

Abstract
The secretin receptor (SCTR), a prototypical class B G protein-coupled receptor (GPCR), exerts its effects mainly by activating Gαs proteins upon binding of its endogenous peptide ligand secretin. SCTRs can be found in a variety of tissues and organs across species including pancreas, stomach, liver, heart, lung, colon, kidney and brain. Beyond that, modulation of SCTR-mediated signaling has therapeutic potential for the treatment of multiple diseases, such as heart failure, obesity and diabetes. However, no ligands other than secretin and its peptide analogs have been described to regulate SCTRs, probably due to inherent challenges in family B GPCR drug discovery. Here we report creation of a testing funnel that allowed targeted detection of SCTR small molecule activators. Pursuing the strategy to identify positive allosteric modulators (PAMs), we established a unique primary screening assay employing a mixture of three orthosteric stimulators that was compared in a screening campaign testing 12,000 small molecule compounds. Beyond that, we developed a comprehensive set of secondary assays, such as a radiolabel-free target engagement assay and a NanoBiT (NanoLuc binary technology)-based approach to detect β-arrestin-2 recruitment, all feasible in a high-throughput environment as well as capable of profiling ligands and hits regarding their effect on binding and receptor function. This combination of methods enabled the discovery of five promising scaffolds, four of which have been validated and further characterized with respect to their allosteric activities. We propose that our results may serve as starting points for developing the first in vivo active small molecules targeting SCTRs.
Keywords: High-throughput screening, secretin receptor, positive allosteric modulator, TR-FRET binding, G protein-coupled receptor
Introduction
In 1902, Bayliss and Starling discovered secretin (Sec-FL (full-length)), a 27 amino acid hormone, in the duodenal mucosa stimulating the secretion of bicarbonate, enzymes and potassium ions from the pancreas1. Sec-FL exerts its physiological effects by activating the secretin receptor (SCTR), which was cloned in 1991 as the first member of the class B family of G protein-coupled receptors (GPCRs)2. Thus, secretin and its receptor formed the backbone of this highly important family of therapeutic targets, which are crucially involved in hormonal homeostasis3. Beyond its secretory effect, Sec-FL is implicated in a number of physiological and pathological conditions involving the heart, lung, liver, brain and gastrointestinal (GI) system4. According to Grossini et al.5, intracoronary infusion of secretin in animal models or human patients demonstrated beneficial cardiac responses, such as positive inotropic, chronotropic and vasodilating effects, without changing the blood pressure. Furthermore, SCTRs have been shown to stimulate meal-induced brown fat thermogenesis (BAT) resulting in satiation and short-term reduction of food intake6. A recent report highlighted elevated postprandial secretin plasma concentrations after Roux-en-Y gastric bypass (RYGB) surgery, which unveiled glucose-sensitive S (secretin)-cells in the distal small intestine7. In addition, SCTR activation might be of therapeutic value for functional dyspepsia as indicated in a small clinical study in humans8. Altogether, modulating SCTR signaling outlines a unique strategy to develop novel therapeutics with potential benefits for the treatment of comorbid conditions, such as obesity, diabetes, abnormal gastric accommodation and heart failure. However, to date, no ligands other than secretin peptide and closely related analogs have been identified to interact with SCTRs9–11. Beyond that, synthetic Sec-FL represents the only clinically utilized SCTR ligand for diagnostic purposes12–14. This might be due to the practical clinical limitations of Sec-FL, such as its short half-life (~2–4 min) and the need for intravenous administration8. Moreover, the development of potent small molecule ligands for class B GPCRs remains challenging, likely due to the complex binding mechanism of orthosteric endogenous ligands within the N-domain and the highly open seven-transmembrane region of these receptors3, 15. Nonetheless, recent diabetes research has led the way by identifying a vast number of small molecule modulators targeting the glucagon-like peptide-1 receptor (GLP-1R)16–21, a close relative to the SCTR. Although no orally available GLP-1R drug has been approved for human use21, the pace of emerging small molecule GLP-1R ligands expands drug discovery for class B GPCRs. The quest for ligands that interact via allosteric binding sites, contributed largely to this breakthrough. This class of compounds, which binds to structurally distinct receptor pockets, offers an opportunity to selectively modulate GPCR function in a diverse and rich way22. Positive allosteric modulators (PAMs) are able to elevate the natural effect of hormones and endogenous ligands either by increasing potency or by maximizing efficacy and may additionally display intrinsic activity (ago-PAMs)22. Furthermore, PAMs may induce functional selectivity for signaling pathways not inherent to the natural ligand22. Another characteristic of allosteric modulators is their saturable effect, which has the potential to fine-tune receptor signaling, as well as minimizing risks such as drug overdosing.22 Thus, the search for allosteric scaffolds provides a novel opportunity to identify the first orally bioavailable drug candidates modulating SCTRs.
Here, we outline the development of a four-stage testing funnel with the capability to detect, validate and characterize biologically relevant SCTR small molecule modulators. Our approach includes the design of a diverse selection of PAM primary screening assays not only differing from their detection method but also from their orthosteric stimulus response, which have been compared by screening a 12,000-compound (cmpd) small molecule library. We also established a set of secondary assays to validate and characterize promising scaffolds regarding target engagement and functional selectivity. The results demonstrate how the combination of assays arranged in the testing funnel led to the discovery of the first low molecular-weight, non-peptidyl SCTR-specific PAMs.
Materials
Peptides and ligands:
Sec-FL (full length human secretin (1–27), #4031250), GLP-1 (glucagon-like peptide-1 trifluoroacetate salt, #4030663) and AVP ((Arg8)-vasopressin trifluoroacetate salt, #4012215) were obtained from Bachem AG (Bubendorf, Switzerland). Sec(1–23) (HSDGTFTSELSRLREGARLQRLL-OH) and Sec(3–27) (DGTFTSELSRLREGARLQRLLQGLV-NH2) were custom synthesized by Biopeptide, San Diego, CA, USA. GLP-1(9–36) (GLP-1(9–36) amide, #AS-65070) was obtained from Anaspec (Fremont, CA, USA). BETP (4-(3-(Benzyloxy)phenyl)-2-(ethylsulfinyl)-6-(trifluoromethyl)pyrimidine) was purchased from Sigma Aldrich (St. Louis, MO, USA, #SML0558, purity ≥98% (HPLC)).
Cells and culture reagents:
Chinese Hamster Ovary (CHO-K1) cells and Human Embryonic Kidney (HEK)-293(T) cells were obtained from ATCC (Manassas, VA, USA). CHO-K1 cells were maintained in the CHO cell growth media (Ham’s F-12K ((Kaighn’s modification), Corning Life Sciences, Tewksbury, MA, USA, Cellgro #10-025-CV), 5% Fetal Bovine Serum (FBS) Clone II (GE Healthcare Life Sciences, Marlborough, MA, USA, Hyclone #SH30066.03), 1% penicillin (10,000 units)/ streptomycin (10 mg) (Pen/Strep, Thermo Fisher Scientific, Waltham, MA, USA, Gibco #15140122), 1% L-glutamine (200 mM) (Gibco #25030081). HEK-293(T) cells were maintained in HEK cell growth media (Dulbecco’s Modified Eagles Medium (DMEM) Corning Life Sciences, Cellgro #10-013-CV), 10% FBS (Omega Scientific, Tarzana, CA, USA, #FB-12), 1% Pen/Strep, 1% L-Glutamine). Cells were detached using TrypLE Express (Gibco #12605036). Antibiotics used for stable cell selection were Hygromycin (Hygromycin B, Omega Scientific, #HG-80), G418 (Omega Scientific, #GN-04) and Blasticidin (InvivoGen, #ant-bl-1, San Diego, CA, USA).
The chemical library
The chemical library was obtained from BioAscent Discovery Ltd. (Newhouse, Lanarkshire, UK) consisting of 12,000 cluster centroids providing diversity representatives of expanded 125,000 small molecule compound BioAscent collection. Each of the scaffolds in the expanded library contains 10–30 analogs that help to rapidly validate scaffolds and establish nascent structure-activity relationships (SAR) through cherry pick orders of hits and analogs as liquid stocks. The library was stored as 2 mM stocks in 100% DMSO.
Methods
All assays were performed at the Conrad Prebys Center for Chemical Genomics (CPCCG) High-Throughput Screening Facility.
cAMP (cyclic adenosine monophosphate) accumulation assays
Cyclic AMP assays were performed using frozen stocks of parental CHO-K1 cells, SCTR- or AVP2R-overexpressing CHO-K1 cells or GLP-1R-overexpressing HEK-293T cells, all derived from a single cell clone at a low passage number. After reaching 80–90% confluency, cells were harvested using TrypLE Express, centrifuged and re-suspended in freeze media (10% DMSO in growth media). Final concentration of cell stocks was 20 million cells/mL. Corresponding ligand standard curves were recorded for each cell batch to obtain adequate EC20 and EC95 concentrations.
General procedure:
The TR-FRET-based cAMP accumulation assays were performed according to manufacturer’s instructions with a few modifications. In brief, orthosteric stimulator dilutions were prepared freshly in DMSO and transferred to Echo Qualified 384-well low dead volume (384LDV) microplates (Labcyte, San Jose, CA, USA) via CAPP 16-channel pipette (CAPP, Nordhausen, Germany). Compounds and ligands were dispensed onto dry microplates with an Echo liquid handler (Labcyte). Final assay compound concentration was 10 μM for primary screening and hit confirmation (in triplicates) or 12.5 uM for 1,025 hits & analogs (in triplicates). Frozen cell stocks were thawed quickly in a 37°C water bath and diluted in stimulation buffer (HBSS (Hank’s Balanced Salt Solution with Ca2+ and Mg2+, Gibco™ #24020117), 5 mM HEPES (hydroxyethyl piperazineethanesulfonic acid), 0.075% BSA (7.5% DTPA-purified Bovine Serum Albumin, PerkinElmer, Inc., Waltham, MA, USA, #CR84-100) and 0.5 mM IBMX (3-isobutyl-1-methylxanthine, Sigma-Aldrich, St. Louis, MO, USA, #I5879) to obtain desired cell density. Cells were dispensed using a Multidrop Combi dispenser (Thermo Fisher Scientific), centrifuged at 1000 rpm for 1 min, covered with a lid and kept at room temperature (RT) for 30 min. cAMP standard dilutions (4-fold, 0 – 1 μM final) were prepared in the stimulation buffer and transferred to designated wells using a CAPP 16-channel pipette. 0.1% of Antifoam (Antifoam SE-15, Sigma-Aldrich, #A8582) was added to the cAMP detection buffer, which was subsequently filtered through a 40 μm cell strainer. Detection reagents were diluted according to manufacturer’s manual (specific dilutions in supplementary information) and dispensed using a Combi dispenser. The plates were centrifuged at 1000 rpm for 1 min, covered with a lid and read on a Pherastar Plus microplate reader (BMG Labtech, Ortenberg, Germany) with the HTRF (homogeneous time resolved fluorescence) module after 30–60 min at RT. Data were uploaded and analyzed on CBIS (Chemical and Biology Information System software, ChemInnovation Software, Inc., San Diego, CA, USA). EC20 of ligand was set as the negative control (neg.ctrl.), EC95 as the positive control (pos.ctrl.). Further characterization was conducted using the TIBCO Spotfire software (PerkinElmer). Compounds with high fluorescence in the TR-FRET donor (reference) channel in comparison to the negative control wells were eliminated from further studies. Specific procedures are described in the Supplementary Information.
CRE-Luc2P reporter assay
Luciferase reporter assays were performed as described previously23 with a few modifications. In brief, HEK-293 SCTR CRELuc cells were seeded in two T225 cell culture flasks and grown in HEK cell growth media (DMEM, 10% FBS, 1% Pen/Strep, 1% L-glutamine). After two days cells were detached using TrypLE Express, re-suspended in PBS (phosphate-buffered saline) and centrifuged at 300 g for 4 min. The cell pellet was re-suspended in DMEM + 10% FBS. A cell suspension of 0.25 million cells/mL was seeded into TC-treated 384-well microplates (Greiner Bio-One small volume 784080) via a Multidrop Combi dispenser at 5 μL/well. Plates were centrifuged at 500 rpm for 15 sec and incubated over night at 37 °C and 5% CO2. The next day orthosteric stimulator dilutions were freshly prepared in DMSO (positive control: 156 pM Sec-FL final, negative control and compound wells: 5 pM Sec-FL final) and transferred to Echo Qualified 384-well low dead volume (384LDV) microplates via a CAPP 16-channel pipette. Compounds (25 nL/well) and ligands (5 nL/well) were dispensed onto microplates with Labcyte Echo followed by incubation at 37 °C and 5% CO2. Final DMSO concentration was 0.60%. After 4 h, plates and Steady-Glo (Promega) detection reagent were brought to RT for 15 min. Detection reagent was added via a Multidrop Combi dispenser (5 μL/well) and plates were centrifuged 500 rpm for 15 sec. Thereafter, assay plates were kept at RT protected from light for 15 min and luminescence was detected via ViewLux ultra HTS Microplate Imager (PerkinElmer, 5 sec read). Data was uploaded and analyzed using CBIS (Chemical and Biology Information System software). EC20 of ligand was set as negative control, EC95 as positive control. Further characterization was conducted via TIBCO Spotfire. Cell line generation and clonal selection of HEK-293 SCTR CRELuc cells are described in the Supplementary Information.
TR-FRET SNAP-SCTR binding assay
Binding experiments were performed as previously described24–27, with the following modifications:
Competition binding – Allosteric modulator titration
Selected compounds A1, A9, B1, C1 and D1 were stored in 384LDV microplates in a desiccator as 16-point 2-fold dilutions in DMSO. Stock concentrations ranged from 0 to 10 mM. Fluo-Sec and Sec-FL were diluted in DMSO and dispensed into a 384LDV plate. Ligand titrations were prepared in DMSO in adjacent wells. Using Labcyte Echo Fluo-Sec was transferred (25 nL, 6 nM final) into all test wells of a 1536-well plate (Corning #3725), DMSO (25 nL, pos. ctrl.), Sec-FL (25 nL, 5 μM final, neg. ctrl.) or ligand/compound titrations (25 nL, varying concentrations) were dispensed on top. Using a dounce homogenizer thawed HEK-293 SNAP-SCTR membranes labeled with Lumi-4 Terbium cryptate (Cisbio Tag-lite) were diluted in binding buffer (10 mM HEPES, pH 7.4, 100 mM NaCl, 10 mM MgCl2, 1 mM ascorbic acid, 0.2% BSA) to a final concentration of 5 μg/mL. Membrane solution was added via a Multidrop Combi dispenser at 5 μL/well. The plate was centrifuged 1000 rpm for 1 min and incubated at RT for 2 h. Competition binding/ allosteric modulator titration was recorded by Pherastar FSX (LanthaScreen 520/490 module). Data was uploaded and analyzed via CBIS as well as via GraphPad Prism 8.4.0 applying the equation “One site – Fit Ki” to determine equilibrium dissociation constants Ki of orthosteric ligands. Allosteric modulators were analyzed using the equation “Allosteric modulator titration” to obtain equilibrium dissociation constants Kb and cooperativity factors α. α =1 indicates neutral cooperativity, 0< α<1 indicates negative modulation and α>1 supports positive cooperativity. Experiments were performed in duplicate in at least three independent experiments. Procedures for cell line generation, clonal selection and membrane preparation of HEK-293 SNAP-SCTR as well as for saturation and dissociation binding experiments are described in the Supplementary Information.
Calcium flux assay with FLIPR Calcium 6 dye
SCTR-CHO-K1 cells were grown in the CHO cell growth media. After reaching 80–90% confluency, cells were harvested using TrypLE Express, re-suspended in growth media and seeded into a 384-well plate (Greiner Bio-One #781091) (5000 cells/50 μL/well). Plates were covered with lids, centrifuged at 500 rpm for 1 min and incubated at 37 °C and 5% CO2 overnight. Next day, growth media was removed by an upside-down spin (300 rpm for 5 sec). Immediately, 20 μL FLIPR Calcium 6 dye (membrane-permeable Ca2+ indicator, Molecular Devices, San Jose, CA, USA) in assay buffer (HBSS with Ca2+ and Mg2+ containing 20 mM HEPES, 0.1% BSA and 2.5 mM probenecid (Sigma-Aldrich, #P-7861)) were added per well. The plate was centrifuged at 500 rpm 5 sec and subsequently incubated at 37 °C and 5% CO2 for 2 h. Test ligand titrations were prepared on a 384LDV plate and then transferred to a 384-well NBS plate (Non-Binding Surface, Corning #3655). Assay buffer was added to the compound source plate using a Multidrop Combi dispenser to a final volume of 40 μL/well. Cell plates were equilibrated at RT for 30 min. Hamamatsu FDSS 7000 (Functional Drug Screening System, Hamamatsu Photonics, Hamamatsu City, Japan) was used for liquid dispenses and fast kinetic reads. 10 μL of ligand solution was added to 20 μL cells in dye media while monitoring fluorescence (3 min read, addition at 10th read interval, normal exposure FLUO3/4). All experiments were performed in duplicate in at least three independent experiments. Curve fitting analysis was conducted by GraphPad Prism 8.4.0 (GraphPad Software Inc., San Diego, CA).
NanoBiT (NanoLuc binary technology) β-arrestin-2 recruitment assay
β-Arrestin-2 recruitment assays were developed and performed as reported previously28, with a few modifications:
NanoBiT β-arrestin-2 recruitment assay in transiently expressing HEK-293 cells
HEK-293 cells were seeded into a 6-well plate at a cell density of 0.3 M/well and were incubated overnight at 37°C and 5% CO2. Following manufacturer’s manual pFC220K-SCTR-SmBiT & LgBiT-ARRB2, pFC220K-SCTR-SmBiT & ARRB2-LgBiT, or pFC220K-AVP2R-SmBiT & LgBiT-ARRB2 were transfected using TransIT-LT1 transfection reagent (Mirusbio, Madison, WI, USA, #MIR2300) delivering 0.5 μg DNA of each construct per well. After 24 h, cells were harvested using TrypLE Express, re-suspended in growth media, centrifuged at 300 g for 3 min and re-suspended in assay buffer (HBSS with Mg2+ and Ca2+, 5 mM HEPES, 0.1% BSA) to a final cell density of 0.4 million/mL. Cell suspension was dispensed in an AlphaPlate-384 (light gray, shallow well; Perkin Elmer #6008350) using a Multidrop Combi dispenser. After centrifugation at 500 rpm for 15 sec, 3 μL NanoBiT detection reagent was added per well via Multidrop Combi. Plate was covered, centrifuged at 1000 rpm for 1 min and monitored with Pherastar FSX (luminescence, kinetic mode, 0.2 sec read) for 2 h or until baseline appeared to stabilize. Ligand (Sec-FL, Sec(1–23), Sec(3–27) or AVP) titrations were prepared in DMSO in 384LDV plates. Utilizing Echo liquid dispenser ligand titrations (40 nL/well) were transferred into cell plate. In case of characterization of PAMs, DMSO (20 nL) or CMPD A1/B1 (20 nL, 12.5 μM final) were dispensed into sample wells before Sec-FL titration. Plate was covered, centrifuged at 1000 rpm for 1 min and β-arrestin-2 recruitment was recorded via Pherastar FSX (luminescence, kinetic mode, 0.2 sec read) for 30 min. Experiments were performed in duplicate or triplicate in at least three independent experiments and curves were fitted using GraphPad Prism 8.4.0. Plasmids, construct generation and clonal selection of HEK-293 SCTR-SmBiT LgBiT-ARRB2 are described in the Supplementary Information.
Results
Development of a four-stage testing funnel to detect PAMs targeting SCTRs
To enhance our chances for discovering orally bioavailable drug candidates targeting SCTRs, we focused our efforts on the identification of positive allosteric modulators (PAMs), which may not only potentiate the effect of secretin and related peptides, but also selectively channel activation of signaling pathways (Fig. 1A). We fortified our drug discovery program by developing a panel of diverse assays that build four essential stages for our screening campaign. Starting with HTS (High-throughput Screening) and hit confirmation (Fig. 1B), it was our aim to develop a robust and efficient primary screening assay based on Gαs protein signaling, which represents the dominant physiological effect upon SCTR activation and leads to increased cAMP (cyclic adenosine monophosphate) levels. It is well known in the HTS field that different assays detecting the same cellular processes would inadvertently produce distinct sets of hits; some of them may be enhanced by the detection approach used in the assay, while others may be enhanced by unique settings specific to each of these assays29. Therefore, we compared three different cAMP detection methods (Perkin Elmer LANCE Ultra (LU), Cisbio Gs Dynamic (CB GsD) and Promega CRELuc2P reporter (CRELuc) technology23) by performing pilot screens of a small molecule library consisting of 12,000 compounds. Since PAMs may display reduced or no activity without an orthosteric ligand present30–32, we developed PAM screening assays using a fixed concentration (EC10-EC20; based on TR-FRET ratios or RLUs) of agonist as basal orthosteric stimulator response in compound and negative control wells. Since PAM assays are able to detect both, PAMs and (allosteric) agonists, we additionally characterized compounds in agonist mode assays performed in the absence of peptide ligands to determine their intrinsic activity. The EC95 concentration of the peptide agonist served as a positive control in both cases. Similar approaches have been described for recent PAM screening efforts against other GPCRs21, 33.
Figure 1: Strategies for the development of a testing funnel to detect PAMs targeting SCTRs:
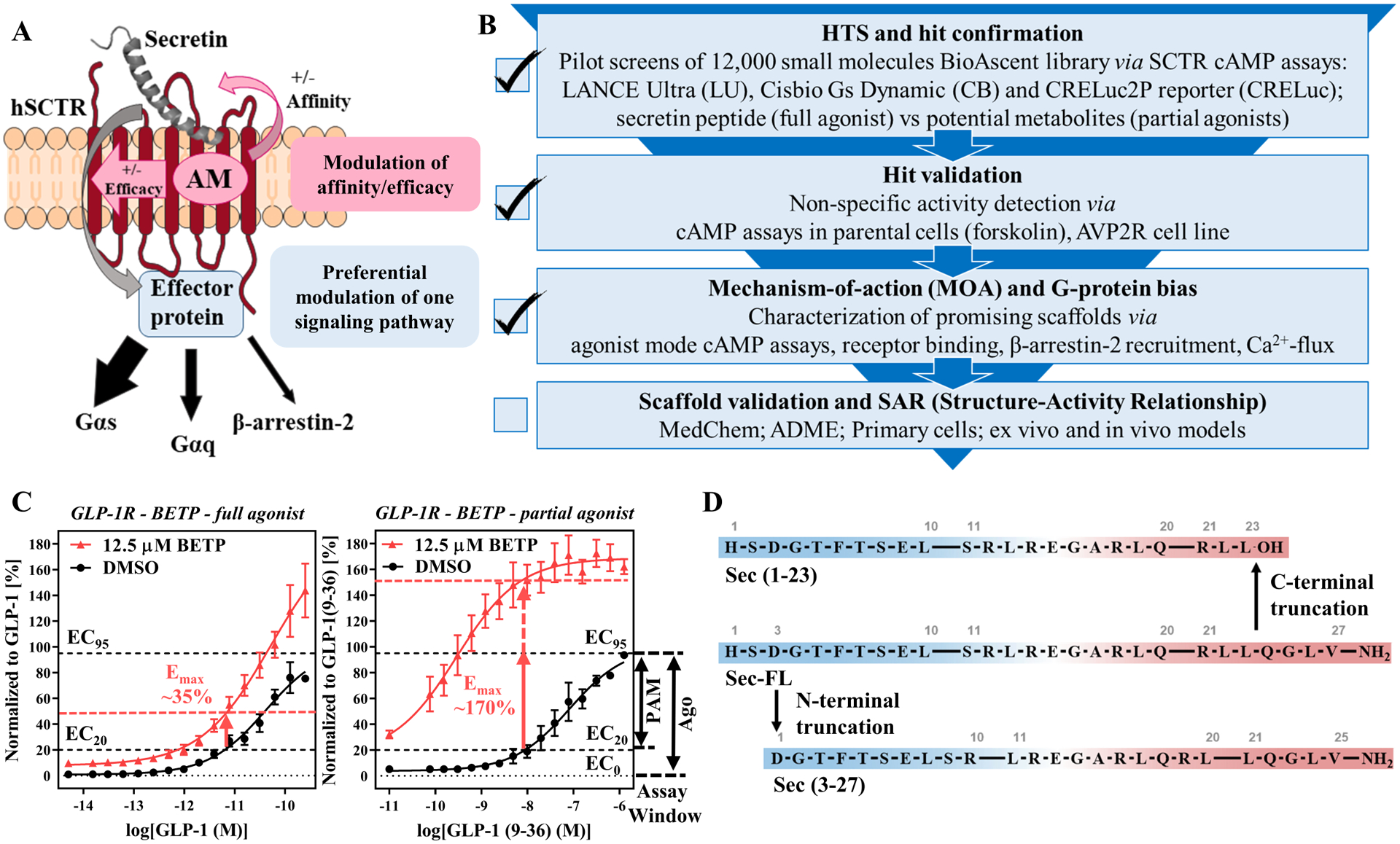
(A) Advantages of allosteric modulator signaling and display of SCTR signaling pathways (adopted from ref.51). (B) Development of biological assays for the establishment of a testing funnel to identify biologically relevant SCTR small molecule modulators consisting of four levels: HTS and hit confirmation, hit validation, MOA and G-protein bias as well as scaffold validation and SAR studies; Check marks indicate stages that have been completed successfully. (C) GLP-1R PAM BETP18, 34 as a model demonstrating substantial probe-dependency and corresponding effect on cAMP accumulation assay sensitivity; left panel: DMSO (black) vs BETP (12.5 μM, red) treated GLP-1 full-length (full agonist) predicting primary screening response around 35%; right panel: DMSO (black) vs BETP (12.5 μM, red) treated GLP-1(9–36) (partial agonist) predicting primary screening response well beyond 100%; TR-FRET ratios (relative fluorescence units (RFU)) normalized to corresponding peptide agonist and plotted using GraphPad Prism; data points shown as mean ± SEM. Depiction of assay windows of PAM mode (neg. ctrl: EC20, pos. ctrl: EC95 of ligand) and agonist mode (neg. ctrl: no ligand, pos. ctrl: EC95 of ligand), which served as the basis of SCTR screening assays. (D) Secretin and its truncated analogs mimicking potential metabolites of secretin (from top down): Sec(1–23), product of C-terminal truncation (carboxylic acid (-OH)); Sec-FL (full-length, amide (-NH2)), endogenous peptide acting on SCTRs; Sec(3–27) amide, product of N-terminal truncation.
Beyond that, we evaluated the screening efficiency comparing full agonists and partial agonists. This was based on the observation of substantial probe dependency of GLP-1R PAMs, such as BETP18, 34, toward the partial agonist GLP-1(9–36), which is generated via N-terminal truncation of GLP-1 by DPP4 (dipeptidyl-peptidase-4)35. Figure 1C illustrates benefits of using a partial agonist in the PAM assay. The extent of response from BETP in the presence of EC20 concentration of GLP-1 (Fig. 1C, left panel) is significantly lower than observed with partial agonist GLP-1(9–36) (Fig. 1C, right panel). Intriguingly, BETP would be considered a weak hit with 35% response using the full agonist as basal stimulator, but reach activity well beyond 100% when used with the partial agonist. Inspired by this increase of sensitivity we explored potential secretin peptide metabolites. Sec-FL, like other peptide hormones, is known to be metabolized and cleared rapidly (half-life ~ 2–4 min8). Although there is no clear description of physiologic Sec-FL degradation products, we mimicked metabolites after known truncated versions of endogenous ligands targeting other class B GPCRs, which could serve as useful tools to evaluate assay sensitivity and probe dependency of potential PAMs. For example, Sec-FL was shown to be degraded by NEP (neutral endopeptidase) 24.11, but no specific cleavage products were reported36. Another in vitro study suggested the formation of secretin 1–23 (Sec(1–23), Fig.1D, top) by VIP-degrading endoprotease through C-terminal cleavage of Sec-FL (Fig.1D, middle)37. To explore effects of C- and N-terminal truncation of Sec-FL we acquired Sec(1–23) and secretin 3–27 (Sec(3–27), Fig.1D, bottom). In stage two of our testing funnel (Fig. 1B) we validated hits by eliminating non-specific activators. To that end, we translated our developed cAMP detection methods to cell lines not expressing SCTRs, such as parental cells and type 2 arginine vasopressin receptor (AVP2R)-bearing cells. AVP2R was selected, since it also couples to Gαs proteins while exerting physiologic effects contradictory to SCTR activation38. Besides, being a class A GPCR, AVP2R should not be affected by SCTR targeting hits.
Development of secondary assays to enable broader scaffold validation and compound profiling
To strengthen our ability to validate and profile promising scaffolds regarding their mechanism-of-action (MOA), we developed and optimized secondary assays to evaluate target engagement and functional selectivity of potential PAMs. Inspired by previously reported TR-FRET based binding assays24, 25, 39, we inserted a SNAP-tag to the N-domain of SCTR and created stably expressing HEK-293 cell clones. The construct was compared to wildtype (WT) receptor by monitoring agonist response in cAMP accumulation assays to ensure functional integrity (Fig. S1C). Additionally, we introduced a fluorescein molecule to the C-terminal end of Sec-FL via a diethylene glycol linker presenting a lysine residue (Fig. S1A). Of note, the absence of basic amino acids in the sequence of secretin enabled the use of lysine instead of an oxidation-sensitive cysteine residue to attach the probe. cAMP accumulation experiments in CHO-SCTR cells confirmed that the introduction of the fluorescein-tag did not hamper agonist activity (Fig. S1B). Being assured our constructs maintained WT-like biological behavior, we prepared membranes of Lumi4-Tb (Lumi-4 terbium cryptate, Cisbio Tag-lite) labeled SNAP-SCTR cell clones. Depending on the assay format, membranes were incubated with fluorescein-labeled secretin (Fluo-Sec) in addition to orthosteric or allosteric ligands, resulting in TR-FRET signaling that can be quantified to assess the fraction of Fluo-Sec bound to SNAP-SCTR (Fig. 2A). To determine the dissociation constant (Kd) saturation binding experiments were performed (Fig. 2B), which served as the basis to conduct further experiments like competition binding (Fig. 2C, 3B, 6C) and dissociation binding assays (Fig. 6D). Intriguingly, the homogeneous format, simplicity and lack of radioactivity allows time-dependent measurements in a high-throughput format with minimal use of resources (time, ligand, compounds, plates, etc.) in contrast to standard radioisotope binding assays.
Figure 2: Development of GPCR-specific secondary assays to enable immediate scaffold validation and compound characterization:
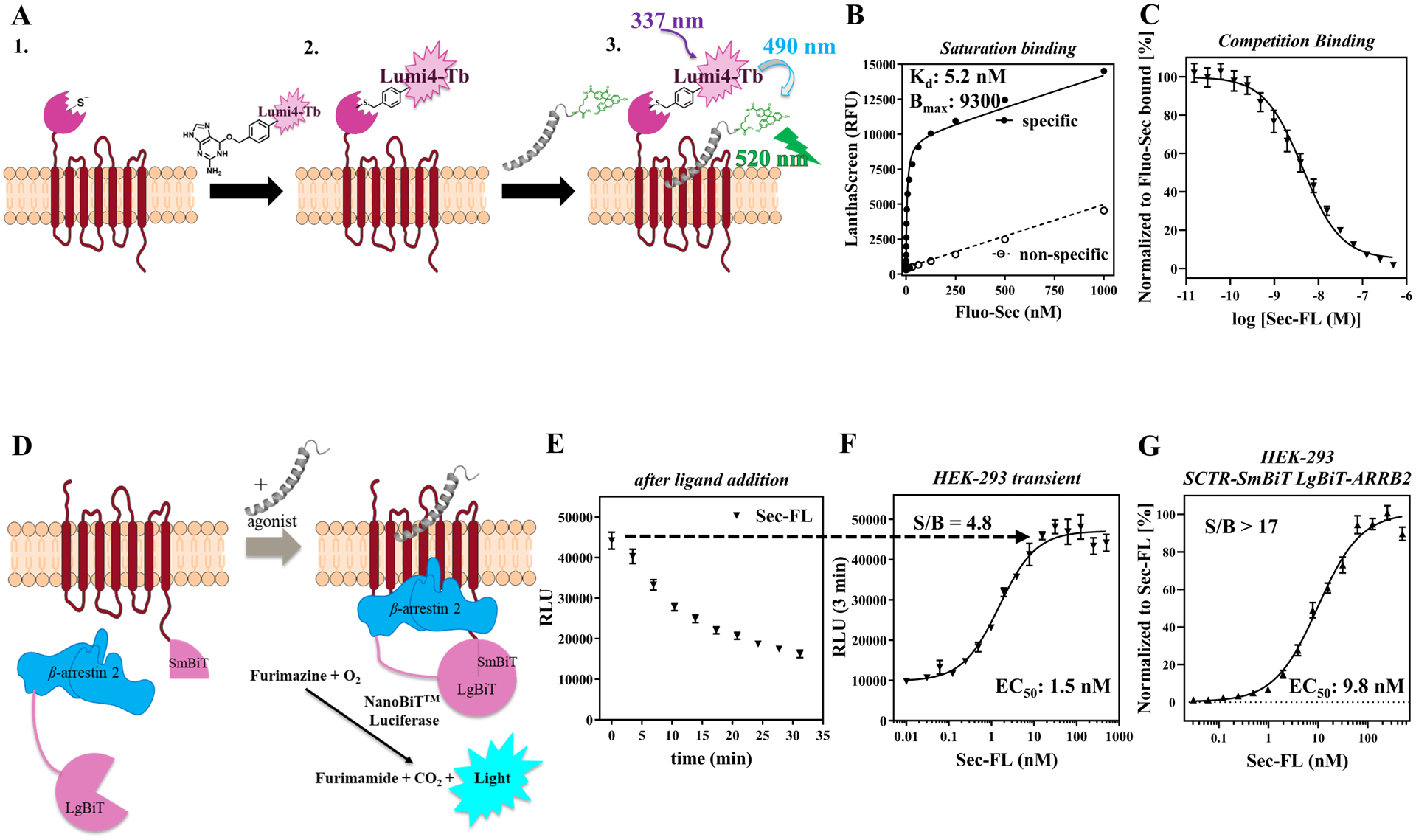
(A) Design and mechanism of TR-FRET based binding assay employing SNAP-tag technology and fluorescein-labeled secretin peptide (Fluo-Sec); (B) Saturation binding curve of Fluo-Sec determining dissociation constant Kd; (C) Competition binding curve of Sec-FL; LanthaScreen ratios normalized to Fluo-Sec bound. (D) Design and mechanism of β-arrestin-2 recruitment assay exploiting NanoBiT (Promega) technology; (E) Real-time luminescence after ligand addition to evaluate best time point for endpoint read; (F) Dose-response curve of Sec-FL at 3 min (transiently expressing HEK-293 cells); (G) Normalized dose-response curve of Sec-FL (HEK-293 SCTR-SmBiT LgBiT-ARRB2 cell clone, n = 4), raw data signal-to-background ratio (S/B) over 17. Relative luminescence units (RLU) normalized to Sec-FL; graphs plotted using GraphPad Prism; data points shown as mean ± SEM.
Figure 3: C-terminal truncation of secretin leads to low affinity, low potency but fully efficacious agonist whereas N-terminal cleavage drastically reduces efficacy among investigated signaling pathways:
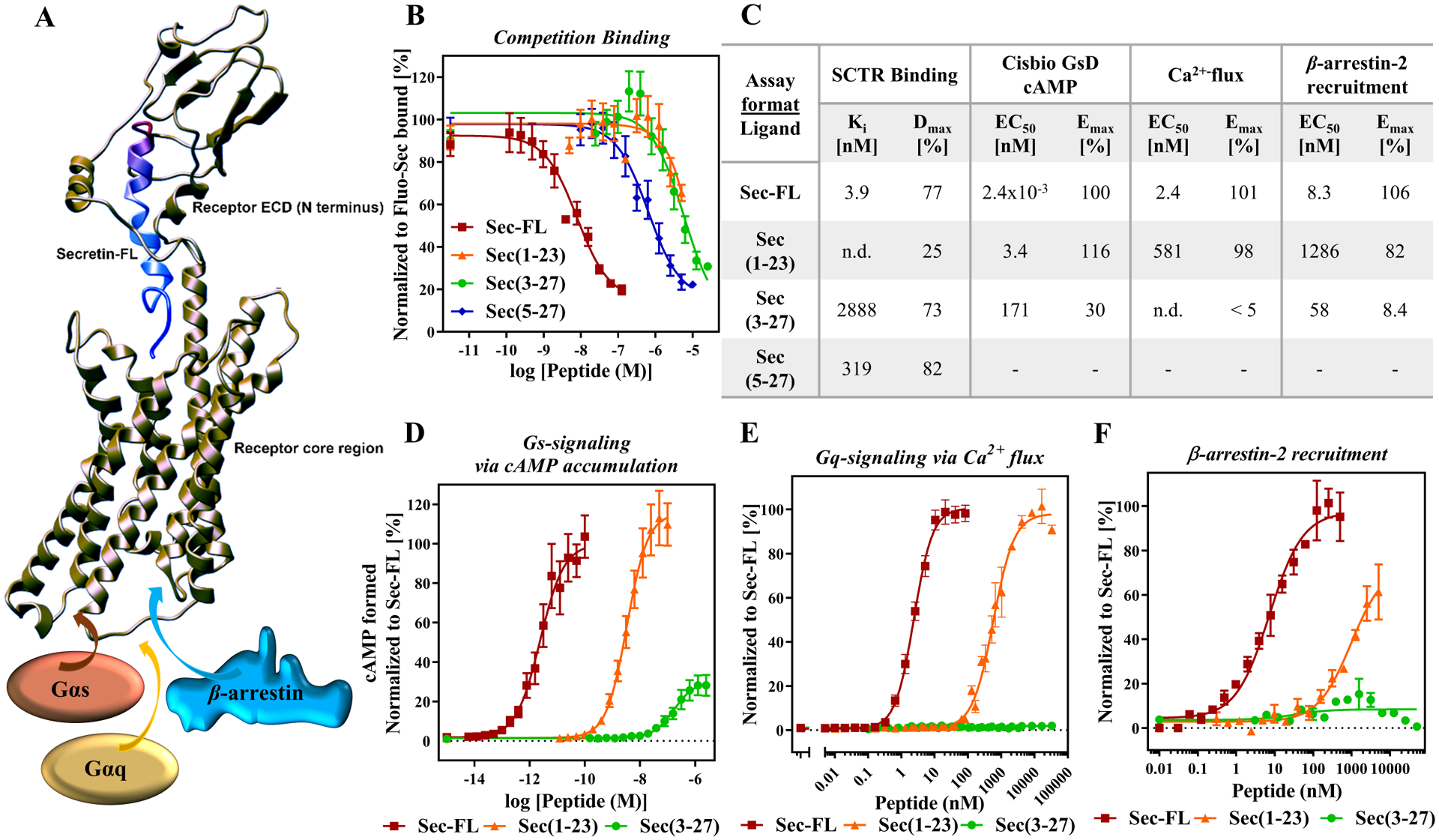
(A) 3D-model of secretin bound to SCTR (adopted from reference46) with illustration of two peptide binding sites: C-terminal end of secretin (red) binding to receptor N-domain (also known as extracellular domain (ECD)); N-terminal tail of secretin (blue) positioning in receptor core helical bundle. Upon secretin binding, recruitment of effector proteins including Gαs and Gαq proteins as well as β-arrestin. (B) Competition binding of Fluo-Sec with secretin and truncated analogs: Sec-FL (red), Sec(1–23) (orange), Sec(3–27) (green) and Sec(5–27) (blue) to determine binding affinities Ki [nM] and fractions displaced (Dmax [%]); LanthaScreen ratios normalized to Fluo-Sec bound. (C) Summary of affinities, potencies and efficacies of Sec-FL analogs in performed assays in table format. Activity of Sec-FL (red), Sec(1–23) (orange), Sec(3–27) (green) inducing (D) cAMP accumulation, (E) Ca2+ flux and (F) β-arrestin-2 recruitment; (D) TR-FRET ratios converted to cAMP concentrations [nM], (E) Cytosolic Ca2+ F/F0 ratio and (F) RLU, (D-F) normalized to Sec-FL; graphs plotted using GraphPad Prism; experiments performed in duplicate or triplicate; (B), (D) and (E) combined and (F) representative graph of three independent experiments; data points shown as Mean ± SEM.
Figure 6: Validation and characterization of scaffold A, B, C and D:
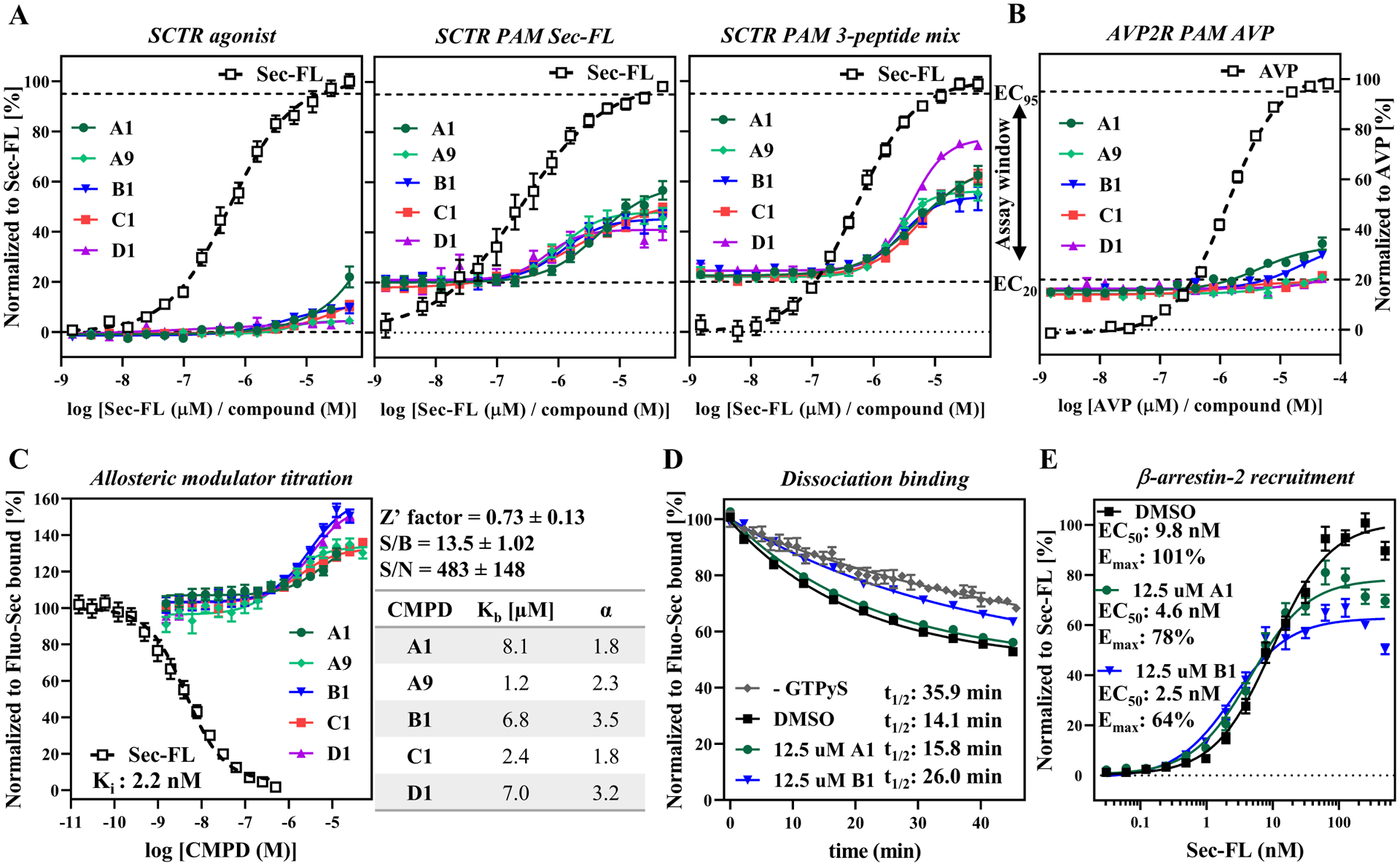
from left to right: Compound titration on (A) SCTR-bearing CHO-K1 cells detected by Cisbio GsD agonist (no stimulant), Cisbio GsD Sec-FL (stimulant EC20 Sec-FL), Cisbio GsD 3 pep mix (stimulant EC10 Sec-FL, Sec(1–23) and Sec(3–27)) or on (B) AVP2R-expressing CHO-K1 cells via Cisbio GsD kit (stimulant EC20 AVP); (C) Allosteric modulator titration using TR-FRET binding assay in high-throughput mode (assay performance data right to graph) and subsequent analysis in GraphPad Prism yielding allosteric activity parameters Kb (equilibrium dissociation constant of PAM) and α (cooperativity factor). (D) CMPD B1 demonstrating effect on Fluo-Sec dissociation comparable to presence of G protein (− GTPγS). (E) CMPD A1 and B1 slightly enhance potency but diminish efficacy of Sec-FL for recruitment of β-arrestin-2. (A, B) TR-FRET ratios resulting from cAMP accumulation normalized to full agonist, (C, D) LanthaScreen ratios normalized to Fluo-Sec bound or (E) RLU normalized to Sec-FL. (A) - (E) CMPD A1 (dark green), A9 (light green), B1 (blue), C1 (red) and D1 (purple); graphs plotted using GraphPad Prism; experiments performed in duplicate or triplicate in at least three independent experiments; data points shown as mean ± SEM.
Since functional selectivity has been shown to provide potential therapeutic benefits relevant for GPCRs40–44, we designed a β-arrestin-2 recruitment assay applying Promega’s NanoBiT technology45. As reported in a similar study on cannabinoid receptors31, we made use of an 11 amino acid peptide (Small BiT, SmBiT) attached to the C-terminal end of the GPCR and the 17.6 kDa Large BiT (LgBiT) tag introduced either on the N- or C-terminal end of β-arrestin-2 (ARRB2). To validate our procedure, we generated and tested constructs for SCTR and AVP2R (Fig. S2). The latter served as a control described by Promega’s Brochure for NanoBiT® Technology. Upon agonist activation, GPCRs get phosphorylated followed by β-arrestin-2 recruitment. This event leads to complementation of the inactive SmBiT and LgBiT subunits, resulting in luminescent signal by restoring catalytic activity of the very bright NanoLuc luciferase (Fig. 2D). The engagement of GPCR and β-arrestin-2 can be monitored in real-time revealing the best suitable time point for endpoint reads. As described by Promega, addition of AVP resulted in a sustained luminescence signal peaking after about ten minutes (Fig S2B, S2C). Interestingly, in the case of HEK-293 cells expressing SCTR-SmBiT and LgBiT-ARRB2, the luminescence signal peak occurs immediately upon Sec-FL addition, decreasing steadily over time (Fig. 2E). Therefore, we chose to analyze concentration-response curves after three minutes of ligand addition (Fig. 2F). In transiently overexpressing HEK-293 cells, dose-response was recorded with a signal-to-background (S/B) of 4.8 (Fig. S2D) while the selection of a single cell clone expressing SCTR-SmBiT and LgBiT-ARRB2 constructs improved S/B to over 17 (Fig. 2G). Of note, co-transfection of SCTR-SmBiT with ARRB2-LgBiT resulted in a similar kinetic profile but over 10-fold lower RLU (relative luminescence units) values; hence, it was the less favorable option for assay optimization (Fig. S2B).
C-terminal cleavage of secretin leads to low affinity, low potency, but fully efficacious agonist, whereas N-terminal truncation drastically reduces efficacy among investigated signaling pathways
After establishing a strong panel of assays, we validated the developed assays by characterizing our truncated secretin analogs Sec(1–23) and Sec(3–27). Several examples in recent studies revealed a two-site binding mode of hormones to class B GPCRs, which is also depicted in Figure 3A illustrating a three-dimensional (3D) model46 of secretin-bound SCTR. As shown previously15, 46, 47, the C-terminal end of secretin peptide binds tightly to the large extracellular domain (ECD) of the receptor while directing the N-terminal end of secretin toward the helical core bundle, inducing intramolecular arrangements that lead to the activation of effector proteins, such as Gαs and Gαq proteins or β-arrestins. Considering this binding model, we were not surprised that compared to Sec-FL (red, Ki = 3.9 nM), Sec(1–23) (orange, Ki not determined) suffered from a slightly greater loss of affinity than Sec(3–27) (green, Ki = 2.9 μM). Interestingly, further cleavage of the N-terminal tail resulting in Sec(5–27) led to higher binding affinity (Ki = 319 nM) compared to other truncated analogs (Fig. 3B, C). In agreement with the 3D model, Sec(1–23) displays an up to 3-log loss of potency (cAMP formed EC50 = 3.4 nM, Ca2+-flux EC50 = 581 nM, β-arrestin-2 EC50 = 1.3 μM) compared to Sec-FL (cAMP formed EC50 = 2.4 pM, Ca2+-flux EC50 = 2.4 nM, β-arrestin-2 EC50 = 8.3 nM) but retains full efficacy among performed functional assays (Fig 3D, E, F). Sec(3–27) displayed additional loss of potency (cAMP formed EC50 = 171 nM) and significantly decreased efficacy (Emax = 30%) in cAMP accumulation assays while lacking any activity in experiments investigating Ca2+ flux or β-arrestin-2 recruitment. This dramatic loss of function due to two missing amino acids at the N-terminal end of the natural peptide is consistent with the two-site binding model and with reports on other class B GPCRs33. Of note, the inability of Sec(3–27) to induce a Gαq-mediated response or β-arrestin-2 recruitment might indicate a potential bias toward Gαs-signaling that we plan to investigate further in future studies.
Stage 1.1: Pilot Screen of BioAscent library using LANCE Ultra cAMP assay revealed probe dependency of hits towards secretin or its truncated forms
Our screening efforts began with a pilot screen utilizing Perkin Elmer LANCE Ultra cAMP detection kit (Fig. 4A) against the 12,000-compound BioAscent library at a 10 μM concentration. Deploying Sec-FL as orthosteric basal stimulator in PAM mode, we obtained 361 hits (3% hit rate). Hit confirmation studies (EC20 Sec-FL, 10 μM compound in triplicate) led to 106 confirmed hits. Purchase of these 106 hits and 919 analogs (5–15 analogs/scaffold, 1,025 compounds total) selected to provide nascent SAR for identified hits as liquid stocks from BioAscent and subsequent evaluation in hit confirmation format (EC20 Sec-FL, 10 μM compound in triplicate) yielded 368 hits (90 reconfirmed original hits and 278 active analogs). Eleven of these hits were eliminated by screening in parental CHO-K1 cells using the same detection method. To investigate effects of truncated secretin analogs, we performed in parallel three screens of 1,025 hits & analogs in PAM format with either Sec-FL, Sec(1–23) or Sec(3–27) as orthosteric basal stimulator (EC20 for each peptide). These efforts resulted in a pool of 34 confirmed hits, and by elucidating the chemical structures we were able to identify five common scaffolds (SCFLD A-E). In-depth analysis of 34 confirmed hits via scatterplot and pie chart (Fig. 4B) revealed probe dependency of hits toward certain peptides. Two of 34 molecules have been confirmed with all three secretin analogs, whereas 30 hits were obtained from screens with individual peptides. The pie chart illustrates the distribution of scaffolds among the individual peptides and indicates scaffolds B and E could be found by employing either one of the peptides, whereas the confirmation of scaffolds A, C, and D was dependent on the orthosteric stimulator applied. It should be pointed out that scaffolds A, B and C have related chemical structures mainly differing from their R2 substituents at the pyrimidine ring (Fig. 4C), strengthening their credibility as hits. While implementing and performing these assays, we observed high day-to-day variability in the detected level of stimulation with the same concentration of agonist. For example, screening the same compound set of 1,025 hits & analogs in Sec-FL PAM mode resulted in two very different hit rates, 368 hits in the first (indicated as squares in scatterplot) and 21 hits in the second run. After in-depth attempts to troubleshoot, we established that minor experimental variances in cell density and concentration of agonist resulted in disproportionally high variation of signal due to the high slope of the standard curve resulting in narrow dynamic range. We addressed this issue by comparing calibration curves of cAMP either detected by LANCE Ultra or the Cisbio GsD cAMP kit (Fig. 4D), which is advertised to perform with a greater dynamic range. The assay dynamic range was determined by the cAMP concentration range between IC10 and IC90 of the standard curve. Indeed, in contrast to LANCE Ultra (slope ~ 1.5; dynamic range = 0.18–3.52 nM cAMP), the CisBio GsD detection kit demonstrated close to unity slope and much broader dynamic range (slope ~ 0.9; dynamic range = 0.22–38.2 nM cAMP). Greater sensitivity and steeper slope was also observed in the dose-response curves of secretin peptides detected with the LANCE Ultra cAMP kit (Fig. 4E). We decided that the small dynamic range of the LANCE Ultra kit is a major drawback for performing larger screens in PAM mode, since it is crucial to assure consistent baseline stimulation.
Figure 4: Pilot Screen of BioAscent library employing LANCE Ultra cAMP assay revealed probe dependency of hits towards secretin or its truncated forms:
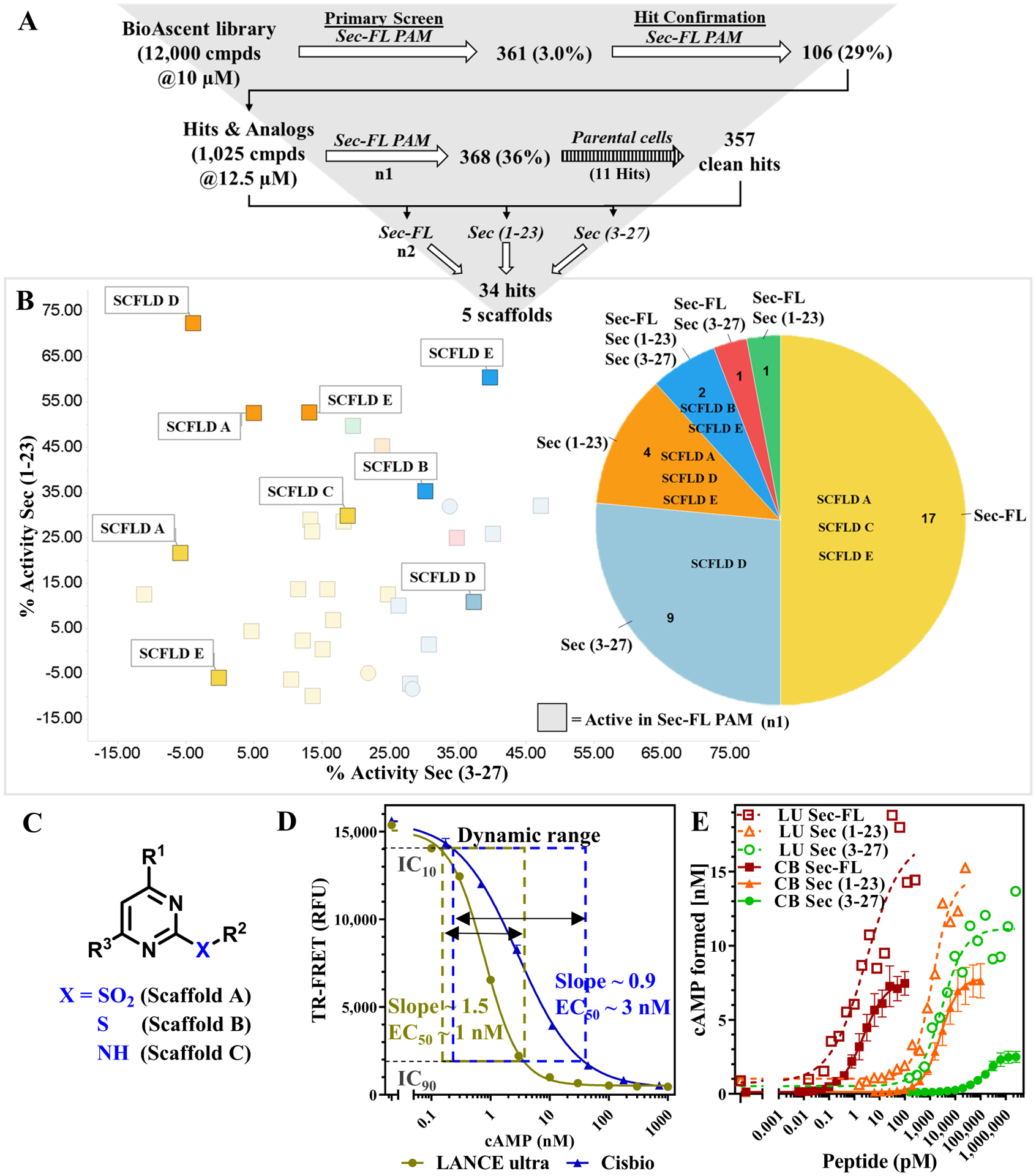
(A) Pilot Screen of BioAscent library using LANCE Ultra cAMP kit and EC20 of Sec-FL as stimulant: primary screen and hit confirmation yielded 106 hits; 1,025 liquid stocks comprising of hits and corresponding analogs were purchased and subsequently validated in primary assay; counter screen with parental cells eliminated 11 compounds; 1,025 hits & analogs were screened with individual secretin peptides yielding 34 hits and 5 prevalent scaffolds. (B) Breakdown of 34 hits into 6 groups: confirmed hit by Sec-FL (yellow, 17), Sec(3–27) (light blue, 9), Sec(1–23) (orange, 4), all three peptides (dark blue, 2), Sec-FL & Sec(3–27) (red, 1) or Sec-FL & Sec(1–23) (green, 1). Scatterplot showing % activity with Sec(3–27) (x-axis) in correlation to % activity with Sec(1–23) (y-axis), squares indicating hit confirmation in first round of screen with Sec-FL; pie chart depicting fraction of hits and detected scaffolds per peptide/peptide mixture; scatterplot and pie chart created using TIBCO Spotfire. (C) Scaffold A, B and C contain closely related chemical structures. (D) Comparison of cAMP standard curves using LANCE Ultra (beige) or Cisbio GsD (blue) detection kit confirmed lower sensitivity but broader dynamic range for Cisbio GsD cAMP detection; assay dynamic range determined by IC10 – IC90 of cAMP standard curve: LANCE Ultra 0.18–3.52 nM cAMP; Cisbio GsD 0.22–38.2 nM cAMP. (E) cAMP formation of secretin analogs applying LANCE Ultra (dotted lines) compared to Cisbio GsD (solid lines) technology: Sec-FL (red), Sec(1–23) (orange) and especially Sec(3–27) (green) demonstrate greater responsiveness but lower reproducibility with LANCE Ultra (LANCE Ultra representative of three experiments, Cisbio GsD combined results of three experiments). Graphs plotted using GraphPad Prism. Data points shown as mean ± SEM.
Stage 1.2: Pilot screens comparing three different detection methods and two sets of ligand probes elucidate strength of combining Sec-FL, Sec(1–23) and Sec(3–27) (3-peptide mix) as orthosteric stimulator
To explore further primary screening options, we tested the 12,000-compound BioAscent collection in three further formats (Fig. 5A). Taking into account the probe dependency and potentially increased sensitivity for stimulating with truncated peptides, we decided to perform in parallel a screen with full agonist Sec-FL and a screen with a mixture of EC10 concentrations of all three peptides (3-peptide mix, 3-pep mix), which resulted in a basal stimulation comparable to EC20 of Sec-FL alone. To obtain better reproducibility and a greater dynamic range, we used the Cisbio GsD cAMP kit as detection method. We were also interested in applying a technology that is distinct from TR-FRET detection of intracellular cAMP. Hence, we selected CRELuc2P reporter (CRELuc) system with a luminescence-based detection system for our third pilot screen employing Sec-FL as basal stimulator. All four methods performed well in the primary screen, having decent to good Z’-factors (0.54–0.61) and reasonable S/B ratios (Table S1). We utilized Z-score for selection of hits in primary HTS with slightly different cutoff criteria for each assay to obtain a similar number of hits. Selecting hits using Z-score is not optimal for hit confirmation studies, thus, we employed NZ-score of (−) 3 as cutoff criteria to compare the four screening assays in the hit confirmation stage (Fig. 5A). The NZ-score is calculated as the difference of sample mean and the mean of negative control divided by the standard deviation of negative control wells in the plate. Hits demonstrate negative NZ-scores in loss-of-signal cAMP TR-FRET assays and positive NZ-scores in gain-of-signal CRELuc assays. In total, 466 hits were confirmed. Comparing the three detection methods using Sec-FL as orthosteric stimulator, we found that CRELuc confirmed the highest number of hits (115), followed by LANCE Ultra with 113 hits. In contrast, Cisbio GsD was only able to confirm 42 hits when Sec-FL was deployed as agonist, probably due to the lower sensitivity of this detection kit. However, 3-pep mix Cisbio GsD assay outnumbered the other three detection methods by confirming 284 hits, which indicates significantly increased assay sensitivity by utilizing a mixture of full and partial agonists. We created a pie chart depicting the fractions of compounds found in individual assays as well as compounds that have been detected in more than one assay (Fig. 5B, left pie chart). We further analyzed the 70 hits confirmed in multiple assays by assigning them to their originating method (Fig. 5B, right pie chart). Intriguingly, 68 of the 70 compounds were hits confirmed by Cisbio GsD 3-pep mix, whereas only 21 of overlapping hits derived from the CRELuc screening. A closer look into activities of overlapping hits via scatterplot revealed two molecules (scaffold A and D) that had been detected by individual peptide screening with LANCE Ultra detection (Fig. S3).
Figure 5: Pilot screens comparing three different detection methods and two sets of ligand probes elucidate strength of combining Sec-FL, Sec(1–23) and Sec(3–27), i.e. 3-peptide mix as orthosteric stimulator:
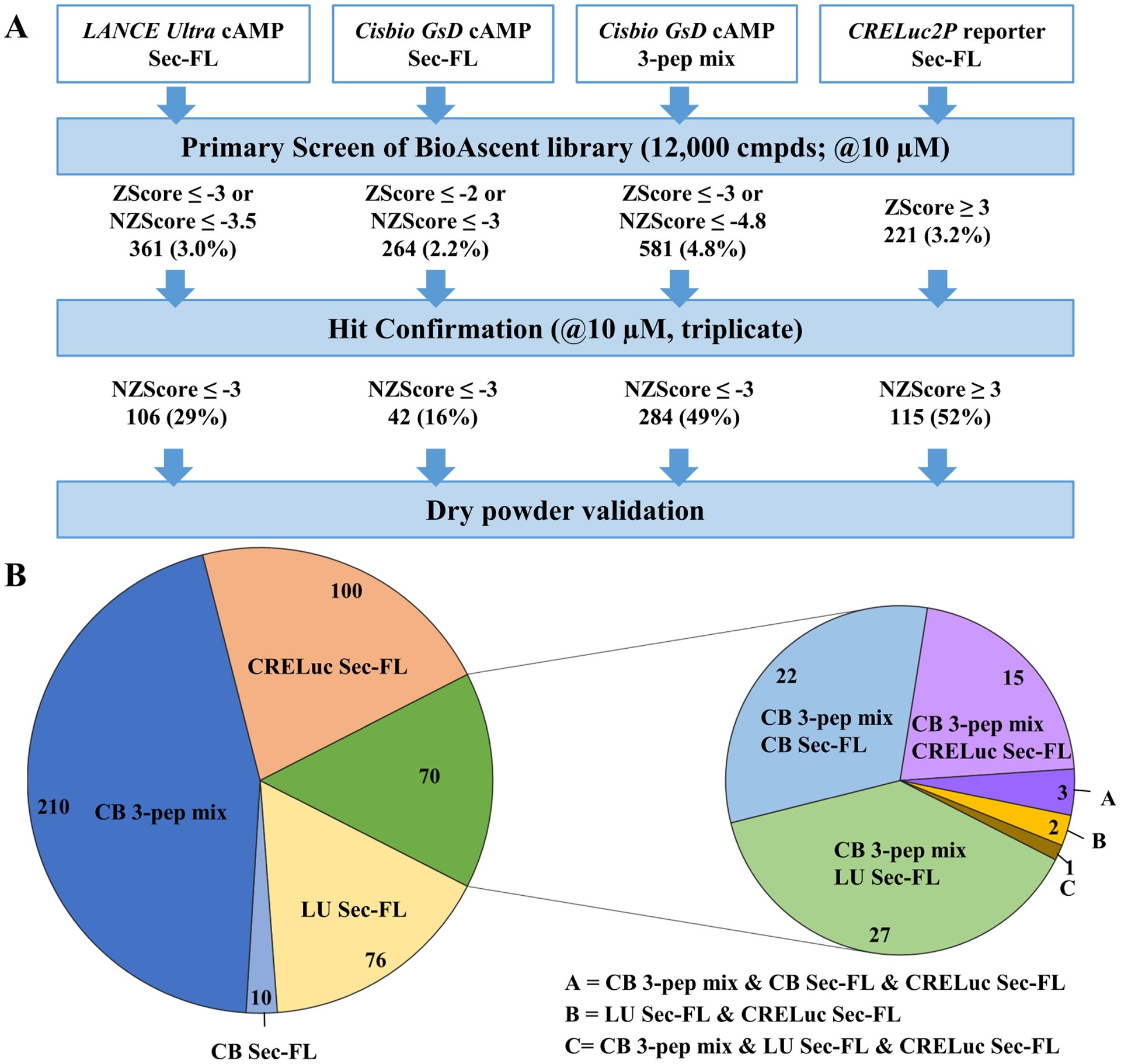
(A) Pilot Screens using LANCE Ultra cAMP (stimulant EC20 Sec-FL), Cisbio GsD Sec-FL (stimulant EC20 Sec-FL), Cisbio GsD 3-pep mix (stimulant EC10 Sec-FL, Sec(1–23) and Sec(3–27)) or CRELuc2P reporter (stimulant EC10 Sec-FL) technologies. Hit confirmation criteria of NZ-Score ≥ (≤ -) 3 was applied to all four formats. (B) Pie chart breaking down total confirmed hits (466): Confirmed via LANCE Ultra (light yellow, 76), Cisbio GsD Sec-FL (light blue, 10), Cisbio GsD 3-pep mix (dark blue, 210), CRELuc (light orange, 100) or via multiple assays (green, 70), whereby 68 of 70 compounds were confirmed by Cisbio GsD 3-pep mix. Pie chart created using Micrsoft Excel.
Stage 2 & 3: Molecules with scaffolds A, B, C and D are SCTR-selective PAMs
Moving into stages two and three of our testing funnel (Fig. 1B), we purchased dry powders of 110 compounds for hit validation. In the course of this study, we focused on two compounds (A1, A9) containing scaffold A, two compounds (B1, C1) with closely related structures scaffold B and C, respectively, and one compound (D1) incorporating scaffold D. To explore intrinsic activity, we recorded dose-response curves in SCTR-CHO cells in agonist mode (Fig. 6A, left panel). In this setting, we detected negligible activity for compounds A1, B1 and C1. Performing the same assay except stimulating cells with an EC20 concentration of Sec-FL (Fig. 6A, middle panel), we were able to measure dose-response curves for all five compounds with potencies in the single digit micro molar range and efficacies ranging from 20% (D1, purple) to 40% (A1, dark green) (Table S2 & S3). We then utilized the 3-peptide mix as orthosteric stimulator (Fig. 6A, right panel), which led to similar ranges in potencies and efficacies for compounds with scaffold A (e.g. 44% for A1 (dark green)) but significantly higher response in case of compound D1 (purple, 53%) indicating probe dependency. To evaluate non-specific activities, we performed the same assay in AVP2R-expressing CHO-K1 cells and AVP as stimulating agent (Fig. 6B). Compared to their PAM activity on SCTRs, A1 (dark green) and B1 (blue) demonstrated negligible activity in the AVPR2 PAM assay. For further discrimination of their allosteric activity, we studied compounds in newly developed secondary assays.
Binding assays are useful tools not only to dissect orthosteric from allosteric ligands but also to determine allosteric activity parameters15. In a competition binding-type experiment we detected a dose-dependent increase of Fluo-Sec binding for all five compounds supporting an allosteric binding site on SCTRs (Fig. 6C, Table S3). Experiment was carried out in a 1536-well plate, thus, consistent with HTS, and demonstrated robust behavior with a Z’-factor of 0.73 and S/B ratio around 13.5. The data was subjected to an allosteric modulator titration analysis, which generated equilibrium dissociation constants Kb and cooperativity factors α for each compound (Fig. 6C, table). Similar to potencies in cAMP assays, the Kb values were within the micro molar range. Interestingly, compound B1 (blue) and D1 (purple) displayed the highest cooperativity to Fluo-Sec, with α-values 3.5 and 3.2, respectively. The best tool to distinguish between orthosteric and allosteric binders is a dissociation binding experiment48 (Fig. 6D). To accelerate Fluo-Sec dissociation, we added GTPγS to our dissociation buffer, which is known to shift the equilibrium of receptors into the G protein-uncoupled state and thereby attenuates agonist binding49. Addition of 12.5 μM A1 (dark green) only slightly decelerated Fluo-Sec dissociation, whereas equal concentration of B1 (blue) was able to slow down dissociation of Fluo-Sec comparable to presence of G proteins (minus GTPγS, grey). Beyond that, we tested A1 (dark green) and B1 (blue) regarding their PAM activity for Sec-FL stimulated β-arrestin-2 recruitment (Fig. 6E). Both compounds slightly enhanced potency, but attenuated efficacy of Sec-FL to recruit β-arrestin-2.
Discussion
Comparison of methods reveals key features for a successful screen to identify PAMs
Here we report the development and results of a testing funnel that incorporates a structured comprehensive toolbox to identify, validate and characterize low-molecular-weight non-peptidyl SCTR modulators. To date, no other small molecule compounds have been described to interact with SCTRs. Hence, it was crucial to develop and select a robust and global primary screening method. By not only comparing three different detection methods (LANCE Ultra, Cisbio GsD and CRELuc), but also the effect of individual as well as mixed full and partial peptide agonists deployed as orthosteric basal stimulators, we were able to evaluate the most critical aspects for successful implementation of a SCTR PAM screening assay based on pilot screens comprising 12,000 compounds. Our goal was to establish a primary screening assay with great assay sensitivity, reproducibility and hit detection range. Initial efforts were conducted applying LANCE Ultra cAMP technology stimulating with Sec-FL. While demonstrating great assay sensitivity and good assay performance in the one-day primary screen, the approach suffered from a lack of day-to-day reproducibility, likely due to a narrow dynamic range of the detection kit. Hence, we decided to test orthogonal approaches relying on intracellular cAMP accumulation, such as Cisbio GsD and CRELuc detection systems. Despite having a broader dynamic range, Sec-FL Cisbio GsD assay demonstrated lower assay sensitivity, which led to a significant reduction in the number of confirmed hits. Utilizing the luminescence-based CRELuc reporter system resulted in higher assay sensitivity, improved S/B ratio and a higher number of hits. These factors beyond the lower cost of detection reagents make it attractive for large-scale HTS campaigns. However, the overlap of CRELuc-derived hits and hits confirmed by TR-FRET based methods was only around 30%, indicating substantial differences in the nature of hits.
The application of 3-pep mix is novel, resulted in higher assay sensitivity and allowed the identification of SCTR PAMs with distinct probe dependencies
The profiling of 1,025 hits & analogs via LANCE Ultra cAMP employing Sec-FL or truncated secretin peptides Sec(1–23) and Sec(3–27) revealed that the response of potential PAMs is dependent on the nature of the orthosteric stimulator. Although this phenomenon has already been described for PAMs acting on other GPCRs34, there is no such pattern described for SCTRs. This might be due to the poorly understood metabolism of Sec-FL, which might vary depending on its site of expression. However, potential secretin metabolites are likely to be cleavage products from either end of the full-length peptide, thus, we focused our studies on one analog truncated at the C-terminal tail37 (Sec(1–23)) and one analog designed as a hypothetical N-terminal degradation product (Sec(3–27)). Due to the development of a comprehensive set of SCTR binding and functional assays, we were able to thoroughly characterize our metabolite models, revealing that C-terminal peptide truncation leads to a lower affinity, but fully functional analog whereas N-terminal cleavage results in an analog exerting only partial SCTR activation. These distinct activity profiles in combination with the modification of receptor occupancy and restricted binding space can be utilized to increase the sensitivity and effectiveness of PAM screening assays33. Looking ahead, PAMs able to enhance activity of truncated partially active versions of Sec-FL might be particularly useful for therapeutic applications by extending the functional response of the natural ligand after its degradation and inactivation21, 31, 33. Our studies with individual secretin peptides in the LANCE Ultra PAM screening assay elucidated that each peptide finds distinct sets of compounds. Since the nature of physiological metabolites remains unclear, we decided to combine all three analogs into one orthosteric stimulator probe (3-peptide mix) to identify PAMs that are likely to work with any potential metabolite. Due to the broader dynamic range, we employed the Cisbio GsD detection kit to conduct the screen of the BioAscent library in 3-peptide mix format. The primary screen worked well with good Z’-factor and decent S/B ratio around 3. Moreover, Cisbio GsD 3-pep mix was not only able to identify the largest number of hits but was also empowered to detect 97% of overlapping hits, which were found utilizing Sec-FL in LANCE Ultra, Cisbio GsD or CRELuc assays. Hence, Cisbio GsD 3-pep mix assay is a robust, reproducible, sensitive and effective primary screening assay.
Secondary assays reveal that identified scaffolds exert different MOAs
The pool of overlapping hits provided five interesting scaffolds, four of which were investigated in detail. Dry powders of compound A1, A9, B1, C1 and D1 were validated in dose-response studies on SCTRs regarding their ability to accumulate cAMP with no orthosteric ligand, Sec-FL or 3-peptide mix basal stimulation. In general, compounds showed no to marginal intrinsic activity in agonist mode, but significant responses in PAM modes. Intriguingly, compounds A1 and A9 demonstrated comparable activities in both, Sec-FL and 3-pep mix cAMP assays, whereas D1 exerted greater effects with mixed agonists, therefore suggesting distinct MOAs. A1 and B1 appeared to produce slight responses in CHO-AVP2R cells stimulated with AVP, however, compound B1 displayed 18-fold selectivity towards SCTRs. Of note, structural analog A9 was inactive in the AVP2R-PAM cAMP assay proposing that specificity is achievable through structural modification. The development of a fluorescence-based target engagement assay enabled the characterization of compounds on SCTR-binding in a high-throughput environment. We were excited to see that all five compounds were able to increase Fluo-Sec binding in a dose-dependent manner. Resulting cooperativity factors α revealed further subtle differences between investigated scaffolds. A1 displayed lower α values in the allosteric modulator titration and only slightly decreased Fluo-Sec dissociation rates in SNAP-SCTR dissociation binding experiments. However, compound B1, exhibiting stronger positive cooperativity to Fluo-Sec, was able to exert comparable deceleration of Fluo-Sec dissociation like coupled G proteins, the most widely studied endogenous PAM targeting GPCRs. To explore potential signaling bias, we also subjected A1 and B1 to Sec-FL stimulated β-arrestin-2 recruitment studies. Both compounds reduced efficacy but were able to slightly enhance Sec-FL potency. If this phenomenon is due to functional selectivity of PAMs or a non-specific interaction with the assay will be addressed in future studies.
Testing funnel identifies the first small molecule modulators described for SCTRs
The combination of a diverse selection of primary screens, intensive comparison of their primary screen performance, design of GPCR-specific secondary assays and consequential arrangement of funnel stages resulted in the discovery of PAMs targeting SCTRs. One of the main novelties of our testing funnel was the development of the 3-peptide mix stimulator that not only substantially enhanced assay sensitivity and effectiveness, but also allowed the detection of probe-dependent hits. Another unique advantage was the development and implementation of GPCR-specific secondary assays, which led to immediate profiling and validation of distinct sets of hits with respect to target engagement and functional selectivity. This is the first report, to our knowledge, describing small-molecular weight compounds that not only interact with SCTRs but also significantly enhance the binding and response of secretin peptides. Encouragingly, discovered hits cover effects on both natural peptide ligand and metabolite models, considerably increasing chances for potential therapeutic application.
Future Directions- testing funnel stage 4
In this study we discovered bona fide SCTR PAM hits. We continue working with identified scaffolds in further hit validation and optimization studies by extending structural variety through analog-by-catalog and medicinal chemistry efforts. Like many HTS-suitable cell-based assays, the use of receptor overexpressing cell lines may result in overly amplified or distorted signaling. Thus, hits will be confirmed using cell models with endogenous SCTR expression and signaling. Hit-to-lead studies involving further optimization of MOA, pharmacokinetics and toxicity profiles is expected to identify analogs suitable for in vivo testing. Beyond that, future work will include the expansion of GPCR selectivity screens not only to detect possible off-target effects but also to explore the potential of class B GPCR polypharmacology50, which might provide additional benefits for the treatment of metabolic disorders.
Supplementary Material
Acknowledgements
We acknowledge support from the US National Institutes of Health (NIH) National Heart, Lung, and Blood Institute (NHLBI) grant R01HL133501. We would like to thank personnel of the Conrad Prebys Center for Chemical Genomics (CPCCG) at the Sanford Burnham Prebys Medical Discovery Institute (SBP) for help in diverse aspects of this project.
Footnotes
Declaration of Conflicting Interests
We declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Bayliss WM; Starling EH The mechanism of pancreatic secretion. The Journal of Physiology 1902, 28, 325–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishihara T; Nakamura S; Kaziro Y; et al. Molecular cloning and expression of a cDNA encoding the secretin receptor. The EMBO journal 1991, 10, 1635–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Graaf C; Song G; Cao C; et al. Extending the Structural View of Class B GPCRs. Trends Biochem Sci 2017, 42, 946–960. [DOI] [PubMed] [Google Scholar]
- 4.Afroze S; Meng F; Jensen K; et al. The physiological roles of secretin and its receptor. Annals of Translational Medicine 2013, 1, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grossini E; Molinari C; Morsanuto V; et al. Intracoronary secretin increases cardiac perfusion and function in anaesthetized pigs through pathways involving beta-adrenoceptors and nitric oxide. Exp Physiol 2013, 98, 973–87. [DOI] [PubMed] [Google Scholar]
- 6.Li Y; Schnabl K; Gabler S-M; et al. Secretin-Activated Brown Fat Mediates Prandial Thermogenesis to Induce Satiation. Cell 2018, 175, 1561–1574.e12. [DOI] [PubMed] [Google Scholar]
- 7.Modvig IM; Andersen DB; Grunddal KV; et al. Secretin release after Roux-en-Y gastric bypass reveals a population of glucose-sensitive S cells in distal small intestine. Int J Obes (Lond) 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brandler J; Miller LJ; Wang XJ; et al. Secretin effects on gastric functions, hormones and symptoms in functional dyspepsia and health: randomized crossover trial. Am J Physiol Gastrointest Liver Physiol 2020, 318, G635–G645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Witteloostuijn SB; Dalboge LS; Hansen G; et al. GUB06–046, a novel secretin/glucagon-like peptide 1 co-agonist, decreases food intake, improves glycemic control, and preserves beta cell mass in diabetic mice. J Pept Sci 2017, 23, 845–854. [DOI] [PubMed] [Google Scholar]
- 10.Wu N; Meng F; Invernizzi P; et al. The secretin/secretin receptor axis modulates liver fibrosis through changes in transforming growth factor-beta1 biliary secretion in mice. Hepatology 2016, 64, 865–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh K; Senthil V; Arokiaraj AW; et al. Structure-Activity Relationship Studies of N- and C-Terminally Modified Secretin Analogs for the Human Secretin Receptor. PLoS One 2016, 11, e0149359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chey WY; Chang TM Secretin, 100 years later. J Gastroenterol 2003, 38, 1025–35. [DOI] [PubMed] [Google Scholar]
- 13.Imamura M; Takahashi K; Adachi H; et al. Usefulness of selective arterial secretin injection test for localization of gastrinoma in the Zollinger-Ellison syndrome. Ann Surg 1987, 205, 230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang YM; Kuen DS; Chung Y; et al. Galpha12/13 signaling in metabolic diseases. Exp Mol Med 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoare SRJ Allosteric Modulators of Class B G-Protein-Coupled Receptors. Current Neuropharmacology 2007, 5, 168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morris LC; Nance KD; Gentry PR; et al. Discovery of (S)-2-cyclopentyl-N-((1-isopropylpyrrolidin2-yl)-9-methyl-1-oxo-2,9-dihydro-1H-py rrido[3,4-b]indole-4-carboxamide (VU0453379): a novel, CNS penetrant glucagon-like peptide 1 receptor (GLP-1R) positive allosteric modulator (PAM). J Med Chem 2014, 57, 10192–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knudsen LB; Kiel D; Teng M; et al. Small-molecule agonists for the glucagon-like peptide 1 receptor. Proceedings of the National Academy of Sciences 2007, 104, 937–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sloop KW; Willard FS; Brenner MB; et al. Novel small molecule glucagon-like peptide-1 receptor agonist stimulates insulin secretion in rodents and from human islets. Diabetes 2010, 59, 3099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.King K; Lin NP; Cheng YH; et al. Isolation of Positive Modulator of Glucagon-like Peptide-1 Signaling from Trigonella foenum-graecum (Fenugreek) Seed. J Biol Chem 2015, 290, 26235–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bueno AB; Showalter AD; Wainscott DB; et al. Positive Allosteric Modulation of the Glucagon-like Peptide-1 Receptor by Diverse Electrophiles. J Biol Chem 2016, 291, 10700–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris LC; Days EL; Turney M; et al. A Duplexed High-Throughput Screen to Identify Allosteric Modulators of the Glucagon-Like Peptide 1 and Glucagon Receptors. J Biomol Screen 2014, 19, 847–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Congreve M; Oswald C; Marshall FH Applying Structure-Based Drug Design Approaches to Allosteric Modulators of GPCRs. Trends Pharmacol Sci 2017, 38, 837–847. [DOI] [PubMed] [Google Scholar]
- 23.Cheng Z; Garvin D; Paguio A; et al. Luciferase Reporter Assay System for Deciphering GPCR Pathways. Current chemical genomics 2010, 4, 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emami-Nemini A; Roux T; Leblay M; et al. Time-resolved fluorescence ligand binding for G protein-coupled receptors. Nat Protoc 2013, 8, 1307–20. [DOI] [PubMed] [Google Scholar]
- 25.Senthil Vijayalakshmi, L. J, Vaudry David, Chow Billy Kwok Chong. Fluorescence Resonance Energy Transfer Competitive Binding Assay for Secretin Receptor (Class B-GPCR). Journal of Pharmacy and Pharmacology2 2014, 295–303. [Google Scholar]
- 26.Sykes DA; Moore H; Stott L; et al. Extrapyramidal side effects of antipsychotics are linked to their association kinetics at dopamine D2 receptors. Nat Commun 2017, 8, 763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sykes DA; Stoddart LA; Kilpatrick LE; et al. Binding kinetics of ligands acting at GPCRs. Mol Cell Endocrinol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cannaert A; Storme J; Franz F; et al. Detection and Activity Profiling of Synthetic Cannabinoids and Their Metabolites with a Newly Developed Bioassay. Anal Chem 2016, 88, 11476–11485. [DOI] [PubMed] [Google Scholar]
- 29.Sills MA; Weiss D; Pham Q; et al. Comparison of Assay Technologies for a Tyrosine Kinase Assay Generates Different Results in High Throughput Screening. Journal of Biomolecular Screening 2002, 7, 191–214. [DOI] [PubMed] [Google Scholar]
- 30.Laprairie RB; Kulkarni PM; Deschamps JR; et al. Enantiospecific Allosteric Modulation of Cannabinoid 1 Receptor. ACS Chem Neurosci 2017, 8, 1188–1203. [DOI] [PubMed] [Google Scholar]
- 31.Wootten D; Savage EE; Valant C; et al. Allosteric modulation of endogenous metabolites as an avenue for drug discovery. Molecular pharmacology 2012, 82, 281–90. [DOI] [PubMed] [Google Scholar]
- 32.Kenakin T The Quantitative Characterization of Functional Allosteric Effects. Curr Protoc Pharmacol 2017, 76, 9 22 1–9 22 10. [DOI] [PubMed] [Google Scholar]
- 33.Nakane A; Gotoh Y; Ichihara J; et al. New screening strategy and analysis for identification of allosteric modulators for glucagon-like peptide-1 receptor using GLP-1 (9–36) amide. Anal Biochem 2015, 491, 23–30. [DOI] [PubMed] [Google Scholar]
- 34.Nolte WM; Fortin JP; Stevens BD; et al. A potentiator of orthosteric ligand activity at GLP-1R acts via covalent modification. Nat Chem Biol 2014, 10, 629–31. [DOI] [PubMed] [Google Scholar]
- 35.Mulvihill EE; Drucker DJ Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr Rev 2014, 35, 992–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hupe-Sodmann K; McGregor GP; Bridenbaugh R; et al. Characterisation of the processing by human neutral endopeptidase 24.11 of GLP-1(7–36) amide and comparison of the substrate specificity of the enzyme for other glucagon-like peptides. Regulatory Peptides 1995, 58, 149–156. [DOI] [PubMed] [Google Scholar]
- 37.Gwang-Ho Jeohn KT Purification and Characterization of a Vasoactive Intestinal Polypeptide-degrading Endoprotease from Porcine Antral Mucosal Membranes J. Biol. Chem 1995, 270, 7809–7815. [DOI] [PubMed] [Google Scholar]
- 38.Juul KV; Bichet DG; Nielsen S; et al. The physiological and pathophysiological functions of renal and extrarenal vasopressin V2 receptors. Am J Physiol Renal Physiol 2014, 306, F931–40. [DOI] [PubMed] [Google Scholar]
- 39.Cottet M; Faklaris O; Zwier JM; et al. Original Fluorescent Ligand-Based Assays Open New Perspectives in G-Protein Coupled Receptor Drug Screening. Pharmaceuticals 2011, 4, 202–214. [Google Scholar]
- 40.Kenakin T; Barker EL Biased Receptor Signaling in Drug Discovery. Pharmacological Reviews 2019, 71, 267–315. [DOI] [PubMed] [Google Scholar]
- 41.Manglik A; Lin H; Aryal DK; et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gurevich VV; Gurevich EV Biased GPCR signaling: Possible mechanisms and inherent limitations. Pharmacol Ther 2020, 107540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bermudez M; Nguyen TN; Omieczynski C; et al. Strategies for the discovery of biased GPCR ligands. Drug Discov Today 2019. [DOI] [PubMed] [Google Scholar]
- 44.Ilter M; Mansoor S; Sensoy O Utilization of Biased G Protein-Coupled ReceptorSignaling towards Development of Safer andPersonalized Therapeutics. Molecules 2019, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dixon AS; Schwinn MK; Hall MP; et al. NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem Biol 2016, 11, 400–8. [DOI] [PubMed] [Google Scholar]
- 46.Miller LJ; Dong M; Harikumar KG Ligand binding and activation of the secretin receptor, a prototypic family B G protein-coupled receptor. Br J Pharmacol 2012, 166, 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dong M; Koole C; Wootten D; et al. Structural and functional insights into the juxtamembranous amino-terminal tail and extracellular loop regions of class B GPCRs. Br J Pharmacol 2014, 171, 1085–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klein MT; Vinson PN; Niswender CM Approaches for probing allosteric interactions at 7 transmembrane spanning receptors. Prog Mol Biol Transl Sci 2013, 115, 1–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeVree BT; Mahoney JP; Velez-Ruiz GA; et al. Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature 2016, 535, 182–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sloop KW; Briere DA; Emmerson PJ; et al. Beyond Glucagon-like Peptide-1: Is G-Protein Coupled Receptor Polypharmacology the Path Forward to Treating Metabolic Diseases? ACS Pharmacology & Translational Science 2018, 1, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ortiz Zacarias NV; Lenselink EB; AP IJ; et al. Intracellular Receptor Modulation: Novel Approach to Target GPCRs. Trends Pharmacol Sci 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.