

Abstract
INTRODUCTION:
Nucleosomes—the primary, repeating unit of chromatin—package and protect the genome while also transmitting various regulatory signals through, in part, posttranslational modifications of histones. These histone modifications (for example, acetylation, ubiquitination, or methylation) affect critical processes such as transcription, replication, recombination, and repair, often forming complicated networks to ensure finely tuned signaling for chromatin enzymes. One such enzyme—evolutionarily conserved disruptor of telomeric silencing (Dot1)—catalyzes mono-, di-, and trimethylation of histone H3 lysine 79 (H3K79). H3K79 methylation by Dot1 is a prominent example of trans-histone cross-talk, a process in which one histone and its modification affects the modification of another histone. In mammals, the human homolog Dot1L plays critical roles in embryogenesis and hematopoiesis. Although recent advances have provided insights into Dot1L stimulation through histone H2B ubiquitination, how other modifications mechanistically regulate Dot1 activity is not known. We were particularly interested in how histone lysine acetylation contributes to the regulation of chromatin enzymes. Lysine acetylation plays pivotal roles in chromatin decondensation, transcriptional activation, and maintenance of euchromatin, serving as a general antisilencing mark in eukaryotes. Here, we present mechanistic studies that show how histone acetylation regulates the activity of Dot1.
RATIONALE:
Histone H4 acetylation, histone H2B ubiquitination, and H3 methylation are conserved cotranscriptional histone modifications that work together to ensure the appropriate regulation of chromatin structure during transcription. The mechanisms of cross-talk between these modifications and enzymes that deposit them are crucial for understanding transcription. Advances in cryo–electron microscopy (cryo-EM), the ability to make specifically acetylated and ubiquitinated nucleosomes, and established in vitro and in vivo assays allowed us to determine the detailed mechanisms of yeast Dot1 regulation through histone acetylation and ubiquitination.
RESULTS:
We tested nucleosomes with different acetylation states of histone H4 in vitro, and we show that Dot1 is allosterically stimulated by acetylation of H4 and that this effect is specific to lysine 16 (H4K16ac). The other known acetylation targets on histone H4 (H4K5ac, H4K8ac, and H4K12ac) do not stimulate the activity of Dot1, which highlights the distinctive role of H4K16 acetylation in regulating chromatin structure. We also show that the effect of H4K16 acetylation is direct and further enhanced by H2B ubiquitination (H2BUb), resulting in an optimal catalytic rate for Dot1. To gain mechanistic insights into stimulation by H4K16ac and its coordination with H2BUb, we determined two cryo-EM structures: one of Dot1 in complex, with nucleosomes bearing both H4K16ac and H2Bub, and the second of Dot1, with a nucleosome bearing only H2BUb. Upon examining our cryo-EM dataset of Dot1 bound to the nucleosome containing unacetylated H4, the particles classified into two main three-dimensional (3D) classes. In the first class, Dot1 is bound to the nucleosome in a catalytic conformation. In the second class, it is bound in a noncatalytic conformation. This is different from the dataset in which Dot1 is bound to an H4K16 acetylated nucleosome. When H4K16ac is present, the cryo-EM data is more homogeneous, and Dot1 is bound to the nucleosome predominantly in a catalytic conformation. This suggests a model in which acetylation of the H4 tail restricts the sampling space of Dot1, resulting in an active conformation leading to increased activity. We therefore propose that H2BUb partially restricts the conformation of yeast Dot1 on the nucleosome and that H4K16ac further restricts and stabilizes the active conformation. Comparing both of these cryo-EM structures allowed us to identify residues that are critical for Dot1 stimulation by H4K16ac and H2BUb. Site-directed mutagenesis of Dot1 coupled with enzymatic assays on nucleosomes revealed the details of these interfaces. These results show that the allosteric stimulation of Dot1 by H4K16ac and H2BUb plays a crucial role in H3K79 di- and trimethylation.
CONCLUSION:
This work demonstrates how Dot1 is regulated by histone acetylation and how H4K16ac coordinates with H2BUb to regulate Dot1. H4K16ac plays a critical role in opening chromatin structure by counteracting the binding of silencing proteins, while simultaneously stimulating an enzyme that is important for transcription. We provide an example in which the activity of the fundamental methyltransferase Dot1 is modulated through cross-talk between distinct histone modifications to ensure optimal maintenance and propagation of an epigenetic state. Cross-talk such as this may represent a general property of chromatin enzymes.
Graphical Abstract
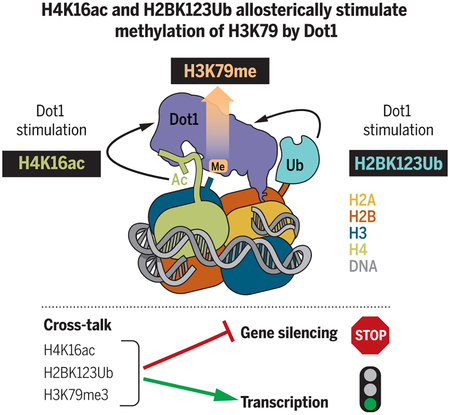
H3K79 methylation by Dot1 is allosterically stimulated by H4K16 acetylation and by H2BK123 ubiquitination. Cross-talk between different histone posttranslational modifications can orchestrate distinct chromatin states to regulate transcription and gene silencing.
Dot1 (disruptor of telomeric silencing-1), the histone H3 lysine 79 (H3K79) methyltransferase, is conserved throughout evolution, and its deregulation is found in human leukemias. Here, we provide evidence that acetylation of histone H4 allosterically stimulates yeast Dot1 in a manner distinct from but coordinating with histone H2B ubiquitination (H2BUb). We further demonstrate that this stimulatory effect is specific to acetylation of lysine 16 (H4K16ac), a modification central to chromatin structure. We provide a mechanism of this histone cross-talk and show that H4K16ac and H2BUb play crucial roles in H3K79 di- and trimethylation in vitro and in vivo. These data reveal mechanisms that control H3K79 methylation and demonstrate how H4K16ac, H3K79me, and H2BUb function together to regulate gene transcription and gene silencing to ensure optimal maintenance and propagation of an epigenetic state.
Histones are subject to a vast array of posttranslational modifications (PTMs) that influence chromatin structure and function by altering interactions between nucleosomes or acting as docking sites for recruitment of effector proteins (1). Modifications such as acetylation of histones H3 and H4 (H3ac and H4ac), methylation of H3K79 (H3K79me), or monoubiquitination of histone H2B (H2BUb) are associated with active transcription (2–4). Regulation of these modifications and the enzymes that deposit them is critical to transcription, and disruption of their deposition leads to aberrant gene expression and disease (5).
Histone acetylation is perhaps the most extensively studied PTM (6–8). There are many lysines that can be acetylated, and they play critical roles in gene regulation (8–10). Acetylation of lysines in H3 and H4 N-terminal tails correlates positively with gene transcription, peaking sharply at active promoters and present in gene bodies, with the levels of acetylation being proportional to the transcription rates (7, 11, 12). Acetylation neutralizes positive charges of lysines, which results in the opening of the chromatin, allowing greater access to transcription factors, and facilitating passage of the RNA polymerase II (13, 14). Acetylated histones are easier to displace from DNA both in vivo (15) and in vitro (16). In particular, acetylation of histone H4 lysine 16 (H4K16ac) inhibits the formation of compact 30-nm fibers (17).
The conserved factor Dot1 (disruptor of telomeric silencing-1) is the only known methyltransferase that catalyzes mono-, di-, and trimethylation of H3K79 (H3K79me1, -me2, and -me3) (18–20). Genome-wide analyses of H3K79me in yeast, fly, mouse, and human have demonstrated a high correlation between this modification and transcriptional activity (21–25). In Saccharomyces cerevisiae, for example, ~90% of the genome is methylated at H3K79 (20). In mammals, the homologous Dot1L (Dot1-like) is essential for embryogenesis, hematopoiesis, and cardiac function (26, 27). Aberrant transcriptional activation through Dot1L is found in leukemias that result from oncogenic chromosomal translocations involving the MLL gene (28–31). A prerequisite for efficient H3K79me2 and -me3 by Dot1 and Dot1L is monoubiquitination of histone H2B lysine K123 in yeast and K120 in humans (hereafter H2BUb) (32–36). Recent structural studies by us and other groups found that interaction of Dot1L with a hydrophobic patch on ubiquitin (Ub) reduces sampling space of Dot1L on the nucleosome, resulting in higher methylation states (37–41).
The activity of Dot1 has also been linked to histone acetylation. For example, the activity of the histone deacetylase Rpd3L restricts H3K79me3 at its euchromatic targets in budding yeast, and inactivation of the Rpd3 homolog HDAC1 in mouse thymocytes leads to an increase in H3K79me (42). H4K16ac regulates the Dot1-mediated distribution of H3K79me on euchromatin (43). H4K16A and H4K16R mutations decrease the global level of H3K79me3 (43). Genome-wide H3K79me2 and -me3 were decreased in a null mutant of the H4K16-specific acetyltransferase Sas2p (sas2Δ) (43). These observations suggest a possible existence of evolutionarily conserved cross-talk between histone acetylation and H3K79me by Dot1 and Dot1L. It is unknown whether histone acetylation stimulates Dot1 and Dot1L directly, and the mechanism of such putative cross-talk is unknown.
Dot1 was originally implicated in the silencing of genes in yeast telomeres (44). Telomeric silencing is established through the recruitment and binding of SIR (silent information regulator) complex (Sir2, −3, and −4) to chromatin (45, 46). Activity of Sir2, nicotinamide adenine dinucleotide (NAD+)–dependent H4K16 deacetylase, is essential to create a nucleosomal binding site for Sir3 (47–49). Nucleosome-Sir3 binding is maximally perturbed when H4K16 is acetylated and H3K79 is methylated (50, 51). Overexpression of Dot1 spreads H3K79me into silent chromatin, displacing Sir proteins, and mutation of H3K79 or deletion of Dot1 compromises telomeric silencing by mislocalizing the Sir complex (20). Furthermore, it has been shown that a basic patch on the histone H4 tail is critical for Dot1 binding and H3K79 methylation (52, 53) and that Sir3 competes with Dot1 for this site (53, 54).
Given the strong correlation between H4 acetylation, H2B ubiquitination, and H3K79 methylation and the critical role of Dot1, we sought to determine the nature of this putative cross-talk. The cross-talk between H4K16ac and Dot1 could involve (i) an indirect effect, through structural changes to the nucleosome caused by acetylation; (ii) an interaction between Dot1 and H4K16ac mediated by another accessory protein; or (iii) direct interaction and stimulation of Dot1 by H4K16ac. To distinguish among these different mechanisms, we used a combination of biochemical, structural, and functional approaches. We demonstrate that direct interaction of H4K16ac with Dot1 stimulates its catalytic activity. We also show that Dot1 H3K79me activity on the doubly modified substrate (H4K16ac-H2Bub nucleosome) is higher when compared with that on the singly modified nucleosome. We define the structural basis of H4K16ac recognition by Dot1 that results in stimulation of its H3K79me activity and propose a molecular mechanism for this trans-histone cross-talk. Last, we assess the biological impact of our proposed biochemical mechanism for Dot1 stimulation by H4K16ac and H2BUb. On the basis of our structural, biochemical, and in vivo functional data, we propose a direct interaction model that describes the rules of Dot1 stimulation by the combined action of histone acetylation and ubiquitination. An extended view of the implications of our data suggests a scheme for how H3K79me by Dot1 regulates the kinetics of gene silencing in yeast.
Results
Acetylation of H4K16 directly stimulates the catalytic activity of Dot1 on nucleosomes
Four invariant lysine residues in the histone H4 N terminus (K5, K8, K12, and K16) can be acetylated in eukaryotes (Fig. 1A, top). To test whether global acetylation of histone H4 stimulates Dot1, we measured activity of the catalytic domain of yeast Dot1 (residues 158 to 582) using semisynthetic tetraacetylated (H4K5acK8acK12acK16ac) “designer” nucleosomes as substrates (Fig. 1A, bottom). We observed a twofold stimulation of Dot1 activity by tetraacetylated H4 nucleosomes as compared with unmodified nucleosomes (“Unmod nuc”). To understand whether any specific H4 residue underlies stimulation, we performed the enzymatic reactions using designer nucleosomes acetylated at individual sites (H4K5ac, H4K8ac, H4K12ac, or H4K16ac) (Fig. 1A, bottom). Of all individually acetylated tested residues, only acetylated lysine 16 showed a substantial (sevenfold) stimulation of Dot1 catalytic activity (Fig. 1A, bottom). This observation established that acetylation of H4K16 directly stimulates catalytic activity of Dot1 and suggested that the modification of either K5, K8, or K12 may inhibit this stimulatory effect in the context of nucleosome tetraacetylated on H4 tail. This result is in line with the biological roles of K5, K8, and K12, which are acetylated during chromatin assembly, and K16 acetylation, which regulates chromatin opening and gene activation (17).
Fig. 1. . Acetylation of H4K16 directly stimulates the catalytic activity of Dot1 on nucleosome substrates.
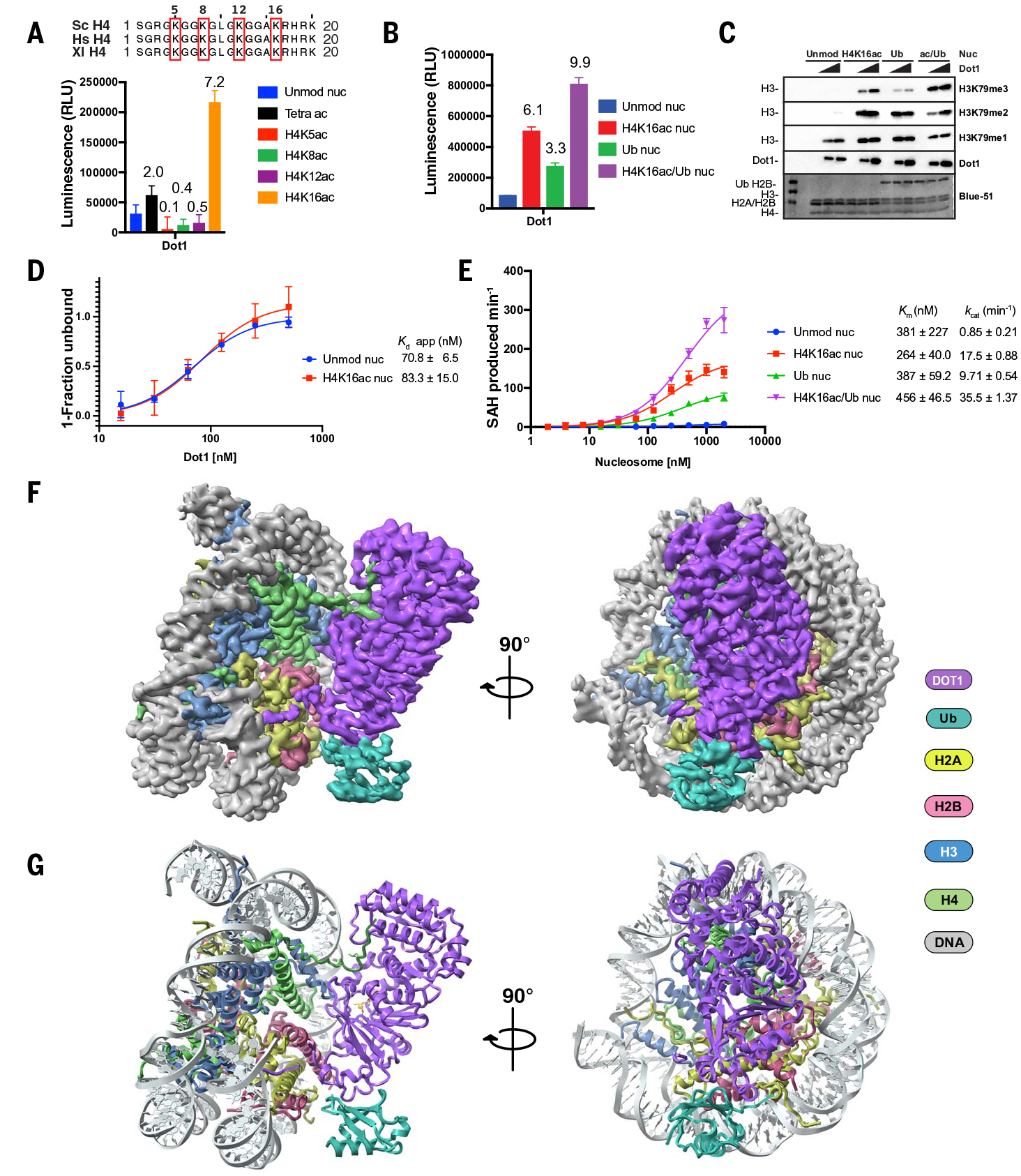
(A) (Top) Multiple sequence alignment of the H4 tail showing the conservation of the N-terminal region. Residues acetylated for biochemistry experiments are indicated with numbers. (Bottom) Endpoint methyltransferase assay for yeast Dot1 using Unmod nuc, tetraacetylated (K5acK8acK12acK16ac), or monoacetylated (H4Kxac) nucleosomes. Numbers above the columns are ratios between Dot1 activity on acetylated H4 and unmodified nucleosome substrates. (B) Representative endpoint methyltransferase assay for Dot1 by using Unmod nuc, H4K16ac nuc, Ub nuc, or ac/Ub nuc substrates. Each data point and error bar indicate the mean ± SD from three independent experiments. (C) Representative HMT (histone methyltransferase) assay measuring activity of Dot1 on different nucleosome substrates. HMT assays were performed with an increasing amount of Dot1 in the presence of Unmod nuc, H4K16ac nuc, Ub nuc, or ac/Ub nuc substrates, and reaction products were identified by using Western blot. (D) Binding curves of Dot1 to Unmod nuc (Kd = 70.8 nM) or H4K16ac nuc (Kd = 83.3 nM) measured with EMSA. Each data point and error bar indicate the mean ± SD from three independent experiments. The standard errors of dissociation constants (Kd) are indicated. (E) Michaelis–Menten saturation curves for Dot1 on Unmod nuc or modified nucleosomes. The Km and kcat values of the fitted data are reported in the graph. Each data point and error bar represent the mean ± SD from three independent experiments. The reported errors of the fitted kcat and Km correspond to the standard error. (F) 3.1-Å cryo-EM reconstruction of Dot1-H4K16ac structure displayed in two separate views related by 90°. (G) Structural model of Dot1-H4K16ac complex. The Dot1 catalytic domain is depicted in purple, ubiquitin in cyan, DNA in gray, histone H2A in pale yellow, histone H2B in red salmon, histone H3 in marine blue, and histone H4 in lime green.
H2BUb stimulates both yeast and human Dot1 activity (32, 34, 55–57). To understand how this well-described stimulation compares with that of H4K16 acetylation, we reconstituted singly modified H4K16ac and H2BUb nucleosome substrates (fig. S1) and measured methyltransferase activity of Dot1 (Fig. 1B). Dot1 stimulation by singly modified H4K16ac nucleosomes is higher (roughly twofold) than that by control H2BUb-only nucleosomes (Fig. 1B). We asked whether the combination of these two histone modifications on a single nucleosome results in further stimulation of Dot1 activity. We reconstituted H4K16ac/H2BUb (“doubly modified”; ac/Ub) nucleosomes and used them as a substrate (Fig. 1B and fig. S1). We observed that the doubly modified substrate increased the stimulation effect on Dot1 compared with single modifications (Fig. 1B). Then, we performed enzymatic assays probing levels of H3K79 methylation (me1, me2, and me3) on Unmod nuc, H4K16ac (H4K16ac nuc), H2BUb (Ub nuc), and doubly modified (ac/Ub nuc) nucleosomes by using immunoblot (Fig. 1C). Although unmodified nucleosomes can be monomethylated, the higher-level methylation states (me2 and me3) can only be achieved when stimulatory histone modifications, such as H4K16ac or H2BUb or both, coreside on nucleosomes (Fig. 1C). We further confirmed these results using full-length yeast Dot1 (fig. S2).
Then, we tested whether stimulation by H4K16ac is due to higher binding affinity of Dot1 by using electrophoretic mobility shift assays (EMSAs) with unmodified and H4K16ac nucleosomes (Fig. 1D and fig. S3). We did not observe any difference in affinity as a function of H4K16ac [Dissociation constant (Kd) on unmod nuc, 70.8 nM; on H4K16ac nuc, 83.3 nM]. To further understand the mechanism, we performed enzyme kinetics using unmodified, H4K16ac, H2BUb, and doubly modified nucleosomes (Fig. 1E). We observed that both acetylated and ubiquitinated nucleosomes result in a higher apparent uni-molecular rate constant (kcat) compared with that of unmodified nucleosomes and that this effect is even stronger on doubly modified nucleosomes, whereas the Michaelis constant (Km) for methyl transfer is similar for all assayed substrates (average Km = ~375 nM) (Fig. 1E). Binding and kinetic data for H4K16ac suggest an allosteric role of this modification in stimulating Dot1.
Structural basis for Dot1 stimulation by H4K16 acetylation
To gain mechanistic insight of Dot1 stimulation by the combined action of H4K16ac and H2BUb modifications, we determined the cryo–electron microscopy (cryo-EM) structure of the catalytic domain of yeast Dot1 (residues 158 to 582) bound to the H4K16ac/H2BUb/H3K79M nucleosome (hereafter “Dot1-H4K16ac structure”) (Fig. 1F). We used the H3K79M mutation because it was shown to increase the affinity of methyltransferases for nucleosomes (58, 59). We reconstituted the complex, cross-linked it with glutaraldehyde using the GraFix method (60), froze grids, and collected cryo-EM data (fig. S4 and S6) on a Titan Krios (300 kV). This resulted in a 3.1 Å–resolution map of the complex, which we used to unambiguously model the structures of the nucleosome, catalytic domain of yeast Dot1, and ubiquitin (Fig. 1, F and G). The structure revealed Dot1 (residues 176 to 580) bound to the nucleosome in a catalytic conformation, with clearly resolved nucleosome-Dot1, Ub-Dot1, and H4-Dot1 interfaces (Fig. 1, F and G, and fig. S7). The cryo-EM map is of high quality and allows unambiguous interpretation of key interactions (Fig. 1F and figs. S7, S8, and S9). The understanding of the mechanism of stimulation by H4K16ac and H4K16ac/H2BUb nucleosomes required a control structure of Dot1 bound to nucleosomes with unacetylated histone H4K16 (hereafter “Dot1-unacetylated H4 structure”) (fig. S5) and followed the same experimental procedures to determine the cryo-EM structure of this complex at a comparable resolution of 3.2 Å (figs. S5, S6, and S9). These structures allowed us to make detailed comparisons to understand the structural changes underlying Dot1 stimulation by H4K16ac.
Upon careful examination of cryo-EM data, we found that in the Dot1-unacetylated H4 dataset, most of the particles classified into two main three-dimensional (3D) classes (fig. S6). In the first class, Dot1 is bound to the nucleosome in a catalytic conformation, and in the second class, it is bound in a noncatalytic conformation, resembling the “poised state” previously described for Dot1L (figs. S6 and S10) (38, 40). By contrast, in the Dot1-H4K16ac dataset, only the catalytic conformation can be observed, leading us to propose a model in which acetylation of the H4 tail restricts the sampling space of Dot1, resulting in an active conformation and leading to increased activity. To investigate the mechanisms of this stabilization, we compared the Dot1-unacetylated H4 and Dot1-H4K16ac structures in catalytically competent conformations. Overall, both structures are very similar (Fig. 2, A and B, and fig. S7), but with two major differences in histone H4 and in the Acetyl-Gate loop (“AcG loop”; residues 252 to 264) of Dot1. These differences imply a stabilization of the Dot1-H4 tail interface when K16 is acetylated. In the Dot1-H4K16ac structure, the H4 tail is ordered starting from the backbone of residue K12 (Fig. 2A and figs. S11 and S12), whereas in the Dot1-unacetylated H4 structure, the tail density is only visible from the backbone of residue K16 (Fig. 2B and figs. S11 and S12). Similarly, in the Dot1-H4K16ac structure, residues 255 to 262 of the AcG loop of Dot1 are ordered, whereas the density for this region is spurious in the Dot1-unacetylated H4 structure (figs. S11 and S12). In both structures, histidine H355 in Dot1 makes potential H-bonds between main-chain carbonyls of H4R17 and H4K16 and between H347 in Dot1 and the main-chain carbonyl of H4K16 (Fig. 2, A and B, and fig. S11). In the Dot1-H4K16ac structure, we observed an additional potential H-bond between the main chain carbonyl of A15 of H4 and Dot1 H347 (Fig. 2A). In the Dot1-H4K16ac structure, a clear density of the H4K16ac side chain allowed us to analyze the key interactions (fig. S11). The carbonyl component of the acetyl group is within H-bonding distance (~3.5 Å) of the imidazole group of H355 (Fig. 2A). Additionally, the methyl component of the acetyl group establishes van der Waals interactions with the side chain of Dot1 I261, stabilizing its backbone. The I261 main chain carbonyl forms a H-bond with Dot1 H355, resulting in overall stabilization of the AcG loop of Dot1 (Fig. 2A). By contrast, the side chain of H4K16 is not visible in the structure of the Dot1-unacetylated H4 nucleosome (Fig. 2B and figs. S11 and S12). On the basis of this, we hypothesize that in the unacetylated nucleosome, the imidazole of H347 and H355 of Dot1 may repel the positively charged, protonated amino group of the lysine H4K16, preventing stabilization of I216, which in turn results in destabilization of the AcG loop in Dot1 and H4 N terminus (residues 12 to 16). Acetylation of H4K16 would neutralize its charge, thereby favoring stabilizing interactions.
Fig. 2. Structural basis for Dot1 stimulation by H4K16 acetylation.

(A and B) Side-by-side overview and close-up of interactions of Dot1 with the H4 tail in the (A) Dot1-H4K16ac structure and (B) Dot1-unacetylated H4 structure. The structure is color-coded as in Fig. 1. The yellow dashed lines show distances of ≤3.5 Å. (C) Close-up of interactions of Dot1 with H3K79M at the catalytic site (blue arrow), the position of residues in H4 tail, and SAM. The red square indicates the region of Dot1 and H4 that is stabilized by H4K16 acetylation as shown in (A). (D) Representative endpoint methyltransferase assay for Dot1 and mutants (H347E-H355E, H335E, and I261E) in the presence of Unmod nuc or H4K16ac nuc or Ub nuc or ac/Ub nuc substrates. Reactions were performed with 0.125 and 0.250 pmol of Dot1. Each data point and error bar indicate the mean ± SD from three independent experiments. (E) Representative HMT assay measuring activity of Dot1 and its mutants on different nucleosome substrates. HMT assays were performed with an increasing amount of Dot1 mutants (H347E-H355E, H335E, and I261E) in the presence of Unmod nuc or H4K16ac nuc or Ub nuc or ac/Ub nuc substrates, and reaction products were identified by using Western blot.
We examined whether stabilization of H4 and Dot1 as a function of H4K16 acetylation affects the Dot1 active site (Fig. 2C, blue arrow). The active site of Dot1 consists of hydrophobic and aromatic residues of Dot1, cofactor S-adenosyl methionine (SAM), and residues of the H4 amino tail (Fig. 2C). H3K79 is inserted into the hydrophobic active site and anchored through van der Waals interactions of residues including W543, F481, V371, and L482 of Dot1. Furthermore, W543, F481, and F367 are present in loops that are stabilized upon binding of H3 substrate and critical for activity (40). In particular, W543 might be involved in establishing cation-π interactions with K79 and its different methyl states (61, 62). Dot1 binding induces a conformational change in loop L1 of histone H3 residues Q76, F78, K79, T80, and D81, reorienting the side chain of K79 (mutated in this work to methionine) ~95° and directing it ~10 Å away of the nucleosome, which moves it closer to the SAM cofactor and into the Dot1 active site (Fig. 2C and fig. S13). The position of hydrophobic residues in the Dot1 active site and the contacts of this region with nucleosome are similar in the Dot1-unacetylated H4 and Dot1-H4K16ac structures and are conserved in human Dot1L (fig. S13). W543 and F367 of Dot1 and the H4 tail residues R17, H18, and R19 play critical roles in the remodeling of H3 (40). H4R19 can make contacts with the backbone of H3 residues T80, K79, and Q76 and is also in proximity to contact the carbonyl of the main chain of the W543 that is part of the hydrophobic pocket (Fig. 2C and fig. S13). H4H18 contacts S542 of Dot1, probably helping to stabilize the loop of W543, and is also close in distance to establish a stacking interaction with Dot1 Y372 that is part of the SAM binding site. H4R17 is well defined in the Dot1-H4K16ac structure. It establishes several contacts with Dot1, including interactions with E374 and N404. Previous studies established a critical role of H4 tail overall and for residues 17 to 19 in particular in regulating Dot1 in vivo and in vitro (52, 53). Substitution of R17 or R19 for neutral or negative charges retained only wild-type levels of monomethylation, abolishing higher methylation states (53). Our structures suggest that because of direct contacts of R17, H18, and R19 of H4 in the active site, any conformational change in H4 such as that resulting from the stabilization by H4K16ac would directly affect the structure at the active site in a manner that increases catalysis (Fig. 2, A to C).
To further characterize Dot1 stimulation by H4K16ac, we mutated the residues at the Dot1: H4K16ac interface and measured Dot1 enzymatic activity (Fig. 2D and fig. S14). Double mutation of H347E-H355E and point mutation of H355E resulted in almost complete loss of Dot1 activity on unmodified and H4K16ac nucleosomes and a decreased activity on H2BUb and doubly modified nucleosomes (Fig. 2D and fig. S14, alanine mutants). Mutation of AcG loop residue I261 resulted in decreased stimulation by H4K16ac, whereas stimulation by H2BUb was not affected (Fig. 2D and fig. S14, alanine mutant). Immunoblot analysis is consistent with the essential role of H347 and H355 in histone H4 binding, in which their mutations resulted in loss of all methylation states on unmodified and H4K16ac nucleosomes (Fig. 2E and fig. S14, alanine mutants). The analysis of methylation patterns in the I261E mutant confirmed that this mutant is active, whereas stimulation by H4K16ac is reduced (Fig. 2E and fig. S14, alanine mutant). These results support our structural data, showing that interaction between I261 with acetylated H4K16 leads to stabilization of the H4-Dot1 interface.
Interactions of Dot1 with H2BUb and the acidic patch
Yeast Dot1 uses a different general mode of interaction with ubiquitin, resulting in a distinct interface from that observed with human Dot1L (Fig. 3, A and B, and fig. S15). Unlike human Dot1L, which interacts with a non-canonical “I36 patch” on ubiquitin, we observed interactions of the canonical “I44 patch” on ubiquitin (residues L8, I44, H68, and V70), as defined in (63) with hydrophobic residues in yeast Dot1 (Fig. 3C and fig. S15). Specifically, the conserved F564 of Dot1 interacts with ubiquitin residues H68, L8, and V70, and Dot1 V559 and V522 interact with ubiquitin residue I44 (Fig. 3C). Dot1 I517 establishes potential interactions with ubiquitin residues V70 and L8 (Fig. 3C). In addition to the canonical I44 patch, Dot1 F519 establishes a cation-π interaction with ubiquitin residue R72. We tested the relevance of the Dot1-Ub interface using site-directed mutagenesis coupled with enzymatic assays. We showed that this surface is indeed critical for the observed stimulatory role of H2BUb, and its mutation mainly disrupts effects by Ub (with only a marginal effect on H4K16ac stimulation) (Fig. 3, D and E). The Dot1 point mutant V522D is active and shows a decrease in stimulation on H2BUb nucleosomes, with only small effects on stimulation by H4K16ac (Fig. 3, D and E, and fig. S14, alanine mutant). The analysis of methylation by using immunoblot confirmed that V522 mutations reduce the stimulation by H2BUb, as evidenced by loss of di- and trimethylation of H3K79, whereas monomethylation was not affected (Fig. 3E and fig. S14, alanine mutant).
Fig. 3. Interactions of Dot1 with H2BUb and the acidic patch.
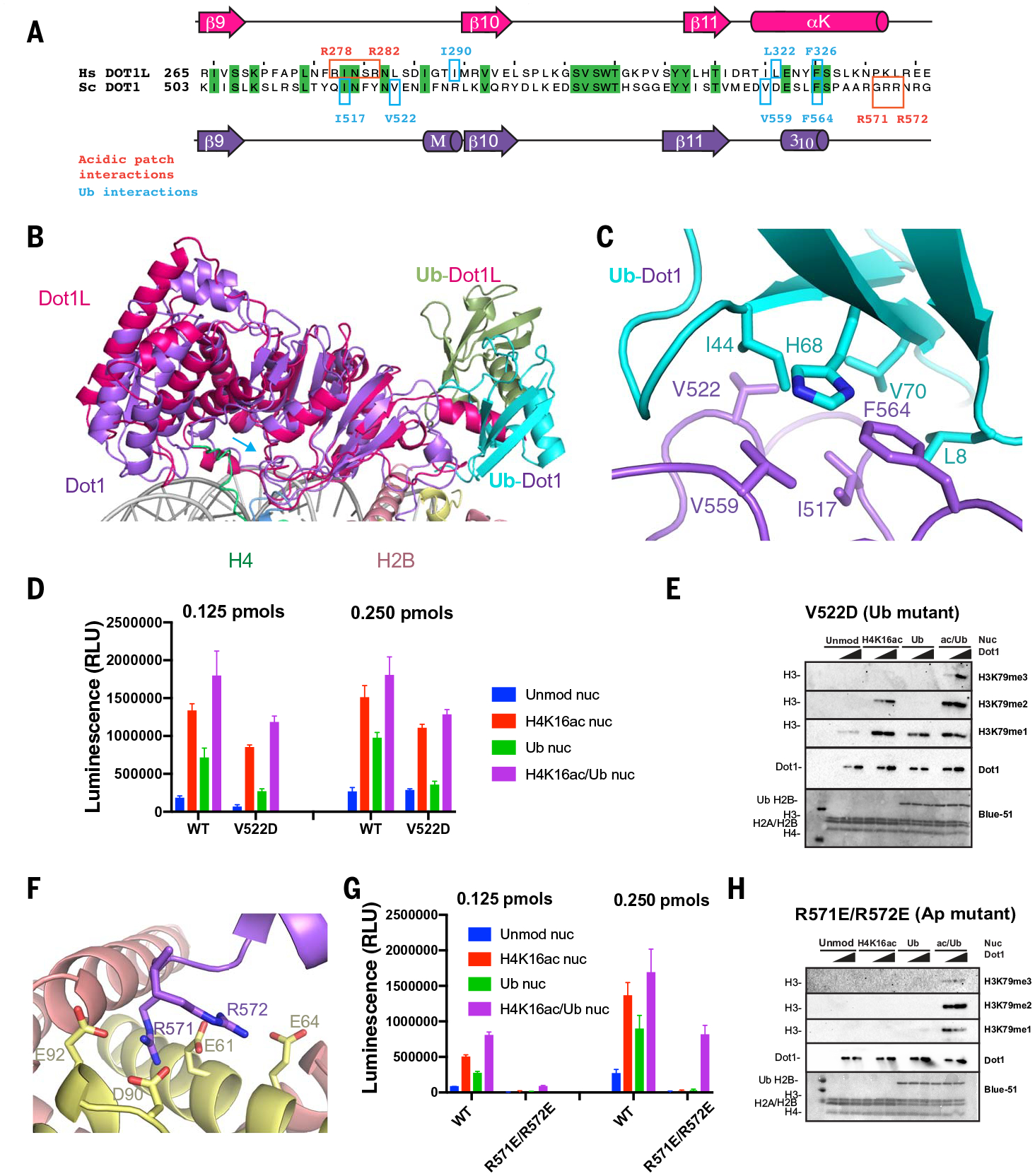
(A) Sequence alignment of budding yeast Dot1 (purple) and human Dot1L (red) showing primary and secondary structure of the catalytic domain and residues interacting with the ubiquitin (cyan) and the acidic patch (red). (B) Superposition of yeast Dot1 (purple) and Dot1L (red) [PDB ID 6NJ9 (40)] nucleosome structures showing the different surface of interaction with ubiquitin (shown in cyan in the yeast Dot1 structure and in green in the Dot1L structure). (C) Detailed view of interactions between Dot1 (purple) and ubiquitin (cyan). (D) Representative endpoint methyltransferase assay for Dot1 and V522D mutant in the presence of Unmod nuc or H4K16ac nuc or Ub nuc or ac/Ub nuc substrates. Reactions were performed with 0.125 and 0.250 pmol of Dot1. Each data point and error bar indicate the mean ± SD from three independent experiments. (E) Representative HMT assay measuring activity of Dot1 and V522D mutant on different nucleosome substrates. HMT were performed with an increasing amount of Dot1 mutant (V522D) in the presence of Unmod nuc or H4K16ac nuc or Ub nuc or ac/Ub nuc substrates, and reaction products were identified by using Western blot. (F) Detailed view of interactions between Dot1 (purple) and nucleosome acidic patch (H2A, yellow; H2B, red). (G) Representative endpoint methyltransferase assay for Dot1 and (R571E-R572E) mutant in the presence of Unmod nuc or H4K16ac nuc or Ub nuc or ac/Ub nuc substrates. Reactions were performed with 0.125 pmol and 0.250 pmol of Dot1. Each data point and error bar indicate the mean ± SD from three independent experiments. (H) Representative HMT assay measuring activity of Dot1 and mutant on different nucleosome substrates. HMT assays were performed with an increasing amount of Dot1 mutant (R571E-R572E) in the presence of Unmod nuc, H4K16ac nuc, Ub nuc, or ac/Ub nuc substrates, and reaction products were identified by using Western blot.
There is an unconventional, extended, bipartite interface between Dot1 and H2A-H2B dimer in the nucleosome (fig. S15). One part of this interface is formed by interactions between Dot1 residues E485 and T514 with residues in the αC helix of H2B (fig. S15). Other canonical interactions are established through salt bridges between positively charged residues in Dot1 and the nucleosome acidic patch (Fig. 3F). Interactions with the acidic patch involve two arginines: a stable one between R571 and H2A residues E92, D90, and E61 and a more flexible one between R572 and H2A E61 and possibly E64 (Fig. 3F and fig. S15). The relevance of these residues is highlighted by a loss of activity of the R571E-R572E double mutant in enzymatic assays on unmodified and singly modified nucleosomes (Fig. 3G). This mutant is active only on doubly modified (H4K16ac/H2BUb) nucleosomes (Fig. 3G), where it is able to perform mono-, di-, and trimethylation of H3K79 (Fig. 3H). Both in yeast and in human Dot1, regions that interact with ubiquitin and with the acidic patch are in close spatial proximity (Fig. 3, A and B). In the main interface with Ub, Dot1 residues V559 and F564 are inside and flank the acidic motif 557–561 EDVDE (fig. S15). On the basis of our structure, we predict that deletion of this motif would affect the interaction of Dot1 with Ub and/or with the acidic patch, which explains previous data that show that deletion of these residues limits catalysis by Dot1 to only monomethylation (52).
Regulation of H3K79 methyltransferase activity of Dot1 by extended cross-talk in vivo.
Having provided the structural and biochemical mechanism for Dot1 stimulation by H4K16ac and H2BUb, we decided to test the effects of our mutants in vivo (Fig. 4, A and B). We used an established approach for assessing the biological impact of Dot1 mutants in S. cerevisiae (64). We introduced single-copy plasmids containing Dot1 mutants to dot1Δ cells containing a telomeric URA3 reporter gene. Loss of H3K79 methylation affects URA3 silencing and leads to growth toxicity on 5–fluoroorotic acid (5-FOA) (64), although the exact mechanism is not well understood (65, 66). As a control, we included Dot1 mutants known to reduce H3K79 methylation (G401A and G401R) (Fig. 4A). The mutants affected URA3 silencing by two to four orders of magnitude (Fig. 4A). To confirm these genetic observations by examining in vivo methylation status, we assessed with immunoblotting global H3K79 mono-, di-, and trimethylation (Fig. 4B). Consistent with the growth assays, the mutants disrupted di- and trimethylation of H3K79 (Fig. 4B). Dot1 mutants H355E and V522D, which retain residual methylation activity, lead to URA3 silencing similar to that by the catalytically dead G401R mutant. This echoes previous work that showed that catalytically reduced Dot1 G401A also affects in vivo URA3 silencing similarly to that by G401R (64). These results suggest that acetylation of lysine 16 and ubiquitination of histone H2B are both required for efficient methylation of H3K79 in yeast.
Fig. 4. Regulation of H3K79 methyltransferase activity of Dot1 through extended cross-talk in vivo.
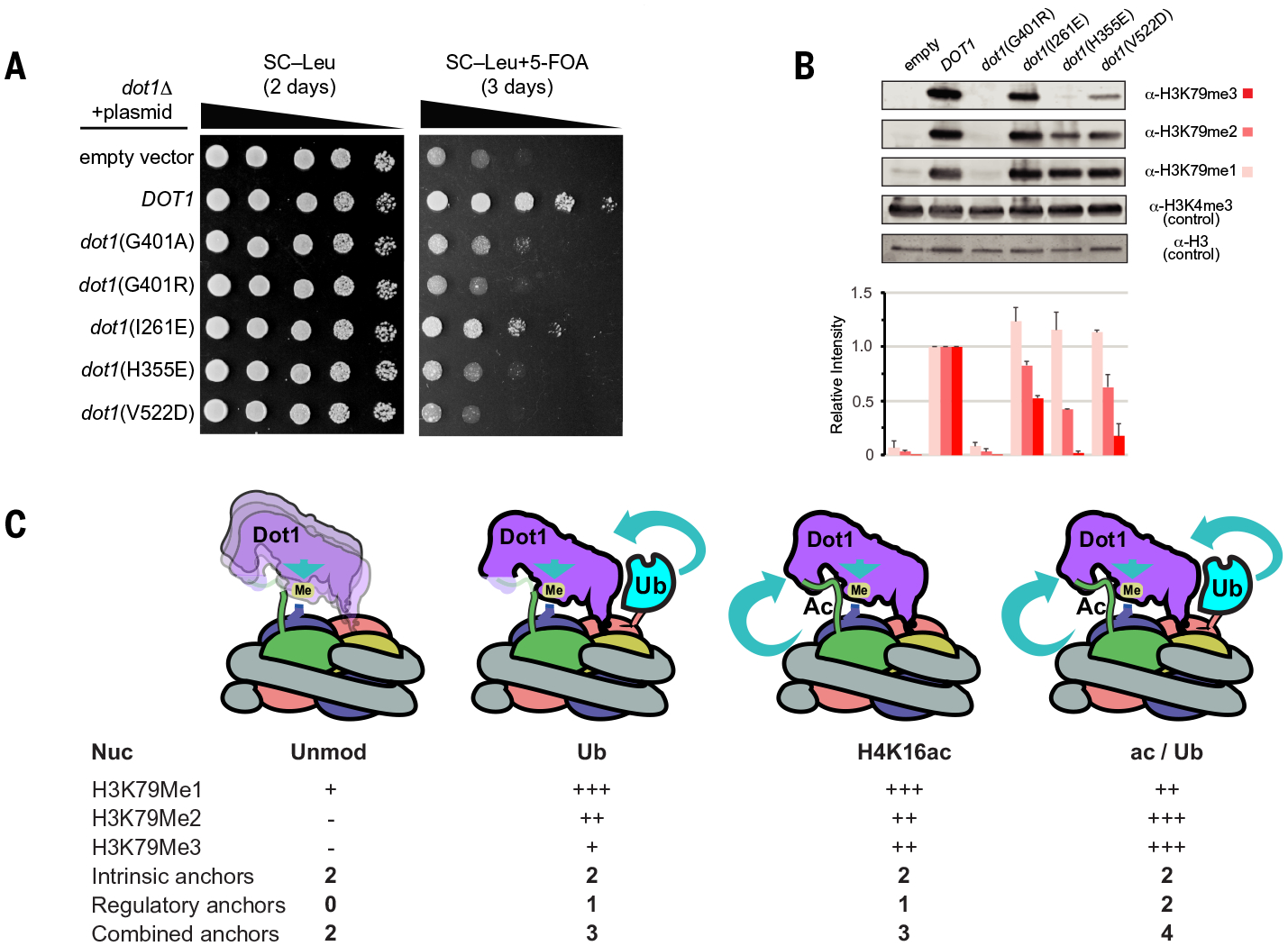
(A) Tenfold serial dilutions of UCC1783 yeast containing Dot1 mutants on media with or without 5-FOA (n = 3 experiments). Proper silencing of a URA3 gene on telomere VIIIL confers resistance to 5-FOA. (B) (Top) Representative Western blot showing H3K79 methylation by Dot1 mutants. (Bottom) Quantitative image analysis of Western blot in (B) based on n = 2 blots. Errors bars show standard deviation. (C) General model of how Dot1 activity is regulated by histone posttranslational modifications. Intrinsic anchors represent interactions between Dot1 and histone H4 and acidic patch. These anchors allow Dot1 to monomethylate H3K79. Regulatory anchors are provided when histone H4K16 is acetylated or when H2B is ubiquitinated. The expression anchor is used here to denominate an interaction that stabilizes a productive conformation of Dot1 on the nucleosome. The illustration is color-coded as in Fig. 1.
On the basis of our structural, biochemical, and in vivo functional data, we propose a model that describes the rules of Dot1 stimulation by histone acetylation and ubiquitination (Fig. 4C). The outcome of enzymatic reaction by Dot1 is directly linked to its ability to interact with the nucleosome using several “anchors” that stabilize its conformation for optimal activity. The most relevant intrinsic anchor is established through the interaction of Dot1 with the acidic patch. This anchor, together with the Dot1–histone H4 interaction, suffices to allow Dot1-mediated monomethylation of H3K79. H4K16ac and H2BUb individually provide critical regulatory anchors that allow Dot1 to perform di- and trimethylation (Fig. 4C). Nucleosomes that are both H4K16 acetylated and H2B ubiquitinated allow optimal positioning of Dot1 on the nucleosome, restricting its orientation and allowing mostly productive conformations, resulting in increased trimethylation (Fig. 4C).
Discussion
Our findings suggest a possible mechanism for how H3K79 methylation by Dot1 regulates the kinetics of gene silencing in yeast. In this process, SIR complex binds to nucleosomes to form a repressive chromatin structure. Specifically, the NAD+–dependent deacetylase Sir2 removes the acetyl group from H4K16ac, allowing binding of Sir3 and thus promotion of chromatin silencing (67, 68). There are data that show that methylation of lysine 79 by Dot1 slows down kinetics of establishment of silent chromatin in yeast (69). Competition for access to H4K16ac between deacetylase Sir2 and Dot1 could be central to this mechanism, especially at regions distal to SIR recruitment sites. Acetylated H4K16 stimulates H3K79 methylation, and once this happens, nucleosomes that harbor both H4K16ac and H3K79me3 could prevent SIR complex from binding to chromatin. Because H2BUb has also been shown to reduce silencing in yeast (24, 70), nucleosomes modified at multiple sites would be predicted to have an even stronger effect. Additionally, Dot1 binds to the acidic patch, a nucleosome region with key functions in chromatin structure (71). It has been shown that the H4 tail interacts with the acidic patch of another nucleosome through charge complementarity (72). Acetylation of H4K16 neutralizes the charge and prevents the formation of chromatin fiber (17, 73). Therefore, acetylation of H4K16 would (i) obstruct contacts of this tail with the acidic patch, (ii) block contacts of this tail with Sir3, and (iii) allow Dot1 to bind and (iv) stimulate its catalytic activity, and (v) Dot1 binding would further occlude the acidic patch from the binding of silencing proteins such as Sir3.
What could be the mechanism of Dot1 stimulation by H4K16ac? Our data point to H4K16ac playing a role in restricting the conformation of Dot1. This is based on the comparison of Dot1-unacetylated H4 and Dot1-H4K16ac structures where in the latter, stabilization of Dot1 and histone H4 is observed. Conformational restriction has been proposed as a mechanism for stimulation of human Dot1L by H2Bub (33, 40). On the basis of our data, H2BUb acts in the similar way, but perhaps less efficiently in yeast. The presence of a 3D class in which Dot1 is in the noncatalytic position exclusively in the Dot1-unacetylated H4 dataset supports the role of H4K16 acetylation in further restricting Dot1 in the productive configuration, which results in the increased catalytic rate. Given the substantial structural differences between Dot1-H4K16ac and Dot1-unacetylated H4 datasets, and the functional experiments that support our structures, it is unlikely that the presence of H3K79M masked the mechanisms of stimulation by H4K16ac. There is no major change of Kd or Km for Dot1 on H4K16ac nucleosomes. This is also in line with the stimulation of human Dot1L by the H2BK120Ub nucleosome, for which it was postulated that the energy from Ub binding is used on conformational changes to bring the active site into catalytically compatible conformation (33, 40). In the structure of Dot1L bound to the H2BK120Ub nucleosome in the catalytically active conformation, it was speculated in (40) that the energetic cost of the H3 distortion is paid off by energy gained by binding to ubiquitin. This is supported in our structure, in which the distortion around H3K79 was also observed, and there are additionally changes in the AcG loop of Dot1 and histone H4.
How can the PTMs change the outcome of methylation by Dot1? A recent review raised the possibility that perhaps histone modifications render human Dot1L processive through multivalent interactions with chromatin, such as was shown for other chromatin enzymes (74). In this Dot1 binding scenario, stimulatory histone modifications would facilitate a search for Dot1’s lysine substrate, and Dot1 would methylate H3K79 in a processive manner, after which it would dissociate. If Dot1 is a distributive enzyme, as has been shown before (75), at each round of methylation there would be costs associated with the conformational sampling of the different positions on the nucleosome. In such a case, at each round of methylation Dot1 would bind nucleosome, and H4K16ac and H2BUb would help Dot1 reach the active conformation. Because there is no substantial increase in affinity in the presence of H2BUb and H4K16ac, the reduction of the conformational sampling space would still be predicted to be the most relevant contribution of these histone modifications. These PTMs would together optimally position Dot1 to allow a more robust deposition of higher-order methyl marks.
As stated, our current structure is different in terms of ubiquitin recognition from that by Dot1L but similar to that by MLL1/3 subcomplexes. Dot1L uses the I36 patch on ubiquitin, and Dot1 and MLL1/3 use the I44 patch (fig. S15) (40, 76). The recognition of ubiquitin by the COMPASS subcomplex shares both patches of recognition I36 and I44 (fig. S15) (77). These findings highlight the plasticity of ubiquitin recognition, as summarized before (74, 77). Why ubiquitin is differently recognized by these proteins and complexes remains an unresolved question that will require further work.
There are several examples of trans-histone cross-talk described in chromatin biology (78, 79). The concept of positive- and negative-feedback loops is also quite well established. We hypothesize that during transcription, histone modifications—as well as enzymes that catalyze their deposition—are tightly regulated through trans-histone cross-talk to provide positive feed-forward loops that result in an optimal chromatin environment. There are many examples in which this happens. H2BUb regulates both MLL complex deposition of H3K4 methylation and Dot1 deposition of H3K79 methylation. In turn, Dot1 helps to catalyze histone H2B ubiquitination (80). In this work, we show that acetylation of lysine 16, perhaps the most critical modification on histone H4, provides the means to stimulate K79 methylation. It would be interesting to see whether H3K79 methylation in turn stimulates histone acetylation, providing a self-sustaining chromatin state maintenance circuit.
Methods summary
Yeast Dot1 (158 to 582) and its mutants were expressed in Escherichia coli ONESHOT BL21(DE3) and purified by using nickel affinity chromatography, ion exchange, and size-exclusion liquid chromatography. Ubiquitin (Ub) G76C was expressed as soluble protein in E. coli SoluBL21 and purified by using nickel affinity chromatography (81). Wild-type and H2BK120C Xenopus histones were expressed in E. coli BL21(DE3) pLysS cells, extracted from inclusion bodies (82), purified by size exclusion chromatography, and lyophilized. Ubiquitination of histone H2BK120C was performed by using a previously published protocol (81). Ub G76C and histone H2BK120C were mixed at the ratio of 2:1 and cross-linked by using 1,3-dichloroacetone. The ubiquitinated histone H2BK120C (H2BK120Ub) was purified by using nickel affinity chromatography and lyophilized. The genetically encoded histone H4 acetyl K16 (H4K16ac) was produced according to Wilkins et al. (83). Plasmid pASB567_9 g7 (amber codon expression vector) for expression of the histone H3(Δ93–98)-H4 K16ac fusion and pAcKRS-3 plasmid containing acetyl-lysyl-tRNA synthetase/tRNACUA pair were transformed into E. coli C321.Δ A.exp cells (Addgene), and protein expression was induced by the addition of 0.2% arabinose in presence of Nε-Acetyl-l-lysine. The histone 6xHis-H3(Δ93–98)-H4K16ac fusion was purified from inclusion bodies by nickel affinity purification and H4 peptide was excised with TEV protease and lyophilized.
Nucleosome substrates were assembled as described (54, 82). Octamers were reconstituted by mixing equimolar amounts of each lyophilized histone and dialysis into refolding buffer. Nucleosomes were assembled by combining equimolar ratios of purified Widom 601 DNA and histone octamers and dialyzing the mix overnight with gradient salt dialysis. Designer, individually acetylated (H4K5ac, H4K8ac, H4K12ac, or H4K16ac), or tetraacetylated nucleosomes were purchased from EpiCypher.
For the nucleosome binding assay, a twofold serial dilution of Dot1 (50 to 1000 nM) was incubated with 12 nM recombinant nucleosomes at room temperature for 30 min and analyzed by using native polyacrylamide gels [6% polyacrylamide gel electrophoresis (PAGE), 0.2× tris-boric acid–EDTA buffer], stained with SYBR Gold (Thermo Fisher), imaged in a Typhoon Trio+ scanner, and quantified with the program ImageQuant 5.2v (Molecular Dynamics).
The Dot1 global methyltransferase activity and Michaelis-Menten kinetic analysis were determined by monitoring the production of S-(5′-adenosyl)-l-homocysteine (SAH) with MTase-Glo methyltransferase kit (Promega) on nucleosome substrates. The Dot1 global methyltransferase activity (Endpoint methylation assay) reactions were performed by mixing 50 or 100 nM of wild-type or mutant Dot1 proteins with 1 μM unmodified or modified nucleosomes [in the presence of 20 μM S-(5′-adenosyl)-l-methionine (SAM)] at 30°C for 30 min and stopped with 5 μl of 0.5% TFA (trifluoroacetic acid). The Michaelis-Menten kinetic analysis of Dot1 methyltransferase activity on nucleosome was performed by mixing 10 nM Dot1 containing 20 μM SAM with a twofold dilution series of nucleosomes (4 to 4000 nM). The reaction was incubated for 5 min at 30°C and stopped on ice by the addition of 4 μl of 0.5% TFA. The kinetic parameters Km and kcat of Dot1 methyltransferase activity were determined by fitting the initial velocities into the Michaelis-Menten equation using the enzymatic kinetics analysis (kcat analysis) in Prism 8 (GraphPad).
For the histone methyltransferase assay (HMT) by use of Western blot, reactions of Dot1 (0.250 pmol at the highest concentration) and nucleosomes (10 pmol) in presence of 20 μM SAM were incubated at 30°C for 1 hour and stopped by the addition of 5 μl of 5× SDS buffer. The products were resolved in a 15% SDS-PAGE gel and transferred to polyvinylidene difluoride membrane. The histone methylation level was determined by incubating the membranes with anti-H3K79Me3 (Abcam Ab2621), anti-H3K79Me2 (Abcam Ab3594), or anti-H3K79Me1 (sera 58) antibody. The Western blots were developed by using the ECL reagent (ThermoFisher).
Gradient Fixation (GraFix) of Dot1 with the nucleosome for structural studies was performed according to (60). Nucleosomes were mixed with purified Dot1 protein and supplemented with 5× molar ratio of SAM. The sample was applied to a 10 to 30% glycerol and 0 to 0.1% glutaraldehyde gradient and centrifuged for 16 hours at 30,000 revolutions per minute (rpm) (Beckman Coulter Optima XL-100K). Selected fractions were quenched, dialyzed into dialysis buffer (20 mM Tris 7.0, 50 mM KCl, 1 mM MgCl2, 1 mM dithiothreitol), and concentrated.
Cryo-EM grids of the Dot1-H4K16ac complex and Dot1-unacetylated H4 complex were prepared following an established protocol (84): 3.0 μl of the samples at 0.45 mg/ml were applied to glow-discharged Quantifoil gold grids (400 mesh, 1.2-μm hole size). The grids were then blotted for 3 s at 4°C and 100% humidity and plunge-frozen by using Vitrobot Mark IV (FEI Company). All sample images were recorded on FEI Titan Krios operated at 300 kV by using a Gatan K2 Summit direct electron detector camera in counting mode at a nominal magnification of 130,000× (calibrated pixel size of 1.035 Å per pixel). Total accumulated electron exposure was 74.5 electrons per Å2 for the Dot1-H4K16ac complex and 52 electrons per Å2 for the Dot1-unacetylated H4 complex.
Images were motion corrected by using UCSF MotionCor2 v1.2.1 (85); GCTF (86) was used to calculate CTF, Gautomatch (www.mrc-lmb.cam.ac.uk/kzhang) for particle picking, Relion3 (87) for particle extraction, cryoSPARC (88) for Ab Initio reconstruction and 3D refinement, and cisTEM (89) for the final reconstruction. This procedure led to obtaining reconstructions of the Dot1-H4K16ac complex at 3.1 Å and Dot1-unacetylated H4 complex at 3.2 Å (Fourier shell correlation = 0.143). To validate that the cryo-EM reconstructions and our interpretation were not distorted by an overrepresentation of certain views in the particle datasets, we conducted additional distribution analysis and normalization that is described in the supplementary materials.
For the model building of the Dot1-H4K16ac complex, we used the following available x-ray crystal structures for rigid body fit into the 3.1 Å reconstruction: for the nucleosome, PDB IDs 3TU4 (54) and 5AV5 (90); for catalytic domain of Dot1, 1U2Z (91); and for ubiquitin, 1UBQ (92). We used 1NW3 (93) to inform the placement of SAM. We then used Coot (94) for local adjustments of secondary elements and side chains, and the complete model was refined by using real-space refinement implemented in PHENIX (95). For the Dot1-unacetylated H4 complex, we used the cryo-EM reconstruction at 3.2 Å and the PDB of the Dot1-H4K16ac structure as a starting point, removing the missing atoms and applying the same protocol of refinement.
For in vivo experiments, we used yeast spot assays performed with Dot1 deletion yeast strain (UCC7183), as described in (64). Yeast strains were cultured in synthetic complete medium (SC). UCC7183 was transformed with Dot1 wild-type and mutant plasmids containing pRS315 and selected for 2 days at 30°C on SC–Leu plates. Spot assays were performed by preparing 10-fold dilutions of overnight cultures (A600 of ~10) and then spotted on both SC–Leu and SC–Leu+5-FOA plates.
Histone H3K79 modification in yeast was tested with protein Western blotting. Histone protein fractions were prepared by growing Dot1 yeast strains in SC–Leu to an A600 of ~1 to 1.2. Cells were disrupted in presence of 0.25 M HCl, histones were solubilized in 0.25 M HCL/97.5% ethanol and precipitated with acidified acetone. Solubilized protein pellet in 1X NuPAGE LDS loading buffer (Invitrogen) was electrophoresed on 12% Bis-Tris NuPAGE gels. Immunoblots were performed as described (96) by using the Licor Odyssey system. Commercial primary antibodies used were mouse anti H3 (Abcam) and mouse anti H3K4me3 (Abcam). H3K79 and yeast Dot1 primary antibodies were a gift from F. Van Leeuwen (64): rabbit anti H3K79me1 (sera #58), rabbit anti H3K79me2 (sera #30), rabbit anti H3K79me3 (sera #34), and rabbit anti Dot1 (affinity purified). Secondary antibodies were IRDye 800 goat anti mouse and IRDye 680 goat anti rabbit (LiCor).
Supplementary Material
ACKNOWLEDGMENTS
We thank W. Rice and B. Wang for helping with data collection at the New York University (NYU) cryo-EM Shared Resource. We thank the staff at the NYU Microscopy Laboratory for helping with negative stain microscopy. We thank M. Costantino and HPC Core at NYU Langone Health for computer access and support. We thank C. Yun for helping with luminescence assays at the NYUMC High Throughput Biology Laboratory. We thank F. Van Leeuwen for sharing the rabbit anti-H3K79me1 (sera 58), rabbit anti-H3K79me2 (sera 30), rabbit anti-H3K79me3 (sera 34), and rabbit anti-Dot1. We thank the members of the Boeke and Armache laboratories for critical comments and discussion. We thank R. E. Kingston, J. Cochrane, D. Smith, and A. Armache for comments and critical review of this manuscript.
Funding:
The work in the Boeke laboratory is supported by NSF grant MCB-1921641. The work in Armache laboratory is supported by grants from the David and Lucile Packard Foundation and the National Institutes of Health (5R01GM115882).
Footnotes
The list of author affiliations is available in the full article online.
Competing interests: J.D.B. is a founder and director of CDI Labs, a founder of Neochromosome, a founder of and consultant to ReOpen Diagnostics, and serves or served on the Scientific Advisory Board of the following: Sangamo, Modern Meadow, Sample6, and the Wyss Institute.
Data and materials availability: All data are available in the manuscript or the supplementary materials. The structure models and the cryo-EM density maps have been deposited in the Protein Data Bank (PDB) and Electron Microscopy Data Bank (EMDB) with accession codes 7K6Q, 7K6P, EMD-22691, EMD-22692, EMD-22693, EMD-22694, and EMD-2265. Further inquiries and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, K.-J.A. (karim-jean.armache@nyulangone.org).
SUPPLEMENTARY MATERIALS
science.sciencemag.org/content/371/6527/eabc6663/suppl/DC1
Materials and Methods
Supplementary Text
Figs. S1 to S19
Tables S1 to S3
MDAR Reproducibility Checklist
REFERENCES AND NOTES
- 1.Bannister AJ, Kouzarides T, Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011). doi: 10.1038/cr.2011.22; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berger SL, The complex language of chromatin regulation during transcription. Nature 447, 407–412 (2007). doi: 10.1038/nature05915; [DOI] [PubMed] [Google Scholar]
- 3.Schübeler D et al. , The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. 18, 1263–1271 (2004). doi: 10.1101/gad.1198204; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Z et al. , Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet 40, 897–903 (2008). doi: 10.1038/ng.154; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Audia JE, Campbell RM, Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol 8, a019521 (2016). doi: 10.1101/cshperspect.a019521; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner BM, O’Neill LP, Histone acetylation in chromatin and chromosomes. Semin. Cell Biol 6, 229–236 (1995). doi: 10.1006/scel.1995.0031; [DOI] [PubMed] [Google Scholar]
- 7.Görisch SM, Wachsmuth M, Tóth KF, Lichter P, Rippe K, Histone acetylation increases chromatin accessibility. J. Cell Sci 118, 5825–5834 (2005). doi: 10.1242/jcs.02689; [DOI] [PubMed] [Google Scholar]
- 8.Kuo MH, Allis CD, Roles of histone acetyltransferases and deacetylases in gene regulation. BioEssays 20, 615–626 (1998). doi: ; [DOI] [PubMed] [Google Scholar]
- 9.Clarke DJ, O’Neill LP, Turner BM, Selective use of H4 acetylation sites in the yeast Saccharomyces cerevisiae. Biochem. J 294, 557–561 (1993). doi: 10.1042/bj2940557; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kurdistani SK, Tavazoie S, Grunstein M, Mapping global histone acetylation patterns to gene expression. Cell 117, 721–733 (2004). doi: 10.1016/j.cell.2004.05.023; [DOI] [PubMed] [Google Scholar]
- 11.Zhang W, Bone JR, Edmondson DG, Turner BM, Roth SY, Essential and redundant functions of histone acetylation revealed by mutation of target lysines and loss of the Gcn5p acetyltransferase. EMBO J. 17, 3155–3167 (1998). doi: 10.1093/emboj/17.11.3155; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pokholok DK et al. , Genome-wide map of nucleosome acetylation and methylation in yeast. Cell 122, 517–527 (2005). doi: 10.1016/j.cell.2005.06.026; [DOI] [PubMed] [Google Scholar]
- 13.Shahbazian MD, Grunstein M, Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem 76, 75–100 (2007). doi: 10.1146/annurev.biochem.76.052705.162114; [DOI] [PubMed] [Google Scholar]
- 14.Li B, Carey M, Workman JL, The role of chromatin during transcription. Cell 128, 707–719 (2007). doi: 10.1016/j.cell.2007.01.015; [DOI] [PubMed] [Google Scholar]
- 15.Reinke H, Hörz W, Histones are first hyperacetylated and then lose contact with the activated PHO5 promoter. Mol. Cell 11, 1599–1607 (2003). doi: 10.1016/S1097-2765(03)00186-2; [DOI] [PubMed] [Google Scholar]
- 16.Chandy M, Gutiérrez JL, Prochasson P, Workman JL, SWI/SNF displaces SAGA-acetylated nucleosomes. Eukaryot. Cell 5, 1738–1747 (2006). doi: 10.1128/EC.00165-06; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shogren-Knaak M et al. , Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847 (2006). doi: 10.1126/science.1124000; [DOI] [PubMed] [Google Scholar]
- 18.Feng Q et al. , Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol 12, 1052–1058 (2002). doi: 10.1016/S0960-9822(02)00901-6; [DOI] [PubMed] [Google Scholar]
- 19.Lacoste N, Utley RT, Hunter JM, Poirier GG, Côte J, Disruptor of telomeric silencing-1 is a chromatin-specific histone H3 methyltransferase. J. Biol. Chem 277, 30421–30424 (2002). doi: 10.1074/jbc.C200366200; [DOI] [PubMed] [Google Scholar]
- 20.van Leeuwen F, Gafken PR, Gottschling DE, Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 109, 745–756 (2002). doi: 10.1016/S0092-8674(02)00759-6; [DOI] [PubMed] [Google Scholar]
- 21.Kouskouti A, Talianidis I, Histone modifications defining active genes persist after transcriptional and mitotic inactivation. EMBO J. 24, 347–357 (2005). doi: 10.1038/sj.emboj.7600516; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krogan NJ et al. , The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: Linking transcriptional elongation to histone methylation. Mol. Cell 11, 721–729 (2003). doi: 10.1016/S1097-2765(03)00091-1; [DOI] [PubMed] [Google Scholar]
- 23.Steger DJ et al. , DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol. Cell. Biol 28, 2825–2839 (2008). doi: 10.1128/MCB.02076-07; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ng HH, Ciccone DN, Morshead KB, Oettinger MA, Struhl K, Lysine-79 of histone H3 is hypomethylated at silenced loci in yeast and mammalian cells: A potential mechanism for position-effect variegation. Proc. Natl. Acad. Sci. U.S.A 100, 1820–1825 (2003). doi: 10.1073/pnas.0437846100; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wood K, Tellier M, Murphy S, DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules 8, 11 (2018). doi: 10.3390/biom8010011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones B et al. , The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLOS Genet. 4, e1000190 (2008). doi: 10.1371/journal.pgen.1000190; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McLean CM, Karemaker ID, van Leeuwen F, The emerging roles of DOT1L in leukemia and normal development. Leukemia 28, 2131–2138 (2014). doi: 10.1038/leu.2014.169; [DOI] [PubMed] [Google Scholar]
- 28.Bernt KM et al. , MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 20, 66–78 (2011). doi: 10.1016/j.ccr.2011.06.010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen CW, Armstrong SA, Targeting DOT1L and HOX gene expression in MLL-rearranged leukemia and beyond. Exp. Hematol 43, 673–684 (2015). doi: 10.1016/j.exphem.2015.05.012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krivtsov AV et al. , H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 14, 355–368 (2008). doi: 10.1016/j.ccr.2008.10.001; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okada Y et al. , hDOT1L links histone methylation to leukemogenesis. Cell 121, 167–178 (2005). doi: 10.1016/j.cell.2005.02.020; [DOI] [PubMed] [Google Scholar]
- 32.Briggs SD et al. , Gene silencing: Trans-histone regulatory pathway in chromatin. Nature 418, 498 (2002). doi: 10.1038/nature00970; [DOI] [PubMed] [Google Scholar]
- 33.McGinty RK, Kim J, Chatterjee C, Roeder RG, Muir TW, Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature 453, 812–816 (2008). doi: 10.1038/nature06906; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ng HH, Xu RM, Zhang Y, Struhl K, Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated methylation of histone H3 lysine 79. J. Biol. Chem 277, 34655–34657 (2002). doi: 10.1074/jbc.C200433200; [DOI] [PubMed] [Google Scholar]
- 35.Nakanishi S et al. , Histone H2BK123 monoubiquitination is the critical determinant for H3K4 and H3K79 trimethylation by COMPASS and Dot1. J. Cell Biol 186, 371–377 (2009). doi: 10.1083/jcb.200906005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu B et al. , Monoubiquitination of human histone H2B: The factors involved and their roles in HOX gene regulation. Mol. Cell 20, 601–611 (2005). doi: 10.1016/j.molcel.2005.09.025; [DOI] [PubMed] [Google Scholar]
- 37.Valencia-Sánchez MI et al. , Structural Basis of Dot1L Stimulation by Histone H2B Lysine 120 Ubiquitination. Mol. Cell 74, 1010–1019.e6 (2019). doi: 10.1016/j.molcel.2019.03.029; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anderson CJ et al. , Structural Basis for Recognition of Ubiquitylated Nucleosome by Dot1L Methyltransferase. Cell Rep. 26, 1681–1690.e5 (2019). doi: 10.1016/j.celrep.2019.01.058; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jang S et al. , Structural basis of recognition and destabilization of the histone H2B ubiquitinated nucleosome by the DOT1L histone H3 Lys79 methyltransferase. Genes Dev. 33, 620–625 (2019). doi: 10.1101/gad.323790.118; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Worden EJ, Hoffmann NA, Hicks CW, Wolberger C, Mechanism of Cross-talk between H2B Ubiquitination and H3 Methylation by Dot1L. Cell 176, 1490–1501.e12 (2019). doi: 10.1016/j.cell.2019.02.002; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yao T et al. , Structural basis of the crosstalk between histone H2B monoubiquitination and H3 lysine 79 methylation on nucleosome. Cell Res. 29, 330–333 (2019). doi: 10.1038/s41422-019-0146-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vlaming H et al. , Conserved crosstalk between histone deacetylation and H3K79 methylation generates DOT1L-dose dependency in HDAC1-deficient thymic lymphoma. EMBO J. 38, e101564 (2019). doi: 10.15252/embj.2019101564; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee S et al. , Dot1 regulates nucleosome dynamics by its inherent histone chaperone activity in yeast. Nat. Commun 9, 240 (2018). doi: 10.1038/s41467-017-02759-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singer MS et al. , Identification of high-copy disruptors of telomeric silencing in Saccharomyces cerevisiae. Genetics 150, 613–632 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grunstein M, Gasser SM, Epigenetics in Saccharomyces cerevisiae. Cold Spring Harb. Perspect. Biol 5, a017491 (2013). doi: 10.1101/cshperspect.a017491; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hecht A, Laroche T, Strahl-Bolsinger S, Gasser SM, Grunstein M, Histone H3 and H4 N-termini interact with SIR3 and SIR4 proteins: A molecular model for the formation of heterochromatin in yeast. Cell 80, 583–592 (1995). doi: 10.1016/0092-8674(95)90512-X; [DOI] [PubMed] [Google Scholar]
- 47.Kimura A, Umehara T, Horikoshi M, Chromosomal gradient of histone acetylation established by Sas2p and Sir2p functions as a shield against gene silencing. Nat. Genet 32, 370–377 (2002). doi: 10.1038/ng993; [DOI] [PubMed] [Google Scholar]
- 48.Oppikofer M et al. , A dual role of H4K16 acetylation in the establishment of yeast silent chromatin. EMBO J. 30, 2610–2621 (2011). doi: 10.1038/emboj.2011.170; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suka N, Luo K, Grunstein M, Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine16 and spreading of heterochromatin. Nat. Genet 32, 378–383 (2002). doi: 10.1038/ng1017; [DOI] [PubMed] [Google Scholar]
- 50.Behrouzi R et al. , Heterochromatin assembly by interrupted Sir3 bridges across neighboring nucleosomes. eLife 5, e17556 (2016). doi: 10.7554/eLife.17556; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goodnight D, Rine J, S-phase-independent silencing establishment in Saccharomyces cerevisiae. eLife 9, e58910 (2020). doi: 10.7554/eLife.58910; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fingerman IM, Li HC, Briggs SD, A charge-based interaction between histone H4 and Dot1 is required for H3K79 methylation and telomere silencing: Identification of a new trans-histone pathway. Genes Dev. 21, 2018–2029 (2007). doi: 10.1101/gad.1560607; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Altaf M et al. , Interplay of chromatin modifiers on a short basic patch of histone H4 tail defines the boundary of telomeric heterochromatin. Mol. Cell 28, 1002–1014 (2007). doi: 10.1016/j.molcel.2007.12.002; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Armache KJ, Garlick JD, Canzio D, Narlikar GJ, Kingston RE, Structural basis of silencing: Sir3 BAH domain in complex with a nucleosome at 3.0 Å resolution. Science 334, 977–982 (2011). doi: 10.1126/science.1210915; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wood A, Schneider J, Dover J, Johnston M, Shilatifard A, The Paf1 complex is essential for histone monoubiquitination by the Rad6-Bre1 complex, which signals for histone methylation by COMPASS and Dot1p. J. Biol. Chem 278, 34739–34742 (2003). doi: 10.1074/jbc.C300269200; [DOI] [PubMed] [Google Scholar]
- 56.Sun ZW, Allis CD, Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature 418, 104–108 (2002). doi: 10.1038/nature00883; [DOI] [PubMed] [Google Scholar]
- 57.Wood A et al. , Bre1, an E3 ubiquitin ligase required for recruitment and substrate selection of Rad6 at a promoter. Mol. Cell 11, 267–274 (2003). doi: 10.1016/S1097-2765(02)00802-X; [DOI] [PubMed] [Google Scholar]
- 58.Jayaram H et al. , S-adenosyl methionine is necessary for inhibition of the methyltransferase G9a by the lysine 9 to methionine mutation on histone H3. Proc. Natl. Acad. Sci. U.S.A 113, 6182–6187 (2016). doi: 10.1073/pnas.1605523113; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lewis PW et al. , Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861 (2013). doi: 10.1126/science.1232245; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stark H, GraFix: Stabilization of fragile macromolecular complexes for single particle cryo-EM. Methods Enzymol. 481, 109–126 (2010). doi: 10.1016/S0076-6879(10)81005-5; [DOI] [PubMed] [Google Scholar]
- 61.Beaver JE, Peacor BC, Bain JV, James LI, Waters ML, Contributions of pocket depth and electrostatic interactions to affinity and selectivity of receptors for methylated lysine in water. Org. Biomol. Chem 13, 3220–3226 (2015). doi: 10.1039/C4OB02231A; [DOI] [PubMed] [Google Scholar]
- 62.Kamps JJ et al. , Chemical basis for the recognition of trimethyllysine by epigenetic reader proteins. Nat. Commun 6, 8911 (2015). doi: 10.1038/ncomms9911; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Komander D, Rape M, The ubiquitin code. Annu. Rev. Biochem 81, 203–229 (2012). doi: 10.1146/annurev-biochem-060310-170328; [DOI] [PubMed] [Google Scholar]
- 64.Frederiks F et al. , Nonprocessive methylation by Dot1 leads to functional redundancy of histone H3K79 methylation states. Nat. Struct. Mol. Biol 15, 550–557 (2008). doi: 10.1038/nsmb.1432; [DOI] [PubMed] [Google Scholar]
- 65.Takahashi YH et al. , Dot1 and histone H3K79 methylation in natural telomeric and HM silencing. Mol. Cell 42, 118–126 (2011). doi: 10.1016/j.molcel.2011.03.006; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rossmann MP, Luo W, Tsaponina O, Chabes A, Stillman B, A common telomeric gene silencing assay is affected by nucleotide metabolism. Mol. Cell 42, 127–136 (2011). doi: 10.1016/j.molcel.2011.03.007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Imai S, Armstrong CM, Kaeberlein M, Guarente L, Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 (2000). doi: 10.1038/35001622; [DOI] [PubMed] [Google Scholar]
- 68.Martino F et al. , Reconstitution of yeast silent chromatin: Multiple contact sites and O-AADPR binding load SIR complexes onto nucleosomes in vitro. Mol. Cell 33, 323–334 (2009). doi: 10.1016/j.molcel.2009.01.009; [DOI] [PubMed] [Google Scholar]
- 69.Osborne EA, Dudoit S, Rine J, The establishment of gene silencing at single-cell resolution. Nat. Genet 41, 800–806 (2009). doi: 10.1038/ng.402; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Emre NC et al. , Maintenance of low histone ubiquitylation by Ubp10 correlates with telomere-proximal Sir2 association and gene silencing. Mol. Cell 17, 585–594 (2005). doi: 10.1016/j.molcel.2005.01.007; [DOI] [PubMed] [Google Scholar]
- 71.McGinty RK, Tan S, Recognition of the nucleosome by chromatin factors and enzymes. Curr. Opin. Struct. Biol 37, 54–61 (2016). doi: 10.1016/j.sbi.2015.11.014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ, Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260 (1997). doi: 10.1038/38444; [DOI] [PubMed] [Google Scholar]
- 73.Dorigo B, Schalch T, Bystricky K, Richmond TJ, Chromatin fiber folding: Requirement for the histone H4 N-terminal tail. J. Mol. Biol 327, 85–96 (2003). doi: 10.1016/S0022-2836(03)00025-1; [DOI] [PubMed] [Google Scholar]
- 74.Worden EJ, Wolberger C, Activation and regulation of H2B-Ubiquitin-dependent histone methyltransferases. Curr. Opin. Struct. Biol 59, 98–106 (2019). doi: 10.1016/j.sbi.2019.05.009; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stulemeijer IJ et al. , Dot1 histone methyltransferases share a distributive mechanism but have highly diverged catalytic properties. Sci. Rep 5, 9824 (2015). doi: 10.1038/srep09824; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xue H et al. , Structural basis of nucleosome recognition and modification by MLL methyltransferases. Nature 573, 445–449 (2019). doi: 10.1038/s41586-019-1528-1; [DOI] [PubMed] [Google Scholar]
- 77.Worden EJ, Zhang X, Wolberger C, Structural basis for COMPASS recognition of an H2B-ubiquitinated nucleosome. eLife 9, e53199 (2020). doi: 10.7554/eLife.53199; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee JS, Smith E, Shilatifard A, The language of histone crosstalk. Cell 142, 682–685 (2010). doi: 10.1016/j.cell.2010.08.011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Suganuma T, Workman JL, Crosstalk among Histone Modifications. Cell 135, 604–607 (2008). doi: 10.1016/j.cell.2008.10.036; [DOI] [PubMed] [Google Scholar]
- 80.van Welsem T et al. , Dot1 promotes H2B ubiquitination by a methyltransferase-independent mechanism. Nucleic Acids Res. 46, 11251–11261 (2018). doi: 10.1093/nar/gky801; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Long L, Furgason M, Yao T, Generation of nonhydrolyzable ubiquitin-histone mimics. Methods 70, 134–138 (2014). doi: 10.1016/j.ymeth.2014.07.006; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dyer PN et al. , Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 375, 23–44 (2004). doi: 10.1016/S0076-6879(03)75002-2; [DOI] [PubMed] [Google Scholar]
- 83.Wilkins BJ et al. , Genetically encoding lysine modifications on histone H4. ACS Chem. Biol 10, 939–944 (2015). doi: 10.1021/cb501011v; [DOI] [PubMed] [Google Scholar]
- 84.Li X et al. , Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–590 (2013). doi: 10.1038/nmeth.2472; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zheng SQ et al. , MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). doi: 10.1038/nmeth.4193; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang K, Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12 (2016). doi: 10.1016/j.jsb.2015.11.003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zivanov J et al. , New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). doi: 10.7554/eLife.42166; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA, cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). doi: 10.1038/nmeth.4169; [DOI] [PubMed] [Google Scholar]
- 89.Grant T, Rohou A, Grigorieff N, cisTEM, user-friendly software for single-particle image processing. eLife 7, e35383 (2018). doi: 10.7554/eLife.35383; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wakamori M et al. , Intra- and inter-nucleosomal interactions of the histone H4 tail revealed with a human nucleosome core particle with genetically-incorporated H4 tetra-acetylation. Sci. Rep 5, 17204 (2015). doi: 10.1038/srep17204; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sawada K et al. , Structure of the conserved core of the yeast Dot1p, a nucleosomal histone H3 lysine 79 methyltransferase. J. Biol. Chem 279, 43296–43306 (2004). doi: 10.1074/jbc.M405902200; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vijay-Kumar S, Bugg CE, Cook WJ, Structure of ubiquitin refined at 1.8 A resolution. J. Mol. Biol 194, 531–544 (1987). doi: 10.1016/0022-2836(87)90679-6; [DOI] [PubMed] [Google Scholar]
- 93.Min J, Feng Q, Li Z, Zhang Y, Xu RM, Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell 112, 711–723 (2003). doi: 10.1016/S0092-8674(03)00114-4; [DOI] [PubMed] [Google Scholar]
- 94.Emsley P, Cowtan K, Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132 (2004). doi: 10.1107/S0907444904019158; [DOI] [PubMed] [Google Scholar]
- 95.Adams PD et al. , PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221 (2010). doi: 10.1107/S0907444909052925; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Truong DM, Boeke JD, Resetting the yeast epigenome with human nucleosomes. Cell 171, 1508–1519.e13 (2017). doi: 10.1016/j.cell.2017.10.043; [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.