

Abstract
Mitochondria control life and death in eukaryotic cells. Harboring a unique circular genome, a by-product of an ancient endosymbiotic event, mitochondria maintain a specialized and evolutionary divergent protein synthesis machinery, the mitoribosome. Mitoribosome biogenesis depends on elements encoded in the mitochondrial genome (the RNA components) and the nuclear genome (all ribosomal proteins and assembly factors). Recent cryo-EM structures of mammalian mitoribosomes have illuminated their composition and provided hints regarding their assembly and elusive mitochondrial translation mechanisms. A rising body of literature involves the mitoribosome in inherited primary mitochondrial disorders. Mutations in genes encoding mitoribosomal RNAs, proteins, and assembly factors, impede mitoribosome biogenesis, causing protein synthesis defects that lead to respiratory chain failure and mitochondrial disorders such as encephalo- and cardiomyopathy, deafness, neuropathy, and developmental delays. In this article, we will review the current fundamental understanding of mitoribosome assembly and function and the clinical landscape of mitochondrial disorders driven by mutations in mitoribosome components and assembly factors, to portrait how the synergistic lessons obtained from the two sources are helping us to better understand both mitochondrial biology and medicine.
Keywords: Mitochondrial ribosome, Mitochondrial translation, Mitoribosome assembly, OXPHOS deficiency, Mitochondrial disease
1. INTRODUCTION
The discovery in the late 1950s of a protein synthesis activity within mitochondria [1,2] that was found sensitive to antibiotics such as chloramphenicol provided an argument for the existence of mitochondrial ribosomes (mitoribosomes) [3]. In the mid-1960s, the observation by light and electron microscopy of filamentous DNA components and ribosome-like particles containing RNA within mitochondria of yeast, mouse, and human HeLa cells offered visual proof for their occurrence [4]. The subsequent isolation and characterization of the fungal [5] and mammalian mitoribosomes [6,7] in the late 1960s were critical milestones in the history of biology. Nearly 50 years later, in another remarkable scientific breakthrough, researchers used cutting-edge cryo-electron microscopy (cryo-EM) to unravel the structure of the large subunit of yeast mitoribosome at 3.2 Å resolution [8]. This work was the flag of the so-called “resolution revolution” [9] and laid the foundations for the structure determination of the complete yeast mitoribosome [10] and its porcine and human mitochondrial counterparts [11–15]. These discoveries have shaped the history of mitochondrial biology and contributed to adding a chapter to the mitochondrial medicine book.
Mitoribosomes drive the synthesis of a small set of proteins encoded in the mitochondrial DNA (mtDNA) [16], a vestige of the genome from the free-living α-proteobacterium mitochondrial ancestor [17]. In mammals, the mtDNA codes for 13 proteins that are essential membrane components of the oxidative phosphorylation (OXPHOS) enzymatic complexes. The mammalian mitoribosome is a 55S ribonucleoprotein complex, formed by a 39S large subunit (mt-LSU) with 52 mitoribosomal proteins (MRPs), a 16S rRNA, and a structural tRNA (tRNAVal in human cells), and a 28S small subunit (mt-SSU) with 30 MRPs and a 12S rRNA. The 55S ribosomes are only 25–30% RNA compared to bacterial and eukaryotic cytoplasmic ribosomes, which are ~60% RNA [11,13]. All MRPs are encoded in the nuclear genome (nDNA), synthesized on cytoplasmic ribosomes, and imported into the mitochondrial matrix to be assembled with the subunit-specific RNAs, which are encoded in the mtDNA. The assembly of the mitoribosome involves a growing number of non-ribosomal proteins, including RNA processing and modification enzymes, guanosine triphosphatases (GTPases), DEAD-box RNA helicases, and kinases [18,19]. They act as assembly factors to guide the processing and modification of mitoribosomal components and their temporal association to form pre-ribosomal particles during the assembly of individual subunits and subunit joining to form the monosome [20,21]. Once the monosome is assembled, its function in mRNA translation and protein synthesis involves the mtDNA-encoded tRNAs as well as translation initiation, elongation, and termination factors, mitoribosome recycling factors, mitochondrial aminoacyl tRNA synthetases (ARS), and mt-tRNA modification enzymes, all of which are encoded in the nuclear genome [22].
Defects of mitochondrial protein synthesis due to mutations in most mtDNA-encoded tRNAs and the 12S rRNA, as well as in nuclear genes encoding mitoribosomal proteins and assembly factors, translation initiation, and elongation factors, cause a subset of mitochondrial disorders typically associated with decreased activities of multiple OXPHOS enzymes in the affected tissues. Therefore, these diseases are genetically heterogeneous and can present with a broad spectrum of clinical manifestations. In general, they are infantile, severe, and often fatal multisystemic diseases, such as Leigh’s syndrome, sensorineural hearing loss, encephalomyopathy, and hypertrophic cardiomyopathy [23–27]. Furthermore, the deregulation of mitoribosome components and assembly factors is often associated with cancer development and progression [28]. Finally, mitoribosomes are sensitive to antibiotics used as frontline therapy against microbial infectious diseases and can cause side effects in humans [29].
Excellent reviews on impaired mitochondrial translation in human disease have been reported elsewhere. They describe mutations affecting mitochondrial tRNAs, tRNA modification factors, formylation of the mitochondrial methionine tRNA (Met-tRNAMet), aminoacyl-tRNA synthetases, and translation initiation, elongation, and release factors [23–25,30,31]. This article will focus on updating the growing list of disorders resulting from mutations in mitoribosome structural components and assembly factors. We will describe recent progress in fundamental aspects of mitoribosome biogenesis, how they help us to understand disease mechanisms, and, in turn, what we are learning from studying patients with mitoribosome defects.
2. OVERVIEW OF MITOCHONDRIAL TRANSLATION
The mitochondrial translation system evolved from that of the bacterial ancestor of mitochondria. As a consequence, the catalytic properties of mitochondrial and bacterial ribosomes are similar. Translation factors are conserved, and several mitochondrial factors can functionally replace their homologs in bacteria [32]. Nevertheless, the evolution of the mitochondrial system resulted in deviations in the genetic code [33,34] and significant differences in the actual process of translation [22].
Mitochondrial translation follows the canonical four steps of initiation, elongation, termination, and recycling [35], represented in Figure 1 as in their bacterial and cytosolic counterparts. Translation initiation is the rate-limiting step of protein synthesis that in mitoribosomes differs from bacterial or cytosolic translation systems. Key differences are seen in the interaction of the mitoribosome with the mRNAs, the tRNAs, and the mitochondrial translational factors. Most human mitochondrial mRNAs lack 5′ leader sequences to promote their binding to the ribosome through the potential action of translation activators as it occurs in yeast mitochondria [36–38]. Moreover, mitochondria contain a single tRNAMet that fulfills the dual role of the initiator and elongator tRNAMet. A fraction of met-tRNAMet is formylated by mitochondrial methionyl-tRNA formyltransferase (MTFMT) to generate N-formylmethionine- tRNAMet (fMet-tRNAMet), which is used for translation initiation [39,40]. Also, mitochondrial translation initiation lacks initiation factor 1 (IF1), which is essential in all other translation systems [41], and only involves mtIF2 and mtIF3 [42–44]. Cryo-EM structures of the mammalian mt-SSU with mt-IF2 and mt-IF3, and the complete mitochondrial initiation complex with the joint mitoribosomal subunits have been recently obtained [45–47] and revealed important molecular details of mitochondrial translation initiation specific to this system.
Figure 1. The mitochondrial translation process.
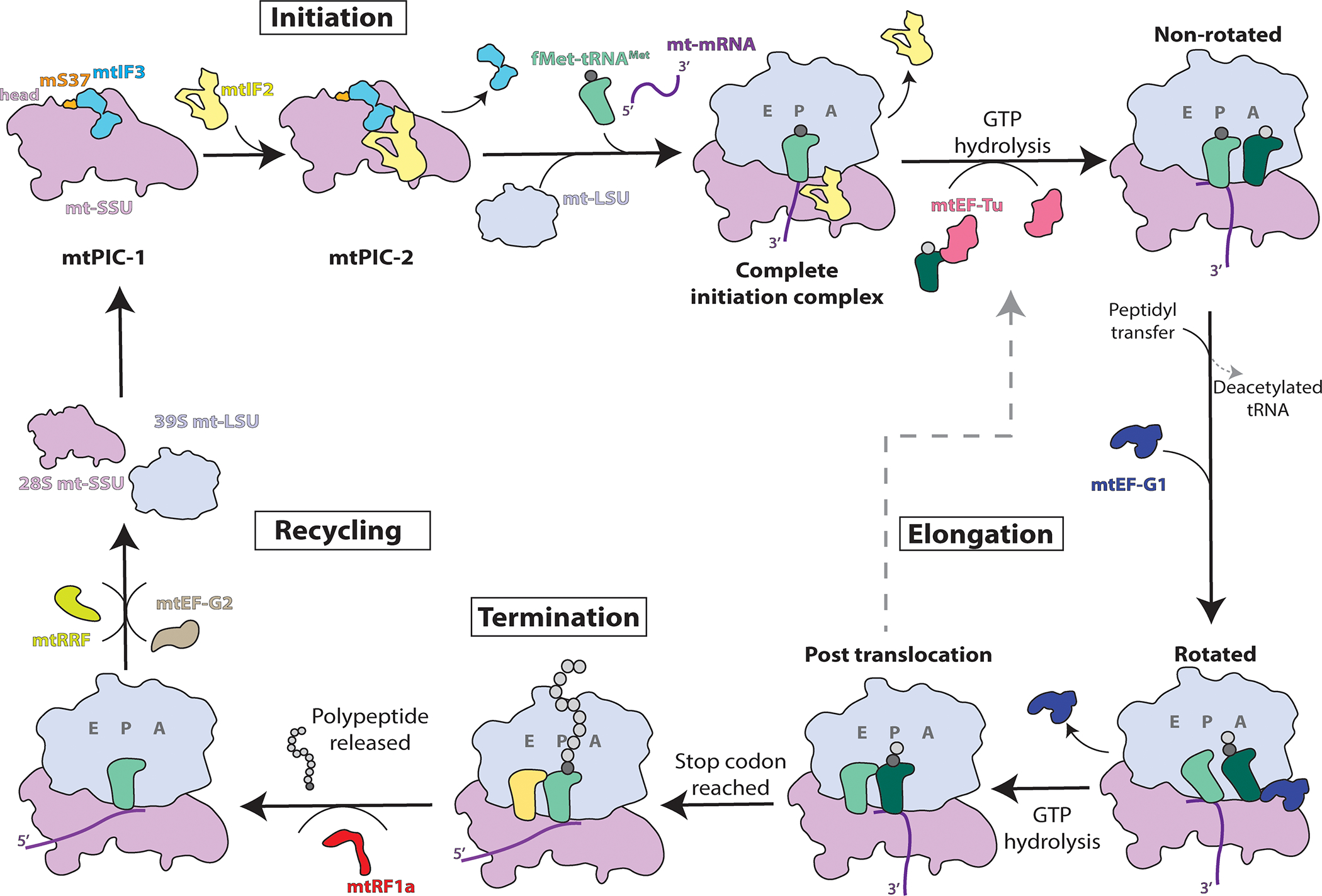
Schematic overview of mitochondrial translation indicating the major steps of initiation, elongation, termination, and recycling. Mitochondrial pre-initiation complexes are denoted mtPIC-1 and mtPIC-2. See full explanation in the text.
The first steps of mitochondrial translation initiation have been recently characterized in detail by a combination of cryo-EM and fluorescence techniques [47]. The study revealed the existence of two mt-SSU pre-initiation states (termed mtPIC-1 and mtPIC-2) that lead to the formation of the complete initiation complex. In mtPIC-1, mt-IF3 binds the mt-SSU, and interactions with mS37 (located in the head region) maintain the mt-SSU in a conformation competent to accommodate mt-IF2 in the subsequent mtPIC-2 [47]. However, the role of mt-IF3 is dispensable for the translation of most mitochondrial transcripts in human HeLa cells, with the single exception of ATP6 mRNA [44]. The specific effect is probably related to the fact that the ATP6 ORF has a 46 nucleotide overlap of its 5′-end with the 3′-end of the preceding ATP8 ORF in a bicistronic transcript [44].
A mitochondrion-specific extension of mt-IF2 blocks the premature binding of initiator tRNA to the mitoribosomal A-site [47], mimicking the function of the bacterial IF1. mtPIC-2 is competent for recruiting the initiator tRNA (fMet-tRNAMet) in the P-site, the mRNA, and allowing joining of the mt-LSU, upon the release of mt-IF3. However, contrary to what occurs in bacteria, wherein most initiation events, binding of the canonical mRNA Shine-Dalgarno sequence to the SSU is a requirement for subunits joining and formation of the complete initiation complex [48,49], no stable binding to the mtSSU was observed by cryo-EM [47]. On the contrary, the complete mitochondrial translation initiation complex was observed only with the joint mitoribosomal subunits [45,47]. Furthermore, experiments based on optical tweezers and confocal microscopy using a leaderless COX2 mRNA construct have shown that this mRNA forms a stable complex with the monosome during translation initiation [47].
A cryo-EM structure of the human mitoribosome at ~3.0 Å resolution has revealed the mtSSU pentatricopeptide-repeat (PPR)-containing protein mS39 engaged with MTCO3 mRNA at the entrance of the mt-mRNA channel [15]. Notably, mS39 associates via its PPR motifs with the transacting protein module formed by the Leucine-Rich Pentatricopeptide Repeat Containing protein (LRPPRC) and the SRA stem-Loop Interacting RNA binding Protein (SLIRP) [15]. The LRPPRC-SLIRP module is proposed to deliver mt-mRNAs to the mt-SSU, thereby facilitating the subsequent threading of the mRNA into the mRNA channel for start codon–anticodon interaction [15]. Therefore, the mitoribosome, which lacks proteins that in bacteria assist with mRNA unfolding during translation (full uS4 and some domains of uS3 and bS1), has evolved a system for mRNA delivery based on transacting proteins and mS39 acting as a linker [15].
Once the monosome is formed, elongation of the nascent chain proceeds by cycles of aminoacyl-tRNAs binding, peptide bond formation, and displacement of deacylated tRNAs. This process is catalyzed by the mitochondrial Elongation Factor Tu (mtEF-Tu), with the assistance of Elongation Factor Ts (mtEF-Ts), a nucleotide exchange factor, and the Elongation Factor G1 (mtEF-G1). mtEF-Tu participates in the formation of the ternary complex that includes GTP, and aminoacyl–tRNA, which delivers the aminoacyl–tRNA to the acceptor site of the ribosome using energy supplied by GTP hydrolysis [50]. mtEF-G1 hydrolyzes GTP to catalyze peptidyl–tRNA translocation from the ribosomal-acceptor site to the peptidyl site after peptide-bond formation, whereas the concurrent movement of mRNA exposes the next codon in the acceptor site. Recent cryo-EM structures of the human mitochondrial ribosome bound to mtEF-G1 in three distinct conformational states [51] and a series of eight structures of mitoribosome functional complexes with mt-mRNA, mt-tRNAs, recycling factor, and additional trans-acting factors [15], have revealed distinct features of mitochondrial translation elongation (Fig 2). Through the different mt-tRNA movement stages, mitochondrion-specific proteins mL40, mL48, and mL64 undergo specific conformational changes, which support the progress of mt-tRNA from A- to E-site [15,51,52] (Fig 2A–B). Release of the deacetylated tRNA from the E-site is facilitated by conformational changes in mL64 and uS7m [15]. It has been noted that in the non-translating mitoribosome, the polypeptide exit tunnel contains the N-terminal tail of the mitochondrion-specific protein mL45, that contacts with uL23m and uL24m and must retract during polypeptide delivery [45]. When the translation machinery encounters a termination codon, protein synthesis is complete. The stop codon is recognized by a GTP-independent class I ribosome release factor (mt-RF1a) that induces the release of the newly-synthesized polypeptide from the mt-LSU [53,54]. Following the action of the release factor, two ribosomal recycling factors, mtRRF [15,55,56] and mtEF-G2 [57] promote the disruption of the mitoribosome post-termination complex by enabling the dissociation of the ribosomal subunits and the release of mt-mRNA and deacylated mt-tRNA. Following the release of the recycling factors from the mt-LSU, the translation cycle can reinitiate.
Figure 2. Structure of mt-tRNAs in the translating mitoribosome.
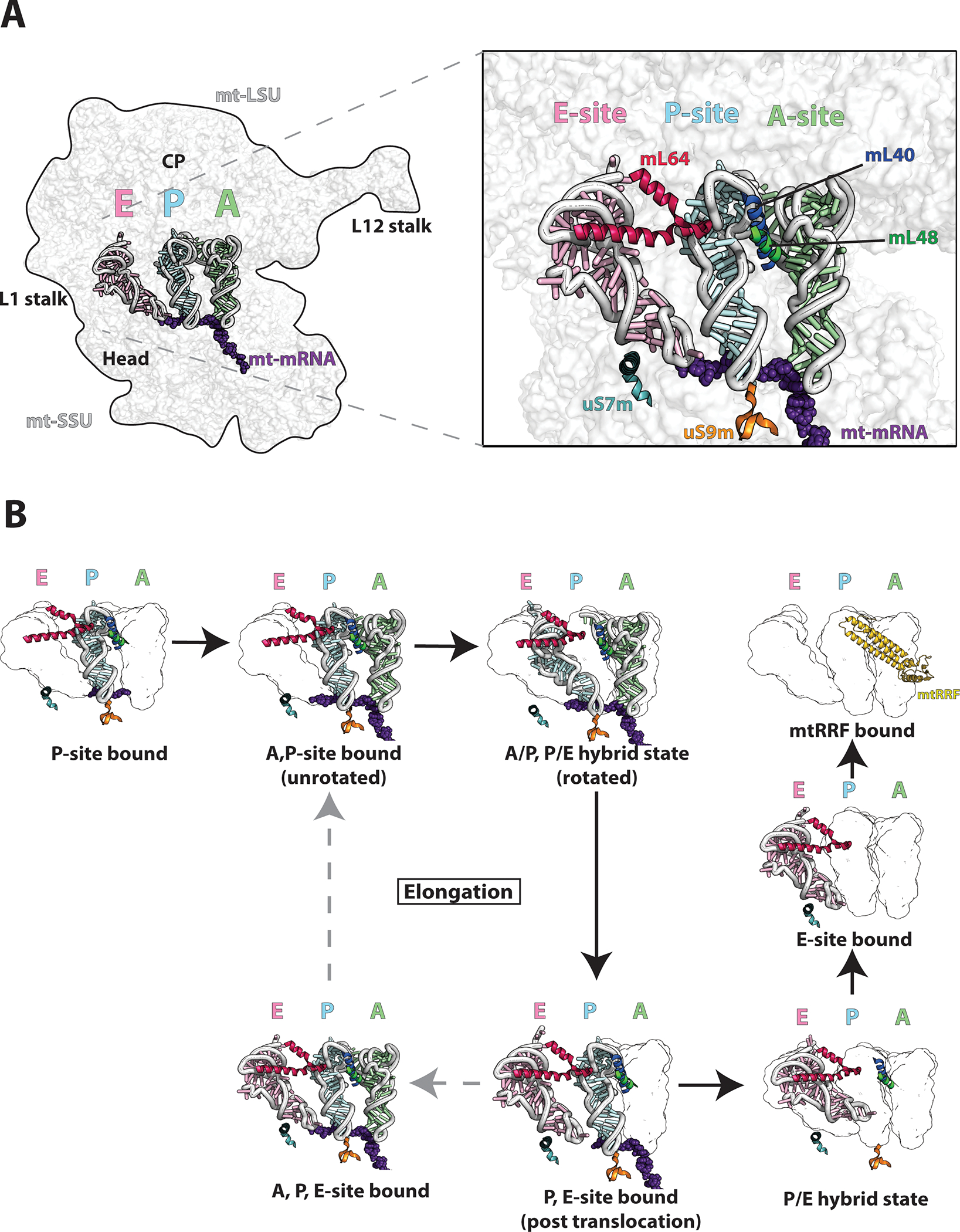
A) The cryo-EM structure (PDB-6ZS9) [15] is used to depict the mitoribosome bound to A,P, and E-site tRNAs highlighting key structural features (left). Detailed view of A,P,E-site tRNAs with bound mt-mRNA. mL40 N-terminal helix (blue) interacts with both A and P-site tRNA elbows with supporting hydrophobic interactions from the mL48 C-terminal helix (green). uS9m C-terminal tail (orange) binds the P-site tRNA anticodon stem loop. C-terminal helix of mL64 (red) interacts with the E-site tRNA elbow. uS7m C-terminal helix (teal) stabilizes the E-site tRNA through alignment with the major groove of the E-site tRNA anticodon stem. B) Eight representative cryo-EM structures [15] indicating tRNA translocation during elongation and termination/recycling. The figures were prepared using PYMOL and Adobe Illustrator software.
3. MITORIBOSOME STRUCTURAL FEATURES
The proteins and RNA domains of mitochondrial and bacterial ribosomes that contribute to decoding and peptide bond formation share a high degree of similarity, supporting the conservation of their catalytic properties. However, evolution led to the formation of mitoribosomes that differ significantly in structure and composition, compared not only to their bacterial relatives but also among different species [58,59,60], which has been attributed to divergent structural patching [59,60].
Cryo-EM analyses of human and porcine mitoribosomes have shown that their catalytic region at the subunit interface is largely conserved [11,13]. However, whereas the bacterial ribosome has a ~2:1 RNA:protein ratio, the value is ~1:2 for the mammalian mitoribosome, in which significant amounts of rRNA and several bacterial proteins (uS4, uS8, uS13, uS19, and bS20) were lost, conserved homologs of bacterial proteins frequently acquired N- or C- terminal extensions, and 36 mitochondrion-specific proteins, out of a total of 82 MRPs, were recruited [58]. These mitochondrion-specific proteins are mainly peripherally distributed over the solvent-accessible surface. In the mt-LSU, they form clusters at the central protuberance, the L7/L12 stalk, and adjacent to the polypeptide exit site. In the mt-SSU, two of these proteins form the head (mS29) and the foot (mS27), giving an elongated aspect to the subunit [11,13]. Like most protein extensions, the mitochondrion-specific proteins accommodate novel positions rather than compensate for the missing rRNA [14]. Some of these proteins were recruited to stabilize mitoribosome structures, including the rRNA (mS27, mS30, mS38, and mL37), or the L7/L12 stalk (mL63). Other proteins play relevant roles during translation, including pre-initiation (mS37), mRNA binding (mS39), tRNA translocation (mL40, mL48, and mL64), and elongation (mL53) [15].
A ~3.0 Å resolution cryo-EM structure of the human mitoribosome, including the L7/L12 stalk, has been recently reported [15]. It shows six copies of bL12m N-terminal domain that bridge interactions with uL10m and mL53, whereas mL54 connects with uL11m, thus explaining how the human mitoribosome functional L7/L12 is stabilized [15].
The human mitoribosome interface is rich in protein-mediated contacts, with three protein-protein and six protein-RNA bridges out of a total of fifteen [13]. This feature is different from bacterial and eukaryotic ribosomes that typically contain RNA-RNA intersubunit connections [61]. Eight intersubunit bridges are mitochondria-specific, several of which are established by mitochondrion-specific proteins or extensions of proteins conserved in bacteria [13].
The recent reconstructions of the 55S human [13–15] and porcine [11,12] mitoribosome by cryo-EM have allowed distinguishing unique features in the mammalian mt-SSU. A significant structural remodeling is observed at the mRNA entrance of the mt-SSU compared to the bacterial SSU. The mRNA channel, an RNA-rich tunnel that encompasses the neck region of the small subunit and houses the A and P sites for decoding and t-RNA binding [62]. In the bacterial ribosome, the mRNA channel entrance is formed by ribosomal proteins uS3, uS4, and uS5, in which basic residues from uS3 and uS4 confer a helicase-like activity responsible for unwinding secondary structures of the mRNA to be translated [62,63]. However, as mentioned earlier, in the human mitoribosome, the uS3 homolog uS3m lacks the C-terminal domain, and a homolog for uS4 is not present [15]. Instead, there are mitochondria-specific extensions of uS5m, which widen the opening of the channel from 9Å to 15Å, and the mitoribosome has acquired the PPR protein mS39 that resides near the mRNA entrance and serves as a binding platform for mRNA bound to the LRPPRC/SLIRP complex [15]. The mRNA channel exit (bS1m, bS21m, and mS37 in humans) has also experienced significant structural rearrangement [15]. Mitochondrial transcripts lack Shine Dalgarno sequences, and the 3’ end of the 12S rRNA stably interacts with mS37, suggesting this region is not involved in translation [15]. Whereas in bacteria, bS1 has six oligonucleotide-binding (OB) fold domains that serve to unfold mRNA being translated in the channel, its human homolog bS1m has only one OB fold [15]. Although the conserved position of this protein in the exit tunnel still supports a role for bS1m in RNA binding, the mechanism by which the start codon is selected remains to be discovered.
Although the mechanism of decoding is well conserved in the human mitoribosome, there is strong evidence of structural adaption in the aminoacyl (A), peptidyl (P), and exit (E) tRNA binding sites in the mt-LSU [15]. Unlike typical tRNAs, mitochondrial tRNAs are highly variable in the elbow region due to varying deletions in the D and/or T-loops [64]. To this effect, the elbow regions of tRNA binding sites of the mitoribosome have become more versatile by the loss of some ribosomal elements [14,65]. The A-site of the mitoribosome is missing bL25 and a portion of 16S rRNA h38, the P-site lacks uL5 and h84 known to stabilize its elbow region in the bacterial ribosome, and the E-site lacks h76 and h77 of the L1 stalk that stabilize the elbow of tRNAs [14,65]. These unique structural adaptations allow the accommodation of human tRNAs. Furthermore, the so-called P-site finger, unique to the mammalian ribosome, compensates for the missing mt-tRNA binding sites [66]. In the human mitoribosome, mL40 and mL48 form the P-site finger [15]. Anchored to the central protuberance through interactions with the structural tRNAVal, bL31m, and mL46, the N-terminal helix of mL40 extends to allow a series of positively charged residues to stabilize the A-site and P-site tRNA elbows [15]. This interaction is made possible by a hydrophobic interface created by the C-terminal helix of mL48 that supports the N-terminal helix of mL40 (Fig 2) [15].
The mammalian mt-LSU CP is characterized by the absence of 5S rRNA, but the presence of a structural tRNAVal in humans [15] and tRNAPhe in porcine [11]. Analyses of additional species have shown that although each mammal favors one of these two mt-tRNA species in all tissue types, at least the human mitoribosome shows a high degree of plasticity to incorporate mt-tRNAPhe when the availability of mt-tRNAVal is compromised [67]. Notably, in the mtDNA, these tRNA genes flank the 12S rRNA and are transcribed as part of a polycistronic transcript generated from the mtDNA heavy strand that starts with tRNAPhe-12SrRNA-tRNAVal-16S-rRNA and can terminate either after the contiguous tRNALeu or extend to almost the complete heavy strand sequence [68]. The arrangement of the mitoribosome structural tRNA genes in the mtDNA is reminiscent of the incorporation of the 5S rRNA into the same bacterial rRNA operon as the major rRNA species [69].
Like in other ribosomes, the CP of the mitoribosome mediates interactions with the small subunit through intersubunit bridges and contains sites for tRNA binding [70]. A unique feature of mitoribosomes is that intersubunit bridges connecting the CP to the head region of the mt-SSU are mediated by an intrinsic GTPase mS29 [15]. mS29 coordinates two mitochondrion-specific intersubunit bridges with mL40, mL46, and mL48 of the CP [15]. Through its GTPase activity, mS29 may mediate subunit joining or assist in the dynamic ratcheting of the head region during translation elongation [15]. Interestingly, recent findings from the fungi translating mitoribosome structure uncovered a role for the typically disordered N-terminal loop of mS29 [52]. Upon subunit rotation, the N-terminal loop of mS29 becomes ordered to stabilize the tRNA in the P/E state, which is then disrupted in the following unrotated state [52]. In the human mitoribosome, the N-terminal region of mS29 also represents an unstructured loop region [15], albeit mammalian mS29 has a shorter N-terminus than the fungal counterpart [15,52]. The GTPase activity of mS29 might be coupled to the N-terminal loop restructuring during subunit ratcheting; however, a role for the N-terminus of mS29 in mammals has not been observed.
The polypeptide exit tunnel is adapted to the transit and delivery of hydrophobic nascent peptides [12,14]. The tunnel exit site is formed by proteins conserved in bacteria, namely bL23m, bL29m, bL22m, bL24m, and bL17m, which form a ring around the exit site. Surrounding the exit tunnel is the mitochondrion-specific mL45, which tethers the mitoribosome to the inner mitochondrial membrane [12,14]. Membrane anchoring aligns the polypeptide delivery site with the OXA1L translocon to facilitate co-translational membrane insertion of newly-synthesized proteins [8,71].
Another unusual characteristic of mitoribosomes is the presence of several zinc-binding motifs in which a protein pair coordinates the binding of a single zinc ion. Such interactions in the mammalian mitoribosome occur between mL66 and uL10m near the L7/L12 stalk [13,15,65]. Two other zinc-binding motifs are present in the small subunit -one between bacterial homolog proteins bS18m and bS6m and the other between bS16m and mS25 [13,15,65]. Interestingly, pathogenic mutations in both bS16m and mS25 have been reported and result in small subunit instability [72–74]. Although the role of these zinc-binding motifs in the mitoribosome remains unknown, zinc-binding likely serves to stabilize the binding and overall structure of the mitoribosome.
4. MITORIBOSOME ASSEMBLY PATHWAY AND FACTORS INVOLVED
Mitoribosome biogenesis follows a maturation pathway involving, for each subunit, the processing and maturation of rRNAs [75–78] and the cooperative incorporation to the rRNA of MRP sets, forming structural clusters and preassembled modules [20,21,79–81]. Structural insights into mitoribosomal assembly have been provided by cryo-EM analyses of mitoribosomes from human HEK293T cells [82] and the human parasite Trypanosoma brucei [80,81] in native states of assembly. The structures have indicated that assembly proceeds through large-scale conformational changes in rRNA coupled with the successive incorporation of MRPs. The biogenetic process requires numerous trans-acting factors, some of which are conserved in bacterial systems, acting in all steps of the process.
Several approaches have been used to advance our understanding of the mitoribosome assembly pathway in mammalian cells. A study performed in human HeLa cells analyzed the order of MRP assembly using stable isotope pulse-chase labeling in cell culture (pulse-chase SILAC) and mass spectrometry analysis of the 55S monosome [21]. The approach, based on the model that the kinetics of incorporation of different MRPs into 55S mitoribosomes indicates their relative assembly order, provided a useful albeit low-resolution draft of the assembly pathway by defining sets of early, intermediate, and late assembly proteins [21]. The study showed that, for each mitoribosomal subunit, the protein components are synthesized in excess and imported into mitochondria, where their stoichiometric accumulation is regulated by degradation of the non-assembled free protein fractions [21]. To identify potential mitoribosome assembly factors, some studies have relied on searching for mitochondrial RNA-binding proteins [83,84], or proteomics analyses of the mitoribosome interactome [19,75,85,86]. Other studies have taken advantage of the better characterized bacterial ribosome assembly pathway [87,88] and screens in the amenable Saccharomyces cerevisiae yeast model [18,89,90]. These assembly factors have been subsequently characterized in human cultured cells or mouse models [76,91,92]. Furthermore, structures of the human HEK293 cells mitochondrial ribosome in native states of assembly have revealed insights into the timing of rRNA folding and protein incorporation during the final steps of ribosomal maturation [82]. Finally, studies in patients with mitochondrial translation efficiency disorders have allowed identifying new assembly factors and the relevance of MRPs for mitoribosome assembly across tissues [23,31,93,94].
Together, the information obtained from these studies is emerging as a working model of the assembly pathway, depicted in Figures 3 and 4. This model is largely based on the SILAC study in HeLa cells [21]. However, a comparison with the assembly pathways described for ribosomes from bacteria [95,96] and mitochondria from S. cerevisiae [20] and Trypanosoma brucei [79–81] have revealed some substantial differences that seem to be independent of the biological system. They could perhaps be attributed to the lack of granularity of the SILAC study in HeLa cells and the fact that it follows the incorporation of proteins into the monosome rather than into each subunit before their joining [21]. The most relevant differences will be discussed below in the sections dedicated to mt-SSU and mt-LSU assembly.
Figure 3. Mitoribosome SSU assembly.
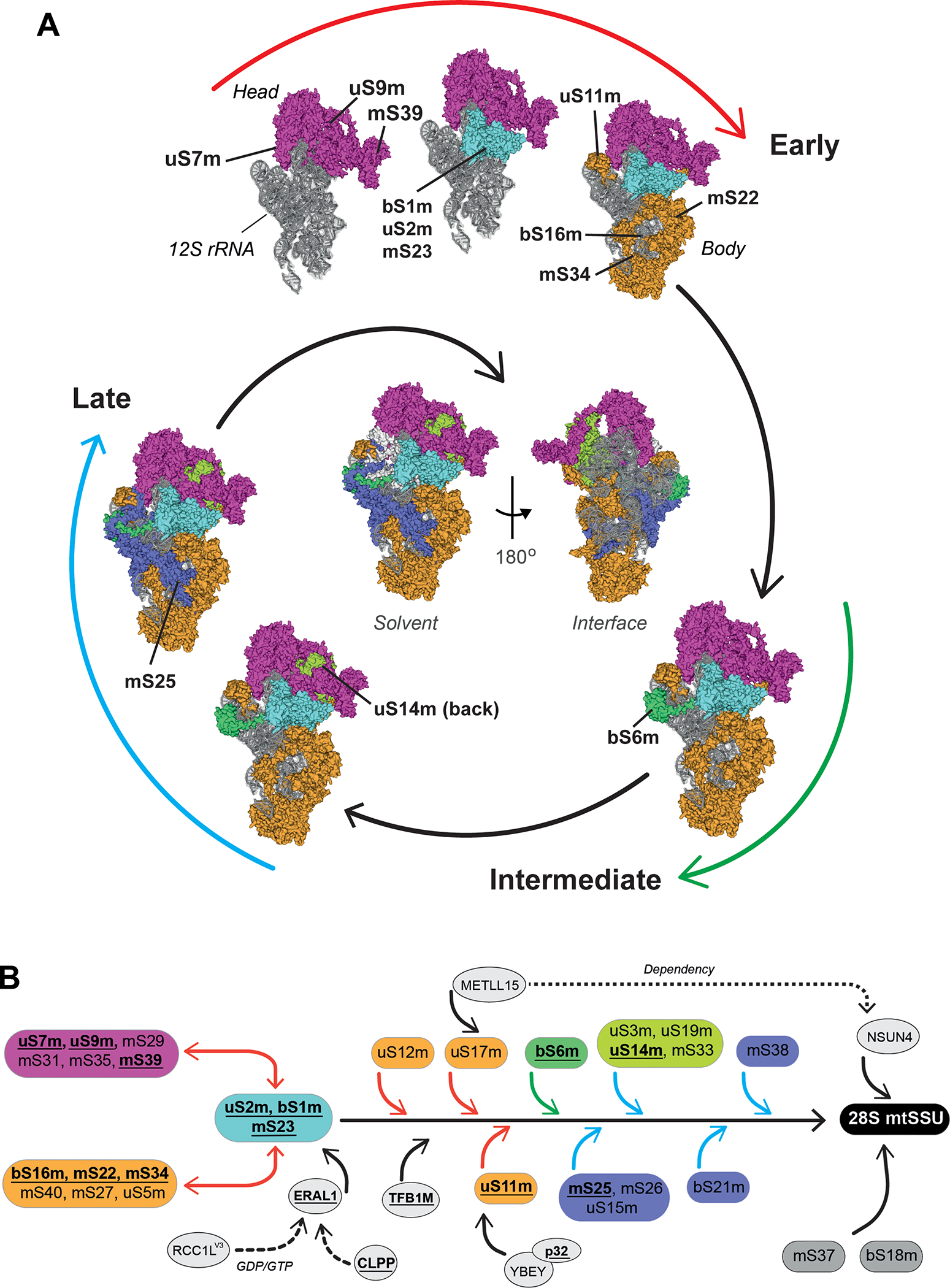
(A) Model of human 28S mt-SSU biogenesis depicting a hierarchical and module-based protein assembly pathway as reported [21]. All modules are color-coded. The 12S rRNA is shown in grey. The assembly process is divided in three stages: early, intermediate, and late (see explanation in the text). Solvent-facing views of the cryo-EM structure (PDB-3J9M) [13] are used to depict the assembly pathway. The localization of mt-SSU protein components that have been found mutated in patients suffering from primary mitochondrial disorders is indicated. In the center of the spiral scheme, complete solvent-facing and interface-facing views of the 28S mt-SSU are presented. The figures were prepared using PYMOL and Adobe Illustrator software. (B) Human 28S mt-SSU assembly pathway as determined by SILAC-proteomics [21], including known assembly factors at their approximate stage of incorporation. Boxes, highlighted with the same color used in the spiral assembly, represent different protein clusters at different assembly stages: early (red arrows), intermediate (green arrows) late (blue arrows). Proteins in gray boxes were not assigned to any assembly stage [21]. Disease-driven mitoribosome proteins and assembly factors are in bold and underlined. Continuous arrows highlight the activity of assembly factors during the assembly process while dotted arrows indicate dependency or extended interaction until the mt-SSU formation.
Figure 4. Mitoribosome LSU assembly.
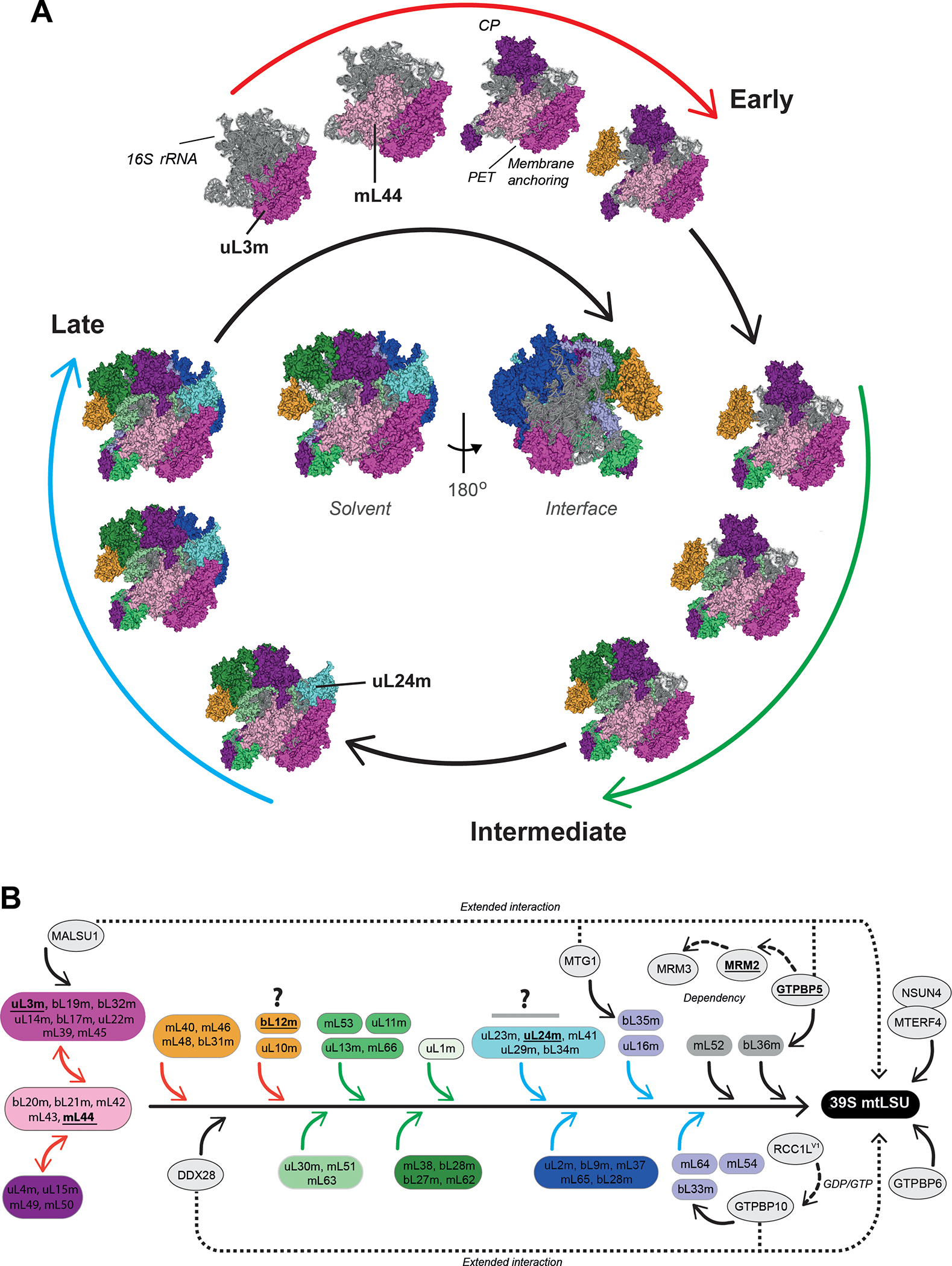
(A) Model of human 39S mt-LSU biogenesis depicting a hierarchical and module-based protein assembly pathway as reported [21]. All modules are color-coded. The 16S rRNA is shown in grey. The assembly process is divided in three stages: early, intermediate, and late (see explanation in the text). Solvent-facing views of the cryo-EM structure (PDB-3J9M) [13] are used to depict the assembly pathway. The localization of mt-SSU protein components that have been found mutated in patients suffering from primary mitochondrial disorders is indicated. In the center of the spiral scheme, complete solvent-facing and interface-facing views of the 28S mt-SSU are presented. The figures were prepared using PYMOL and Adobe Illustrator software. CP, central protuberance; PET, polypeptide exit tunnel. (B) Human 39S mt-LSU assembly pathway as determined by SILAC-proteomics [21], incorporating known assembly factors at their approximate stage of incorporation. Boxes, highlighted with the same color used in the spiral assembly, represent different protein clusters at different assembly stages: early (red arrows), intermediate (green arrows) late (blue arrows). Proteins in gray boxes were not assigned to any assembly stage [21]. Disease-driven mitoribosome proteins and assembly factors are in bold and underlined. Continuous arrows highlight the activity of assembly factors during the assembly process while dotted arrows their dependency or extended interaction until the mt-LSU formation. Question marks highlight proteins whose proposed assembly kinetics [21] differ from bacterial and yeast mitoribosome systems (see explanation in the text).
Although presented as separated assembly lines in Figures 3 and 4, the biogenesis of the two mitoribosome subunits is coordinated. It starts co-transcriptionally, with a subset of 27 mt-LSU proteins forming a subcomplex on an unprocessed RNA containing the 16S rRNA, whose formation is required for precursor RNA processing by the mitochondrial ribonuclease P (RNase P) and the mitochondrial RNase Z known as ELAC2, and liberation of the 12S rRNA as a condition for mt-SSU protein incorporation [76]. The identity of these mt-LSU proteins is uncertain since those reported in a mouse KO for the RNase P MRPP3 component [76] are a mix of early, intermediate, and late-assembly proteins as detected by the SILAC study [21], which could suggest an off-pathway subassembly. In support of this model, silencing of the mt-LSU assembly factor MPV17L2 causes not only a decrease in mt-LSU without accumulation of assembly intermediates but also a severe mt-SSU depletion and accumulation of mt-SSU proteins in aggregated nucleoids. It has been proposed that MPV17L2 is required for early mt-LSU assembly steps needed to facilitate rRNA precursor processing and release from the mtDNA nucleoids, where transcription occurs, in order to proceed with its maturation within the mitochondrial RNA granule compartment [21,93].
4.1. mt-SSU Assembly
According to the SILAC proteomics study, mt-SSU protein assembly (Fig 3A) proceeds by the early incorporation of two large protein modules [21]. One module formed by proteins (uS5m, bS16m, mS22, mS27, mS34, mS40) binds to the mt-SSU lower body/foot contacting with the 5’ and 3’ rRNA domains, and the other, formed by proteins (uS7m, uS9m, mS29, mS31, mS35, mS39), localizes to the head, extending through the major 3’ domain in the 12S rRNA. A smaller set of proteins (mS23, uS2m, and bS1m) interacts with both modules. To complete the early assembly process, another three proteins (uS11m, uS17m, and uS12m) bind independently [21]. These early proteins bind to the outer surface of the mt-SSU and are important for recruiting the late assembly proteins.
A major discrepancy with the bacterial assembly pathway is the incorporation of SSU proteins binding to the major 3′ domain of 12S rRNA in the head region (uS7m and uS9m), which in bacteria occurs at intermediate-late stages [95].
Several assembly factors sustain early mt-SSU biogenesis (Fig 3B). The human GTPase ERAL1 (Era G-protein-like 1) acts as an RNA chaperone to protect 12S rRNA from degradation before maturation and assembly. It co-immunoprecipitates with early assembly proteins mS22 and mS31 [97] and binds to helix 45 at the 3’ terminus of the 12S mt-rRNA [91], which contains two highly conserved adenines that undergo subsequent methylation catalyzed by TFB1M (mitochondrial transcription factor B) [98]. ERAL1 levels are tightly controlled by the ATP-dependent protease CLPP to avoid excess accumulation that prevents mitoribosome formation [99]. ERAL1 was also found to interact with the endoribonuclease YBEY, although probably due to the GTP-binding dependence of this interaction, their labile association was only observed in situ by Fluorescence lifetime imaging microscopy (FLIM) combined with Förster Resonance Energy Transfer (FRET), which provides high spatial (nanometer) and temporal (nanoseconds) resolution of protein-protein interactions [100]. In contrast, YBEY stably binds to the multifunctional protein p32 (also known as Complement component 1 Q subcomponent-binding protein or C1QBP) and is required for uS11m incorporation into the assembly pathway [100]. The N4-methylcytidine (m4C) methyltransferase METTL15 methylates position C839 in the 12S rRNA, which is required for the incorporation of uS12m and uS17m, and the subsequent assembly of late proteins such as bS21m and mS38 [101]. Notably, the assembly factors seem to promote the incorporation of non-modular, individually assembled MRPs (Fig 3B). The action of METTL15 probably enables the recruitment of the m(5)C methyltransferase NSUN4 to methylate C911 in the 12S rRNA during late assembly. The GTPase MTG3 (also known as NOA1 or C4ORF14) also participates in mt-SSU assembly [102,103], perhaps at the early stages as its bacterial counterpart [103], but its precise role remains unknown. It has been recently reported that RCC1L (regulator of chromatin condensation 1 like, also known as WBSCR16), a putative GDP/GTP exchange factor, interacts with the mitoribosome. Two different alternative splicing isoforms, RCC1LV1, and RCC1LV3, associate with the mt-LSU and mt-SSU, respectively. ERAL1, MTG3, and the mS29 MRP could be the targets of RCC1LV3 [104].
Late-binding proteins cluster into two groups. One binds in the head region (uS14m, uS10m, uS3m, and mS33) in association with the early uS7m-mS29 group, and the second (uS15m, mS25, and mS26), near the early bS16m-mS22 cluster. Most late-binding proteins localize to the interface with the mt-LSU [21]. The late assembly of uS15m is intriguing since uS15 assembles early with uS15 in bacteria [95].
Several other factors are required for mt-SSU biogenesis, although their functions remain ill-defined and could be indirect. Among these proteins, the G-rich sequence binding factor 1 (GRSF1) is a bona fide component of mitochondrial RNA granules where it interacts with RNAseP to modulate the cleavage of primary polycistronic transcripts [84]. Silencing of GRSF1 also resulted in abnormal mt-SSU assembly, decreased amounts of the mt-LSU, and then in a reduction in monosome formation [83]. These defects could be a consequence of poor precursor RNA processing. However, GRSF1 was found to stabilize the 12S rRNA [83,84], and a small fraction of it co-sediments with the mt-SSU [86]. Furthermore, GRSF1 silencing causes the formation of an mt-SSU subassembly with aberrant conformation because it sediments as a 22S subunit but contains all the canonical 28S mt-SSU proteins [83], which suggests a direct role for GRSF1 in mt-SSU assembly.
4.2. mt-LSU Assembly
According to the SILAC proteomics study, mt-LSU assembly (Fig 4A) is complex and involves protein modules and individual proteins incorporating during three defined stages, early, intermediate, and late [21].
During the early phase, three big modules, including 24 proteins, were identified to assemble with similar kinetics, suggesting a possible coordinated binding. These proteins mainly localize in a region encompassing the 5’ rRNA domain. A first module, formed by rRNA binding (uL3m, bL19m) and other proteins (uL14m, bL17m, uL22m, and bL32m) serve to anchor mL39, and then mL45. The protein mL45 could tether the mt-LSU at the inner membrane during subsequent assembly steps as it occurs in yeast with the membrane facing protuberance mt-LSU proteins [20]. The assembly of this module involves the action of one trans-acting factor (Fig 4B). MALSU1 (Mitochondrial Assembly of ribosomal Large SUbunit 1) assists the insertion of uL14m [105,106]. Although in yeast, the mAAA protease complex is responsible for bL32m precursor processing to enable its incorporation into the mt-LSU assembly line [107], this mechanism does not operate in mammalian tissues [108]. The DEAD-box RNA helicase DDX28 [85,86] and the Fas-activated serine/threonine (FAST) kinase family protein FASTKD2 [85,109] bind and stabilize the 16S rRNA, suggesting an early action, although their precise roles remain unknown. DDX28 remains bound to the growing mt-LSU particle until late maturation stages [86]. A second early MRP module includes the mRNA-binding bL20m protein and bL21m, mL42, mL43, and mL44. A third module is formed by the mRNA binding heterodimer uL4m-uL15m, which recruits mL49 and mL50. The fourth early-assembly module includes proteins associated with the tRNAVal in the mt-LSU central protuberance (mL40, mL46, and mL48).
At the intermediate stage, the fourth early module facilitates incorporating a second group of tRNAVal surrounding proteins: mL38, uL18m, and bL27m. How tRNAVal is recruited remains unknown. Showing unique plasticity, however, when mt-tRNAVal is limited, human mitoribosomes can instead integrate mt-tRNAPhe to assemble a functional ribosome [110]. At this stage, the dimer uL13m-mL66, and uL11m, bind the uL10m stalk through interactions with RNA and early-binding proteins. The protein uL12m was included in the early group based on the kinetic proteomics data in HeLa cells [21]. However, it is intriguing that it can incorporate before uL11m and uL10m, which locate at the base of the L12 stalk. In fact, uL12m assembles late in ribosomes from both bacteria [95,96] and yeast mitochondria [20]. Subsequently, a large module of intermediate-late proteins (mL41, uL23m, uL24m, uL29m, and bL34m) is proposed to be recruited to form the polypeptide exit tunnel. Also here, the timing regarding the assembly of the polypeptide exit tunnel differs from what has been found in bacteria and yeast mitochondria, where uL22m, uL23m, and uL24m are assembled early [20,95,96]. This is particularly relevant for mitoribosomes since the exit tunnel is surrounded by mitochondria-specific proteins that in yeast form the membrane-facing protuberance (mL44 and mL50) and, in human cells, the membrane-anchoring site (mL45), which also assemble early [20,21].
At the late stages, proteins located at the interface with the mt-SSU are incorporated, including a large module formed by proteins uL2m, uL28m, uL29m, mL37, and mL65, some of which form intersubunit bridges. This data is in agreement with Cryo-EM studies showing that the intersubunit interface is well organized only at a late stage of assembly [82]. Several assembly factors act at this stage to finalize the maturation of the mt-LSU particle and establish several quality-control checkpoints during the formation of the mt-LSU catalytic site, the peptidyl transferase center (PTC). The late assembly factors include at least four GTPases: GTPBP7/MTG1 (homolog of bacterial RbgA), GTPBP5/MTG2, and GTPBP10 (two homologs of bacterial Obg), and GTPBP6 (homolog of bacterial HflX) (reviewed in [111]). Proteomics, biochemical, and structural studies have suggested sequential recruitment of assembly factors. GTPBP10 is recruited first, binds to the 16S rRNA, and its absence prevents the incorporation of bL33m and bL34m [19]. GTPBP10 may be the target of the GTP/GDP exchange factor RCC1LV1 [104].
GTPBP7/MTG1 interacts with domain VI helices in the 16S rRNA and with bL19m, which induces a conformational change and remodeling of the bL19m-containing domain, thereby facilitating the incorporation of the late assembly proteins bL36m and bL35m to complete the formation of mature mt-LSU [112]. GTPBP7/MTG1 remains bound to the mt-LSU until maturation is completed. Only when subunit joining is about to occur, GTPBP7/MTG1 interacts with the mt-SSU protein mS27, a putative guanosine triphosphate exchange factor (GEF) that catalyzes fast GDP-GTP exchange to enable the release of GTPBP7/MTG1 from the ribosome and facilitate the formation of the mB6 intersubunit bridge between bL19m and mS27 [112]. MALSU1 also plays a quality control role during the late stages of mt-LSU assembly [82,105]. It remains bound to early-assembled uL14m, probably blocking the formation of intersubunit bridge B8, and forms a complex with the mitochondrial ACP (acyl carrier protein), usually involved in fatty acid synthesis, and the LYRM (leucine-tyrosine-arginine motif) family protein L0R8F8 (also known as AltMiD51 or MIEF1) [82,113]. This module prevents the premature association of the 28S and 39S ribosomal subunits [82]. GTPBP5 binds to the 16S rRNA and interacts with several mt-LSU proteins and assembly factors, including the methyltransferase MRM2 [114] that catalyzes the 2’-O-methyl modification at position U1369 of the 16S rRNA A-loop, an essential component of the peptidyl transferase center (PTC) [78,115]. Following 16S rRNA maturation by MRM2 and MRM3-catalyzed methylation, and probably also its pseudouridylation by the pseudouridine synthase RPUSD4 [75,116], bL36m can be recruited to the maturing mt-LSU to finalize its assembly [114]. During these late stages, the mitochondrial transcription termination factor (MTERF) family protein mTERF4 forms a complex with NSUN4 that associates with a late matured mt-LSU and acts as an assembly checkpoint during monosome formation [92,117]. Yet, another GTPase, GTPBP6, incorporates to the very late mtLSU particle that already contains all of the 52 mitoribosome proteins, including bL36m, and has several late-acting mtLSU biogenetic factors such as MALSU1, GTPBP5, GTPBP7, GTPBP10, and NSUN4-MTERF4 bound to the complex [118]. Remarkably, GTPBP6 has been demonstrated to promote ribosome dissociation into subunits [118], a role that has also been suggested for GTPBP5, GTPBP7, and GTPBP10, whose overexpression is deleterious to monosome accumulation [19,112,114,119]. Therefore, all these factors are required for the very final tuning steps of mtLSU maturation, potentially acting as quality-control and anti-association factors to prevent premature subunit joining.
5. THE MITORIBOSOME IN PRIMARY MITOCHONDRIAL DISORDERS
Taking into consideration the universal involvement of the mitochondrial OXPHOS system in human cell bioenergetics, it is not surprising that the machineries involved in its biogenesis are deeply implicated in the etiology, progression, and treatment of multiple diseases. Among them, mutations in the mitochondrial translation apparatus, the mitoribosome structural components and assembly factors, cause primary mitochondrial OXPHOS disorders. These are multisystemic mitochondrial diseases such as Leigh syndrome, cardio- and encephalo-myopathies, liver disease, and Perrault syndrome.
5.1. Mutations in mitoribosome RNA components
Numerous mutations have been identified in the 12S rRNA, the 16S rRNA, and the tRNAVal, associated with a variety of mitochondrial disorders (reviewed in [23]). Since these three RNAs are encoded in the mtDNA, the clinical manifestations in each patient depend on several genetic determinants such as level of heteroplasmy (percentage of mtDNA molecules carrying a given mutation), threshold effect (minimum level of heteroplasmy required to manifest in a biochemical defect), mitotic segregation, and maternal inheritance [120]. While most mutations in the rRNAs have been presented in clinical reports that lack deep mechanistic insight, estimating their disruptive effect by heterologous inferential analysis has been informative. By this approach, conservational information is combined with functional and structural data acquired from heterologous ribosomal sources to deliver a high predictive power [121,122]. Here, we will discuss some of the mutations found in patients.
12S rRNA:
Mutations in the 12S rRNA are a frequent cause of hearing loss. Specifically, the homoplasmic A1555G and C1494T mutations at the highly conserved decoding site of the 12S rRNA gene are the major contributors to aminoglycoside-induced [123,124] and non-syndromic hearing loss [125] in many families across ethnic backgrounds. Aminoglycosides, such as gentamicin, kanamycin, and streptomycin are used as antibacterial therapeutic agents that interact with the A-site of the SSU rRNA to inhibit protein synthesis. The mutations in the human 12S rRNA contribute to creating a binding site for aminoglycoside drugs [126]. Consequently, exposure to aminoglycosides can induce or aggravate hearing loss in individuals carrying one of these mutations. Clinical expression of the deafness-associated homoplasmic A1555G mutation varies from profound congenital hearing loss to normal hearing [123], due to the complex inheritance of several nuclear-encoded modifier genes. Such modifiers include the mitochondrial transcription factor B1 (TFB1M) [127], which methylates adenine residues in the 12S rRNA loop adjacent to the A1555G mutation, as well as MTO1 and GTPBP3 [128], which form a heterodimer complex that interacts with the mt-SSU and optimizes translation by a proof-reading mechanism [129]. Another source of variability associated with the 1555A>G mutation comes from its combination with additional mutations. For example, a patient harboring an additional 12S rRNA 4309G>A mutation suffered from hearing-loss, progressive external ophthalmoplegia, and exercise intolerance [130]. In another family with the 1555A>G mutation, some of the members suffered from Leigh syndrome in addition to hearing loss, which has been attributed to unidentified environmental (e.g., intrauterine exposure to aminoglycosides) or nDNA-encoded factors [131].
16S rRNA:
Only three disease-driven mutations have been identified in the 16S rRNA. A near homoplasmic substitution at a conserved position, G3090A, was found in muscle of a young woman with severe myopathy and profound combined deficiency of OXPHOS complexes in muscle. The mutation was not detected in the patient’s mother, suggesting a de novo origin probably during embryonic development [132]. This mutation lies in helix h93 that could affect the correct positioning of tRNAs in the PTC and disrupt ribosome function [122]. A homoplasmic T2336C mutation was associated with hypertrophic cardiomyopathy in the four related patients [133]. The mutation is predicted to disturb the T2336-A2438 base pairing in the first position of a stem-loop in the 16S rRNA domain III. Studies in both patient iPSC-derived cardiomyocytes and trans-mitochondrial cybrid lines have shown that the mutation decreases the stability of the 16S rRNA and some MRPs (bL19m and uL23m) [134]. The third mutation, C2835T, has been reported to be associated with the pediatric neurological disorder Rett syndrome [135], chronic progressive external ophthalmoplegia (CPEO) plus muscle weakness, and cerebellar dysfunctions [136]. C2835T maps to the neighborhood of the P-loop, which may explain its disruptive character.
tRNAVal:
The tRNAVal is a hotspot for mutations causing mitochondrial disorders. Variants in the MT-TV gene have been reported, associated with heterogeneous clinical manifestations that have been discussed elsewhere [23,137]. The reported mutations include a heteroplasmic G1606A change that affects the acceptor stem of tRNAVal, associated with an adult-onset complex neurologic syndrome (hearing loss, cataracts, ataxia, myoclonus, and dementia). A homoplasmic C1624T mutation affecting a base pair in the dihydrouridine loop destabilizes tRNAVal and caused several neonatal deaths and one surviving child with Leigh syndrome. A T1658C mutation alters the T loop structure of mitochondrial tRNAVal was associated, in heteroplasmy, with chronic progressive external ophthalmoplegia (CPEO). A G1644A change that destabilizes tRNAVal has been found in several patients, presenting in homoplasmy associated with adult-onset progressive encephalo-cardiomyopathy or heteroplasmy associated with either adult-onset Leigh syndrome or MELAS. Other mutations were associated with encephalo-cardiomyopathy (homoplasmic C1628T), MNGIE-like gastrointestinal dysmotility, and cachexia (heteroplasmic A1630G), and MELAS syndrome (mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes) (heteroplasmic G1642A). Studies of the pathogenic mechanism of the G1642A mutation in trans-mitochondrial cybrids allowed to understand the effect of reduced mt-tRNAVal levels on mitoribosome assembly. In this scenario, most available mt-tRNAVal was preferentially used in elongation, whereas mt-tRNAPhe was recruited to build the mitoribosome, thus demonstrating an example of exceptional structural plasticity [67].
5.2. Mutations in mitoribosomal proteins
Mutations in mitoribosome proteins are a frequent cause of disorders owing to mitochondrial protein synthesis deficiencies ([74,138–141] and reviewed in [23,142]). Up to date, disease-causing mutations have been found in 13 mt-SSU proteins (bS1m, uS2m, bS6m, uS7m, uS9m, uS11m, uS14m, bS16m, mS22, mS23, mS25, mS34, and mS39) and 4 mt-LSU proteins (uL3m, bL12m, uL24m, and mL44). Most of these proteins participate at early stages in mitoribosome assembly, which may facilitate disrupting the mitoribosome structure and subsequent assembly steps [141].
5.2.1. Mutations in mt-SSU proteins
5.2.1.1. mt-SSU early assembly module [uS7m, uS9m, m29, mS31, mS35, mS39]
Disease-causing mutations have been found in three components of the [uS7m, uS9m, mS29, mS31, mS35, mS39] early assembly module [21], uS7m, uS9m, and mS39, which bind to the mt-SSU head region extending through the major 3’ domain in the 12S rRNA [15].
uS7m.
This protein contacts the 12S rRNA and mS37 to bridge the mt-SSU head and body regions [15]. To date, two siblings have presented with clinical manifestations of sensorineural deafness, lactic acidemia, and combined OXPHOS complex deficiencies driven by a homozygous mutation: c.550A>G (p.M184V) in uS7m [143]. One of the patients developed more severe conditions at the age of 14, suffering from liver failure and renal dysfunction [143]. The M184V mutation affects a highly conserved methionine positioned in the hydrophobic core of uS7m, where several interactions function to stabilize the protein’s structure [143]. In patient fibroblasts, mRNA, and steady-state protein levels of uS7m were decreased, indicating that the mutation destabilizes the uS7m transcript [143]. This resulted in 12S rRNA instability without affecting the mt-LSU 16S rRNA. Consequently, mitochondrial translation was attenuated, resulting in decreased steady-state levels of mitochondrion-encoded OXPHOS subunits and their host complexes [143].
uS9m.
It neighbors uS7m in the head region and mediates substantial interactions with the major 3’ domain of the 12S rRNA, and directly binds the GTPase mS29 [15]. Genomic microarray analysis in a patient presenting with intellectual disability and development delay revealed a cryptic microdeletion in chromosome band 2q12.1 [144]. This deletion spans 360kb and results in the loss of both MRPS9 (uS9m) and POU3F3 genes [144]. POU3F3 is a transcription factor expressed in the central nervous system, and heterozygous variants of POU3F3 have been associated with neurodevelopmental disorders, including intellectual disability and developmental delay [145]. This suggests that the effects of the microdeletion are likely due to the loss of POU3F3 and not the MRPS9 gene, although the mitochondrial function was not characterized.
mS39.
Adjacent to the entry of the mRNA channel, mS39 binds the mRNA-bound LRPPRC/SLIRP complex and may, in this way, assist in delivering leaderless mitochondrial mRNAs during translation initiation [15]. One patient has been reported to carry two genetic variants, c.415–2A>G affecting splicing, and c.1747_1748insCT (p.F583Sfs*3) in the PTCD3 (mS39) gene associated with abnormal brain development at birth and infantile-onset Leigh syndrome [139]. Further analyses in patient fibroblasts revealed a general decrease in most mt-SSU protein levels, mitochondrial translation, and steady-state levels of OXPHOS subunits from CI and CIV [139].
5.2.1.2. mt-SSU early assembly module [uS5m, bS16m, mS22, mS27, mS34, mS40]
Disease-causing mutations have been found in three components of this early assembly module, bS16m, mS22, and mS34, which binds to the mt-SSU lower body/foot contacting with the 5’ and 3’ rRNA domains of the 12S rRNA.
bS16m.
In support of its early assembly, bS16m and mS40 have extensive contacts with the 5’ domain of the 12S rRNA, providing the foundation for overall mt-SSU assembly [13,15,21]. The bS16m-mS22-mS40 cluster forms a scaffold for the foot module mS27 and mS34, just before the assembly of uS5m in the mRNA entry channel [21]. Additionally, bS16m coordinates the binding of a single zinc ion with mS25 [13,15,65] that could play a structural role. So far, a single patient has presented with the homozygous mutation c.331C>T (p.Arg111*), encoding for a premature stop codon in bS16m [72]. Clinically, the patient suffered from agenesis of corpus callosum diagnosed during gestation, dysmorphism, and ultimately fatal lactic acidosis three days after birth [72]. Biochemically, it had a combined OXPHOS enzymatic deficiency in liver and muscle [72]. In patient fibroblasts, the bS16m transcript was unstable, full bS16m protein was absent, and the truncated bS16m fragment was not detected [73]. The absence of bS16m resulted in low levels of 12S rRNA, whereas the 16S rRNA was unaffected [72]. The mt-SSU failed to assemble without bS16m, although some proteins, such as uS2m, were found particularly stable [73], suggesting that the [mS23, uS2m, bS1m] module can pre-assemble even in the absence of 12S rRNA.
mS22.
The mitochondrion-specific mt-SSU protein mS22 is a unique early assembly protein in that it does not have any contacts with the 12S rRNA, but instead interacts with bS16m and mS40 [15]. To date, many disease-driving mutations have been described for mS22, leading to a host of different disease phenotypes (Table 1). The first reported case was of three patients harboring the homozygous missense mutation c.509G>A (p.R170H), all born to the same consanguineous parents who were heterozygous for the mutation [146]. All patients suffered from cardiomyopathy, renal tubulopathy, antenatal skin edema, and muscle hypotonia [146], associated with multiple OXPHOS enzymatic deficiencies and decreased 12S rRNA levels in muscle mitochondria [73,146]. In patient fibroblasts, the steady-state levels of uS11m and bS16m were drastically decreased, whereas uS2m was found stable, as for fibroblasts depleted of bS16m [73].
Table 1. Mutations in mitoribosome elements and human disease.
AD, Alzheimer’s disease. AMDF, Ataxia, myoclonus and deafness. CPEO, chronic progressive external ophthalmoplegia. LVNC, left ventricular noncompaction. MELAS, mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. MERRF, myoclonus epilepsy and ragged red muscle fibers. MNGIE, Myo-, neuro-, gastrointestinal encephalopathy. PD, Parkinson’s disease. POAG, Primary open-angle glaucoma. SNP, Single nucleotide polymorphism. For most mutations, only one or a few references were included. A complete set of references can be found in the MITOMAP database.
| Gene | Mutation | Clinical Manifestations | Ref. |
|---|---|---|---|
| Mutations in mitoribosomal RNAs | |||
| 12S rRNA | T669C, A735G, A745G, C792T, A801G, A827G, A839G, A856G, T921C, C960del, C960CC, T961delT+/-C(n)ins, T961G, T961TC, G988A, T990C, T1005C, A1027G, T1095C, A1116G, T1180G, C1192A, C1192T, C1226G, T1291C, C1310T, A1331G, T1349G, A1374G, T1420G, T1452C, A1453G, A1492C, C1494T, A1517C, C1537T, A1544T, A1546T, G1554A, A1555G, T1575G, T1577G | Non-syndromic antibiotic induced hearing loss | [123,200–203] |
| G652del, G653GG, A663G | Atherosclerosis risk | [204–206] | |
| T721C, T850C, T961C | Possibly LVNC associated | [207] | |
| C869T, T1391C, C1556T | Hypertrophic cardiomyopathy | [208] | |
| A750A, A1438A | Schizophrenia associated | [209] | |
| 16S rRNA | T2158C | Reduced risk PD | [210] |
| T2336C | Hypertrophic cardiomyopathy | [133] | |
| T2352C, G2361A, A2755G | Possibly LVNC associated | [207] | |
| C2639A | POAG associated | [211] | |
| T2648C, C2835T | Rett Syndrome | [135,212] | |
| A2706A | Increased risk of type 2 diabetes | [213] | |
| G3010A A3111T |
CVS and migraine Migraine |
[135] | |
| G3090A | Mitochondrial encephalomyopathy | [132] | |
| C3093G | MELAS | [214] | |
| G3196A | AD/PD associated | [215] | |
| MT-TV | G1606A | AMDF | [216] |
| T1607C | Suspected mitochondrial disease | [217] | |
| A1616G, G1642A | MELAS | [218,219] | |
| C1624T | Leigh syndrome | [220] | |
| A1630G | MNGIE | [221] | |
| A1643G | fatal infantile mitochondrial disease | [222] | |
| G1644A | Leigh syndrome, MELAS, hypertrophic cardiomyopathy | [223] | |
| G1644T | Adult-onset Leigh syndrome | [224] | |
| T1659C | Movement disorder | [225] | |
| Mutations in mitoribosomal proteins | |||
| MRPS28 (bS1m) | c.356A>G (p.Lys119Arg) | Intrauterine growth retardation, facial dysmorphism, sensorineural hearing loss, and development delay | [138] |
| MRPS2 (uS2m) | Compound heterozygous c.328C>T (p.Arg110Cys) c.340G>A (p.Asp114Asn) Homozygous c.413G>A (p.Arg138His) |
Sensorineural hearing loss, developmental delay, and hypoglycemia | [141] |
| MRPS6 (bS6m) | SNP (rs9982601) | Increased risk factor for myocardial infarction | [155,156] |
| MRPS7 (uS7m) | Homozygous c.550A>G (p.Met184Val) |
Congenital sensorineural deafness, progressive hepatic and renal failure, and lactic acidemia | [143] |
| MRPS9 (uS9m) | Included in cryptic microdeletion in chromosome band 2q12.1 | Intellectual disability and development delay | [144] |
| MRPS11 (uS11m) | Ankylosing spondylitis | [154] | |
| MRPS14 (uS14m) | Homozygous c.322C>T (p.Arg108Cys) |
Hypertrophic cardiomyopathy with neonatal lactic acidosis, growth retardation, dysmorphic features, and neurological involvement | [158] |
| MRPS16 (bS16m) | Homozygous c.331C>T (p.Arg111*) |
Fatal neonatal lactic acidosis | [72,73] |
| MRPS22 (mS22) | Homozygous c.509G>A (p.Arg170His) |
Cardiomyopathy, renal tubulopathy, antenatal skin oedema, and muscle hypotonia | [146] |
| Homozygous c.644T>C (p.Leu215Pro) |
Cornelia de Lange-like dysmorphic features, encephalocardiomyopathy | [147] | |
| Homozygous c.1032_1035dup (p.Leu346Asnfs*21) |
Fatal lactic acidosis, cardiomyopathy, and brain abnormalities | [148] | |
| Homozygous c.339+5G>A |
Dysmorphism, hypotonia, developmental delay, and Leigh syndrome-like lesions affecting the medulla oblongata and brain stem | [226] | |
| Homozygous c.404G>A (p.Arg135Gln) Homozygous c.605G>A (p.Arg202His) |
Primary ovarian insufficiency | [149] | |
| MRPS23 (mS23) | Homozygous c.119C>G (p.Pro40Arg) |
Hepatic Disease | [153] |
| MRPS25 (mS25) | Homozygous c.215C>T (p.Pro72Leu) |
Mitochondrial encephalomyopathy | [74] |
| MRPS34 (mS34) | Homozygous c.321+1G>T (p.Val100_Gln107del) Homozygous c.322–10G>A (p.Asn108Leufs*12) (p.Asn108Glyfs*50) Compound heterozygous c.37G>A (p.Glu13Lys) c.94C>T (p.Gln32*) |
Leigh syndrome | [151] |
| MRPS39 (mS39) | Compound heterozygous c.415–2A>G c.1747_1748insCT (p.Phe583Serfs*3) |
Leigh syndrome | [139] |
| MRPL3 (uL3m) | Compound heterozygous c.950C>G (p.Pro317Arg)/large deletion |
Hypertrophic cardiomyopathy and psychomotor retardation | [159] |
| MRPL12 (bL12m) | Homozygous c.542C>T (p.Ala181Val) |
neurological deterioration and growth retardation | [164] |
| MRPL24 (uL24m) | Homozygous c.272C>T (p.Leu91Pro) |
Movement disorder and intellectual disability | [140] |
| MRPL44 (mL44) | Homozygous c.467T>G (p.Leu156Arg) Compound heterozygous c.233G>A (p.Arg78Gln) c.467T>G (p.Leu156Arg) |
Infantile-onset hypertrophic cardiomyopathy | [160,161] |
Another patient presented with a p.L215P homozygous mutation that led to Cornelia de Lange-like dysmorphic features, brain abnormalities, hypertrophic cardiomyopathy, and combined OXPHOS deficiency [147]. Patient fibroblasts revealed trace levels of mS22 protein, enough to sustain a moderate mitochondrial protein synthesis capacity [147]. In a separate case, a single patient, also born from consanguineous parents, carried a homozygous 4-bp duplication c.1032_1035dup, (p.L346Nfs*21) that predicts a frameshift resulting in a change of the last 15 mS22 amino acids [148]. This newborn patient had fatal lactic acidosis, cardiomyopathy, brain abnormalities such as agenesis of corpus callosum, periventricular cysts, and combined OXPHOS deficiency [148]. Although at ~50% decreased levels, the mutant mS22 protein was still detectable [148], suggesting that it is probably incorporated into the mt-SSU. In the human mitoribosome structure, the mS22 C-terminus does not interact with any protein or rRNA domains [15]. Yet, the aberrant C-terminal tail of the mutant protein could cause some conformational distortion preventing proper mt-SSU assembly and function.
Most recently, four patients from two different consanguineous families carried the homozygous missense mutations c.404G>A (p.R135Q) and c.605G>A (p.R202H), leading to primary ovarian insufficiency [149]. Unlike the previous mutations described, these mS22 mutations produce a far less severe phenotype. Analysis of patient-derived cells revealed no effect on the steady-state protein levels of mS22 or the levels of 12S and 16S rRNA, indicating a tissue-specific defect [149]. To assess the function of mS22 in ovarian development, a Drosophila model was used in which the fly ortholog of mS22 (mRpS22) was knocked down by shRNA [149]. Whole-body knockdown (KD) of mRpS22 led to larval death [149]. Interestingly, however, KD in germ cells of the ovary caused infertility in the flies, whereas KD in the ovarian somatic cells did not [149]. This cell-autonomous effect on female fertility implies that mS22 may have moonlighting functions in the regulation of reproductive development.
mS34.
Forming the protein-dense foot region of the mt-SSU, mS34 binds the mitochondrion-specific protein and GEF mS27 [13,15,112]. mS34 is part of the early assembly cluster containing bS16m and mS22, and makes direct interactions with neighboring proteins mS26 and mS40, as well as the 5’ and 3’ minor domains of the 12S rRNA [13,21]. mS34 is expected to be important for mt-SSU stability and mitochondrial translation, as suggested by the effects of an L68P mutation studied in mice [150]. Although not embryonically lethal, mice homozygous for the c.203T>C (p.L68P) mutation in mS34 have severely decreased amounts of translating ribosomes and heterogeneous pathology of the heart and liver [150]. Four different pathogenic mutations have been reported in human mS34. Two of the variants were homozygous splice-site mutations, and the others owing to a subject with compound heterozygous mutations [151]. All mutations led to developmental delay and Leigh or Leigh-like syndrome (Table 1), associated with drastic decreases in the mS34 levels, mitochondrial translation impairment, and combined OXPHOS deficiency [151]. The splicing mutations involved a truncation of exon 1, resulting in an in-frame deletion of 8 amino acids (p.V100_Q107del) [151]. Although the mutant mRNA was stable, mS34 was barely detectable, and levels of most mt-SSU proteins were decreased, whereas the mt-LSU was relatively unaffected, further supporting a role for mS34 in stabilizing the 12S rRNA and promoting subsequent steps of mt-SSU assembly [151].
5.2.1.3. mt-SSU early assembly module [bS1m, uS2m, mS23]
The [mS23, uS2m, bS1m] module proteins are located at the exit from the mRNA channel [15]. The module interacts with and connects the two RNA binding modules described earlier that locate in the head region and the body of the mt-SSU [15,21] (Fig 1A). Patients carrying mutations in mS23, uS2m, or bS1m have been identified.
bS1m.
A disease-causing mutation (K119R) in bS1m has been identified in a patient with severe intrauterine growth retardation, facial dysmorphism, sensorineural hearing loss, and development delay [138]. The mutation renders bS1m unstable and disrupts mt-SSU assembly and combined OXPHOS enzymatic deficiency [138]. Mitochondrial extracts from patient fibroblasts showed decreased levels of 12S rRNA, bS1m, and other mt-SSU proteins. Although mt-LSU levels were normal, the 55S monosome was virtually absent, and protein synthesis was severely impaired [138].
uS2m.
Bi-allelic mutations in uS2m have been described in two unrelated subjects suffering from sensorineural hearing loss, developmental delays, hypoglycemia, lactic acidemia, and combined OXPHOS complex deficiencies [141]. Exome sequencing of one subject revealed two heterozygous mutations, c.328C>T (p.R110C) and c.340G>A (p.D114N), while the second subject carried the homozygous mutation c.413G>A (p.R138H) [141]. The mutations at highly conserved amino acids R110, R114, and R138 lead to decreased steady-state levels of uS2m and other mt-SSU proteins, including the structurally adjacent protein bS1m, in patient fibroblasts [141]. Furthermore, 12S rRNA levels were significantly decreased, whereas the 16S rRNA and mt-LSU proteins remained unaffected [141]. Of particular note in one subject was the presence of a previously identified seven-protein assembly intermediate, containing several early assembly proteins (bS16m, uS17m, uS11m, mS22, mS26, mS27, and mS34) [141,152]. Additionally, mitochondrial translation was drastically decreased, resulting in a reduction of fully assembled OXPHOS complexes I and IV [141]. Kinetic assembly data has placated uS2m as an early assembly protein, and the above-described mutations destabilize uS2m, preventing SSU assembly and leading to deficits in mitochondrial translation and OXPHOS complex assembly [21].
mS23.
One patient has been reported with the homozygous mS23 mutation c.119C>G (p.P40R), leading to liver disease and combined OXPHOS deficiency [153]. The patient’s parents were non-consanguineous and both heterozygous for the mutation [153]. In patient’s cells, 12S rRNA levels were decreased, indicative of defective mitoribosome assembly and translation [153]. The structure of mS23 in the fully assembled mitoribosome shows that the P40 residue is most likely interacting with Y234 of uS2m [13]. The mutation may disrupt the incorporation of mS23 and neighboring proteins, resulting in instability of the 12S rRNA. Alternatively, the mitoribosome may fully assemble, although the mutation in mS23 may distort the structure or conformation of nearby mRNA entry tunnel, causing protein synthesis defects.
5.2.1.4. mt-SSU early assembly independent binders
From the three early assembly proteins proposed to bind as single units, uS11m, uS17m, and uS12m, disease-causing mutations have been found in uS11m.
uS11m:
uS11m joins the mt-SSU body early during assembly and interacts with the central domain of the 12S rRNA and neighboring proteins uS7m, bS18m, bS21m, and mS37 [13,15,21]. A meta-analysis conducted on a dataset of genes differentially expressed in ankylosing spondylitis revealed MRPS11 (coding for uS11m) to be the top downregulated gene [154], although genes coding for mL40 (MRPL40) and bL27m (MRPL27) as well as for several respiratory chain subunits were also significantly downregulated [154]. Taken together, this suggests a general role for mitochondrial translation and OXPHOS in the pathogenesis of ankylosing spondylitis.
5.2.1.5. mt-SSU intermediate/late assembly independent binders
Proteins bS6m and mS38 were identified as independent proteins with variable association with the late mS26-containing cluster. Pathogenic mutations have been found in bS6m, which joins the mt-SSU during the intermediate stage of assembly.
bS6m.
Joining the mt-SSU, bS6m forms mitochondria-specific intersubunit bridge mB2 with uL2m of the mt-LSU [13,15,21]. As a result of a genome-wide association study, the polymorphism rs9982601 (C>T) in the non-coding region between SLC5A3/MRPS6 and KCNE2 genes was found to be an increased risk factor for early-onset myocardial infarction [155,156]. This observation further supports the role of mitochondrial translation in cardiac function. Of particular note, bS6m is one of the six zinc-binding proteins in the mammalian mitoribosome, coordinating a single zinc ion with bS18m [13,15,65]. Interestingly, the bS18m gene has been identified as a suppressor of a pathogenic variant in the MT-ND1 gene that leads to epileptic encephalopathy [157].
5.2.1.6. mt-SSU late assembly module [uS15m, mS25, mS26]
The [uS15m, mS25, mS26] module binds near the early bS16m-mS22 cluster and localizes to the interface with the mt-LSU. Incorporation of this cluster has been suggested to be dependent on the presence of the early assembly module containing the mitochondrial disease proteins bS16m and mS22 [21]. Pathogenic mutations have been identified in mS25.
mS25.
A homozygous mutation in mS25, c.215C>T (p.P72L) was identified in a 25-year-old male suffering from mitochondrial encephalomyopathy with dyskinetic cerebral palsy and partial agenesis of the corpus callosum [74]. Studies with the patient’s fibroblasts showed that the steady-state levels of the 12S rRNA, protein mS25 and its binding partners uS17m and mS22, as well as the head module protein mS29, were severely decreased [74]. On the contrary, mt-LSU components were equal or slightly elevated compared to control cells [74]. Fully assembled mt-SSU or 55S monosome were severely reduced, and the overall protein synthesis was decreased, which resulted in multiple OXPHOS complex deficiencies [74]. The P72L mutation in mS25 likely disrupts its interaction with uS17m and compromises the integrity of the whole mt-SSU [13,74]. However, it allows enough residual protein synthesis activity to support life until early adulthood.
5.2.1.7. mt-SSU late assembly module [uS14m, uS10m, uS3m, mS33]
The [uS14m, uS10m, uS3m, mS33] module binds to the mt-SSU head region at the subunit interface, in association with the early uS7m-mS29 group. Disease-causing mutations have been identified in uS14m.
uS14m.
uS14m joins the head region of the 28S mt-SSU during the late stages of assembly at the subunit interface, coordinating substantial 12S rRNA contacts and direct interactions with uS3m of the mRNA entry tunnel, as well as uS10m, mS31, and mS33 [13,15,21]. A single patient harboring the homozygous mutation c.322C>T (p.R108C) in the uS14m gene was born to parents who were both heterozygous for the mutation [158]. Upon birth, the patient showed increased levels of lactate and was diagnosed with moderate hypertrophic cardiomyopathy [158]. By four months of age, Wolff-Parkinson White syndrome was revealed in the patient along with muscle hypotonia, developmental retardation, hyperlactatemia, and failure to thrive [158]. After two years of age, however, the patient’s conditions became less severe, and the hypertrophic cardiomyopathy remains stable [158]. Patient fibroblasts had a combined OXPHOS complex and enzymatic deficiency [158]. As expected for a late assembly protein, the uS14m R108C mutation did not affect the steady-state levels of the 12S rRNA but neither of any other mt-SSU protein, full subunit, or monosome [158]. Therefore, the uS14m mutation does not impair mitoribosome assembly, although it attenuates mitochondrial translation [158]. In-silico modeling of this mutation in the cryo-EM structure of the human mitoribosome initiation complex containing initiation factor mtIF2, tRNA, and mRNA showed that the R108C mutation was unlikely to be involved in coordinating tRNA binding due to distance restrictions [45,158]. Instead, however, R108 is responsible for bridging the 12S rRNA to the head region of the small subunit through coordinating binding with uS10m and A579 of the 12S rRNA [13,15,45,158]. The close proximity of this mutation to the mRNA entry channel may cause distortion of the mRNA tunnel and decoding center, leading to impairment in translation initiation and elongation.
5.2.2. Large subunit proteins
5.2.2.1. mt-LSU early assembly proteins
Pathogenic mutations have been identified in three mt-LSU proteins that incorporate during the early phase of assembly: uL3m, mL44 and bL12m.
uL3m.
uL3m forms extensive interactions with the 16S rRNA and makes contacts with neighboring proteins bL17m, bL19m, bL32m, and mL39 [13–15,21]. Four siblings, all compound heterozygous for the mutation c.950C>G (p.P317R) and a large-scale deletion at the MRPL3 (uL3m) locus, suffered from hypertrophic cardiomyopathy, failure to thrive, and liver dysfunction [159]. Biochemical analysis in patient fibroblasts revealed a severe decrease in steady-state levels of 16S rRNA, and proteins uL3m and bL12m, resulting in attenuated protein synthesis and combined OXPHOS complex deficiency [159]. In the human mitoribosome cryo-EM structure of uL3m, the P317R mutation occurs in a coiled region containing many disordered loops, which may destabilize the protein and subsequent mt-LSU assembly [13,15].
mL44.
mt-LSU protein mL44 also forms part of the largely restructured polypeptide exit tunnel in mammalian mitoribosomes [13–15]. Pathogenic mutations in mL44 have been identified in two families. A homozygous c.467T>G (p.L156R) mutation in mL44 was described in two siblings presenting with infantile-onset hypertrophic cardiomyopathy, liver steatosis, and combined OXPHOS deficiency of CI and CIV in heart and muscle tissue [160]. Interestingly, the defect was milder in patient fibroblasts, in which although levels of 16S rRNA and proteins mL44 and uL13m were decreased, there was only a mild attenuation of assembled mt-LSU levels, monosomes, and translation capacity [160]. Peculiarly, little to no effect on mitochondrial translation was seen in patient fibroblasts [160]. The mL44 Leu156 residue is highly conserved among vertebrates, residing in an alpha-helical RNase III-like domain [160]. It forms numerous interactions with surrounding residues and likely functions to ensure protein folding and stability [12,160]. A more recent case described a patient compound heterozygous for the same L156R mutation and a novel missense variant c.233G>A (p.R78Q) [161]. In addition to the infantile cardiomyopathy reported for the homozygous L156R mutant patient, the compound heterozygous patient presented with Leigh-like lesions on brain MRI, pigmentary retinopathy, hemiplegic migraine, renal insufficiency, and hepatopathy. In the structure of the human mitoribosome, R78 is at the interface between mL44 and mL43, and likely forms several interactions with a β-sheet of mL43. The additional neuro-ophthalmological and hepatic clinical features in the compound heterozygous patient suggests these abnormalities to be specific to the R78Q mutation.
bL12m.
Orthologous to mammalian bL12m, the bacteria L7/L12 stalk is a multimer extending to uL10m and plays essential roles in recruiting GTPase translation factors to the ribosome during translation [162,163]. In the mitoribosome, the L7/L12 stalk is largely remodeled, exhibiting increased stability due to stabilization of uL10m by mL53 and the zinc-binding protein mL66 [13,14]. Recently this region was further resolved to show six copies of bL12m N-terminal domains that bridge interactions with uL10m and mL53 [15]. Three siblings homozygous for the bL12m mutation c.542C>T (p.A181V), born to consanguineous parents, presented with neurological deterioration, growth retardation, and combined OXPHOS deficiency in muscle and liver tissue [164]. Analysis in patient fibroblasts indicated a general decrease in mitochondrial translation, and attenuated OXPHOS complexes I and CIV [164]. Although the steady-state levels of bL12m and other mt-LSU proteins uL3m and mL62 were mildly reduced, assembled mt-LSU and monosomes were severely decreased, without effect on mt-SSU levels [164]. In addition to destabilizing the mt-LSU, the bL12m A181V mutation could also affect the recruitment of translation factors.
5.2.2.2. mt-LSU intermediate/late assembly proteins
uL24m.
uL24m is incorporated as part of a module [mL41, uL23m, uL24m, uL29m, and bL34m] that joins the base of the mt-LSU to contribute to the formation of the polypeptide exit tunnel [13–15,21]. An N-terminal extension and conformational change of uL24m in the mitoribosome compensates for lost helices in the 16S rRNA [14,15]. A single uL24m patient has been reported carrying the homozygous missense mutation c.272C>T (p.L91P). The patient suffered from cerebellar atrophy, choreoathetosis of the limbs and face, intellectual disability, associated with combined OPXHOS deficiency in muscle [140]. Analyses of patient fibroblasts revealed a mild translation defect [140]. Furthermore, decreased steady-state levels of uL24m and other mt-LSU proteins such as mL41 and mL45, drastic decrease in assembled mt-LSU, and accumulation of an mt-LSU assembly intermediate containing at least uL24m, mL41, and mL45 [140]. In the structure of the mitoribosome Leu91 is positioned just upstream of a β-hairpin that coordinates interactions with mL45 and the 16S rRNA [13–15]. A mutation to proline at this position is predicted to induce a conformational change in the downstream β-hairpin secondary structure, which would likely disturb uL24m stability and weaken its interactions [140]. Modeling in zebrafish showed that knockdown of uL24m produces decreased muscle response, mitochondrial dysfunction, and a mild cardiac defect [140]. Notably, complementation with L91P mutant uL24m rescued the heart defect, but locomotion response remained impaired, supporting an effect of this mutation in motor behavior [140].
5.3. Mutations in assembly factors
Disease-causing mutations have so far been found in four rRNA processing, stabilization, and maturation factors (ELAC2, MRPP3, FASTKD2, MRM2, TFB1M), five mitoribosome assembly chaperones (ERAL1, p32, GTPBP5, GTPBP10, DHX30), and one mitochondrial protease (CLPP). All available information about mutations in assembly factors is schematized in Table 2.
Table 2. Mutations in mitoribosome assembly factors associated with human mitochondrial disorders.
MELAS, mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. VACTERL, vertebral anomalies, anal atresia, cardiovascular anomalies, trachea-esophageal fistula, esophageal atresia, renal or radial anomalies, or limb defects. OXPHOS, oxidative phosphorylation. SNP, single nucleotide polymorphism. PEO, progressive external ophtalmoplegia.
| Gene | Mutation | Clinical Manifestations | Ref. |
|---|---|---|---|
| rRNA processing and stability defects | |||
| TRNT10C | Compound heterozygous c.542G>T, (p.R181L) c.814A>G, (p.T272A) Homozygous c.542G>T, (p.R181L) |
-Lactic acidosis -Hypotonia -Feeding problems -Deafness -Defect in RNA processing -Multiple OXPHOS deficiency |
[173] |
| HSD17B10 | Hemizygous X:g.53458504T>C, (p.K212E) |
-Lactic acidosis -Intractable epilepsy -Global developmental delay -Static encephalopathy -Optic atrophy and blindness -Global tRNA processing and maturation |
[178] |
| ELAC2 (RNAse Z) | Compound heterozygous c.631C>T, (p.Arg211*) c.1559C>T, (p.Thr520Ile) Homozygous c.460T>C, (p.Phe154Leu) c.1267C>T, (p.Leu423Phe) |
-Hypertrophic cardiomyopathy -Lactic acidosis -Complex I deficiency |
[165] |
| Homozygous and heterozygous mutations | Early-onset hypertrophic or dilatated cardiomyopathy | [168] | |
| FASTKD2 | Homozygous c.1246C>T, (p.Arg416*) |
-Early-onset developmental delay -Mitochondria encephalomyopathy -Isolated complex IV deficiency in muscle |
[180] |
| Compound heterozygous c.613C > T, (p.Arg205Stop) c.764 T >C, (p.Leu255Pro) |
-MELAS-like syndrome | [181] | |
| Compound heterozygous c.868C>T, (p.R290*) c.1859delT, (p.S621Lfs*14) |
-MELAS-like syndrome -Cerebral atrophy |
[182] | |
| Homozygous: c.808_809ins, (p.L270fs*11) c.868C>T, (p.R290*) |
-Early-onset mitochondrial encephalomyopathy -Lactic acidosis -Hypertrophic cardiomyopathy -Multiple OXPHOS deficiency |
||
| rRNA maturation defects | |||
| MRM2 | Homozygous: c.567G>A, (p.Gly189Arg) |
-MELAS | [183] |
| TFB1M | In combination with12S rRNA mutation A1555G | -Hearing loss | [127] |
| SNP (rs950994) | -Risk factor in type 2 diabetes | [185] [186] | |
| Assembly chaperoning defects | |||
| ERAL1 | Homozygous c.707A>T, (p.Asn236Ile) |
-Perrault syndrome | [189] |
| CLPP | Homozygous and heterozygous mutations | -Perrault syndrome | [190] [191] |
| C1QBP/p32 | Compound heterozygous c.557G>C, (p.Cys186Ser) c.612C>G, (p.Phe204Leu) c.739G>T, (p.Gly247Trp) c.824T>C, (p.Leu275Pro) Homozygous c.823C>T, (p.Leu275Phe) c.562_564delTAT, (p.Tyr188del) |
-Cardiac manifestation -PEO -Combined CI, CIII and CIV OXPHOS deficiency |
[193] |
| DHX30 | c.1478G>A, (p.Arg493His) c.1685A>G, (p.His562Arg) c.2342G>A, (p.Gly781Asp) c.2344C>T, (p.Arg782Trp) c.2353C>T, (p.Arg785Cys) c.2354G>A, (p.Arg785His) |
-Neurodevelopmental disorder -Accumulation of cytosolic stress RNA granule |
[196] |
| GTPBP5 | 0.7 Mb deletion in 20q13.33 | -VACTERL | [196] |
5.3.1. rRNA processing and stability defects
Transcription of the mtDNA generates large polycistronic transcripts that contain rRNAs and mRNAs in most cases punctuated at each end by the mitochondrial tRNAs, which are conventionally cleaved by the RNase P-complex at 5’ end and the RNase Z activity of ELAC2 at the 3’ end. The activity of these enzymes on the 16SrRNA-tRNAVal-12SrRNA-tRNAPhe precursor controls mitoribosome biogenesis [76]. Pathogenic mutations have been identified in ELAC2 and the RNAse P components MRPP1, MRPP2, and MRPP3. They have also been found in the kinase FASTKD2 that binds and stabilizes the 16S rRNA, and other RNAs such as the ND6 mRNAs [85,109], although its precise role in mitoribosome biogenesis remains poorly understood.
ELAC2.
Mutations in ELAC2 were identified by exome sequencing in five individuals from three independent families with infantile hypertrophic cardiomyopathy, lactic acidosis, and complex I deficiency [165]. In one family with unrelated parents, the two patients were compound heterozygous for p.R211* and T520I mutations. The other two families were of consanguineous parents. In one, two children died, and the proband was found to carry a homozygous p.F154I mutation. In the other, two children were found homozygous for a p.L423F variant [165]. The mutations resulted in the accumulation of unprocessed mitochondrial transcripts in muscle and fibroblasts. Analysis of fibroblasts from the R211*/T520I patient showed that although mitochondrial translation was impaired, mature mitochondrial RNA levels were not decreased [165]. It has been suggested that the accumulation of unprocessed transcripts that are not degraded by the RNA degradosome could interfere with mitochondrial protein synthesis [165]. Considering that hierarchical RNA processing is required for mitoribosome biogenesis, one could expect that the ELAC2 mutations could affect this process. However, previous ELAC2 silencing in HeLa cells did not result in mitochondrial translation defects [166], perhaps due to the residual protein being expressed. It could be important to consider that alternative translation initiation of ELAC2 drives the synthesis of two RNase Z isoforms with localizations in mitochondria and the nucleus [167], since the function of nuclear ELAC2, potentially altered in patients, could contribute to their clinical manifestations. A subsequent study in 13 patients identified 16 novel ELAC2 mutations associated with early-onset hypertrophic or dilatated cardiomyopathy [168]. Three of the patients carried a p.R781H variant associated with prostate cancer susceptibility [169] that impairs the mitochondrial enzymatic activity of ELAC2 [168]. However, it has been reported that nuclear ELAC2 becomes up-regulated during malignant transformation in a subset of prostate cancers [170]. Therefore, the links between ELAC2 mutations, nuclear and mitochondrial RNA metabolisms, mitochondrial cardiomyopathy, and tumorigenesis remain to be fully established.
MRPP1.
MRPP1, MRPP2, and MRPP3 form the mitochondrial RNase P complex that cleaves the 5’ ends of mt-tRNAs from polycistronic precursor transcripts [171]. MRPP1 and MRPP2 additionally form a stable complex with m(1)R9 methyltransferase activity on mitochondrial tRNAs, which is essential for their correct folding [172]. Recessive mutations in TRNT10C, encoding the mitochondrial RNase P protein 1 (MRPP1), have been reported to cause defects in mitochondrial RNA processing and multiple respiratory chain deficiencies in two unrelated newborns suffering from lactic acidosis, hypotonia, feeding problems, and deafness who died at 5 months [173]. One of the subjects was compound heterozygous for variants c.542G>T (p.R181L) and c.814A>G (p.T272A), and the second was homozygous for a c.542G>T (p.R181L) variant. These mutations, analyzed in patients’ fibroblasts, affected mitochondrial RNA processing and protein synthesis but did not alter tRNA methylation [173].
MRPP2.
MRPP2 is encoded in HSD17B10, a gene located on the X-chromosome. It was initially called SDR5C1, as a member of the NAD+/NADP-dependent short-chain dehydrogenase/reductase (SDR) family [174]. SDR5C1 deficiency causes a mitochondrial disorder known as ‘HSD10 disease’ [175] that was traditionally diagnosed based on the detection of elevated metabolites from isoleucine breakdown [176]. However, the clinical phenotypes, including progressive neurodegeneration and cardiomyopathy in early childhood [175], differ from classical α-ketothiolase (AKT) deficiencies in which the same metabolites accumulate [177].
HSD17B10 missense mutations are associated with heterogeneous clinical manifestations of different severity and age of onset [175], including a neonatal form (variants p.D86G, p.R226Q, and p.N247S), an infantile form (variants p.L122V, p.R130C, and p.P210S), and a later-onset form (variant p.E249Q). The report of a 9-year-old boy with a novel HSD17B10 p.K212E mutation suffering from chronic lactic acidemia, intractable epilepsy, global developmental delay, static encephalopathy, optic atrophy, and blindness was crucial to linking the “HSD10 disease” to mitochondrial RNAse P [178]. In vitro assays using recombinant purified proteins and reconstituted RNAse P complex showed that the p.K212E mutation only affects the dehydrogenase activity of SDR5C1 mildly but attenuates its ability to form a complex with MRPP1, and causes a global deficiency in mitochondrial tRNA processing and maturation [178].
MRPP3.
Although there are no published reports of disease-causing mutations in KIAA0391, coding for MRPP3, three likely pathogenic variants have been deposited in the ClinVar database. Heterozygous compound mutations S400* and R445Q were associated with persistent lactic acidosis, global developmental delay, hypertonia, diffuse white matter abnormalities, microcephaly, and feeding difficulties. A homozygous A485V substitution was associated with Perrault Syndrome, principally characterized by sensorineural hearing loss. The two reported missense mutations localize in the metallonuclease domain of MRPP3, but only R445 is in proximity to catalytic residues. In the MRPP3 crystal structure (PDB ID: 4xgl), R445 is positioned to form a salt bridge with D479, a residue crucial to maintain the enzymatic activity. Although MRPP3 acts as endonuclease only in the trimeric complex with MRPP1 and MRPP2 that could be necessary to induce a major structural rearrangement in the MRPP3 active site [179], the mutations found in patients are unlikely to disrupt the formation of the complex but rather affect its enzymatic activity.
FASTKD2.
Mutations in FASTKD2 result in heterogeneous clinical manifestations. A loss of function homozygous p.R416* mutation in FASTKD2 was first identified in two siblings suffering from early-onset developmental delay and mitochondrial encephalomyopathy with isolated complex IV deficiency in muscle [180]. Although the patient fibroblasts did not display any respiratory deficiency, modeling a FASTKD2-KO in HEK293T cells resulted in defective mitochondrial translation and combined OXPHOS complex I, III, and IV deficiency [109]. In another family, compound heterozygous mutant variants p.R205* and p. L255P were identified in a patient suffering from milder symptoms in the form of late (adult)-onset MELAS like-syndrome [181]. The Leu255 residue is conserved but far from the catalytic or RNA binding domains, which suggests that the protein retains some function and explains the milder symptoms in the patient. Recently, three new pathological mutations, homozygous p.L270fs, homozygous p.R290*, and compound heterozygous p.R290*/S621Lfs, were identified in patients suffering from early-onset mitochondrial encephalomyopathy presenting with lactic acidosis, hypertrophic cardiomyopathy, or MELAS like syndrome with cerebral atrophy, respectively [182]. Patient-derived immortalized lymphocytes carrying FASTKD2 L270fs or R290* homozygous mutations exhibited 16S rRNA levels decreased by 30% and 54%, and multiple OXPHOS deficiency [182], although mitochondrial protein synthesis was not examined.
5.3.2. rRNA maturation defects
Modifications of rRNA are essential for ribosome biogenesis and function. Ten modified sites have been identified in the mammalian mitochondrial rRNAs whose formation is catalyzed by dedicated enzymes. Pathogenic mutations have been identified in two of them, MRM2 and TFB1M.
MRM2.
MRM2 is the methyltransferase responsible for 2’-O-methyl modification at position U1369 in the 16S rRNA [77,78]. A homozygous G189R mutation in this enzyme has been reported in a patient displaying a MELAS-like clinical syndrome [183]. At eight months of age, the patient displayed development delay and severe movement disorder, and finally died at the age of 7-years after complications caused by liver failure and recurrent epilepsy episodes [183]. Patient fibroblasts had a mild respiratory deficiency, although the extent of 2’-OH modification was not affected. Heterologous complementation studies in an S. cerevisiae mrm2 mutant strain, however, confirmed that contrary to the human MRM2 wild-type version, the p.G189R variant was not able to complement the yeast mutant [183]. In the human MRM2 structure (PDB ID: 2NYU), G189 does not lie near the active site involved in S-adenosyl-methionine binding and catalysis. However, structural analyses have predicted that the mutation could affect the global structure of the protein diminishing its catalytic function [183].
TFB1M.
TFB1M is a dimethyltransferase that methylates two adjacent adenine residues on the conserved stem-loop closed to the 3′ end of mitochondrial 12S rRNA [98], which is necessary for in vivo stability of the mt-SSU in a mouse model [184]. TFB1M has been identified as a modifier gene in two kinds of disorders. First, TFB1M is a modifier for hearing loss associated with the mitochondrial A1555G mutation [127], located in a loop adjacent to two adenine residues targeted by TFB1M. It has been proposed that, in combination with the A1555G mutation, TFB1M variants may cause 12S rRNA hypermethylation leading to aberrant mitochondrial biogenesis [185], activating AMP kinase in a reactive oxygen species-dependent manner, thereby up-regulating E2F1 pro-apoptotic signaling that damages inner hair cells [186]. Second, a common variant of TFB1M (rs950994), which reduced the levels of TFB1M mRNA by 17% per allele, was identified as a risk factor for the development of type 2 diabetes (T2D) and to impair insulin secretion (29% per allele) [187]. Studies in β-cell-specific TFB1M KO mice, which gradually developed diabetes, showed that TFB1M deficiency in β-cells causes mitochondrial dysfunction and, subsequently, diabetes due to a combined loss of β-cell function and cell death via apoptosis and necrosis [188].
5.3.3. Assembly chaperoning defects
Among the mitoribosome assembly chaperones, pathogenic mutations have been identified in three mt-SSU factors, the GTPase ERAL1, its regulatory peptidase CLPP, and its interactor p32, and two mt-LSU factors, the helicase DHX30, and the GTPase GTPBP5.
ERAL1.
A homozygous ERAL1 N236I variant, affecting the GTP binding pocket, was identified in three unrelated females suffering from Perrault syndrome (PS), mainly characterized by sensorineural hearing loss, ovarian dysgenesis in female, and, in a few cases, also by neurological impairment. Analysis of patient fibroblasts showed a 30–40% reduction of assembled mt-SSU proteins and decreased 12S rRNA levels, whereas the mt-LSU levels were as in controls. The ribosome accumulation defect resulted in impaired cellular respiration [189]. Modelling ERAL1 deficiency in C. elegans showed that it compromises fecundity by reducing the number of laid eggs, thus resembling the human clinical manifestation [189].
CLPP.
ERAL1 has been identified as a substrate of the peptidase CLPP, preventing ERAL1 levels from becoming elevated and impairing mitoribosome biogenesis [99]. Several mutations in CLPP have been identified in patients with Perrault syndrome type 3 [190], which can be arranged into two groups depending on the mutated CLPP residues. Some mutations cluster near the docking site for the cognate unfoldase CLPX (i.e., T145P or C147S), and others are adjacent to the peptidase active site (i.e., Y229D) [191]. In vitro assays have shown that each of these CLPP mutant proteins mutations exhibits a specific defect that ranges from protein stability to peptidase activity, or their binding to CLPX [191]. This variability may explain the clinical heterogeneity of the disease, which presents with sensorineural hearing loss in both genders and, in severe cases, additional symptoms such as ataxia, neuropathies, and intellectual disability [192]. Biochemically, defective CLPP functions will result in increased ERAL1 levels, aberrant mitoribosome biogenesis, and mitochondrial OXPHOS function as a common cause of the disease.
C1QBP/p32.
The p32 protein is mostly localized in the mitochondrial matrix and works as a multifunctional chaperone involved in the formation of mitoribosome complexes [193], by interacting with ERAL1 and YBEY [100]. p32 also participates in the regulation of mitochondrial morphology and dynamics by regulating the OMA1-dependent proteolytic processing of OPA1 [194], which may also affect mtDNA maintenance. Studies in mouse embryonic fibroblasts KO for C1QBP have shown that the two mitoribosome subunits accumulate and sediment normally in sucrose gradients but the amount of 55S translating ribosomes, and therefore the mitochondrial translation rate, is significantly reduced in the absence of p32 [193]. On the contrary, no defects were measured in mtDNA levels [193]. Notably, C1QBP/p32 has been found to bind all mitochondrial mRNAs in HeLa cells, presumably to promote their translation [193]. Biallelic mutations in C1QBP have been found in four unrelated patients suffering from cardiac manifestations, progressive external ophtalmoplegia (PEO), and presenting a combined CI, CIII, and CIV OXPHOS deficiency [195]. The protein mutations were compound heterozygous p.C186S/F204L and p.G247W/L275P, and homozygous p.L275F and p.Y188*, all affecting residues located in important domains of the p32 homotrimer doughnut-shaped structure [195]. Although all p32 protein mutant variants are unstable, the age at onset ranged from childhood to adulthood. As a potential contributing factor, multiple mtDNA deletions were identified in muscle tissues from patients with adulthood onset [195]. Mitoribosome assembly or function was not characterized in fibroblasts from these patients.
DHX30.
Six missense homozygous mutations in DHX30 have been identified in 12 independent patients suffering from severe global developmental delay and intellectual disability [196]. Four mutations were recurrent and found each in three (p.R782W, and p.R785C) or two patients (p.R493H, and p.G781D), and two were identified in single patients (p.R785H, and p.H562R). In the protein structure, all residues affected locate within highly conserved helicase motifs, and the pathogenic changes impair either ATPase activity (H562R, G781D, R782W, R785C, R785H) and or RNA recognition (R493H) in recombinant proteins expressed in transiently transfected HEK293T cells and purified by immunoprecipitation [196]. In addition to the potential effect on mitochondrial protein synthesis, which has not been evaluated, DHX30 mutant variants were shown to additionally interfere with global translation by promoting the formation of stress granules [196], which have been frequently associated with neurodevelopmental disorders [197].
GTPBP5.
High-density microarrays on a cohort of 20 patients presenting with VACTERL (vertebral anomalies, anal atresia, cardiovascular anomalies, trachea-esophageal fistula, esophageal atresia, renal or radial anomalies, or limb defects) association of clinical manifestations, identified, in one of the patients, a 0.7 Mb deletion of chromosome 20q13.33 that included the GTPBP5 locus. The patient has tracheo-esophageal fistula with cardiac defects and genitourinary anomalies [198]. VACTERL has been previously associated with mitochondrial defects [199], which suggested that GTPBP5 could also be a primary contributor to the disease
6. CONCLUSIONS AND PERSPECTIVES
The field of ribosome biogenesis and function is rapidly progressing due to technical advances in mass spectrometry, cryo-electron microscopy, and gene editing approaches to modify human cell lines. However, the substrates and precise mechanisms of action for the many auxiliary assembly factors remain, in most cases, poorly understood. Although it is not expected that mitoribosome biogenesis will require some 200 factors as cytoplasmic ribosome biogenesis does (only ~25 are needed in bacteria), the large number of uncharacterized proteins found in the mitoribosome-associated proteome anticipate the identification of new ones. SILAC-mass spectrometry approaches have provided a draft of the protein incorporation map during mitoribosome biogenesis. The assembly kinetics analyses reported need to be extended to multiple intermediate time-points to refine the pathway. Furthermore, investigations on whether and how mitoribosome biogenesis is modified or adapts to changing environmental conditions such as oxygen levels or oxidative stress, relevant to human disorders, warrant future research efforts. Importantly, the tools are now available to ultimately decipher the sophisticated details of mitoribosome biogenesis.
By synthesizing essential OXPHOS complex proteins, mitoribosomes are essential to sustain aerobic life. Although the biomedical relevance of the mitoribosome and mitochondrial protein synthesis has been appreciated for decades, the accessible and fast full-genome sequencing approaches currently at hand have made a more manageable task the identification of pathogenic alleles in mitoribosome components and assembly factors in patients suffering from mitochondrial disorders. Fundamental research helps to understand the pathogenic mechanisms of known factors and uncovers new factors as candidates when screening genetic causes of mitochondrial disorders associated with mitochondrial translation defects and multiple OXPHOS enzyme deficiencies. In turn, mitochondrial medicine provides subjects carrying mutations for structure-function relationship studies, contributing to shaping our understanding of mitoribosome biogenesis.
Figure 5. Defective mitochondrial ribosome assembly and human disease.
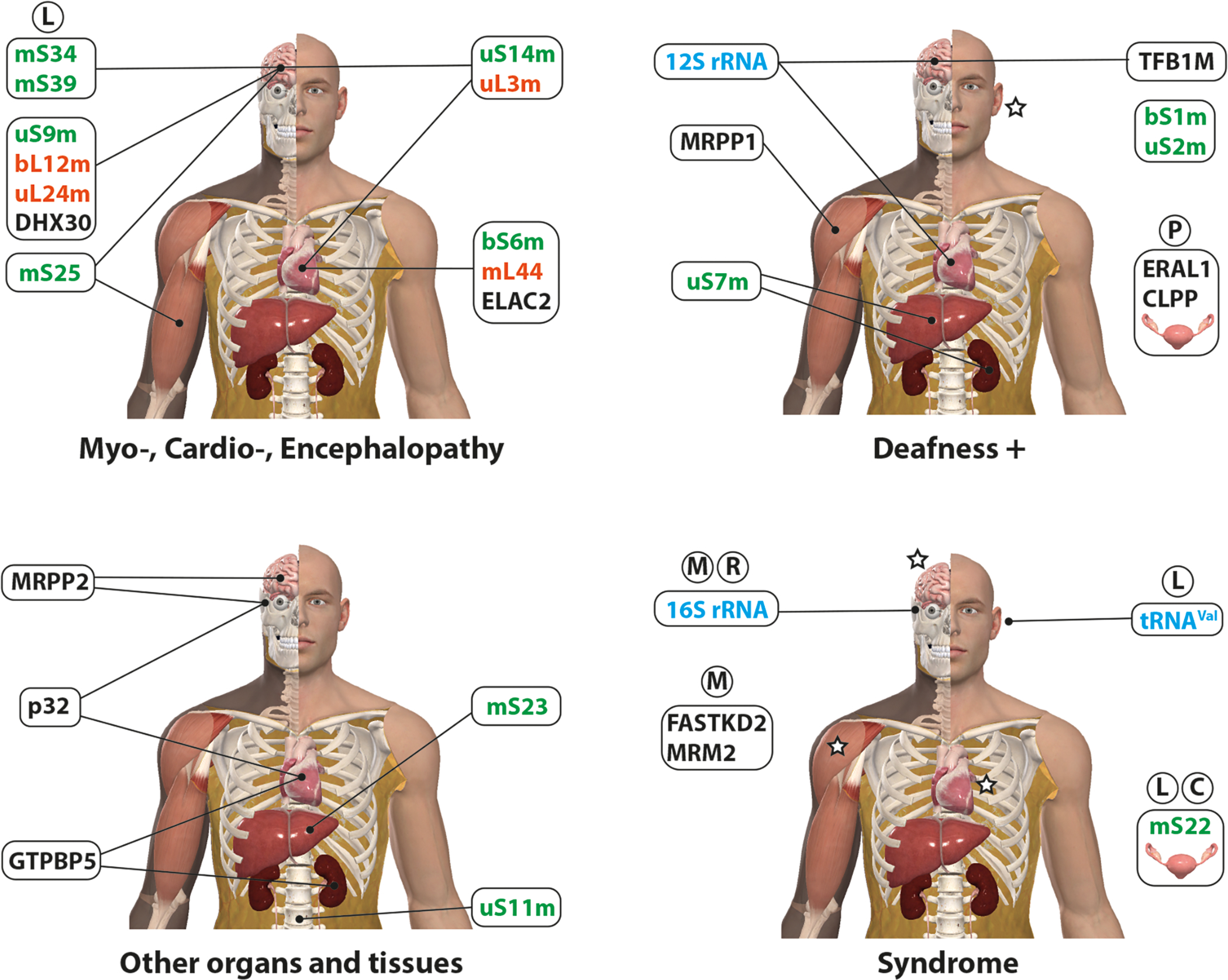
Schematic representation of organs and tissues affected by mutations in mitoribosome components: assembly factors (black), tRNA or rRNA (light blue), mt-SSU proteins (green) and mt-LSU proteins (orange). For clarity, four human body models, generated by using the Complete Anatomy software, are presented.= indicate organs or tissues commonly affected by all the mitoribosome components in the same group, C = Cornelia de Lange syndrome, L = Leigh syndrome, M = MELAS, P= Perrault Syndrome, R = Rett Syndrome.
ACKNOWLEDGEMENTS
Our research is supported by a NIGMS-MIRA award [R35GM118141 to A.B.], and a Muscular Dystrophy Association Research Grant [MDA-381828 to A.B.].
Footnotes
Conflict of interest statement.
None declared.
REFERENCES
- [1].McLean JR, Cohn GL, Brandt IK and Simpson MV (1958). Incorporation of labeled amino acids into the protein of muscle and liver mitochondria. J. Biol. Chem. 233, 657–63. [PubMed] [Google Scholar]
- [2].Rendi R (1959). On the occurrence of intramitochondrial ribonucleoprotein particles. Exp. Cell Res. 17, 585–7. [DOI] [PubMed] [Google Scholar]
- [3].Kalf GF (1963). The incorporation of leucine-1-C-14 into the protein of rat heart sarcosomes: an investigation of optimal conditions. Arch. Biochem. Biophys. 101, 350–59. [DOI] [PubMed] [Google Scholar]
- [4].Swift H (1965). Nucleic Acids of Mitochondria and Chloroplasts. The American Naturalist 99, 201–214. [Google Scholar]
- [5].Küntzel H and Noll H (1967). Mitochondrial and cytoplasmic polysomes from Neurospora crassa. Nature 215, 1340–5. [DOI] [PubMed] [Google Scholar]
- [6].O’Brien TW and Kalf GF (1967). Ribosomes from rat liver mitochondria. I. Isolation procedure and contamination studies. J. Biol. Chem. 242, 2172–9. [PubMed] [Google Scholar]
- [7].O’Brien TW and Kalf GF (1967). Ribosomes from rat liver mitochondira. II. Partial characterization. J. Biol. Chem. 242, 2180–5. [PubMed] [Google Scholar]
- [8].Amunts A et al. (2014). Structure of the yeast mitochondrial large ribosomal subunit. Science 343, 1485–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kühlbrandt W (2014). Biochemistry. The resolution revolution. Science 343, 1443–4. [DOI] [PubMed] [Google Scholar]
- [10].Desai N, Brown A, Amunts A and Ramakrishnan V (2017). The structure of the yeast mitochondrial ribosome. Science 355, 528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Greber BJ, Bieri P, Leibundgut M, Leitner A, Aebersold R, Boehringer D and Ban N (2015). Ribosome. The complete structure of the 55S mammalian mitochondrial ribosome. Science 348, 303–8. [DOI] [PubMed] [Google Scholar]
- [12].Greber BJ, Boehringer D, Leibundgut M, Bieri P, Leitner A, Schmitz N, Aebersold R and Ban N (2014). The complete structure of the large subunit of the mammalian mitochondrial ribosome. Nature 515, 283–6. [DOI] [PubMed] [Google Scholar]
- [13].Amunts A, Brown A, Toots J, Scheres SHW and Ramakrishnan V (2015). The structure of the human mitochondrial ribosome. Science 348, 95–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Brown A, Amunts A, Bai XC, Sugimoto Y, Edwards PC, Murshudov G, Scheres SH and Ramakrishnan V (2014). Structure of the large ribosomal subunit from human mitochondria. Science 346, 718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Aibara S, Singh V, Modelska A and Amunts A (2020). Structural basis of mitochondrial translation. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Anderson S et al. (1981). Sequence and organization of the human mitochondrial genome. Nature 290, 457–65. [DOI] [PubMed] [Google Scholar]
- [17].Bertgen L, Mühlhaus T and Herrmann JM (2020). Clingy genes: Why were genes for ribosomal proteins retained in many mitochondrial genomes? Biochim. Biophys. Acta Bioenerg. 1861, 148275. [DOI] [PubMed] [Google Scholar]
- [18].De Silva D, Fontanesi F and Barrientos A (2013). The DEAD-Box protein Mrh4 functions in the assembly of the mitochondrial large ribosomal subunit. Cell Metab. 18, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Maiti P, Kim HJ, Tu YT and Barrientos A (2018). Human GTPBP10 is required for mitoribosome maturation. Nucleic Acids Res. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zeng R, Smith E and Barrientos A (2018). Yeast mitoribosome large subunit assembly proceeds by hierarchical incorporation of protein clusters and modules on the inner membrane. Cell Metab. 27, 645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bogenhagen DF, Ostermeyer-Fay AG, Haley JD and Garcia-Diaz M (2018). Kinetics and mechanism of mammalian mitochondrial ribosome assembly. Cell Rep. 22, 1935–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mai N, Chrzanowska-Lightowlers ZM and Lightowlers RN (2017). The process of mammalian mitochondrial protein synthesis. Cell Tissue Res. 367, 5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].De Silva D, Tu YT, Amunts A, Fontanesi F and Barrientos A (2015). Mitochondrial ribosome assembly in health and disease. Cell Cycle 14, 2226–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Boczonadi V, Ricci G and Horvath R (2018). Mitochondrial DNA transcription and translation: clinical syndromes. Essays Biochem 62, 321–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rotig A (2011). Human diseases with impaired mitochondrial protein synthesis. Biochim. Biophys. Acta. 1807, 1198–205. [DOI] [PubMed] [Google Scholar]
- [26].Jacobs HT and Turnbull DM (2005). Nuclear genes and mitochondrial translation: a new class of genetic disease. Trends Genet. 21, 312–4. [DOI] [PubMed] [Google Scholar]
- [27].Perez-Martinez X, Funes S, Camacho-Villasana Y, Marjavaara S, Tavares-Carreon F and Shingu-Vazquez M (2008). Protein synthesis and assembly in mitochondrial disorders. Curr. Top. Med. Chem. 8, 1335–50. [DOI] [PubMed] [Google Scholar]
- [28].Kim HJ, Maiti P and Barrientos A (2017). Mitochondrial ribosomes in cancer. Semin. Cancer Biol. 47, 67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Singh R, Sripada L and Singh R (2014). Side effects of antibiotics during bacterial infection: mitochondria, the main target in host cell. Mitochondrion 16, 50–4. [DOI] [PubMed] [Google Scholar]
- [30].Rudler DL, Hughes LA, Viola HM, Hool LC, Rackham O and Filipovska A (2020). Fidelity and coordination of mitochondrial protein synthesis in health and disease. J. Physiol. 25. [DOI] [PubMed] [Google Scholar]
- [31].Gopisetty G and Thangarajan R (2016). Mammalian mitochondrial ribosomal small subunit (MRPS) genes: A putative role in human disease. Gene 589, 27–35. [DOI] [PubMed] [Google Scholar]
- [32].Spremulli LL, Coursey A, Navratil T and Hunter SE (2004). Initiation and elongation factors in mammalian mitochondrial protein biosynthesis. Prog. Nucleic Acid Res. Mol. Biol. 77, 211–61. [DOI] [PubMed] [Google Scholar]
- [33].Osawa S, Ohama T, Jukes TH, Watanabe K and Yokoyama S (1989). Evolution of the mitochondrial genetic code. II. Reassignment of codon AUA from isoleucine to methionine. J. Mol. Evol. 29, 373–80. [DOI] [PubMed] [Google Scholar]
- [34].Jukes TH and Osawa S (1990). The genetic code in mitochondria and chloroplasts. Experientia. 46, 1117–26. [DOI] [PubMed] [Google Scholar]
- [35].Dawitz H, Schäfer J, Schaart JM, Magits W, Brzezinski P and Ott M (2019). Rcf1 modulates cytochrome c oxidase activity especially under energy-demanding conditions. Front. Physiol. 10, 1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Poutre CG and Fox TD (1987). PET111, a Saccharomyces cerevisiae nuclear gene required for translation of the mitochondrial mRNA encoding cytochrome c oxidase subunit II. Genetics 115, 637–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fontanesi F (2013). Mechanisms of mitochondrial translational regulation. IUBMB Life. 65, 397–408. [DOI] [PubMed] [Google Scholar]
- [38].Seshadri SR, Banarjee C, Barros MH and Fontanesi F (2020). The translational activator Sov1 coordinates mitochondrial gene expression with mitoribosome biogenesis. Nucleic Acids Res. 48, 6759–6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kuzmenko A, Atkinson GC, Levitskii S, Zenkin N, Tenson T, Hauryliuk V and Kamenski P (2014). Mitochondrial translation initiation machinery: conservation and diversification. Biochimie 100, 132–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tucker EJ et al. (2011). Mutations in MTFMT underlie a human disorder of formylation causing impaired mitochondrial translation. Cell Metab. 14, 428–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Atkinson GC et al. (2012). Evolutionary and genetic analyses of mitochondrial translation initiation factors identify the missing mitochondrial IF3 in S. cerevisiae. Nucleic Acids Res. 40, 6122–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Spencer AC and Spremulli LL (2005). The interaction of mitochondrial translational initiation factor 2 with the small ribosomal subunit. Biochim. Biophys. Acta 1750, 69–81. [DOI] [PubMed] [Google Scholar]
- [43].Koc EC and Spremulli LL (2002). Identification of mammalian mitochondrial translational initiation factor 3 and examination of its role in initiation complex formation with natural mRNAs. J. Biol. Chem. 277, 35541–9. [DOI] [PubMed] [Google Scholar]
- [44].Chicherin IV, Baleva MV, Levitskii SA, Dashinimaev EB, Krasheninnikov IA and Kamenski P (2020). Initiation Factor 3 is dispensable for mitochondrial translation in cultured human cells. Sci. Rep. 10, 7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kummer E, Leibundgut M, Rackham O, Lee RG, Boehringer D, Filipovska A and Ban N (2018). Unique features of mammalian mitochondrial translation initiation revealed by cryo-EM. Nature 560, 263–267. [DOI] [PubMed] [Google Scholar]
- [46].Koripella RK, Sharma MR, Haque ME, Risteff P, Spremulli LL and Agrawal RK (2019). Structure of human mitochondrial Translation Initiation Factor 3 bound to the small ribosomal subunit. iScience 12, 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Khawaja A, Itoh Y, Remes C, Spåhr H, Yukhnovets O, Höfig H, Amunts A and Rorbach J (2020). Distinct pre-initiation steps in human mitochondrial translation. Nat. Commun. 11, 2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Höfig H, Yukhnovets O, Remes C, Kempf N, Katranidis A, Kempe D and Fitter J (2019). Brightness-gated two-color coincidence detection unravels two distinct mechanisms in bacterial protein translation initiation. Commun. Biol. 2, 459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kaempfer R (1969). Ribosomal subunit exchange in the cytoplasm of a eukaryote. Nature 222, 950–3. [DOI] [PubMed] [Google Scholar]
- [50].Cai YC, Bullard JM, Thompson NL and Spremulli LL (2000). Interaction of mammalian mitochondrial elongation factor EF-Tu with guanine nucleotides. Protein Sci. 9, 1791–800. [PMC free article] [PubMed] [Google Scholar]
- [51].Koripella RK et al. (2020). Structures of the human mitochondrial ribosome bound to EF-G1 reveal distinct features of mitochondrial translation elongation. Nat. Commun. 11, 3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Itoh Y, Naschberger A, Mortezaei N, Herrmann JM and Amunts A (2020). Analysis of translating mitoribosome reveals functional characteristics of translation in mitochondria of fungi. Nat. Commun. 11, 5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Soleimanpour-Lichaei HR et al. (2007). mtRF1a is a human mitochondrial translation release factor decoding the major termination codons UAA and UAG. Mol. Cell 27, 745–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Schmeing TM, Huang KS, Strobel SA and Steitz TA (2005). An induced-fit mechanism to promote peptide bond formation and exclude hydrolysis of peptidyl-tRNA. Nature 438, 520–4. [DOI] [PubMed] [Google Scholar]
- [55].Rorbach J et al. (2008). The human mitochondrial ribosome recycling factor is essential for cell viability. Nucleic Acids Res. 36, 5787–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Koripella RK, Sharma MR, Risteff P, Keshavan P and Agrawal RK (2019). Structural insights into unique features of the human mitochondrial ribosome recycling. Proc. Natl. Acad. Sci. U. S. A. 116, 8283–8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tsuboi M, Morita H, Nozaki Y, Akama K, Ueda T, Ito K, Nierhaus KH and Takeuchi N (2009). EF-G2mt is an exclusive recycling factor in mammalian mitochondrial protein synthesis. Mol. Cell 35, 502–10. [DOI] [PubMed] [Google Scholar]
- [58].Smits P, Smeitink JA, van den Heuvel LP, Huynen MA and Ettema TJ (2007). Reconstructing the evolution of the mitochondrial ribosomal proteome. Nucleic Acids Res 35, 4686–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Petrov AS, Wood EC, Bernier CR, Norris AM, Brown A and Amunts A (2019). Structural Patching Fosters Divergence of Mitochondrial Ribosomes. Mol. Biol. Evol. 36, 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].van der Sluis EO et al. (2015). Parallel structural evolution of mitochondrial ribosomes and OXPHOS complexes. Genome Biol. Evol. 7, 1235–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Chen J, Tsai A, O’Leary SE, Petrov A and Puglisi JD (2012). Unraveling the dynamics of ribosome translocation. Curr. Opin. Struct. Biol. 22, 804–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Yusupova GZ, Yusupov MM, Cate JH and Noller HF (2001). The path of messenger RNA through the ribosome. Cell 106, 233–41. [DOI] [PubMed] [Google Scholar]
- [63].Takyar S, Hickerson RP and Noller HF (2005). mRNA helicase activity of the ribosome. Cell 120, 49–58. [DOI] [PubMed] [Google Scholar]
- [64].Helm M, Brulé H, Friede D, Giegé R, Pütz D and Florentz C (2000). Search for characteristic structural features of mammalian mitochondrial tRNAs. RnNA 6, 1356–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Greber BJ and Ban N (2016). Structure and function of the mitochondrial ribosome. Annu. Rev. Biochem. 85, 103–32. [DOI] [PubMed] [Google Scholar]
- [66].Sharma MR, Koc EC, Datta PP, Booth TM, Spremulli LL and Agrawal RK (2003). Structure of the mammalian mitochondrial ribosome reveals an expanded functional role for its component proteins. Cell 115, 97–108. [DOI] [PubMed] [Google Scholar]
- [67].Rorbach J, Gao F, Powell CA, D’Souza A, Lightowlers RN, Minczuk M and Chrzanowska-Lightowlers ZM (2016). Human mitochondrial ribosomes can switch their structural RNA composition. Proc. Natl. Acad. Sci. U. S. A. 113, 12198–12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Gustafsson CM, Falkenberg M and Larsson NG (2016). Maintenance and Expression of Mammalian Mitochondrial DNA. Annu Rev Biochem [DOI] [PubMed] [Google Scholar]
- [69].Liao D (2000). Gene conversion drives within genic sequences: concerted evolution of ribosomal RNA genes in bacteria and archaea. J. Mol. Evol. 51, 305–17. [DOI] [PubMed] [Google Scholar]
- [70].Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH and Noller HF (2001). Crystal structure of the ribosome at 5.5 A resolution. Science 292, 883–96. [DOI] [PubMed] [Google Scholar]
- [71].Greber BJ et al. (2014). Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature 505, 515–9. [DOI] [PubMed] [Google Scholar]
- [72].Miller C, Saada A, Shaul N, Shabtai N, Ben-Shalom E, Shaag A, Hershkovitz E and Elpeleg O (2004). Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Ann. Neurol. 56, 734–8. [DOI] [PubMed] [Google Scholar]
- [73].Emdadul Haque M, Grasso D, Miller C, Spremulli LL and Saada A (2008). The effect of mutated mitochondrial ribosomal proteins S16 and S22 on the assembly of the small and large ribosomal subunits in human mitochondria. Mitochondrion 8, 254–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bugiardini E et al. (2019). MRPS25 mutations impair mitochondrial translation and cause encephalomyopathy. Hum. Mol. Genet. 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Antonicka H, Choquet K, Lin ZY, Gingras AC, Kleinman CL and Shoubridge EA (2017). A pseudouridine synthase module is essential for mitochondrial protein synthesis and cell viability. EMBO Rep. 18, 28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Rackham O et al. (2016). Hierarchical RNA processing is required for mitochondrial ribosome assembly. Cell Rep. 16, 1874–90. [DOI] [PubMed] [Google Scholar]
- [77].Lee KW and Bogenhagen DF (2014). Assignment of 2’-O-methyltransferases to modification sites on the mammalian mitochondrial large subunit 16 S ribosomal RNA (rRNA). J Biol Chem. 289, 24936–42. doi: 10.1074/jbc.C114.581868. Epub 2014 Jul 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Rorbach J et al. (2014). MRM2 and MRM3 are involved in biogenesis of the large subunit of the mitochondrial ribosome. Mol. Biol. Cell 9, 01–0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Tobiasson V, Gahura O, Aibara S, Baradaran R, Zíková A and Amunts A (2020). Interconnected assembly factors regulate the biogenesis of mitoribosomal large subunit. bioRxiv, 2020.06.28.176446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Saurer M et al. (2019). Mitoribosomal small subunit biogenesis in trypanosomes involves an extensive assembly machinery. Science 365, 1144–1149. [DOI] [PubMed] [Google Scholar]
- [81].Jaskolowski M et al. (2020). Structural insights into the mechanism of mitoribosomal large subunit biogenesis. Mol. Cell, DOI: 10.1016/j.molcel.2020.06.030. [DOI] [PubMed] [Google Scholar]
- [82].Brown A, Rathore S, Kimanius D, Aibara S, Bai XC, Rorbach J, Amunts A and Ramakrishnan V (2017). Structures of the human mitochondrial ribosome in native states of assembly. Nat. Struct. Mol. Biol. 24, 866–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Antonicka H, Sasarman F, Nishimura T, Paupe V and Shoubridge EA (2013). The mitochondrial RNA-binding protein GRSF1 Localizes to RNA granules and Is required for posttranscriptional mitochondrial gene expression. Cell Metab. 17, 386–98. [DOI] [PubMed] [Google Scholar]
- [84].Jourdain AA, Koppen M, Wydro M, Rodley CD, Lightowlers RN, Chrzanowska-Lightowlers ZM and Martinou JC (2013). GRSF1 Regulates RNA Processing in Mitochondrial RNA Granules. Cell Metab. 17, 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Antonicka H and Shoubridge EA (2015). Mitochondrial RNA granules are centers for posttranscriptional RNA processing and ribosome biogenesis. Cell Rep. 10, 920–932. [DOI] [PubMed] [Google Scholar]
- [86].Tu YT and Barrientos A (2015). The human mitochondrial DEAD-box protein DDX28 resides in RNA granules and functions in mitoribosome assembly. Cell Rep. 10, 854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Shajani Z, Sykes MT and Williamson JR (2011). Assembly of bacterial ribosomes. Annu. Rev. Biochem. 80, 501–26. [DOI] [PubMed] [Google Scholar]
- [88].Britton RA (2009). Role of GTPases in bacterial ribosome assembly. Annu. Rev. Microbiol. 63, 155–176. [DOI] [PubMed] [Google Scholar]
- [89].Barrientos A, Korr D, Barwell KJ, Sjulsen C, Gajewski CD, Manfredi G, Ackerman S and Tzagoloff A (2003). MTG1 codes for a conserved protein required for mitochondrial translation. Mol. Biol. Cell 14, 2292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Paul MF, Alushin GM, Barros MH, Rak M and Tzagoloff A (2012). The putative GTPase encoded by MTG3 functions in a novel pathway for regulating assembly of the small subunit of yeast mitochondrial ribosomes. J. Biol. Chem. 287, 24346–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Dennerlein S, Rozanska A, Wydro M, Chrzanowska-Lightowlers ZM and Lightowlers RN (2010). Human ERAL1 is a mitochondrial RNA chaperone involved in the assembly of the 28S small mitochondrial ribosomal subunit. Biochem. J. 430, 551–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Metodiev MD et al. (2014). NSUN4 is a dual function mitochondrial protein required for both methylation of 12S rRNA and coordination of mitoribosomal assembly. PLoS Genet. 10, e1004110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Dalla Rosa I et al. (2014). MPV17L2 is required for ribosome assembly in mitochondria. Nucleic Acids Res. 42, 8500–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Boczonadi V and Horvath R (2014). Mitochondria: impaired mitochondrial translation in human disease. Int. J. Biochem. Cell Biol. 48, 77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Chen SS and Williamson JR (2013). Characterization of the ribosome biogenesis landscape in E. coli using quantitative mass spectrometry. J. Mol. Biol. 425, 767–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Davis JH, Tan YZ, Carragher B, Potter CS, Lyumkis D and Williamson JR (2016). Modular Assembly of the Bacterial Large Ribosomal Subunit. Cell 167, 1610–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Uchiumi T, Ohgaki K, Yagi M, Aoki Y, Sakai A, Matsumoto S and Kang D (2010). ERAL1 is associated with mitochondrial ribosome and elimination of ERAL1 leads to mitochondrial dysfunction and growth retardation. Nucleic Acids Res. 38, 5554–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Seidel-Rogol BL, McCulloch V and Shadel GS (2003). Human mitochondrial transcription factor B1 methylates ribosomal RNA at a conserved stem-loop. Nat. Genet. 33, 23–4. [DOI] [PubMed] [Google Scholar]
- [99].Szczepanowska K et al. (2016). CLPP coordinates mitoribosomal assembly through the regulation of ERAL1 levels. EMBO J. 35, 2566–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Summer S et al. (2020). YBEY is an essential biogenesis factor for mitochondrial ribosomes. Nucleic Acids Res. 48, 9762–9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Van Haute L et al. (2019). METTL15 introduces N4-methylcytidine into human mitochondrial 12S rRNA and is required for mitoribosome biogenesis. Nucleic Acids Res. 47, 10267–10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Kolanczyk M et al. (2011). NOA1 is an essential GTPase required for mitochondrial protein synthesis. Mol. Biol. Cell 22, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].He J et al. (2012). Human C4orf14 interacts with the mitochondrial nucleoid and is involved in the biogenesis of the small mitochondrial ribosomal subunit. Nucleic Acids Res. 40, 6097–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Reyes A, Favia P, Vidoni S, Petruzzella V and Zeviani M (2020). RCC1L (WBSCR16) isoforms coordinate mitochondrial ribosome assembly through their interaction with GTPases. PLoS Genet. 16, e1008923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Fung S, Nishimura T, Sasarman F and Shoubridge EA (2013). The conserved interaction of C7orf30 with MRPL14 promotes biogenesis of the mitochondrial large ribosomal subunit and mitochondrial translation. Mol. Biol. Cell 24, 184–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Rorbach J, Gammage PA and Minczuk M (2012). C7orf30 is necessary for biogenesis of the large subunit of the mitochondrial ribosome. Nucleic Acids Res. 40, 4097–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Nolden M, Ehses S, Koppen M, Bernacchia A, Rugarli EI and Langer T (2005). The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell 123, 277–89. [DOI] [PubMed] [Google Scholar]
- [108].Almajan ER et al. (2012). AFG3L2 supports mitochondrial protein synthesis and Purkinje cell survival. J. Clin. Invest. 122, 4048–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Popow J, Alleaume AM, Curk T, Schwarzl T, Sauer S and Hentze MW (2015). FASTKD2 is an RNA-binding protein required for mitochondrial RNA processing and translation. RNA 21, 1873–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Chrzanowska-Lightowlers Z, Rorbach J and Minczuk M (2017). Human mitochondrial ribosomes can switch structural tRNAs - but when and why? RNA Biol. 14, 1668–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Maiti P, Lavdovskaia E, Barrientos A and Richter-Dennerlein R (2020). Role of GTPases in driving mitoribosome assembly. Trends in Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Kim H-J and Barrientos A (2018). MTG1 couples mitoribosome large subunit assembly and intersubunit bridge formation. Nucleic Acid Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Dibley MG et al. (2020). The mitochondrial acyl-carrier protein interaction network highlights important roles for LYRM family members in complex i and mitoribosome assembly. Mol. Cell Proteomics 19, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Maiti P, Antonicka H, Gingras A-C, Shoubridge EA and Barrientos A (2020). Human GTPBP5 (MTG2) fuels mitoribosome large subunit maturation by facilitating 16S rRNA methylation. Nucleic Acids Res. in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Lee KW, Okot-Kotber C, Lacomb JF and Bogenhagen DF (2013). Mitochondrial rRNA methyltransferase family members are positioned to modify nascent rrna in foci near the mtDNA nucleoid. J. Biol. Chem. 288, 31386–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Zaganelli S et al. (2017). The pseudouridine synthase RPUSD4 Is an essential component of mitochondrial RNA granules. J. Biol. Chem. 292, 4519–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Camara Y et al. (2011). MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab. 13, 527–39. [DOI] [PubMed] [Google Scholar]
- [118].Lavdovskaia E, Denks K, Nadler F, Steube E, Linden A, Urlaub H, Rodnina MV and Richter-Dennerlein R (2020). Dual function of GTPBP6 in biogenesis and recycling of human mitochondrial ribosomes. Nuc. Acids Res. DOI: 10.1093/nar/gkaa1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Lavdovskaia E, Kolander E, Steube E, Mai MM, Urlaub H and Richter-Dennerlein R (2018). The human Obg protein GTPBP10 is involved in mitoribosomal biogenesis. Nucleic Acids Res. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].DiMauro S and Schon EA (2003). Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 348, 2656–68. [DOI] [PubMed] [Google Scholar]
- [121].Smith PM et al. (2014). The role of the mitochondrial ribosome in human disease: searching for mutations in 12S mitochondrial rRNA with high disruptive potential. Hum. Mol. Genet 23, 949–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Elson JL, Smith PM, Greaves LC, Lightowlers RN, Chrzanowska-Lightowlers ZM, Taylor RW and Vila-Sanjurjo A (2015). The presence of highly disruptive 16S rRNA mutations in clinical samples indicates a wider role for mutations of the mitochondrial ribosome in human disease. Mitochondrion 25, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Prezant TR et al. (1993). Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat. Genet. 4, 289–94. [DOI] [PubMed] [Google Scholar]
- [124].Ding Y, Leng J, Fan F, Xia B and Xu P (2013). The role of mitochondrial DNA mutations in hearing loss. Biochem. Genet. 51, 588–602. doi: 10.1007/s10528-013-9589-6. Epub 2013 Apr 21. [DOI] [PubMed] [Google Scholar]
- [125].Estivill X et al. (1998). Familial progressive sensorineural deafness is mainly due to the mtDNA A1555G mutation and is enhanced by treatment of aminoglycosides. Am. J. Hum. Genet. 62, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Hamasaki K and Rando RR (1997). Specific binding of aminoglycosides to a human rRNA construct based on a DNA polymorphism which causes aminoglycoside-induced deafness. Biochemistry 36, 12323–8. [DOI] [PubMed] [Google Scholar]
- [127].Bykhovskaya Y, Mengesha E, Wang D, Yang H, Estivill X, Shohat M and Fischel-Ghodsian N (2004). Human mitochondrial transcription factor B1 as a modifier gene for hearing loss associated with the mitochondrial A1555G mutation. Mol. Genet. Metab. 82, 27–32. [DOI] [PubMed] [Google Scholar]
- [128].Bykhovskaya Y, Mengesha E, Wang D, Yang H, Estivill X, Shohat M and Fischel-Ghodsian N (2004). Phenotype of non-syndromic deafness associated with the mitochondrial A1555G mutation is modulated by mitochondrial RNA modifying enzymes MTO1 and GTPBP3. Mol. Genet. Metab. 83, 199–206. [DOI] [PubMed] [Google Scholar]
- [129].Li X, Li R, Lin X and Guan MX (2002). Isolation and characterization of the putative nuclear modifier gene MTO1 involved in the pathogenesis of deafness-associated mitochondrial 12 S rRNA A1555G mutation. J. Biol. Chem. 277, 27256–64. [DOI] [PubMed] [Google Scholar]
- [130].Campos Y et al. (2002). Cosegregation of the mitochondrial DNA A1555G and G4309A mutations results in deafness and mitochondrial myopathy. Muscle Nerve 25, 185–8. [DOI] [PubMed] [Google Scholar]
- [131].Habbane M et al. (2020). Leigh Syndrome in a pedigree harboring the m.1555A>G mutation in the mitochondrial 12S rRNA. Genes (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Coulbault L et al. (2007). A novel mutation 3090 G>A of the mitochondrial 16S ribosomal RNA associated with myopathy. Biochem. Biophys. Res. Commun. 362, 601–5. [DOI] [PubMed] [Google Scholar]
- [133].Liu Z et al. (2014). The novel mitochondrial 16S rRNA 2336T>C mutation is associated with hypertrophic cardiomyopathy. J. Med. Genet. 51, 176–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Li S et al. (2018). Mitochondrial dysfunctions contribute to hypertrophic cardiomyopathy in patient ipsc-derived cardiomyocytes with mt-RNR2 mutation. Stem Cell Reports 10, 808–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Tang J, Qi Y, Bao XH and Wu XR (1997). Mutational analysis of mitochondrial DNA of children with Rett syndrome. Pediatr Neurol 17, 327–30. [DOI] [PubMed] [Google Scholar]
- [136].Lv ZY et al. (2017). Mitochondrial mutations in 12S rRNA and 16S rRNA presenting as chronic progressive external ophthalmoplegia (CPEO) plus: A case report. Medicine (Baltimore) 96, e8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Arredondo JJ et al. (2012). Mitochondrial tRNA valine as a recurrent target for mutations involved in mitochondrial cardiomyopathies. Mitochondrion 12, 357–62. [DOI] [PubMed] [Google Scholar]
- [138].Pulman J, Ruzzenente B, Bianchi L, Rio M, Boddaert N, Munnich A, Rötig A and Metodiev MD (2019). Mutations in the MRPS28 gene encoding the small mitoribosomal subunit protein bS1m in a patient with intrauterine growth retardation, craniofacial dysmorphism and multisystemic involvement. Hum. Mol. Genet. 28, 1445–1462. [DOI] [PubMed] [Google Scholar]
- [139].Borna NN et al. (2019). Mitochondrial ribosomal protein PTCD3 mutations cause oxidative phosphorylation defects with Leigh syndrome. Neurogenetics 20, 9–25. [DOI] [PubMed] [Google Scholar]
- [140].Di Nottia M et al. (2020). A homozygous MRPL24 mutation causes a complex movement disorder and affects the mitoribosome assembly. Neurobiol Dis. 141, 104880. [DOI] [PubMed] [Google Scholar]
- [141].Gardeitchik T et al. (2018). Bi-allelic Mutations in the Mitochondrial Ribosomal Protein MRPS2 Cause Sensorineural Hearing Loss, Hypoglycemia, and Multiple OXPHOS Complex Deficiencies. Am. J. Hum. Genet. 102, 685–695. doi: 10.1016/j.ajhg.2018.02.012. Epub 2018 Mar 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].O’Brien TW, O’Brien BJ and Norman RA (2005). Nuclear MRP genes and mitochondrial disease. Gene 354, 147–51. [DOI] [PubMed] [Google Scholar]
- [143].Menezes MJ et al. (2015). Mutation in mitochondrial ribosomal protein S7 (MRPS7) causes congenital sensorineural deafness, progressive hepatic and renal failure, and lactic acidemia. Hum. Mol. Genet 2. [DOI] [PubMed] [Google Scholar]
- [144].Dheedene A, Maes M, Vergult S and Menten B (2014). A de novo POU3F3 deletion in a boy with Intellectual disability and dysmorphic features. Mol. Syndromol. 5, 32–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Snijders Blok L et al. (2019). De novo variants disturbing the transactivation capacity of POU3F3 cause a characteristic neurodevelopmental disorder. Am. J. Hum. Genet. 105, 403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Saada A, Shaag A, Arnon S, Dolfin T, Miller C, Fuchs-Telem D, Lombes A and Elpeleg O (2007). Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J. Med. Genet. 44, 784–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Smits P et al. (2011). Mutation in mitochondrial ribosomal protein MRPS22 leads to Cornelia de Lange-like phenotype, brain abnormalities and hypertrophic cardiomyopathy. Eur. J. Hum. Genet. 19, 394–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Baertling F et al. (2015). MRPS22 mutation causes fatal neonatal lactic acidosis with brain and heart abnormalities. Neurogenetics 16, 237–40. [DOI] [PubMed] [Google Scholar]
- [149].Chen A et al. (2018). Mutations in the mitochondrial ribosomal protein MRPS22 lead to primary ovarian insufficiency. Hum. Mol. Genet. 27, 1913–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Richman TR et al. (2015). Mutation in MRPS34 Compromises Protein Synthesis and Causes Mitochondrial Dysfunction. PLoS Genet. 11, e1005089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Lake NJ et al. (2017). Biallelic Mutations in MRPS34 Lead to Instability of the Small Mitoribosomal Subunit and Leigh Syndrome. Am. J. Hum. Genet. 101, 239–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Wessels HJ et al. (2013). Analysis of 953 human proteins from a mitochondrial HEK293 fraction by complexome profiling. PLoS One 8, e68340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Kohda M et al. (2016). A comprehensive genomic analysis reveals the genetic landscape of mitochondrial respiratory chain complex deficiencies. PLoS Genet. 12, e1005679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Lee YH and Song GG (2015). Meta-analysis of differentially expressed genes in ankylosing spondylitis. Genet. Mo.l Res. 14, 5161–70. [DOI] [PubMed] [Google Scholar]
- [155].Kathiresan S et al. (2009). Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat. Genet. 41, 334–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Szpakowicz A et al. (2015). The rs9982601 polymorphism of the region between the SLC5A3/MRPS6 and KCNE2 genes associated with a prevalence of myocardial infarction and subsequent long-term mortality. Pol. Arch. Med. Wewn. 125, 240–8. [DOI] [PubMed] [Google Scholar]
- [157].Rodríguez-García ME, Cotrina-Vinagre FJ, Carnicero-Rodríguez P and Martínez-Azorín F (2017). An innovative strategy to clone positive modifier genes of defects caused by mtDNA mutations: MRPS18C as suppressor gene of m.3946G>A mutation in MT-ND1 gene. Hum. Genet. 136, 885–896. [DOI] [PubMed] [Google Scholar]
- [158].Jackson CB et al. (2019). A variant in MRPS14 (uS14m) causes perinatal hypertrophic cardiomyopathy with neonatal lactic acidosis, growth retardation, dysmorphic features and neurological involvement. Hum. Mol. Genet. 28, 639–649. [DOI] [PubMed] [Google Scholar]
- [159].Galmiche L et al. (2011). Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum. Mutat. 32, 1225–31. [DOI] [PubMed] [Google Scholar]
- [160].Carroll CJ et al. (2013). Whole-exome sequencing identifies a mutation in the mitochondrial ribosome protein MRPL44 to underlie mitochondrial infantile cardiomyopathy. J. Med. Genet. 50, 151–9. [DOI] [PubMed] [Google Scholar]
- [161].Distelmaier F et al. (2015). MRPL44 mutations cause a slowly progressive multisystem disease with childhood-onset hypertrophic cardiomyopathy. Neurogenetics, 24. [DOI] [PubMed] [Google Scholar]
- [162].Kavran JM and Steitz TA (2007). Structure of the base of the L7/L12 stalk of the Haloarcula marismortui large ribosomal subunit: analysis of L11 movements. J. Mol. Biol. 371, 1047–59. [DOI] [PubMed] [Google Scholar]
- [163].Carlson MA, Haddad BG, Weis AJ, Blackwood CS, Shelton CD, Wuerth ME, Walter JD and Spiegel PC Jr. (2017). Ribosomal protein L7/L12 is required for GTPase translation factors EF-G, RF3, and IF2 to bind in their GTP state to 70S ribosomes. FEBS J. 284, 1631–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Serre V, Rozanska A, Beinat M, Chretien D, Boddaert N, Munnich A, Rotig A and Chrzanowska-Lightowlers ZM (2013). Mutations in mitochondrial ribosomal protein MRPL12 leads to growth retardation, neurological deterioration and mitochondrial translation deficiency. Biochim. Biophys. Acta 1832, 1304–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Haack TB et al. (2013). ELAC2 mutations cause a mitochondrial RNA processing defect associated with hypertrophic cardiomyopathy. Am. J. Hum. Genet. 93, 211–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Sanchez MI et al. (2011). RNA processing in human mitochondria. Cell Cycle 10, 2904–16. [DOI] [PubMed] [Google Scholar]
- [167].Rossmanith W (2011). Localization of human RNase Z isoforms: dual nuclear/mitochondrial targeting of the ELAC2 gene product by alternative translation initiation. PLoS One 6, e19152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Saoura M et al. (2019). Mutations in ELAC2 associated with hypertrophic cardiomyopathy impair mitochondrial tRNA 3’-end processing. Hum. Mutat. 40, 1731–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Tavtigian SV et al. (2001). A candidate prostate cancer susceptibility gene at chromosome 17p. Nat. Genet. 27, 172–80. [DOI] [PubMed] [Google Scholar]
- [170].Schroeder C et al. (2019). Nuclear ELAC2 overexpression is associated with increased hazard for relapse after radical prostatectomy. Oncotarget 10, 4973–4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Holzmann J, Frank P, Loffler E, Bennett KL, Gerner C and Rossmanith W (2008). RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 135, 462–74. [DOI] [PubMed] [Google Scholar]
- [172].Vilardo E, Nachbagauer C, Buzet A, Taschner A, Holzmann J and Rossmanith W (2012). A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase--extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res. 40, 11583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Metodiev MD et al. (2016). Recessive mutations in TRMT10C cause defects in mitochondrial RNA processing and multiple respiratory chain deficiencies. Am. J. Hum. Genet. 98, 993–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Kallberg Y, Oppermann U, Jörnvall H and Persson B (2002). Short-chain dehydrogenases/reductases (SDRs). Eur. J. Biochem. 269, 4409–17. [DOI] [PubMed] [Google Scholar]
- [175].Zschocke J (2012). HSD10 disease: clinical consequences of mutations in the HSD17B10 gene. J. Inherit. Metab. Dis. 35, 81–9. [DOI] [PubMed] [Google Scholar]
- [176].Korman SH (2006). Inborn errors of isoleucine degradation: a review. Mol. Genet. Metab. 89, 289–99. [DOI] [PubMed] [Google Scholar]
- [177].Fukao T, Scriver CR and Kondo N (2001). The clinical phenotype and outcome of mitochondrial acetoacetyl-CoA thiolase deficiency (beta-ketothiolase or T2 deficiency) in 26 enzymatically proved and mutation-defined patients. Mol. Genet. Metab. 72, 109–14. [DOI] [PubMed] [Google Scholar]
- [178].Falk MJ et al. (2016). A novel HSD17B10 mutation impairing the activities of the mitochondrial RNase P complex causes X-linked intractable epilepsy and neurodevelopmental regression. RNA Biol. 13, 477–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Reinhard L, Sridhara S and Hällberg BM (2015). Structure of the nuclease subunit of human mitochondrial RNase P. Nucleic Acids Res. 43, 5664–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Ghezzi D, Saada A, D’Adamo P, Fernandez-Vizarra E, Gasparini P, Tiranti V, Elpeleg O and Zeviani M (2008). FASTKD2 nonsense mutation in an infantile mitochondrial encephalomyopathy associated with cytochrome c oxidase deficiency. Am J Hum Genet. 83, 415–23. doi: 10.1016/j.ajhg.2008.08.009. Epub 2008 Sep 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Yoo DH, Choi YC, Nam DE, Choi SS, Kim JW, Choi BO and Chung KW (2017). Identification of FASTKD2 compound heterozygous mutations as the underlying cause of autosomal recessive MELAS-like syndrome. Mitochondrion 35, 54–58. [DOI] [PubMed] [Google Scholar]
- [182].Wei X et al. (2020). Mutations in FASTKD2 are associated with mitochondrial disease with multi-OXPHOS deficiency. Hum. Mutat. 41, 961–972. [DOI] [PubMed] [Google Scholar]
- [183].Garone C et al. (2017). Defective mitochondrial rRNA methyltransferase MRM2 causes MELAS-like clinical syndrome. Hum. Mol. Genet. 26, 4257–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Metodiev MD et al. (2009). Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab. 9, 386–97. [DOI] [PubMed] [Google Scholar]
- [185].Cotney J, McKay SE and Shadel GS (2009). Elucidation of separate, but collaborative functions of the rRNA methyltransferase-related human mitochondrial transcription factors B1 and B2 in mitochondrial biogenesis reveals new insight into maternally inherited deafness. Hum. Mol. Genet. 18, 2670–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Raimundo N et al. (2012). Mitochondrial stress engages E2F1 apoptotic signaling to cause deafness. Cell 148, 716–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Koeck T et al. (2011). A common variant in TFB1M is associated with reduced insulin secretion and increased future risk of type 2 diabetes. Cell Metab. 13, 80–91. [DOI] [PubMed] [Google Scholar]
- [188].Sharoyko VV et al. (2014). Loss of TFB1M results in mitochondrial dysfunction that leads to impaired insulin secretion and diabetes. Hum. Mol. Genet. 23, 5733–49. [DOI] [PubMed] [Google Scholar]
- [189].Chatzispyrou IA et al. (2017). A homozygous missense mutation in ERAL1, encoding a mitochondrial rRNA chaperone, causes Perrault syndrome. Hum. Mol. Genet. 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Jenkinson EM et al. (2013). Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am. J. Hum. Genet. 92, 605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Brodie EJ, Zhan H, Saiyed T, Truscott KN and Dougan DA (2018). Perrault syndrome type 3 caused by diverse molecular defects in CLPP. Sci. Rep. 8, 12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Marlin S et al. (2008). Perrault syndrome: report of four new cases, review and exclusion of candidate genes. Am. J. Med. Genet. A. 146a, 661–4. [DOI] [PubMed] [Google Scholar]
- [193].Yagi M, Uchiumi T, Takazaki S, Okuno B, Nomura M, Yoshida S, Kanki T and Kang D (2012). p32/gC1qR is indispensable for fetal development and mitochondrial translation: importance of its RNA-binding ability. Nucleic Acids Res. 40, 9717–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Noh S, Phorl S, Naskar R, Oeum K, Seo Y, Kim E, Kweon HS and Lee JY (2020). p32/C1QBP regulates OMA1-dependent proteolytic processing of OPA1 to maintain mitochondrial connectivity related to mitochondrial dysfunction and apoptosis. Sci. Rep. 10, 10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Feichtinger RG et al. (2017). Biallelic C1QBP Mutations Cause Severe Neonatal-, Childhood-, or Later-Onset Cardiomyopathy Associated with Combined Respiratory-Chain Deficiencies. Am. J. Hum. Genet. 101, 525–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Lessel D et al. (2017). De novo missense mutations in DHX30 impair global translation and cause a neurodevelopmental disorder. Am. J. Hum. Genet. 101, 716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Wolozin B and Ivanov P (2019). Stress granules and neurodegeneration. Nat. Rev. Neurosci. 20, 649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Solomon BD et al. (2011). De novo deletion of chromosome 20q13.33 in a patient with tracheo-esophageal fistula, cardiac defects and genitourinary anomalies implicates GTPBP5 as a candidate gene. Birth Defects Res. A. Clin. Mol. Teratol. 91, 862–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Solomon BD, Patel A, Cheung SW and Pineda-Alvarez DE (2011). VACTERL association and mitochondrial dysfunction. Birth Defects Res. A. Clin. Mol. Teratol. 91, 192–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Fischel-Ghodsian N, Prezant TR, Bu X and Oztas S (1993). Mitochondrial ribosomal RNA gene mutation in a patient with sporadic aminoglycoside ototoxicity. Am. J. Otolaryngol. 14, 399–403. [DOI] [PubMed] [Google Scholar]
- [201].Zhao H et al. (2004). Maternally inherited aminoglycoside-induced and nonsyndromic deafness is associated with the novel C1494T mutation in the mitochondrial 12S rRNA gene in a large Chinese family. Am. J. Hum. Genet. 74, 139–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Li R, Xing G, Yan M, Cao X, Liu XZ, Bu X and Guan MX (2004). Cosegregation of C-insertion at position 961 with the A1555G mutation of the mitochondrial 12S rRNA gene in a large Chinese family with maternally inherited hearing loss. Am. J. Med. Genet. A. 124A, 113–7. [DOI] [PubMed] [Google Scholar]
- [203].Lu J et al. (2010). Mitochondrial 12S rRNA variants in 1642 Han Chinese pediatric subjects with aminoglycoside-induced and nonsyndromic hearing loss. Mitochondrion 10, 380–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Sazonova MA et al. (2017). Role of mitochondrial genome mutations in pathogenesis of carotid atherosclerosis. Oxid. Med. Cell Longev. 2017, 6934394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Sazonova MA et al. (2018). Mitochondrial genome mutations associated with myocardial Infarction. Dis. Markers 2018, 9749457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Sawabe M et al. (2011). Mitochondrial haplogroups A and M7a confer a genetic risk for coronary atherosclerosis in the Japanese elderly: an autopsy study of 1,536 patients. J. Atheroscler. Thromb. 18, 166–75. [DOI] [PubMed] [Google Scholar]
- [207].Tang S, Batra A, Zhang Y, Ebenroth ES and Huang T (2010). Left ventricular noncompaction is associated with mutations in the mitochondrial genome. Mitochondrion 10, 350–7. [DOI] [PubMed] [Google Scholar]
- [208].Prasad GN, Vanniarajan A, Emmanuel C, Cherian KM, Singh L and Thangaraj K (2006). Novel mitochondrial DNA mutations in a rare variety of hypertrophic cardiomyopathy. Int. J. Cardiol. 109, 432–3. [DOI] [PubMed] [Google Scholar]
- [209].Rollins B et al. (2009). Mitochondrial variants in schizophrenia, bipolar disorder, and major depressive disorder. PLoS One 4, e4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Hudson G et al. (2013). Two-stage association study and meta-analysis of mitochondrial DNA variants in Parkinson disease. Neurology 80, 2042–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Collins DW et al. (2016). Association of primary open-angle glaucoma with mitochondrial variants and haplogroups common in African Americans. Mol. Vis. 22, 454–71. [PMC free article] [PubMed] [Google Scholar]
- [212].Aldosary M et al. (2020). Rett syndrome, a neurodevelopmental disorder, whole-transcriptome, and mitochondrial genome multiomics analyses identify novel variations and disease pathways. Omics 24, 160–171. [DOI] [PubMed] [Google Scholar]
- [213].Jiang W, Li R, Zhang Y, Wang P, Wu T, Lin J, Yu J and Gu M (2017). Mitochondrial DNA mutations associated with Type 2 diabetes mellitus in Chinese Uyghur population. Sci. Rep. 7, 16989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Hsieh RH, Li JY, Pang CY and Wei YH (2001). A novel mutation in the mitochondrial 16S rRNA gene in a patient with MELAS syndrome, diabetes mellitus, hyperthyroidism and cardiomyopathy. J. Biomed. Sci. 8, 328–35. [DOI] [PubMed] [Google Scholar]
- [215].Shoffner JM et al. (1993). Mitochondrial DNA variants observed in Alzheimer disease and Parkinson disease patients. Genomics 17, 171–84. [DOI] [PubMed] [Google Scholar]
- [216].Tiranti V, D’Agruma L, Pareyson D, Mora M, Carrara F, Zelante L, Gasparini P and Zeviani M (1998). A novel mutation in the mitochondrial tRNA(Val) gene associated with a complex neurological presentation. Ann. Neurol 43, 98–101. [DOI] [PubMed] [Google Scholar]
- [217].Tang S et al. (2013). Transition to next generation analysis of the whole mitochondrial genome: a summary of molecular defects. Hum. Mutat. 34, 882–93. [DOI] [PubMed] [Google Scholar]
- [218].Toyoshima Y, Tanaka Y and Satomi K (2017). MELAS syndrome associated with a new mitochondrial tRNA-Val gene mutation (m.1616A>G). BMJ Case Rep. 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Taylor RW, Chinnery PF, Haldane F, Morris AA, Bindoff LA, Wilson J and Turnbull DM (1996). MELAS associated with a mutation in the valine transfer RNA gene of mitochondrial DNA. Ann. Neurol. 40, 459–62. [DOI] [PubMed] [Google Scholar]
- [220].McFarland R, Clark KM, Morris AA, Taylor RW, Macphail S, Lightowlers RN and Turnbull DM (2002). Multiple neonatal deaths due to a homoplasmic mitochondrial DNA mutation. Nat. Genet. 30, 145–6. [DOI] [PubMed] [Google Scholar]
- [221].Horvath R et al. (2009). Heteroplasmic mutation in the anticodon-stem of mitochondrial tRNA(Val) causing MNGIE-like gastrointestinal dysmotility and cachexia. J.Neurol. 256, 810–5. [DOI] [PubMed] [Google Scholar]
- [222].del Mar O’Callaghan M et al. (2012). New mitochondrial DNA mutations in tRNA associated with three severe encephalopamyopathic phenotypes: neonatal, infantile, and childhood onset. Neurogenetics 13, 245–50. [DOI] [PubMed] [Google Scholar]
- [223].Fraidakis MJ et al. (2014). Phenotypic diversity associated with the MT-TV gene m.1644G>A mutation, a matter of quantity. Mitochondrion 15, 34–9. [DOI] [PubMed] [Google Scholar]
- [224].Chalmers RM, Lamont PJ, Nelson I, Ellison DW, Thomas NH, Harding AE and Hammans SR (1997). A mitochondrial DNA tRNA(Val) point mutation associated with adult-onset Leigh syndrome. Neurology 49, 589–92. [DOI] [PubMed] [Google Scholar]
- [225].Blakely EL, Poulton J, Pike M, Wojnarowska F, Turnbull DM, McFarland R and Taylor RW (2004). Childhood neurological presentation of a novel mitochondrial tRNA(Val) gene mutation. J. Neurol. Sci. 225, 99–103. [DOI] [PubMed] [Google Scholar]
- [226].Kılıç M, Oğuz KK, Kılıç E, Yüksel D, Demirci H, Sağıroğlu M, Yücel-Yılmaz D and Özgül RK (2017). A patient with mitochondrial disorder due to a novel mutation in MRPS22. Metab. Brain Dis. 32, 1389–1393. [DOI] [PubMed] [Google Scholar]