of cellular genes, and its dysregulation often results in disease. Not surprisingly, many drugs have been developed, or found, to modulate chromatin epigenetics; many more are at different stages of development. The potential use of these drugs in antiviral therapy against latent viruses such as HSV-1 has also been explored.
Fig. 1.
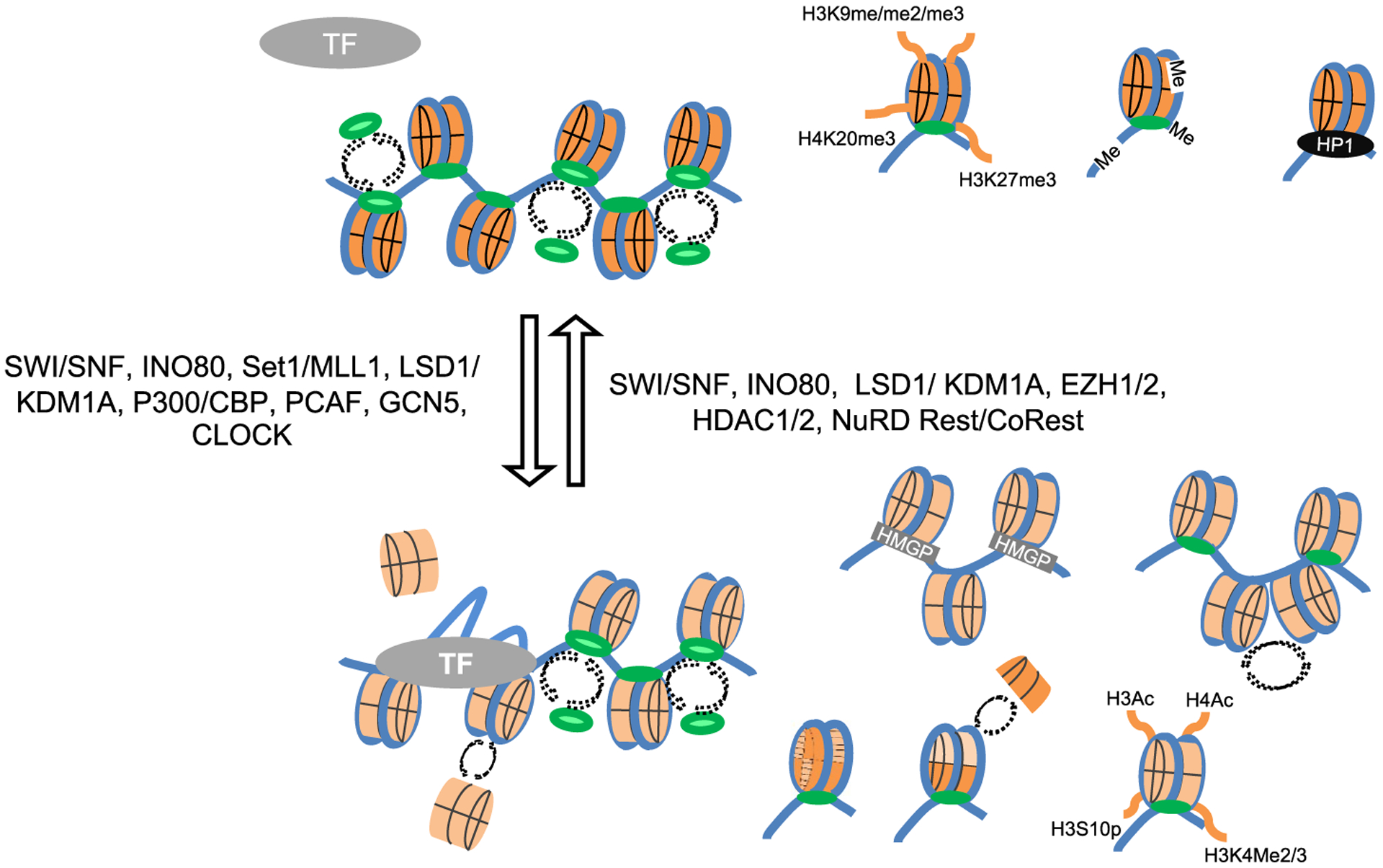
Epigenetic chromatin regulation. Top, silenced chromatin inaccessible to transcription factors and some of its typical epigenetic modifications; Bottom, active chromatin accessible to transcription factors with some of its typical epigenetic modifications. Selected chromatin modifiers regulating the transitions between the two are presented at the sides of the arrows. Blue, DNA; orange, core nucleosome; green, linker histone H1 (darker shade for the least dynamic, lighter shade for the most dynamic); black oval, heterochromatin protein 1; gray oval, transcription factor TF; Gray rectangle, high mobility group proteins; dash arrows, disassembly and reassembly. Me/2/3, methylation, di- or tri-; Ac, acetylation; P, phosphorylation.
The earliest approaches to use epigenetic modulation in antiviral therapy focused on inducing a massive and coordinated reactivation of all integrated HIV progenomes in the presence of highly active antiretroviral therapy (HAART). The goal would be to re-induce expression of viral proteins from the integrated proviruses, such that the cells harboring them are killed by viral replication, while preventing the infection of other cells with highly effective antiviral drugs. This is an obviously risky approach that requires both a very efficient coordinated induction of reactivation of all latent proviral genomes and very efficient approaches to block infection of other cells. These approaches have not reached the clinics as yet, although improvements continue to be described (Nixon et al., 2020).
The converse approach is to irreversibly silence latent genomes to prevent the resumption of the replicative cycle. Although intrinsically less risky, this approach must still overcome two major hurdles. Firstly, it may require life-long continuous treatment - unless the latent viral genomes were eventually lost in the continuous absence of any reactivation or after early abortive reactivations (Doll et al., 2020). However, HSV-1 or 2 reactivate upon physical or physiological stress and some patients go through long periods of clinically uninterrupted latency. One could envision discontinuous treatment concomitantly with, or ensuing, specific stresses or even prophylactically when such stresses are predictable. The high frequency of subclinical reactivation (Mark et al., 2008) suggests that this approach may minimize recrudescence of disease but is likely to have only limited impact on transmission.
The second hurdle may appear to pose an even bigger challenge. Most models propose that HSV-1 and 2 are epigenetically regulated by the same mechanisms that govern cellular epigenetic regulation (Bloom et al., 2010; Cliffe and Wilson, 2017; Knipe et al., 2017; Kristie, 2015; Lieberman, 2016; Nicoll et al., 2012; Singh and Tscharke, 2020; Washington et al., 2019). The obvious expectation from these models is that any epigenetic drug would equally affect viral and cellular chromatin, regardless of continuous or discontinuous use. Any epigenetic drug modulating viral reactivation would thus be expected to likely have side effects through its modulation of cellular gene expression. However, this apparent major limitation would only be so if viral and cellular chromatins were indeed regulated by the same mechanisms and in the same manner. Although much evidence is consistent with such models, some recent data challenges them and suggests that viruses may instead be epigenetically regulated through somewhat distinct mechanisms.
This review will focus on the similarities and differences between the epigenetic regulation of the HSV-1 or cellular chromatins, in the context of whether there are different enough as to support the exploration of epigenetic modulators as potential antiviral drugs.
2. HSV-1, 2 lytic and latent cycles
2.1. Lytic infection
HSV-1 or 2 primary infections occur in epithelia, most commonly in the orofacial and genital area. The virus first replicates in the basal layer cells, albeit not in those entering mitosis (Drayman et al., 2017; Schang, 2018), inducing their lysis.
During lytic infections, transcription of the so called immediate early (IE) genes is activated by constitutively expressed cellular proteins and a structural virion protein, VP16 (Figs. 2 and 3). The early (E) genes are not expressed until the IE proteins are synthesized. The IE proteins, however, do not activate transcription through binding to the promoters of the E genes and recruiting transcription proteins. Instead, they act as anti-silencers, activating the expression of genes that would otherwise be silenced, promoting ongoing transcription after the polymerase has cleared the promoter, or promoting RNA processing and export. The E genes encode for the proteins required for DNA replication, among other functions, including the seven directly involved in viral DNA replication (DNA polymerase catalytic and processive subunits, the three-subunit helicase primase, the origin binding protein, and the single stranded DNA binding protein), as well as many proteins involved in nucleic acid metabolism (thymidine kinase, ribonucleotide reductase, uracyl-nglycosylase, and alkaline nuclease, among others). Current anti-herpetic antivirals target mostly proteins involved in DNA replication. The viral genomes are replicated after the E proteins are expressed. Transcription of the strictly late (L) genes is activated only after the genomes are replicated, whereas the early/late genes are transcribed to only very low levels before DNA replication. The L proteins participate in the production of newly infectious virions.
Fig. 2.
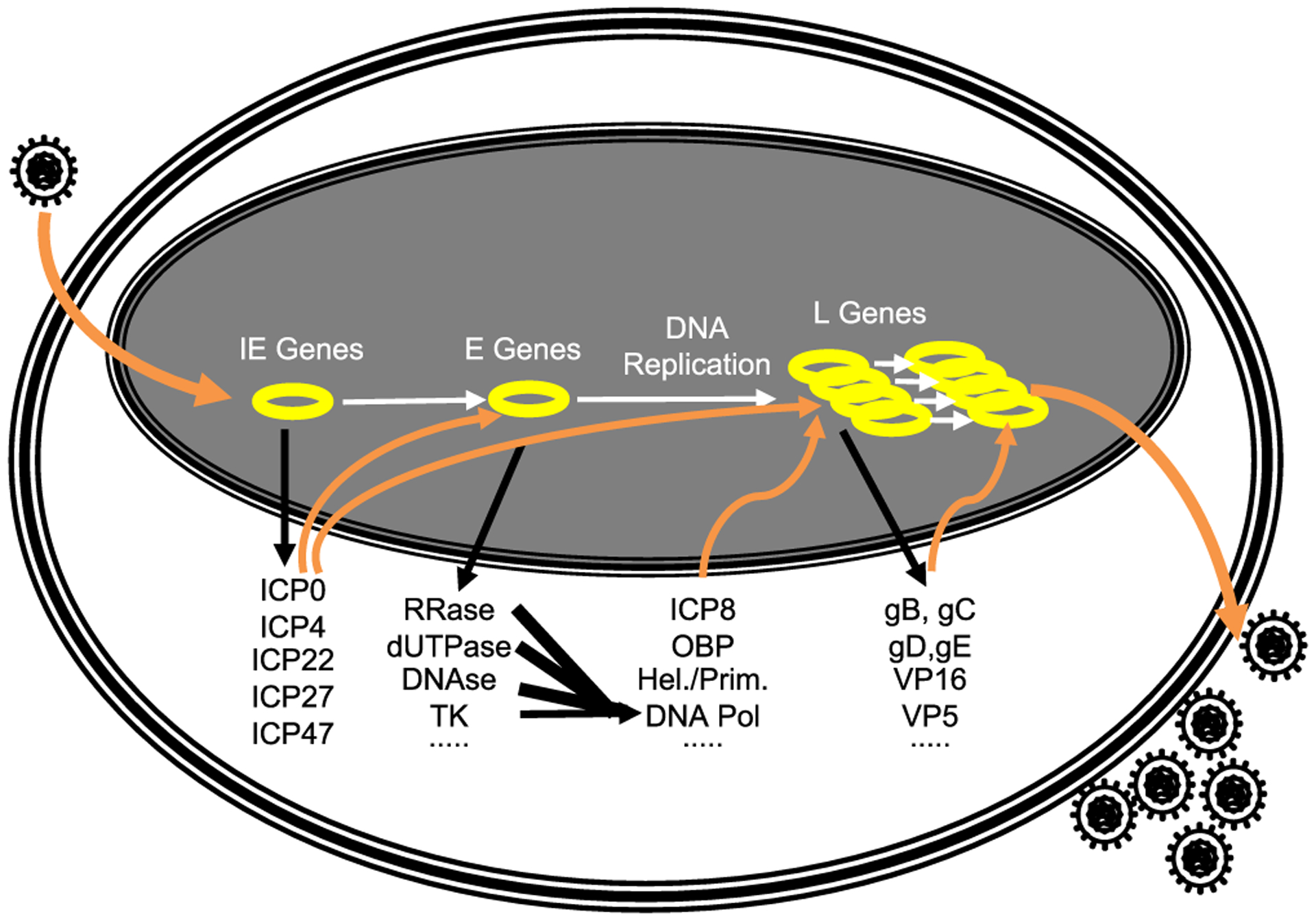
The HSV-1 lytic replication cycle. Yellow circles, nuclear HSV-1 genomes.
Fig. 3.
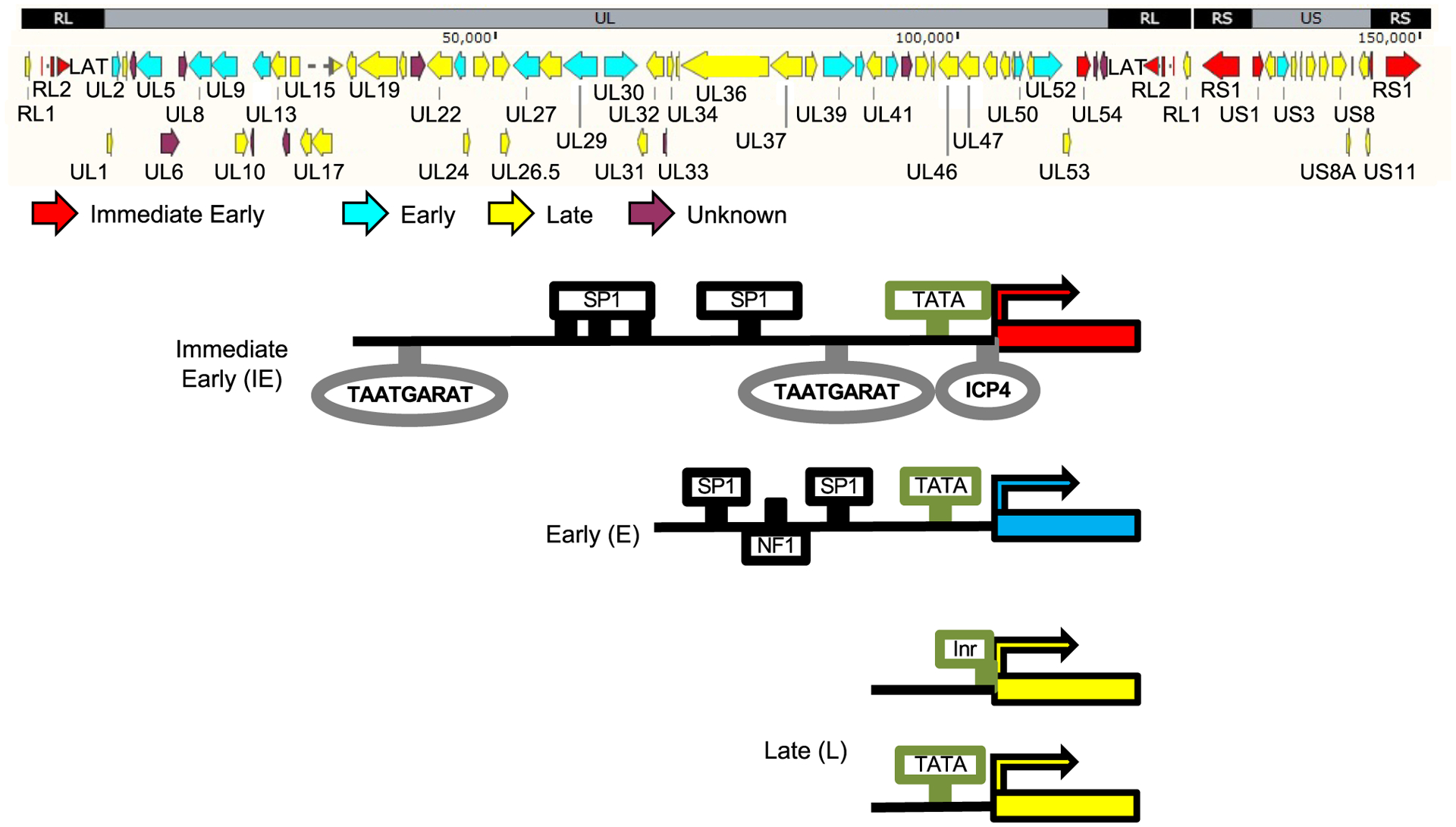
Typical structures of the promoters of HSV-1 IE, E and L genes. Cartoon on top, HSV-1 genome and ORF. Red, immediate early genes; Blue, early genes; Yellow, late genes; Gray rectangles and ovals, cognate sequences for the VP16/HCF-1/Oct1 complex or ICP4, which are recognized by viral proteins (alone or in complexes); Black rectangles, cognate sequences for cellular transcription factors. Green rectangles and ovals, TATA box or Inr elements. See the text for details and discussion.
2.1.1. Regulation of lytic gene expression
Despite the well described kinetics of gene expression, and the many excellent studies on the functions of the participating proteins, the actual regulation of HSV-1 transcription is still only partially understood. Activation of the IE promoters is understood best. As recently discussed (Hu et al., 2019), these promoters are rather complex in comparison to the E and L ones (albeit not so in comparison with cellular promoters), encompassing multiple binding sites for the potent transactivation complex formed by VP16/Oct-1/HCF-1, and multiple binding sites for other cellular transcription activators (Fig. 3). VP16 forms a complex with HCF-1, which gets recruited by the particular conformation of Oct-1 when bound to the TAATGARAT sequences in the IE promoters. HCF-1 and VP16 in this complex recruit multiple chromatin modifiers, including the histone demethylase (HDM), the histone lysin methyltransferase (KMT) Set1 (Liang et al., 2009), and the histone acetyltransferases (HAT) p300/CBP (Herrera and Triezenberg, 2004), as well as the cellular RNA polymerase II transcription complex (Herrera and Triezenberg, 2004).
The promoters of the E genes typically include only a couple of binding sites for constitutively expressed cellular proteins and a TATA box (Fig. 3). Activation of these promoters requires previous expression of the IE proteins ICP0 and ICP4, whereas ICP22 and ICP27 act co- and post-transcriptionally. ICP0 is an E3 ubiquitin ligase that induces the degradation of many cellular proteins involved in antiviral responses, including PML and the centromeric repressive histone H3 variants CENP-A, CENP-B, CENP-C, CENP-I, CENP-H, and CENP-N (Everett et al., 1999; Gross et al., 2012; Lomonte and Morency, 2007; Lomonte et al., 2001; Morency et al., 2007). ICP0 disrupts the function of hDaxx1, a histone chaperone that assembles silencing chromatin with histone H3.3 (Lukashchuk and Everett, 2010). It also interacts with BMAL1, a partner and regulator of circadian HAT CLOCK (Kawaguchi et al., 2001), displaces histone deacetylase (HDAC) 1/2 from the REST/CoREST repressor complex (Gu et al., 2005; Gu and Roizman, 2007), promotes histone acetylation on viral chromatin (Cliffe and Knipe, 2008), and heterochromatin removal on viral DNA (Lee et al., 2016).
ICP4 is a large DNA binding protein with preference for HSV-1 DNA (Dremel and DeLuca, 2019); it has no known enzymatic activity. It also binds to, or disrupts, epigenetic modifiers and transcription proteins, such as CLOCK (Kalamvoki and Roizman, 2010), the chromatin remodelers NuRD and INO80, and the BRM and BRG-1 helicase ATPase subunits from the SWI/SNF family (Wagner and DeLuca, 2013). ICP4 is necessary and sufficient to enhance the dynamics of all histones (Gibeault et al., 2016).
The promoters of the L genes are minimalistic, encompassing one or no binding sites for any transcription factors and sometimes a TATA box or an Initiator element (Fig. 3). It is not yet entirely clear why L promoters do not drive transcription to high levels until after the start of DNA replication. It is also unclear why the IE promoters are the most complex and have the most binding sites for transcription factors, whereas the L are the least complex and have the least binding sites (Fig. 3).
2.2. Latent infection
2.2.1. Establishment of latency
The virions produced in the epithelial cells infect other epithelial cells and the sensory axons. The later eventually reach the neuronal bodies, in the sensory ganglia, and the viral genomes enter the nucleus (Fig. 4). Although there is transient virus replication in the ganglia, infection of neurons mostly leads to latency. During latency, viral transcription is severely restricted, there is no significant expression of any viral proteins, or viral DNA replication, and no infectious virions are produced. The mechanisms that result in the silencing of the viral genomes are likely multifactorial and remain incompletely characterized, but there is a general consensus in that chromatin epigenetics play a major role (Bloom et al., 2010; Cliffe and Wilson, 2017; Knipe and Cliffe, 2008; Lieberman, 2016; Nicoll et al., 2012; Singh and Tscharke, 2020; Washington et al., 2019); recently reviewed in (Kobiler and Afriat, 2021).
Fig. 4.
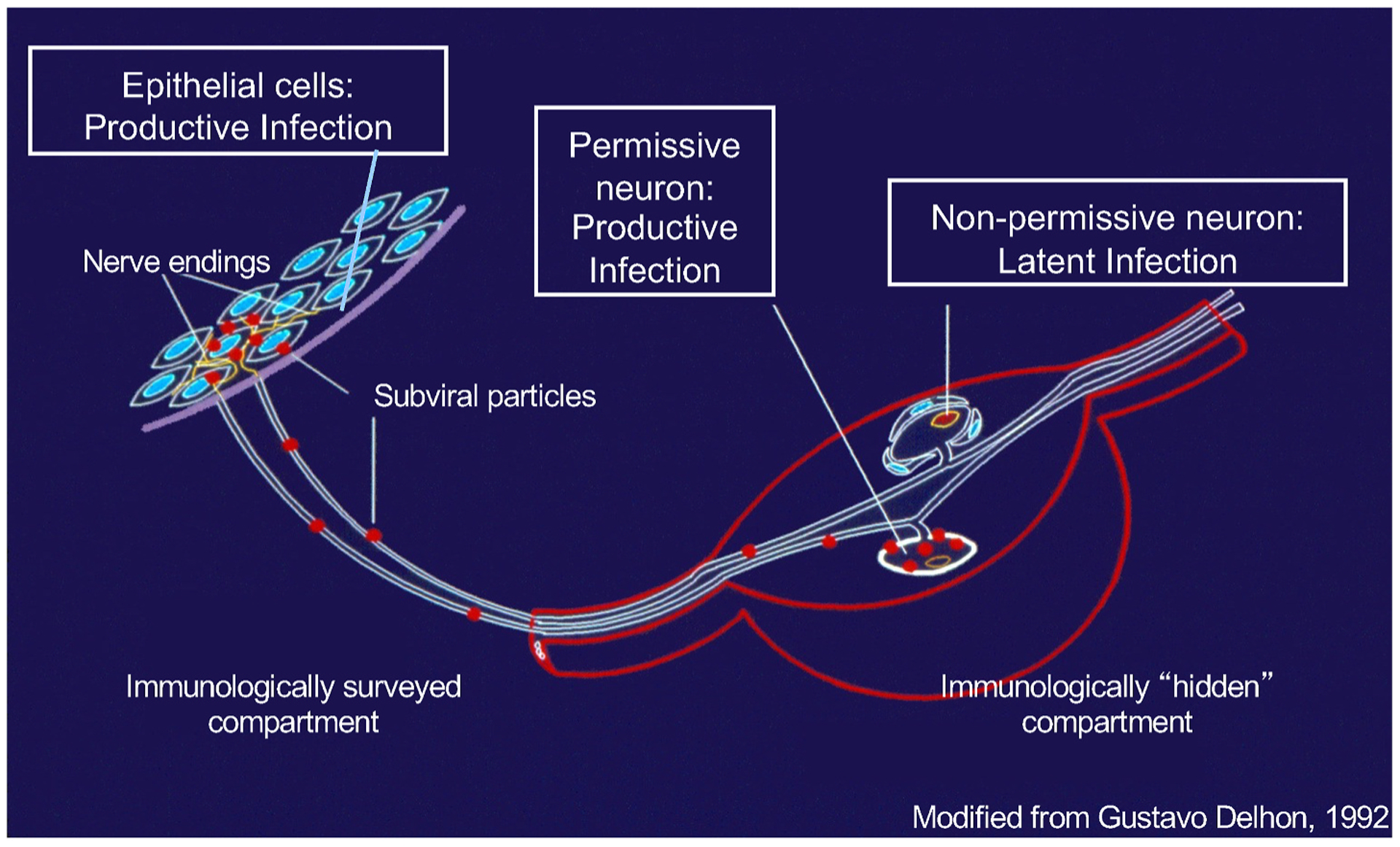
The HSV-1 infection cycle in humans depicting lytic and latent infections; for details see the main text.
2.2.2. Maintenance of latency
The restriction of transcription may not be absolute, and some low levels of transcription may occur during latency, at least periodically (Kramer and Coen, 1995; Ma et al., 2014; Russell and Tscharke, 2016; Singh and Tscharke, 2020). Whether this transcription is driven by specific promoters or results in mature mRNAs that are then translated into actual proteins is still unclear. CD8 lymphocytes infiltrate latent ganglia and cluster around latently infected neurons (Theil et al., 2003; van Lint et al., 2005); this infiltrate may result from abortive reactivations (Doll et al., 2020). Latent HSV-1 transcription modulates gene expression by the infected neuron (Ma et al., 2014), suggesting expression of viral proteins. Some neurons in which late promoters have been activated survive (Ma et al., 2014; Russell and Tscharke, 2016), whereas abortive reactivations in other cases result in neuronal death (Doll et al., 2020).
2.2.3. Reactivation from latency
Latent virus is reactivated by stressing stimuli, resulting in generalized transcription, protein expression, and genome replication. It eventually results in the release of infectious virions at the axon ends, which re-infect epithelial cells and produce recrudescence of disease (clinical reactivation) or are shed without overt signs or symptoms (subclinical reactivation) (Fig. 4). The processes that initiate reactivation are still poorly understood, but cellular, not viral, proteins most likely trigger and initiate it. The only viral transcripts that accumulates to high levels in a subset of latently infected neurons, the latency associated transcripts (LATs), regulate the apparent efficiency of reactivation in a variety of animal or ex vivo models (Bloom, 2004; Nicoll et al., 2012; Watson et al., 2018),. However, this phenotype may result from differences in the establishment of latency (Perng et al., 2000; Thompson and Sawtell, 2001). LATs are not essential in animal models; their importance in humans is unknown. Peripheral replication of re-activated virus re-stimulates the immune system, which promptly controls the ensuing lytic replication (Mark et al., 2008). Any lesions resolve, and shedding is stopped. The latent genomes persist for life, however, resulting in a constant source for recrudescence of disease and infection spread. For a recent comprehensive review of latency, please see (Sawtell and Thompson, 2020).
2.2.4. Antiviral drugs are not effective during HSV-1 latency
The available, and effective, antivirals against HSV-1 inhibit lytic replication with good potency and low toxicity (James and Prichard, 2014). Activity of the acyclic nucleotides, however, requires the expression of the viral thymidine kinase (TK) to activate them, and polymerase UL30, which is their target. Activity of pritelivir, which is in phase III clinical trials, requires expression of the helicase primase (James and Prichard, 2014; Quenelle et al., 2018). These drugs consequently have no effect on the latent genomes, which express no TK, polymerase, or helicase primase, and which do not replicate. They cannot preclude reactivation either, as their activity requires previous expression of their targets. They therefore cannot entirely block pathogenesis or transmission, although they do minimize shedding and curtail the symptomatic period by about a day (Chen et al., 2017; Whitley and Roizman, 2001).
3. Lytic and latent HSV-1 chromatin
3.1. Classic analyses of HSV-1 chromatin
The HSV-1 chromatin started to be probed back in the late 1980’s, using nuclease protection assays. Digestion of chromatinized cellular DNA with non-sequence specific endonucleases, such as micrococcal nuclease (MCN), results in DNA fragments with lengths of multiples of nucleosomes, the so called “nucleosomal ladder”. This protection is a direct result of the much longer residency times of the core nucleosome over that of the linker histones, resulting in preferential cleavage of the far more often exposed “linker DNA” (Fig. 5). In classic studies, MCN digestion of encapsidated DNA failed to show the nucleosomal ladder, leading to the conclusion that encapsidated HSV-1 DNA was not chromatinized (Leinbach and Summers, 1980). The negative charges of the DNA phosphate backbone are instead partially neutralized by spermidine (Gibson and Roizman, 1971). More recent proteomic studies have detected no histones, or any other chromatin protein, in the virions (Lippe, 2020; Loret et al., 2008).
Fig. 5.
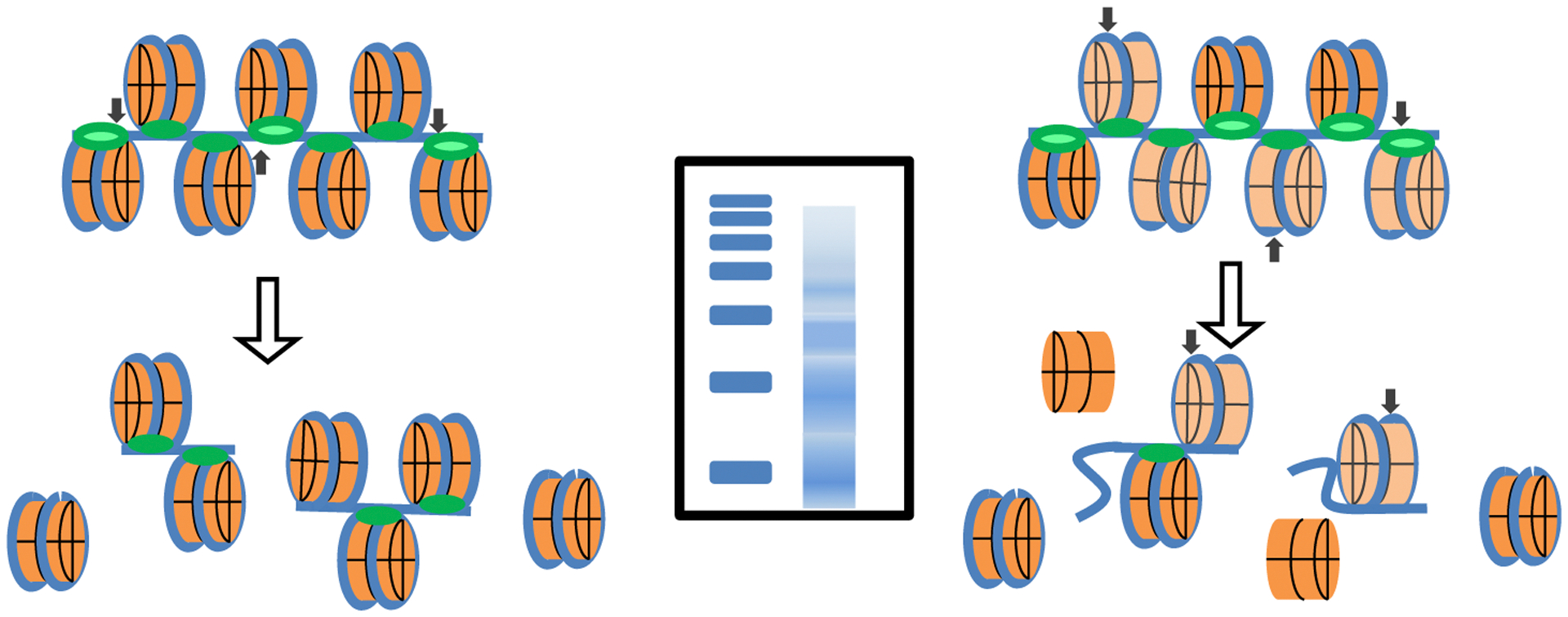
Chromatin protection assays. Left, protection of a regularly chromatinized DNA template and the expected DNA products. Right, protection of a dynamically chromatinized DNA template in which the residency time of the core nucleosome approaches that of the linker histones. Middle, resolution of the protected DNA fragments expected from each chromatin type. Blue, DNA; orange, core nucleosome; green, linker histone H1 (shades as in Fig. 1); Black arrows, cleavage sites; Blue rectangles, protected DNA fragments sizes, as resolved by agarose gel electrophoresis.
Digestion of intracellular HSV-1 DNA of lytically infected cells or mice also failed to produce the nucleosomal ladder pattern, whereas the HSV-1 DNA in latently infected trigeminal ganglia did (Deshmane and Fraser, 1989; Leinbach and Summers, 1980; Muggeridge and Fraser, 1986). These results were for many years understood as indicating that the intracellular HSV-1 DNA was not associated with nucleosomes during lytic infections, but it did become chromatinized during latency.
3.2. Modern analyses of HSV-1 chromatin
A new approach to test chromatin accessibility was developed in 2013 taking advantage of a highly active retrotransposon system, ATAC-seq (Buenrostro et al., 2013). The retrotransposase is linked to a DNA tag which is recombined with the DNA being probed, resulting in cleavage and tagging of the cleaved DNA, a process that has been called tagmentation. The tagmented DNA is then sequenced and analyzed. Although the probe is different, the principle is the same as for nuclease protection: linker DNA is exposed far more frequently than core nucleosome DNA because the linker histone H1 exchanges at a much faster rate than the core nucleosome. Therefore, like nuclease protection, tagmentation also produces tagged DNA with lengths of multiple of the nucleosome size. Consistently with the patterns of protection from endonucleases, the HSV-1 genomes in lytic infections do not yield the clear repeated peaks of protected DNA at multiples of nucleosome sizes (McSwiggen et al., 2019). Instead, HSV-1 DNA produced one peak almost consistent with the size of nucleosome DNA, albeit somewhat shorter and not nearly as sharp as that produced by cellular DNA, and then broader and less marked peaks of sizes of multiples of nucleosome DNA (McSwiggen et al., 2019). Tagmentation to completion also produced DNA protected to about nucleosome size (Dremel and DeLuca, 2019), but no DNA protected to di-, or any longer poly-nucleosomal size. Moreover, tagmentation of the viral DNA was about 2- to 100-fold more efficient than that of the cellular DNA (Dremel and DeLuca, 2019). These results were interpreted as evidence that the HSV-1 genomes were not in nucleoprotein complexes, or that they were in nucleoprotein complexes with the viral protein ICP4 rather than histones (Dremel and DeLuca, 2019).
The results from protection assays of the HSV-1 genomes in lytic infections are also consistent with an alternative chromatin model in which the core nucleosomes in the viral chromatin have a much shorter residency time than those in most cellular chromatin, approaching that of linker histone H1 (Conn et al., 2008, 2011, 2013; Conn and Schang, 2013; Gibeault et al., 2016; Hu et al., 2019; Lacasse and Schang, 2010, 2012). Under this model, the linker DNA would not be much more exposed than the core nucleosomal DNA, resulting in random, and highly efficient, cleavage (Fig. 5) or tagmentation of both.
3.3. Chromatin epigenetics in latent and lytic HSV-1 infections
2004 was a key year in our current understanding of HSV-1 epigenetics. A series of studies from Drs. Bloom, Neumann, and Hill groups starting that year re-affirmed the chromatinization of the latent viral genomes by chromatin immunoprecipitation (ChIP) analyses (Amelio et al., 2006a; Kubat et al., 2004a, 2004b; Neumann et al., 2007a). Also in 2004, the inhibition of transcription driven by the ICP0 promoter by an inhibitor of cellular cyclin-dependent protein kinases, roscovitine, was found to be dependent on the genome in which the promoter was placed, not on the promoter sequences (Diwan et al., 2004). The same ICP0 promoter recombined into the genome of the infected cells still activated transcription in the presence of roscovitine, whereas the native copy in the viral promoter did not (Diwan et al., 2004). The simplest explanation was a then unknown epigenetic regulation (Fig. 6). The first direct evidence supporting a role for chromatin in HSV-1 lytic infections was also reported in 2004 by the groups of Drs. Berger, Fraser, and Triezenberg (Herrera and Triezenberg, 2004; Kent et al., 2004). In parallel studies, HSV-1 DNA in lytically infected cells was co-immunoprecipitated with histone H3. Moreover, the levels of histone H3 acetylation at K9 and K14 in the ICP0, TK, and VP16 promoters, and of H3K9me3 at the genes, was correlated to some extent to their respective levels of transcription (Huang et al., 2006; Kent et al., 2004). Deletion of the transcription activation acidic domain of VP16 resulted in higher association of unacetylated histone H3 in the promoters of the IE ICP4, ICP0, and ICP27, the E TK, and the L VP16 and gC, as well as the ICP27 gene (Herrera and Triezenberg, 2004). These studies challenged for the first time the classic interpretation lytic HSV-1 or 2 genomes were not chromatinized and presented the first pieces of evidence that HSV-1 transcription could be epigenetically regulated during lytic infections as well as during latency.
Fig. 6.
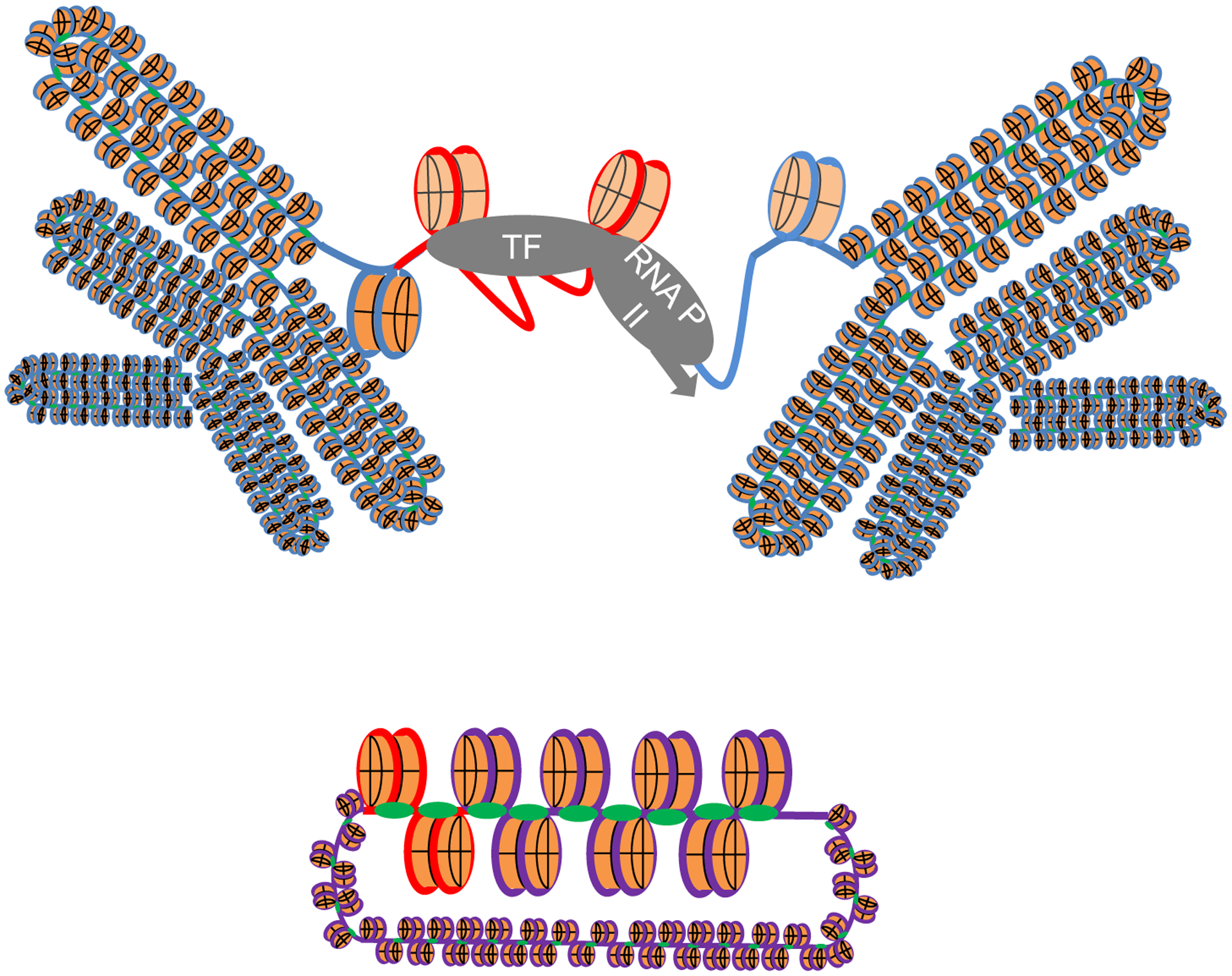
Roscovitine inhibits transcription driven by the ICP0 promoter in the HSV-1 genome but not by the same promoter recombined in the cellular genome. The ICP0 promoter (red line) recombined in cellular genome (blue line) remains accessible to transcription proteins in cells treated with roscovitine (top), whereas the same promoter (red line) in the viral genome (purple line) is inaccessible in the same cells (bottom). Blue line, cellular DNA; Purple line, viral DNA; light blue line, cellular promoters; red line, viral promoter.
4. The HSV-1 epigenome
4.1. Latent HSV-1 chromatin
4.1.1. Silencing of HSV-1 gene expression during the establishment of latency
The establishment of latency is still poorly understood, in no small part due to the intrinsic limitations of the models available. True latency can only be established in animal models, in which only a small number of sensory ganglia neurons become latently infected. Moreover, the establishment of latency is asynchronous, such that the virus is actively replicating in some neurons while establishing latency in others and already latent in others. As an alternative model, a latency-like state is also established in different types of cultured neurons, using low multiplicity of infection and acyclovir or interferon. The latently infected neurons in these cultures, like those in vivo, express no detectable viral proteins, but transcribe LAT and harbor reactivable genomes (Thellman and Triezenberg, 2017). Reactivation can be induced pharmacologically, resulting in the fairly simultaneous expression of all viral genes and the production of infectious virions (Kobayashi et al., 2012a).
According to most current models, the HSV-1 genomes must become silenced at some time during early neuronal infection. However, it is still unclear whether this process occurs only in neurons that have never expressed the lytic proteins, or already started gene expression is shut down to establish latency. Expression of lytic genes and HSV-1 DNA replication is invariably lethal to infected cells in culture. Elegant experiments performed using ROSA reporter mice and CRE expression driven from the viral genome by promoters of different kinetic classes strongly suggest that neurons that express IE, E, or even L genes often survive and become latently infected (Ma et al., 2014; Russell and Tscharke, 2016). In contrast, abortive reactivations are often lethal (Doll et al., 2020). It is as yet also unclear whether transcriptional silencing is a direct result of the specific anatomy of the neurons, active shut down by viral factors, differential gene expression, signaling, protein localization, neuronal type, the terminally differentiated state of the neurons, or a variety of other factors (Cabrera et al., 2018; Chen et al., 1997; Flowerdew et al., 2013; Hafezi et al., 2012; Harrison et al., 2020; Kobayashi et al., 2012b; Kristie et al., 1999; Margolis et al., 2007; Maroui et al., 2016; Nicoll et al., 2016; Zhao et al., 2020). In summary, silencing at the establishment of latency is still the least understood stage in latency.
4.1.2. Histone PTM in the latent chromatin
As the HSV-1 DNA is not methylated in either lytic or latent infections (Kubat et al., 2004b), the studies on epigenetic regulation have focused on chromatin epigenetics. Latent genomes are quantitatively chromatinized with PTM and variants of silenced chromatin, such as H3K9me2, H3K9me3, H3K27me3, and H4K20me3, and macroH2A (Cliffe et al., 2009; Kwiatkowski et al., 2009; Wang et al., 2005). In contrast, the transcriptionally active LAT locus is associated with PTM characteristic of active transcription, such as H3K4me2, H3K4me3, H3K9ac and H3K14ac (Amelio et al., 2006a; Creech and Neumann, 2010; Kubat et al., 2004a, 2004b; Neumann et al., 2007a; Wang et al., 2005) (Fig. 7). Considering the high gene density of the HSV-1 genome with the consequent close proximity of the territories with PTM characteristic of transcription or silencing, it is perhaps not unexpected that insulator elements separate the chromatin rich in acetylated H3K9 and K14 at the transcribed LAT locus from the hypoacetylated chromatin in the promoters of the non-transcribed ICP4 and UL59 genes (Amelio et al., 2006b; Lee et al., 2018). These sequences are recognized by CTCF.
Fig. 7.
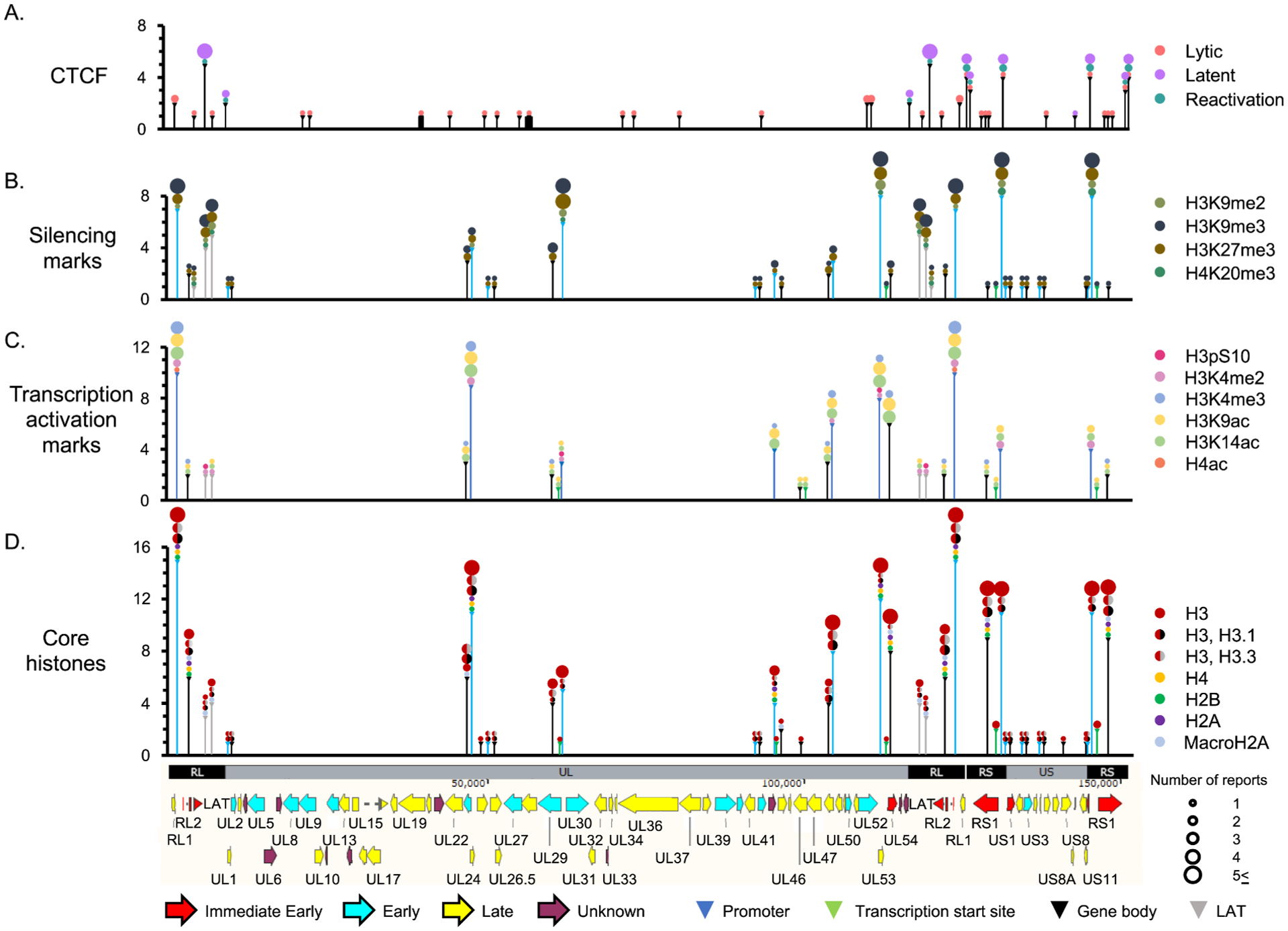
Interactions between HSV-1 DNA, core histones, histone H3 or H4 with PTM associated with transcription or silencing, or CTCF. X-axis, HSV-1 genome; Y axes, number of publications describing any association with any given histone, PTM, or CTCF for that particular genome locus. Vertical lines, the regions of the HSV-1 genome analyzed, to scale (i.e., the coverage); Colors of lines (and triangle heads on top), type of infection analyzed (for CTCF), or region of the gene analyzed (promoter, TSS, gene, or LAT) for all others; Colors and sizes of circles on top of each line, histone or PTM analyzed or number of studies analyzing each particular association, respectively. H4ac, H4 acetylated in K5, K8, K12 or K16. Note that several of the loci evaluated are in the repeated regions.
4.1.3. Insulator elements in latency and reactivation
CTCF is a ubiquitously expressed and highly conserved 11-zinc-finger sequence specific DNA binding protein (Klenova et al., 1993; Lobanenkov et al., 1990). Binding to its cognate sequences serves as insulator boundary element separating adjacent domains of active and inactive chromatin by restricting both interactions between enhancers and promoters and heterochromatin spread (Bell et al., 1999; Chung et al., 1993; Hark et al., 2000; Kim et al., 2007). In concerted action with cohesin, CTCF aids in the three dimensional genome folding through ‘loop extrusion’. This process creates physically separated chromosome loops, also known as topologically associated domains (TADs), or loop domains. Only convergently oriented pairs of CTCF sites form CTCF-anchored loops, by acting as barriers to the loop extrusion process (Beagan and Phillips-Cremins, 2020; Li et al., 2020).
Computational analysis predicted seven unique CTCF-binding sites in the HSV-1 genome, named CTRL1, CTRL2, CTa’m, CTRS1 and 2 (separated by only 100 bp, and usually referred together as CTRS1/2), CTRS3, and CTUS1, six of which are in the repeat regions encompassing the ICP0, ICP4, and LAT loci; these sites were bound by CTCF during latency (Amelio et al., 2006b). Twenty-five unique CTCF binding sites were later identified by ChIP analyses in lytic infections (Lang et al., 2017). Binding to CTRL2, CTa’m and, most prominently, CTRS3 decreased early after induction of reactivation with sodium butyrate (NaBu) (Washington et al., 2018a, 2018b), when the H3K9me3 and H3K27me3 PTM, characteristic of silencing, are replaced by the H3K4me3, H3K9ac and H3K14ac PTM, characteristic of active transcription, in the IE genes. Suz12, a component of the polycomb-repressive complex 2 (PCR2), is recruited to CTRS3, CTRS1/2, CTa’m, and CTRL1, and deletion of the LAT promoter decreased its binding to CTRS3 (Washington et al., 2018b). As that of CTCF, Suz12 binding to CTa’m, CTRS1/2, and CTRS3 also decreased during NaBu-induced reactivation.
Depletion of CTCF through adeno associated virus (AAV)-8 delivery of a CTCF-targeting siRNA to latently infected mouse trigeminal ganglia induced transcription of the ICP0 gene (Washington et al., 2018b), which is flanked by CTRL2 and CTa’m. An HSV-1 mutant lacking 135 bp including the CTRL2 sequences had increased virulence and was less efficient at establishing latency (Washington et al., 2018b). This phenotype may well be a result of its enhanced replication in the infected neurons which are thus killed during the acute infection. Transcription of ICP0 and ICP27 was not properly suppressed in trigeminal ganglia and less H3K27me3 was detected on the chromatin of the IE locus (Washington et al., 2019), supporting an insulator role for CTCF. A different 370 bp deletion mutant including the CTRL2 sequences from within the 2.0 Kbp LAT intron, in a different strain, showed no defect in the lytic cycle or the establishment and maintenance of latency in mice (Lee et al., 2018). However, H3K9me3 and H3K27me3, which are normally restricted to the adjacent ICP0 and UL54 genes during latency, increased at the LAT locus, and explant-induced reactivation was inhibited (Lee et al., 2018). The CTRL2 may help preventing heterochromatin spreading into the LAT locus, thus maintaining parts of the viral genome in a more open state poised for reactivation.
Rad21, a component of the cohesin complex, also bound to CTRL1, CTRL2, CTa’m and CTRS3 during latency in mouse. Rad21 binding to the HSV-1 mutant lacking the 135 bp including the CTRL2 sequences was reduced at 7 and 14 days after infection, and there were about 5-fold fewer latent HSV-1 genomes at day 28 (Singh and Neumann, 2021). These results indicate that CTCF binding sites most likely results in the formation of functional chromatin loops.
Expression of a transgene flanked by CTCF binding sites from HSV-1 vectors containing non-functional VP16, ICP0, or ICP4 was extended to up to 28 days into latency, but it still became eventually silenced (Han et al., 2018). Multiple CTCF binding sites may need to act together, or the position of the CTCF binding sites in the viral genome may be critical to allow transcription from a locus in the midst of the mostly silenced latent viral genomes.
4.1.4. Changes in HSV-1 chromatin during reactivation
The mechanisms triggering chromatin remodeling during reactivation are not fully elucidated. Nonetheless, a variety of tissue culture latency models indicate that the first phase of reactivation involves an early methyl/phospho switch (Cliffe et al., 2015) and a general disruption of silencing which is followed approximately 24 h later with specific transcription of the lytic genes of the different kinetic classes (Kim et al., 2012; Kobayashi et al., 2012a; Pourchet et al., 2017). Consistently with these models, much older results had already hinted that the early transcription during explant induced reactivation did not follow the typical IE-E-L cascade. Rather, low levels of transcription of all genes were identified at approximately the same time (Kosz-Vnenchak et al., 1993).
The disruption of CTCF binding to the insulator elements, the changes in global PTM, and in particular the methyl/phospho switch, are among the first events in the reactivation process (Cliffe et al., 2015; Lee et al., 2018; Washington et al., 2018a). All these changes are consistent with the models proposing that de-silencing plays a critical role early in the reactivation process.
4.2. Lytic HSV-1 chromatin
4.2.1. Histone PTM during lytic infections
A still growing body of literature describes a number of histone PTM and their relevant modulators in HSV-1 infections. Briefly summarizing, histone PTM associated with transcription (e.g., H3 K9ac, K14ac, K4me3) have been consistently reported to associate with transcribed HSV-1 genes during lytic infections and histone PTM normally associated with silencing (e.g., H3K9me2/3 or K27me3), with genes transcribed to only low levels or not at all (Fig. 7) (recently reviewed in Kobiler and Afriat, 2021). VP16 recruits to the IE promoters the HDM LSD1/KDM1A to remove H3K9me3, which is associated with silencing. It also recruits the histone KMT Set/MLL1 to trimethylate H3K4 (Liang et al., 2009) and the HAT p300/CBP to introduce acetylate histones. These last two modifications are normally associated with transcription. Moreover, VP16 recruits the chromatin remodeler SWI/SNF (Herrera and Triezenberg, 2004). However, downregulation, or lack of expression, of some chromatin modifiers such as the SWI/SNF remodeler helicase subunits BRM or BRG1, or the HAT p300/CBP, PCAF, or GCN5, does not inhibit IE gene transcription (Kutluay et al., 2009). In contrast, small molecule inhibition of histone acetyl-transferases (HATs) including p300/CBP, inhibits the number of HSV-1 genomes that start replication (Shapira et al., 2016). The bases for these apparently discordant results regarding p300/CBP have not been resolved, but there is a major intrinsic difference in the experimental designs. Knockdown of the epigenetic modifiers is performed before infection, whereas small molecule inhibition is performed after infection. As discussed (Conn and Schang, 2013), the former affects the epigenetic landscape before the viral genomes can establish their transcription program, influencing the pool of histones available to chromatinize incoming viral genomes, whereas the latter are added only after the viral genomes are already chromatinized, which occurs almost immediately after infection (Johnson et al., 2014; Sanders et al., 2015).
The results from DNA protection studies had often been concluded to indicate that the HSV-1 DNA in lytic infections is not chromatinized, or that it has sporadic, randomly distributed, nucleosomes, or that only a minority of the genomes are chromatinized whereas the vast majority are not (Dremel and DeLuca, 2019; Knipe and Cliffe, 2008; Kristie, 2015; Kutluay and Triezenberg, 2009; Oh et al., 2015). Either of these models is difficult to reconcile with the recruitment of a number of epigenetic regulators to HSV-1 genomes in lytic infections (Dembowski and DeLuca, 2015). They are also difficult to reconcile with any epigenetic changes modulating viral gene expression. The epigenetic modifications associated with transcribed or non-transcribed genes act by eventually recruiting (or not) transcription activating or repressing proteins in the context of a regularly chromatinized template. It is not obvious how these modifications could also regulate transcription if most of the DNA to be transcribed was not chromatinized. Similarly, if most of the lytic HSV-1 DNA was not chromatinized, one could expect that these genomes would be widely accessible and transcribed. As a counterargument, the natural template for transcription is regularly chromatinized DNA and therefore the minority of properly chromatinized genomes could be the ones actually transcribed. Moreover, abortive infections, either using viral mutants or by sorting infected cells with non-replicating genomes, consistently show regular chromatinization of the quiescent genomes (Cohen et al., 2020; Ferenczy and DeLuca, 2009). The body of the evidence thus is the most consistent with models under which there is a functionally relevant chromatinization of sorts of the viral genome during lytic infections.
4.2.2. CTCF insulation during lytic infections
CTCF binding could separate the HSV-1 genomes into differentially regulated transcriptional territories. As detected through ChIP-Seq, CTCF interacts with 25 unique HSV-1 loci during lytic infections, some of which are in the repeated sequences (Fig. 7). Eight unique sites where further analyzed through ChIP-qPCR. No binding to CTRL2 or CTRS3, which are occupied by CTCF during latency or at early times of infection, was detected at this time. Knocking down CTCF decreased the occupancy of Pol II on viral genes and increased the association of H3K27me3 and H3K9me3 on the promoter and genes of ICP0, ICP4 (IE genes), ICP8, and TK (E genes) (Lang et al., 2017). Transcription of viral genes of all kinetic classes was consequently inhibited, and three components of the cohesin complex, SMC1(structural maintenance of chromosomes protein 1), SMC3 and Rad21 localized to the viral replication compartments. Knocking down SMC1 or Rad21 (from 48 h before infection) decreased viral yield by about 1,000-fold. H3K27me3 was enriched on the promoter and gene of ICP0, VP16 and TK under these conditions (Li et al., 2021). These results also suggest that chromatin looping plays an important role in HSV-1 lytic infections. As a caveat, knocking down CTCF or cohesin is expected to have global effects on host chromatin and gene expression, which both could indirectly affect viral functions.
Based on the analyses of chromatin epigenetic modifications on selected genes of different kinetic classes (Fig. 7), the PTM on the histones in the HSV-1 chromatin at a given gene have been often observed to generally correlate with the transcription level of that same gene. Several epigenetic inhibitors have thus been explored for their potential antiviral activities.
5. Antiviral activity of epigenetic drugs against HSV-1
5.1. Brief introduction to epigenetic drugs
Considering the importance of epigenetic regulation in physiology and pathology, it is not surprising that a number of epigenetic inhibitors have been developed with the goal of exploring therapeutic uses. Several different approaches have been undertaken, such as inhibition of the enzymatic activities or blocking specific protein-protein interactions. In many instances, existing molecules were found to have epigenetic activities. For example, the HDM LSD1 is in the mono-amino oxidase superfamily and uses FAD as a cofactor. It is consequently sensitive to some clinical psychotropic mono-amino oxidase inhibitors. Although these compounds are most useful reagents in research, their therapeutic uses are likely to be constrained by their psychotropic activities.
Another approach to develop epigenetic inhibitors is to target the domains that recognize specific histone modifications. Many epigenetic regulators have bromo- or chromo-domains, which recognize acetylated or methylated histones, respectively, through binding pockets proven to be amenable to drug targeting. Many such inhibitors have been developed with potential clinical uses in mind. We will discuss their known antiviral activities against HSV-1.
5.2. Antiviral activities of epigenetic drugs in lytic and latent HSV-1 infections
5.2.1. Histone deacetylase inhibitors
The first evaluations of epigenetic drugs on HSV-1 started already in the 1990’s, when it was first shown that generic inhibitors of histone deacetylases, like valproic acid (VPA) and trichostatin (TSA), induced reactivation of latent or quiescent HSV-1 and enhanced the replication defect of HSV-1 mutants in the gene encoding for the IE protein ICP0 (Hobbs and DeLuca, 1999). A single intraperitoneal 1,200 mg/kg dose of the generic HDAC NaBu sufficed to induce reactivation in 75–100% of trigeminal ganglia latently infected with either of two different HSV-1 strains (Neumann et al., 2007b). However, this treatment also resulted in the occasional death, which precludes its use to flush latent genomes (Neumann et al., 2007b). These compounds did not induce a degree of reactivation that could raise any expectation of flushing all latent genomes, and induced lethality. Although most useful as research tools, these experiments were not aimed at developing therapeutics and it is not easy to foresee how such generic inhibitors could be used in antiviral therapy.
NaBu and the also generic HDAC inhibitor TSA also activated quiescent HSV-1 genomes in PC12 cells, inducing their reactivation (Danaher et al., 2005). H3K9me3, which is associated with silencing, was enriched on the promoters of ICP0 genes during quiescent infections whereas H4ac, which is associated with transcription, was partly depleted (Coleman et al., 2008). Quiescent HSV-1 genomes could also be reactivated by superinfections with wild-type HSV-1, superinfections that resulted in lower levels of H3K9me3 and higher levels of H3ac and H4ac on the ICP0, LAT, and gC genes (Coleman et al., 2008). TSA also enhanced viral replication, increasing ICP27 mRNA levels by about 100-fold at 400 nM (Du et al., 2013). NaBu enhanced reactivation 6-fold at 50 mM (Danaher et al., 2005). TSA (1.32 μM), suberohydroxamic acid (SBX - 40 μM) or 4 mM VPA, in contrast, decreased the number of expressed viral genomes by 15–25% at an moi of 100 (Shapira et al., 2016). Other HDAC (suberoylanilide hydroxamic - SAHA) or HAT inhibitors (anacardic acid - AA) had no major effects (at 10 and 250 μM, respectively), regardless of moi (Shapira et al., 2016). TSA (1.32 μM) increased PML, ATRX and hDaxx levels by about 1.8–2.5- fold in infected HFF cells, or of only PML, but by 6-fold, in U2OS cells (Shapira et al., 2016); histone acetylation levels were not evaluated. Two other HDAC inhibitors, 20 μM MS275 or 10 μM MC1568, also induced reactivation by 10-fold, increasing ICP27 mRNA by about 50-fold, (Du et al., 2013). Pharmacological modulation of histone acetyl levels, thus, affects viral replication and gene expression but not always in the expected manner.
5.2.2. Specific epigenetic compounds
The second wave of testing epigenetic inhibitors against HSV-1 evolved from the many studies showing specific epigenetic modifications on the chromatin of HSV-1 genes. A series of studies from the Kristie lab using inhibitors of the HDM LSD1 or JMJD2 provided the first proof of concept that specific epigenetic modulation can indeed inhibit HSV-1 replication in cultured cells and in mice and reactivation from latency in animal models (Hill et al., 2014; Liang et al., 2009, 2013). Following on those pioneering studies, a number of specific epigenetic inhibitors have been tested as potential anti-HSV-1 agents.
5.2.2.1. Histone demethylase inhibitors.
Inhibition of LSD1 by 3 mM pargyline or 2 mM tranylcypromine (TCP) increased the levels of H3K9me and H3K9me2 on the ICP0 promoter by 2- to 10-fold (Liang et al., 2009), and those of H3K9me3 by about 60-fold. In latently infected mice, time-released TCP pellets also increased the levels of H3K9me3 on the ICP4 promoter by 20-fold, while decreasing those of H3K9me2 and H3K27me3 to about half to a third on average, respectively, on the ICP0, ICP27 and ICP8 loci (Hill et al., 2014).
Inhibition of JMJD2 family proteins with 2.5 mM dimethyloxalylglycine (DMOG) decreased viral yields by 5,000-fold, and the levels of ICP27 mRNA by about 10-fold (at 2 mM) (Liang et al., 2013). The levels of H3K9me3, and total histone H3 on the promoters of ICP27, ICP4, and ICP0, increased by 3- to 4-fold, or 1.5- to 2-fold, respectively. A more potent JMJD2 inhibitor, ML324, also resulted in lower levels of mRNA levels of ICP4, ICP27, and ICP22, to less than 80% at 50 μM, and 10,000-fold lower viral yields, at 50 μM; levels of histone methylation were not tested (Liang et al., 2013). Modulation of histone methylation with specific HDM inhibitors thus results in the predicted alterations in HSV-1 chromatin and gene expression.
5.2.2.2. Histone methyltransferase inhibitors.
The EZH2/1 inhibitors GSK126 (30 μM), GSK343 (40 μM), or UNC1999 (15 μM) all decreased ICP4, ICP22 and ICP27 mRNA levels by about 60% (Arbuckle et al., 2017). GSK126 also decreased the levels of HCF-1, RNAPII, and H3K4me3 on the ICP0 gene promoter by 70, 50 and 30% respectively (Arbuckle et al., 2017). The authors concluded that the antiviral activities resulted from their ability to activate immune responses, and the levels of PML, IFN-α, and IL-8 were indeed increased by 2- to 10-fold, respectively, by 30 μM GSK126 (Arbuckle et al., 2017).
The protein methylation inhibitor 5′-deoxy-5′-methylthioadenosine (MTA), at 1 mM, induced a 50% decrease in the levels of H3K4me3, but not of H3K4me2, on the ICP0, TK and VP16 genes. It also resulted in a similar reduction in mRNA levels of selected IE, E and L genes and of HSV-1 genome copy number (by 5-fold). Knockdown of the histone KMT Set1 had a similar effect (Huang et al., 2006).
The effects of histone KMT inhibitors in HSV-1 gene expression are thus not always consistent with what would be predicted by extrapolation from their activities on the regulation of cellular gene expression. Moreover, several of the effects on viral transcription may be an indirect result of the modulation of cellular gene expression.
5.2.2.3. Bromodomain targeted inhibitors.
The bromodomain and extra-terminal motif (BET) inhibitors JQ1, PFI-1, I-BET-762, TG-101348 and OTX-015 all promoted viral replication. JQ1 (0.5 μM) enhanced viral yield by 100-fold, but ICP4 mRNA levels by only 2-fold. At higher concentrations (1 μM), JQ1 increased the levels of elongating RNAPII (RNAP-S2p), SEC (AFF4), and P-TEFb (CDK9) associated with the ICP4 promoter and transcription start site (TSS), by about 8-fold (Alfonso--Dunn et al., 2020). PFI-1 (0.5 μM) or I-BET-762 (1 μM) enhanced viral yield by 10-fold, while the latter increased ICP4 mRNA levels by only about 2-fold. TG-101348 enhanced viral yield by 10-fold at 3 μM. The authors concluded that these effects were mediated by BRD4 inhibition (Ren et al., 2016). However, BRD4 inhibition by 1 μM JQ1 (added concomitantly with the infecting virions) had no effect on transcription of another alphaherpesvirus, pseudorabies virus, in porcine kidney cells in another study, although viral replication was not analyzed (Wang et al., 2020). Treatment of HEK293 cells with JQ-1, OTX-015 or I-BET 151 decreased H3K9, H3K27, H4K8, H4K12 and H4K16, but not H3K56, acetylation, but these effects were only evaluated in non-infected cells (Wang et al., 2020). BRD4 inhibition was proposed to induce genomic instability, increasing the levels of cytoplasmic DNA, which in turn would promote the secretion of type I interferon and proinflammatory cytokines through cGAS/STING/TBK1/IRF3 signaling (Wang et al., 2020). This mechanism would result in broad antiviral activity through inhibition of viral attachment.
Like those of the histone KMT inhibitors, those of the molecules targeting bromodomains were not always consistent with what would be expected by extrapolating from their effects on cellular gene expression, and often the effects on viral transcription are secondary to the alterations in cellular gene expression.
6. Uniqueness of the HSV-1 chromatin
6.1. The distinct epigenetic landscape of the HSV-1 chromatin
6.1.1. Current models of HSV-1 chromatin and epigenetic regulation
All these small molecule epigenetic modulators were tested for their effects on HSV-1 under the models postulating similar mechanisms of regulation for viral or cellular chromatin, and the effects were most often consistent with the effects predicted under these models - with some notable exceptions (Arbuckle et al., 2017; Wang et al., 2020). However, the chromatinization of the HSV-1 DNA is clearly distinct from that of cellular DNA, regardless of whether the transcribed viral DNA is chromatinized in highly dynamic nucleosomes with very short residency times or partly or mostly non-chromatinized. The HSV-1 genome is also gene-rich with minimal to no intergenic regions other than promoters, 5′ or 3’ untranslated regions. IE, E and L genes are not segregated, and in many instances promoters of different kinetic classes are only a few base pairs apart. For example, the ORF of the divergent E UL9 and L UL10 genes overlap by 56 bp, and those of the E TK and L UL24, by 66 bp. The start of the ORF of the also divergent L UL49A and E UL50 are separated only by 16 bp, and those of the L UL51 and E UL52, by 36. Those of the E US3 and L US2, are separated by only 293 bp, and the L UL41 and E UL42, by 475. The normal cellular distribution of epigenetic PTM characteristic of transcription or silencing spans from about 1 to 2 Kbp for H3K4me1, for example, to about 3 Kbp for H3K4me2 or 3, 10 Kbp for H3K9me1 or H3 or H4 acetylation, to up to tens of thousands bp for H3K27me3 (Barski et al., 2007; Heintzman et al., 2007). It is thus difficult to envision how such epigenetic modifications could differentially regulate the expression of each individual HSV-1 gene with no or very short intergenic regions. In some instances, two or more entire genes of different kinetic classes are encoded in only a few thousand base pairs. UL23 (E) and UL24 (L) are encoded in 2,300 bp, for example, whereas US1 (IE), US2 (L) and US3 (E), in 4,117, and UL51 (L) and UL52 (E), in 4,060. It is difficult to envision how could each of these genes be differentially regulated by chromatin modifications that typically span regions that would cover the entirety of the differentially expressed genes. It is also difficult to envision how the chromatin modifications known to modulate access to regularly chromatinized cellular DNA would modulate the access to a DNA template that is either partially chromatinized, chromatinized with sporadic nucleosomes, or chromatinized in highly dynamic nucleosomes. It appears that the mechanisms that participate in the epigenetic regulation of viral gene expression would have to differ to some extent from those that regulate cellular gene expression.
6.1.2. Recent results suggest novel models of HSV-1 chromatin and epigenetic regulation
A series of recent publications have started to provide evidence that the HSV-1 DNA is uniquely chromatinized and regulated by somewhat different epigenetic mechanisms than most cellular DNA (Table 1). ChIP-seq was used to map the interactions between HSV-1 DNA and total histone H3, or H3 with specific PTM characteristic of transcription or silencing (Gao et al., 2020). The levels of viral DNA that co-immunoprecipitated with total H3, or with H3 with the different PTM, varied with infection time. Early in infection, from 1 to 2 h, the interactions with histone H3 bearing PTM associated with non-transcribed genes, such as H3K27me3 and H3K9me3, were enriched, while those associated with transcribed genes, like H3K27ac, were depleted. The former two PTM were relatively depleted later on, from 8 to 12 h, to be replaced with PTM characteristic of highly transcribed genes, such as H3K27ac or, to a lesser extent, H3K4me3 (Gao et al., 2020).
Table 1.
Genome wide analyses of HSV-1 chromatin.
| Model | Technique | Nucleosome/Histone | PTM | Reference | |
|---|---|---|---|---|---|
| Cell culture | Lytic | MNase-microarray | Nucleosome | N/A | Oh et al. (2015) |
| Cell culture | Lytic | aniPOND-Proteomics | H1.0, H1.1, H1.2, Hist1H2AC, Hist2H2AB, H2AFX, H2AFY, H2AFY2 | N/A | Dembowskiand deLuaca, 2015 |
| Cell culture | Lytic | MNase-Frac-Seq | Nucleosome/H3 | N/A | Huet al.(2019) |
| Cell culture | Lytic | ChIP-Seq | H3 | K4me3, K9me3, K27me3, K27ac | Dremelet al.(2019) |
| Cell culture | Lytic | ATAC-Seq | Nucleosome | N/A | McSwiggen et al. (2019) |
| Cell culture | Lytic | ATAC-Seq | Nucleosome | N/A | Dremeland DeLuca.2019 |
| Cell culture | Lytic | ChIP-Seq | H3 | K4me3, K9me3, K27me3, K27ac | Gaoet al.(2020) |
| Mouse TG | Latent | ChIP-Seq | H3 | K27me3 | Washington et al. (2019) |
The levels of each different modification did not correlate to the levels of transcription of any given HSV-1 gene. Moreover, the enrichment on the different PTM characteristic of active or repressed transcription on the viral genome did not correlate with other PTM characteristic of the same transcription activity, or counter-correlate with the opposite ones. In contrast, and as expected, the PTM on the cellular genome correlated with other PTM characteristic of the same transcription state and counter-correlated with those of the opposed transcription state. Transcription and PTM levels in any HSV-1 gene resolved and clustered according to time after infection, not individual gene transcription level (Gao et al., 2020). The changes in the histone PTM thus occur synchronously through the entire HSV-1 genome, and regardless of the levels of transcription of any individual gene, as the infection progresses. In contrast, the histone PTM typically change in the cellular chromatin according to the level of transcription of the individual gene.
6.2. The peculiar dynamics of the HSV-1 chromatin
6.2.1. HSV-1 chromatin has distinct biophysical properties
In contrast to cellular, HSV-1 DNA is protected from MCN digestion mostly to very large sizes, heterogeneously sized, or mono-nucleosome sized fragments even after very short digestions. Moreover, whereas the percentage of cellular soluble chromatin DNA digested to mono-nucleosome size increases with digestion time, as expected, approximately 20% of the viral DNA in the soluble chromatin is protected in mono-nucleosome complexes at any time (Lacasse and Schang, 2010, 2012). This highly unexpected protection pattern is the most consistent with the models in which the viral chromatin is so dynamic that the residency time of the core nucleosomes is of the same scale as that of the linker histones, and thus the mono-nucleosomes are degraded at basically the same rate at which they are released from the long poly-nucleosome chains in the insoluble chromatin. Then, cleavage, or tagmentation, would occur at random in the core nucleosome or linker DNA, yielding the observed variety of fragments of heterogeneous sizes migrating as a smear in the agarose gel.
The forty-one chromatin proteins and epigenetic modifiers detected to interact with lytic HSV-1 genomes by iPOND or aniPOND also supports a highly dynamic state for the viral chromatin (Dembowski and DeLuca, 2015). Such a dynamic chromatin would also result in inefficient cross-linking, as the core nucleosomes would not interact with any given sequence of DNA for long, and thus the commonly observed low ChIP efficiency of HSV-1 DNA.
The nuclease protection assays were thus modified to minimize the digestion of any unstable poly-nucleosome intermediate. In brief, digestion is performed while centrifuging to separate insoluble and soluble chromatin. Every 5 min, the soluble chromatin is recovered and the MCN in this fraction is immediately quenched, whereas the still insoluble chromatin is resuspended in fresh MCN to continue the digestion. Any poly-nucleosome already released into the soluble chromatin fraction is no longer exposed to MCN and thus cannot be further degraded regardless of the dynamics of the nucleosomes. All soluble chromatin is then pooled and resolved by hydrodynamic ratio in sucrose gradients, which resolve native poly-nucleosome chains. Under this modified protection, named “serial” digestion, the viral DNA is quantitatively protected to sizes corresponding to those of mono-to poly-nucleosomes in complexes that have the hydrodynamic ratio of cellular mono-to poly-nucleosomes (Lacasse and Schang, 2010, 2012), in which it interacts with histone H3 as shown by ChIP of the HSV-1 DNA in all chromatin fractions (Hu et al., 2019). The simplest model for these complexes is that HSV-1 forms highly dynamic nucleosomes during lytic infections. Such highly dynamic nucleosomes are also consistent with the unique patterns of protection to nucleases or tagmentation (Dremel and DeLuca, 2019; Lacasse and Schang, 2010, 2012; McSwiggen et al., 2019), as well as the commonly observed low efficiency of co-immunoprecipitation of HSV-1 DNA with histones.
6.2.2. The major HSV-1 transcription activator, ICP4, modulates chromatin dynamics
The dynamics of the viral chromatin could well be related to transcription, in that chromatin generally regulates the accessibility of DNA to transcription proteins. Interestingly, the major essential activator of HSV-1 gene expression, ICP4, is necessary and sufficient to regulate the dynamics of all types of histones, and particularly so in the viral replication compartments (Gibeault et al., 2016). ICP4 interacts widely with the viral genome, by recognizing loosely defined binding sites (Dremel and DeLuca, 2019). The authors concluded that those interactions replaced nucleosomes (Dremel and DeLuca, 2019), but it is also possible that ICP4 binding to the viral DNA with low affinity may well contribute to modulate the residency time of the core nucleosome, resulting in the highly dynamic chromatin (Conn et al., 2008, 2011, 2013; Conn and Schang, 2013; Gibeault et al., 2016; Hu et al., 2019; Lacasse and Schang, 2010, 2012; McSwiggen et al., 2019). The enrichment in epigenetic modifiers interacting with the HSV-1 genome detected by iPOND and aniPOND is also consistent with a highly dynamic viral chromatin (Dembowski and DeLuca, 2015).
6.2.3. HSV-1 chromatin dynamics and transcription
The dynamics of the viral chromatin could well be related to transcription levels. One could envision that the genes in the most dynamic chromatin would be more often accessible and therefore transcribed to higher levels than those in less dynamic chromatin, dynamics which could be modulated by PTM.
The model proposing transcriptional regulation by chromatin dynamics was tested by analyzing the DNA in each fraction after the serial digestion followed by sucrose gradient fractionation and deep sequencing, “MCN-Frac-Seq” (Hu et al., 2019). Accessibility of the DNA in the chromatin is probed by the restricted serial MCN digestion, resolving the released chromatin fragments through a sucrose gradient, and deep sequencing the DNA in each fraction (Hu et al., 2019). If accessibility of each individual gene dictated its transcription, then all highly transcribed genes should be equally accessible, and thus enriched in the fractions containing the shortest poly-nucleosomes, and all poorly transcribed genes should be equally inaccessible, and thus enriched in the largest poly-nucleosome fractions. Instead, the accessibility of the entire genome was related to its global level of transcription (Hu et al., 2019). In infections treated with the protein synthesis inhibitor cycloheximide (CHX), conditions in which only the IE genes are transcribed, for example, the IE genes were not more (or less) accessible than the rest of the genome. However, the entire genome was far less accessible than in untreated infections (Hu et al., 2019). Roscovitine, the (indirect) inhibitor of global HSV-1 transcription (Diwan et al., 2004; Schang et al., 1998, 1999) resulted in similar global inaccessibility of the HSV-1 genome. Phosphonoacetic acid (PAA-which inhibits DNA replication and thus the transcription of only the strictly L genes) did not result in less accessibility to the L genes, and the (largely transcribed) viral genomes had similar accessibility than in the untreated infections (Hu et al., 2019). As a caveat, population analyses cannot resolve any small subpopulation of transcribed genomes from a vast majority of non-transcribed ones and some populations of genomes in the same infected cell are transcribed whereas other are not (Cohen and Kobiler, 2016; Kobiler and Afriat, 2021; Kobiler et al., 2010, 2011). Nonetheless, the simultaneous changes in accessibility and general transcription activity are the most consistent with any model proposing that chromatin accessibility regulates the transcriptional competency of the HSV-1 genomes, whereas the transcription level of any given gene is regulated by promoter-specific factors, likely in only a minority of the genomes in the cell (Hu et al., 2019).
ATAC-seq or MCN protection assays have also shown no preferential accessibility to transcribed over non-transcribed genes (Dremel and DeLuca, 2019; McSwiggen et al., 2019), and ChIP-seq has shown no good correlation between specific histone PTM and the level of transcription of a given gene, but rather global changes in PTMs along the entire viral genomes that correlate with the global level of transcription of the genomes (Dremel and DeLuca, 2019; Gao et al., 2020). Using high resolution imaging, Dr. O’Hare has shown that the HSV-1 genomes must decondense for transcription to occur (Sekine et al., 2017), which is consistent with them becoming competent by increased chromatin dynamics.
7. The unique characteristics of the HSV-1 chromatin and its epigenetic regulation indicate a potential role for epigenetic targets in antiviral therapy
Chromatin epigenetic PTM associated with transcription or silencing are present in more or less transcribed viral genomes, respectively, and several epigenetic inhibitors modulate viral transcription and replication, strongly supporting that epigenetics regulate HSV-1 transcription. More specifically, chromatin epigenetics appears to regulate the transcriptional competency of the viral genomes, not the actual transcription levels of individual genes. Cellular transcription competency is also regulated epigenetically, but the mechanisms appear to be divergent. Whereas in cellular chromatin competency is often marked by specific epigenetic PTM at regions spanning the promoters of the regulated genes, which are sometimes marked with ambivalent or poised PTM (Barski et al., 2007; Blanco et al., 2020; Sen et al., 2016), the competency of the viral chromatin appears to be determined by the overall dynamics of the viral chromatin on the entire genomes (Fig. 8) (Hu et al., 2019); recently reviewed in (Kobiler and Afriat, 2021). These dynamics themselves appear to be in part determined by global enrichment in particular PTM in the viral chromatin, in that the PTM change along infection as HSV-1 DNA accessibility does (Gao et al., 2020; Hu et al., 2019).
Fig. 8.
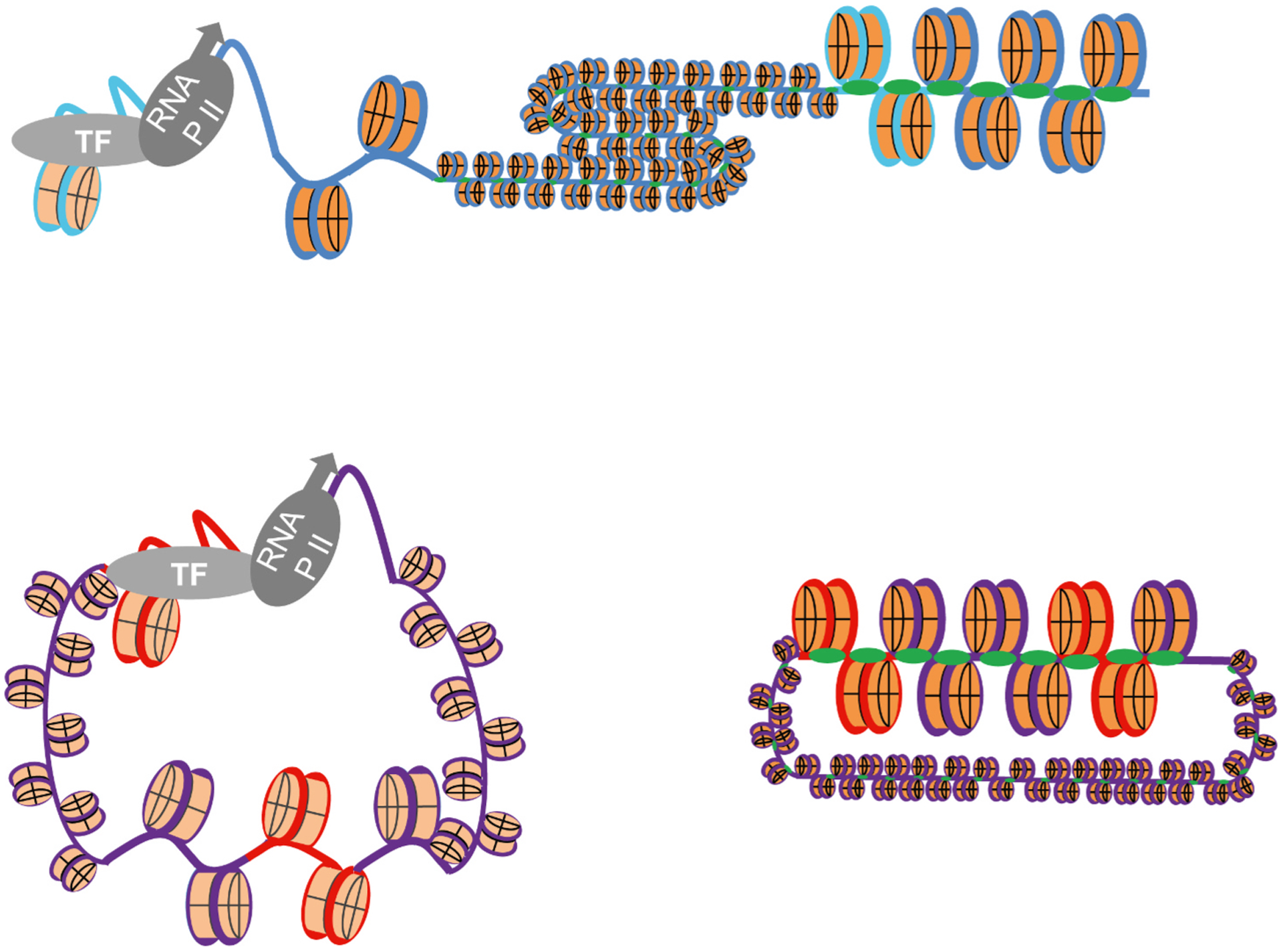
Chromatin dynamics mediated regulation of cellular or viral transcription. Epigenetic regulation of a specific cellular gene with PTM characteristic of active transcription increases its accessibility to transcription proteins and therefore activates transcription, whereas another gene in the same chromosome remains tightly chromatinized with PTM characteristic of repression and transcriptionally inactive (top). Epigenetic regulation of HSV-1 genomes, in contrast, dictates the accessibility to entire genomes (bottom). HSV-1 genomes in dynamic and accessible chromatin, enriched in PTM associated with transcription, are accessible to transcription proteins and therefore transcriptionally competent although only the gene on top is actually transcribed at this time (left). HSV-1 genomes in less dynamic or accessible chromatin, enriched in PTM characteristic of silencing, are not accessible to transcription proteins and therefore the entire genomes are transcriptionally incompetent (right). Blue line, cellular DNA; Purple line, viral DNA; light blue line, cellular promoters; red line, viral promoters.
These global dynamic changes are consistent with the architecture of the viral promoters. The IE promoters are activated early on, when the viral chromatin is less dynamic and has PTM associated with less accessibility. These promoters are the ones that have the most binding sites for transcription factors (Fig. 3), which would allow them to be accessed even in less dynamic chromatin. The E promoters have only a couple of binding sites for transcription factors (Fig. 3) and would thus not be easily bound by them until the global viral chromatin dynamics increase. The minimalistic L promoters, with basically only a TATA box or an initiator element, would only be accessible to transcription proteins when the viral chromatin is the most dynamic (Hu et al., 2019).
No cellular chromatin is currently known to have similar dynamics to the transcriptionally competent HSV-1 chromatin, or to be globally regulated by chromatin dynamics as the viral chromatin appears to be. Cellular promoters typically have many more binding sites for a variety of transcription factors, which would allow their access in less dynamic chromatin.
The differences between the viral and cellular chromatin and their regulation are so marked that perhaps unique names are needed for the viral nucleosome- and chromatin-like structures. It is theoretically possible to exploit the uniqueness of the viral chromatin to develop antiviral approaches targeting the factors that determine its extreme dynamics, thus minimizing the risk of using epigenetic approaches to episodically treat life-long latent HSV-1 infections. For this potential to be materialized, however, the mechanisms leading to the unique properties of the viral chromatin must be identified and characterized, such that suitable targets can be identified. Many of the multiple epigenetic drugs being developed are likely to eventually show not to have much of an effect i in humans. Combination of such epigenetic drugs targeting relevant targets particularly involved in the regulation of the highly dynamic viral chromatin could open new avenues toward the development of innovative antivirals.
Acknowledgements
The LMS lab is supported by NIH/NIAID 1R01AI153396, NIH/NINSD 5R21NS111416, a Fast Grant, NIH 3UM1HG009393, and Cornell University. Fig. 4 is based on an original cartoon by Gustavo Delhon, Ph. D. (University of Nebraska-Lincoln).
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Alfonso-Dunn R, Arbuckle JH, Vogel JL, Kristie TM, 2020. Inhibition of the super elongation complex suppresses herpes simplex virus immediate early gene expression, lytic infection, and reactivation from latency. mBio 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amelio AL, Giordani NV, Kubat NJ, O’Neil JE, Bloom DC, 2006a. Deacetylation of the herpes simplex virus type 1 latency-associated transcript (LAT) enhancer and a decrease in LAT abundance precede an increase in ICP0 transcriptional permissiveness at early times postexplant. J. Virol 80, 2063–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amelio AL, McAnany PK, Bloom DC, 2006b. A chromatin insulator-like element in the herpes simplex virus type 1 latency-associated transcript region binds CCCTC-binding factor and displays enhancer-blocking and silencing activities. J. Virol 80, 2358–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbuckle JH, Gardina PJ, Gordon DN, Hickman HD, Yewdell JW, Pierson TC, Myers TG, Kristie TM, 2017. Inhibitors of the histone methyltransferases EZH2/1 induce a potent antiviral state and suppress infection by diverse viral pathogens. mBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert M, Madden EA, Loprieno M, DeSilva Feelixge HS, Stensland L, Huang ML, Greninger AL, Roychoudhury P, Niyonzima N, Nguyen T, Magaret A, Galleto R, Stone D, Jerome KR, 2016. In vivo disruption of latent HSV by designer endonuclease therapy. JCI Insight 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert M, Strongin DE, Roychoudhury P, Loprieno MA, Haick AK, Klouser LM, Stensland L, Huang ML, Makhsous N, Tait A, De Silva Feelixge HS, Galetto R, Duchateau P, Greninger AL, Stone D, Jerome KR, 2020. Gene editing and elimination of latent herpes simplex virus in vivo. Nat. Commun 11, 4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Trang P, Li H, Kim K, Zhou T, Liu F, 2008. Effective inhibition in animals of viral pathogenesis by a ribozyme derived from RNase P catalytic RNA. Proc. Natl. Acad. Sci. U. S. A 105, 10919–10924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K, 2007. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837. [DOI] [PubMed] [Google Scholar]
- Beagan JA, Phillips-Cremins JE, 2020. On the existence and functionality of topologically associating domains. Nat. Genet 52, 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell AC, West AG, Felsenfeld G, 1999. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 98, 387–396. [DOI] [PubMed] [Google Scholar]
- Blanco E, Gonzalez-Ramirez M, Alcaine-Colet A, Aranda S, Di Croce L, 2020. The bivalent genome: characterization, structure, and regulation. Trends Genet. 36, 118–131. [DOI] [PubMed] [Google Scholar]
- Bloom DC, 2004. HSV LAT and neuronal survival. Int. Rev. Immunol 23, 187–198. [DOI] [PubMed] [Google Scholar]
- Bloom DC, Giordani NV, Kwiatkowski DL, 2010. Epigenetic regulation of latent HSV-1 gene expression. Biochim. Biophys. Acta 1799, 246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ, 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera JR, Charron AJ, Leib DA, 2018. Neuronal subtype determines herpes simplex virus 1 latency-associated-transcript promoter activity during latency. J. Virol 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Xu H, Liu J, Cui Y, Luo X, Zhou Y, Chen Q, Jiang L, 2017. Efficacy and safety of nucleoside antiviral drugs for treatment of recurrent herpes labialis: a systematic review and meta-analysis. J. Oral Pathol. Med 46, 561–568. [DOI] [PubMed] [Google Scholar]
- Chen SH, Kramer MF, Schaffer PA, Coen DM, 1997. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J. Virol 71, 5878–5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JH, Whiteley M, Felsenfeld G, 1993. A 5’ element of the chicken beta-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell 74, 505–514. [DOI] [PubMed] [Google Scholar]
- Cliffe AR, Arbuckle JH, Vogel JL, Geden MJ, Rothbart SB, Cusack CL, Strahl BD, Kristie TM, Deshmukh M, 2015. Neuronal stress pathway mediating a histone methyl/phospho switch is required for herpes simplex virus reactivation. Cell Host Microbe 18, 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Garber DA, Knipe DM, 2009. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J. Virol 83, 8182–8190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Knipe DM, 2008. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J. Virol 82, 12030–12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Wilson AC, 2017. Restarting lytic gene transcription at the onset of herpes simplex virus reactivation. J. Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen EM, Avital N, Shamay M, Kobiler O, 2020. Abortive herpes simplex virus infection of nonneuronal cells results in quiescent viral genomes that can reactivate. Proc. Natl. Acad. Sci. U. S. A 117, 635–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen EM, Kobiler O, 2016. Gene expression correlates with the number of herpes viral genomes initiating infection in single cells. PLoS Pathog. 12, e1006082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman HM, Connor V, Cheng ZSC, Grey F, Preston CM, Efstathiou S, 2008. Histone modifications associated with herpes simplex virus type 1 genomes during quiescence and following ICP0-mediated de-repression. J. Gen. Virol 89, 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn KL, Hendzel MJ, Schang LM, 2008. Linker histones are mobilized during infection with herpes simplex virus type 1. J. Virol 82, 8629–8646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn KL, Hendzel MJ, Schang LM, 2011. Core histones H2B and H4 are mobilized during infection with herpes simplex virus 1. J. Virol 85, 13234–13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn KL, Hendzel MJ, Schang LM, 2013. The differential mobilization of histones H3.1 and H3.3 by herpes simplex virus 1 relates histone dynamics to the assembly of viral chromatin. PLoS Pathog. 9, e1003695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn KL, Schang LM, 2013. Chromatin dynamics during lytic infection with herpes simplex virus 1. Viruses 5, 1758–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creech CC, Neumann DM, 2010. Changes to euchromatin on LAT and ICP4 following reactivation are more prevalent in an efficiently reactivating strain of HSV-1. PloS One 5, e15416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danaher RJ, Jacob RJ, Steiner MR, Allen WR, Hill JM, Miller CS, 2005. Histone deacetylase inhibitors induce reactivation of herpes simplex virus type 1 in a latency-associated transcript-independent manner in neuronal cells. J. Neurovirol 11, 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembowski JA, DeLuca NA, 2015. Selective recruitment of nuclear factors to productively replicating herpes simplex virus genomes. PLoS Pathog. 11, e1004939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmane SL, Fraser NW, 1989. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J. Virol 63, 943–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwan P, Lacasse JJ, Schang LM, 2004. Roscovitine inhibits activation of promoters in herpes simplex virus type 1 genomes independently of promoter-specific factors. J. Virol 78, 9352–9365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll JR, Hoebe K, Thompson RL, Sawtell NM, 2020. Resolution of herpes simplex virus reactivation in vivo results in neuronal destruction. PLoS Pathog. 16, e1008296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drayman N, Karin O, Mayo A, Danon T, Shapira L, Rafael D, Zimmer A, Bren A, Kobiler O, Alon U, 2017. Dynamic proteomics of herpes simplex virus infection. mBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dremel SE, DeLuca NA, 2019. Herpes simplex viral nucleoprotein creates a competitive transcriptional environment facilitating robust viral transcription and host shut off. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du T, Zhou G, Roizman B, 2013. Modulation of reactivation of latent herpes simplex virus 1 in ganglionic organ cultures by p300/CBP and STAT3. Proc. Natl. Acad. Sci. U. S. A 110, E2621–E2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Earnshaw WC, Findlay J, Lomonte P, 1999. Specific destruction of kinetochore protein CENP-C and disruption of cell division by herpes simplex virus immediate-early protein Vmw110. EMBO J. 18, 1526–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferenczy MW, DeLuca NA, 2009. Epigenetic modulation of gene expression from quiescent herpes simplex virus genomes. J. Virol 83, 8514–8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flowerdew SE, Wick D, Himmelein S, Horn AK, Sinicina I, Strupp M, Brandt T, Theil D, Hufner K, 2013. Characterization of neuronal populations in the human trigeminal ganglion and their association with latent herpes simplex virus-1 infection. PloS One 8, e83603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Chen L, Tang SB, Long QY, He JL, Zhang NA, Shu HB, Chen ZX, Wu M, Li LY, 2020. The epigenetic landscapes of histone modifications on HSV-1 genome in human THP-1 cells. Antivir. Res 176, 104730. [DOI] [PubMed] [Google Scholar]
- Gibeault RL, Conn KL, Bildersheim MD, Schang LM, 2016. An essential viral transcription activator modulates chromatin dynamics. PLoS Pathog. 12, e1005842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson W, Roizman B, 1971. Compartmentalization of spermine and spermidine in the herpes simplex virion. Proc. Natl. Acad. Sci. U. S. A 68, 2818–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross S, Catez F, Masumoto H, Lomonte P, 2012. Centromere architecture breakdown induced by the viral E3 ubiquitin ligase ICP0 protein of herpes simplex virus type 1. PloS One 7, e44227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Liang Y, Mandel G, Roizman B, 2005. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc. Natl. Acad. Sci. U. S. A 102, 7571–7576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Roizman B, 2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc. Natl. Acad. Sci. U. S. A 104, 17134–17139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafezi W, Lorentzen EU, Eing BR, Muller M, King NJ, Klupp B, Mettenleiter TC, Kuhn JE, 2012. Entry of herpes simplex virus type 1 (HSV-1) into the distal axons of trigeminal neurons favors the onset of nonproductive, silent infection. PLoS Pathog. 8, e1002679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han F, Miyagawa Y, Verlengia G, Ingusci S, Soukupova M, Simonato M, Glorioso JC, Cohen JB, 2018. Cellular antisilencing elements support transgene expression from herpes simplex virus vectors in the absence of immediate early gene expression. J. Virol 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM, 2000. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405, 486–489. [DOI] [PubMed] [Google Scholar]
- Harrison KS, Zhu L, Thunuguntla P, Jones C, 2020. Herpes simplex virus 1 regulates beta-catenin expression in TG neurons during the latency-reactivation cycle. PloS One 15, e0230870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B, 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet 39, 311–318. [DOI] [PubMed] [Google Scholar]
- Herrera FJ, Triezenberg SJ, 2004. VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J. Virol 78, 9689–9696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JM, Quenelle DC, Cardin RD, Vogel JL, Clement C, Bravo FJ, Foster TP, Bosch-Marce M, Raja P, Lee JS, Bernstein DI, Krause PR, Knipe DM, Kristie TM, 2014. Inhibition of LSD1 reduces herpesvirus infection, shedding, and recurrence by promoting epigenetic suppression of viral genomes. Sci. Transl. Med 6, 265ra169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs WE 2nd, DeLuca NA, 1999. Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. J. Virol 73, 8245–8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Depledge DP, Flores Cortes E, Breuer J, Schang LM, 2019. Chromatin dynamics and the transcriptional competence of HSV-1 genomes during lytic infections. PLoS Pathog. 15, e1008076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Kent JR, Placek B, Whelan KA, Hollow CM, Zeng PY, Fraser NW, Berger SL, 2006. Trimethylation of histone H3 lysine 4 by Set1 in the lytic infection of human herpes simplex virus 1. J. Virol 80, 5740–5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James SH, Prichard MN, 2014. Current and future therapies for herpes simplex virus infections: mechanism of action and drug resistance. Curr Opin Virol 8, 54–61. [DOI] [PubMed] [Google Scholar]
- Johnson KE, Bottero V, Flaherty S, Dutta S, Singh VV, Chandran B, 2014. IFI16 restricts HSV-1 replication by accumulating on the hsv-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLoS Pathog. 10, e1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalamvoki M, Roizman B, 2010. Circadian CLOCK histone acetyl transferase localizes at ND10 nuclear bodies and enables herpes simplex virus gene expression. Proc. Natl. Acad. Sci. U. S. A 107, 17721–17726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Tanaka M, Yokoymama A, Matsuda G, Kato K, Kagawa H, Hirai K, Roizman B, 2001. Herpes simplex virus 1 alpha regulatory protein ICP0 functionally interacts with cellular transcription factor BMAL1. Proc. Natl. Acad. Sci. U. S. A 98, 1877–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney JL, Guinness ME, Reiss M, Lacy J, 2001. Antisense to the Epstein-Barr virus (EBV)-encoded latent membrane protein 1 (LMP-1) sensitizes EBV-immortalized B cells to transforming growth factor-beta and chemotherapeutic agents. Int. J. Canc 91, 89–98. [DOI] [PubMed] [Google Scholar]
- Kent JR, Zeng PY, Atanasiu D, Gardner J, Fraser NW, Berger SL, 2004. During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J. Virol 78, 10178–10186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC, 2012. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of latent HSV-1 in neurons. PLoS Pathog. 8, e1002540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B, 2007. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 128, 1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenova EM, Nicolas RH, Paterson HF, Carne AF, Heath CM, Goodwin GH, Neiman PE, Lobanenkov VV, 1993. CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c-myc gene, is an 11-Zn-finger protein differentially expressed in multiple forms. Mol. Cell Biol 13, 7612–7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipe DM, Cliffe A, 2008. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol 6, 211–221. [DOI] [PubMed] [Google Scholar]
- Knipe DM, Raja P, Lee J, 2017. Viral gene products actively promote latent infection by epigenetic silencing mechanisms. Curr Opin Virol 23, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Kim JY, Camarena V, Roehm PC, Chao MV, Wilson AC, Mohr I, 2012a. A primary neuron culture system for the study of herpes simplex virus latency and reactivation. J Vis Exp 62, e3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Wilson AC, Chao MV, Mohr I, 2012b. Control of viral latency in neurons by axonal mTOR signaling and the 4E-BP translation repressor. Genes Dev. 26, 1527–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobiler O, Afriat A, 2021. The fate of incoming HSV-1 genomes entering the nucleus. Curr. Issues Mol. Biol 41, 221–266. [DOI] [PubMed] [Google Scholar]
- Kobiler O, Brodersen P, Taylor MP, Ludmir EB, Enquist LW, 2011. Herpesvirus replication compartments originate with single incoming viral genomes. mBio 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobiler O, Lipman Y, Therkelsen K, Daubechies I, Enquist LW, 2010. Herpesviruses carrying a Brainbow cassette reveal replication and expression of limited numbers of incoming genomes. Nat. Commun 1, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosz-Vnenchak M, Jacobson J, Coen DM, Knipe DM, 1993. Evidence for a novel regulatory pathway for herpes simplex virus gene expression in trigeminal ganglion neurons. J. Virol 67, 5383–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MF, Coen DM, 1995. Quantification of transcripts from the ICP4 and thymidine kinase genes in mouse ganglia latently infected with herpes simplex virus. J. Virol 69, 1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristie TM, 2015. Dynamic modulation of HSV chromatin drives initiation of infection and provides targets for epigenetic therapies. Virology 479–480, 555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristie TM, Vogel JL, Sears AE, 1999. Nuclear localization of the C1 factor (host cell factor) in sensory neurons correlates with reactivation of herpes simplex virus from latency. Proc. Natl. Acad. Sci. U. S. A 96, 1229–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubat NJ, Amelio AL, Giordani NV, Bloom DC, 2004a. The herpes simplex virus type 1 latency-associated transcript (LAT) enhancer/rcr is hyperacetylated during latency independently of LAT transcription. J. Virol 78, 12508–12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubat NJ, Tran RK, McAnany P, Bloom DC, 2004b. Specific histone tail modification and not DNA methylation is a determinant of herpes simplex virus type 1 latent gene expression. J. Virol 78, 1139–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutluay SB, DeVos SL, Klomp JE, Triezenberg SJ, 2009. Transcriptional coactivators are not required for herpes simplex virus type 1 immediate-early gene expression in vitro. J. Virol 83, 3436–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutluay SB, Triezenberg SJ, 2009. Role of chromatin during herpesvirus infections. Biochim. Biophys. Acta 1790, 456–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski DL, Thompson HW, Bloom DC, 2009. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J. Virol 83, 8173–8181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacasse JJ, Schang LM, 2010. During lytic infections, herpes simplex virus type 1 DNA is in complexes with the properties of unstable nucleosomes. J. Virol 84, 1920–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacasse JJ, Schang LM, 2012. Herpes simplex virus 1 DNA is in unstable nucleosomes throughout the lytic infection cycle, and the instability of the nucleosomes is independent of DNA replication. J. Virol 86, 11287–11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang F, Li X, Vladimirova O, Hu B, Chen G, Xiao Y, Singh V, Lu D, Li L, Han H, Wickramasinghe JM, Smith ST, Zheng C, Li Q, Lieberman PM, Fraser NW, Zhou J, 2017. CTCF interacts with the lytic HSV-1 genome to promote viral transcription. Sci. Rep 7, 39861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Raja P, Knipe DM, 2016. Herpesviral ICP0 protein promotes two waves of heterochromatin removal on an early viral promoter during lytic infection. mBio 7 e02007–02015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Raja P, Pan D, Pesola JM, Coen DM, Knipe DM, 2018. CCCTC-binding factor Acts as a heterochromatin barrier on herpes simplex viral latent chromatin and contributes to poised latent infection. mBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinbach SS, Summers WC, 1980. The structure of herpes simplex virus type 1 DNA as probed by micrococcal nuclease digestion. J. Gen. Virol 51, 45–59. [DOI] [PubMed] [Google Scholar]
- Li X, Yu Y, Lang F, Chen G, Wang E, Li L, Li Z, Yang L, Cao X, Fraser NW, Zhou J, 2021. Cohesin promotes HSV-1 lytic transcription by facilitating the binding of RNA Pol II on viral genes. Virol. J 18, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Haarhuis JHI, Sedeno Cacciatore A, Oldenkamp R, van Ruiten MS, Willems L, Teunissen H, Muir KW, de Wit E, Rowland BD, Panne D, 2020. The structural basis for cohesin-CTCF-anchored loops. Nature 578, 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Vogel JL, Arbuckle JH, Rai G, Jadhav A, Simeonov A, Maloney DJ, Kristie TM, 2013. Targeting the JMJD2 histone demethylases to epigenetically control herpesvirus infection and reactivation from latency. Sci. Transl. Med 5, 167ra165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM, 2009. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat. Med 15, 1312–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman PM, 2016. Epigenetics and genetics of viral latency. Cell Host Microbe 19, 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippe R, 2020. Characterization of extracellular HSV-1 virions by proteomics. Methods Mol. Biol 2060, 279–288. [DOI] [PubMed] [Google Scholar]
- Lobanenkov VV, Nicolas RH, Adler VV, Paterson H, Klenova EM, Polotskaja AV, Goodwin GH, 1990. A novel sequence-specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5’-flanking sequence of the chicken c-myc gene. Oncogene 5, 1743–1753. [PubMed] [Google Scholar]
- Lomonte P, Morency E, 2007. Centromeric protein CENP-B proteasomal degradation induced by the viral protein ICP0. FEBS Lett. 581, 658–662. [DOI] [PubMed] [Google Scholar]
- Lomonte P, Sullivan KF, Everett RD, 2001. Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J. Biol. Chem 276, 5829–5835. [DOI] [PubMed] [Google Scholar]
- Loret S, Guay G, Lippe R, 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol 82, 8605–8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashchuk V, Everett RD, 2010. Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. J. Virol 84, 4026–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JZ, Russell TA, Spelman T, Carbone FR, Tscharke DC, 2014. Lytic gene expression is frequent in HSV-1 latent infection and correlates with the engagement of a cell-intrinsic transcriptional response. PLoS Pathog. 10, e1004237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis TP, Imai Y, Yang L, Vallas V, Krause PR, 2007. Herpes simplex virus type 2 (HSV-2) establishes latent infection in a different population of ganglionic neurons than HSV-1: role of latency-associated transcripts. J. Virol 81, 1872–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark KE, Wald A, Magaret AS, Selke S, Olin L, Huang ML, Corey L, 2008. Rapidly cleared episodes of herpes simplex virus reactivation in immunocompetent adults. J. Infect. Dis 198, 1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroui MA, Calle A, Cohen C, Streichenberger N, Texier P, Takissian J, Rousseau A, Poccardi N, Welsch J, Corpet A, Schaeffer L, Labetoulle M, Lomonte P, 2016. Latency entry of herpes simplex virus 1 is determined by the interaction of its genome with the nuclear environment. PLoS Pathog. 12, e1005834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSwiggen DT, Hansen AS, Teves SS, Marie-Nelly H, Hao Y, Heckert AB, Umemoto KK, Dugast-Darzacq C, Tjian R, Darzacq X, 2019. Evidence for DNA-mediated nuclear compartmentalization distinct from phase separation. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morency E, Sabra M, Catez F, Texier P, Lomonte P, 2007. A novel cell response triggered by interphase centromere structural instability. J. Cell Biol 177, 757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muggeridge MI, Fraser NW, 1986. Chromosomal organization of the herpes simplex virus genome during acute infection of the mouse central nervous system. J. Virol 59, 764–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann DM, Bhattacharjee PS, Giordani NV, Bloom DC, Hill JM, 2007a. In vivo changes in the patterns of chromatin structure associated with the latent herpes simplex virus type 1 genome in mouse trigeminal ganglia can be detected at early times after butyrate treatment. J. Virol 81, 13248–13253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann DM, Bhattacharjee PS, Hill JM, 2007b. Sodium butyrate: a chemical inducer of in vivo reactivation of herpes simplex virus type 1 in the ocular mouse model. J. Virol 81, 6106–6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll MP, Hann W, Shivkumar M, Harman LE, Connor V, Coleman HM, Proenca JT, Efstathiou S, 2016. The HSV-1 latency-associated transcript functions to repress latent phase lytic gene expression and suppress virus reactivation from latently infected neurons. PLoS Pathog. 12, e1005539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll MP, Proenca JT, Efstathiou S, 2012. The molecular basis of herpes simplex virus latency. FEMS Microbiol. Rev 36, 684–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon CC, Mavigner M, Sampey GC, Brooks AD, Spagnuolo RA, Irlbeck DM, Mattingly C, Ho PT, Schoof N, Cammon CG, Tharp GK, Kanke M, Wang Z, Cleary RA, Upadhyay AA, De C, Wills SR, Falcinelli SD, Galardi C, Walum H, Schramm NJ, Deutsch J, Lifson JD, Fennessey CM, Keele BF, Jean S, Maguire S, Liao B, Browne EP, Ferris RG, Brehm JH, Favre D, Vanderford TH, Bosinger SE, Jones CD, Routy JP, Archin NM, Margolis DM, Wahl A, Dunham RM, Silvestri G, Chahroudi A, Garcia JV, 2020. Systemic HIV and SIV latency reversal via non-canonical NF-kappaB signalling in vivo. Nature 578, 160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh HS, Neuhausser WM, Eggan P, Angelova M, Kirchner R, Eggan KC, Knipe DM, 2019. Herpesviral lytic gene functions render the viral genome susceptible to novel editing by CRISPR/Cas9. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Sanders IF, Chen EZ, Li H, Tobias JW, Isett RB, Penubarthi S, Sun H, Baldwin DA, Fraser NW, 2015. Genome wide nucleosome mapping for HSV-1 shows nucleosomes are deposited at preferred positions during lytic infection. PloS One 10, e0117471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng GC, Jones C, Ciacci-Zanella J, Stone M, Henderson G, Yukht A, Slanina SM, Hofman FM, Ghiasi H, Nesburn AB, Wechsler SL, 2000. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 287, 1500–1503. [DOI] [PubMed] [Google Scholar]
- Pourchet A, Modrek AS, Placantonakis DG, Mohr I, Wilson AC, 2017. Modeling HSV-1 latency in human embryonic stem cell-derived neurons. Pathogens 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenelle DC, Birkmann A, Goldner T, Pfaff T, Zimmermann H, Bonsmann S, Collins DJ, Rice TL, Prichard MN, 2018. Efficacy of pritelivir and acyclovir in the treatment of herpes simplex virus infections in a mouse model of herpes simplex encephalitis. Antivir. Res 149, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren K, Zhang W, Chen X, Ma Y, Dai Y, Fan Y, Hou Y, Tan RX, Li E, 2016. An epigenetic compound library screen identifies BET inhibitors that promote HSV-1 and −2 replication by bridging P-TEFb to viral gene promoters through BRD4. PLoS Pathog. 12, e1005950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell TA, Tscharke DC, 2016. Lytic promoters express protein during herpes simplex virus latency. PLoS Pathog. 12, e1005729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders I, Boyer M, Fraser NW, 2015. Early nucleosome deposition on, and replication of, HSV DNA requires cell factor PCNA. J. Neurovirol 21, 358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM, Thompson RL, 2020. Alphaherpesvirus latency and reactivation with a focus on herpes simplex virus. Curr. Issues Mol. Biol 41, 267–356. [DOI] [PubMed] [Google Scholar]
- Schang LM, 2018. Timing is everything. mBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schang LM, Phillips J, Schaffer PA, 1998. Requirement for cellular cyclin-dependent kinases in herpes simplex virus replication and transcription. J. Virol 72, 5626–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schang LM, Rosenberg A, Schaffer PA, 1999. Transcription of herpes simplex virus immediate-early and early genes is inhibited by roscovitine, an inhibitor specific for cellular cyclin-dependent kinases. J. Virol 73, 2161–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine E, Schmidt N, Gaboriau D, O’Hare P, 2017. Spatiotemporal dynamics of HSV genome nuclear entry and compaction state transitions using bioorthogonal chemistry and super-resolution microscopy. PLoS Pathog. 13, e1006721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S, Block KF, Pasini A, Baylin SB, Easwaran H, 2016. Genome-wide positioning of bivalent mononucleosomes. BMC Med. Genom 9, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira L, Ralph M, Tomer E, Cohen S, Kobiler O, 2016. Histone deacetylase inhibitors reduce the number of herpes simplex virus-1 genomes initiating expression in individual cells. Front. Microbiol 7, 1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Tscharke DC, 2020. Herpes simplex virus latency is noisier the closer we look. J. Virol 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Neumann DM, 2021. Cohesin subunit Rad21 binds to the HSV-1 genome near CTCF insulator sites during latency in vivo. J. Virol 95 e00364–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theil D, Derfuss T, Paripovic I, Herberger S, Meinl E, Schueler O, Strupp M, Arbusow V, Brandt T, 2003. Latent herpesvirus infection in human trigeminal ganglia causes chronic immune response. Am. J. Pathol 163, 2179–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thellman NM, Triezenberg SJ, 2017. Herpes simplex virus establishment, maintenance, and reactivation: in vitro modeling of latency. Pathogens 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Sawtell NM, 2001. Herpes simplex virus type 1 latency-associated transcript gene promotes neuronal survival. J. Virol 75, 6660–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lint AL, Kleinert L, Clarke SR, Stock A, Heath WR, Carbone FR, 2005. Latent infection with herpes simplex virus is associated with ongoing CD8+ T-cell stimulation by parenchymal cells within sensory ganglia. J. Virol 79, 14843–14851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner LM, DeLuca NA, 2013. Temporal association of herpes simplex virus ICP4 with cellular complexes functioning at multiple steps in PolII transcription. PloS One 8, e78242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Li GL, Ming SL, Wang CF, Shi LJ, Su BQ, Wu HT, Zeng L, Han YQ, Liu ZH, Jiang DW, Du YK, Li XD, Zhang GP, Yang GY, Chu BB, 2020. BRD4 inhibition exerts anti-viral activity through DNA damage-dependent innate immune responses. PLoS Pathog. 16, e1008429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QY, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM, 2005. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. U. S. A 102, 16055–16059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington SD, Edenfield SI, Lieux C, Watson ZL, Taasan SM, Dhummakupt A, Bloom DC, Neumann DM, 2018a. Depletion of the insulator protein CTCF results in herpes simplex virus 1 reactivation in vivo. J. Virol 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington SD, Musarrat F, Ertel MK, Backes GL, Neumann DM, 2018b. CTCF binding sites in the herpes simplex virus 1 genome display site-specific CTCF occupation, protein recruitment, and insulator function. J. Virol 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington SD, Singh P, Johns RN, Edwards TG, Mariani M, Frietze S, Bloom DC, Neumann DM, 2019. The CCCTC binding factor, CTRL2, modulates heterochromatin deposition and the establishment of herpes simplex virus 1 latency in vivo. J. Virol 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson ZL, Washington SD, Phelan DM, Lewin AS, Tuli SS, Schultz GS, Neumann DM, Bloom DC, 2018. In vivo knockdown of the herpes simplex virus 1 latency-associated transcript reduces reactivation from latency. J. Virol 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber ND, Aubert M, Dang CH, Stone D, Jerome KR, 2014. DNA cleavage enzymes for treatment of persistent viral infections: recent advances and the pathway forward. Virology 454–455, 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley R, Baines J, 2018. Clinical Management of Herpes Simplex Virus Infections: Past, Present, and Future. F1000Res 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley RJ, Roizman B, 2001. Herpes simplex virus infections. Lancet 357, 1513–1518. [DOI] [PubMed] [Google Scholar]
- Zhao J, Zhu L, Wijesekera N, Jones C, 2020. Specific akt family members impair stress-mediated transactivation of viral promoters and enhance neuronal differentiation: important functions for maintaining latency. J. Virol 94. [DOI] [PMC free article] [PubMed] [Google Scholar]