

Key Points
SARS-CoV-2 surface proteins in isolation activate inflammatory responses.
Lung inflammation depends on alveolar epithelial cell type I IFN signaling.
Abstract
SARS-CoV-2, the virus that has caused the COVID-19 pandemic, robustly activates the host immune system in critically ill patients. Understanding how the virus engages the immune system will facilitate the development of needed therapeutic strategies. In this study, we demonstrate both in vitro and in vivo that the SARS-CoV-2 surface proteins spike (S) and envelope (E) activate the key immune signaling IFN pathway in both human and mouse immune and epithelial cells independent of viral infection and replication. These proteins induce reactive oxidative species generation and increases in human- and murine-specific, IFN-responsive cytokines and chemokines, similar to their upregulation in critically ill COVID-19 patients. Induction of IFN signaling is dependent on canonical but discrepant inflammatory signaling mediators, as the activation induced by S is dependent on IRF3, TBK1, and MyD88, whereas that of E is largely MyD88 independent. Furthermore, these viral surface proteins, specifically E, induced peribronchial inflammation and pulmonary vasculitis in a mouse model. Finally, we show that the organized inflammatory infiltrates are dependent on type I IFN signaling, specifically in lung epithelial cells. These findings underscore the role of SARS-CoV-2 surface proteins, particularly the understudied E protein, in driving cell specific inflammation and their potential for therapeutic intervention.
Introduction
The SARS-CoV-2 pandemic has profoundly impacted human health globally, leading to more than 125 million cases and 2,700,000 deaths as of March 25, 2021. The ensuing illness, termed COVID-19, predominantly manifests as a respiratory disease that disproportionately affects the older population and those with comorbidities. Many critically ill patients with COVID-19 develop respiratory failure characterized by poor gas exchange and damaging lung inflammation (1, 2).
This novel virus was quickly identified as a β-coronavirus that has 79.5% genetic similarity with severe acute respiratory syndrome coronavirus (SARS-CoV) and 50% with Middle East respiratory syndrome (MERS) (3–5). SARS-CoV-2 also shares a host receptor with SARS-CoV for cell entry, namely angiotensin-converting enzyme 2 (ACE2), via the binding of its surface protein spike (S) (4, 6, 7). The S protein of SARS-CoV-2 binds ACE2 more avidly than that of SARS-CoV, although these two S proteins share similar tertiary structures (8). Genomic comparison of SARS-CoV-2 with SARS-CoV shows there are 27 changes in the amino acid sequence of S, and the majority of these substitutions occur outside of the ACE2 binding domain (9). However, mutations in key S epitopes may contribute to conformational changes that increase ACE2 affinity, influence antigenicity, and/or affect the ability of SARS-CoV-2 to activate immune responses (10).
Although the S protein interaction with ACE2 has been the focus of vaccine design, other structural proteins likely play key roles in disease pathogenesis. The coronaviral genomes also encode structural proteins nucleocapsid (N), envelope (E), and membrane (9, 11). However, little is known about these structural proteins’ roles in immune activation and pathogenesis. The N protein has been shown to have an immunomodulatory function in SARS-CoV infection (12, 13). Interestingly, the SARS-CoV and SARS-CoV-2 E proteins have no amino acid substitutions. SARS-CoV E is essential for viral morphology, budding, and tropism (14, 15). Importantly, the SARS-CoV E was found to enhance inflammasome activation (16–19). Therefore, the conserved E protein and its engagement of the host immune response could prove to be a potent therapeutic intervention point useful for targeting multiple coronaviruses if its mechanistic actions are clearly understood (20).
During acute infection, COVID-19 patients are in a seemingly hyperinflammatory state with a dysregulated immune response (21). Similar to other RNA-viral infections, the pulmonary disease of COVID-19 is likely a combination of direct viral damage and this hyperactivated host immune response. Although lymphopenia has been a consistent finding in COVID-19 (9, 22, 23), many patients also exhibit a cytokine storm that is associated with disease severity and outcome (7–9, 21, 24–27). These patients demonstrate an increase in number of inflammatory monocytes and elevated serum levels of proinflammatory chemokines and cytokines including IL-2, IL-7, IL-10, IL-6, G-CSF, IP-10, MIP-1α, MCP-1, and TNF-α (1, 2, 7, 21, 25, 26, 28–34). Although these chemokines and cytokines attract immune cells to mount an antiviral defense, the resulting cytokine storm and cellular infiltration have been implicated in lung cell damage and disease pathogenesis.
Given the key roles of the innate immune response in both viral clearance and disease pathogenesis, understanding how SARS-CoV-2 structural proteins elicit host immunity is necessary for designing optimal therapeutic strategies. Therefore, we sought to investigate the innate immune response to SARS-CoV-2 Ags independent of viral infectivity and nuclei acid replication. In this report, we demonstrate that the purified structural proteins of SARS-CoV-2 alone activate inflammatory pathways in immune and epithelial cells and induce localized lung pathology dependent on IFN signaling in epithelial cells. These findings implicate the contribution of the viral surface proteins to driving inflammation in a cell type–specific manner and highlight their potential for therapeutic intervention.
Materials and Methods
Mice
All mice were originally obtained from The Jackson Laboratory (Bar Harbor, ME) and subsequently maintained at Washington University under specific pathogen–free conditions and were bred in-house. Adult (8–16-wk-old male and female) mice were anesthetized with isoflurane and intranasally administered 10 µg of truncated E protein (E-Trunc) (3531P; ProSci), S (40589-V08B1; Sino Biological, Beijing, China), or water (HyClone) in 50 µl total volume (25 µl/nostril). Mice were sacrificed on day 3 postadministration, and lung specimens were isolated and evaluated by histology. For the IFN-depleting experiments, mice were injected i.p. with 2 mg Ab in 500 µl volume (anti-Ifnar [I-401] or isotype control [I-443]; Leinco Technologies) 6 d prior and 0.5 mg Ab in 500 μl volume 2 d prior to intranasal administration of protein E. All animal protocols used in this study were approved by the Washington University’s Animal Studies Committee (19-0768), which approved these methods. Humane sacrifice of animals occurred with isoflurane administration and cervical dislocation.
Peritoneal and alveolar macrophage harvest and treatment
Peritoneal macrophages were collected via the mouse peritoneal cavity after lavage with 10 ml PBS (Sigma-Aldrich). Alveolar macrophages were isolated from the lungs after 0.7 ml of PBS was flushed serially via the trachea and fluid recollected. The isolated cells were pelleted at 6000 rpm for 4 min and then resuspended in DMEM media (with 10% FBS) (Sigma-Aldrich). Cells were seeded at 1 × 105 cells per 96 wells and incubated with SARS-CoV-2 S, E-Trunc, N protein (40588-V08B; Sino Biological), SARS-CoV-1 S (40634-V08B; Sino Biological), or MERS S (40069-V08B; Sino Biological) at 2 µg/ml or equal volume of water as control.
Reactive oxygen species activity measurement
Isolated cells above were stimulated with zymosan at 20 µg/ml (Sigma-Aldrich). A total of 50 µM luminol (Sigma-Aldrich) in 0.1 M NaOH (Sigma-Aldrich) (4 µl of 50 nM), and 1.6 U of HRP (Sigma-Aldrich) were added to each well. Chemiluminescence was measured in a SpectraMax L plate reader (Molecular Devices) for 2 h at 37°C.
Quantitative PCR analysis
Total mRNA was isolated from THP1 cells using RNeasy plus Mini Kit (74104; QIAGEN, Hilden, Germany) as per manufacturer’s instructions. The cDNA was made using 1 μg of RNA and iScript cDNA synthesis kit (1708890; Bio-Rad Laboratories). The quantitative PCR (qPCR) reaction was performed in triplicates using qPCR-specific primers (β2-microglobulin [β2M]–forward [F]: 5′-TGCTGTCTCCATGTTTGATGTATCT-3′; β2M–reverse [R]: 5′- TCTCTGCTCCCCACCTCTAAGT-3′; CCL5-F: 5′-CCTGCTGCTTTGCCTACATTGC-3′; CCL5-R: 5′-ACACACTTGGCGGTTCTTTCGG-3′; TNF-α–F: 5′- ATGGGCTACAGGCTTGTCACTC-3′; and TNF-α–R: 5′-CTCTTCTGCCTGCTGCACTTTG-3′) using TB green qPCR premix (639676; TaKaRa, Otsu, Japan) on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories). The fold change expression (−ΔΔCt) was calculated after normalization with β2M expression.
Cell lines
The cell lines A549-Dual (adenocarcinoma human alveolar basal epithelial cells a549-nfis; InvivoGen), RAW-Lucia ISG (RAW-mouse macrophagesrwal-isg; InvivoGen) and RAW-Dual KO-TLR4 (RAW-mouse macrophagesrawd-kotlr4; InvivoGen) were cultured in DMEM (Sigma-Aldrich). The growth media was supplemented with 10% FBS (Sigma-Aldrich), 1% (v/v) of penicillin/streptomycin, 100 µg/ml Normocin/Zeocin (InvivoGen). The A459 cells were also supplemented with 100 µg/ml Blasticidin (InvivoGen). The THP1-Dual, THP1-Dual KO-IRF3, THP1-Dual KO-TBK1, and THP1-Dual KO-MyD (thpd-koirf3/thpd-kotbk/thpd-komyd; InvivoGen) human lung monocytes were cultured in RPMI 1640 (Sigma-Aldrich) medium supplemented with 10% heat-inactivated FBS, 2 mM l-glutamine, 25 mM HEPES (Sigma-Aldrich), 1% (v/v) of penicillin/streptomycin, and 100 µg/ml of Normocin/Zeocin/Blasticidin (InvivoGen). The test media for A549 and THP1 cells excluded Zeocin and Blasticidin from their respective growth media.
Reporter cell assays
A549-Dual, THP1-Dual, THP1-Dual KOs (IRF3/TBK1/MyD88), RAW-Lucia ISG, and RAW-Dual KO-TLR4 cells stably express an IFN regulatory factor–inducible Lucia luciferase reporter construct. Cells were seeded at 1 × 106 or 1 × 105 cells per well in a 6- or 96-well plate, respectively. Cells were then incubated with SARS-CoV-2 S, E-Trunc, full-length E (E-Full), N protein, SARS-CoV-1 S, or MERS S protein at 2 µg/ml or equal volume of water as control, and after 24 h, culture supernatant was collected to measure luciferase. Polymyxin B (Pb; catalog tlrl-pmb; InvivoGen) at 10 µg/ml was added to all experimental conditions except those with RAW-Dual KO-TLR4 cells. QUANTI-Luc (rep-qlc1 and rep-qlc2; InvivoGen) was used to detect the level of luciferase by adding to culture supernatant and reading immediately with a plate reader (Infinite M200 Pro; Tecan Life Sciences, Zurich, Switzerland) at a 0.1-s reading time. QUANTI-Blue (rep-qbs, rep-qbs2, and rep-qbs3; InvivoGen) was used to detect the level of secreted embryonic alkaline phosphatase (SEAP) by adding to culture supernatant and incubating for 1 h and reading with a plate reader (Infinite M200 Pro; Tecan Life Sciences) at 650 nm.
Chemokine and cytokine analyses
Chemokine and cytokine protein quantification were performed using Proteome Human and Mouse Cytokine Array kits (R&D Systems, San Diego, CA) as per the manufacturer’s instructions. Dot arrays were quantified for pixel density with ImageJ (https://imagej.nih.gov/ij/).
Lung tissue preparation for histology
Lungs were inflated with formalin at the time of sacrifice and harvested into formalin containing conical tubes. The tissue was serially washed with PBS, 30% ethanol, and 50% ethanol 48 h after harvesting and stored in 70% ethanol until processed for paraffin embedding, sectioning, and staining. Ag retrieval was performed via boiling with Trilogy solution (920P-09; Cell Marque) for 20 min. The samples were incubated overnight at 4°C with primary Ab (Anti-Mouse CD-45 Ab [dilution 1:300]-550539; BD Pharmingen). The samples were incubated at room temperature with secondary Ab (dilution 1:300). The immunofluorescent staining for CD64 and GFP was done using Opal Multiplex IHC staining kit (NEL791001KT; PerkinElmer) as per the manufacturer’s instructions (three-plex immunohistochemistry in formalin paraffin-embedded tissue). The samples were incubated with primary Ab (CD64 [dilution 1:1000]-AF2074; R&D Systems; GFP Ab (dilution 1:1000)-ab13970; Abcam) for 30 min. With the secondary Ab (dilution 1:200), the samples were incubated for 30 min. The RNA in situ hybridization for assessment of ISG-15 expression (RNAscope probe-Mm-Isg15-01; ACDBio) was done using ACDBio RNAscope 2.5 HD Assay RED (322360) following the manufacturer’s instructions.
Software
ZEN 3.1 blue edition (Zeiss, Oberkochen, Germany) was used to visualize and image immunofluorescence staining of lung sections (https://www.zeiss.com/microscopy/us/products/microscope-software.html).
Statistics
GraphPad Prism (San Diego, CA) version 7.02 software was used to perform all statistical analyses as described.
Results
Purified SARS-CoV-2 proteins induce reactive oxygen species generation and proinflammatory chemokine and cytokine production
Increased reactive oxygen species (ROS) generation has been detected in clinical COVID-19 sputum samples (35), although it is unclear to what extent active infection or inflammatory stimulation contribute to this finding. To examine the role of the SARS-CoV-2 surface proteins in directly (i.e., the absence of infectious virus) activating this innate immune cell effector function, we investigated the ability of S and E Ags to induce ROS generation in macrophages. Although SARS-CoV-2 does not infect wild-type (wt) mice in vivo (36), S protein and an N-terminal 10-aa E-Trunc potently enhanced zymosan-induced ROS generation in ex vivo–isolated wt murine peritoneal macrophages after overnight incubation compared with control samples by 2.09 ± 0.35-fold and 2.63 ± 0.95-fold, respectively. (Fig. 1A). Alveolar macrophages also demonstrated increased zymosan-induced ROS production in response to E-Trunc (1.71 ± 0.19-fold), but not in response to S (Fig. 1B).
FIGURE 1.
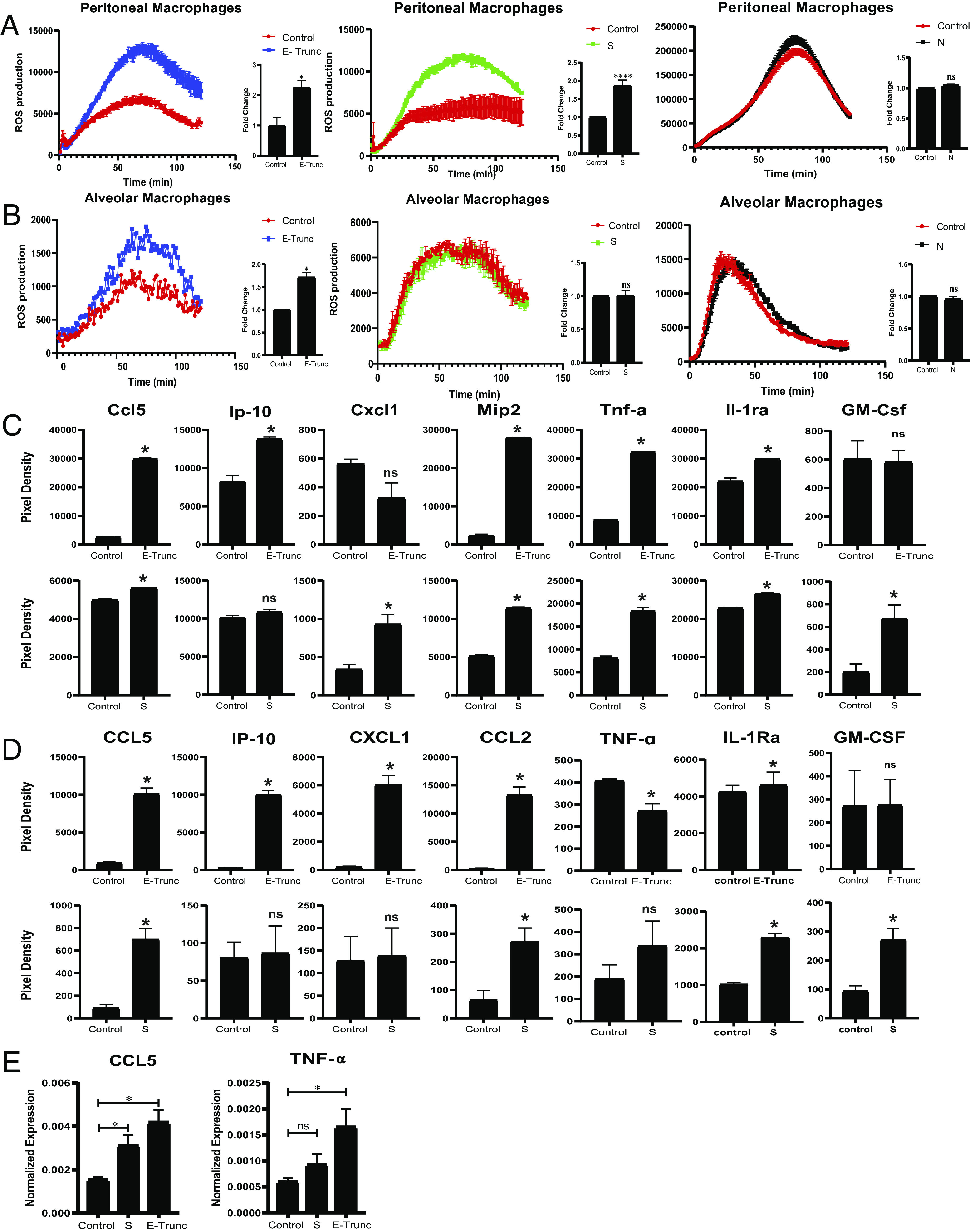
SARS-CoV-2 Ags induce macrophages to produce ROS and express proinflammatory chemokines and cytokines. (A and B) ROS production in peritoneal (A) and alveolar (B) macrophages after 20 µg/ml zymosan stimulation and incubation with SARS-CoV-2 peptides E-Trunc, S, and N. Representative figures for ROS production and area under the curve with n = 2 experiments: 6 biological and 6 technical for E-Trunc and N; n = 5 experiments: 15 biological and 15 technical for S for peritoneal macrophages; n = 2 experiments: 4 biological and 4 technical for E-Trunc and N; and n = 3 experiments: 6 biological and 6 technical for S for alveolar macrophages. (C and D) Detection of chemokines and cytokines in the culture supernatant of RAW (C) and THP1 (D) cells incubated with control, E-Trunc, or S at 2 µg/ml for 24 h. The graphs show measurements of the pixel density (n = 2 biological samples for each condition with 2 technical replicates per sample). (E) Expression of CCL5 and TNF-α RNA from THP1 cells incubated with control, S, or E-Trunc at 2 µg/ml for 3 h (n = 2 experiments: 4 biological and 4 technical replicates per sample). Graphs depict average with SEM. Mann–Whitney U test was used for statistical analysis in (A)–(D) and one-way ANOVA in (E). *p < 0.05, ****p < 0.0001. ns, not statistically significant.
We also tested the structural SARS-CoV-2 N protein for its ability to enhance zymosan-induced ROS generation. Of note, the N structural protein is contained inside the virion, whereas E and S are displayed on the viral surface. This protein also serves as an important control, as the N protein was purified and obtained in an identical manner as SARS-CoV-2 S protein. However, N did not increase ROS production in peritoneal or alveolar macrophages (Fig. 1A, 1B). To assess the specificity of SARS-CoV-2 surface proteins to enhance ROS production, we also analyzed the response to SARS-CoV-1 and MERS S proteins. Neither SARS-CoV-1 nor MERS S protein led to enhanced zymosan-induced ROS generation in ex vivo–isolated murine peritoneal and alveolar macrophages (Supplemental Fig. 1A). Of note, these S proteins were also purified and obtained in the same manner as SARS-CoV-2 S and N.
Given the finding that viral surface proteins induced an increase in an innate immune effector function in mouse cells, we also examined the induction of specific chemokines and cytokines in murine myeloid reporter cells. Similar to induction of ROS generation in primary cells, E-Trunc and S induced increases in chemokines and cytokines when incubated with IFN reporter RAW cells (RAW-Lucia ISG). Both E-Trunc and S enhanced the following chemokines and cytokines: Ccl5/CCL5, Mip-2, CCL2, Tnf-α, and IL-1rα/IL-1Rα (Fig. 1C). E-Trunc peptide independently increased the expression of Ip-10/IP-10, whereas the S protein increased Cxcl1 and GM-Csf/GM-CSF. This distinct induction of specific chemokines and cytokines indicates that these viral proteins likely induce host inflammatory responses by different mechanisms.
To determine if human myeloid cells similarly responded to the SARS-CoV-2 structural proteins, we next incubated human monocyte THP1 reporter cells (THP1-Dual) with E-Trunc and S Ags for 24 h. Indeed, E-Trunc and S induced both shared and distinct increases in inflammatory mediators in human monocytes (Fig. 1D). E-Trunc dramatically increased the expression of CCL5 (10-fold), IP-10 (41-fold), CXCL1 (30-fold), and MIP-1α (57-fold). S protein similarly increased the expression of CCL5 (7.6-fold) and MIP-1α (4.2-fold), albeit to a lesser magnitude than increased by E-Trunc. The increase in CCL5 transcript expression was confirmed by quantitative real time–PCR (Fig. 1E). Interestingly, S protein alone specifically increased IL-1Rα (2.3-fold) and GM-CSF (2.9-fold). These findings underscore shared and distinct immune responses to specific coronavirus surface Ags and imply unique mechanisms of activation.
Increased serum TNF-α has been found during COVID-19 infection (9, 25, 26, 31). Previous work has also shown a specific increase in TNF-α expression in mouse macrophages by the SARS-CoV S protein (37). Likewise, we also found that TNF-α increased in mouse monocytes incubated with SARS-CoV-2 proteins E-Trunc or S (Fig. 1C). However, in human myeloid cells, our results were inconsistent; there was no difference in the levels of TNF-α after exposure to S and a small decrease (0.7-fold) in protein, but increased mRNA transcript after incubation with E-Trunc (Fig. 1D, 1E). These results highlight key commonalities and differences in inflammatory responses between human and mouse cells. This knowledge bears critical attention as we rely on animal models to investigate SARS-CoV-2 mechanistically and test new therapeutic strategies.
Purified SARS-CoV-2 proteins induce inflammatory signaling
To verify that our findings were not due to contamination of the protein preparations, we tested for LPS specifically using the limulus amebocyte lysate assay and found minimal LPS (<0.4 ng/ml) in our viral protein preparations, consistent with the manufacturer’s report.
As our findings above showed that E-Trunc and S proteins upregulate multiple chemokines and cytokines known to be IFN responsive, we directly asked whether these Ags activate IFN induction. To further assure against LPS contamination, we performed the following experiments in the presence of 10 µg/ml Pb, a potent LPS-neutralizing agent that inhibited LPS induction of both IFN and NF-κB signaling at 1 ng/ml (Supplemental Fig. 2A).
We incubated IFN reporter cell lines, which harbor tandem IFN-stimulated response elements inducing luciferase expression, with E-Trunc and S as well as the SARS-CoV-2 structural protein N, E-Full, SARS-CoV-1 S, and MERS S proteins individually. After 24 h, IFN induction was enhanced by E and S in both murine and human monocytes, most robustly by E-Full in the human THP1 cells and E-Trunc in the murine RAW cells (Fig. 2A). E-Full enhanced luciferase expression by 3-fold in RAW cells and 6.5-fold in THP1 cells, whereas E-Trunc led to 5.5-fold and 2.8-fold increases, respectively. Protein S enhanced IFN signaling in these cells to a lesser extent by 1.3-fold in RAW cells and 1.5-fold in THP1 cells. Similar to our ROS findings, the SARS-CoV-2 structural nonsurface protein N did not induce IFN signaling in any of the cell lines tested. Neither SARS-CoV-1 S nor MERS S elicited IFN signaling in human (Supplemental Fig. 1B) or murine macrophages (Supplemental Fig. 1C).
FIGURE 2.
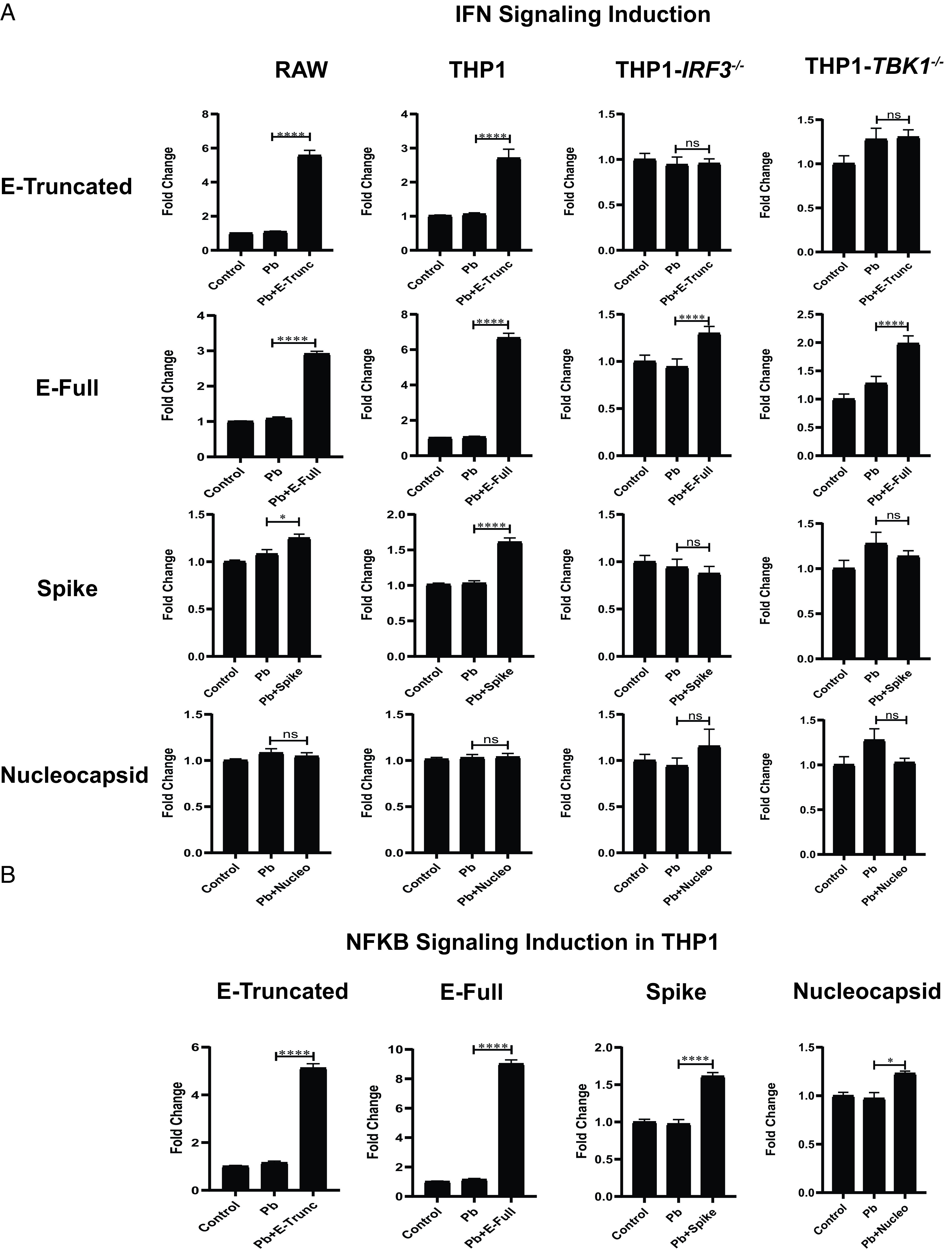
SARS-CoV-2 Ags induce IFN and NF-κB signaling. (A) Fold change in IFN reporter activity in RAW, THP1, THP1-IRF3−/−, or THP1-TBK1−/− cells treated with control or polymyxin (Pb) at 10 µg/ml or SARS-CoV-2 Ags (E-Trunc, E-Full, S, or N) at 2 µg/ml and Pb at 10 µg/ml for 24 h (n = 2 experiments: 6 biological and 9 technical replicates for RAW with each viral Ag; n = 3 experiments: 9 biological and 6–12 technical replicates for THP1 with each viral Ag; and n = 2 experiments: 9 biological and 9 technical replicates for THP1-IRF3−/−and THP1-TBK1−/−cells with each viral Ag). (B) Fold change in NF-κB reporter activity in THP1 cells treated with control or Pb at 10 µg/ml or SARS-CoV-2 Ags (E-Trunc, E-Full, S, or N) at 2 µg/ml and Pb at 10 µg/ml for 24 h (n = 3 experiments: 9 biological and 6–12 technical replicates for THP1 with each viral Ag). Graphs depict average with SEM. *p < 0.05, ****p < 0.0001. ns, not statistically significant, by Mann–Whitney U test.
To gain further mechanistic insight into how viral Ags independently activate IFN signaling, we investigated the role of known mediators of IFN induction. THP1 reporter cells deficient in IRF3 and TBK1 did not exhibit IFN induction in response to E-Trunc or S and had a dramatic decrease in response to E-Full (Fig. 2A), demonstrating key dependence on these well-described IFN induction mediators. As MyD88 also modulates IFN responses, we also tested THP1 cells deficient in this mediator. Indeed, E-Trunc and E-Full showed partial decreases in IFN induction in THP1-Dual KO-MyD88 cells, whereas the response to S was abolished (Supplemental Fig. 2B).
We found that E and S proteins maintained an increase in IFN signaling induction in cells deficient in Tlr4 (Supplemental Fig. 2C), also indicating our results are not due to LPS contamination. In further confirmation, we assessed the responsiveness of our reporter cells lines to low doses of LPS (0.1–0.5 ng/ml) and found minimal responsiveness comparatively in THP-1 and RAW cells (Supplemental Fig. 2D).
Given that these THP1 cells are also capable of reporting NF-κB induction, we investigated the ability of these viral Ags to induce NF-κB signaling. NF-κB induction was also increased in response to SARS-CoV-2 proteins, with the most striking response to E-Full (8.5-fold) and least induction to N protein (1.2-fold) (Fig. 2B). Overall, these responses were dependent on MyD88, consistent with its well-described role in NF-κB signaling, as THP1-MyD−/− cells had decreased NF-κB induction in response to E proteins and no response to S or N (Supplemental Fig. 2B).
Purified SARS-CoV-2 peptides E and S induce lung inflammation and pathology in a mouse model
The critical events that follow an acute pulmonary SARS-CoV-2 infection are injurious viral infection and exuberant immune responses with resultant lung inflammation. Given our findings that E and S can induce similar inflammatory activation pathways in murine cells in vitro, we next studied the direct effect of these viral surface Ags in vivo. We administered E-Trunc and S intranasally to C57BL/6J wt mice and examined the effect on lung histology 3 d later. Cross-sections of lungs showed significant organized peribronchial and medium-sized airway pathology in those mice exposed to E-Trunc or S compared with control-treated mice (Fig. 3A, 3D). We used a graded scoring system to quantify the degree of pathology across specimens (Supplemental Fig. 3). Immunostaining for CD45 demonstrated the hematopoietic origin of these inflammatory infiltrates (Fig. 3B). Furthermore, animals exposed to E-Trunc and S also showed significant vascular pathology with evidence of vasculitis (Fig. 3C), a finding that has been uniquely highlighted in patients with COVID-19 disease (38). These observations demonstrate a striking and direct role of the viral surface proteins in induction of SARS-CoV-2–mediated pathology independent of active viral infection and replication.
FIGURE 3.
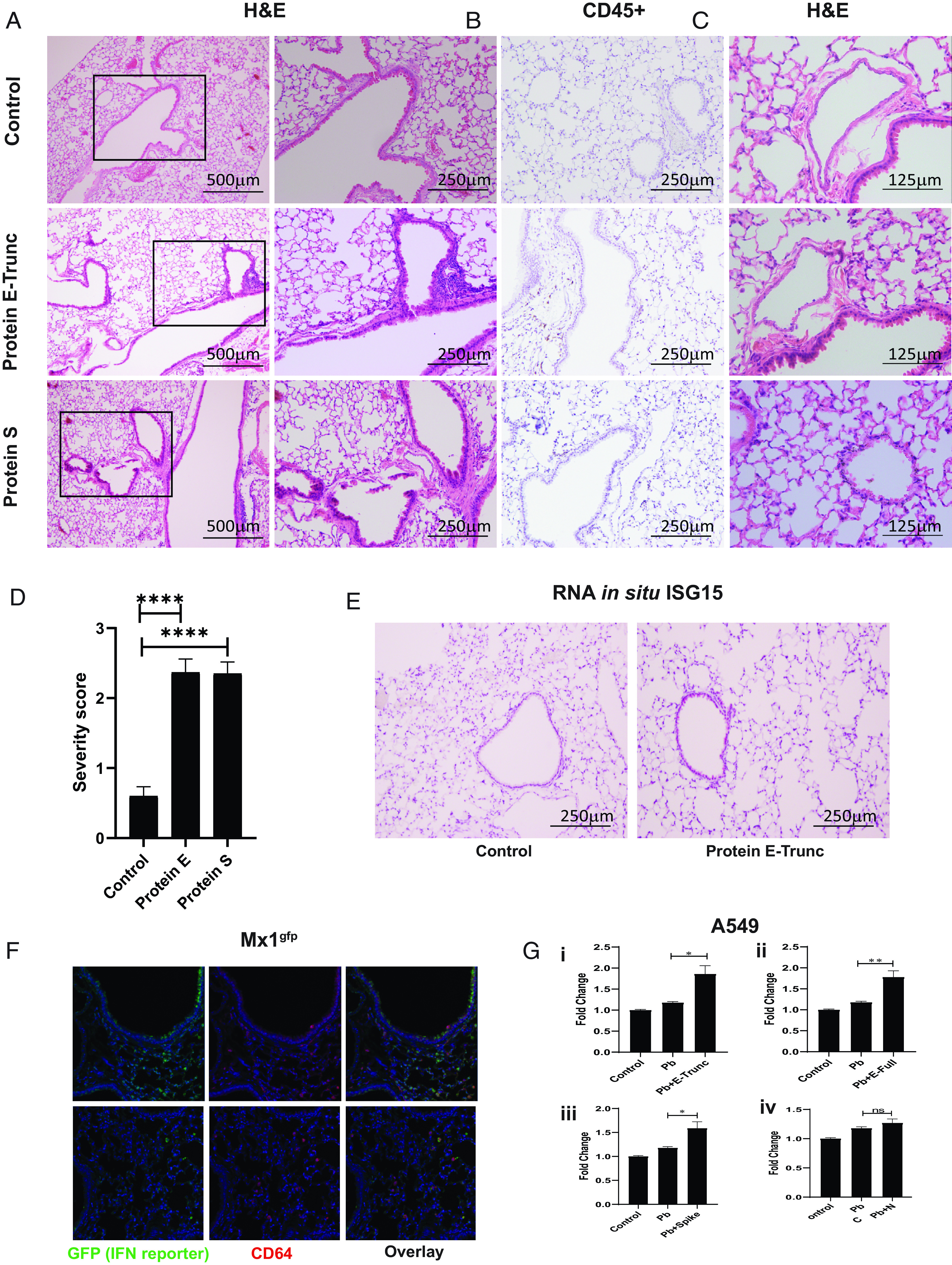
SARS-CoV-2 viral Ags induce lung inflammation and vasculitis in mice. (A) Representative images of lung cross-sections from mice sacrificed 3 d after intranasal delivery of control, E-Trunc, or S at 10 µg. H&E-stained sections are shown. Boxed areas on the left are magnified adjacently. (B) Representative images of the lung cross-sections immunostained for CD45 expression. (C) Representative images of lung cross-sections depicting blood vessel pathology in each condition. Scale bars depicted in each picture. (D) Quantification of percentage of lobes with inflammatory infiltrates in lungs harvested in each condition (n = 3 mice per condition). (E) Representative images of the lung cross-sections stained for Isg15 by RNA in situ (n = 3 mice per condition). (F) Representative immunofluorescent images of the lung cross-sections immunostained for GFP and CD64 expression per above conditions; original magnification ×200 (n = 2 mice per condition). (G) Fold change in IFN reporter activity in A549 cells treated with control, Pb at 10 µg/ml, or SARS-CoV-2 Ags [2 µg/ml E-Trunc (i), E-Full (ii), S (iii), or N (iv)] and Pb at 10 µg/ml for 24 h (n = 2 experiments: 6 biological and 9–21 technical replicates for each viral Ag). Graphs depict average with SEM. One-way ANOVA in (D) and Mann–Whitney U test in (G) used for statistical analysis. *p < 0.05, **p < 0.01, ****p < 0.0001. ns, not statistically significant.
Further investigation revealed IFN activation in vivo, as E-Trunc–treated animals showed evidence of IFN-stimulated gene responses by scattered Isg15 staining by RNA in situ as compared with control-treated animals (Fig. 3E). Notably, Isg15 staining was demonstrated in medium-sized airways as well as scattered peripherally in terminal alveolar spaces. Therefore, we next investigated which cell types respond to viral peptide–mediated IFN signaling in vivo using the Mx1gfp reporter mouse (39). Three days after protein E-Trunc intranasal administration, fluorescent GFP+ staining of lung cross-sections demonstrated IFN responses in patchy epithelial cells of medium-sized airways and CD64+ cells (monocytes and macrophages) in response to viral peptide (Fig. 3F).
In light of this airway epithelial IFN responsiveness, we sought to determine the cell-intrinsic induction of inflammatory signaling by coronavirus proteins in A549 reporter cells. SARS-CoV-2 E-Trunc, E-Full, and S enhanced IFN signaling in reporter A549 pulmonary epithelial cells by ∼1.5-fold each; N and SARS-CoV-1 S did not induce IFN signaling, whereas MERS S had a 1.1-fold effect (Fig. 3G, Supplemental Fig. 4A). None of the SARS-CoV-2 proteins induced NF-κB signaling in A549 cells (there was a small response to MERS S, but not SARS-CoV-1 S) (Supplemental Fig. 4A, 4B), although these cells are responsive to LPS at low doses (Supplemental Fig. 4C).
SARS-CoV-2 E protein–mediated organized inflammation is dependent on type I IFN in pulmonary epithelial cells
To determine the role of type I IFN signaling in SARS-CoV-2 surface protein induction of pulmonary pathology, we rendered the type I IFN signaling pathway defective genetically via the type I IFN receptor (Ifnar−/−) or using an Ifnar blocking mAb (40) prior to viral peptide treatment. Ifnar−/− animals or those that received the blocking Ab demonstrated an altered inflammatory infiltrative pattern, as demonstrated on lung histological cross-sections compared with wt littermates or isotype-treated controls, respectively (Fig. 4A, 4B). These IFN-deficient animals had similar scattered immune cells readily apparent in the alveoli spaces, whereas the control animals exhibited the previously seen organized infiltrates surrounding medium to large airways.
FIGURE 4.
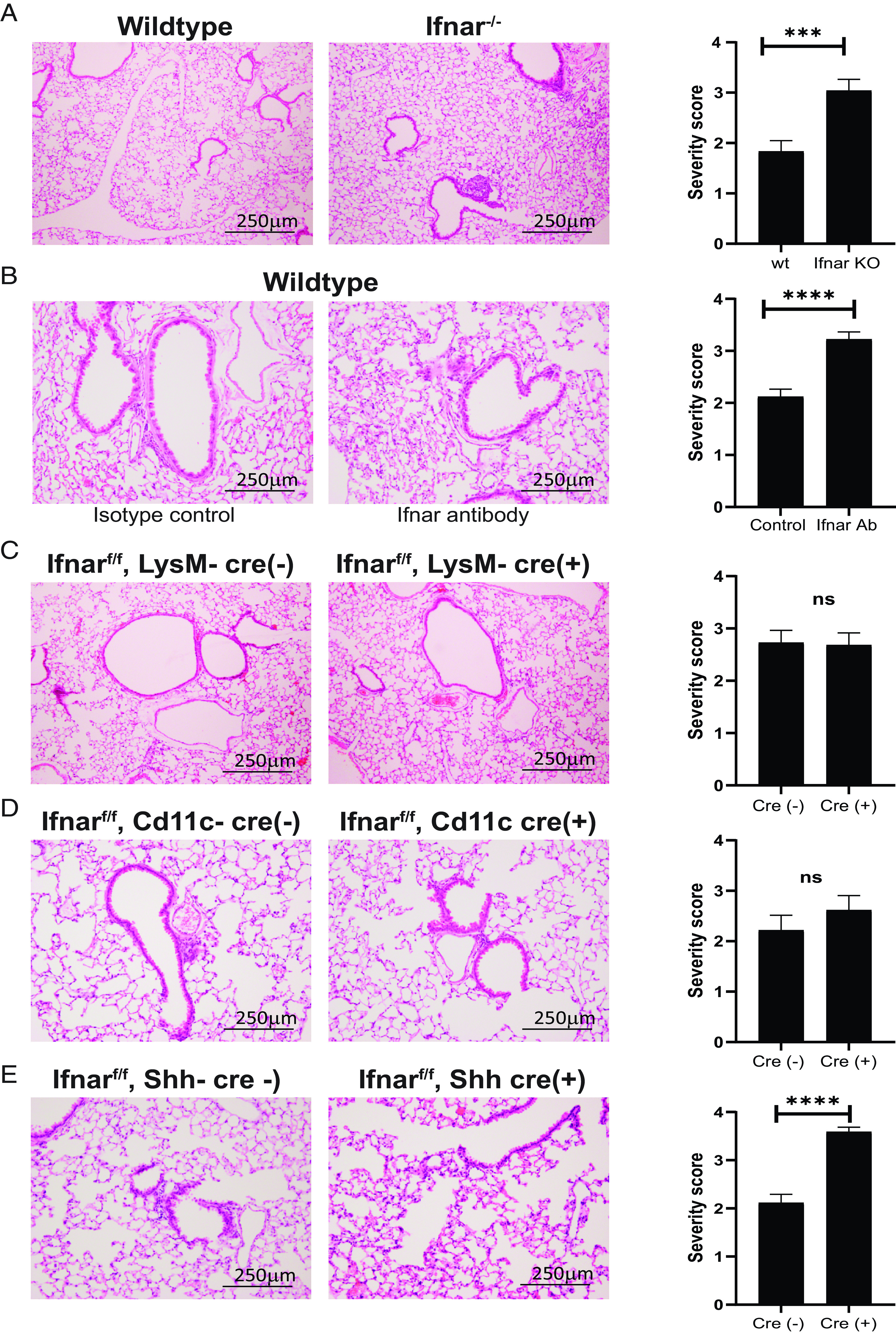
SARS-CoV-2 viral Ag E induces airway epithelial IFN-dependent inflammation. (A) Representative images of lung cross-sections from Ifnar−/− mice sacrificed 3 d after intranasal delivery of control or E-Trunc at 10 µg. H&E-stained sections are shown (n = 3 with 4–5 mice per condition). (B) Representative images of lung cross-sections from wt mice sacrificed 3 d after intranasal delivery of E-Trunc at 10 µg subsequent to Ifnar-depleting or isotype control Ab administration. H&E-stained sections are shown (n = 2 with 6 mice per condition). (C) Representative images of lung cross-sections from Ifnarf/f;LysM-Cre(+/–) mice sacrificed 3 d after intranasal delivery of control or E-Trunc at 10 µg. H&E-stained sections are shown (n = 3 mice per condition). (D) Representative images of lung cross-sections from Ifnarf/f;Cd11c-Cre(+/–) mice sacrificed 3 d after intranasal delivery of control or E-Trunc at 10 µg. H&E-stained sections are shown (n = 2 mice per condition). (E) Representative images of lung cross-sections from Ifnarf/f;Shh-Cre(+/–) mice sacrificed 3 d after intranasal delivery of control or E-Trunc at 10 µg. H&E-stained sections are shown (n = 3 with 8–9 mice per condition). Scale bars depicted in each picture. Severity scores per lobe are quantified to the right of each experimental condition in (A)–(E). Graphs depict average with SEM. ***p < 0.001, ****p < 0.0001, by Mann–Whitney U test. ns, not statistically significant.
Given the dependence on IFN signaling to induce organized inflammation in response to SARS-CoV-2 structural peptides, we used a genetic conditional deletion of the type I IFN receptor (Ifnarf/f) (41) to determine in which specific cell types IFN signaling determines pulmonary pathology. We targeted the myeloid lineage broadly using LysM-Cre (42) as well as alveolar macrophages and dendritic cells using Cd11c-Cre transgenic mice (43) crossed to Ifnarf/f mice. SARS-CoV-2 E-Trunc peptide–induced pulmonary pathology was unaffected in Ifnarf/f;LysM-Cre(+) and Ifnarf/f;Cd11c-Cre(+) compared with their littermate Cre(–) controls (Fig. 4C, 4D). Organized immune infiltrates surrounding medium to large airways was indistinguishable between these groups on lung histological cross-sections. However, mice with type I IFN signaling genetically abolished specifically in pulmonary epithelial cells (44) [Ifnarf/f;Shh-Cre(+)] exhibited similar pathology to global blockade of IFN signaling in response to E-Trunc (Fig. 4E), whereas the control Ifnarf/f;Shh-Cre(–) lungs exhibited the afore-observed wt pathology. These findings implicate the importance of IFN signaling in the pulmonary epithelium as the necessary driver of organized medium to large airway inflammation in response to SARS-CoV-2 surface Ags.
Discussion
The novel coronavirus SARS-CoV-2 that has caused the COVID-19 global health crisis necessitates a thorough investigation of the host immune response to develop effective therapeutic strategies. The innate immune system is the first line of defense that is critical for viral pathogen clearance, and at the same time, it is also implicated in the pathogenesis of many viral disease processes (45). Studies have shown that a hyperinflammatory state and a dysregulated immune response may underlie COVID-19 pathogenesis (1, 2, 7–9, 21, 24–27). COVID-19 patients experience a characteristic cytokine storm with sharply high levels of proinflammatory mediators that is directly proportional to viral load and severity of illness (1, 2, 7, 21, 25, 26, 29–34).
Although viral nucleic acid sensing is the predominantly accepted mechanism for virus detection by pathogen-recognition receptors, viral surface proteins may also directly activate the innate immune system independent of virus uncoating and replication. This knowledge is crucial to understand the initiation of the inflammatory response and the mechanism of viral engagement of the immune system. In addition, this mechanism is important to consider given its implications for noninfected cells to induce an immune response, as recognition of viral Ags may occur independently of viral uncoating and replication and, thus, may not be restricted to cells or tissues permissive to infection.
Hence, we evaluated the ability of isolated SARS-CoV-2 structural Ags to activate IFN signaling, a key innate immune pathway that bridges to adaptive immune responses. Our findings demonstrate that the SARS-CoV-2 surface peptides E and S independently activate IFN signaling in both immune and epithelial cells. We show that these viral Ags individually alter the expression of key chemokines and cytokines, including many regulated by IFN, in both human and murine cell lines. Distinctly, the truncated E peptide enhanced the levels of human CCL5, IP-10, CXCL1, and MIP-1α, which are associated with neutrophil and monocyte recruitment. These findings are essential in light of in vivo infection, as COVID-19 patients often have a high ratio of neutrophils to lymphocytes (30). In murine cells, the E protein led to increased levels of TNF-α, which is also markedly increased in human SARS-CoV-2 infection (1, 21). Furthermore, we demonstrate that in vivo delivery of these peptides, particularly E-Trunc, to mice induces peribronchial and medium-sized airway inflammation and vasculitis, which are recapitulated in human disease specimens (38, 46). This inflammatory recruitment is dependent on IFN signaling in epithelial cells as specific genetic IFN signaling deficiency in pulmonary epithelial cells abolished organized inflammation, although alveolar inflammatory infiltrates persisted. These findings indicate that the pulmonary epithelium can induce IFN signaling and localized inflammation in response to SARS-CoV-2 viral surface protein recognition. Similarly, the SARS-CoV E protein induced severe lung pathology, including profuse hemorrhage and cellular infiltration with elevation of cytokines (47). The significance to disease pathogenesis of these inflammatory responses with distinct pathological patterns warrants further assessment in genetically modifiable host–pathogen and SARS-CoV-2 host–susceptible model systems.
Although the S protein is responsible for cell entry via ACE2 and is the focus of numerous therapeutic strategies, the E protein of SARS-CoV-2 is understudied, although recent evidence points to its potential as an ion channel (48). Prior work in other coronaviruses has demonstrated that E protein is indispensable for viral morphogenesis and tropism as well as enhances inflammasome activation (14–18); our work further points to its crucial role on innate immune activation and function. Interestingly, the ability of protein E to induce IFN is markedly reduced in the absence of TLR4, suggesting dependence on this receptor. This potential interaction is bolstered by emerging evidence that S protein of SARS-CoV-2 interacts with TLR4 (49–51). Given that E is highly conserved with SARS-CoV (9, 52), further study is necessary, as E may be a potent target for therapeutic strategies with broader applications, including anticipated emerging coronaviruses. Our observations also highlight the importance of the direct effect of coronavirus surface proteins and will usher investigation of other viral surface proteins as determinants of the host–pathogen interaction.
Finally, this work has broad implications for the pathogen–host immune response; we show that activation of innate immune signaling pathways independent of viral nucleic acid detection by pathogen-recognition receptors engage host immunity similarly to complete infectious virus. Understanding the immune response to independent viral structural proteins is an important step forward in deciphering the interaction of this novel virus, as well as other clinically relevant viruses, with host immunity.
Supplementary Material
This work was supported by the Washington University and Burroughs Wellcome Fund.
The online version of this article contains supplemental material.
- ACE2
- angiotensin-converting enzyme 2
- E
- envelope
- E-Full
- full-length E
- E-Trunc
- truncated E protein
- F
- forward
- β2M
- β2-microglobulin
- MERS
- Middle East respiratory syndrome
- N
- nucleocapsid
- Pb
- polymyxin B
- qPCR
- quantitative PCR
- R
- reverse
- ROS
- reactive oxygen species
- S
- spike
- SARS-CoV
- severe acute respiratory syndrome coronavirus
- wt
- wild-type
Disclosures
The authors have no financial conflicts of interest.
References
- 1. Huang D., Lian X., Song F., Ma H., Lian Z., Liang Y., Qin T., Chen W., Wang S.. 2020. Clinical features of severe patients infected with 2019 novel coronavirus: a systematic review and meta-analysis. Ann. Transl. Med. 8: 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mehta P., McAuley D. F., Brown M., Sanchez E., Tattersall R. S., Manson J. J., HLH Across Speciality Collaboration, UK . 2020. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395: 1033–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Petrosillo N., Viceconte G., Ergonul O., Ippolito G., Petersen E.. 2020. COVID-19, SARS and MERS: are they closely related? Clin. Microbiol. Infect. 26: 729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu C., Chen X., Cai Y., Xia J., Zhou X., Xu S., Huang H., Zhang L., Zhou X., Du C., et al. 2020. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern. Med. 180: 934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim J. M., Chung Y. S., Jo H. J., Lee N. J., Kim M. S., Woo S. H., Park S., Kim J. W., Kim H. M., Han M. G.. 2020. Identification of coronavirus isolated from a patient in Korea with COVID-19. Osong Public Health Res. Perspect. 11: 3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T. S., Herrler G., Wu N. H., Nitsche A., et al. 2020. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181: 271–280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang F., Hou H., Luo Y., Tang G., Wu S., Huang M., Liu W., Zhu Y., Lin Q., Mao L., et al. 2020. The laboratory tests and host immunity of COVID-19 patients with different severity of illness. JCI Insight 5: e137799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gong J., Dong H., Xia Q. S., Huang Z. Y., Wang D. K., Zhao Y., Liu W. H., Tu S. H., Zhang M. M., Wang Q., Lu F. E.. 2020. Correlation analysis between disease severity and inflammation-related parameters in patients with COVID-19: a retrospective study. BMC Infect. Dis. 20: 963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., et al. 2020. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395: 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Korber B., Fischer W. M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E. E., Bhattacharya T., Foley B., et al. Sheffield COVID-19 Genomics Group . 2020. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182: 812–827.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim D., Lee J. Y., Yang J. S., Kim J. W., Kim V. N., Chang H.. 2020. The architecture of SARS-CoV-2 transcriptome. Cell 181: 914–921.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lu X., Pan J., Tao J., Guo D.. 2011. SARS-CoV nucleocapsid protein antagonizes IFN-β response by targeting initial step of IFN-β induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes 42: 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hu Y., Li W., Gao T., Cui Y., Jin Y., Li P., Ma Q., Liu X., Cao C.. 2017. The severe acute respiratory syndrome coronavirus nucleocapsid inhibits type i interferon production by interfering with TRIM25-mediated RIG-I ubiquitination. J. Virol. 91: e02143-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pervushin K., Tan E., Parthasarathy K., Lin X., Jiang F. L., Yu D., Vararattanavech A., Soong T. W., Liu D. X., Torres J.. 2009. Structure and inhibition of the SARS coronavirus envelope protein ion channel. PLoS Pathog. 5: e1000511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arbely E., Khattari Z., Brotons G., Akkawi M., Salditt T., Arkin I. T.. 2004. A highly unusual palindromic transmembrane helical hairpin formed by SARS coronavirus E protein. J. Mol. Biol. 341: 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nieto-Torres J. L., Verdiá-Báguena C., Jimenez-Guardeño J. M., Regla-Nava J. A., Castaño-Rodriguez C., Fernandez-Delgado R., Torres J., Aguilella V. M., Enjuanes L.. 2015. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 485: 330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen I. Y., Moriyama M., Chang M. F., Ichinohe T.. 2019. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front. Microbiol. 10: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shi C. S., Nabar N. R., Huang N. N., Kehrl J. H.. 2019. SARS-coronavirus open reading frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 5: 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. De Maio F., Lo Cascio E., Babini G., Sali M., Della Longa S., Tilocca B., Roncada P., Arcovito A., Sanguinetti M., Scambia G., Urbani A.. 2020. Improved binding of SARS-CoV-2 envelope protein to tight junction-associated PALS1 could play a key role in COVID-19 pathogenesis. Microbes Infect. 22: 592–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schoeman D., Fielding B. C.. 2020. Is there a link between the pathogenic human coronavirus envelope protein and immunopathology? A review of the literature. Front. Microbiol. 11: 2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Giamarellos-Bourboulis E. J., Netea M. G., Rovina N., Akinosoglou K., Antoniadou A., Antonakos N., Damoraki G., Gkavogianni T., Adami M. E., Katsaounou P., et al. 2020. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe 27: 992–1000.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu Z., McGoogan J. M.. 2020. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72 314 cases from the Chinese center for disease control and prevention. JAMA 323: 1239–1242. [DOI] [PubMed] [Google Scholar]
- 23. Guan W. J., Ni Z. Y., Hu Y., Liang W. H., Ou C. Q., He J. X., Liu L., Shan H., Lei C. L., Hui D. S. C., et al. China Medical Treatment Expert Group for Covid-19 . 2020. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 382: 1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ruan Q., Yang K., Wang W., Jiang L., Song J.. 2020. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. [Published erratum appears in 2020 Intensive Care Med. 46: 1294–1297.] Intensive Care Med. 46: 846–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen G., Wu D., Guo W., Cao Y., Huang D., Wang H., Wang T., Zhang X., Chen H., Yu H., et al. 2020. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 130: 2620–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qin C., Zhou L., Hu Z., Zhang S., Yang S., Tao Y., Xie C., Ma K., Shang K., Wang W., Tian D. S.. 2020. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 71: 762–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang Y., Shen C., Li J., Yuan J., Wei J., Huang F., Wang F., Li G., Li Y., Xing L., et al. 2020. Plasma IP-10 and MCP-3 levels are highly associated with disease severity and predict the progression of COVID-19. J. Allergy Clin. Immunol. 146: 119–127.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou Y., Fu B., Zheng X., Wand D., Zhao C., Qi Y., Sun R., Tian Z., Xu X., Wei H.. 2020. Pathogenic T cells and inflammatory monocytes incite inflammatory storm in severe COVID-19 patients. Natl. Sci. Rev. 7: 998–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wölfel R., Corman V. M., Guggemos W., Seilmaier M., Zange S., Müller M. A., Niemeyer D., Jones T. C., Vollmar P., Rothe C., et al. 2020. Virological assessment of hospitalized patients with COVID-2019. [Published erratum appears in 2020 Nature 588: E35.] Nature 581: 465–469. [DOI] [PubMed] [Google Scholar]
- 30. Zhou Z., Ren L., Zhang L., Zhong J., Xiao Y., Jia Z., Guo L., Yang J., Wang C., Jiang S., et al. 2020. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe 27: 883–890.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Diao B., Wang C., Tan Y., Chen X., Liu Y., Ning L., Chen L., Li M., Liu Y., Wang G., et al. 2020. Reduction and functional exhaustion of t cells in patients with coronavirus disease 2019 (COVID-19). Front. Immunol. 11: 827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang M. Q., Wang X. H., Chen Y. L., Zhao K. L., Cai Y. Q., An C. L., Lin M. G., Mu X. D.. 2020. [Clinical features of 2019 novel coronavirus pneumonia in the early stage from a fever clinic in Beijing]. Zhonghua Jie He He Hu Xi Za Zhi 43: 215–218. [DOI] [PubMed] [Google Scholar]
- 33. Wen W., Su W., Tang H., Le W., Zhang X., Zheng Y., Liu X., Xie L., Li J., Ye J., et al. 2020. Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. [Published erratum appears in 2020 Cell Discov. 6: 41.] Cell Discov. 6: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blanco-Melo D., Nilsson-Payant B. E., Liu W. C., Uhl S., Hoagland D., Møller R., Jordan T. X., Oishi K., Panis M., Sachs D., et al. 2020. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181: 1036–1045.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miripour Z. S., Sarrami-Forooshani R., Sanati H., Makarem J., Taheri M. S., Shojaeian F., Eskafi A. H., Abbasvandi F., Namdar N., Ghafari H., et al. 2020. Real-time diagnosis of reactive oxygen species (ROS) in fresh sputum by electrochemical tracing; correlation between COVID-19 and viral-induced ROS in lung/respiratory epithelium during this pandemic. Biosens. Bioelectron. 165: 112435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bao L., Deng W., Huang B., Gao H., Liu J., Ren L., Wei Q., Yu P., Xu Y., Qi F., et al. 2020. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 583: 830–833. [DOI] [PubMed] [Google Scholar]
- 37. Wang W., Ye L., Ye L., Li B., Gao B., Zeng Y., Kong L., Fang X., Zheng H., Wu Z., She Y.. 2007. Up-regulation of IL-6 and TNF-alpha induced by SARS-coronavirus spike protein in murine macrophages via NF-kappaB pathway. Virus Res. 128: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang W., Zhao Y., Zhang F., Wang Q., Li T., Liu Z., Wang J., Qin Y., Zhang X., Yan X., et al. 2020. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): the perspectives of clinical immunologists from China. Clin. Immunol. 214: 108393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Uccellini M. B., García-Sastre A.. 2018. ISRE-reporter mouse reveals high basal and induced type I IFN responses in inflammatory monocytes. Cell Rep. 25: 2784–2796.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sheehan K. C., Lai K. S., Dunn G. P., Bruce A. T., Diamond M. S., Heutel J. D., Dungo-Arthur C., Carrero J. A., White J. M., Hertzog P. J., Schreiber R. D.. 2006. Blocking monoclonal antibodies specific for mouse IFN-alpha/beta receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. J. Interferon Cytokine Res. 26: 804–819. [DOI] [PubMed] [Google Scholar]
- 41. Kamphuis E., Junt T., Waibler Z., Forster R., Kalinke U.. 2006. Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia. Blood 108: 3253–3261. [DOI] [PubMed] [Google Scholar]
- 42. Prinz M., Schmidt H., Mildner A., Knobeloch K. P., Hanisch U. K., Raasch J., Merkler D., Detje C., Gutcher I., Mages J., et al. 2008. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity 28: 675–686. [DOI] [PubMed] [Google Scholar]
- 43. Caton M. L., Smith-Raska M. R., Reizis B.. 2007. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J. Exp. Med. 204: 1653–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Harfe B. D., Scherz P. J., Nissim S., Tian H., McMahon A. P., Tabin C. J.. 2004. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 118: 517–528. [DOI] [PubMed] [Google Scholar]
- 45. Opitz B., van Laak V., Eitel J., Suttorp N.. 2010. Innate immune recognition in infectious and noninfectious diseases of the lung. Am. J. Respir. Crit. Care Med. 181: 1294–1309. [DOI] [PubMed] [Google Scholar]
- 46. Xu Z., Shi L., Wang Y., Zhang J., Huang L., Zhang C., Liu S., Zhao P., Liu H., Zhu L., et al. 2020. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 8: 420–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jimenez-Guardeño J. M., Nieto-Torres J. L., DeDiego M. L., Regla-Nava J. A., Fernandez-Delgado R., Castaño-Rodriguez C., Enjuanes L.. 2014. The PDZ-binding motif of severe acute respiratory syndrome coronavirus envelope protein is a determinant of viral pathogenesis. PLoS Pathog. 10: e1004320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mandala V. S., McKay M. J., Shcherbakov A. A., Dregni A. J., Kolocouris A., Hong M.. 2020. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. Nat. Struct. Mol. Biol. 27: 1202–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bhattacharya M., Sharma A. R., Mallick B., Sharma G., Lee S. S., Chakraborty C.. 2020. Immunoinformatics approach to understand molecular interaction between multi-epitopic regions of SARS-CoV-2 spike-protein with TLR4/MD-2 complex. Infect. Genet. Evol. 85: 104587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Choudhury A., Mukherjee S.. 2020. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 92: 2105–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shirato K., Kizaki T.. 2021. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon 7: e06187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Grifoni A., Sidney J., Zhang Y., Scheuermann R. H., Peters B., Sette A.. 2020. A sequence homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe 27: 671–680.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.