

Structured abstract
Introduction:
Surgical resection is curative for some patients with early lung squamous cell carcinoma. Staging and clinical factors do not adequately predict recurrence risk. We sought to validate discriminative performance of proposed prognostic gene expression signatures at a level of rigor sufficient to support clinical use.
Methods:
Two-stage validation used independent core laboratories, objective quality control standards, locked test parameters, and large multi-institutional specimen/data sets. First stage validation confirmed a signature’s ability to stratify patient survival. Second stage validation determined which signature(s) optimally improved risk discrimination when added to baseline clinical predictors. Participants were prospectively enrolled on institutional (Cohort I) or cooperative group (Cohort II) biospecimen/data collection protocols. All cases underwent central review of clinical, pathologic and biospecimen parameters using objective criteria to determine final inclusion (Cohort I: n=249; Cohort II: n=234). Primary selection required that a signature significantly predict 3-years survival after surgery in Cohort I. Signatures meeting this criterion were further tested in Cohort II, comparing risk prediction using baseline risk factors alone versus in combination with the signature.
Results:
Male sex, advanced age, and higher stage were associated with shorter survival in Cohort I and established a baseline clinical model. Of three signatures validated in Cohort I, one signature was validated in Cohort II and statistically significantly enhanced prognosis relative to the baseline model (C-index difference 0.122; p<0.05).
Conclusions and relevance:
These results represent the first rigorous validation of a test appropriate to direct adjuvant treatment or clinical trials for patients with lung squamous cell carcinoma.
Introduction
Lung cancer is a global health problem with about 228,150 new patients diagnosed annually in the US and leading to about142,670 deaths. Non-small cell lung cancer (NSCLC) represents 87% of all lung cancers, with about 65,000 comprising the squamous cell carcinoma subtype (SC).1, 2 Patients with SC (stage I-IIIA) are treated by surgery with curative intent. Post-operative adjuvant chemotherapy (ACT) improves survival modestly in AJCC 8th edition stages II-III NSCLC,3–6 but selection of appropriate candidates for treatment remains a challenge absent prognostic and predictive molecular tests. While new systemic and biological therapy options guided by molecular biomarkers have become available for patients with adenocarcinoma, there has been little progress in patients with SC other than immune checkpoint inhibitors.7 Thus, even patients with stage IA (1–3), IB and IIA SC after complete surgical resection, experience only 90%, 85%, 80%, 73% and 65% five-year survival, respectively.3
Since the late 1990s, continuously improving methods to determine gene expression signatures have been directed to develop prognostic signatures for a variety of cancers, some of which have been commercialized for clinical use, e.g. Mammaprint for breast cancer.8, 9 Efforts in lung cancer, mostly focused on adenocarcinoma, have resulted in a few commercialized products which have not yet been accepted for reimbursement by Medicare/HCFA.10–12 Several tests that have been proposed specifically for lung SC have not yet been subjected to independent multi-institutional validation.13–18
In 2010, Subramanian and Simon published a seminal review of all significant prognostic tests proposed for NSCLC to that time and concluded that most, if not all, were flawed.19 They furthermore set down their recommendations for the development and validation of a successful prognostic test. Using their paper as a guide, we sought to examine existing (published/non-published) promising signatures for prognosis in lung SC and potentially to derive more robust new ones.
The primary objective of the current study was to validate biomarkers to identify the 10% to 27% of patients with stage I and 35% of patients with stage IIA SC who are likely to recur following surgical resection, so that these patients may be offered enrollment in clinical trials evaluating directed ACT. A secondary objective was to identify patients with stage IIB SC who are unlikely to develop recurrences and might thereby be spared the potential significant toxicity and expense of ACT. Herein we describe the identification and validation of a signature in two independent patient-specimen cohorts, one from a multi-center biorepository and the other from a prospective multi-institutional NCTN lung biobanking protocol, all with clinical annotations.
Materials and Methods
Oversight, Tumor Collection, and Annotation:
A multi-institutional NCI-funded network of investigators (Lung SC Consortium) assembled a large cohort of well-characterized resected frozen tumor specimens from patients with early stage SC to systematically evaluate relevant published and other proposed molecular signatures to select and validate the most accurate gene expression signature(s) in a CLIA environment. Steering, Pathology, Biostatistics, Assay, and Clinical cores were established to prepare and annotate the specimens, as well as to collect and analyze the data.
mRNA Signatures examined:
A review of the literature and preliminary results led to these RNA expression signatures: University of Michigan (UM): Beer and Raponi reported a 50-gene prognostic signature based on Affymetrix U133A mRNA profiling of 129 lung SC.14 Princess Margaret Hospital (PMH): Zhu et. al. developed a 12-gene signature using the UM dataset14 by applying MARSA (Maximizing R Square Algorithm) and further testing in silico in two independent lung cancer data sets.15, 20 The group also independently developed a 15-gene signature using tumor samples of the JBR 10 adjuvant chemotherapy trial involving 482 stage IB-II NSCLC patients.21, 22 Although this was a NSCLC-based prognostic signature, its prognostic value was also validated using the 79 stage IB-II SC patients from the UM dataset,14 indicating that it could also be a SC prognostic classifier. Brigham and Women’s Hospital (BWH): Signatures were developed by Bueno and colleagues using the UM dataset.14 Applying a gene ratio algorithm,23–25 two ratios made up of 3 genes (IL16/C2CD2L; IL16/216857_at) were identified that were validated in the Duke8 (p=0.0041) and Lee20 datasets (p=0.01). Duke Medical Center (DMC): A novel signature was developed based on comparative genomics in genetically-identical mice between quiescent basilar bronchial epithelium and squamous metaplastic cells activated by tracheobronchial injury. The differential gene profile was translated to human equivalent using Chip Comparer and correlated to the DMC data set of 45 clinical stage I SC patients26 to derive a genomic profile of SC tumors that could identify a more aggressive phenotype. Washington University School of Medicine (WU): Govindan and You conducted a meta-analysis of 7 published data sets to identify differentially-expressed genes related to survival.27
Specimens used for the validation cohorts:
Cohort I included 249 specimens from unique patients obtained from six participating sites. All specimens had been collected and fresh frozen with IRB approval from patients who underwent surgery for early stage lung cancer and for whom no neoadjuvant or adjuvant therapy was administered. Full demographic, clinical and outcome data were collected via REDCap and reviewed by the clinical core.
Cohort II consisted of 234 Alliance for Clinical Trials in Oncology (Alliance) /CALGB SC tumors obtained as part of an approved correlative science protocol (CALGB 150807) from the lung cancer tumor bank (CALGB 140202, now part of the Alliance biospecimen resource). This protocol opened in 2004 and accrued 1220 patients from 15 participating centers. Fresh frozen specimens collected at participant sites were shipped to the Alliance biorepository at BWH for quality control and storage. Clinical annotation data including semi-annual outcome assessments were submitted to the CALGB/Alliance Statistical and Data Center. Among available SC tumors, 234 met the criteria for this study.
Experimental design:
For Cohort I, a histological H&E slide was made from frozen material adjacent to the portion from each tumor used to prepare 2 µg of total RNA. The slides were sent to the pathology core at the University of Colorado for review of histology and tumor content, the RNA was robotically prepared by the Duke core lab and sent to the BWH genomic core lab.
For Cohort II, flanking slides and 1–2 µg of total RNA were prepared and sent to the BWH genomic core lab from the Alliance biorepository.
Pathology digital imaging, central review, specimen collection and processing:
Flanking frozen sections and corresponding clinical fixed SC specimens and redacted pathology reports from eligible patients were centrally received and registered at the University of Colorado (UC). On registration, a single representative excellent quality Aperio whole slide scanned image of a H&E stained clinical FFPE section was uploaded into the web-database using the caTissue Core tool. Study entry was based on a histological diagnosis of squamous carcinoma rendered by the local pathologist and confirmed by review of the scanned digital image. Tumors were classified according to WHO nomenclature for squamous carcinoma.28 Specimens were annotated with consensus diagnosis, % tumor cellularity and % necrosis. Specimens chosen for analysis had confirmed diagnosis of SC and utilized regions with ≥40% tumor cellularity for RNA extraction. The original pathology report for each case was also submitted for path core confirmation and REDCap data entry of: number of blocks and slides available; type of specimen; surgical procedure, site of tumor origin; WHO/IASLC histological type and subtype, differentiation grade, lymphovascular invasion, visceral pleura invasion and pathological stage of the tumor. Because the time period from specimen collection to data analysis spanned several editions of the AJCC Cancer Staging Manual, 6th, 7th and 8th edition criteria were applied to source pathology data from all cases in both cohorts. All 3 AJCC editions were used to compute multivariable models for all specimens.
mRNA profiling:
RNA isolation, cRNA synthesis and Affymetrix gene expression profiling was performed as described by Raponi et al.14 Following robotic RNA isolation and quality assessment using the Qiagen Symphony system at the Duke core, total RNA samples (50 ng/microliter) were plated in fully skirted 96 well plates (two wells reserved for internal controls). All microarray analysis for Cohorts I and II was performed in the CLIA approved BWH core lab of the Partners Center for Personalized Genetic Medicine in Cambridge MA. Quality assurance analysis was repeated by the core for all specimens before and after amplification and labeling. The cut-off was RIN of 7. Contributing institutions were asked to replace samples that did not meet QC standards when possible for Cohort I specimens. All samples that passed the QC step were hybridized to U133A microarrays of the same or sequential lots and processed. Cohorts I and II specimens were hybridized and processed sequentially in roughly the same time-line, process, instruments and personnel. Cohort II data were not examined until all final analytic work was completed and locked on Cohort I specimens. Profiling was performed blinded to all patient and outcome information.
Data and Statistical analysis:
Database:
Clinical variables included age, sex, smoking history (never, former, current), tumor location (central/peripheral), tumor size, lymph node involvement, date of surgery, type of surgery, recurrence status (site and date of first recurrence), vital status (cause and date of death), date of last disease assessment and date of last follow-up. Patients were only included if they survived at least three months post-surgery and had at least three years of follow-up (for patients still alive). In addition, raw data from gene expression analyses was submitted to the biostatistical core and entered into the SC database. Clinical, molecular and pathology data were linked within the SC database hosted at the biostatistical core which was originally at the Alliance Statistical Data Center at the Mayo Clinic and subsequently moved to Weill Cornell Medicine.
All elements of the validation analysis, including lab process, the algorithms generating the risk score for each signature and the prediction for binary and survival outcomes were conducted in a lock-down manner with the prediction parameters, models and cutoff points for high vs low risk patients reported in the original signature articles. Also, the REMARK guideline was followed in the reporting.29
Data pre-processing:
An initial quality control check of the array data was performed by the CLIA certified laboratory that generated the data and any arrays failing quality control checks were repeated when possible. Affymetrix gene expression CEL data that passed all quality control checks were normalized with fastlo30 by the statistical core and probeset expression values (i.e. gene expression values) generated with RMA.31
Prognostic signature performance validation:
Each signature was evaluated using Cox regression for its ability to predict patient death within 3 years of treatment according to its originally proposed cutoff. The 3-year time was selected because > 70% of deaths from recurrent SC after surgery for stage I lung cancer occur within 3 years. The overall discriminative ability of each signature was summarized with the Uno C-statistics for censored data.32 The survival curves were constructed using the Kaplan-Meier method. The primary selection criterion was that a signature could significantly predict high or low risk of death within 3 years in the independent test set (Cohort I). All signatures meeting the primary selection criterion were further tested in Cohort II by comparing risk prediction using baseline risk factors alone versus in combination with the signature. Using difference confidence intervals estimated by the Uno method, each signature was evaluated for its ability, when combined with baseline risk factors, to significantly improve prediction of 3-year survival relative to baseline factors alone. All statistical analyses were performed in R (R Foundation for Statistical Computing).
Selection of validated prognostic signatures:
All signatures meeting the primary selection criterion (validation in Cohort I) were referred to as the best validated set(s). The signatures comprising the best validated set(s) were modified to include clinical information (age, stage, gender). The best modified signature was identified, and all other modified signatures were compared to it. The set of best modified signatures were brought forward to validation in Cohort II.
Results
Cohort I:
There were 249 patients with early stage SC meeting all the requirements to constitute Cohort I (Table 1). The median age was 70 years, 161 (65%) were male, and most were former or current smokers. AJCC staging editions 6,33 734 and 835 were examined because each was in clinical use at some point during the timeline of the tumor collection, signature design, and conduct of the current study. Most patients (180) survived at least 3 years after surgery while 69 died within 3 years. Overall median survival was 6.2 years and relatively similar across sites (eFigure 1).
Table 1:
Cohort characteristics
| Cohort I (n = 249) | Cohort II (n = 234) | |
|---|---|---|
| Age, years | ||
| mean ± SD | 69.4 ± 8.7 | 68.5 ± 8.5 |
| median (min, max) | 70 (43.0, 92.0) | 68.0 (46.0, 89.0) |
| Gender, n (%) | ||
| female | 88 (35.3) | 82 (35.0) |
| male | 161 (64.7) | 152 (65.0) |
| Smoking history, n (%) | ||
| never | 4 (1.6) | 4 (1.7) |
| former | 165 (66.3) | 182 (77.8) |
| current | 67 (26.9) | 48 (20.5) |
| unknown | 13 (5.2) | |
| Node status, n (%) | ||
| N0 | 218 (87.6) | 172 (73.5) |
| N1 | 29 (11.6) | 50 (21.4) |
| missing | 3 (1.2) | 12 (5.1) |
| Tumor size, cm | ||
| mean ± SD | 3.67 ± 2.01 | 4.25 ± 2.04 |
| median (min, max) | 3.0 (0.5, 14.0) | 4.00 (1.0, 11.8) |
| Vital status at 3 years, n (%) | ||
| alive | 174 (69.9) | 118 (50.4) |
| dead | 75 (30.1) | 116 (49.6) |
| T classification (AJCC 6th ed.), n (%) | ||
| T1 | 113 (45.4) | 66 (28.2) |
| T2 | 130 (52.0) | 157 (67.1) |
| T3 | 4 (1.6) | 10 (4.3) |
| T4 | 2 (0.8) | 1 (0.4) |
| Stage (AJCC 6th ed.), n (%) | ||
| IA | 107 (43.0) | 55 (23.5) |
| IB | 109 (43.6) | 118 (50.4) |
| IIA | 6 (2.4) | 11 (4.7) |
| IIB | 24 (9.6) | 49 (20.9) |
| IIIA | 1 (0.4) | 0 |
| IIIB | 2 (0.8) | 1 (0.4) |
| T classification (AJCC 7th ed.), n (%) | ||
| T1a | 45 (18) | 24 (10.3) |
| T1b | 68 (27.3) | 42 (19.9) |
| T2a | 83 (33.2) | 92 (39.3) |
| T2b | 34 (13.6) | 46 (19.7) |
| T3 | 19 (7.6) | 30 (12.8) |
| Stage (AJCC 7th ed.), n (%) | ||
| IA | 107 (43.0) | 55 (23.5) |
| IB | 68 (27.2) | 72 (30.8) |
| IIA | 50 (20.0) | 64 (27.4) |
| IIB | 22 (8.8) | 37 (15.8) |
| IIIA | 2 (0.8) | 6 (2.6) |
| T classification (AJCC 8th ed.), n (%) | ||
| T1a | 4 (1.6) | 1 (0.4) |
| T1b | 41 (16.4) | 23 (9.8) |
| T1c | 68 (27.3) | 42 (17.9) |
| T2a | 52 (20.8) | 51 (21.8) |
| T2b | 31 (12.4) | 41 (17.5) |
| T3 | 39 (15.6) | 56 (23.9) |
| T4 | 14 (5.6) | 20 (8.5) |
| Stage (AJCC 8th ed.), n (%) | ||
| IA1 | 4 (1.6) | 1 (0.4) |
| IA2 | 39 (15.6) | 22 (9.4) |
| IA3 | 64 (25.7) | 32 (13.7) |
| IB | 44 (17.6) | 40 (17.1) |
| IIA | 24 (9.6) | 32 (13.7) |
| IIB | 54 (21.6) | 74 (31.6) |
| IIIA | 20 (8.0) | 33 (14.1) |
Three signatures, the BWH 2-gene ratios signature, the PMH 15-gene signature, and the UM signature each separated Cohort 1 patients into two groups with statistically significant survival differences as originally designed (Figure 1). Male sex, advanced age, and higher AJCC T classification and stage were associated with shorter survival in the univariable model and, therefore, were used as covariates to establish a base model to assess whether the signatures provided prognostic information independent of known clinical predictors. When these risk factors were included, only the UM signature significantly enhanced prognosis relative to the base model in all staging editions. Table 2 shows the estimates of the c-index (or concordance statistic) for each signature’s ability, combined with the clinical model using AJCC 8th edition criteria, to predict the vital status of a patient at 3 years in Cohort 1. Similar analyses based on AJCC 6th and 7th edition criteria were also statistically significant (eTable 1).
Figure 1.
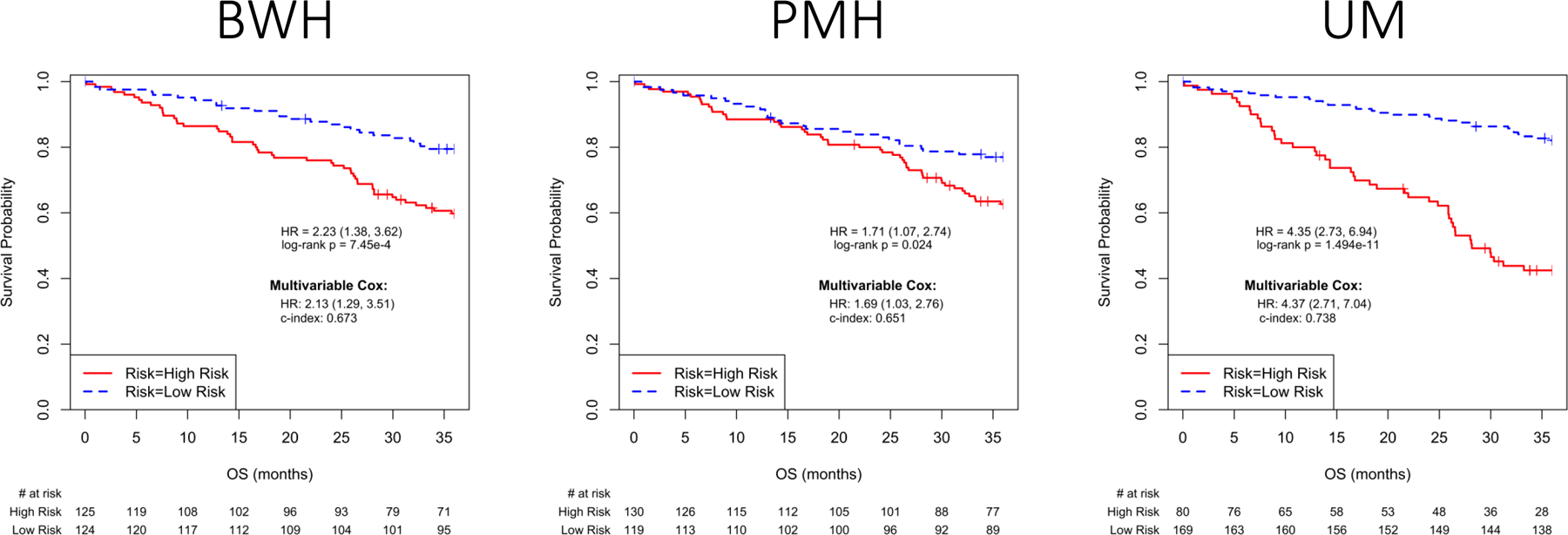
Validated signatures and Risk Model Performance in Cohort I. Kaplan-Meier plots of overall survival associated with the three signatures that met primary selection criteria in Cohort I. Insets in each panel describe univariable and multivariable Hazard Ratios (95% C.I.) for the signature, and C-index (95% C.I.) for the multivariable model. A) BWH signature, B) PMH signature and C) UM signature.
Table 2:
Risk of death at 3 years based on signatures and risk factors in Cohort I
| Cohort I, N=249 | ||||
|---|---|---|---|---|
| C-index (95% CI) of each model | ||||
| Risk factors include: Age + Gender + T_classification (AJCC 8th ed.) | ||||
| Signature | Signature alone (A) | Risk factors alone (B) | Signature + Risk factors (C) | C vs B diff (95% CI) |
| BWH | 0.598 (0.534, 0.661) | 0.653 (0.581, 0.724) | 0.671 (0.606, 0.736) | 0.018 (−0.024, 0.060) |
| PMH | 0.561 (0.484, 0.638) | 0.653 (0.583, 0.723) | 0.658 (0.591, 0.726) | 0.0058 (−0.023, 0.034) |
| UMI | 0.671 (0.617, 0.724) | 0.658 (0.587, 0.729) | 0.744 (0.684, 0.804) | 0.091 (0.028, 0.154) |
| Risk factors include: Age + Gender + stage (AJCC 8th ed.) | ||||
| Signature | Signature alone (A) | Risk factors alone (B) | Signature + Risk factors (C) | C vs B diff (95% CI) |
| BWH | 0.598 (0.534, 0.661) | 0.654 (0.586, 0.721) | 0.672 (0.609, 0.735) | 0.019 (−0.021, 0.058) |
| PMH | 0.561 (0.484, 0.638) | 0.654 (0.589, 0.718) | 0.661 (0.595, 0.726) | 0.0072 (−0.020, 0.034) |
| UMI | 0.671 (0.617, 0.724) | 0.654 (0.587, 0.720) | 0.744 (0.686, 0.802) | 0.090 (0.028, 0.153) |
Cohort II:
Cohort II consisted of 234 eligible patients for whom sufficient, high-quality RNA was available that met QC standards. The median age was 68 and 153 (65%) were male (Table 1). The tumor sizes and stages were somewhat larger and more advanced than in Cohort I. The statistically significant signatures identified in Cohort I, were evaluated in cohort II. The UM signature was the only one to be validated in this dataset (Table 3). Of note, the predicted low risk group had 3-year survival greater than 80%.
Table 3:
Risk of death at 3 years based on signatures and risk factors in Cohort II
| Cohort II, N=234 | ||||
|---|---|---|---|---|
| C-index (95% CI) of each model | ||||
| Risk factors include: Age + Gender + T_classification (AJCC 8th ed.) | ||||
| Signature | Signature alone (A) | Risk factors alone (B) | Signature + Risk factors (C) | C vs B diff (95% CI) |
| BWH | 0.561 (0.489, 0.632) | 0.561 (0.470, 0.652) | 0.590 (0.517, 0.663) | 0.029 (−0.047, 0.104) |
| PMH | 0.510 (0.441, 0.579) | 0.561 (0.474, 0.649) | 0.561 (0.470, 0.653) | 0.00013 (−0.017, 0.017) |
| UMI | 0.649 (0.594, 0.703) | 0.561 (0.470, 0.652) | 0.683 (0.610, 0.756) | 0.122 (0.048, 0.196) |
| Risk factors include: Age + Gender + stage (AJCC 8th ed.) | ||||
| Signature | Signature alone (A) | Risk factors alone (B) | Signature + Risk factors (C) | C vs B diff (95% CI) |
| BWH | 0.561 (0.489, 0.632) | 0.561 (0.465, 0.657) | 0.584 (0.505, 0.664) | 0.023 (−0.119, 0.119) |
| PMH | 0.510 (0.441, 0.579) | 0.561 (0.468, 0.654) | 0.562 (0.468, 0.656) | 0.00061(−0.019, 0.020) |
| UMI | 0.649 (0.594, 0.703) | 0.561 (0.468, 0.654) | 0.683 (0.611, 0.755) | 0.122 (0.044, 0.201) |
We also examined the UM signature in each of the sub-stages including AJCC 8th edition IA, IB, IIA and IIB and IIIA (see Figures 2–3) for both cohorts. The UM signature significantly separates each of the sub-stages based on survival in Cohort I and all substages IA-C and IIA-B in Cohort II. These results were similarly observed utilizing 6th and 7th edition criteria (eTable 2).
Figure 2.
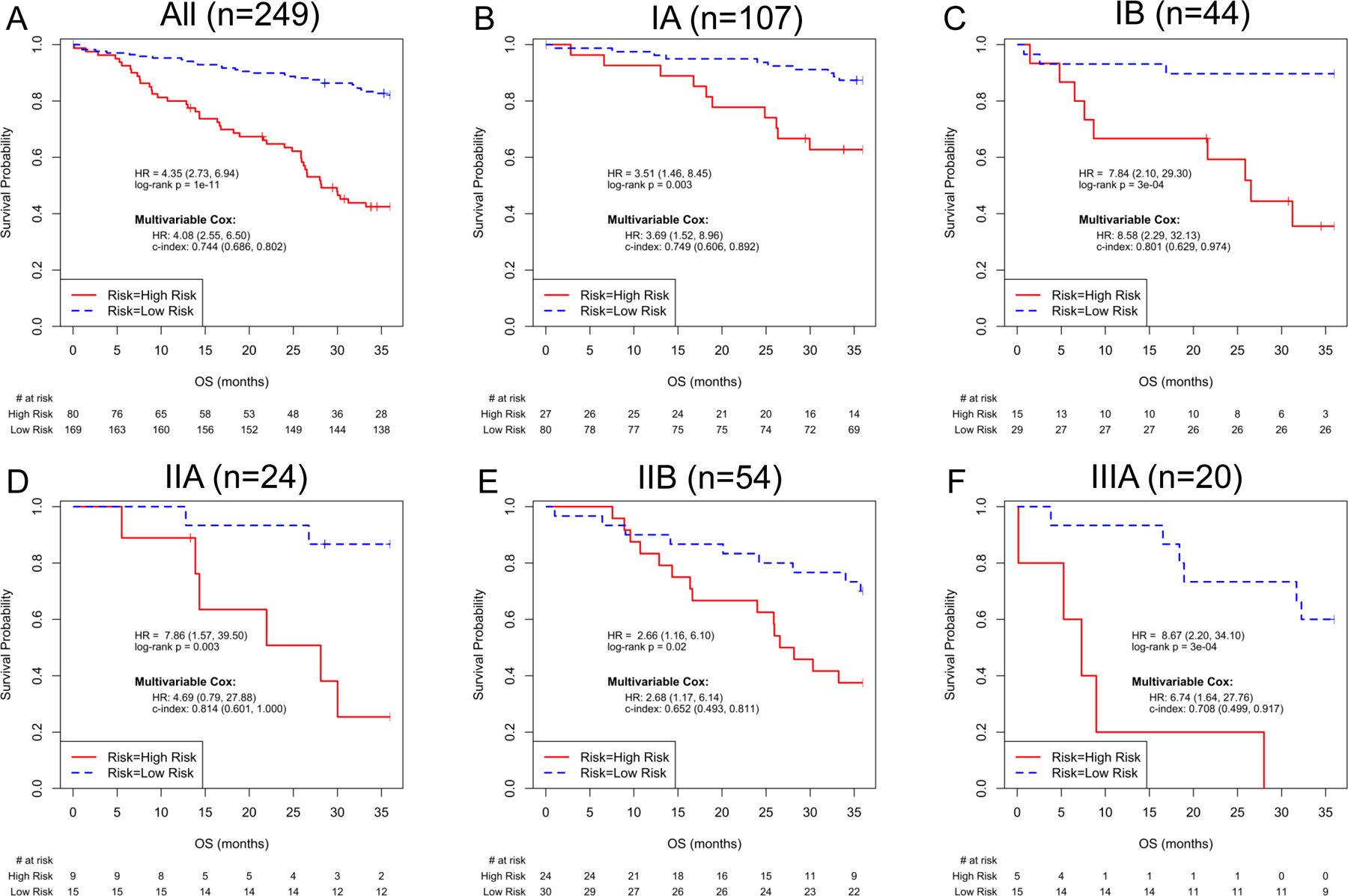
UM signature and Risk Model Performance in Cohort I, for all cases and for subgroups by AJCC 8th edition Stage (IA1, IA2 and IA3 pooled as Stage IA). Kaplan-Meier plots of overall survival based on UM signature predictions for patients in Cohort I. Insets in each panel describe univariable and multivariable Hazard Ratios (95% C.I.) for the UM signature, and C-index (95% C.I.) for the multivariable model. When all cases (n=249) were used, the multivariable Cox analysis included ‘Risk+Age+Gender+Stage’; Cox models of stage-specific subgroups included ‘Risk+Age+Gender’. A) All patients, B) AJCC 8th edition Stage IA, C) AJCC 8th edition Stage IB, D) AJCC 8th edition Stage IIA, E) AJCC 8th edition Stage IIB and F) AJCC 8th edition Stage IIIA.
Figure 3.
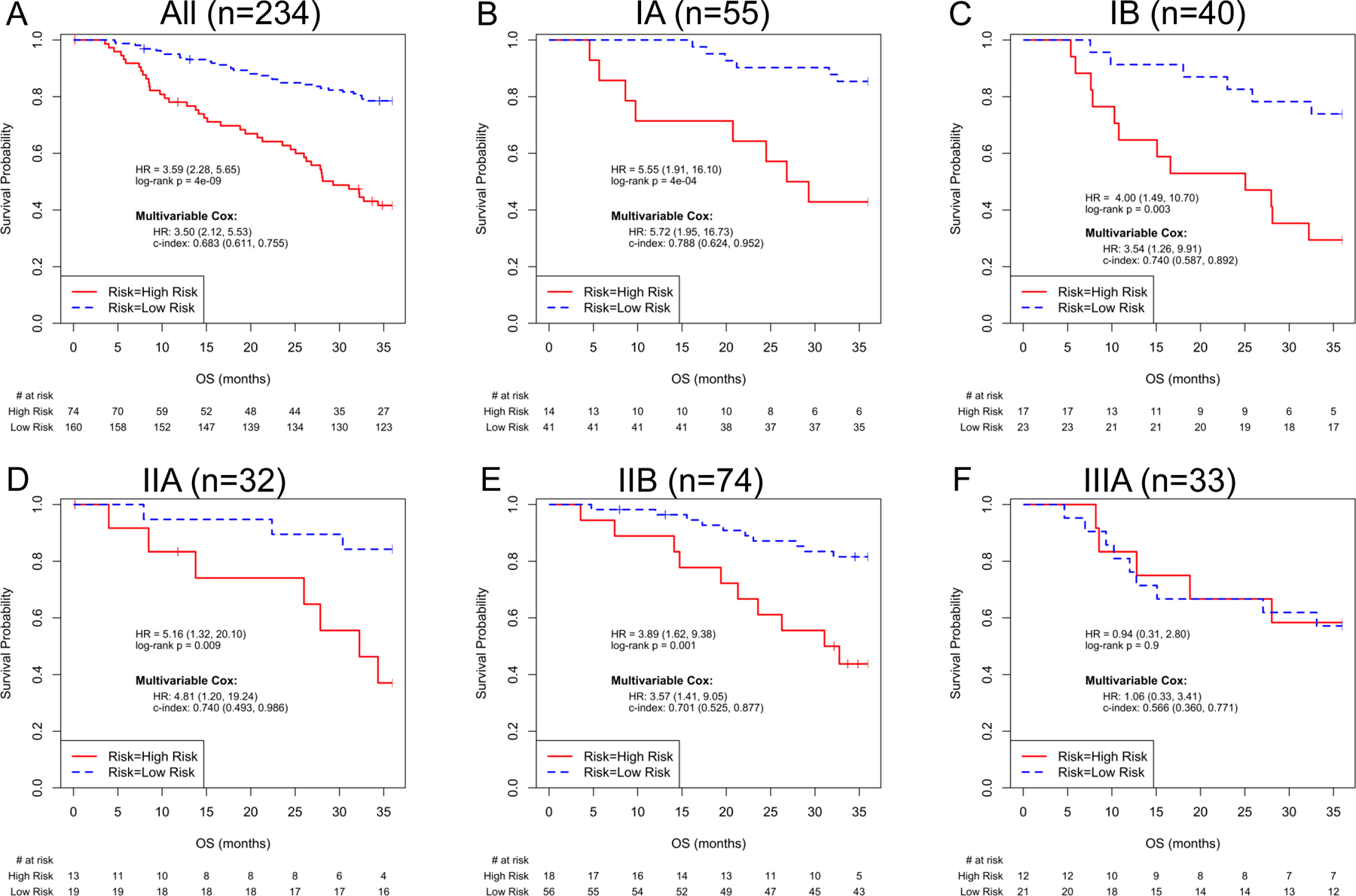
UM signature and Risk Model Performance in Cohort II, for all cases and for subgroups by AJCC 8th edition Stage (IA1, IA2 and IA3 pooled as Stage IA). Kaplan-Meier plots of overall survival based on UM signature predictions for patients in Cohort II. Insets in each panel describe univariable and multivariable Hazard Ratios (95% C.I.) for the UM signature, and C-index (95% C.I.) for the multivariable model. When all cases (n=234) were used, the multivariable Cox analysis included ‘Risk+Age+Gender+Stage’; Cox models of stage-specific subgroups included ‘Risk+Age+Gender’. A) All patients, B) AJCC 8th edition Stage IA, C) AJCC 8th edition Stage IB, D) AJCC 8th edition Stage IIA, E) AJCC 8th edition Stage IIB and F) AJCC 8th edition Stage IIIA.
Two de novo signatures derived from Cohort I data using modified counter-propagation (MCP) clustering methods, incorporating either all genes represented on the microarrays or all genes (pooled) constituting the tested models, each significantly stratified OS in Cohort I, but were not validated in the Cohort II dataset.
Discussion
We report herein the assembly of the Lung SC Consortium and one of its first accomplishments, the validation of the efficacy of a prognostic expression signature for resected early stage SC. We demonstrated that a 50-gene signature developed by UM group in 2006 could be used to significantly predict outcome in each of the early stages (and sub-stages) of SC patients who underwent definitive surgery. This molecular signature was used as part of a classifier which also includes the age, sex and stage (or T classification) which should be easily available for each patient after surgery. The distribution of patients between the risk strata was roughly 1/3 low- and 2/3 high-risk. For patients in stages IA-IIA, to whom ACT is not currently offered, this classifier can be used to support a clinical trial to determine whether ACT may add value to patients at higher predicted risk of death within 3 years of treatment. For patients with stage IIB SC, this strategy can support a trial to determine whether a predicted low-risk subgroup can avoid the need for ACT that is currently a standard of care.
The original publication of the UM signature included the list of 50 prognostic probe sets as well as significantly enriched gene ontology groups (ref 14; Supplemental Tables 4 and 3; http://cancerres.aacrjournals.org/content/suppl/2006/08/02/66.15.7466.DC1). The gene ontology class that showed the strongest membership was GO ID 8544, epidermal differentiation, representing 17 of the signature genes.
We were only able to validate one of the multiple signatures proposed, even though all candidate signatures had been validated locally at time of initial report. Although the PMH 15-gene signature, for example, was developed using multi-institutional and randomized clinical trial samples, it was created using NSCLC instead of SC samples. This emphasizes the need for histology-specific prognostic gene signatures in NSCLC, a heterogeneous cancer. These observations also suggest that there may be inherent biases in some individual sites’ collections and support the notion that multiple broad, multi-institutional biobanks are required for development and definitive validation of reliable prognostic biomarkers for cancer. These findings also support continuous effort in systematic tissue banking, perhaps by the cooperative group mechanisms. Finally, the findings confirm the need for the rigorous experimental design and thorough validation recommended by Subramanian and Simon in their critique of early gene expression signature efforts.19
This study was limited by the need to select cases based on tumor type, specimen quality and patient parameters as required by the experimental design and gene expression assays, such that not all patients from the original specimen banking study provided genetic samples. This could have resulted in a selection bias.
While efforts to move prognostic testing for lung cancer to the clinical arena have faltered because of challenges in validation and insurance coverage, the technology is improving, and the need continues to exist. This study not only documents the validation of a clinically useful existing signature but also shares with the scientific community two valuable datasets that can be used for evaluation of other locally developed signatures or other research. Finally, although this work was performed using this rigorous validation study required fresh frozen tumor specimens and a microarray platform, we believe and expect that it will be possible to obtain the signature from routine fixed pathological materials in most cases using alternative analytic platforms more suited to clinical application. Parallel studies of fixed specimens from these cohorts are underway.
The clinical implications of these findings are particularly important because for the first time we may have a sufficiently validated and transparent test that can be used to direct ACT or clinical trials for SC patients. Also, the validation set samples and outcome data were collected through an NCI funded, CTEP approved process which was independent and multi-institutional. Given the poor survival of patients found to be at high-risk (40% at 3 years), future clinical trials using this test may be useful for improving survival in SC patients.
Supplementary Material
Acknowledgements:
The authors wish to acknowledge the Jannie Moore-Askew and Paul J Novotny for helpful comments on the manuscript. ClinicalTrials.gov Identifier: NCT01517971
Support:
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Numbers U10CA180821 and U10CA180882 (to the Alliance for Clinical Trials in Oncology), U24CA196171, U01CA157715, UG1CA233160, UG1CA233253, UG1CA233324, UG1CA233339, and P30CA046592. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict:
F.R.H. reports advisory board participation for Genentech/Roche, BMS, AstraZeneca, Merck, Novartis, Daiichi, Amgen, OncoCyte, Lilly/Loxo, Boehringer-Ingelheim, outside the submitted work;
R.G. reports personal fees from Inivata, Pfizer, AstraZeneca, Genentech, Millennium Pharmaceuticals, AbbVie, F. Hoffman La-Roche, NeoHealth, Janssen, BMS, Eli Lilly, Roche, Nektar, Merck, Celgene, Janssen, Partner Therapeutics, GSK, Jounce, Amgen, Achilles, GenePlus, outside the submitted work;
D.M. reports grants from Bristol-Myers-Squibb, personal fees from Genentech, personal fees from Roche, outside the submitted work;
R.B. reports grants from Genentech, Roche, Merck, Verastem, Epizyme, MedGenome, Siemens, Gritstone, outside the submitted work; In addition, R.B. has 4 patents through the BWH (no royalties to date) and Equity in a new start-up company, Navigation Sciences, outside the submitted work.
References:
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA: a cancer journal for clinicians 2009;59:225–249. [DOI] [PubMed] [Google Scholar]
- 2.Zappa C, Mousa SA. Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res 2016;5:288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Detterbeck FC, Boffa DJ, Kim AW, et al. The Eighth Edition Lung Cancer Stage Classification. Chest 2017;151:193–203. [DOI] [PubMed] [Google Scholar]
- 4.Ettinger DS, Aisner DL, Wood DE, et al. NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 5.2018. J Natl Compr Canc Netw 2018;16:807–821. [DOI] [PubMed] [Google Scholar]
- 5.Arriagada R, Dunant A, Pignon J-P, et al. Long-term results of the international adjuvant lung cancer trial evaluating adjuvant Cisplatin-based chemotherapy in resected lung cancer. J Clin Oncol 2010;28:35–42. [DOI] [PubMed] [Google Scholar]
- 6.Pignon J-P, Tribodet H, Scagliotti GV, et al. Lung adjuvant cisplatin evaluation: a pooled analysis by the LACE Collaborative Group. Journal of clinical oncology 2008;26:3552–3559. [DOI] [PubMed] [Google Scholar]
- 7.Lee WC, Diao L, Wang J, et al. Multiregion gene expression profiling reveals heterogeneity in molecular subtypes and immunotherapy response signatures in lung cancer. Mod Pathol 2018;31:947–955. [DOI] [PubMed] [Google Scholar]
- 8.Bild AH, Yao G, Chang JT, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 2006;439:353. [DOI] [PubMed] [Google Scholar]
- 9.Nicolini A, Ferrari P, Duffy MJ. Prognostic and predictive biomarkers in breast cancer: Past, present and future. Semin Cancer Biol 2018;52:56–73. [DOI] [PubMed] [Google Scholar]
- 10.Bueno R, Hughes E, Wagner S, et al. Validation of a molecular and pathological model for five-year mortality risk in patients with early stage lung adenocarcinoma. J Thorac Oncol 2015;10:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng Y, Bueno R. Commercially available prognostic molecular models in early-stage lung cancer: a review of the Pervenio Lung RS and Myriad myPlan Lung Cancer tests. Expert Rev Mol Diagn 2015;15:589–596. [DOI] [PubMed] [Google Scholar]
- 12.Kratz JR, Haro GJ, Cook NR, et al. Incorporation of a Molecular Prognostic Classifier Improves Conventional Non-Small Cell Lung Cancer Staging. J Thorac Oncol 2019;14:1223–1232. [DOI] [PubMed] [Google Scholar]
- 13.Raponi M, Dossey L, Jatkoe T, et al. MicroRNA classifiers for predicting prognosis of squamous cell lung cancer. Cancer research 2009;69:5776–5783. [DOI] [PubMed] [Google Scholar]
- 14.Raponi M, Zhang Y, Yu J, et al. Gene expression signatures for predicting prognosis of squamous cell and adenocarcinomas of the lung. Cancer research 2006;66:7466–7472. [DOI] [PubMed] [Google Scholar]
- 15.Zhu CQ, Strumpf D, Li CY, et al. Prognostic gene expression signature for squamous cell carcinoma of lung. Clin Cancer Res 2010;16:5038–5047. [DOI] [PubMed] [Google Scholar]
- 16.Zhang W, Cui Q, Qu W, et al. TRIM58/cg26157385 methylation is associated with eight prognostic genes in lung squamous cell carcinoma. Oncol Rep 2018;40:206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mascaux C, Laes J-F, Anthoine G, et al. Evolution of microRNA expression during human bronchial squamous carcinogenesis. European Respiratory Journal 2009;33:352–359. [DOI] [PubMed] [Google Scholar]
- 18.Zhang J, Bing Z, Yan P, et al. Identification of 17 mRNAs and a miRNA as an integrated prognostic signature for lung squamous cell carcinoma. J Gene Med 2019;21:e3105. [DOI] [PubMed] [Google Scholar]
- 19.Subramanian J, Simon R. Gene expression-based prognostic signatures in lung cancer: ready for clinical use? J Natl Cancer Inst 2010;102:464–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee E-S, Son D-S, Kim S-H, et al. Prediction of recurrence-free survival in postoperative non–small cell lung cancer patients by using an integrated model of clinical information and gene expression. Clinical cancer research 2008;14:7397–7404. [DOI] [PubMed] [Google Scholar]
- 21.Winton T, Livingston R, Johnson D, et al. Vinorelbine plus cisplatin vs. observation in resected non–small-cell lung cancer. New England Journal of Medicine 2005;352:2589–2597. [DOI] [PubMed] [Google Scholar]
- 22.Zhu C-Q, Ding K, Strumpf D, et al. Prognostic and predictive gene signature for adjuvant chemotherapy in resected non–small-cell lung cancer. Journal of clinical oncology 2010;28:4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gordon GJ, Dong L, Yeap BY, et al. Four-gene expression ratio test for survival in patients undergoing surgery for mesothelioma. Journal of the National Cancer Institute 2009;101:678–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gordon GJ, Jensen RV, Hsiao L-L, et al. Translation of microarray data into clinically relevant cancer diagnostic tests using gene expression ratios in lung cancer and mesothelioma. Cancer research 2002;62:4963–4967. [PubMed] [Google Scholar]
- 25.Gordon GJ, Richards WG, Sugarbaker DJ, et al. A prognostic test for adenocarcinoma of the lung from gene expression profiling data. Cancer Epidemiology and Prevention Biomarkers 2003;12:905–910. [PubMed] [Google Scholar]
- 26.Rock JR, Onaitis MW, Rawlins EL, et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proceedings of the National Academy of Sciences 2009;106:12771–12775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu Y, Lemon W, Liu P-Y, et al. A gene expression signature predicts survival of patients with stage I non-small cell lung cancer. PLoS medicine 2006;3:e467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Travis WD, Brambilla E, Muller-Hermelink KH, et al. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of the Lung, Pleura, Thymus and Heart. Lyon: IARC Press; 2004. [Google Scholar]
- 29.McShane LM, Altman DG, Sauerbrei W, et al. Reporting recommendations for tumor marker prognostic studies (REMARK). J Natl Cancer Inst 2005;97:1180–1184. [DOI] [PubMed] [Google Scholar]
- 30.Ballman KV, Grill DE, Oberg AL, et al. Faster cyclic loess: normalizing RNA arrays via linear models. Bioinformatics 2004;20:2778–2786. [DOI] [PubMed] [Google Scholar]
- 31.Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003;4:249–264. [DOI] [PubMed] [Google Scholar]
- 32.Uno H, Cai T, Pencina MJ, et al. On the C-statistics for evaluating overall adequacy of risk prediction procedures with censored survival data. Stat Med 2011;30:1105–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greene FL, Page DL, Fleming ID, et al. AJCC Cancer Staging Manual. New York: Springer; 2002. [Google Scholar]
- 34.Edge SB, Byrd DR, Compton CC, et al. Cancer Staging Manual New York: Springer; 2010. [Google Scholar]
- 35.Amin MB, Edge SB, Byrd DR, et al. AJCC Cancer Staging Manual. New York: Springer International Publishing; 2017. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.