

Abstract
Recently, we demonstrated that Pseudomonas aeruginosa Exotoxin T (ExoT) employs two distinct mechanisms to induce potent apoptotic cytotoxicity in a variety of cancer cell lines. We further demonstrated that it can significantly reduce tumor growth in an animal model for melanoma. During these studies, we observed that melanoma cells that were transfected with ExoT failed to undergo mitosis, regardless of whether they eventually succumbed to ExoT-induced apoptosis or survived in ExoT’s presence. In this report, we sought to investigate ExoT’s antiproliferative activity in melanoma. We delivered ExoT into B16 melanoma cells by bacteria (to show necessity) and by transfection (to show sufficiency). Our data indicate that ExoT exerts a potent antiproliferative function in melanoma cells. We show that ExoT causes cell cycle arrest in G1 interphase in melanoma cells by dampening the G1/S checkpoint proteins. Our data demonstrate that both domains of ExoT; (the ADP-ribosyltransferase (ADPRT) domain and the GTPase activating protein (GAP) domain); contribute to ExoT-induced G1 cell cycle arrest in melanoma. Finally, we show that the ADPRT-induced G1 cell cycle arrest in melanoma cells likely involves the Crk adaptor protein. Our data reveal a novel virulence function for ExoT and further highlight the therapeutic potential of ExoT against cancer.
Graphical Abstract
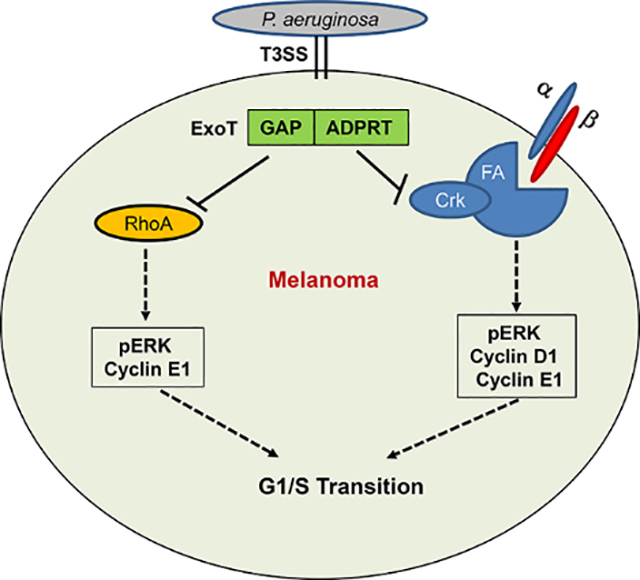
Pseudomonas aeruginosa ExoT causes G1 cell cycles arrest by dampening G1/S transition checkpoint regulators, and both of its domains contribute. The GAP domain likely prevents progression through G1/S checkpoint by targeting RhoA and the ADPRT prevents progression through G1/S checkpoint likely by targeting Crk adaptor protein and by interfering with the anchorage-dependent integrin signaling.
INTRODUCTION
Although, cancer related mortality rates have declined across many cancers in the past decade, cancer continues to have a devastating impact on public health in the United States and across the world (Henley et al., 2020; Mattiuzzi & Lippi, 2020; Siegel, Miller, & Jemal, 2020). The Center for Disease Control and Prevention (CDC) estimates that in 2020, over 1.8 million new cancer cases will be diagnosed, and 606,520 cancer deaths will occur in US alone (Siegel et al., 2020). These daunting statistics highlight the urgent need for new cancer therapeutics.
Due to new technological advances to overcome the barriers in the field of Xenotoxin-based cancer therapeutics, development of bacterial toxin-based immunotoxins, (e.g., Diphtheria toxin (DT) or Pseudomonas Exotoxin (PE) in combination with various immune modalities), has gained significant momentum in the field of cancer therapeutics in recent years with several promising immunotoxins currently in clinical trials (Akbari et al., 2017; Karpiński & Adamczak, 2018; Zahaf & Schmidt, 2017). We have been interested in evaluating the therapeutic potential of Pseudomonas aeruginosa Exotoxin T (ExoT) against cancer. ExoT is a bifunctional virulence factor, possessing an N-terminal GTPase activating protein (GAP) domain activity which inhibits RhoA, Rac1, and Cdc42 small Ras-like GTPases, and a C-terminal ADP-ribosyltransferase (ADPRT) domain which ADP-ribosylates Crk adaptor proteins and phosphoglycerate kinase 1 (PGK1) glycolytic enzyme (Krall, Schmidt, Aktories, & Barbieri, 2000; Sun & Barbieri, 2003).
Recently, we demonstrated that ExoT is capable of inducing potent cytotoxicity in a variety of hard to kill cancer cell lines and its ability to cause significant reduction in tumor volume in an animal model of melanoma (Goldufsky, Wood, Hajihossainlou, et al., 2015). Underlying ExoT’s potent cytotoxicity in cancer is its ability to induce two distinct forms of apoptosis in cancer cells (S. J. Wood, Goldufsky, & Shafikhani, 2015; S. J. Wood, Goldufsky, Bello, Masood, & Shafikhani, 2015). Through its ADPRT domain, ExoT induces anoikis apoptosis by disrupting the integrin-mediated survival signaling (S. J. Wood, Goldufsky, & Shafikhani, 2015), and through its GAP domain, it induces prototypical intrinsic (mitochondrial) apoptosis (S. J. Wood, Goldufsky, Bello, et al., 2015). During these studies, we observed that melanoma cells that were transfected with ExoT failed to undergo mitosis and proliferate, regardless of whether they eventually succumbed to ExoT-induced apoptosis or survived in ExoT’s presence. In this report, we sought to investigate ExoT’s antiproliferative activity in melanoma.
RESULTS
ExoT blocks proliferation in melanoma cells.
To assess the dynamics of ExoT-induced cytotoxicity in melanoma, we transfected B16 melanoma cells (Wellbrock et al., 2008) with the pIRES expression vector expressing ExoT, C-terminally fused to GFP (pExoT), or the control empty vector (pGFP) and assessed cytotoxicity by IF video-microscopy, using propidium iodide (PI) uptake, which stains dead or dying cells red or yellow, depending on transfection with GFP containing vectors (Kaminski et al., 2018; S. Wood et al., 2013; S. J. Wood, Goldufsky, & Shafikhani, 2015; S. J. Wood, Goldufsky, Bello, et al., 2015). These vectors have been described previously and GFP fusion does not alter ExoT’s virulence functions (Shafikhani & Engel, 2006; Shafikhani, Morales, & Engel, 2008; S. Wood, Sivaramakrishnan, Engel, & Shafikhani, 2011). Of note, similar transfection efficiencies were observed for pExoT and pGFP, as assessed by Western blotting and by determination of the percent of GFP-positive cells (Fig. S1). Transient transfection with ExoT resulted in 65.3% cell death which was significantly (p<0.001) more than the 25% cell death that occurred in B16 cells that were transfected with GFP empty vector (Fig. 1A–B, Movies S1–S2, red arrows point to representative transfected cells, succumbing to death). Pre-treatment with Z-VAD pancaspase inhibitor significantly reduced ExoT-induced cytotoxicity in melanoma cells (Fig. 1A–B, Movies S3–S4), indicating that ExoT-induced cytotoxicity in B16 is primarily apoptotic in nature, as we have previously shown for other epithelial cells (Shafikhani et al., 2008; S. J. Wood, Goldufsky, & Shafikhani, 2015; S. J. Wood, Goldufsky, Bello, et al., 2015).
Fig. 1: Impact of ExoT on cell death and proliferation in B16 melanoma cells.
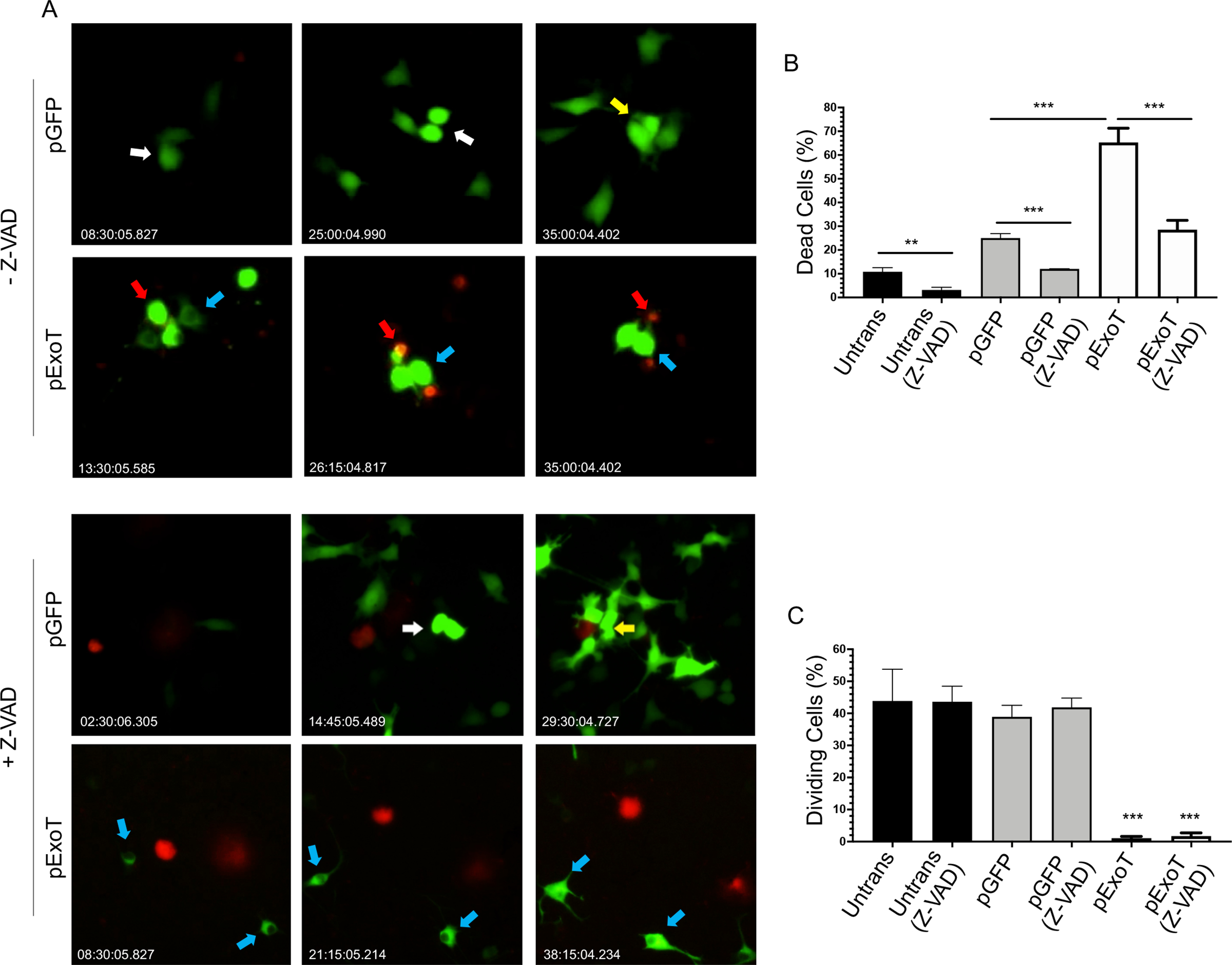
B16 cells were transfected with pIRES mammalian expression vector expressing either wild type ExoT, fused to GFP at the C-terminus (pExoT), or the vector control (pGFP), in the absence or presence of ZVAD (60μM) to block apoptosis. Cell death was analyzed by time-lapse video microscopy in the presence of the impermeant nuclear dye propidium iodide (PI) which stains dead cells (red or yellow). Proliferation was assessed by determining the percent of cells undergoing mitosis. A) Representative frames of videos are shown. Red arrows point to representative transfected cells that succumb to death. White arrows point to non-apoptotic transfected cells that undergo mitosis for the first time after transfection. Yellow arrows point to non-apoptotic transfected daughter cells that undergo mitosis for the second time after transfection. And blue arrows point to representative non-apoptotic transfected cells that fail to undergo mitosis. The tabulated results for cell death are shown as the Mean ± SEM in (B) and for proliferation (cell division) in (C). (N≥3, **p<0.01, ***p<0.001, Student’s t-test).
During these studies, we noted that while ~42% of pGFP-transfected B16 cells underwent mitosis and proliferated, only 1.1% of pExoT-transfected B16 cells underwent mitosis within the same timeframe, regardless of whether these cells eventually succumbed to apoptosis or survived in the presence of ExoT, or whether they were treated with Z-VAD or not (Fig. 1A, 1C Movies S3–S4. White arrows point to representative transfected cells that divide once after transfection and blue arrows point to ExoT-transfected non-apoptotic representative cells that did not undergo mitosis). Interestingly, even some GFP-transfected cells proliferated twice within this timeframe (Movies S3–S4, yellow arrows). Although, Z-VAD treatment significantly protected against ExoT-induced cytotoxicity (Fig. 1A–B), it did not increase the frequency of proliferating cells in pExoT- or pGFP-transfected B16 cells (Fig. 1C), indicating that Z-VAD has no effect on B16 proliferation. Of note, the mean time to proliferation (MTP) in untransfected and GFP-transfected B16 cells were similar (15.39 ± 1.21 hours and 13.26 ± 1.12 hours respectively), indicating that GFP transfection did not alter the dynamics of proliferation in B16 cells. Collectively, these data strongly suggested that ExoT has a potent antiproliferative effect in melanoma cells.
ExoT causes cell cycle arrest in G1 interphase in melanoma cells.
To hone in on the nature of ExoT’s antiproliferative effect in melanoma cells, we next transfected B16 cells with pGFP or pExoT vectors in the presence of Z-VAD, and determined where in the cell cycle, ExoT was blocking B16 progression toward mitosis by flow cytometry, 48h after transfection, (as we described previously (Shafikhani et al., 2008) and in the Experimental Procedures). While untransfected and pGFP-transfected B16 cells exhibited nearly identical distributions in G1, S, and G2 phases in cell cycle, pExoT-transfected B16 cells were significantly accumulated in the G1 interphase (Fig. 2A–B), indicating that ExoT causes G1 cell cycle arrest in B16. To corroborate these data, we transfected B16 cells with pGFP and pExoT vectors, fixed the cells 48h after transfection, and measured B16 entry into the S-phase using EdU incorporation (a marker for DNA synthesis in S-phase (Gupta et al., 2017; Salic & Mitchison, 2008)) by IF microscopy. While approximately 28% of pGFP-transfected B16 cells were EdU positive, indicating that they had entered the S-phase, only ~1% of pExoT-transfected cells stained positive with EdU, indicating that ExoT transfection blocked their entry into S-phase (Fig. 2C–D), confirming the flow cytometry data in Fig. 2A–B. We further corroborated these data on a large scale, using BrdU incorporation assay by ELISA, (as described in (Forrest, McNair, Vincenten, Darlington, & Stone, 2016; Zhang et al., 2015) and in Experimental Procedure), which again showed substantial reduction in BrdU staining, indicating ExoT caused G1 cell cycle arrest (Fig. 2E).
Fig. 2. ExoT causes cell cycle arrest in G1 interphase in melanoma cells.
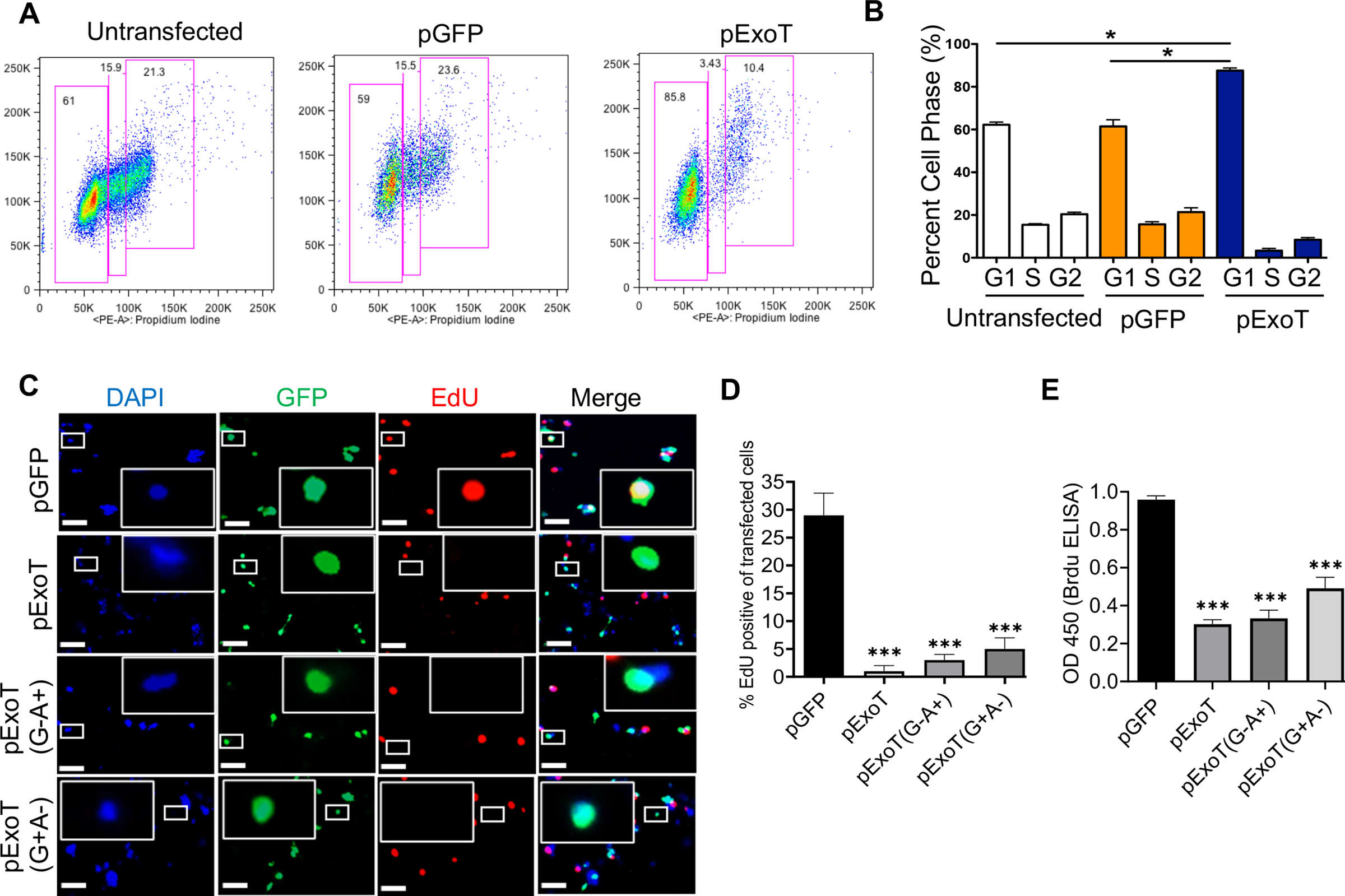
A-B) B16 cells were transfected with indicated expression vectors for 48h, fixed, and assessed for their cell cycle distribution by flow cytometry. Representative histograms are shown in (A) and the tabulated results are shown as the Mean ± SEM in (B). (N=3; *p<0.01; Student’s t-test). C-D) B16 cells were transfected with the indicated expression vectors in the presence of Z-VAD. Cell cycle arrest in G1 was determined by EdU incorporation (red) by IF microscopy, 48h after transfection. C) Representative images are shown after fixation and staining with GFP (green) and EdU (red). Inserts are the magnified images of representative cells. The scale bar is 20μM. (D) The tabulated results are shown as the Mean ± SEM (N=3, ***p<0.001, One-way ANOVA). E) G1 cell cycle arrest in B16 was determined by BrdU Cell Proliferation ELISA Kit, 48h after transfection (N=3, *** p<0.001; One-way ANOVA).
ExoT dampens G1/S checkpoint regulators.
To gain insights into the mechanism(s) underlying ExoT-induced G1 cell cycle arrest in melanoma, we next determined the contribution of the ADPRT and the GAP domains of ExoT in causing G1 arrest in B16 cells. Toward this end, we transfected B16 cells in the presence of Z-VAD with expression vectors harboring either ExoT with functional ADPRT domain but mutant GAP, pExoT(G−A+); or ExoT with functional GAP domain but mutant ADPRT, pExoT(G+A−); both C-terminally fused to GFP. These vectors have been previously described (Shafikhani et al., 2008; S. J. Wood, Goldufsky, & Shafikhani, 2015; S. J. Wood, Goldufsky, Bello, et al., 2015). We then assessed the impact of the ADPRT or the GAP domain activities on B16 cell cycle progression through G1 by EdU incorporation analysis using IF microscopy and by BrdU incorporation analysis using ELISA, 48h after transfection. Data indicated that both ADPRT and the GAP domains were able to cause G1 cell cycle arrest and contributed to ExoT-induced G1 arrest in melanoma (Fig. 2C–E). Of note, similar transfection efficiencies were observed (Fig. S1).
Prominent among the signals that trigger cell cycle progression through G1 are the mitogen signal through growth factors/growth factor receptors and the anchorage-dependent signal through focal adhesion-dependent integrin signaling, both of which result in activation of ERK MAP kinase and upregulation of cyclin D1 and cyclin E, leading to transition through G1 into S-phase (Chang et al., 2003; Coleman, Marshall, & Olson, 2004; Foster, Yellen, Xu, & Saqcena, 2010; Pruitt & Der, 2001; Yoon, Mitrea, Ou, & Kriwacki, 2012). We transfected B16 cells with the aforementioned expression vectors in the presence of Z-VAD and assessed the impact of ExoT, ExoT/GAP, ExoT/ADPRT, or pGFP control empty vector on important G1/S checkpoint regulators, namely, ERK1/2, cyclin D1, and cyclin E1 by Western blotting. Data indicated that both GAP and the ADPRT decreased ERK1/2 activation by nearly 60% without affecting ERK1/2 cellular levels, and cyclin E1 levels by nearly 90% (Fig. 3). Of note, ExoT and the ADPRT domain also caused significant reductions in cyclin D1 levels, whereas the GAP domain of ExoT did not impact cyclin D1 levels, indicating that ADPRT and the GAP domain activities employ different mechanisms to cause G1 cell cycle arrest. Again, transfection and Z-VAD did not affect the expression of the aforementioned G1/S checkpoint regulators, as the expression of G1/S regulators were similar in untransfected and GFP-transfected B16 cells, as determined by Western blotting (Fig. S2).
Fig. 3. Both domains of ExoT contribute to ExoT’s dampening of G1/S checkpoint regulators.
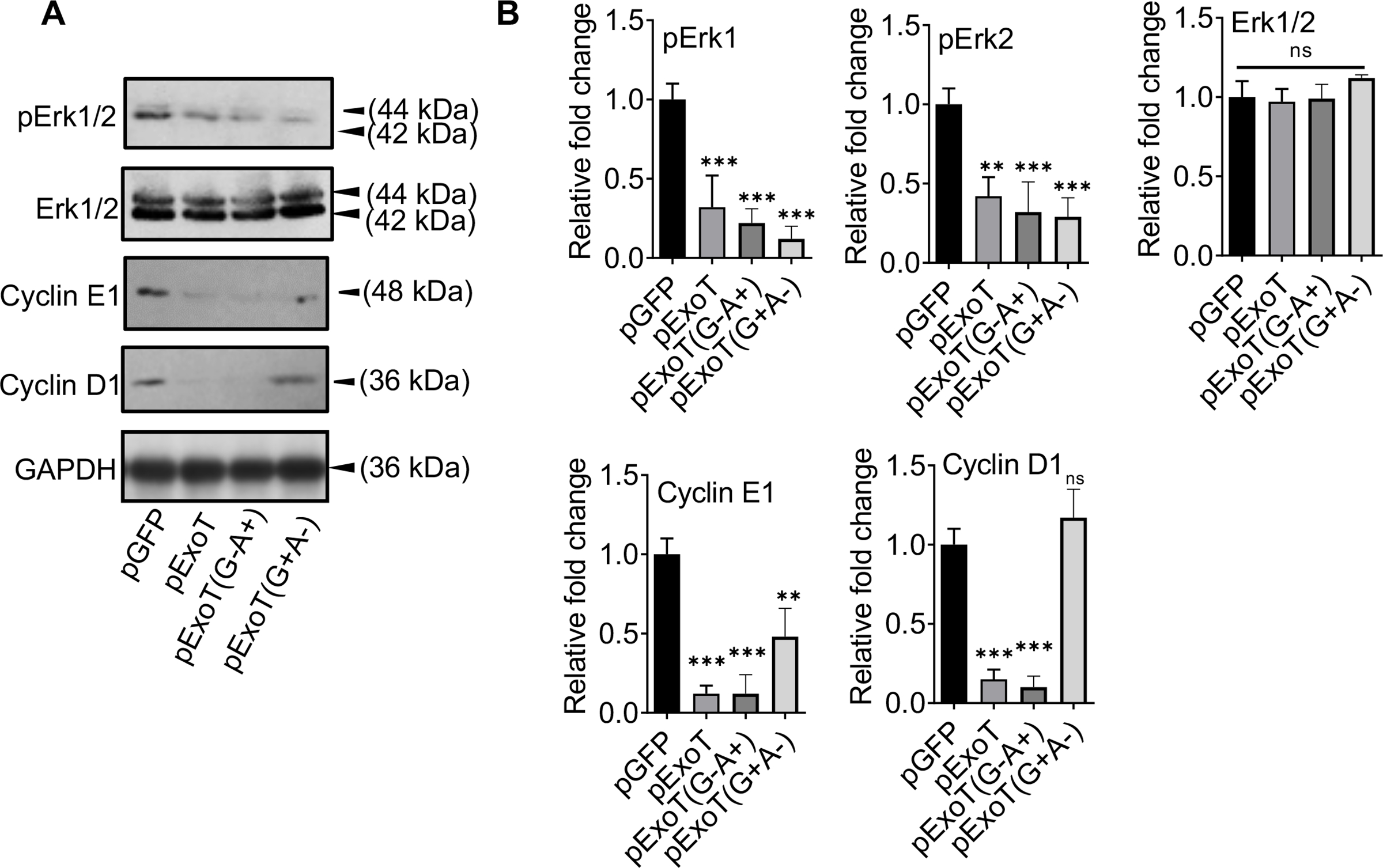
A) B16 cells were transfected with indicated expression vectors in the presence of Z-VAD. Cell lysates were assessed for the indicated G1/S checkpoint proteins, 48h after transfection by Western blotting. B) The corresponding densitometer data from 3 replicates, as compared to pGFP control vector, are shown after normalizing the data to their corresponding GAPDH levels. (N=3; ns, not significant, * p<0.05, ** p<0.01, *** p<0.001; One-way ANOVA).
To assess, the physiological relevance of ExoT-induced G1 cell cycle arrest in P. aeruginosa pathogenesis, we infected B16 cells with ExoU- and ExoT-deleted (ΔUΔT) PA103 strains, which were complemented with the expression vector harboring; ExoT, ExoT(G+A−), ExoT(G−A+), or pUCP20 empty vector. These strains have been previously described (Shafikhani & Engel, 2006; S. Wood et al., 2013; S. J. Wood, Goldufsky, & Shafikhani, 2015; S. J. Wood, Goldufsky, Bello, et al., 2015). We used ΔUΔT strain because ExoU is highly toxic and kills cells in cell culture within 1–2 hours (Rabin & Hauser, 2005; Sato et al., 2003). Five hours post-infection, we assessed the impact of ExoT or its domains on G1/S cell cycle by BrdU incorporation assay by ELISA and on the aforementioned checkpoint regulators by Western blotting. The infection data (Fig. 4) mirrored the transfection data (Fig. 3), indicating that ExoT is necessary to cause G1/S cell cycle arrest during infection and both domains contribute to this ExoT’s virulence activity.
Fig. 4. P. aeruginosa uses ExoT to cause cell cycle arrest in melanoma cells.
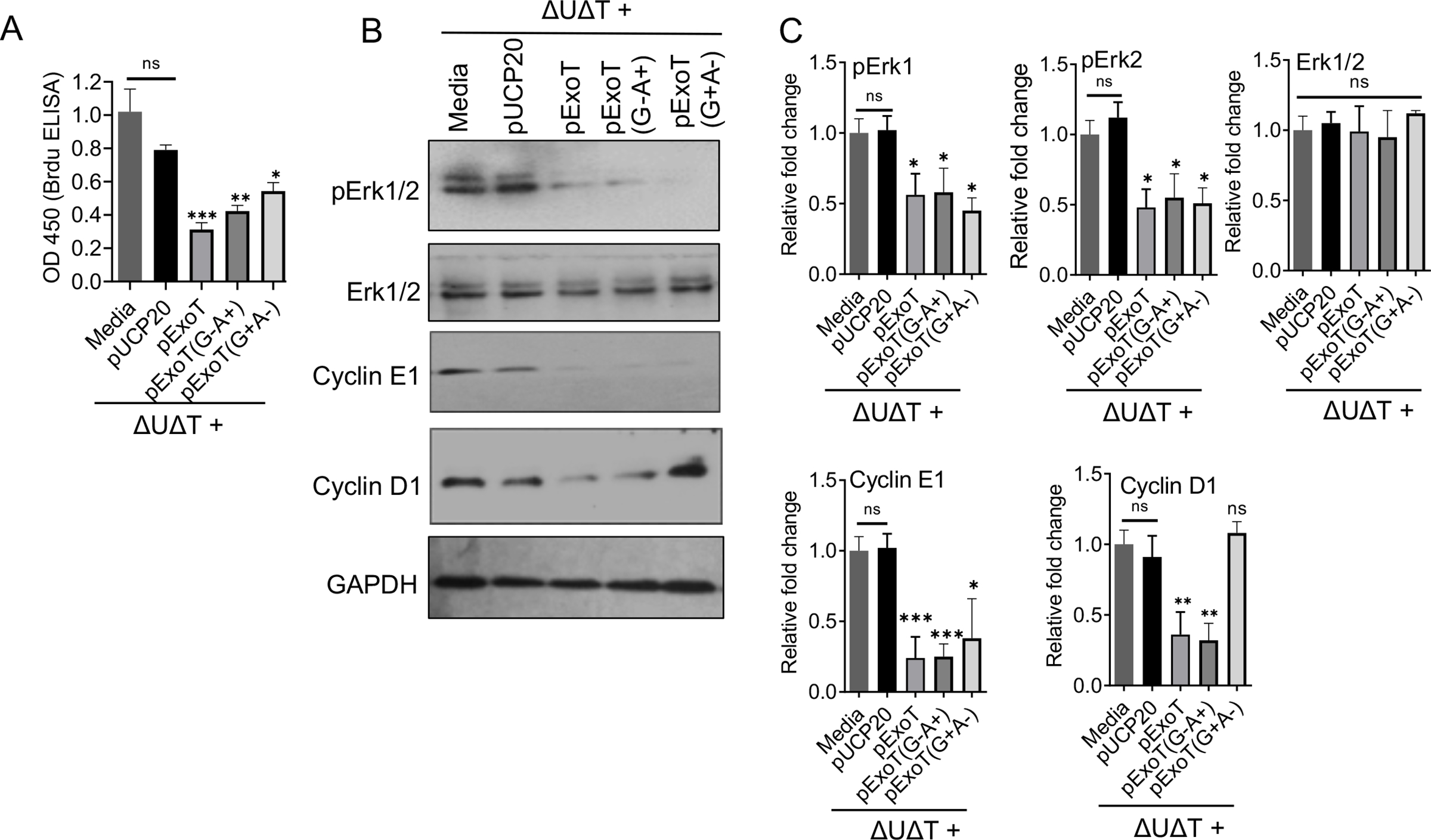
A-C) B16 cells were infected with the indicated bacterial strains. Five hours after infection, cells were harvested and assessed for G1 cell cycle arrest by BrdU Cell Proliferation ELISA Kit (A) and for the expression of the indicated checkpoint proteins by Western blotting (B). C) The corresponding densitometer data of Western blots are shown after normalizing the data with their corresponding GAPDH levels. (N=3; ns, Not significant, * p<0.05, ** p<0.01, *** p<0.001; One-way ANOVA).
ExoT/ADPRT domain induced G1 cell cycle arrest in B16 melanoma cells likely involves Crk.
It was not surprising that the GAP domain of ExoT was causing cell cycle arrest in G1, given that the GAP domain is a potent inhibitor of RhoA and Rac1 small GTPases (Huber, Bouillot, Elsen, & Attree, 2014; Kazmierczak & Engel, 2002), both of which have been shown to be critical for progression through G1/S cell cycle in eukaryotic cells (Coleman et al., 2004; Villalonga, Villalonga, & Ridley, 2006). However, it was not clear how the ADPRT domain of ExoT was causing G1 cell cycle arrest in B16.
Crk adaptor proteins have been demonstrated to localize to focal adhesion sites in eukaryotic cells (Lamorte, Rodrigues, Sangwan, Turner, & Park, 2003; Watanabe et al., 2009). The ADPRT domain of ExoT ADP-ribosylates a conserved arginine residue in the Src Homology 2 (SH2) domain of Crk (Sun & Barbieri, 2003). We have shown that ExoT/ADPRT modification of CrkI transforms this protein into a dominant negative (DN) mutant form which interferes with integrin survival signaling by disrupting focal adhesion sites (S. J. Wood, Goldufsky, & Shafikhani, 2015). Of note, the CrkI mutant harboring an arginine to lysine mutation at the conserved arginine residue in the SH2 domain (CrkI/R38K) phenocopies all ADPRT-associated virulence functions, including disruption of focal adhesion structures and interference with integrin survival signaling (Deng, Sun, & Barbieri, 2005; Gupta et al., 2017; Lamorte, Royal, Naujokas, & Park, 2002; Shafikhani & Engel, 2006; S. J. Wood, Goldufsky, & Shafikhani, 2015).
Given the importance of focal adhesion and integrin-mediated signaling in activating Erk1/2 and for progression through G1/S checkpoint (Chang et al., 2003; Coleman et al., 2004; Foster et al., 2010; Pruitt & Der, 2001; Yoon et al., 2012), and the adverse effects of ExoT/ADPRT and CrkI/R38K on focal adhesion structures and integrin-mediated signaling (S. J. Wood, Goldufsky, & Shafikhani, 2015), we posited that ExoT/ADPRT may involve Crk. To evaluate this possibility, we transfected B16 cells with the expression vectors harboring cellular CrkI or DN CrkI(R38K), C-terminally fused to GFP, or pGFP control vector, and assessed their impacts on cell cycle progression through G1. These vectors have been previously described (Shafikhani & Engel, 2006; Shafikhani et al., 2008; S. J. Wood, Goldufsky, & Shafikhani, 2015). Despite similar transfection efficiencies (Fig. S3), transfection with DN CrkI/R38K caused significant cell cycle arrest in B16, as assessed by EdU and BrdU incorporation using IF microscopy and ELISA respectively (Fig. 5A–C), phenocopying ExoT/ADPRT-induced G1 cell cycle arrest (Figs 3 & 4). Further corroborating these data and similar to ExoT/ADPRT, transfection with CrkI/R38K also dampened ERK activation and caused significant reductions in G1/S transition regulators cyclin D1 and cyclin E1, as assessed by Western blotting (Fig. 5D–E). Interestingly, transfection with cellular CrkI resulted in increased cyclin D1 and cyclin E1 expression, although it did not result in increased transition into the S-phase in B16 melanoma cells.
Fig. 5. ExoT/ADPRT-induced G1 cell cycle arrest in melanoma cells likely involves Crk.
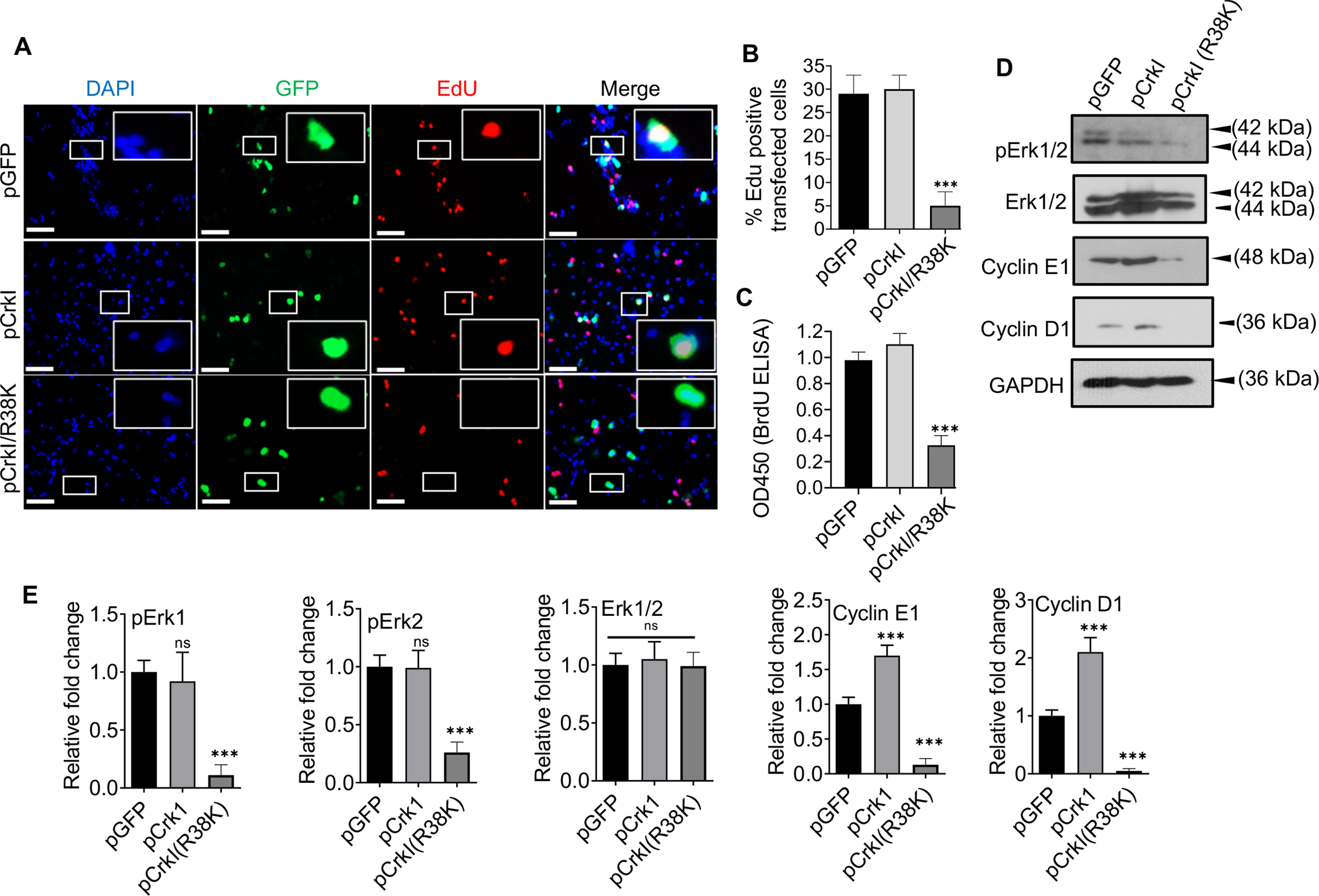
A-B) B16 cells were transfected with the indicated expression vectors in the presence of Z-VAD. Cell cycle arrest in G1 in transfected cells (green) was determined, using EdU DNA incorporation (red), by IF microscopy, 48h after transfection. DAPI was used to stain nucleus (blue). Representative images are shown. Inserts are the magnified images of representative cells. The scale bar is 20μM. B) The tabulated results of (A) are shown as the Mean ± SEM. (N=3, ***p<0.001; One-way ANOVA). C) B16 were transfected with indicated expression vectors in the presence of Z-VAD. Cell cycle arrest in G1 was determined by BrdU Cell Proliferation ELISA Kit, 48h after transfection (N=3; ns, not significant, *** p<0.001; One-way ANOVA). D) B16 were transfected with indicated expression vectors in the presence of Z-VAD. Cell lysates were collected 48h after transfection and assessed for the indicated G1/S checkpoint proteins by Western blotting. (E) the corresponding densitometer data from 3 replicates, as compared to pGFP control vector, are shown, after normalizing the data with their corresponding GAPDH levels. (N=3; ns, not significant, *** p<0.001; One-way ANOVA).
DISCUSSION
Driven by the observation that B16 melanoma cells transfected with ExoT failed to initiate mitosis and proliferate, regardless of whether they eventually succumbed to ExoT-induced apoptosis or survived in its presence, we sought to investigate the nature of P. aeruginosa ExoT’s antiproliferative function in melanoma cells. Our data reveal for the first time that ExoT possesses a potent antiproliferative activity in that it dampens G1/S transition regulators and causes G1 cell cycle arrest in melanoma cells. It is worth noting that ExoT (when delivered by P. aeruginosa) causes cytotoxicity within 20–30h post-infection (Goldufsky, Wood, Hajihossainlou, et al., 2015), whereas ExoT-induced G1/S cell cycle arrest occurred quickly within 5h post-infection, suggesting that ExoT-induced cytotoxicity and ExoT-induced G1/S cell cycle arrest likely involve different mechanisms, or that the mechanisms diverge early prior to caspase activation. In line with this notion, Z-VAD treatment had no effect on ExoT-induced G1/S cell cycle arrest, while it was able to significantly protect B16 against ExoT-induced cytotoxicity.
We further show that both domains of ExoT (GAP and ADPRT) contribute to ExoT-induced G1/S cell cycle arrest in melanoma cells. The molecular mechanisms underlying ExoT/GAP- or ExoT/ADPRT-induced G1/S cell cycle arrest remain unknown and require future investigations. However, we posit that the GAP-induced G1 cell cycle arrest is likely due to its inhibitory effects on RhoA and Rac1 small GTPases (Huber et al., 2014; Kazmierczak & Engel, 2002), given that RhoA and Rac1 functions are critical for progression through G1 into S-phase in mammalian cells (Coleman et al., 2004; Villalonga et al., 2006). We further postulate that the ADPRT-induced G1 cell cycle arrest likely involves Crk adaptor protein. This notion is supported by: (i) reports demonstrating the critical role of focal adhesion-dependent integrin signaling in cell cycle transition from G1 into S-phase in eukaryotic cells (Margadant, van Opstal, & Boonstra, 2007; Oktay, Wary, Dans, Birge, & Giancotti, 1999; Walker & Assoian, 2005); (ii) the finding that ExoT causes focal adhesion disassembly and interferes with integrin-mediated signaling in a manner that is dependent on its ADP-ribosylation of CrkI (S. J. Wood, Goldufsky, & Shafikhani, 2015); and (iii) our data in this communication demonstrating that CrkI (R38K) DN mutant phenocopies ADPRT-induced cell cycle arrest and its dampening of G1/S regulators.
The question that may arise is how ExoT’s anti-proliferative function benefits P. aeruginosa in the context of infection and in the host environment? We posit that ExoT’s antiproliferative effect may further enhance P. aeruginosa’s ability to expand its niche within the host, particularly if some cell types within the host are resistant to ExoT-induced apoptosis and ExoT-induced apoptosis is not available to this pathogen. Consistent with this notion, we found that ~35% of B16 transfected cells were able to survive ExoT-induced apoptosis, but all ExoT-transfected B16 cells failed to undergo mitosis and proliferate, regardless of whether they eventually succumbed to ExoT-induced apoptosis or survived in its presence. More studies are needed to assess the importance of ExoT-induced cell cycle arrest in mediating P. aeruginosa pathogenesis in vivo.
ExoT is the only T3SS effector protein that is expressed in all T3SS-expressing P. aeruginosa (Feltman, Khan, Jain, Peterson, & Hauser, 2001; Feltman, Schulert, et al., 2001; Wolfgang et al., 2003), suggesting a more fundamental role for this virulence factor in the pathogenesis of P. aeruginosa. ExoT-induced cell cycle arrest adds to the growing list of virulence functions that make this toxin so indispensable to P. aeruginosa (Garrity-Ryan et al., 2004; Goldufsky, Wood, Jayaraman, et al., 2015; Gupta et al., 2017; Shafikhani & Engel, 2006; S. J. Wood, Goldufsky, & Shafikhani, 2015; S. J. Wood, Goldufsky, Bello, et al., 2015).
Cancer continues to have a devastating impact on public health worldwide (Henley et al., 2020; Mattiuzzi & Lippi, 2020; Siegel et al., 2020). New technological advances have been developed to overcome the barriers in the field of Xenotoxin-based cancer therapeutics and have now made the use of bacterial toxins in cancer therapy both feasible and attractive, as can be seen by a growing number of promising immunotoxins in various clinical trials (Akbari et al., 2017; Karpiński & Adamczak, 2018; Zahaf & Schmidt, 2017). We propose that ExoT possesses many biological properties that make it an ideal candidate for the development in cancer therapy, including its potent cytotoxicity in various cancers due to its ability to trigger different apoptotic pathways, its ability to prevent apoptotic cancer cells from producing apoptotic compensatory proliferation microvesicles which can stimulate proliferation in other cancer cells, its ability to inhibit cell migration and potentially interfere with the metastatic process, and its anti-proliferative functions, owing it all to the activities of its two domains (ADPRT and GAP) that target multiple non-overlapping cellular substrates (Garrity-Ryan et al., 2004; Goldufsky, Wood, Hajihossainlou, et al., 2015; Gupta et al., 2017; Shafikhani et al., 2008; S. J. Wood, Goldufsky, & Shafikhani, 2015; S. J. Wood, Goldufsky, Bello, et al., 2015).
The primary challenge concerning the formulation of ExoT as a potential cancer therapy is the development of means to safely deliver this toxin and specifically target it to tumors in vivo. We posit that there are several strategies that can accomplish this task. One approach is to conjugate ExoT with tumor-specific receptor ligands, antibodies, or immunoglobulin variable fragment regions which have been shown to enhance the delivery of the toxins (e.g., intravenously) as well as improve their specificity toward tumor cells in vivo (reviewed by (Becker & Benhar, 2012; Chaudhary, FitzGerald, Adhya, & Pastan, 1987; Pastan, Beers, & Bera, 2004). Using this methodology, a number of bacterial-based immunotoxins - (e.g., Moxetumomab pasudotox and Tagraxofusp in which Diphtheria toxin (DT) is fused to either anti-CD22 or IL-3 respectively) - have been approved by Food and Drug Administration (FDA) for the treatment of various cancers (reviewed in (Akbari et al., 2017; Shafiee, Aucoin, & Jahanian-Najafabadi, 2019)). Another promising approach is the use of recombinant viral delivery systems, such as vaccinia virus, which show high inherent tropism toward tumor cells (Baguley, 2010). A viral-based delivery platform has several attractive biological properties that make it ideally suited for delivery and amplification of transgenes within tumors. These include high tropism toward cancerous cells with only minor side-effects observed in cancer patients even at extremely high infection titers (107–109 p.f.u.) (Breitbach et al., 2011) (Park et al., 2008); intravenous stability and the ability to spread to distant tissues (Vanderplasschen, Mathew, Hollinshead, Sim, & Smith, 1998); and preferential accumulation in solid tumors where neovasculature shows increased permeability (Kirn & Thorne, 2009). Another potential approach is to employ image-guided ultrasound-based microbubble technology to deliver and activate ExoT selectively in the tumor microenvironment. Gene therapy using this technology has gained significant momentum in recent years (Chen et al., 2012; A. H. Smith, Fujii, Kuliszewski, & Leong-Poi, 2011; B. Smith & Land, 2012).
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents:
B16 cells were cultured in complete DMEM (Life Technologies) supplemented with 10% FCS, 1% penicillin/streptomycin, and 1% L-glutamine at 37°C in the presence of 5% CO2. For transfection experiments, 0.4μg plasmid DNA was used with Effectene (Qiagen) according to the manufactures protocol.
Antibodies:
All antibodies were obtained from Cell Signaling Technology. The product numbers of antibodies are as follows: anti-Cyclin D1 (#2978), anti-Cyclin E1 (#20808), anti-Phospho-p44/42 MAPK (Erk1/2) (#9101), anti-p44/42 MAPK (Erk1/2) (137F5) (#4695), and anti-GAPDH (#5174), all from Cell Signaling Technology.
Propidium iodide (Sigma; St Louis, MO) was used at 7mg/ml at the time of transfection to identify dead or dying cells. Z-Val-Ala-Asp(OMe)-CH2F (Z-VAD) was obtained from R&D Systems (Minneapolis, MN) and added at 60μM final concentration 1h prior to transfection and video microscopy.
Transient Transfection was performed as previously described (Kaminski et al., 2018; Shafikhani et al., 2008). Briefly, B16 cells were seeded at a density of 4 × 104 cells per well overnight at 37°C with 5% CO2. Effectene transfection reagent was used following manufacturer’s protocol (Qiagen, CA) with 0.4μg plasmid DNA.
Time-lapse Immunofluorescent (IF) videomicroscopy was performed as we described previously (Shafikhani et al., 2008; S. Wood et al., 2013; S. J. Wood, Goldufsky, & Shafikhani, 2015). Briefly, B16 cells were grown in DMEM without phenol red for transfection studies for 24h. These cells were then transfected with indicated expression vectors in the absence or presence of 60μM Z-VAD, as described (Shafikhani et al., 2008; S. Wood et al., 2013; S. J. Wood, Goldufsky, & Shafikhani, 2015). 1h after transfection, cells were given 7μg/ml propidium iodide (PI) which was obtained from Sigma, and then placed on an AxioVert Z1 microscope (Zeiss) fitted with a live-imaging culture box (Pecon) maintaining 37°C in the presence of 5% CO2. Time-lapse videos were taken at 15 min interval, using AxioVision 4.2.8 software.
Mean Time to Death (MTD) determination was performed as we described previously (Shafikhani et al., 2008). Briefly, we used time-lapse video microscopy in the presence of PI to establish the mean time to death (MTD) in cells transfected with pExoT-GFP or pGFP. The time to death was defined as the time between the appearance of GFP (green), an indicator of transfected gene expression, and the time at which the cell membrane integrity was compromised resulting in the uptake of PI (red). We only included cells in our subsequent analyses that could be followed for at least one MTD plus one standard deviation from the mean.
Mean Time to Proliferation (MTP) determination:
We used time-lapse video microscopy to establish the mean time to proliferation (MTP) in transfected and untransfected cells. MTP was defined as the time between two successive mitotic events in the same cell. We only included cells in our subsequent analyses that could be followed for at least one MTP plus one standard deviation from the mean.
Western Blotting was performed as described previously (S. Wood et al., 2013; S. Wood et al., 2011). Briefly, cells were lysed following infection with 1% Triton X-100 containing a protease inhibitor cocktail (Roche Diagnostics), 100mM PMSF, and 100mM Na3VO4. Lysates were mixed with 4X SDS loading buffer and loaded onto 10% SDS-polyacrylamide gels. After resolving, gels were transferred to PVDF membranes, blocked with 5% milk, and probed overnight with primary antibody at 4°C. After washing, blots were probed with HRP-conjugated secondary antibody (Cell Signaling Technologies). Blots were developed with ECL or ECL+ reagent (GE Healthcare). Films were developed with an auto processor. The protein levels were assessed by densitometer using ImageJ and normalized with corresponding GAPDH levels to account for loading inherent errors.
5-Ethynyl-2’-deoxyuridine (EdU) Incorporation Assay was performed as described previously (Gupta et al., 2017). Briefly, 8 × 104 B16 cells were seeded on coverslips, pre-treated with poly-L-lysine and human fibronectin (40μg/mL). The following day cells were transfected as described for 24 h and then treated with 10μM EdU for 2h. Cells were fixed with 3.7% formaldehyde for 15 min and permeabilized with 0.5% Triton X-100 for 20 min at room temperature. Next, cells were washed twice with 3% BSA in PBS, before treatment with Click-iT Plus reaction cocktail (Molecular Probes) for 30min at room temperature protected from light. Following reaction, cells were washed once and blocked with 3% BSA for 1h and then stained with anti-GFP antibody (Abcam; ab5450) overnight followed by anti-goat AF488 secondary antibody (Abcam; ab150129) for 1h. Coverslips were washed three times with PBS and mounted on slides with ProLong Diamond Antifade Mountant with DAPI (Molecular Probes) and imaged using an AxioVert Z1 fluorescent microscope (Zeiss).
Bromodeoxyuridine (BrdU) Incorporation Assay by ELISA was performed using BrdU Cell Proliferation ELISA Kit (ab126556), as described (Zhang et al., 2015). Briefly, B16 cells (104 cells/mL) were cultured in 96 well plates and transfected as described above. BrdU was added to the wells during the final 24 hours of culture. After cell fixation, permeabilization and DNA denaturation, anti-BrdU monoclonal antibody was added for one hr. Followed by a secondary horseradish peroxidase-conjugated antibody after washing cells three times. The amount of incorporated BrdU was estimated using Tetra-MethylBenzidine (TMB) as chromogen. The reaction was monitored at 450nm on a microplate ELISA reader.
Infection Studies:
1 × 105 B16 cells were infected with ExoU- and ExoT-deleted PA103 strain (ΔUΔT), complemented with pUCP20 expressing; ExoT, ExoT(G+A−), ExoT(G−A+), or pUCP20 empty vector. These strains have been previously described (Shafikhani & Engel, 2006; S. Wood et al., 2013; S. J. Wood, Goldufsky, & Shafikhani, 2015; S. J. Wood, Goldufsky, Bello, et al., 2015). We used ΔUΔT strain because ExoU is highly toxic and kills cells in cell culture within 1–2 hours (Rabin & Hauser, 2005; Sato et al., 2003). Infection was done in Opti-MEM +1% FBS at multiplicity of infection (M.O.I) of 20 for 5h. Plates were centrifuged at 800 rpm for 5 min to synchronize infection. Five hours post-infection, we assessed the impact of ExoT or its domains on G1/S cell cycle by BrdU incorporation assay by ELISA and on the aforementioned checkpoint regulators by Western blotting as described above.
Immunofluorescence (IF) static microscopy was performed as described previously (Kaminski et al., 2018; Mahmood et al., 2013; S. Wood et al., 2011). Briefly, cells were fixed 24h following transfection with 4% PFA for 20 mins, blocked and permeabilized in permeabilization buffer (1X PBS + 5% FBS + 0.3% Triton™ X-100) for 60 mins at room temperature, and treated with primary antibodies for GFP, at concentrations as recommended by manufacturer in antibody dilution buffer (1X PBS/1% BSA/0.3% Triton™ X-100) overnight at 4oC. After 3X wash in 1X PBS for 5min each, coverslips were then incubated with fluorochrome-conjugated secondary antibody, Anti-mouse Alexa 488 or Anti-rabbit Alexa 488 procured from Jackson ImmunoResearch, for 2h at room temperature in the dark. Coverslips were then rinsed with 1 X PBS three times for 5min each and visualized under Immunofluorescence microscope.
Cell Cycle Analysis by Flowcytometry was performed as described previously (Fraker, King, Lill-Elghanian, & Telford, 1995; Shafikhani et al., 2008). Briefly, asynchronous B16 cells were transfected with the indicated vectors in the presence of Z-VAD as described above and assessed for their cell cycle distribution profiles by flow cytometry after staining with PI. These experiments were done in triplicates.
Statistical analysis.
Statistical analyses were performed using GraphPad Prism 6.0 software as described previously (S. Wood et al., 2011; S. J. Wood, Goldufsky, Bello, et al., 2015). Comparison between two groups was performed using Student’s t-test. Comparison between more than two groups was performed using One-way ANOVA. To account for error inflation due to multiple testing, the Bonferroni method was used. Statistical significance threshold was set at p ≤ 0.05.
Supplementary Material
Fig. S1. Transfection efficiencies of plasmids. B16 cells were transfected with indicated expression vectors after pre-treatment with Z-VAD pan caspase inhibitor for 1h. A) Cell lysates were assessed for transfection efficiency by Western blotting using anti-GFP antibody. GAPDH was used as the loading control. B) Cells were fixed and stained with anti-GFP antibody and the % of transfected cells (green) was assessed by IF microscopy. The tabulated results are shown as the Mean ± SEM. (N=3; ns, not significant; One-way ANOVA).
Fig. S3. Transfection efficiencies of plasmids. B16 cells were transfected with indicated expression vectors after pre-treatment with Z-VAD pan caspase inhibitor for 1h. A) Cell lysates were assessed for transfection efficiency by Western blot using anti-GFP antibody. GAPDH was used as the loading control. B) Cells were fixed and stained with anti-GFP antibody and the % of transfected cells (green) was assessed by IF microscopy. The tabulated results are shown as the Mean ± SEM. (N=3; ns, not significant; One-way ANOVA).
Fig. S2. Assessment of Z-VAD impact on the expression of G1/S checkpoint proteins. Untransfected B16 cells (in the presence or absence of Z-VAD) or pGFP-transfected B16 cells (in the absence of Z-VAD) were assessed for the expression of the indicated G1/S checkpoint proteins by Western blotting. (For pGFP-transfected cells treated with Z-VAD, see Fig. 3A). Data indicate that Z-VAD has no appreciable effect on the expression of these proteins. GAPDH was used as a loading control.
Movie S1. Impact of ExoT transfection on cellular toxicity and proliferation in B16 melanoma cells. B16 cells were transiently transfected with pExoT-EGFP expression vector (pExoT) in the presence of propidium iodide (PI) to assess dead cells. Video images were captured every 15 min. Arrows in the video point to representative pExoT-transfected B16 cells that succumb to cell death, as indicated by PI uptake (red or yellow).
Movie S2. Impact of GFP transfection on cellular toxicity and proliferation in B16 melanoma cells. B16 cells were transiently transfected with pEGFP control vector in the presence of propidium iodide (PI) to assess dead cells (red or yellow). Video images were captured every 15 min.
Movie S3. Impact of ExoT transfection on cellular toxicity and proliferation in Z-VAD-treated B16 melanoma cells. B16 cells were transiently transfected with pExoT-EGFP expression vector (pExoT) in the presence of Z-VAD to protect against ExoT-induced apoptosis and propidium iodide (PI) to assess dead cells (red or yellow). Video images were captured every 15 min. A cropped portion of the video was used for better magnification. This video demonstrates that while Z-VAD protects against ExoT-induced cell death (lack of PI staining), ExoT-transfected cells that survive, fail to undergo mitosis and cell division.
Movie S4. Impact of GFP transfection on cellular toxicity and proliferation in Z-VAD-treated B16 melanoma cells. B16 cells were transiently transfected with pEGFP control vector in the presence of Z-VAD and propidium iodide (PI) to assess dead cells (red or yellow). Video images were captured every 15 min. A cropped portion of the video was used for better magnification. White arrows point to representative pGFP-transfected cells that undergo mitosis for the first time. Yellow arrows point to representative pGFP-transfected daughter cells that undergo mitosis for the second time.
TAKE AWAY.
ExoT has a potent antiproliferative effect in B16 melanoma cells.
Both GAP and ADPRT domains of ExoT contribute to ExoT-induced G1 cell cycle arrest in melanoma cells.
The ADPRT-induced G1 cell cycle arrest in melanoma cells likely involves Crk.
ACKNOWLEDGEMENTS
We thank all the members in Shafikhani lab for their insightful comments regarding this manuscript. This work was supported by Bears Care Cancer Fund (to SHS), NIH grant R21 AI110685 (to SHS), & Oncology Care Model (OCM) Cancer Award (to SHS).
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors have no conflict of interest to report.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon request.
REFERENCES
- Akbari B, Farajnia S, Ahdi Khosroshahi S, Safari F, Yousefi M, Dariushnejad H, & Rahbarnia L (2017). Immunotoxins in cancer therapy: Review and update. International reviews of immunology, 36(4), 207–219. [DOI] [PubMed] [Google Scholar]
- Becker N, & Benhar I (2012). Antibody-Based Immunotoxins for the Treatment of Cancer. Antibodies, 39–69. doi: 10.3390/antib1010039. [DOI] [Google Scholar]
- Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQ, . . . Kirn DH (2011). Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature, 477(7362), 99–102. doi: 10.1038/nature10358nature10358 [pii]. [DOI] [PubMed] [Google Scholar]
- Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, . . . McCubrey JA (2003). Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia, 17(3), 590–603. doi: 10.1038/sj.leu.24028242402824 [pii]. [DOI] [PubMed] [Google Scholar]
- Chaudhary VK, FitzGerald DJ, Adhya S, & Pastan I (1987). Activity of a recombinant fusion protein between transforming growth factor type alpha and Pseudomonas toxin. Proc Natl Acad Sci U S A, 84(13), 4538–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W-J, Xiong Z-A, Tang Y, Dong P-T, Li P, & Wang Z-G (2012). Feasibility and effect of ultrasound microbubble-mediated wild-type p53 gene transfection of HeLa cells. Experimental and therapeutic medicine, 3(6), 999–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman ML, Marshall CJ, & Olson MF (2004). RAS and RHO GTPases in G1-phase cell-cycle regulation. Nature reviews. Molecular cell biology, 5(5), 355–366. doi: 10.1038/nrm1365nrm1365 [pii]. [DOI] [PubMed] [Google Scholar]
- Deng Q, Sun J, & Barbieri JT (2005). Uncoupling Crk signal transduction by Pseudomonas exoenzyme T. J Biol Chem, 280(43), 35953–35960. doi: 10.1074/jbc.M504901200 [DOI] [PubMed] [Google Scholar]
- Feltman H, Khan S, Jain M, Peterson L, & Hauser A (2001). Type III secretion genotypes of clinical and environmental Pseudmonas aeruginosa isolates. ASM abstracts, D22. [DOI] [PubMed] [Google Scholar]
- Feltman H, Schulert G, Khan S, Jain M, Peterson L, & Hauser AR (2001). Prevalence of type III secretion genes in clinical and environmental isolates of Pseudomonas aeruginosa. Microbiology, 147(Pt 10), 2659–2669. [DOI] [PubMed] [Google Scholar]
- Forrest CM, McNair K, Vincenten MC, Darlington LG, & Stone TW (2016). Selective depletion of tumour suppressors Deleted in Colorectal Cancer (DCC) and neogenin by environmental and endogenous serine proteases: linking diet and cancer. BMC Cancer, 16(1), 772. doi: 10.1186/s12885-016-2795-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DA, Yellen P, Xu L, & Saqcena M (2010). Regulation of G1 Cell Cycle Progression: Distinguishing the Restriction Point from a Nutrient-Sensing Cell Growth Checkpoint(s). Genes Cancer, 1(11), 1124–1131. doi: 10.1177/194760191039298910.1177_1947601910392989 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraker PJ, King LE, Lill-Elghanian D, & Telford WG (1995). Quantification of apoptotic events in pure and heterogeneous populations of cells using the flow cytometer. Methods Cell Biol, 46, 57–76. [DOI] [PubMed] [Google Scholar]
- Garrity-Ryan L, Shafikhani S, Balachandran P, Nguyen L, Oza J, Jakobsen T, . . . Engel JN (2004). The ADP ribosyltransferase domain of Pseudomonas aeruginosa ExoT contributes to its biological activities. Infect Immun, 72(1), 546–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldufsky J, Wood S, Hajihossainlou B, Rehman T, Majdobeh O, Kaufman HL, . . . Shafikhani SH (2015). Pseudomonas aeruginosa exotoxin T induces potent cytotoxicity against a variety of murine and human cancer cell lines. J Med Microbiol, 64(Pt 2), 164–173. doi: 10.1099/jmm.0.000003-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldufsky J, Wood SJ, Jayaraman V, Majdobeh O, Chen L, Qin S, . . . Shafikhani SH (2015). Pseudomonas aeruginosa uses T3SS to inhibit diabetic wound healing. Wound Repair Regen, 23(4), 557–564. doi: 10.1111/wrr.12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta KH, Goldufsky JW, Wood SJ, Tardi NJ, Moorthy GS, Gilbert DZ, . . . Shafikhani SH (2017). Apoptosis and Compensatory Proliferation Signaling Are Coupled by CrkI-Containing Microvesicles. Dev Cell, 41(6), 674–684 e675. doi: 10.1016/j.devcel.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley SJ, Ward EM, Scott S, Ma J, Anderson RN, Firth AU, . . . Kohler BA (2020). Annual report to the nation on the status of cancer, part I: National cancer statistics. Cancer, 126(10), 2225–2249. doi: 10.1002/cncr.32802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber P, Bouillot S, Elsen S, & Attree I (2014). Sequential inactivation of Rho GTPases and Lim kinase by Pseudomonas aeruginosa toxins ExoS and ExoT leads to endothelial monolayer breakdown. Cellular and molecular life sciences, 71(10), 1927–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski A, Gupta KH, Goldufsky JW, Lee HW, Gupta V, & Shafikhani SH (2018). Pseudomonas aeruginosa ExoS Induces Intrinsic Apoptosis in Target Host Cells in a Manner That is Dependent on its GAP Domain Activity. Sci Rep, 8(1), 14047. doi: 10.1038/s41598-018-32491-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpiński TM, & Adamczak A (2018). Anticancer activity of bacterial proteins and peptides. Pharmaceutics, 10(2), 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak BI, & Engel JN (2002). Pseudomonas aeruginosa ExoT acts in vivo as a GTPase-activating protein for RhoA, Rac1, and Cdc42. Infect Immun, 70(4), 2198–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirn DH, & Thorne SH (2009). Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer, 9(1), 64–71. doi:nrc2545 [pii] 10.1038/nrc2545. [DOI] [PubMed] [Google Scholar]
- Krall R, Schmidt G, Aktories K, & Barbieri JT (2000). Pseudomonas aeruginosa ExoT is a Rho GTPase-activating protein. Infect Immun, 68(10), 6066–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamorte L, Rodrigues S, Sangwan V, Turner CE, & Park M (2003). Crk associates with a multimolecular Paxillin/GIT2/beta-PIX complex and promotes Rac-dependent relocalization of Paxillin to focal contacts. Mol Biol Cell, 14(7), 2818–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamorte L, Royal I, Naujokas M, & Park M (2002). Crk adapter proteins promote an epithelial-mesenchymal-like transition and are required for HGF-mediated cell spreading and breakdown of epithelial adherens junctions. Mol Biol Cell, 13(5), 1449–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood F, Hakimiyan A, Jayaraman V, Wood S, Sivaramakrishnan G, Rehman T, . . . Shafikhani SH (2013). A novel human antimicrobial factor targets Pseudomonas aeruginosa through its type III secretion system. J Med Microbiol, 62(Pt 4), 531–539. doi: 10.1099/jmm.0.051227-0. [DOI] [PubMed] [Google Scholar]
- Margadant C, van Opstal A, & Boonstra J (2007). Focal adhesion signaling and actin stress fibers are dispensable for progression through the ongoing cell cycle. J Cell Sci, 120(1), 66–76. [DOI] [PubMed] [Google Scholar]
- Mattiuzzi C, & Lippi G (2020). Cancer statistics: a comparison between World Health Organization (WHO) and Global Burden of Disease (GBD). Eur J Public Health, 30(5), 1026–1027. doi: 10.1093/eurpub/ckz216. [DOI] [PubMed] [Google Scholar]
- Oktay M, Wary KK, Dans M, Birge RB, & Giancotti FG (1999). Integrin-mediated activation of focal adhesion kinase is required for signaling to Jun NH2-terminal kinase and progression through the G1 phase of the cell cycle. The Journal of cell biology, 145(7), 1461–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BH, Hwang T, Liu TC, Sze DY, Kim JS, Kwon HC, . . . Kirn DH (2008). Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol, 9(6), 533–542. doi:S1470–2045(08)70107–4 [pii] 10.1016/S1470-2045(08)70107-4. [DOI] [PubMed] [Google Scholar]
- Pastan I, Beers R, & Bera TK (2004). Recombinant immunotoxins in the treatment of cancer. Methods Mol Biol, 248, 503–518. [DOI] [PubMed] [Google Scholar]
- Pruitt K, & Der CJ (2001). Ras and Rho regulation of the cell cycle and oncogenesis. Cancer Lett, 171(1), 1–10. [DOI] [PubMed] [Google Scholar]
- Rabin SD, & Hauser AR (2005). Functional regions of the Pseudomonas aeruginosa cytotoxin ExoU. Infect Immun, 73(1), 573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salic A, & Mitchison TJ (2008). A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A, 105(7), 2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Frank DW, Hillard CJ, Feix JB, Pankhaniya RR, Moriyama K, . . . Sawa T (2003). The mechanism of action of the Pseudomonas aeruginosa-encoded type III cytotoxin, ExoU. EMBO J, 22(12), 2959–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafiee F, Aucoin MG, & Jahanian-Najafabadi A (2019). Targeted Diphtheria Toxin-Based Therapy: A Review Article. Front Microbiol, 10, 2340. doi: 10.3389/fmicb.2019.02340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafikhani SH, & Engel J (2006). Pseudomonas aeruginosa type III-secreted toxin ExoT inhibits host-cell division by targeting cytokinesis at multiple steps. Proc Natl Acad Sci U S A, 103(42), 15605–15610. doi: 10.1073/pnas.0605949103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafikhani SH, Morales C, & Engel J (2008). The Pseudomonas aeruginosa type III secreted toxin ExoT is necessary and sufficient to induce apoptosis in epithelial cells. Cellular microbiology, 10(4), 994–1007. doi: 10.1111/j.1462-5822.2007.01102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, & Jemal A (2020). Cancer statistics, 2020. CA Cancer J Clin, 70(1), 7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- Smith AH, Fujii H, Kuliszewski MA, & Leong-Poi H (2011). Contrast ultrasound and targeted microbubbles: diagnostic and therapeutic applications for angiogenesis. J Cardiovasc Transl Res, 4(4), 404–415. doi: 10.1007/s12265-011-9282-2. [DOI] [PubMed] [Google Scholar]
- Smith B, & Land H (2012). Anticancer activity of the cholesterol exporter ABCA1 gene. Cell Rep, 2(3), 580–590. doi: 10.1016/j.celrep.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, & Barbieri JT (2003). Pseudomonas aeruginosa ExoT ADP-ribosylates CT10 regulator of kinase (Crk) proteins. J Biol Chem, 278(35), 32794–32800. [DOI] [PubMed] [Google Scholar]
- Vanderplasschen A, Mathew E, Hollinshead M, Sim RB, & Smith GL (1998). Extracellular enveloped vaccinia virus is resistant to complement because of incorporation of host complement control proteins into its envelope. Proc Natl Acad Sci U S A, 95(13), 7544–7549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalonga P, Villalonga P, & Ridley AJ (2006). Rho GTPases and cell cycle control. In: Taylor & Francis. [DOI] [PubMed] [Google Scholar]
- Walker JL, & Assoian RK (2005). Integrin-dependent signal transduction regulating cyclin D1 expression and G1 phase cell cycle progression. Cancer and Metastasis Reviews, 24(3), 383–393. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Tsuda M, Makino Y, Konstantinou T, Nishihara H, Majima T, . . . Tanaka S (2009). Crk adaptor protein-induced phosphorylation of Gab1 on tyrosine 307 via Src is important for organization of focal adhesions and enhanced cell migration. Cell Res, 19(5), 638–650. [DOI] [PubMed] [Google Scholar]
- Wellbrock C, Rana S, Paterson H, Pickersgill H, Brummelkamp T, & Marais R (2008). Oncogenic BRAF regulates melanoma proliferation through the lineage specific factor MITF. PLoS One, 3(7), e2734. doi: 10.1371/journal.pone.0002734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfgang MC, Kulasekara BR, Liang X, Boyd D, Wu K, Yang Q, . . . Lory S (2003). Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A, 100(14), 8484–8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood S, Pithadia R, Rehman T, Zhang L, Plichta J, Radek KA, . . . Shafikhani SH (2013). Chronic alcohol exposure renders epithelial cells vulnerable to bacterial infection. PLoS One, 8(1), e54646. doi: 10.1371/journal.pone.0054646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood S, Sivaramakrishnan G, Engel J, & Shafikhani SH (2011). Cell migration regulates the kinetics of cytokinesis. Cell cycle, 10(4), 648–654. doi:14813 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SJ, Goldufsky J, & Shafikhani SH (2015). Pseudomonas aeruginosa ExoT Induces Atypical Anoikis Apoptosis in Target Host Cells by Transforming Crk Adaptor Protein into a Cytotoxin. PLoS Pathog, 11(5), e1004934. doi: 10.1371/journal.ppat.1004934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SJ, Goldufsky JW, Bello D, Masood S, & Shafikhani SH (2015). Pseudomonas aeruginosa ExoT induces mitochondrial apoptosis in target host cells in a manner that depends on its GAP domain activity. J Biol Chem. doi: 10.1074/jbc.M115.689950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MK, Mitrea DM, Ou L, & Kriwacki RW (2012). Cell cycle regulation by the intrinsically disordered proteins p21 and p27. Biochemical Society Transactions, 40(5), 981–988. doi: 10.1042/BST20120092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahaf N-I, & Schmidt G (2017). Bacterial toxins for cancer therapy. Toxins, 9(8), 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li X, Ciric B, Ma CG, Gran B, Rostami A, & Zhang GX (2015). Therapeutic effect of baicalin on experimental autoimmune encephalomyelitis is mediated by SOCS3 regulatory pathway. Sci Rep, 5, 17407. doi: 10.1038/srep17407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Transfection efficiencies of plasmids. B16 cells were transfected with indicated expression vectors after pre-treatment with Z-VAD pan caspase inhibitor for 1h. A) Cell lysates were assessed for transfection efficiency by Western blotting using anti-GFP antibody. GAPDH was used as the loading control. B) Cells were fixed and stained with anti-GFP antibody and the % of transfected cells (green) was assessed by IF microscopy. The tabulated results are shown as the Mean ± SEM. (N=3; ns, not significant; One-way ANOVA).
Fig. S3. Transfection efficiencies of plasmids. B16 cells were transfected with indicated expression vectors after pre-treatment with Z-VAD pan caspase inhibitor for 1h. A) Cell lysates were assessed for transfection efficiency by Western blot using anti-GFP antibody. GAPDH was used as the loading control. B) Cells were fixed and stained with anti-GFP antibody and the % of transfected cells (green) was assessed by IF microscopy. The tabulated results are shown as the Mean ± SEM. (N=3; ns, not significant; One-way ANOVA).
Fig. S2. Assessment of Z-VAD impact on the expression of G1/S checkpoint proteins. Untransfected B16 cells (in the presence or absence of Z-VAD) or pGFP-transfected B16 cells (in the absence of Z-VAD) were assessed for the expression of the indicated G1/S checkpoint proteins by Western blotting. (For pGFP-transfected cells treated with Z-VAD, see Fig. 3A). Data indicate that Z-VAD has no appreciable effect on the expression of these proteins. GAPDH was used as a loading control.
Movie S1. Impact of ExoT transfection on cellular toxicity and proliferation in B16 melanoma cells. B16 cells were transiently transfected with pExoT-EGFP expression vector (pExoT) in the presence of propidium iodide (PI) to assess dead cells. Video images were captured every 15 min. Arrows in the video point to representative pExoT-transfected B16 cells that succumb to cell death, as indicated by PI uptake (red or yellow).
Movie S2. Impact of GFP transfection on cellular toxicity and proliferation in B16 melanoma cells. B16 cells were transiently transfected with pEGFP control vector in the presence of propidium iodide (PI) to assess dead cells (red or yellow). Video images were captured every 15 min.
Movie S3. Impact of ExoT transfection on cellular toxicity and proliferation in Z-VAD-treated B16 melanoma cells. B16 cells were transiently transfected with pExoT-EGFP expression vector (pExoT) in the presence of Z-VAD to protect against ExoT-induced apoptosis and propidium iodide (PI) to assess dead cells (red or yellow). Video images were captured every 15 min. A cropped portion of the video was used for better magnification. This video demonstrates that while Z-VAD protects against ExoT-induced cell death (lack of PI staining), ExoT-transfected cells that survive, fail to undergo mitosis and cell division.
Movie S4. Impact of GFP transfection on cellular toxicity and proliferation in Z-VAD-treated B16 melanoma cells. B16 cells were transiently transfected with pEGFP control vector in the presence of Z-VAD and propidium iodide (PI) to assess dead cells (red or yellow). Video images were captured every 15 min. A cropped portion of the video was used for better magnification. White arrows point to representative pGFP-transfected cells that undergo mitosis for the first time. Yellow arrows point to representative pGFP-transfected daughter cells that undergo mitosis for the second time.