

Abstract
Epigenetic regulation of gene transcription by chromatin remodeling proteins has recently emerged as an important contributing factor in inner ear development. Pathogenic variants in CHD7, the gene encoding Chromodomain Helicase DNA binding protein 7, cause CHARGE syndrome, which presents with malformations in the developing ear. Chd7 is broadly expressed in the developing mouse otocyst and mature auditory epithelium, yet the pathogenic effects of Chd7 loss in the cochlea are not well understood. Here we characterized cochlear epithelial phenotypes in mice with deletion of Chd7 throughout the otocyst (using Foxg1Cre/+ and Pax2Cre), in the otic mesenchyme (using TCre), in hair cells (using Atoh1Cre), in developing neuroblasts (using NgnCre), or in spiral ganglion neurons (using ShhCre/+). Pan-otic deletion of Chd7 resulted in shortened cochleae with aberrant projections and axonal looping, disorganized, supernumerary hair cells at the apical turn and a narrowed epithelium with missing hair cells in the middle region. Deletion of Chd7 in the otic mesenchyme had no effect on overall cochlear morphology. Loss of Chd7 in hair cells did not disrupt their formation or organization of the auditory epithelium. Similarly, absence of Chd7 in spiral ganglion neurons had no effect on axonal projections. In contrast, deletion of Chd7 in developing neuroblasts led to smaller spiral ganglia and disorganized cochlear neurites. Together, these observations reveal dosage-, tissue-, and time-sensitive cell autonomous roles for Chd7 in cochlear elongation and cochlear neuron organization, with minimal functions for Chd7 in hair cells. These studies provide novel information about roles for Chd7 in development of auditory neurons.
Keywords: development, spiral ganglion, cochlea, mouse genetics
Graphical Abstract
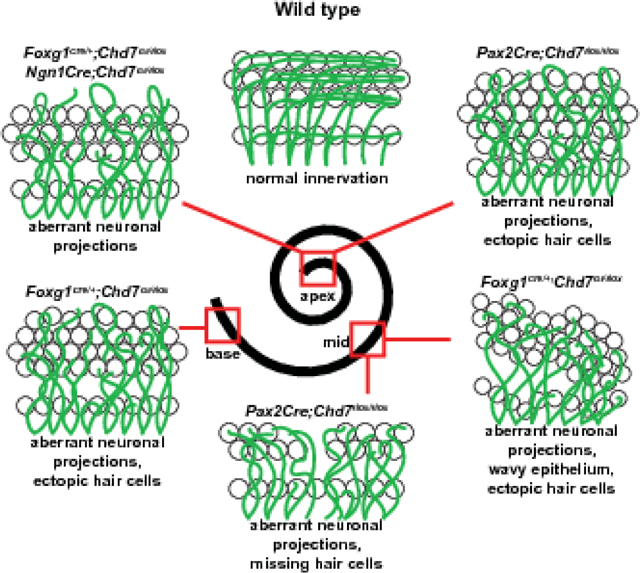
Introduction
The marvelously complex three dimensional structure of the mammalian inner ear arises from a seemingly simple tissue in embryonic development. The otic placodes, identified by Pax2 and Foxg1 expression, invaginate by E9.5 to form a pair of otocysts that develop into the vestibular and auditory organs (Groves and Bronner-Fraser, 2000; Tao and Lai, 1992). The dorsolateral compartment of the otocyst gives rise to the vestibular portion of the ear, containing the semicircular canals, saccule, utricle, and endolymphatic duct. The ventromedial region of the otocyst forms the auditory cochlea, which begins as a protrusion and subsequently elongates and turns to create a spiral. In the mouse, inner ear development proceeds through embryonic development, with mature morphology obtained by postnatal day three (McKenzie et al., 2004).
Vestibular and auditory structures are innervated by the vestibular and spiral ganglia, respectively, which originate from a combined cochleovestibular ganglion (CVG) in embryonic development. The sensory hair cells of the mouse cochlea and the neurons that innervate them are derived from common progenitors in the anterior ventral quadrant of the otocyst, termed proneurosensory cells, demarcated by Sox2 and Lfng expression at E9.5–10.5 (Koundakjian et al., 2007). Proneurosensory cells destined for a neuronal fate, specified by in part by Ngn1, delaminate from the otocyst as neuroblasts to form the CVG (Koundakjian et al., 2007). As the cochlea elongates and curls to form a spiral, Atoh1+ proneurosensory cells fated as hair cells populate the sensory epithelium that will become the organ of Corti within the cochlea (Bermingham et al., 1999).
Cell cycle exit and differentiation of hair cell progenitors follow a tightly orchestrated spatial and temporal pattern, with progenitors exiting the cell cycle in an apical to basal gradient along the length of the cochlea and hair cell differentiation proceeding in the opposite direction (reviewed by (Driver and Kelley, 2020)). Apical hair cells are thus the first to leave the cell cycle and the last to differentiate. While cochlear hair cell differentiation progresses, neurons within the CVG segregate into the vestibular and spiral ganglia and innervate their target cells. The spiral ganglion neurons, most of which express Shh, send neuritic projections to the organ of Corti to synapse onto inner and outer hair cells (Liu et al., 2010). Extrinsic and intrinsic guidance cues govern the trajectory of neurites through the otic mesenchyme to their targets (Coate and Kelley, 2013).
Epigenetic factors are robustly expressed in the otocyst and play important roles in regulating inner ear development (Layman et al., 2013). CHD7 (Chromodomain Helicase Binding Protein 7), an epigenetic regulator and chromatin remodeler, is broadly expressed throughout the developing mammalian otocyst as well as in the cochlea, CVG, and mature spiral ganglion neurons (Bosman et al. 2005, Adams et al. 2007). The critical nature of CHD7 function is highlighted in cases of CHARGE syndrome, a neurodevelopmental disorder caused by CHD7 haploinsufficiency that is characterized by sensorineural hearing loss, semicircular canal dysplasia, cochlear nerve hypoplasia, and shortened cochleae (Green et al. 2014, Choo et al. 2017). The mechanisms by which CHD7 regulates mammalian cochlear and cochlear nerve development are not fully understood. Mice with heterozygous loss of Chd7 display hypoplasia of the lateral and posterior semicircular canals and defects in innervation of the vestibular sensory epithelium (Adams et al. 2007) along with reduced proliferation of neuroblasts in the nascent CVG (Hurd et al. 2012). Conditional deletion of Chd7 in the developing otocyst using Foxg1Cre/+ leads to cochlear hypoplasia and a complete loss of semicircular canals (Hurd et al. 2010).
Here we examined inner ear and neuronal phenotypes in mice with tissue-specific loss of Chd7. Using various Cre lines and conditional Chd7 deletion mice, we investigated the effects of Chd7 deficiency in the otocyst, otic mesenchyme, hair cells, neuroblasts, and differentiating spiral ganglion neurons on cochlear and neuronal development. Our results indicate that Chd7 expression in otic mesenchyme is dispensible for inner ear morphogenesis. In contrast, otocyst-derived cells and maturing neurons appear uniquely sensitive to Chd7 deficiency, providing new and important insights about early development of the mammalian inner ear.
Methods
Mice
Chd7Gt/+ mice were generated as previously described (Hurd et al., 2007) and maintained by backcrossing with B6129SF1/J mice (Jackson Laboratory #101043) to generation N7-N10. Chd7flox/flox mice were generated and genotyped as previously described (Hurd et al., 2010), maintained on a mixed C57BL/6;129S1/SvlmJ background and inbred to generation N6-N8. Foxg1Cre/+ mice (The Jackson Laboratory #004337) were maintained on a Swiss-Webster background. Foxg1Cre/+ mice are haploinsufficient for Foxg1 but do not exhibit overt developmental defects of the inner ear (Hartman et al., 2010). Zsgreen1/Zsgreen1 (The Jackson Laboratory #007906), Ngn1Cre (The Jackson Laboratory #012859), Atoh1Cre (The Jackson Laboratory #011104), Pax2Cre (Ohyama and Groves, 2004) and ShhCre/+ (The Jackson Laboratory #005622) mice were crossed with Chd7flox/flox or Chd7Gt/+ mice to generate Cre;Chd7Gt/+ or Cre;Chd7+/flox mice, which were then mated with Chd7flox/flox or Chd7flox/flox;Zsgreen1/Zsgreen1 mice to generate litters for experimental analysis. Timed pregnancies were established to generate litters of embryos, with the morning of seminal plug detection marking embryonic day 0.5 (E0.5). Control animals for each experiment were selected to allow comparison of genotypes within litters. Controls included Cre positive; Chd7flox/+ when using the Zsgreen reporter and Cre negative, Chd7flox/+, Chd7flox/flox, and Chd7Gt/+ alleles, depending on availability and experimental design. All procedures were approved by The University of Michigan University Institutional Animal Care & Use Committee (IACUC).
Cochlear Wholemount Dissections
Bisected postnatal day 1 (P1) heads were fixed in 4% Paraformaldehyde (PFA; Alfa Aesar Thermo Fisher, Ward Hill, MA) for one hour before being transferred to phosphate buffered saline (PBS; Lonza, Rochester, NY) until the time of dissection. Fixed and bisected P1 heads were dissected in PBS by removing the inner ear using spring scissors and separating the vestibular system from the cochlea. The otic capsule was then peeled away, and both Reissner’s membrane and the tectorial membrane were removed before transferring the cochlea to an Eppendorf tube filled with 1% Bovine Serum Albumin (BSA; Sigma-Aldrich, St. Louis, MO). Cochleae were stored in 1% BSA overnight at 4°C.
Wholemount Immunohistochemistry
P1 cochleae were incubated for one hour in blocking solution (1% BSA; .02% Triton X-100 in 1X PBS), then one hour in primary antibodies (mouse anti-Neurofilament (1:500; clone 2H3, Developmental Studies Hybridoma Bank, Iowa City, IA) and rabbit anti-MYO7A (1:250; 25–6970, Proteus Biosciences Ramona, CA)) diluted in antibody blocking solution (0.5% BSA; .02% Triton X-100 in 1X PBS). Specimens were washed three times in PBS/0.2% Triton X-100 (Sigma-Aldrich, St. Louis, MO) at room temperature, then cochleae were incubated in AlexaFluor 488 or F(ab’) 2 fragment of goat anti-mouse IgG (1:200, Invitrogen, Molecular Probes, Thermo Fisher Waltham, MA) and AlexaFluor 555 goat anti-rabbit IgG (1:200, Invitrogen, Molecular Probes, Thermo Fisher Waltham, MA) secondary antibodies for 45 minutes. Some tissues were incubated for 30 minutes in Phalloidin (Invitrogen, Molecular Probes, Thermo Fisher Waltham, MA) (1:300) in PBS before being rinsed and mounted. Cochleae were mounted in ProLong Gold anti-fade (Invitrogen, Molecular Probes, Thermo Fisher Waltham, MA) on slides with coverslips and stored at 4°C prior to microscopy and imaging. Wholemount cochleae were imaged as Z-stacks at 40x magnification on a Leica SP8 confocal microscope (Buffalo Grove, IL). Stacks were overlaid and compressed on ImageJ software before final processing in Adobe Photoshop.
Immunofluorescence on Frozen Sections
Embryos (E11.5-E17.5) were dissected and fixed in 4% PFA for one hour at room temperature, washed three times in PBS following a 10%−30% sucrose gradient, embedded in OCT (Optimal Cutting Temperature compound; Tissue Tek Sakura Finetek, Torrance, CA) and stored at −80°C until sectioning. Cryosections were cut on a Leica (Buffalo Grove, IL) CM1950 Cryostat at 12 μm or 14 μm (e17.5) in the transverse orientation and stored at −80°C. On the day of staining, sections were removed from the freezer and dried at room temperature for 30–60 minutes. Slides were briefly post-fixed with 4% PFA and washed in PBST (0.1% Tween20, Sigma-Aldrich, St. Louis MO). Sections were blocked in 1% BSA (Sigma-Aldrich, St. Louis, MO) for one hour, then incubated with anti-CHD7 (Cell Signaling Technology #6505, Danvers, MA) at 1:2500 overnight. Sections were incubated in secondary antibody (AlexaFluor 555) goat anti-rabbit IgG (1:200, Invitrogen, Molecular Probes, Thermo Fisher Waltham, MA) at room temperature for one hour, then counter-stained with DAPI (4′,6-diamidino-2-phenylindole; (Invitrogen, Molecular Probes, Thermo Fisher Waltham, MA) 1:20,000 in 1x PBS) and mounted. Sections were imaged using a Leica upright DM5000B microscope and Leica DFC310 FX Digital camera.
Immunofluorescence on Paraffin Sections
Embryos (E14.5-E18.5) were dissected and fixed in 4% PFA for two hours at room temperature, then washed three times in PBS and transferred to 70% EtOH and dehydrated and embedded in paraffin wax using a Tissue Tek (Torrance, CA) embedding machine. Paraffin embedded embryos were stored at room temperature until sectioning. Paraffin sections were cut on a Leica microtome (Buffalo Grove, IL) at 7 μm. For staining, embryos were deparaffinized and rehydrated in a xylene and alcohol gradient. Citric acid (10 mM for three min of boiling followed by 20–30 minutes of cooling in the citrate buffer) was used for antigen retrieval. Sections were blocked in 1% BSA (Sigma-Aldrich St. Louis, MO) for one hour, then incubated with anti-CHD7 (Cell Signaling Technology #6505, Danvers, MA) at 1:2,500 and anti-Neurofilament (1:500; clone 2H3, Developmental Studies Hybridoma Bank, Iowa City, IA) overnight. Sections were incubated in secondary antibody AlexaFluor 555 goat anti-rabbit IgG (1:200, Invitrogen, Molecular Probes, Thermo Fisher, Waltham, MA) and AlexaFluor 488 F(ab’) 2 fragment of goat anti-mouse IgG (1:200, Invitrogen, Molecular Probes, Thermo Fisher, Waltham, MA) at room temperature for one hour, then counter-stained with DAPI (4′,6-diamidino-2-phenylindole; 1:20,000 in 1x PBS; Invitrogen, Molecular Probes, Thermo Fisher, Waltham, MA) and mounted on glass slides. Sections were imaged using a Leica upright DM5000B microscope and Leica DFC310 FX Digital camera.
Ganglion Measurements
Photos of Ngn1Cre;Zsgreen;Chd7+/flox and Ngn1Cre;Zsgreen;Chd7Gt/flox sections from E11.5 ears were processed using Photoshop (Adobe, San Jose, CA) and ImageJ (NIH, Bethesda, MD). One of every six sections throughout the Zsgreen+ cochlear ganglion was outlined for each of three embryos per genotype (six slides per embryo). Average area measurements for each genotype were compared using an unpaired t-test in GraphPad Prism software (GraphPad Software, San Diego, CA).
Cochlear Length Measurements
Images of Pax2Cre;Chd7flox/flox P1 cochleae were captured in wholemount and processed using Photoshop (Adobe, San Jose, CA). For each sample, a longitudinal line was drawn down the middle of the cochlear duct from base to apex and the length of the line measured in ImageJ (NIH, Bethesda, MD). Seven cochleae per genotype (Chd7flox/flox and Pax2Cre;Chd7flox/flox) were measured. Cochlear length measurements were compared between genotype groups using an unpaired t-test in GraphPad Prism software (GraphPad Software, San Diego, CA).
Hair cell quantitation
Whole mount images of anti-MYO7A cochleae were analyzed using ImageJ (NIH, Bethesda, MD). Briefly, a longitudinal line was drawn along the first row of outer hair cells from the base of the cochlea extending to the apex and the length of the line measured. The number of supernumerary outer hair cells were counted, and results presented as number of supernumerary outer hair cells per 100 μm cochlea. Six cochlea were analyzed for Chd7flox/flox and seven for Pax2Cre;Chd7flox/flox. The number of supernumerary outer hair cells were compared between genotypes in the basal, middle and apical part of cochlea using an unpaired t test using GraphPad Prism software (GraphPad Software, San Diego, CA).
Inner Ear Paintfilling
Timed pregnancies were established and embryos collected at E14.5. Heads were bisected and the brain removed to allow access to the inner ear. Embryos were washed in PBS, immersed in Bodian’s fixative (Martin and Swanson, 1993), cleared using methyl salicylate, and filled using 3% Wite-out® (Bic, Shelton, CT) as previously described (Morsli et al., 1998). Paint-filled ears were photographed using Leica MZ75 stereomicroscope and Leica DFC310 FX Digital camera (Buffalo Grove, IL). Digital images were processed with Adobe Photoshop 20.0.7 software.
Results
Prior studies of Chd7 function in inner ear development have revealed important roles in formation of the vestibular system, including semicircular canals, vestibular epithelia, and neuronal projections to the posterior crista ampullaris (Bosman et al., 2005; Hurd et al., 2011; Hurd et al., 2007; Hurd et al., 2012; Hurd et al., 2010). In both humans and mice, heterozygous loss of Chd7 is associated with hearing loss, yet the underlying mechanisms of this loss are not understood. Chd7 is expressed in the inner ear as early as E9.5 in the developing otocyst and cochleovestibular ganglion, and in the cochlear epithelium of mature mice (Hurd et al., 2011; Hurd et al., 2007; Hurd et al., 2010). Therefore, we asked whether Chd7 expression persists throughout embryonic development of the cochlear epithelium. To address this, we co-stained sections of wild-type cochleae at E14.5, E16.5, and E18.5 with anti-CHD7 and anti-Neurofilament antibodies (Fig. 1). At E14.5, there was abundant co-staining between anti-CHD7 and anti-Neurofilament in the cochlear epithelium and in the ganglion (Fig. 1A, D, G). At E16.5, CHD7 was still present in the cochlear epithelium but was restricted to a smaller set of cells in the spiral ganglion (Fig. 1B, E, H). At E18.5, CHD7 was further restricted in Neurofilament-positive spiral ganglion cells (Fig. 1C, F, I), suggesting that CHD7 is high in nascent spiral ganglion neurons but becomes downregulated during mid to late gestation. Analysis of publicly available data from the Shared Harvard Inner Ear Laboratory Database (SHIELD) database of gene expression (Shen et al., 2015) also revealed a gradual reduction of Chd7 mRNA in the mouse spiral ganglion between mid-gestation and birth (Supplementary Fig. 1) (Lu et al., 2011). These observations, along with earlier reports of Chd7 expression in the E9.5-E10.5 otocyst and cochleovestibular ganglion, indicate that temporal Chd7 expression in the auditory system coincides with development of the ganglion and cochlear duct.
Figure 1. Chd7 is dynamically expressed in the developing cochlea.
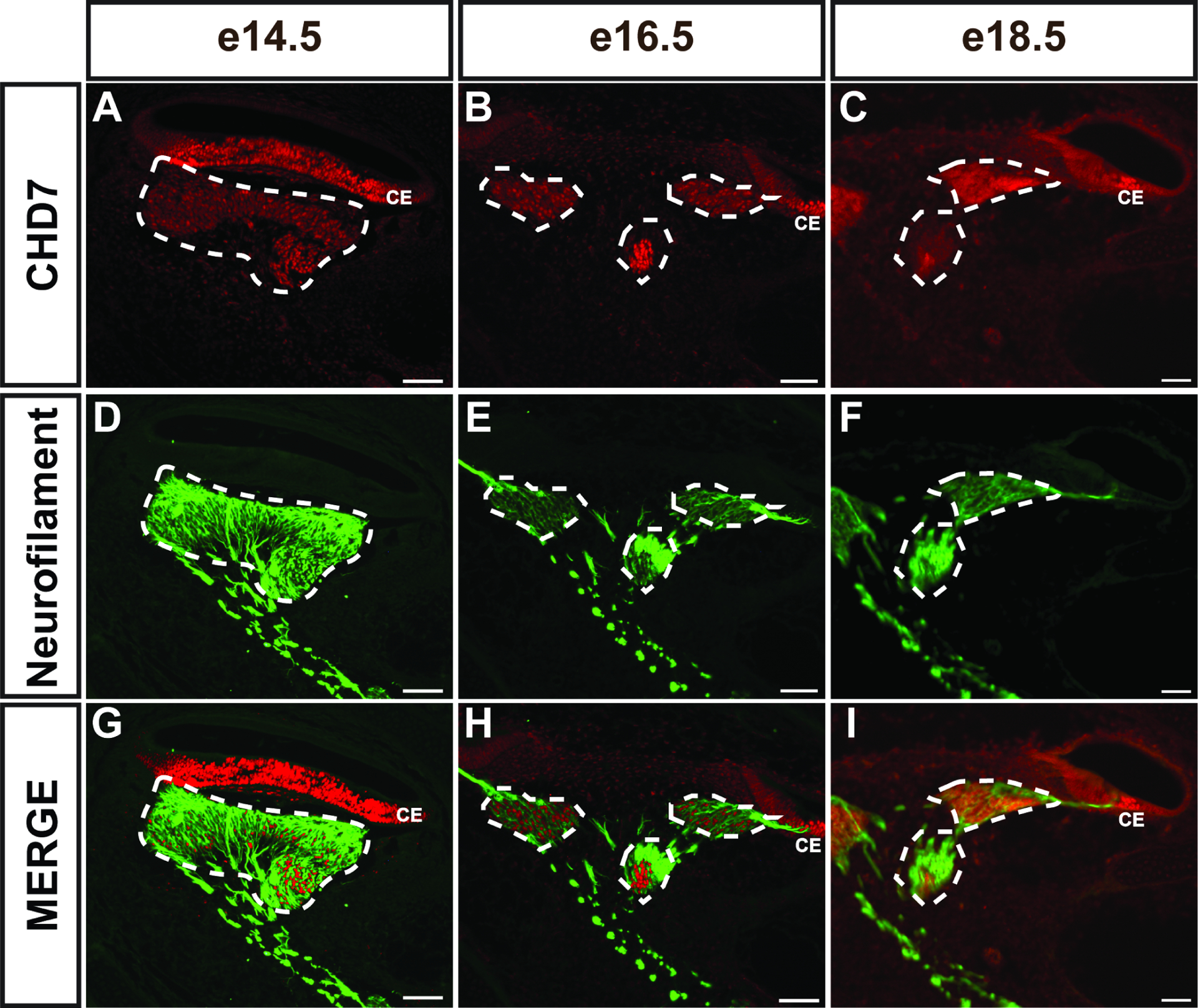
CHD7 stained paraffin sections from E14.5 (A, D, G), E16.5 (B, E, H), and E18.5 (C, F, I) Chd7+/+ embryos show CHD7+ cells present in the spiral ganglion (white dashed outlines) at E14.5 and E16.5, but minimal CHD7+ cells at E18.5. CHD7+ cells are also present in the cochlear epithelium (CE in A, B, C, G, H, I) at these stages. Neurofilament staining of the same sections (D-F) identifies spiral ganglion neurons at each stage. Scale bars = 50 μm.
Loss of Chd7 in the otocyst and surrounding mesenchyme by Foxg1Cre/+ (which expresses Cre throughout the E9.5 otocyst and surrounding mesenchyme, (Hebert and McConnell, 2000) results in a severely hypoplastic cochlea (Hurd et al., 2012). To better define the ultrastructural organization of the auditory epithelium upon Chd7 loss, we stained P1 Foxg1Cre/+;Chd7Gt/flox ears with Phalloidin to identify hair cells and anti-Neurofilament to label neurons. These studies revealed aberrant neuritic looping and supernumerary rows of outer hair cells in the apical cochleae of Foxg1Cre/+;Chd7Gt/flox ears (N=6 ears) compared to Chd7Gt/flox (N=3 ears, Fig. 2). The middle turn of the Foxg1Cre/+;Chd7Gt/flox cochlear epithelium also exhibited aberrant neuritic projections, occasional extra rows of outer hair cells, and an undulating appearance compared to control cochleae (Fig. 2). The basal turn of the Foxg1Cre/+;Chd7Gt/flox cochlea also showed disorganized and misrouted neurites and supernumerary hair cells (Fig. 2L, O, R compared to Fig. 2C, F, I). These findings suggest that CHD7 is necessary of proper guidance of spiral ganglion neurites and cochlear sensory epithelium patterning.
Figure 2. Pan-otic loss of Chd7 leads to morphological defects in the organ of Corti.
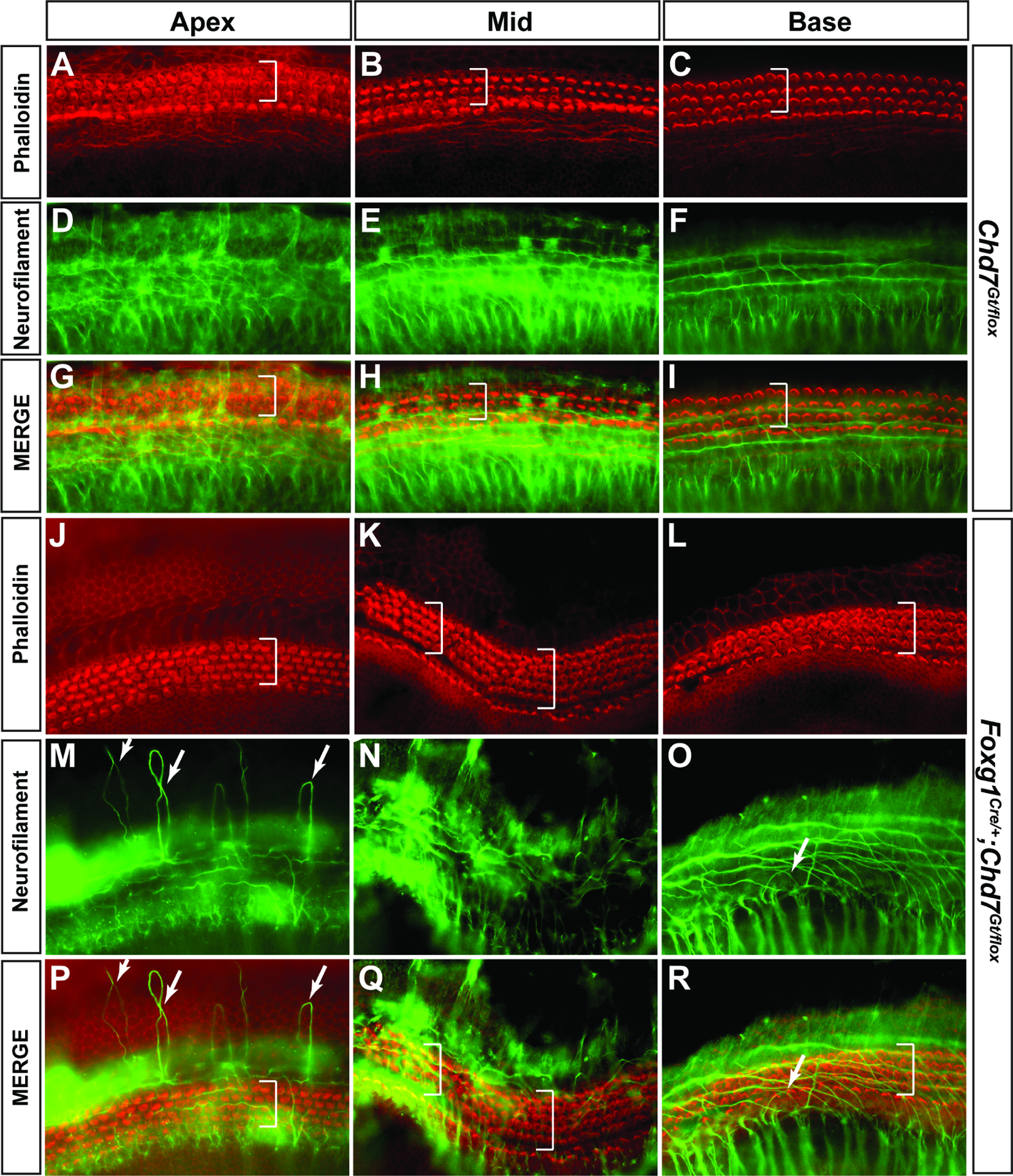
Phalloidin (A-C) and Neurofilament (D-F) staining in P1 Chd7Gt/flox cochleae demonstrates normal hair cell patterning in the organ of Corti and neurites extending away from hair cells toward the spiral limbus. Merged images are shown in panels G-I. White brackets show 3 rows of outer hair cells. Phalloidin (J-L) and Neurofilament (M-O) staining in Foxg1Cre/+;Chd7Gt/flox P1 cochleae reveals aberrant neurite looping in the apex (M, P, white arrows). Additional rows of outer hair cells (white brackets) are present in the apex, middle (K, N, Q) and base (L, O, R) regions of Foxg1Cre/+;Chd7Gt/flox cochleae. Merged images are shown in panels P-R.
Formation of the cochlea is known to depend on tightly regulated gene expression and signaling from growth factors and morphogens in neighboring mesenchymal tissues (Munnamalai and Fekete, 2016). The broad and early expression of Chd7 in the developing inner ear led us to ask whether tissue-specific deletion of Chd7 might result in unique inner ear phenotypes. As all inner ear structures derive from the otocyst proper (with the exception of neural crest-derived glial cells in the spiral ganglion and intermediate cells of the stria vascularis), and Chd7 is broadly expressed in the developing inner ear, we used tissue-specific Cre lines to delete Chd7 from the otocyst, otic mesenchyme, otic neuroblasts, spiral ganglion, hair cells, or a combination of these (Table 1).
Table 1.
Cre mice used in this study and their expression patterns in the inner ear.
| Cre Line | Age | Sites of Expression | Chd7cko | Reference |
|---|---|---|---|---|
| Pax2Cre;Chd7flox/flox | ||||
| TCre | E9.0 | Mesenchyme surrounding otic vesicle; absent from CVG and otic epithelium | TCre;Chd7Gt/flox | (Braunstein et al., 2009; Perantoni et al., 2005) |
| Foxg1Cre/+ | E8.75 | Otic placode | Foxg1Cre/+;Chd7Gt/flox | (Hebert and McConnell, 2000) |
| E12.5 | Sensory epithelial patches of the utricle, saccule, and crista; cochlear staining in the medial wall of the basal and middle turns | |||
| Ngn1Cre | E11.5 | Cochleovestibular ganglion | Ngn1Cre;Chd7Gt/flox | (Quinones et al., 2010) |
| ShhCre/+ | E13.5 | Spiral ganglion | ShhCre/+;Chd7flox/flox | (Harfe et al., 2004) (Liu et al., 2010) |
In a previous study, we showed that Foxg1Cre/+;Chd7Gt/flox mice in which Chd7 is deleted in the otocyst and surrounding mesenchyme exhibit severely hypoplastic semicircular canals and cochlear duct (Hurd et al., 2010). However, Foxg1Cre/+ is expressed in both the otocyst and surrounding mesenchyme, making it difficult to distinguish roles for Chd7 in these tissues. To address this, we compared phenotypes resulting from conditional knockout of Chd7 using Pax2Cre (expressed in the otocyst and hindbrain) and TCre (expressed in the otic mesenchyme) (Fig. 3). Pax2Cre;Chd7Gt/flox mice exhibited absent anterior and lateral semi-circular canals as well as severely malformed and hypoplastic cochleae, similar to previous malformations in Foxg1Cre/+;Chd7Gt/flox ears (Fig. 3) (N=3 ears per genotype) (Hurd et al., 2010; Osborne and Comis, 1991). In contrast, ears from TCre;Chd7Gt/flox mice exhibited normal vestibular and cochlear morphologies (Fig. 3) (N=4 ears per genotype). Taken together, these data suggest that Chd7 is dispensable in the otic mesenchyme but essential in the otocyst for proper cochlear and vestibular development.
Figure 3. Chd7 is required in the membranous labyrinth but not in the mesenchyme for proper formation of the inner ear.
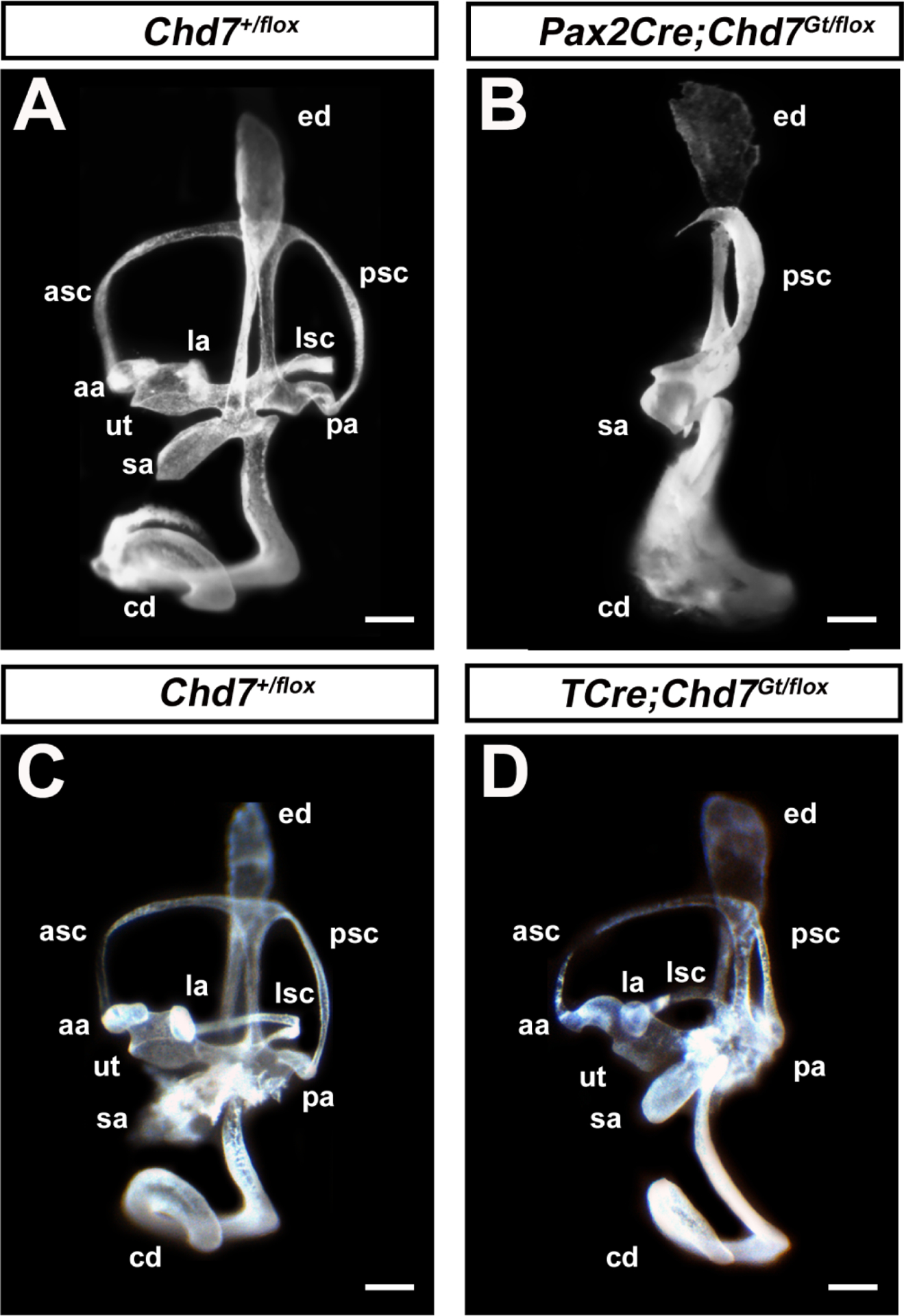
Images of paint-filled inner ears from Chd7+/flox (A) and littermate Pax2Cre;Chd7Gt/flox (B) embryos show enlargement of the endolymphatic duct, hypoplastic cochlear duct (cd), saccule (sa), and posterior canal (psc), absent lateral semicircular canal (lsc), anterior semicircular canal (asc), and absent ampullae associated with lateral (la), posterior (pa), and anterior (aa) semicircular canals. Images of paint-filled inner ears from Chd7+/flox (C) and littermate Tcre;Chd7Gt/flox (D) mice show normal morphology in both vestibular and auditory organs. Scale bars = 50 μm.
The severe cochlear hypoplasia observed in Pax2Cre;Chd7Gt/flox mice led us to examine cochlear morphology and sensory epithelia. To do this, we employed Pax2Cre;Chd7flox/flox mice without the Chd7Gt allele to specifically delete Chd7 in the otic placode. Compared to Chd7flox/flox (N=6 ears), the cochleae of Pax2Cre;Chd7flox/flox P1 mice (N=7 ears) were 57% shorter (1,945 ± 377 μm) than Chd7flox/flox littermate ears (4,544 ± 215 μm, p <0.0001, unpaired t-test) (Fig. 4A–C). We also observed cochlear region-specific defects in innervation and patterning of the sensory epithelium in Pax2Cre;Chd7flox/flox mice. Supernumerary hair cells were abundant in the apex of Pax2Cre;Chd7flox/flox cochleae (Fig. 4D, E–G, N–P, 14.3±4.9 compared to 0.2 ± 0.4 per 100 μm cochlea, p<0.0001, unpaired t-test) but are observed less frequently in the middle (Fig. 4 H–J, Q–S, 2.6 ± 3.1 per 100 μm cochlea compared to none) and rarely in the base (Fig. 4K–M, T–V, 0.3 ± 0.7 per 100 μm cochlea versus none). Disorganized spiral ganglion neurite projections were found in the apex of Pax2Cre;Chd7flox/flox cochleae (Fig. 4E–G versus Fig. 4N–P). Interestingly, the middle region of the Pax2Cre;Chd7flox/flox cochlea exhibited a narrowing of the sensory epithelium with missing outer hair cells (OHCs) and a bifurcation of the direction of neuritic projections, with most neurites projecting towards the apex of the cochlea rather than the base (Fig. 4H–J vs. Q–S). The phenotypes of the Pax2Cre;Chd7flox/flox cochleae were wildly variable in the middle region. While all samples exhibited aberrant neuritic projections and missing cells in the middle region of the cochlear duct, the severity and size of the gaps was variable and some specimens possessed an undulating epithelium (Fig 2 K, N, Q and Supplementary Fig. 2), similar to that observed in Foxg1Cre/+;Chd7 conditional mutants. The base of the cochlea in Pax2Cre;Chd7flox/flox mice contained occasional extra hair cells but was otherwise comparable to Chd7flox/flox cochleae (Fig. 4K–M, T–V). From these experiments, we conclude that Chd7 functions in cell autonomous roles in the otocyst for proper cochlear development and innervation of the sensory epithelium.
Figure 4. Otocyst specific loss of Chd7 by Pax2Cre disrupts cochlear development.
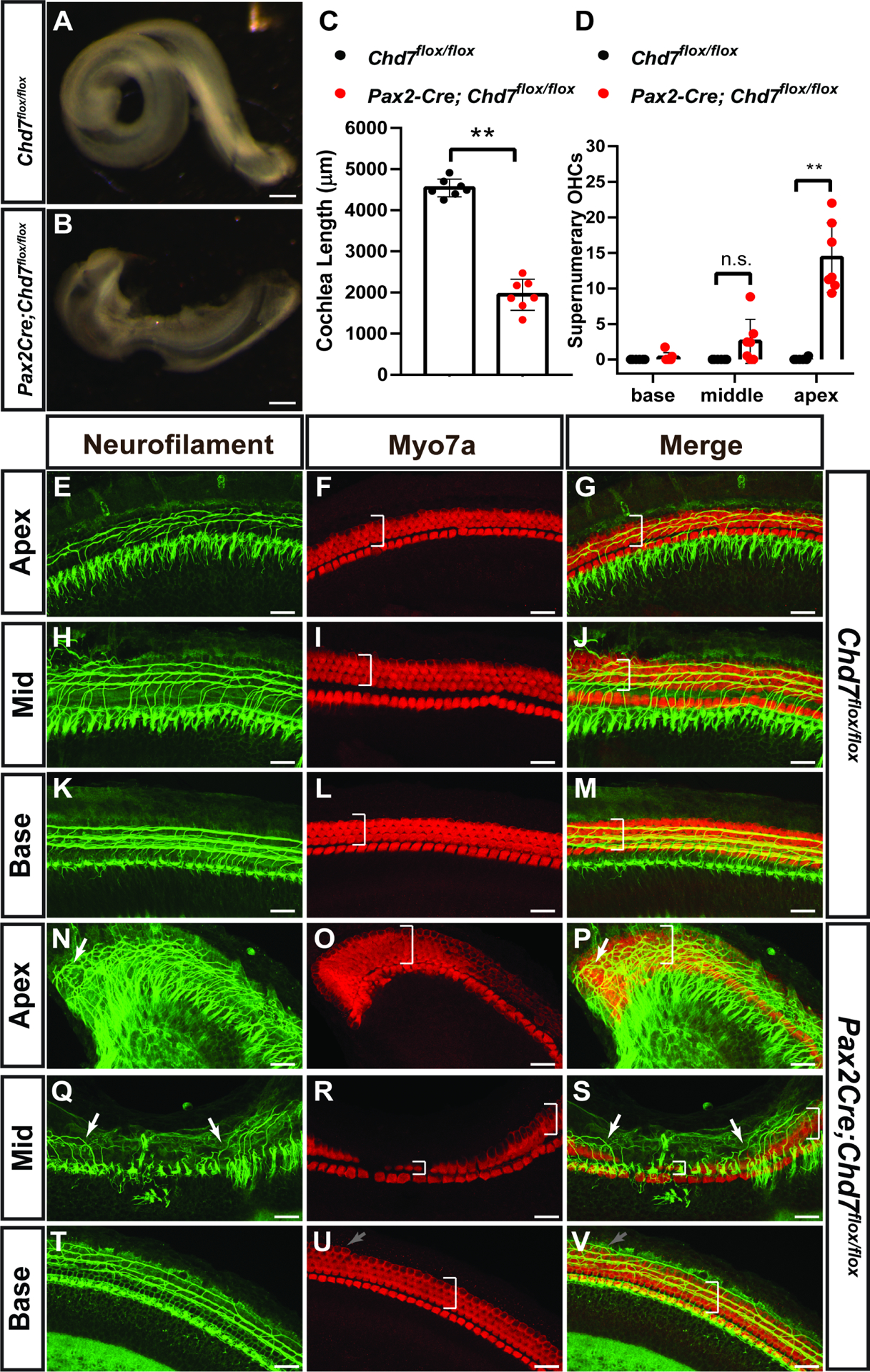
Representative bright field images of cochleae from P1 Chd7flox/flox (A) and Pax2Cre;Chd7flox/flox (B) mice. Cochlear length is significantly reduced (C) in Pax2Cre;Chd7flox/flox mice compared to Chd7flox/flox littermates (**, p< 0.0001, Student’s t-test). The apex of Pax2Cre;Chd7flox/flox cochleae, but not the middle and base regions, exhibits significantly increased supernumerary outer hair cells (D, **, <0.0001, Student’s t-test). Neurofilament and MYO7A staining in the apex, middle, and base regions of the cochlea in Chd7flox/flox mice shows normally organized neuritic projections and hair cell patterning (white brackets) (E-M). Neuritic projections are disorganized in the apex and middle regions of the Pax2Cre;Chd7flox/flox cochlea, but not in the base (N-S, white arrows). Anti-MYO7A staining reveals supernumerary outer hair cells in the apex (O, P, white brackets) and basal turn (U, V, gray arrows) as well as normal basal hair cells (U, V, white brackets) and a narrowing of the organ of Corti epihthelium (white brackets in R, S) in the middle of the Pax2Cre;Chd7flox/flox cochlea. Scale bars A-B = 100 μm; E-V = 25 μm.
Chd7 is broadly expressed in the cochlea at E14.5-E18.5, including the sensory epithelium that harbors hair cells (Fig. 1). To determine if Chd7 regulates the development of hair cells in a cell autonomous manner, we ablated Chd7 expression with Atoh1Cre mice. We also crossed these animals with the Zsgreen1 reporter mouse line to identify co-localization of Atoh1Cre and Chd7 expression. The majority of cochlear Atoh1Cre+ hair cells also express Chd7 (Fig. 5 A, B, C), and this expression was reduced in Atoh1Cre;Chd7flox/flox;Zsgreen cochlear epithelia (N=12 ears) compared to Atoh1Cre;Chd7+/flox;Zsgreen (N=4 ears) (Fig. 5 D, E, F). Hair cells, identified by MYO7A staining, were preserved in the apical, mid, and basal cochlear regions of the Atoh1Cre;Chd7flox/flox cochlea in P1 (N=11 mutant, 3 Cre-negative) (Fig. 5. G, H) ears. From these data, we conclude that the requirement for Chd7 in hair cell development precedes the onset of Atoh1 expression in the prosensory domain.
Figure 5. Chd7 requirement for hair cell development precedes hair cell specification.
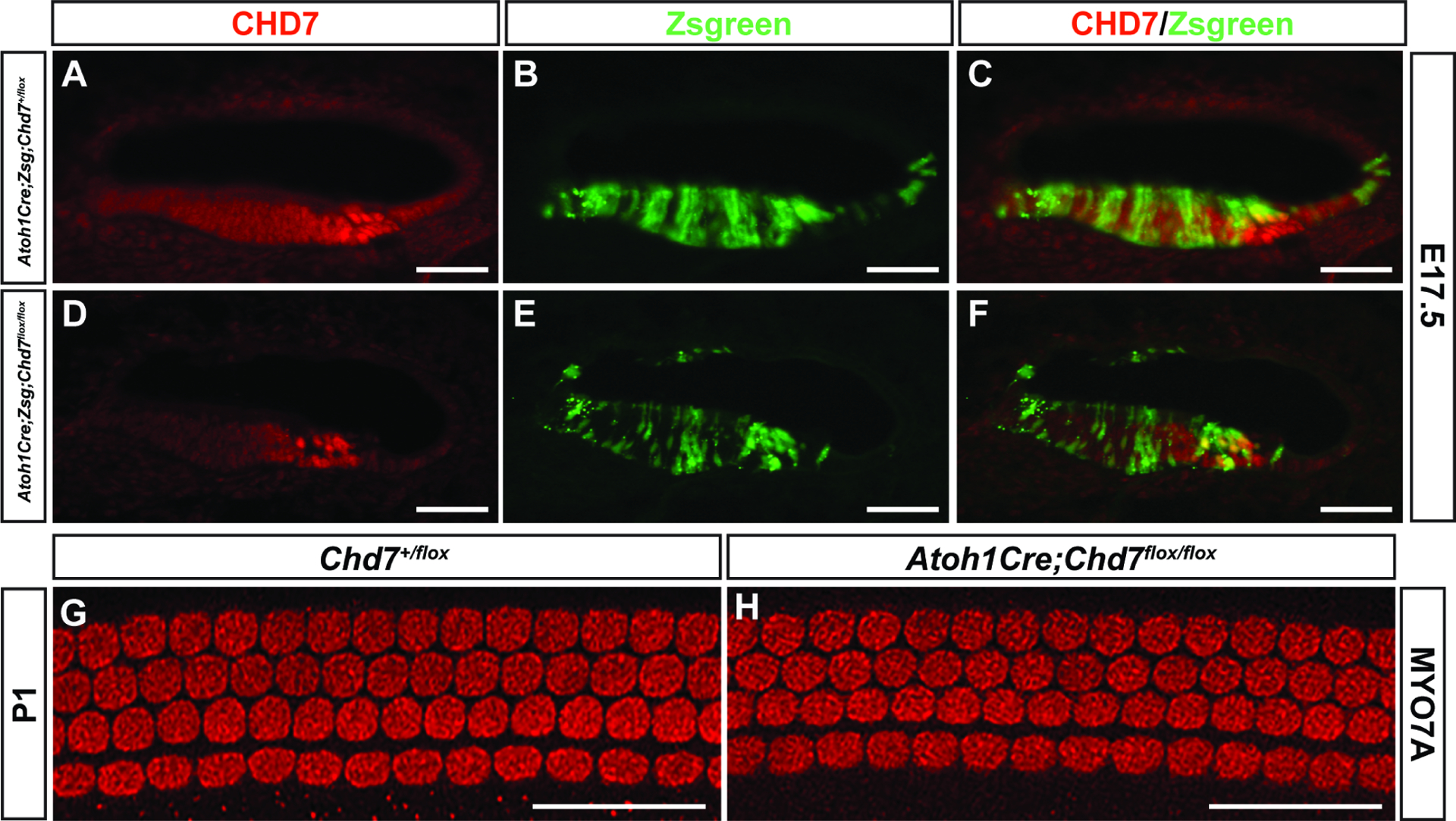
Anti-CHD7 staining of cryosections from E17.5 Atoh1Cre;Zsgreen;Chd7+/flox embryos (A, B, C) shows double positive (Zsgreen+/CHD7+) cells in the cochlear epithelium. Reduced CHD7+ staining is visible in Atoh1Cre;Zsgreen;Chd7flox/flox (D, E, F) cochleae. Whole-mount immunostaining for MYO7A in cochleae from Chd7+/flox (G) or littermate Atoh1Cre;Chd7flox/flox (H) P1 mice shows no differences in cochlear hair cell patterning. Scale bars A-F = 50 μm; G, H = 45 μm.
The neurons innervating the cochlea are derived from neuroblasts that delaminate from the prosensory domain of the otocyst between E10.5–12.5 (Coate and Kelley, 2013). In order to understand the unique requirements for Chd7 in the development of cochlear neurons versus hair cells, we deleted Chd7 from the developing neuroblasts using Ngn1Cre. Using the Zsgreen1 mouse reporter line, we compared Ngn1Cre;Chd7+/flox;Zsgreen embryos to Ngn1Cre;Chd7Gt/flox;Zsgreen knockout embryos in order to determine whether or not the loss Chd7 would affect the developing CVG. In both Ngn1Cre;Chd7+/flox;Zsgreen (Fig. 6 A–C) and Ngn1Cre;Chd7Gt/flox;Zsgreen (Fig. 6 D–F) ears, Chd7 is expressed in some, but not all, Ngn1Cre-expressing cells in the E11.5 CVG. There was a reduction in the total area of the spiral ganglion in P1 Ngn1Cre;Chd7Gt/flox;Zsgreen ears (N=5) compared to Ngn1Cre;Chd7+/flox;Zsgreen (N=6 ears; Fig. 6G). Ngn1Cre;Chd7+/flox;Zsgreen ears exhibited normal organization of hair cells throughout the cochlea (Fig. 6 H, I). In contrast, the apical region of Ngn1Cre;Chd7Gt/flox;Zsgreen cochleae exhibited aberrant axonal projections that crossed, looped, and extended beyond the cochlear epithelium (Fig. 6 I’). From these experiments, we conclude that Chd7 functions in a cell autonomous fashion to regulate the development of CVG neuroblasts and the architecture of neurites projecting to the cochlear sensory epithelium.
Figure 6. Deletion of Chd7 in developing neuroblasts leads to reduced cochleovestibular ganglion size and aberrant neurites.
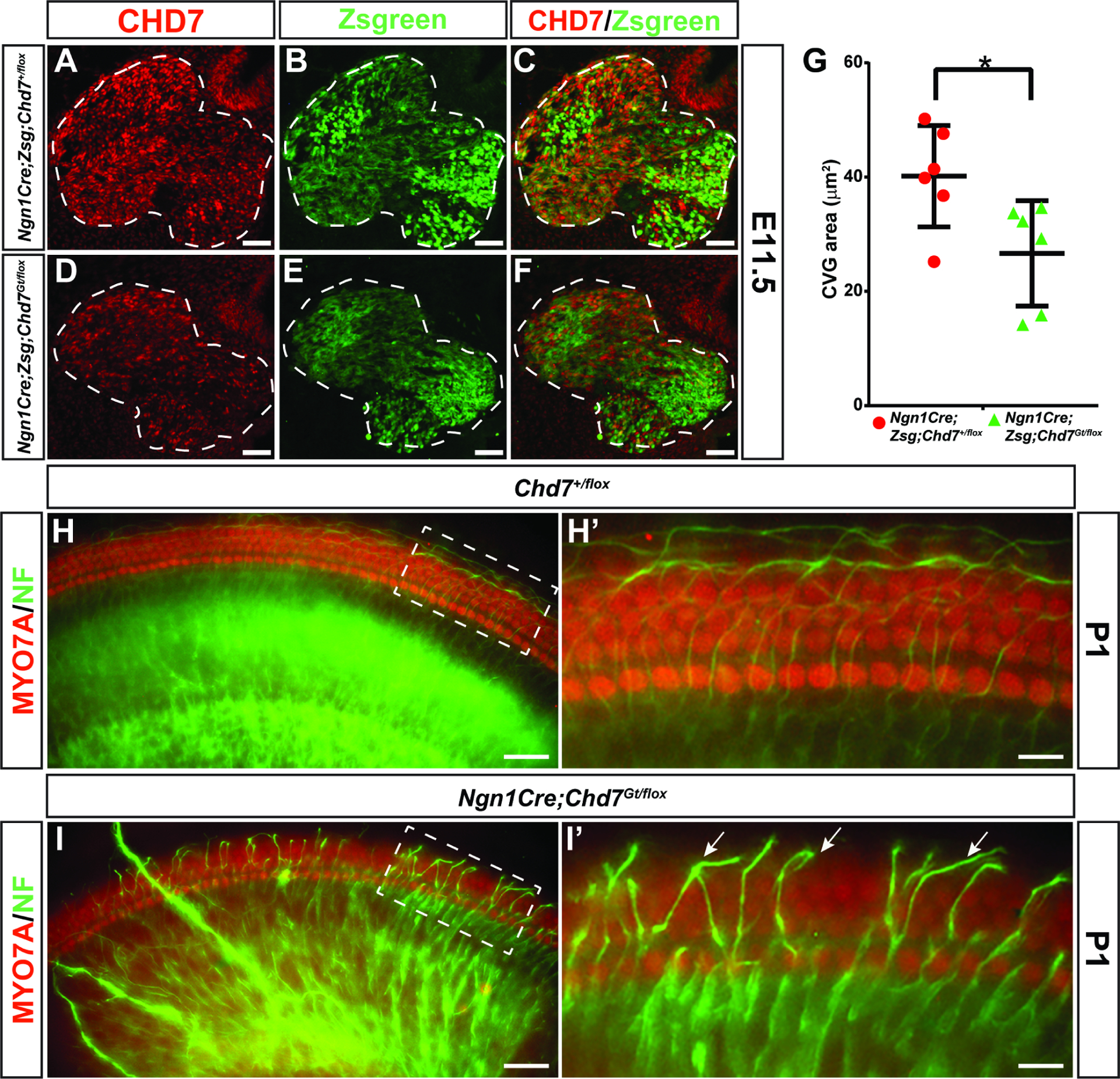
Anti-CHD7 staining and Zsgreen reporter fluorescence overlap in the E11.5 CVG in Ngn1Cre;Zsgreen;Chd7+/flox ears (A-C). CHD7 is reduced in the ganglion of Ngn1Cre;Zsgreen;Chd7Gt/flox ears (D-F). The ganglion area is significantly smaller in Ngn1Cre;Zsgreen;Chd7Gt/flox ears compared to Ngn1Cre;Zsgreen;Chd7+/flox controls (* denotes p-value = 0.0271, Student’s t-test) (G). Whole-mount cochleae from P1 Chd7+/flox or Ngn1Cre;Chd7Gt/flox mice stained with anti-Neurofilament antibody (H, I). Neuronal projections from the apical turn of the Chd7+/flox cochlea extend only to the edge of the epithelium (H’), whereas neurites in the Ngn1Cre;Chd7Gt/flox cochlea extend far beyond the epithelium (arrows in I’). Scale bars A-F = 50 μm; H-I’ = 45 μm.
Given the developmental requirement for Chd7 in the organization and abundance of cochlear neurons, we next investigated whether Chd7 is necessary to maintain ganglion structure and/or neuronal organization using ShhCre/+. Shh expression in the inner ear is restricted to the spiral ganglion and is expressed four days after Chd7 is first expressed in the ear (E13.5 compared to E9.5). Despite this delay in Shh expression, Shh is essential for formation of the cochlea and dynamically expressed in the spiral ganglion (Bok et al., 2013). We observed Zsgreen1 reporter expression in the E13.5 ShhCre/+ spiral ganglion, coincident with CHD7 expression (data not shown). At E15.5, Chd7 expression was reduced in spiral ganglion in ShhCre/+;Zsgreen1/+;Chd7flox/flox ears compared to ShhCre/+; Zsgreen1/+;Chd7flox/+ (Fig. 7). P1 whole-mount cochleae stained with anti-MYO7A and anti-Neurofilament showed normal hair cells and neurites in both Chd7flox/flox (N=4) and ShhCre/+;Chd7flox/flox (N=14) mice (Fig. 7). Together, these observations suggest that Chd7 is not required for maintenance of spiral ganglion neurites, despite important roles in their initial formation and organization.
Figure 7. Loss of Chd7 in the spiral ganglion does not impair neurite organization.
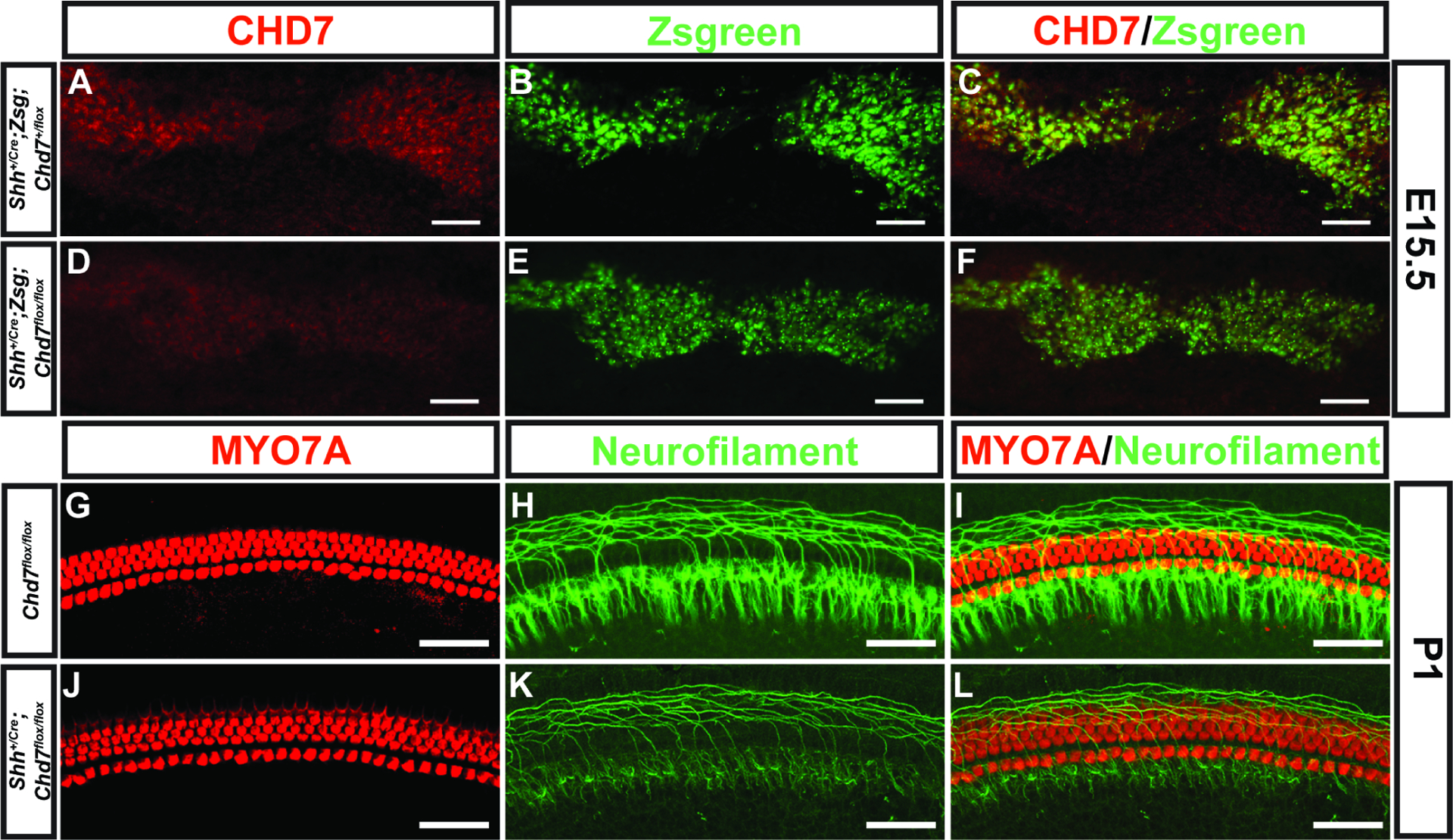
Anti-CHD7 staining and Zsgreen reporter fluorescence overlap in the spiral ganglion of E15.5 of Shh+/Cre;Chd7+/flox;Zsgreen (A-C) mice. CHD7 is reduced in the ganglion of E15.5 Shh+/Cre;Chd7flox/flox;Zsgreen (D-F) ears. Whole-mount cochleae from P1 Chd7+/flox and Shh+/Cre;Chd7flox/flox mice stained with anti-Neurofilament (H, I, K, L) and anti-Myo7A (G, I, J, L) shows normal appearing hair cells and neurites. Scale bars A-F = 50 μm; G-L = 45 μm.
Discussion
Through analysis of this series of Chd7 conditional knockout mice, we made several important observations about the function of CHD7 in mammalian inner ear development. We found that deletion of Chd7 in the early otocyst via Foxg1Cre/+ or Pax2Cre results in severe defects in inner ear structure, cochlear hair cell number and patterning, and neuronal architecture. Similarly, deletion of Chd7 in newly specified neuroblasts via Ngn1Cre leads to reduced cochleovestibular ganglion size and aberrant neurite trajectories. Interestingly, deletion of Chd7 later in inner ear development, either in hair cells (~ E12.5) or spiral ganglion neurons (~E13.5) by Atoh1Cre or ShhCre/+, respectively, has no major effects inner ear development. Additionally, deletion of Chd7 in the otic mesenchyme via TCre has no impact on inner ear morphology. Taken together, this analysis demonstrates critical tissue- and temporal-specific requirements of Chd7 during inner ear development (Fig. 8).
Figure 8. Summary of Chd7 conditional knockout mouse models and key findings.
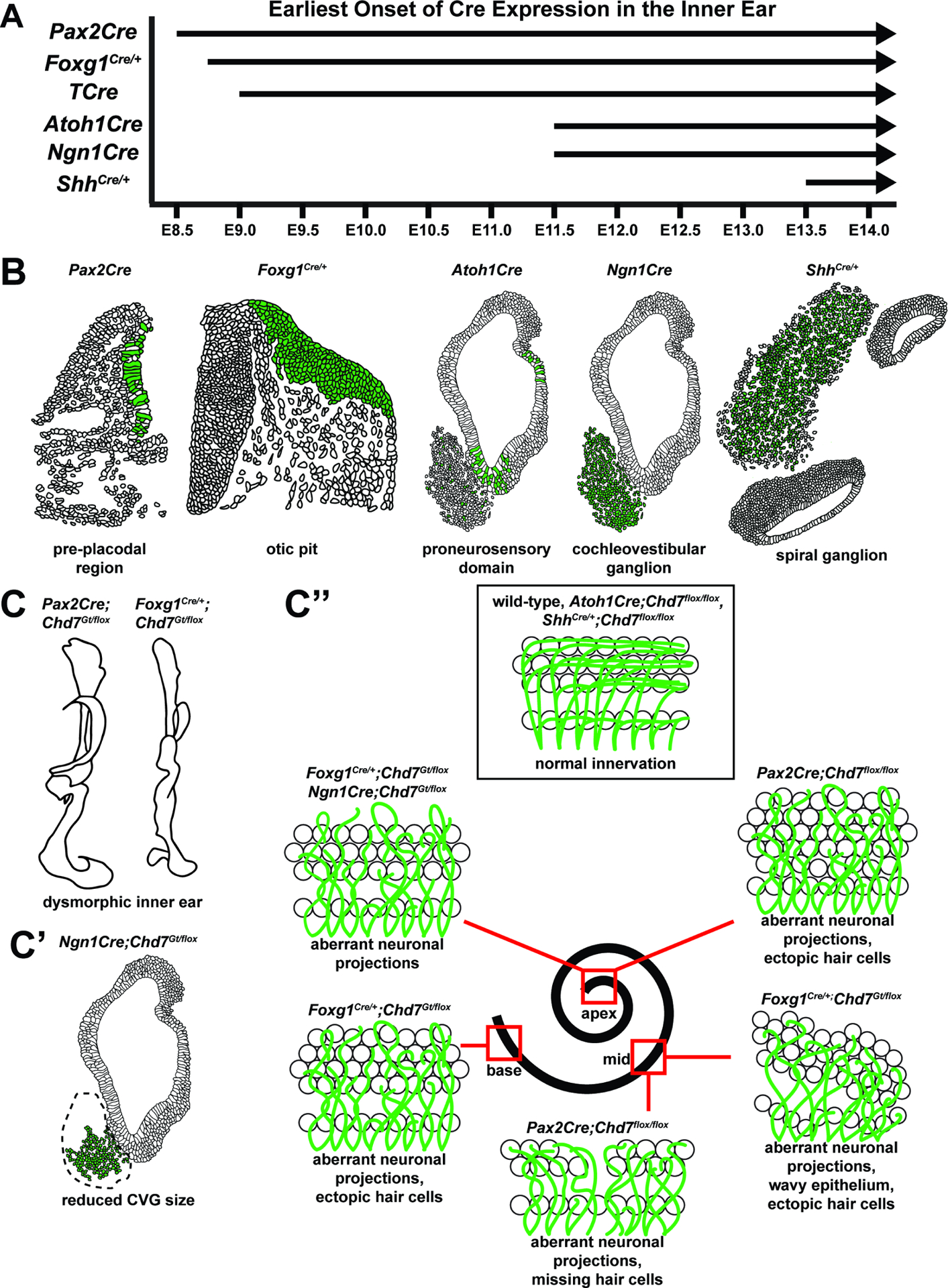
(A) Timeline of Cre activation in the inner ear in mouse embryonic development. (B) Spatial localization of Cre activity at the time points indicated for each Cre mouse line in A. Green indicates Cre expression. (C-C”) Phenotypes observed in Chd7 conditional knockout mice. (C) Gross morphological defects of the inner ear in Pax2Cre;Chd7Gt/flox and Foxg1Cre/+;Chd7Gt/flox mice. (C’) Reduced cochleovestibular ganglion size in Ngn1Cre;Chd7Gt/flox mice. (C”) Aberrant neuronal projections and dysmorphic inner and outer hair cells in various regions of the cochlea (apex, middle, base) in Pax2Cre;Chdfloxt/flox , Foxg1Cre/+;Chd7Gt/flox, and Ngn1Cre;Chd7Gt/flox mice. Normal hair cell morphology and innervation are observed in wild-type mice, as well as Atoh1Cre;Chd7flox/flox and ShhCre/+;Chd7flox/flox animals.
The severity of cochlear morphological defects observed in Foxg1Cre/+ and Pax2Cre conditional knockout mice illustrates the importance of CHD7 in the developing otocyst. Both Foxg1Cre/+ and Pax2Cre-mediated Chd7 conditional knockout mouse lines exhibit shortened, malformed cochleae, indicative of defective cochlear patterning and elongation. In mice, cochlear elongation is regulated by highly orchestrated patterning events that control spatial axes and rates of cell proliferation and differentiation via Wnt, Shh, and Bmp signaling (Braunstein et al., 2009; May-Simera, 2016; Muthu et al., 2019) and secretion of retinoic acid (Kelley et al., 1993). As with reduced Chd7 dosage, loss or reduction of various components of these signaling pathways results in short or hypoplastic cochleae, potentially via effects on cochlear patterning and cell proliferation. Mice with mutations in critical cell cycle regulators, such as p27kip, similarly display truncated cochleae (Chen and Segil, 1999). We demonstrated previously that CHD7 regulates cell proliferation in the inner ear and other neural systems, and it possible that similar defects occur in the developing cochlea (Hurd et al. 2010, Layman et al. 2009, (Micucci et al., 2014).
In addition to the function of Chd7 in regulating cochlear morphogenesis, we also observed spatial- and temporal-specific requirements for Chd7 in regulation of hair cell patterning. Foxg1Cre/+ and Pax2Cre Chd7 conditional knockout mice both display abnormal hair cell numbers in distinct regions of the cochlea. While Foxg1Cre/+;Chd7Gt/flox mutants have supernumerary outer hair cells throughout the length of the cochlea and an undulating sensory epithelium in the middle region, Pax2Cre;Chd7floxflox mutants have supernumerary hair cells in the apex, missing hair cells and narrowed epithelium in the middle cochlear turn, and minimal defects in the base. Differences in hair cell patterning between Foxg1Cre/+ and Pax2Cre Chd7 conditional knockout mice may related to subtle differences in Cre recombinase activity or haploinsufficiency for Foxg1 in Foxg1Cre/+ mice. While Pax2Cre and Foxg1Cre/+ Chd7 conditional knockout mice demonstrate an early requirement for Foxg1Cre/+ in hair cell patterning, Atoh1Cre;Chd7flox/flox mutants display no apparent hair cell or sensory epithelial defects. This observation suggests Chd7 functions early during development of the otocyst, prior to hair cell specification.
We also uncovered critical functions for Chd7 in spiral ganglion neuritic projections to the sensory epithelium. Foxg1Cre/+ and Pax2Cre Chd7 conditional knockout mutants exhibit aberrant neuritic projections throughout the entire length of the cochlea, with looping and disorganized neurites. The spatial- and temporal-specific nature of Chd7 function is highlighted by comparing Foxg1Cre/+ and Pax2Cre Chd7 conditional mutants to Ngn1Cre;Chd7Gt/flox mutants, as the latter mice display neuritic defects only in the apex of the cochlea. The apex of the cochlear sensory epithelium is the first to become innervated by spiral ganglion neuritic projections, while the middle turn and base are innervated soon thereafter (Coate and Kelley, 2013).
Interestingly, conditional deletion of Chd7 via ShhCre/+ in differentiating spiral ganglion neurons does not result in neuritic pathfinding defects. Similar to the hair cell patterning defects described above, Chd7 appears to be uniquely required in early developmental processes and less critical in later processes or differentiated cell types. A recent study showed that loss of Chd7 in mouse embryonic stem cell-derived neurons also impairs their ability to form neurites (Yao et al., 2018). The precise mechanisms by which Chd7 regulates neuritic pathfinding are unknown, but CHD7 may acts by regulating expression of proneural transcription factor genes. Previous research identified Gata3 as an essential regulator of spiral ganglion neuron differentiation, with early deletion resulting in misguided peripheral projections that are unable to form proper radial bundles (Appler et al., 2013). Notably, E10.5 Foxg1Cre/+;Chd7Gt/flox otocysts exhibit reduced Gata3 expression following Chd7 deletion (Hurd et al., 2012). In addition to transcriptional regulation by Gata3, proper formation of neural projections in the inner ear depends upon guidance cues that attract or repel growing axons. For example, Eph/ephrin signaling in the otic mesenchyme plays an important role in regulating axon guidance from the spiral ganglion to the cochlear epithelium (Defourny, 2019). Loss of Efnb2 results in fasciculation defects similar to those that we observed in Pax2Cre;Chd7flox/flox ears (Coate et al., 2012). In addition, disruption of ephrin B/eph B interactions in the mouse cochlea leads to abnormal innervation patterns in the organ of Corti, including neural projections that extend beyond the third row of outer hair cells (Zhou et al., 2011). Additional studies are needed to determine whether disrupted eph/ephrin signaling contributes to the abnormal neurites observed in Pax2Cre, Foxg1Cre, and Ngn1Cre conditional knockout ears.
Cochlear elongation, sensory epithelial patterning, hair cell morphogenesis, and spiral ganglion neuritic outgrowth are all regulated by planar cell polarity pathways (Montcouquiol and Kelley, 2003; Montcouquiol et al., 2003). Mouse mutants of Prickle1, as well as Vangl2 and Dvl1/2, have markedly similar phenotypes to Foxg1Cre/+, Pax2Cre, Ngn1Cre Chd7 conditional knockouts, including truncated cochleae, supernumerary hair cells, and aberrant apical neurite patterning (Montcouquiol et al., 2003), (Mao et al., 2011; Wang et al., 2006; Yang et al., 2017). Future experiments will determine if CHD7 directly or indirectly targets components of the planer cell polarity pathway to regulate cochlear development.
Some phenotypes in Pax2Cre;Chd7cko mice resemble ears with aberrant Notch signaling. Missing OHCs in the middle region of the cochlea are also observed in mouse Jag1 mutants (Kiernan et al., 2001; Tsai et al., 2001), and loss of RBPjκ, which encodes the transcriptional mediator of canonical Notch signaling, leads to shortened cochleae (Basch et al., 2011). Excess proliferation and a raft of supernumerary hair cells occurs in Dll1+/−;Jag2−/− and Dll1hyp/−;Jag2−/− mutants (Kiernan et al., 2005). Our previous work implicated Notch signaling defects in early otic lineages with loss of Chd7 (Durruthy-Durruthy et al., 2018), and Jag1 is reduced with Chd7 haploinsufficiency (Hurd et al., 2012). Closer examination of inner ears with conditional loss of Chd7 will be helpful in determining the contributions of various Notch signaling pathway components contribute to cochlear development.
In summary, these studies show that Chd7 exhibits time and tissue specificity in promoting inner ear development. Chd7 is necessary in the otocyst and neuroblasts for early events that eventually control cochlear elongation, patterning, hair cell organization, and neurite development. In contrast, later expression of Chd7 in differentiated hair cells or spiral ganglion neurons appears dispensable for their overall morphology or maintenance. These time and space constraints on Chd7 will inform the development of interventions and therapeutics for neurosensory cell regeneration in the mammalian ear, with implications for human hearing and balance dysfunction.
Supplementary Material
Highlights.
Chd7 is required in the mouse otocyst for cochlear elongation
Chd7 is required in the mouse otocyst for hair cell patterning
Chd7 is not required in peri-otic mesenchyme for inner ear morhphogenesis
Spiral ganglion neurons and cochlear hair cells are sensitive to Chd7 dosage
Acknowledgements
D.M.M. is supported by NIH R01DC014456 and R01DC018404, the Taubman Medical Institute, and the Ravitz Foundation Professorship in Pediatrics and Communicable Diseases. K.E.R. is supported by NIH T32 DC000011.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Appler JM, Lu CC, Druckenbrod NR, Yu WM, Koundakjian EJ, Goodrich LV, 2013. Gata3 is a critical regulator of cochlear wiring. J Neurosci 33, 3679–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basch ML, Ohyama T, Segil N, Groves AK, 2011. Canonical Notch signaling is not necessary for prosensory induction in the mouse cochlea: insights from a conditional mutant of RBPjkappa. J Neurosci 31, 8046–8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermingham NA, Hassan BA, Price SD, Vollrath MA, Ben-Arie N, Eatock RA, Bellen HJ, Lysakowski A, Zoghbi HY, 1999. Math1: an essential gene for the generation of inner ear hair cells. Science 284, 1837–1841. [DOI] [PubMed] [Google Scholar]
- Bok J, Zenczak C, Hwang CH, Wu DK, 2013. Auditory ganglion source of Sonic hedgehog regulates timing of cell cycle exit and differentiation of mammalian cochlear hair cells. Proc Natl Acad Sci U S A 110, 13869–13874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosman EA, Penn AC, Ambrose JC, Kettleborough R, Stemple DL, Steel KP, 2005. Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum Mol Genet 14, 3463–3476. [DOI] [PubMed] [Google Scholar]
- Braunstein EM, Monks DC, Aggarwal VS, Arnold JS, Morrow BE, 2009. Tbx1 and Brn4 regulate retinoic acid metabolic genes during cochlear morphogenesis. BMC Dev Biol 9, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Segil N, 1999. p27(Kip1) links cell proliferation to morphogenesis in the developing organ of Corti. Development 126, 1581–1590. [DOI] [PubMed] [Google Scholar]
- Coate TM, Kelley MW, 2013. Making connections in the inner ear: recent insights into the development of spiral ganglion neurons and their connectivity with sensory hair cells. Semin Cell Dev Biol 24, 460–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coate TM, Raft S, Zhao X, Ryan AK, Crenshaw EB 3rd, Kelley MW, 2012. Otic mesenchyme cells regulate spiral ganglion axon fasciculation through a Pou3f4/EphA4 signaling pathway. Neuron 73, 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defourny J, 2019. Eph/ephrin signalling in the development and function of the mammalian cochlea. Dev Biol 449, 35–40. [DOI] [PubMed] [Google Scholar]
- Driver EC, Kelley MW, 2020. Development of the cochlea. Development 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durruthy-Durruthy R, Sperry ED, Bowen ME, Attardi LD, Heller S, Martin DM, 2018. Single Cell Transcriptomics Reveal Abnormalities in Neurosensory Patterning of the Chd7 Mutant Mouse Ear. Front Genet 9, 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves AK, Bronner-Fraser M, 2000. Competence, specification and commitment in otic placode induction. Development 127, 3489–3499. [DOI] [PubMed] [Google Scholar]
- Harfe BD, Scherz PJ, Nissim S, Tian H, McMahon AP, Tabin CJ, 2004. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 118, 517–528. [DOI] [PubMed] [Google Scholar]
- Hartman BH, Reh TA, Bermingham-McDonogh O, 2010. Notch signaling specifies prosensory domains via lateral induction in the developing mammalian inner ear. Proc Natl Acad Sci U S A 107, 15792–15797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert JM, McConnell SK, 2000. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol 222, 296–306. [DOI] [PubMed] [Google Scholar]
- Hurd EA, Adams ME, Layman WS, Swiderski DL, Beyer LA, Halsey KE, Benson JM, Gong TW, Dolan DF, Raphael Y, Martin DM, 2011. Mature middle and inner ears express Chd7 and exhibit distinctive pathologies in a mouse model of CHARGE syndrome. Hear Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd EA, Capers PL, Blauwkamp MN, Adams ME, Raphael Y, Poucher HK, Martin DM, 2007. Loss of Chd7 function in gene-trapped reporter mice is embryonic lethal and associated with severe defects in multiple developing tissues. Mamm Genome 18, 94–104. [DOI] [PubMed] [Google Scholar]
- Hurd EA, Micucci JA, Reamer EN, Martin DM, 2012. Delayed fusion and altered gene expression contribute to semicircular canal defects in Chd7 deficient mice. Mech Dev 129, 308–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd EA, Poucher HK, Cheng K, Raphael Y, Martin DM, 2010. The ATP-dependent chromatin remodeling enzyme CHD7 regulates pro-neural gene expression and neurogenesis in the inner ear. Development 137, 3139–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley MW, Xu XM, Wagner MA, Warchol ME, Corwin JT, 1993. The developing organ of Corti contains retinoic acid and forms supernumerary hair cells in response to exogenous retinoic acid in culture. Development 119, 1041–1053. [DOI] [PubMed] [Google Scholar]
- Kiernan AE, Ahituv N, Fuchs H, Balling R, Avraham KB, Steel KP, Hrabe de Angelis M, 2001. The Notch ligand Jagged1 is required for inner ear sensory development. Proc Natl Acad Sci U S A 98, 3873–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan AE, Cordes R, Kopan R, Gossler A, Gridley T, 2005. The Notch ligands DLL1 and JAG2 act synergistically to regulate hair cell development in the mammalian inner ear. Development 132, 4353–4362. [DOI] [PubMed] [Google Scholar]
- Koundakjian EJ, Appler JL, Goodrich LV, 2007. Auditory neurons make stereotyped wiring decisions before maturation of their targets. J Neurosci 27, 14078–14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layman WS, Sauceda MA, Zuo J, 2013. Epigenetic alterations by NuRD and PRC2 in the neonatal mouse cochlea. Hear Res 304, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Owen T, Zhang L, Zuo J, 2010. Dynamic expression pattern of Sonic hedgehog in developing cochlear spiral ganglion neurons. Dev Dyn 239, 1674–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CC, Appler JM, Houseman EA, Goodrich LV, 2011. Developmental profiling of spiral ganglion neurons reveals insights into auditory circuit assembly. J Neurosci 31, 10903–10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Mulvaney J, Zakaria S, Yu T, Morgan KM, Allen S, Basson MA, Francis-West P, Irvine KD, 2011. Characterization of a Dchs1 mutant mouse reveals requirements for Dchs1-Fat4 signaling during mammalian development. Development 138, 947–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P, Swanson GJ, 1993. Descriptive and experimental analysis of the epithelial remodellings that control semicircular canal formation in the developing mouse inner ear. Dev Biol 159, 549–558. [DOI] [PubMed] [Google Scholar]
- Matei V, Pauley S, Kaing S, Rowitch D, Beisel KW, Morris K, Feng F, Jones K, Lee J, Fritzsch B, 2005. Smaller inner ear sensory epithelia in Neurog 1 null mice are related to earlier hair cell cycle exit. Dev Dyn 234, 633–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May-Simera H, 2016. Evaluation of Planar-Cell-Polarity Phenotypes in Ciliopathy Mouse Mutant Cochlea. J Vis Exp, 53559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie E, Krupin A, Kelley MW, 2004. Cellular growth and rearrangement during the development of the mammalian organ of Corti. Dev Dyn 229, 802–812. [DOI] [PubMed] [Google Scholar]
- Micucci JA, Layman WS, Hurd EA, Sperry ED, Frank SF, Durham MA, Swiderski DL, Skidmore JM, Scacheri PC, Raphael Y, Martin DM, 2014. CHD7 and retinoic acid signaling cooperate to regulate neural stem cell and inner ear development in mouse models of CHARGE syndrome. Hum Mol Genet 23, 434–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montcouquiol M, Kelley MW, 2003. Planar and vertical signals control cellular differentiation and patterning in the mammalian cochlea. J Neurosci 23, 9469–9478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montcouquiol M, Rachel RA, Lanford PJ, Copeland NG, Jenkins NA, Kelley MW, 2003. Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature 423, 173–177. [DOI] [PubMed] [Google Scholar]
- Morsli H, Choo D, Ryan A, Johnson R, Wu DK, 1998. Development of the mouse inner ear and origin of its sensory organs. J Neurosci 18, 3327–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munnamalai V, Fekete DM, 2016. Notch-Wnt-Bmp crosstalk regulates radial patterning in the mouse cochlea in a spatiotemporal manner. Development 143, 4003–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthu V, Rohacek AM, Yao Y, Rakowiecki SM, Brown AS, Zhao YT, Meyers J, Won KJ, Ramdas S, Brown CD, Peterson KA, Epstein DJ, 2019. Genomic architecture of Shh-dependent cochlear morphogenesis. Development 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyama T, Groves AK, 2004. Generation of Pax2-Cre mice by modification of a Pax2 bacterial artificial chromosome. Genesis 38, 195–199. [DOI] [PubMed] [Google Scholar]
- Osborne MP, Comis SD, 1991. Preparation of inner ear sensory hair bundles for high resolution scanning electron microscopy. Scanning Microsc 5, 555–564. [PubMed] [Google Scholar]
- Perantoni AO, Timofeeva O, Naillat F, Richman C, Pajni-Underwood S, Wilson C, Vainio S, Dove LF, Lewandoski M, 2005. Inactivation of FGF8 in early mesoderm reveals an essential role in kidney development. Development 132, 3859–3871. [DOI] [PubMed] [Google Scholar]
- Quinones HI, Savage TK, Battiste J, Johnson JE, 2010. Neurogenin 1 (Neurog1) expression in the ventral neural tube is mediated by a distinct enhancer and preferentially marks ventral interneuron lineages. Dev Biol 340, 283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Scheffer DI, Kwan KY, Corey DP, 2015. SHIELD: an integrative gene expression database for inner ear research. Database (Oxford) 2015, bav071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao W, Lai E, 1992. Telencephalon-restricted expression of BF-1, a new member of the HNF-3/fork head gene family, in the developing rat brain. Neuron 8, 957–966. [DOI] [PubMed] [Google Scholar]
- Tsai H, Hardisty RE, Rhodes C, Kiernan AE, Roby P, Tymowska-Lalanne Z, Mburu P, Rastan S, Hunter AJ, Brown SD, Steel KP, 2001. The mouse slalom mutant demonstrates a role for Jagged1 in neuroepithelial patterning in the organ of Corti. Hum Mol Genet 10, 507–512. [DOI] [PubMed] [Google Scholar]
- Wang J, Hamblet NS, Mark S, Dickinson ME, Brinkman BC, Segil N, Fraser SE, Chen P, Wallingford JB, Wynshaw-Boris A, 2006. Dishevelled genes mediate a conserved mammalian PCP pathway to regulate convergent extension during neurulation. Development 133, 1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Kersigo J, Wu S, Fritzsch B, Bassuk AG, 2017. Prickle1 regulates neurite outgrowth of apical spiral ganglion neurons but not hair cell polarity in the murine cochlea. PLoS One 12, e0183773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Hill SF, Skidmore JM, Sperry ED, Swiderski DL, Sanchez GJ, Bartels CF, Raphael Y, Scacheri PC, Iwase S, Martin DM, 2018. CHD7 represses the retinoic acid synthesis enzyme ALDH1A3 during inner ear development. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou CQ, Lee J, Henkemeyer MJ, Lee KH, 2011. Disruption of ephrin B/Eph B interaction results in abnormal cochlear innervation patterns. Laryngoscope 121, 1541–1547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.