

Abstract
The application of next generation sequencing (NGS) technologies in cancer research has accelerated the discovery of somatic mutations; however, progress in the identification of germline variation associated with cancer risk is less clear. We conducted a systematic literature review of cancer genetic susceptibility studies that used NGS technologies at an exome/genome-wide scale to obtain a fuller understanding of the research landscape to-date and to inform future studies. The variability across studies on methodologies and reporting was considerable. Most studies sequenced few high-risk (mainly European) families, used a candidate analysis approach, and identified potential cancer-related germline variants or genes in a small fraction of the sequenced cancer cases. This review highlights the importance of establishing consensus on standards for the application and reporting of variants filtering strategies. It also describes the progress in the identification of cancer related germline variation to-date. These findings point to the untapped potential in conducting studies with appropriately sized and racially diverse families and populations, combining results across studies, and expanding beyond a candidate analysis approach to advance the discovery of genetic variation that accounts for the unexplained cancer heritability.
Keywords: whole exome/genome sequencing, cancer genetic susceptibility, germline population studies
INTRODUCTION
Since 2005, the volume of publications enabled by sequencing approaches has grown at an astonishing rate. Several reviews have described the sequencing technology platforms (1) and advancements made in next-generation sequencing (NGS) over the past decade (2). The widespread availability of NGS technologies—including whole genome sequencing (WGS) and whole exome sequencing (WES)—has not only led to its applications in cancer research, but also for use in the clinical setting (3,4).
Use of NGS has accelerated the discovery of somatic mutations (5) and germline mutations in Mendelian diseases (6). Approximately 60% of Mendelian disease projects have successfully identified disease gene mutations (7) using sequencing technologies, improving upon classical approaches for gene discovery (e.g., linkage analysis). Additionally, the application of NGS technologies is revealing complex somatic mutational signatures associated with different types of cancer, a disease that is, by definition a result of somatic mutations (8, 9).
Given these successes, there is hope that sequencing studies may also aid the identification of genes accounting for the expected cancer heritability. The estimated cancer heritability is 33% for overall cancer (10) and its unexplained component has remained high, where a study by Susswein et al. (11) reported that 91% of cancer cases tested negative for known mutations in a large gene-panel testing study. In addition to the heritability hidden in current array-based studies and likely detectable with larger sample sizes, it has been hypothesized that the missing familial heritability may reside in rare variants of high or moderate/low penetrance that are potentially tractable by NGS technologies (12). As NGS technologies continue to evolve, NGS will play an increasing role in cancer research in the foreseeable future.
We conducted a systematic literature review and evaluated the degree of success and limitations in identifying germline cancer susceptibility variants using NGS technologies at a genome-wide scale, i.e., through WES and WGS, with the goal of learning from past efforts and obtaining a fuller understanding of the NGS-related research landscape to-date. Given the transition of genomic discovery research from candidate genes (historically of limited success) to WES/WGS studies, the high cost of WES/WGS methods, and their specific challenge with sifting through millions of variants, this review focuses on the effectiveness of WES/WGS studies –and not on other NGS gene targeted approaches– to identify novel variants and genes involved in cancer risk. The present review provides selected study-related characteristics, technologies, and methodologic details for 186 WES/WGS-related publications with the goal of informing the design of future studies. We also discuss the research needs and opportunities that could further advance the discovery of cancer susceptibility genes or variants. It should be noted that, although the reviewed articles were not selected based on their focus on rare versus common variants nor on their focus on low versus moderate/high penetrant variants, more cost-efficient approaches, based on genome wide genotyping assays, exist to study common variants, while NGS technologies are necessary to study rare variants.
METHODOLOGY
We followed the methodology for systematic literature review according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines (13).
Search strategy
For this report, PubMed and Embase were searched for the period between January 1, 2000 through December 31, 2018 using various search terms for exome/genome sequencing and germline susceptibility and cancer (see Supplemental Figure S1). Both searches were restricted to articles written in English. The references of the reviewed articles were also checked for the presence of additional in scope articles that may had been missed by the above key words searches.
Exclusion/inclusion criteria
Articles were included in this literature review if they had used genome-wide (i.e., whole exome or whole genome) sequencing to generate germline DNA data in at least two cancer cases (even if within the same family) with the purpose of identifying cancer susceptibility genes or variants (see exclusion criteria in Supplemental Figure S1). We excluded papers that sequenced only one cancer case because they were often case-reports without a variant/gene identification research focus. Notably, we did not exclude publications that restricted the analysis to a priori or candidate genetic regions (referred in this manuscript as “candidate analysis approach”), due to the large methodological diversity across such studies; instead, we opted to include all studies that used WES/WGS independent of their use of a candidate analytical approach, and captured the prior knowledge used to select variants/genes to allow for sensitivity analysis by candidate analysis approach. The eligibility of each abstract/full-text article was assessed independently in a standardized manner by three reviewers. A fourth reviewer confirmed all inclusions and performed quality control on one third of the exclusions. The exclusion criteria were applied in no particular order, and the first reason noted by the coder was recorded, even if a paper could be excluded for multiple reasons.
Data abstraction
We looked for four broad components or phases in each paper: 1) a required discovery phase, and optional phases of 2) technical validation, 3) independent replication, or 4) functional evaluation.
The discovery phase refers to the component of the paper where germline DNA of more than one cancer case (and possibly controls) were sequenced by WES or WGS with the goal of identifying cancer susceptibility variants and genes.
The technical validation attempts to confirm some of the variants observed in the discovery phase using an alternative sequencing technology.
The independent replication phase attempts to replicate some of the variants or genes observed in the discovery phase in independent cases.
The functional evaluation phase characterizes through in silico and/or laboratory functional experiments some of the variants or genes observed in the discovery phase.
Data abstracted (see list and definitions of coding fields in Table 1 and Supplemental Table S2) included publication and general study information, numbers and characteristics of cases, controls, and families used in each phase, sequencing technique, data filtering and analysis methods, in silico and experimental functional assessment, and key conclusions. We extracted information on family history of cancer, early age at diagnosis and/or multiple primaries, which are the National Comprehensive Cancer Network (NCCN) guidelines criteria for genetic cancer risk assessment. Of note, we defined ‘familial studies’ as those studies that sequenced samples from cancer cases (here referred to as ‘familial cases’) belonging to a family in which multiple cancer cases of the studied cancer type had been diagnosed (also referred to in the literature as ‘high-risk families’). We defined ‘unselected studies’ as those studies that sequenced samples from cancer cases (here referred to as ‘unselected cases’) who were unselected for family history of cancer (also referred to in the literature as ‘sporadic cases’). For each authors’ selected variants and genes, we recorded the nomenclature and minor allele frequency (MAF), as reported by the authors, the number of families for which cancer status fully segregated or not with the variant, the number of unselected or familial cases, unaffected relatives or unrelated controls that carried the variant, and the number of families, familial cases, unselected cases, or controls carrying any prioritized variant in the same gene. For quality control purposes, 50% of the papers were also reviewed by a second abstractor and discrepancies between coders were resolved by consensus. Information in the articles that was unclear was coded based on best interpretation of the reviewers.
Table 1.
Coding fields used for reviewing papers.
| Topic | Coding fields |
|---|---|
| Publication | Pubmed ID; Journal; Year; Author; Title; Abstract; |
| Study general | Goal; Study design; Source of individuals; Cancer type; Ethnicity; Sequencing Center; Data repository; |
| Number of individuals sequenced in discovery | Number of cases, controls, families, and cases per family sequenced in discovery phase; |
| Sequencing technique | Samples type; Exome and/or genome; Capture kit; Sequencer; Coverage/depth; |
| Processing of raw-data | Aligner; Reference genome; Variant caller and calling QC; Annotation software and sources; |
| Technical validation | Yes/No; Validation technology; Number of cases, controls, families, and cases per family sequenced in validation phase; variants/genes validated; |
| Independent replication | Yes/No; Replication technology and analysis; Number of cases, controls, families, and cases per family sequenced in replication phase; variants/genes replicated; |
| Functional validation | In silico functional analyses; Experimental functional study; |
| Variants and genes data analysis | Candidate analysis approach; Filtering strategy overall; Analytical methods; |
| Variants and genes identification | Yes/No; Identified variants and genes; Number of cases, controls, families carrying the identified variants and genes; |
| Authors comments and conclusions | Challenges; Suggested next steps; Conclusions; |
| Derived filtering criteria/categories shown in Figure 1 | f_1: variant passing quality control metrics |
| f_2: heterozygous variant | |
| f_3: homozygous variant | |
| f_4: variant located in a coding region | |
| f_5: nonsynonymous or splice variant | |
| f_6: variant damaging according to in silico algorithms | |
| f_7: truncating variant | |
| f_8: variant altering protein properties according to molecular modeling | |
| f_9: not hypervariable gene | |
| f_10: variant absent from Minor Allele Frequency (MAF) databases | |
| f_11: variant rare in MAF databases | |
| f_12: variant segregating with disease status in the family | |
| f_13: variant present in multiple families or independent cases | |
| f_14: variant enriched in cases compared to controls | |
| f_15: gene mutated in multiple families or independent cases | |
| f_16: gene enriched in cases compared to controls | |
| f_17: variant present in disease related databases | |
| f_18: gene known to be linked to disease | |
| f_19: genetic region known to be linked to disease | |
| f_20: biological or molecular pathway known to be linked to disease | |
| f_21: pathway analysis indicating a gene-disease link | |
| f_22: variant confirmed through technical validation | |
| f_23: variant or gene replicating in independent cases | |
| f_24: variant loss of heterozygosity (LOH) observed in tumor | |
| f_25: relevant somatic mutations observed in tumor | |
| f_26: gene-disease link supported by functional experiments | |
| f_27: variant splicing supported by experiment | |
| f_28: variant-disease link supported by functional experiment |
RESULTS
Article selection
The search yielded a total of 6,339 unique articles (see PRISMA flowchart in Supplemental Figure S1) which were evaluated for inclusion and exclusion criteria (Supplemental Table S1). After full-text review, 186 papers met the inclusion criteria and are listed with the derived coding variables in Supplemental Table S2–S3. The distribution of the 186 reviewed articles by publication year shows an increase from 2 to 40 articles/year between 2011 and 2015, followed by a plateau in 2016-2018 (Supplemental Figure S2).
Study design and population characteristics
In the discovery phase, 86% of articles used familial cases (11% of which were in combination with early-age of onset and/or unselected cases), 12% of studies were conducted in unselected cases, and 2% of articles used early-age of onset cases. Fifty-five percent of the studies included controls in the discovery phase, of which 67% were unaffected relatives of the cancer cases. Only 57% of the reviewed articles also attempted some type of replication in an independent group of cancer cases. However, only 17% of the replication phase used the same study design as the discovery phase (Supplemental Figure S3). For example, 16% of the family-based articles that included a replication phase did not include familial cancer cases. Moreover, controls were included more often in the replication phase (80%) than in the discovery phase (55%). Likewise, unselected cases were used more frequently in the replication phase than in the discovery phase (30% compared to 12%).
One third of the studies sequenced only 2 or 3 cancer cases in the discovery phase and only 28 (15%) of the articles sequenced more than 100 cancer cases. Technical validation of some of the variants was performed in 85% of the articles, although most of these were conducted in studies with small sample size (i.e., less than 50 cases). The replication phase had generally larger sample size, including more than 100 cancer cases. Almost half (n=73) of the 160 family-based studies sequenced a single family and the majority (n=115) sequenced one or two familial cancer cases (Table 2).
Table 2.
Number and percentage of cancer cases and families studied per article and by study phase.
| Number (%) of articles | |||
|---|---|---|---|
| Discovery | Independent replication | Technical validation | |
| Number of cancer cases | |||
| 2-3 | 62 (33%) | 0 (0%) | 41 (22%) |
| 4-10 | 38 (20%) | 3 (2%) | 48 (26%) |
| 11-50 | 39 (21%) | 17 (9%) | 35 (19%) |
| 51-100 | 19 (10%) | 10 (9%) | 4 (2%) |
| 101-1000 | 22 (12%) | 43 (23%) | 3 (2%) |
| >1000 | 6 (3%) | 34 (18%) | 1 (1%) |
| Not stated | 0 (0%) | 0 (0%) | 26 (14%) |
| Total* | 186 (100%) | 107 (57%) | 158 (85%) |
| Number of high-risk families | |||
| 1 | 73 (39%) | 3 (2%) | |
| 2-10 | 27 (15%) | 6 (3%) | |
| 11-50 | 40 (22%) | 23 (12%) | |
| 51-100 | 10 (5%) | 8 (4%) | |
| 101-1000 | 6 (3%) | 23 (12%) | |
| >1000 | 2 (1%) | 5 (3%) | |
| Not stated | 2 (1%) | 4 (2%) | |
| Total* | 160 (86%) | 72 (39%) | |
| Average number of sequenced cases per family | |||
| 1 | 49 (26%) | 56 (30%) | |
| 2 | 66 (35%) | 10 (5%) | |
| 3 | 27 (15%) | 1 (1%) | |
| 4 | 10 (5%) | 1 (1%) | |
| 5 | 4 (2%) | 0 (0%) | |
| 6-7 | 3 (2%) | 0 (0%) | |
| Not stated | 1 (1%) | 5 (2%) | |
| Total* | 160 (86%) | 72 (39%) | |
Some totals do not add to 100% because: 15% of articles did not perform technical validation; 43% of articles did not perform independent replication; 14% of articles did not include familial cases in the discovery phase; 61% of articles did not perform independent replication in familial cases.
The most commonly studied cancer types were breast cancer (15%), followed by hematological malignancies (15%, which included pediatric cases), colorectal cancer (10%), melanoma (7%), lung cancer (7%), and prostate cancer (5%) (Supplemental Figure S4). Information on race, ethnicity, or country of origin was reported in 85% of the articles reviewed and mostly referred to the region or country of origin (Supplemental Figure S5), with only few studies reporting sequencing derived ancestry. Over half of the studies were conducted in Caucasians or individuals from Europe (59%), followed by individuals from Asia (13%), the Middle East (7%), of African-descent (3%), from Latin America (2%) and Australia (2%).
Sequencing technologies, read alignment, variant calling and annotation
Sequencing was performed in DNA extracted from blood (65%), formalin-fixed paraffin-embedded –generally non-tumor– tissues (8%) and/or saliva (5%). However, a notable proportion did not state the DNA source (23%). Moreover, the amount of DNA used for sequencing (~1-3ug per sample) was reported only in 33% of the articles (Supplemental Table S2).
Ten articles (5%) analyzed WGS only, seven studies (4%) conducted both WGS and WES, and 169 studies (91%) analyzed WES only. The reviewed studies used 28 different capture methods and 14 different sequencing platforms (Supplemental Figure S6), whereas 12% and 8% did not report the capture or sequencer used, respectively. Sequencing coverage information was reported for only 71% of the articles, as average depth (52%) and/or percentage of the target genome covered at 10x or higher thresholds (42%) (Supplemental Figure S7). For most articles it was unclear if the reported coverage statistics referred to the targeted or actual coverage and to pre-QC or post-QC coverage. In addition, there was no correlation between the number of samples sequenced and the reported coverage depth.
Sequencing reads were aligned to human genome references, where 79% of the studies reported using hg19 (also known as NCBI build 37 or GRCh37) and 3% used hg18 (a.k.a., NCBI build 36). The most widely used aligner (52%) was the Burrows-Wheeler Aligner (BWA) (14). Reference genome and alignment algorithm used were not specified in 18% and 12% of articles, respectively. Over half of the articles used Genome Analysis Toolkit (GATK) (15) variant calling algorithms. One fourth of the studies used more than one algorithm to call variants, which can improve call quality, and 8% of papers did not report their variant calling method. Various quality metrics were applied to screen the sequencing reads (e.g., removal of PCR duplicates, unmapped or non-uniquely mapped or out of target reads) and the called variants (e.g., removal of variants with quality or coverage below a preset threshold) (Supplemental Table S2). In addition, 26% of the articles used additional control sequencing datasets generated in-house via the same technology and pipeline as the study dataset to control for technical artifacts.
The annotation software used to annotate the called variants was not specified in 30% of the articles and included ANNOVAR (16) in 36% of the articles. Eighty-nine percent of the articles used allele frequency information from publicly available databases, mainly from the 1000 Genomes Project (17) (64%), the NHLBI Exome Sequencing Project (ESP) (18) (47%), dbSNP (19) (46%), and the Exome Aggregation Consortium (ExAC) (20) (32%). Several other annotation tools and source databases were used (Supplemental Table S2).
Criteria used to filter variants and genes
Figure 1 shows which criteria or filters (see the f_1, …, f_28 variables described in Table 1) were used in each reviewed article. Filtering criteria were generally used to prioritize/select a variant/gene over others and/or to seek evidence in support of a selected variant/gene. Data in Figure 1 show that the criteria used to identify variants and genes with a role in cancer susceptibility are disparate across articles, and consequently, results cannot be directly compared across studies. Below we examine these filtering/selection criteria and summarize their use and outcomes by grouping them into seven broader themes: 1) Variant quality; 2) Variant effect; 3) Variant rarity; 4) Mode of inheritance and genetic-disease association; 5) Candidate analysis approaches; 6) Independent replication; and 7) Functional validation. Two general observations hold true: i) lack of sensitivity analyses to assess variability in results by changes in variant/gene selection strategy, and ii) no or minimal reporting of a justification for the choice of criteria and thresholds used.
Figure 1.
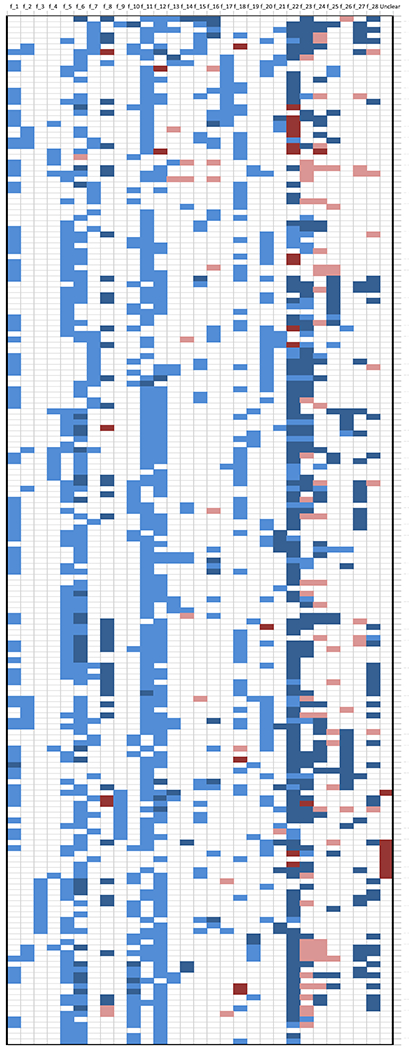
Summary across the 186 reviewed articles of the methods used to conclude if a variant or gene was plausibly involved in a causal pathway to cancer. The y-axis displays the PMID of the 186 coded papers; the x-axis displays 29 broad categories of filtering criteria described in Table 1. White color indicates that the criterion was not used by the authors to identify a variant/gene as possibly linked to the cancer under investigation. Light blue indicates that the criterion was used as a selection filter. Dark blue indicates that the criterion was used as increased evidence of variant/gene-cancer association. Pink color indicates that the criterion was used as decreased evidence of variant/gene-cancer association. Dark red indicates that the information related to that criterion was unclear. The last category on the right “unclear” indicates that some other not clearly stated criteria were used. The order of the papers along the y-axis is based on a computed correlation between values 2 (for supporting evidence), 1 (for used selection criteria), 0 (for not used criteria), −1 (for unsupporting evidence) and −2 (for unclear), i.e., papers using similar filtering criteria sets are shown next to each other.
1). Variant quality (f_1, f_9, f_22).
Approximately half of the reviewed articles explicitly described the use of variant call metrics to exclude low-quality variants from the analyses, e.g., manual inspection using the Integrative Genomics Viewer (21), removing variants in paralogs or repeats regions, and/or with Phred-scaled quality scores or coverage below a given threshold (Supplemental Table S2). Approximately 85% of the articles reported technically validating variants, where most used Sanger sequencing. The technical validation success rate was about 80% for studies that tested over 50 variants versus above 90% for studies that tested fewer variants (Supplemental Figure S8).
2). Variant effect (f_4, f_5, f_6, f_7, f_8).
Most articles (n=168 or 90%) required the variants to be in coding regions, and more specifically, to be non-synonymous or in splice sites or frameshift (n=163 or 88%). A subset of these articles also required the selected variants to be functionally impactful (e.g., ‘deleterious’, ‘damaging’, or ‘pathogenic’) according to various in silico algorithms (n=91 or 49%) or to be truncating (n=38 or 20%). Supplemental Table S2 lists for each article the adopted in silico pathogenicity predictors (e.g., references 22–29) which were often used in combination.
3). Variant rarity (f_10, f_11).
Most articles required the selected variants to be absent/not described (25%) or rare (62%) based on a preset MAF threshold (0-0.1 range, Supplemental Figure S9) in internal or publicly available control datasets, such as 1000 Genomes Project, dbSNP, ESP, or others (Supplemental Table S2).
4). Mode of inheritance (f_2, f_3, f_12) and genetic-disease association (f_13, f_14, f_15, f_16, f21).
Only 10 (5%) and 17 (9%) papers restricted their search to homozygous and heterozygous variants according to a recessive or a dominant mode of inheritance, respectively. Since the remaining papers describe only heterozygous variants in their findings, a dominant inheritance hypothesis can be assumed for all but 10 articles. The majority of articles (n=108 or 58%) required the variant to fully or partially segregate with disease status in at least one family whose members were sequenced in the discovery phase. Only two of the reviewed articles looked for de novo variants. In several studies, the selected genes were required to be mutated in more than one family (n=19 or 10%) or in multiple independent cases (n=5 or 3%), or to be enriched in cases compared to controls according to burden tests (n=18 or 10%). Fewer articles required the same selected variant to be present in more than one family (n=10 or 5%), in multiple independent cases (n=6 or 3%), or to be statistically enriched in cases compared to controls (n=8 or 4%). Finally, 12 articles used pathway analysis techniques to identify biological or molecular functions that were enriched with mutated genes.
5). Candidate analysis approaches (f_17, f_18, f_19, f_20).
Two thirds of the reviewed articles (n=121, 65%) used existing information from the literature and curated databases to restrict the discovery analysis to: variants present in disease-related databases such as ClinVar (30) (n=14 or 7%); genes known to be linked to disease such as those listed in OMIM (31) or reported in the literature (n=59 or 32%); genetic region known to be linked to disease through genome wide association studies (GWAS) or linkage studies (n=14 or 7%); and/or biological or molecular pathways known to be linked to disease, such as DNA repair pathways (n=51 or 27%).
6). Independent replication (f_23).
Only 107 (57%) articles attempted replication of variants/genes in an independent set of cancer cases. Overall, 79 (42%) reported various degrees of confirmatory evidence. In some cases, the authors reported the presence of other pathogenic variants in the same gene, whereas in others, a statistically significant burden test in cases compared to controls for that gene was reported. In a few studies, the exact variant(s) initially found in the discovery phase were found in additional cancer cases in the replication phase.
7). Functional validation (f_24, f_25, f_26, f_28).
In those studies that evaluated function of the identified variants/genes (70% of 186), 60 (32%) tested for loss of heterozygosity in tumor samples (58% of which tested positive); 36 (19%) looked for somatic mutations in the same gene (69% of which were found); 22 (12%) looked for gene/methylation expression changes supporting a link with disease (86% with positive results); 35 (19%) checked for variant splicing (86% of which were verified); and 45 (24%) carried out in vitro experiments or other functional assays on the identified variants, 80% of which showed results consistent with the hypothesized function for these variants.
Variants and genes identified
About 95% (n=176) of reviewed articles indicated that they identified variants or genes (listed in Table 3 with PMIDs by cancer type) with various degrees of certainty. Only eight (4%) articles clearly stated that they were not able to identify variants or genes in the studied cases, and the remaining two (1%) articles pointed to molecular or functional pathways of possible relevance to the studied cancer type. The 176 articles indicated as primary findings (Supplemental Table S3) a total of ~2,095 variants (average 11, median 3, range 1 to 222 per article) and ~1,215 (~954 unique) genes (average 6, median 1, range 1 to <222 per article). An exact count of variants and genes identified was not feasible due to incomplete counts and/or variant nomenclature in some of the articles. For the 99 articles that studied more than one high-risk family, and reported the information, the identified variants/genes accounted on average for 25% of the families evaluated in discovery and replication phases combined (we excluded from this analysis 43 articles that studied only a single family and did not attempt replication, Supplemental Figure S10). Regarding the prevalence of the identified variants among controls, 27 (16% of 176) articles did not sequence these variants in controls, 50 (28%) sequenced only some unaffected relative of the cases, 37 (21%) did not report how many of the sequenced controls carried the investigated variants; in the remaining articles the variants’ frequency in unrelated controls averaged 0.015 with median 0 (Supplemental Table S3).
Table 3.
List and counts of identified genes and PubMed IDs by cancer type.
| Cancer Type | Authors’ identified genes± [or pathways] | # genes [unique] | PMID¶ (references) {number of articles} |
|---|---|---|---|
| Biliary | BRCA1, BRCA2, RAD51D, MLH1, MSH2, POLD1, POLE, TP53, ATM; | 9 [9] |
29360550 (32); {1} |
| Blood | none; none; ACAN; ATM; CBL; CEBPA; CORO1A; DICER1; ETV6; FAAP24; HAVCR2; ITGB2; KDR; MLL; PAX5; POT1; PRDM9; TP53; TYK2; LAPTM5, HCLS1; SLC26A6, FAM107A; POT1, ACD, TERF2IP; ZXDC, ATN1, LRRC3; FNCP/SLX4, FANCA, GEN1; POLK, PRKCB, ZNF676, PRRC2B, PCDHGB6, GNL3L, TTC36, OTOG, OSGEPL1, RASSF9; MLL3; JAK2, MPL, TYK2, BCL2, BCL3, KIT, SH2B3, MCM3AP, PEAR1, TET2; | 52 [50] |
23379653; 27141497; 25715982; 28652578; 25939664; 26721895; 23522482; 29708584; 26522332; 27473539; 30374066; 27629550; 27365461; 23457195; 24013638; 29693246; 23222848; 23255406; 27733777; 26903547; 29755658; 27528712; 28427458; 26201965; 30097855; 24301523; 30553997; (33–59) {27} |
| Bone | ANO5; EXT2; T; | 3 [3] |
27216912; 27636706; 23064415; (60–62) {3} |
| Brain & nervous system | CASP9; GALNT14; JMJD1C; PMS2; POT1; SMARCE1; SUFU; TP53, ATRX; TP53, MSH4, LATS1; CNL2, ANKRD65, TAS1R2, PINK1, DCAF6, F5, TNN, CFH, DDX59, IGFN1, CHI3L1, CR1, OR2B11, TRIM58, RRM2, ANKRD53, CAPG, IMMT, THNSL2, PIKFYVE, COL4A3, SUMF1, CEP120, ARAP3, ARHGEF37, TCOF1, FAT2, OR2V2, HIVEP1, PMS2, ADAM22, C7orf62, TRPA1, TG, ART1, TRIM22, IGSF22, CD6, CD5, SLC22A24, CD248, MYEOV, KIAA1731, PIK3C2G, LRMP, OVCH1, TPPP2, RPGRIP1, TTLL5, KCNK10, C14orf159, ACSBG1, ACAN, C15orf42, KIF7, ZNF23, ZNF469, ANKRD11, STARD3, ERBB2, AKAP1, FAM20A, ABCA6, ABCA10, DNAH17, LAMA1, DOPEY2, MYO18B, NEFH, CCDC157, RNF215, SMTN, APOL3; | 85 [83] |
27935156; 26309160; 24896186; 28805995; 25482530; 23377182; 22958902; 29602769; 25041856; 25537509; (63–72) {10} |
| Breast | none; none; APOBEC3B; ATM; ERCC3; FANCM; GPRC5A; KAT6B; PALB2; RCC1; RECQL; RECQL; RINT1; RNASEL; XRCC2; BRCA2, STK11; FANCC, BLM; ERCC6, BRCA2, BRCA1; BRCA1, TP53, PTEN, PALB2, RAD51C; XCR1, DLL1, TH, ACCS, SPPL3, CCNF, SRL; BRCA1, BRCA2, PALB2, ATM, CHEK2, BARD1, BRIP1, XRCC2; FANCM, WNT8A, SLBP, CNTROB, AXIN1, TIMP3, PTPRF, UBA3, MAPKAP1, TNFSF8, S1PR3; HSD3B1, CFTR, PBK, ITIH2, MMS19, PABPC3, PPL, DNAH3, LRRC29, CALCOCO2, ZNF677, RASSF2; NOTCH2, DNAH7, RAF1, MST1R, LAMB4, NIN, SLX4, ERCC1, SLC22A16, PTPRD, ARHGEF12, ERBB2; ABCC12, APC, ATM, BRCA1, BRCA2, CDH1, ERCC6, MSH2, POLH, PRF1, SLX4, STK11, TP53; ABCA10, CHST15, GRIP1, LOC100129697, LOC388813, NBPF10, PABPC3, C16orf62, KRTAP21-3, NPIPB11, PDE4DIP, CCDC7, CFAP46, CXorf23, SMIM13, GAGE2A, PHIP, SLC15A5, ZNF750, ATP10B, PIGN, PRR14L; ABL1, ADRA2A, DUX2, GATA3, GPRIN1, JAKMIP3, KAT6B, LIG1, LIG4, NANP, NFKBIZ, NFRKB, NOTCH2, PHKB, PINK1, POLK, POLQ, PPFIA4, PRKCQ, RAD23B, SMG1, TNK2, UBE2L3; ABCC11, ADARB1, ADPRH, ATG4C, ATM, ATXN3L, BANK1, BIRC8, BRIX1, BTN2A3, C10orf68, C14orf38, C17orf57, C1orf168, C20orf186, C2orf63, C5orf52, C7orf46, CA12, CASP5, CAT, CCDC99, CDKL2, CFHR5, CGRRF1, CHEK2, CHRNB3, CIB1, CPAMD8, CRYBG3, CXCL6, DCD, DCHS2, DDX60L, DNMT3A, ECEL1, EEF2K, EIF2B4, ENDOD1, ENTPD4, EPS8L1, EPSTI1, ERCC3, EYS, FAM40B, FAP, FETUB, FILIP1, FLG2, FLOT2, FOLH1B, FTMT, GIMAP6, GPRC5A, GTPBP5, HEATR7B2, IFIT2, IGSF22, IL25, ITGB3, KIAA1919, LOC440563, LOC647020, LRRC69, MAGEF1, MBIP, MCAT, ME1, MLL4, MMP3, MTERF, NDUFA10, NEIL1, NLRP7, NOD2, NOX1, NPL, NUP188, OTOA, OXSM, PAFAH2, PARK2, POLQ, POLR3GL, PPEF2, PRMT7, PRSS3, PRSS7, PTCHD3, RBKS, RPS6KC1, SAMD9L, SERPINI2, SGOL2, SIGLEC1, SLAMF6, SLC26A10, SLC4A1AP, SLC6A5, SLCO1B3, SMARCD2, SPATA4, SSX9, STAP2, TAOK1, TIMD4, TMEM56, TNFAIP6, TNFAIP8, TTC21A, USP45, VPS13B, WFDC8, WRN, ZIM2, ZNF311, ZNF451, ZNF491, ZNF582, ZNF599, ZSCAN29; [cell-cell and cell-extracellular matrix adhesion processes;] | 253 [229] |
21858661; 23383274; 28062980; 27913932; 27655433; 25288723; 24470238; 23800003; 27648926; 29363114; 25915596; 25945795; 25050558; 29422015; 22464251; 26576347; 23028338; 25923920; 29325031; 26157685; 25330149; 23409019; 29879995; 29868112; 28202063; 28076423; 24969172; 22527104; 26969729; (73–101) {29} |
| Colorectal | BRF1; FAN1; MUTYH; RPS20; SETD6; CENPE, KIF23; POLE, POLD1; WRN, ERCC6; EMR3, PTPN12, LRP6; TTF2/TRIM45/VTCN1/MAN1A2; BRCA2 (FANCD1), BRIP1 (FANCJ), FANCC, FANCE, REV3L (POLZ); FANCM, LAMB4, LAMC3, PTCHD3, TREX2; IL12RB1, LIMK2, POLE2, POT1, MRE11; CDKN1B, XRCC4, EPHX1, NFKBIZ, SMARCA4, BARD1; ACSL5, ADAMTS4, ARHGAP12, ATM, CYTL1, DONSON, INTS5, MCTP2, ROS1, SYNE1; UACA, SFXN4, TWSG1, PSPH, NUDT7, ZNF490, PRSS37, CCDC18, PRADC1, MRPL3, AKR1C4; DDX20, ZFYVE26, PIK3R3, SLC26A8, ZEB2, TP53INP1, SLC11A1, LRBA, CEBPZ, ETAA1, SEMA3G, IFRD2, FAT1; MLH1, MUTYH, MSH6, MSH2, LRP5, ATM, RYR3, EIF2AK4, PRDM1, RYR2, BUB1, RPS6KB2, DAAM1, TCF7, TSC2, LIG3, MCC, MAX, PARP1, ETV4, FZD10, MAST2, MTOR; | 96 [94] |
28912018; 26052075; 24691292; 24941021; 28973356; 23637064; 23263490; 26344056; 26901136; 29396139; 27165003; 23585368; 27329137; 25058500; 25749350; 24146633; 29844832; 25892863; (102–119) {18} |
| Esophageal | KCNJ12/KCNJ18, GPRIN2; PTEN, SMAD7; DNAH9, GKAP1, NFX1, BAG1, FUK, DDOST; NR4A2, IL6ST, FZD3, SBNO2, NOTCH3, STAT3, CD3D, MUC16, LIN28A, STIL, ANK1, CD3E, PARN, GDF15, ITGA6, LCK, THBS2, ALX4, NOD2, KALRN, EGF, DLK2, HDAC2, AKT1, FBXW11, MET, SFRP1; | 38 [38] |
29405996; 25554686; 28165652; 28459198; (120–123) {4} |
| Gastric | none; ATP4A; CTNNA1; INSR, FBXO24, DOT1L; PALB2, BRCA1, RAD51C; PALB2, MSH2, ATR, NBN, RECQL5; DZIP1L, PCOLCE2, TAS2R7, TRIOBP, IGSF10, NOTCH1, SF3A1, GAL3ST1, NOTCH1, OR13C8, EPB41L4B, SEC16A; | 25 [23] |
28875981; 25678551; 23208944; 25576241; 28024868; 29706558; 26872740; (124–130) {7} |
| Glands | MAX; HOXA11, NCOA4, PCSK7, CENPQ, GALC, GOLGA5, GUCA1C, RHCE, DDX12, FAM22F, FAM71F1, MDN1, MLLT4, SYNE1; IGSF3, STXBP5L, PCDHB12, HAVCR1, SMPD2, OPRM1, GTF21RD2/GTF2IRD2B, TAS2R43, KMT5A, NCOR2, BTBD6, PHBDF1, TSC2, RAMP2, CST2, SHANK3; | 32 [32] |
21685915; 23707928; 28210977; (131–133) {3} |
| Head and neck | MLL3; MST1R; EXO1, HLTF, TDP2, RAD52, PER1, MSH2, ERCC5, MSH6, RAD17, PMS2, NBN, DCLRE1C, ALKBH3, LIG4, XRCC3, FANCM, PALB2, BRCA1, BRIP1, TP53, XRCC1, CHEK2, FAAP20, WDR48, FANCD2, FANCA, SHPRH, MMS19, ALKBH3, PARP4, RECQL5, NUDT1, PARP2, POLE2; | 36 [35] |
26014803; 26951679; 29747023; (134–136) {3} |
| Intestinal | IPMK; MUTYH; | 2 [2] |
25865046; 28634180; (137–138) {2} |
| Kidney | BAP1; CTR9; REST; DICER1; | 4 [4] |
23684012; 25099282; 26551668; 26566882; (139–142) {4} |
| Liver | DICER1; | 1 [1] |
28012864; (143) {1} |
| Lung | CHEK2; HER2; IBSP; MET; PARK2; YAP1; PROM1, CRTC2; CACNB2, CENPE, LCT, MAST1; ARHGEF5, ANKRD20A2, ZNF595, ZNF812, MYO18B; BPIFB1, CHD4, PARP1, NUDT1, RAD52, MFI2; FANCL, FANCG, FANCC, FANCF, BRCA2, PALB2, FANCA, BRCA1, RAD51C, BRIP1; BAG6, SPEN, WISP3, JAK2, TCEB3C, NELFE, TAF1B, EBLN2, GON4L, NOP58, RBMX, KIAA2018, ZNF311; TP53, BRCA1, BRCA2, ERCC4, EXT1, HNF1A, PTCH1, SMARCB1, ABCC10, ATP7B, CACNA1S, CFTR, CLIP4, COL6A1, COL6A6, GCN1, GJB6, RYR1, SCN7A, SEC24A, SP100, TTN, USH2A; | 69 [67] |
27900359; 24317180; 26717996; 28294470; 25640678; 26056182; 24484648; 26178433; 29054765; 29667179; 30425093; 24954872; 30032850; (144–156) {13} |
| Melanoma | none; BAP1; GOLM1; POLE; POT1; POT1; RAD51B; TP53AIP1; VPS41; CDKN2A, BAP1, EBF3; MC1R, MITF, BRCA2, MTAP; | 15 [13] |
29764119; 23977234; 29659923; 26251183; 24686846; 24686849; 25600502; 29359367; 25303718; 29522175; 29317335; (157–167) {11} |
| Multiple familial* | ERCC3; BAP1; POT1; MLL3; POLE; POLE; HNRNPA0, WIF1; | 8 [7] |
28911001; 22889334; 28389767; 23429989; 24788313; 25860647; 25716654; (168–174) {7} |
| Multiple types** | BRCA1, BRCA2, ATM, BRIP1, PALB2, CNKSR1, EME2, MRE11A, MSH6, PIK3C2G, RAD51C, RAD51D, XRCC2; APC, ATM, BAP1, BRCA1, BRCA2, BUB1B, FH, HFE, MAX, MET, MSH6, NF1, PALB2, PTEN, RET, SDHA, SDHB, SDHD, TP53, VHL, WRN; | 34 [29] |
26689913; 29625052; (175–176) {2} |
| Pediatric | APC, DICER1, TP53, BRCA2, CHEK2, BAP1, BUB1, ETV6, ACTB, ARID1A, EP300, EZH2, APOB, SCN2A, SPRED1, KDM3B, TYK2; TP53, APC, BRCA2, NF1, PMS2, RB1, RUNX1, ALK, BRCA1, CDH1, KRAS, MSH2, MSH6, NF2, NRAS, PALB2, PTCH1, RET, SDHA, SDHB, VHL; | 39 [36] |
29351919; 26580448; (177–178) {2} |
| Ovarian | FANCM; SMARCA4; RAD51D, ATM, FANCM; BRCA1, TP53, NF1, MAP3K4, CDKN2B, MLL3 and other ~220 genes not stated; | ~230 | 28881617; 24658002; 28591191; 24448499; (179–182) {4} |
| Ovarian & Breast | ATM, MYC, PLAU, RAD1, RRM2B; ATM, PALB2, CHEK2, MSH6, TP53, RAD51C; | 11 [10] |
27782108; 30128536; (183–184) {2} |
| Pancreatic | ATM; BRCA2; ATM, PALB2; FAN1, NEK1, RHNO1; ATM, CDKN2A, PALB2, BRCA2, BUB1B, CPA1, FANCC, FANCG; ATM, BRCA1, BRCA2, CFTR, MSH2, MSH6, PALB2, PMS2, TP53; | 24 [17] |
22585167; 29074453; 23561644; 26546047; 26658419; 27449771; (185–190) {6} |
| Prostate | none; BTNL2; BRCA2, ATM, NBN; BRCA2, HOXB13, TRRAP, ATP1A1, BRIP1, FANCA, FGFR3, FLT3, HOXD11, MUTYH, PDGFRA, SMARCA4, TCF3; HOXB13, TANGO2, OR5H14, CHAD; LRCC46, PARP2, BLM, KIF2B, CYP3A43; ATM, ATR, BRCA2, FANCL, MSR1, MUTYH, RB1, TSHR, WRN; CYP3A43, HEATR5B, GPR124, HKR1, PARP2, PCTP, MCRS1, DOK3, ATRIP, PLEKHH3; TET2, CEP63, ELK4, NUBP2, SPRR3, TRBV7-7, BRCA1, LAMB3, MSH6, PARP2, DLEC1, ZSWIM2; ACACA, AKR1C1, ALG13, ALG6, APTX, ASXL1, ATP6V0A2, B3GAT3, BGLAP, BLM, BRCA1, CRISP3, CTBP1, CYP1B1, DDB2, DOLK, EFCAB6, ELAC2, FANCA, FANCL, FBXW7, FLT3, GSTA1, HDAC9, HSD3B1, IDE, KDR, MBD5, MGAT2, MSH3, MYH14, NCOR2, NEIL3, NOTCH2, NRIP1, PALB2, PAPSS2, PIAS3, RAD51D, RAD54L2, RNASEL, SCN11A, SP1, SULT1E1, TGIF1, TLN1, TP53BP1, TSC1, BE2D3, UBE2V2; | 107 [95] |
26604137; 23833122; 29915322; 27701467; 27902461; 25111073; 27084275; 26585945; 27486019; 26485759; (191–200) {10} |
| Sarcoma*** | POT1; SMARC4; STAT4; CDKN2A, PDGFRA; APC, BLM, BRCA1, BRIP1, ERCC3, EXT2, FANCC, FANCD2, FANCM, FLCN, MITF, PMS2, POLE, PTCH2, PTPN11, RAD51, RAD51D, RET, SLX4, TINF2, TP53, WRAP53; | 27 [27] |
26403419; 23775540; 25492914; 28592523; 28125078; (201–205) {5} |
| Testicular | none; PKD1; PLEC, DNAH7, EXO5; DNAAF1, LRRC6, CNTRL, DRC1, DYNC2H1, CEP290, MAP4; | 11 [11] |
29433971; 27577987; 29761480; 27996046; (206–209) {4} |
| Thyroid | C14orf93 (RTFC); HABP2; MAP2K5; MET; PARP4; SRRM2; USF3; [extracellular matrix organization and DNA repair;] | 7 [7] |
27864143; 26222560; 30132833; 29219214; 26699384; 26135620; 28011713; 28402931; (210–217) {8} |
The definition of “identified” is based on the authors’ choice and emphasis in presenting their results and not on an evaluation of the quality of the results. Bolded gene symbols indicate genes reported in more than one article, in black if only within cancer type, in blue if both within and across cancer types, in red if only across cancer types.
Articles are referenced in the same order as the results separated by semicolons.
Single families with multiple cancer types: 28911001 (adrenocortical, breast [LF]); 22889334 (melanoma, paraganglioma); 28389767 (melanoma, thyroid, breast); 23429989 (colon, blood); 24788313 (colon, ovary, endometrium, brain); 25860647 (colon, ovary, pancreas, small intestine); 25716654 (prostate, breast, colon, pancreas, melanoma);
Large studies of the genetic landscape across cancer types: 26689913 (across 12 cancer types); 29625052 (across 33 cancer types).
Sarcoma types: soft tissue (28592523), Ewing (28125078), cardiac (26403419), Kaposi (25492914), rhabdoid (23775540).
Overall, 106 genes were identified in two or more articles (see bolded gene symbols in Table 3). The five genes reported by more than 10 articles are well-established cancer susceptibility genes (i.e., ATM, BRCA2, BRCA1, TP53, and PALB2 were observed in 12%, 12%, 9%, 9% and 8% of the articles, respectively). When the analysis was restricted to the articles that used a fully agnostic, not candidate, analytical approach, these genes were observed less frequently (6%, 4%, 7%, 0%, 2%, respectively) and other less established genes were more frequently observed (>4%) (i.e., PMS2, IGSF22, ABCA10, ACAN, and PABPC3). We also observed 43 variants in 22 genes which were independently identified in two or three articles (Table 4). While some of the observed pleiotropic effects are well established (e.g., PALB2 and BRCA1/2 for breast, ovarian, prostate and pancreatic cancer), others are potentially novel, such as BRCA2 for melanoma and head and neck cancers, MUTYH for prostate and small intestine cancers, and KDR for prostate cancer and Hodgkin lymphoma.
Table 4.
Germline variants identified in two or more of the 186 reviewed articles.
| Gene | Variant | Allele Frequency |
Articles’ PMID | Cancer type |
|---|---|---|---|---|
| PALB2 | chr16, c.172_175delTTGT, p.Q60fs, rs1214293842 | 0.000042 | 25330149, 30128536 | Breast, Ovarian & Breast |
| PALB2 | chr16, c.509_510delGA, p.R170fs, rs863224790 | 0.000037 | 25330149, 30128536 | Breast, Ovarian & Breast |
| PALB2 | chr16, c.1240C>T, p.R414*, rs180177100 | 0.000009 | 27449771, 30128536 | Pancreatic, Ovarian & Breast |
| PALB2 | chr16, c.3256C>T, p.R1086*, rs587776527 | 0.000009 | 23561644, 30128536 | Pancreatic, Ovarian & Breast |
| PALB2 | chr16, c.3004_3007delGAAA, p.E1002Tfs | 0 | 26485759, 30128536 | Prostate, Ovarian & Breast |
| PALB2 | chr16, c.3549C>A, p.Y1183*, rs118203998 | 0.000009 | 26689913, 30128536 | Multiple_12, Ovarian & Breast |
| PALB2 | chr16, c.424A>T, p.K142* | 0 | 26689913, 30128536 | Multiple_12, Ovarian & Breast |
| BRCA2 | chr13, c.658_659delGT, p.V220fs, rs876660049 | 0.000028 | 26689913, 28202063 | Multiple_12, Breast |
| BRCA2 | chr13, c.6275_6276delTT, p.Leu2092fs, rs11571658 | 0.000065 | 25330149, 29915322 | Breast, Prostate |
| BRCA2 | chr13, c.9246_9247insA, p.T3085fs, rs80359752 | 0 | 25330149, 29915322 | Breast, Prostate |
| BRCA2 | chr13, c.865A>C, p.N289H, rs766173 | 0.052597 | 29317335, 29747023 | Melanoma, Head and Neck |
| BRCA2 | chr13, c.9294C>G, p.Y3098*, rs80359200 | 0.000009 | 26580448, 29625052 | Pediatric, Multiple_33 |
| ATM | chr11, c.5071A>C, p.S1691R, rs1800059 | 0.001937 | 28202063, 28652578 | Breast, Blood (CLL) |
| ATM | chr11, c.170G>GA, p.W57* | 0 | 22585167, 30128536 | Pancreatic, Ovarian & Breast |
| ATM | chr11, c.6095G>GA, p.R2032K, rs139770721 | 0.000027 | 22585167, 29625052 | Pancreatic, Multiple_33 |
| ATM | chr11, c.6100C>T p.R2034*, rs532480170 | 0.000009 | 27913932, 30128536 | Breast, Ovarian & Breast |
| ATM | chr11, g.108155008_delG, p.E1267fs | 0 | 28652578, 29625052 | Blood (CLL), Multiple_33 |
| BRCA1 | chr17, c.1067A>G, p.Q356R, rs1799950 | 0.045162 | 25923920, 26485759 | Breast, Prostate |
| BRCA1 | chr17, c.4065_4068delTCAA, p.N1355fs, rs886040195 | 0.000018 | 26689913, 29625052 | Multiple_12, Multiple_33 |
| BRCA1 | chr17, c.1054G>T, p.E352*, rs80357472 | 0.000009 | 26689913, 29625052 | Multiple_12, Multiple_33 |
| BRCA1 | chr17, c.68_69delAG, p.E23fs, rs80357410 | 0.000175 | 26689913, 29625052 | Multiple_12, Multiple_33 |
| TP53 | chr17, c.733C>T, p.G245S, rs28934575 | 0 | 26580448, 29351919, 29602769 | Pediatric, Pediatric, Brain |
| TP53 | chr17, c.524G>A, p.R175H, rs28934578 | 0 | 26580448, 30128536 | Pediatric, Ovarian & Breast |
| TP53 | chr17, c.743G>A, p.R248Q, rs11540652 | 0.000009 | 26580448, 30128536 | Pediatric, Ovarian & Breast |
| FANCM | chr14, c.5101C>T, p.Q1701*, rs147021911 | 0.001530 | 25288723, 28881617 | Breast, Ovarian |
| FANCM | chr14, c.5791C>T, p.R1931*, rs144567652 | 0.001181 | 28591191, 28881617 | Ovarian, Ovarian |
| KAT6B | chr10, c.4546G>T, p.D1516Y | 0 | 23800003, 24969172 | Breast, Breast |
| KAT6B | chr10, c.4729C>T, p.R1577C | 0 | 23800003, 24969172 | Breast, Breast |
| POT1 | chr7, c.1851_1852delTA, p.D617fs, rs758673417 | 0.000009 | 25482530, 27329137 | Glioma, Colorectal |
| MSH6 | chr2, c.3261delC, p.F1088fs | 0 | 26689913, 30128536 | Multiple_12, Ovarian & Breast |
| CHEK2 | chr22, c.1100delC, p.T367fs | 0 | 22527104, 29351919, 30128536 | Breast, Pediatric, Breast/Ovarian |
| RAD51D | chr17, g.33433425G>A, p.R206*, rs387906843 | 0.000017 | 26689913, 28591191 | Multiple_12, Ovarian |
| FANCC | chr9, c.C553C>T, p.R185*, rs121917783 | 0.000064 | 23028338, 28125078 | Breast, Sarcoma (Ewing) |
| MUTYH | chr1, c.1187G>A, p.G396D | 0 | 27084275, 28634180 | Prostate, Intestine (small) |
| BLM | chr15, c.1933C>T, p.Q645*, rs373525781 | 0.000018 | 23028338, 28125078 | Breast, Sarcoma (Ewing) |
| TYK2 | chr19, c.2279C>T, p.P760L | 0 | 27733777, 29351919 | Blood (ALL), Pediatric |
| MAX | chr14, c.223C>T, p.R75* | 0 | 21685915, 29625052 | Pheochromocytoma, Multiple_33 |
| NOTCH2 | chr1, c.3625T>G, p.F1209V, rs147223770 | 0.003217 | 26485759, 29868112 | Prostate, Breast |
| XRCC2 | chr7, c.96delT, p.F32fs, rs774296079 | 0.000075 | 25330149, 26689913 | Breast, Multiple_12 |
| RET | chr10, c.2370G>C, p.L790F, rs75030001 | 0.000009 | 26580448, 28125078 | Pediatric, Sarcoma (Ewing) |
| GPRC5A | chr12, c.183delG, p.R61fs, rs527915306 | 0.002113 | 22527104, 24470238 | Breast, Breast |
| KDR | chr4, c.3193G>A, p.A1065T, rs56302315 | 0.000324 | 26485759, 27365461 | Prostate, Blood (HL) |
| MITF | chr3, c.952G>A, p.E318K, rs149617956 | 0.001330 | 28125078, 29317335 | Sarcoma (Ewing), Melanoma |
DISCUSSION
Methodological variability across reviewed articles
One major observation from this review was that the criteria used to identify variants and genes presented by the authors as having a role in cancer susceptibility varied dramatically across studies (Figure 1). In addition, most reviewed articles lacked sensitivity analyses to assess the variability in results by changes in variant/gene selection strategy or a justification for the choice of criteria and thresholds adopted. For example, although restricting the analysis to rare variants is justified in principle by the fact that high-risk/high-penetrance variants are very rare in the general population, no clear justification (e.g., based on disease penetrance estimates) was usually given for the exact choice of MAF thresholds, which can impact both false positives and negatives. In addition, requiring that the selected variants be completely absent from internal or publicly available control datasets (25% of articles) may also lead to false negatives, given that known disease related variants are observed in these datasets. Moreover, differences in methodologies—such as, study design, sequencing technologies, depth of coverage, human genome reference used, annotation software, variant calling methods, in silico prediction tools—have been reported to lead to differences in findings (218–219). Similarly, although the choice of transcript set and annotation software have been quantified to have a substantial effect on variant annotation and impact on the analysis of genome sequencing studies (220), none of the reviewed articles examined or discussed these potential effects. Although this literature review spanned a decade wherein underlying technologies, costs and bioinformatic pipelines evolved significantly, we note that 74% of the reviewed articles were published in the years 2015-2018. When restricting the analyses to this group of recent papers, we observed similar results.
Importance of developing consensus on standards
The observed wide variation and inconsistencies in approaches and strategies underscore the importance of establishing a consensus on standards for filtering strategies and rationale for variant identification (e.g., justification for the criteria and thresholds used). The disadvantage of using different methodologies to identify germline susceptibility genes is that it limits the ability to compare results across studies. While initiatives by the American College of Medical Genetics and Genomics (221) and the National Institutes of Health (222) have developed standards to assign a pathogenicity status to a given variant based on the available literature and annotations, to our knowledge, there has not been an attempt to set standards or a framework for agnostic searches of susceptibility variants or genes. Based on the present systematic review, we would recommend that articles in this field: i) report information for all the relevant components described in Table 1 and Supplemental Table S2; ii) include a complete list of identified variants/genes and a count of the individuals carrying those in a format similar to Supplemental Table S3; iii) report and explain the choice of variants/genes filtering criteria and thresholds, including sensitivity analysis when warranted.
Variants and genes identified in the reviewed articles
Approximately 95% of the reviewed studies reported identifying susceptibility variants or genes in the studied cancer cases. However, this observation may reflect general publication bias. Overall, about 2,000 variants and 1,000 unique genes were reported as primary findings by the authors. Breast cancer studies reported the highest number of genes, possibly reflective of the large proportion of published studies rather than the underlying genetic architecture. Notably, one hundred genes were found in more than one article (see bolded gene symbols in Table 3), indicating that results are recurrent within each cancer type and suggestive of pleiotropic effects across cancer types. Some of these observations may also be due to chance and/or to the wide adoption of variant/gene selection approaches based on known candidate variants, genes, or pathways. Indeed, when restricting the analysis to the articles that used a fully agnostic analysis approach, we observed a decrease in the relative frequency of reporting of these genes and an increase in relative frequency for less established genes. This observation may illustrate that more novel genes could be discovered by using a more expansive analysis approach. In addition, we found that 43 variants (Table 4) were each identified in two or more articles across cancer types. The identified variants/genes accounted on average for 25% of the families evaluated in both discovery and replication, suggesting that the fraction of families explained by the genes identified through exome/genome-wide sequencing may have increased since previous linkage analysis and candidate gene sequencing results (10-25% of families depending on the cancer type (223)). The results collectively show that important progress has been made in the identification of cancer susceptibility genes and that pleiotropy is a common phenomenon in genetic cancer susceptibility. Nevertheless, the progress made to-date is not without caveats.
Challenges limiting progress in variant/gene identification
The present review reveals scientific gaps and challenges in the body of literature. Of note, many (especially rare) cancer types remain understudied (or under published) and over 75% of cancer-prone families remain unexplained. While the lack of identification of mutations for cancer in heavily loaded families could reflect a polygenic or omnigenic architecture in these families, several additional challenges may have limited further progress in identifying germline variants associated with cancer, as indicated by the suspected publication bias (95% articles reported positive findings) and by the limited number of articles identified (only 186 articles across ten years and all cancer types). First, through careful review of the literature, we observed variation in study design and case selection, even within studies. For example, many of the studies included in this review used familial cases in the discovery phase before switching to unselected cases in the replication phase (perhaps due to a lack of additional familial samples or funds), which may introduce etiological heterogeneity (e.g., familial cases may carry different and/or more penetrant variants/genes than unselected cases) and may in part explain the lack of replication for some of the reviewed studies. In addition, including suitable control populations is important to ascertain magnitude of risk, whereas the frequency of the identified variants in such controls was reported only in one third of the articles. Second, focusing exclusively on the exome (only 10 of the articles were WGS) may be a limitation in complex trait genetics for which noncoding genetic variation is believed to play a larger role than in Mendelian genetics (224, 225) – a hypothesis that still needs to be verified for rare variants specifically. A third aspect that may have limited progress is the widespread use of candidate analysis approaches that focus the discovery analysis on known variants or genes or pathways by leveraging relevant existing information to select the resulting variants/genes. Although articles that used a candidate sequencing approach were excluded, the use of candidate analysis approaches was reported in 65% of the reviewed articles. These challenges (lack of genome-wide and agnostic studies) may be due to the fact that researchers do not yet have the tools to examine agnostically the whole exome or genome effectively. Alternatively, the researchers may have had specific reasons to focus on candidate regions of interest. Whatever the origin, an important consequence should be acknowledged: much of the human exome (and genome) remains unexplored or untested for cancer. Lastly, authors frequently stated a need for additional research to replicate their findings in larger and more homogeneous (e.g., by race/ethnicity or cancer histology) study populations. Indeed, although the majority of the reviewed articles used in the discovery phase a familial study design (which does not require cancer case numbers as large as unselected case-control study designs), 39% of articles exome/genome sequenced only a single family and 26% only a single member per family. Increasing the number of sequenced families and of cancer cases within each family may be an important avenue for future studies.
Technical considerations
From a technical point of view, the differences in utilization of the various technologies are dependent on the timing of their development and subsequent replacement by the next capture kit or sequencer version. We showed that the reviewed articles reported a technical validation rate of about 80% for studies that tested over fifty variants versus over 90% for studies that tested fewer variants; the difference may be due to pre-validation manual inspection steps (e.g., through IGV) being more feasibly applied to a limited number of variants. This observation suggests the importance for researchers using current sequencing technologies at a genome-wide scale to technically validate any observed variants. Another technical limitation stems from within study aggregation of samples across multiple sequencing experiments, as this approach can generate biases in variant detection and false positives/negatives in variant-cancer associations, particularly for WES datasets which vary also in capture efficiency. Several strategies to control for biological and technical heterogeneity and to minimize calling discordance and erroneous findings were described in the reviewed articles, including checking for comparable depths and rare variants detection and performing alignment and variant calling of all samples simultaneously. One notable point, reinforced by the observed lack of data sharing for over 80% of the reviewed articles, is the importance of saving, storing and being able to access BAM or CRAM files of the studied datasets for the wider research community, both for publicly and privately sponsored datasets. Advancements in long range sequencing and other new technologies may also help address some of the described technical shortcomings in the future and influence near term approaches to genomic analyses (226).
Importance of functional validation
Finally, most reviewed articles acknowledged the importance of functional validation (e.g., through in vitro and in vivo models) to determine whether the function of the mutated gene product is consistent with the cancer of interest and to inform the interpretation of the reported findings. Even though 65% of the articles did attempt some type of experimental validation for the final selection of variants/genes, the reported functional results were usually not considered definitive by the authors. In fact, most articles described the need for additional functional studies to determine whether the identified genes or variants play a causal role in carcinogenesis and to describe the mechanisms for these variants to impact disease.
Limitation and strengths of this literature review
Limitations of our literature review include: the lack of access to primary data, and consequent inability to systematically evaluate how different filtering choices would lead to different results in these studies; likely publication bias towards non-null results; use of the authors definition of “identified gene or variant” which varied greatly across the reviewed articles; the exclusion of articles in which only a single cancer case was sequenced, which although usually case reports, can also in principle lead to the identification of novel cancer susceptibility genes (e.g., PALB2 (227), or NPAT (228)). Strengths of our review are the systematic inclusion/exclusion approach, the comprehensive key term search, and thorough data abstraction.
Conclusions
In conclusion, the findings from this review indicate a growth in usage of NGS technologies at the exome/genome scale to identify genes associated with cancer risk. Nevertheless, progress has been limited by a range of challenges inherent in the field. The review highlights several important next steps including establishing consensus on standards for use and reporting of filtering strategies, describing rationale for variant identification, developing analytical methods that truly mine the whole exome/genome, improving the accuracy and cross-studies interoperability of current sequencing technologies, sharing of the primary data with the research community, and performing extensive variant functional validation. It also points to the untapped potential in conducting studies with more/larger families and in more diverse populations and cancers types, harmonizing results across studies, and expanding searches beyond a candidate analysis approach.
Supplementary Material
Financial Support:
This research was supported by the National Cancer Institute, National Institutes of Health at the Division of Cancer Control and Populations Sciences, the Intramural Research Program of the Division of Cancer Epidemiology and Genetics, and the Scientific Consulting Group [N.I.S.] (contract number HHSN261201400011I).
Footnotes
Disclosure of Potential Conflicts of Interest: The authors have no conflicts to disclose.
References
- 1.Meztker ML. Sequencing technologies—the next generation. Nat Rev Genet 2010; 11:31–46. [DOI] [PubMed] [Google Scholar]
- 2.Levy SE, Myers RM. Advancements in next-generation sequencing. Annu Rev Genom Hum Genet 2016; 17:95–115. Doi: 10/1146/annurev-genom-083115-022413. [DOI] [PubMed] [Google Scholar]
- 3.Bertier G, Hetu M, Joly Y. Unsolved challenges of clinical whole-exome sequencing: a systematic literature review of end-users’ views. BMC Med Genomics 2016; 9:52 doi: 10.1186/s12920-016-0213-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stadler ZK, Schrader KA, Vijai J, Robson ME, Offit K. Cancer genomics and inherited risk. J Clin Oncol 2014; 32:687–698. Doi: 10.1200/JCO.2013.49.7271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexandrov LB, Stratton MR. Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr Opin Genet Dev 2014; 24:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rabbani B, Mahdieh N, Hosomichi K, Nakaoka H, Inoue I. Next-generation sequencing: impact of exome sequencing in characterizing Mendelian disorders. J Hum Genet 2012; 10:621–632. Doi: 10.1038/jhg.2012.91. [DOI] [PubMed] [Google Scholar]
- 7.Gilissen C, Hoischen A, Brunner HG, Veltman JA. Disease gene identification strategies for exome sequencing. Eur J Hum Genet 2012; 20:490–497. Doi: 10.1038/ejhg.2011.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alexandrov LB, Nik-Zianal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013; 500:415–421. Doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet 2014; 15:585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mucci LA, Hjelmborg JB, Harris JR, Czene K, Havelick DJ, Scheike T, et al. , Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 2016; 315(1):68–76. Doi: 10.1001/jama.2015.17703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Susswein LR, Marshall ML, Nusbaum R, Vogel Postula KJ, Weissman SM, Yackowski L, et al. Genet Med 2016, 18(8):823–32. doi: 10.1038/gim.2015.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009. October 8;461(7265):747–53. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group (2009) Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med 6(7): e1000097. 10.1371/journal.pmed.1000097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H and Durbin R Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. GENOME RESEARCH 2010. 20:1297–303. Doi: 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data Nucleic Acids Research, 38:e164, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.The 1000 Genomes Project Consortium. A global reference for human genetic variation, Nature 2015526, 68–74; doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu W, O’Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 2012, 493, 216–220. Doi: 10.1038/nature11690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001. January 1;29(1):308–11. Doi: 10.1093/nar/29.1.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lek M, Karczewski KJ, and the Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536: 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative Genomics Viewer. Nature Biotechnology 2011; 29: 24–26. Doi: 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Research 2012; 40: W542–7. Doi: 10.1093/nar/gks539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7(4):248–249. Doi: 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014;11(4):361–2. Doi: 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- 25.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ward LD and Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Research 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Research 2012, 22(9):1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: Protein structure and function prediction. Nature Methods 2015; 12: 7–8. Doi: 10.1038/nmeth.3213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Webb B, Sali A. Comparative Protein Structure Modeling Using Modeller. Current Protocols in Bioinformatics 2016; 54: 5.6.1–5.6.37. doi: 10.1002/cpbi.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014. January;42(Database issue):D980–5. doi: 10.1093/nar/gkt1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKusick VA Mendelian Inheritance in Man. A Catalog of Human Genes and Genetic Disorders. Baltimore: Johns Hopkins University Press, 1998. (12th edition). [Google Scholar]
- 32.Wardell CP, Fujita M, Yamada T, Simbolo M, Fassan M, Karlic R, et al. Genomic characterization of biliary tract cancers identified driver genes and predisposing mutations. J Hepatol 2018; 68:959–969. Doi: do.1016/j.hep2018.01.009 [DOI] [PubMed] [Google Scholar]
- 33.Chang VY, Basso G, Sakamoto KM, Nelson SF. Identification of somatic and germline mutations using whole exome sequencing of congenital acute lymphoblastic leukemia. BMC Cancer 2013; 13:55. Doi: 10.1186/1471-2407-13-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sabri S, Keyhani M, Akbari MT. Whole exome sequencing of chronic myeloid leukemia patients. Iran J Public Health 2016; 45:346–352. [PMC free article] [PubMed] [Google Scholar]
- 35.Ristolainen H, Kilpivaara O, Kamper P, Taskinen M, Sarrinen S, Leppa S, et al. Identification of homozygous deletion in ACAN and other candidate variants in familial classical Hodgkin lymphoma by exome sequencing. Br J Haematol 2015; 170:428–431. Doi: 10.111/bjh.13295 [DOI] [PubMed] [Google Scholar]
- 36.Tiao G, Improgo MR, Kasar S, Poh W, Kamburov A, Landau DA, et al. Rare germline variants in ATM are associated with chronic lymphocytic leukemia. Leukemia 2017; 31:2244–2247. Doi: 10.1038/leu.2017.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pathak A, Pemov A, McMaster ML, Dewan R, Ravichandran S, Pak E, et al. Juvenile myelomonocytic leukemia due to a germline CBL Y371C mutation: 35-year follow-up of a large family. Hum Genet 2015; 137:775–787. Doi: 10.1007/s00438-015-1550-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pathak A, Seipel K, Pemov A, Dewan R, Brown C, Ravichandran S, et al. Whole exome sequencing reveals a C-terminal germline variant in CEBPA-associated acute myeloid leukemia: 45-year follow up of a large family. Haematologica 2016; 101:846–852. Doi: 10.3342/haematol.2015.130799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moshous D, Martin E, Carpentier W, Lim A, Callebaut I, Canioni D, et al. Whole-exome sequencing identified Coronin-1A deficiency in 3 siblings with immunodeficiency and EBV-associated B-cell lymphoproliferation. J Allergy Clin Immunol 2013; 13: 1594–1603. Doi: 10.1016/j.jaci.2013.01.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bandapali OR, Paramasivam N, Giangiobbe S, Kumar A, Benisch W, Engert A, et al. Whole genome sequencing reveals DICER1 as a candidate predisposing gene in familial Hodgkin lymphoma. Int J Cancer 2018; 143:2076–2078. Doi: 10.1002.ijc.31576 [DOI] [PubMed] [Google Scholar]
- 41.Moriyama T, Metzger ML, Wu G, Nishii R, Qian M, Devidas M, et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: a systematic genetic study. Lancet Oncol 2015. 16:1659–1666. Doi: 10.1016/S1470-2045(15)00361-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Daschkey S, Bienemann K, Schuster V, Kreth HW, Linka RM, Honscheid A, et al. Fatal lymphoproliferative disease in two siblings lacking functional FAAP24. J Clin Immunol 2016; 36:684–692. Doi: 10.1007/s10875-016-0317-y [DOI] [PubMed] [Google Scholar]
- 43.Gayden T, Sepulveda FE, Khuong-Quang DA, Pratt J, Valera ET, Garrigue A, et al. Germline HAVCR2 mutations altering TIM-3 characterize subcutaneous panniculitis-like T cell lymphomas with hemaphagocytic lymphohistiocytic syndrome. Nat Genet 2018; 50:1650–1657. Doi: 10.1038/s41588-018-0251-4 [DOI] [PubMed] [Google Scholar]
- 44.Goldin LR, McMaster ML, Rotunno M, Herman SE, Jones K, Zhu B, et al. Whole exome sequencing in families with CLL detects a variant in Integrin β 2 associated with disease susceptibility. Blood 2016; 128: 2261–2263. Doi: 10.1182/blood-2016-02-697771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rotunno M, McMaster ML, Boland J, Bass S, Zhang X, Burdett L, et al. Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene. Haematologica 2016; 101:853–860. Doi: 10.3324/haematol.2015.135475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saarinen S, Kaasinen E, Karjalainen-Lindsberg ML, Vesanen K, Aavikko M, Katainen R, et al. Primary mediastinal large B-cell lymphoma segregating in a family: exome sequencing identified MLL as a candidate predisposition gene. Blood 2013; 121:3428–3430. Doi: 10.1182/blood-2012-06-437210 [DOI] [PubMed] [Google Scholar]
- 47.Shah S, Schrader KA, Waanders E, Timms AE, Vijai J, Meithing C, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet 2013; 45:1226–1231. Doi: 10.1038/ng.2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McMaster ML, Sun C, Landi MT, Savage SA, Rotunno M, Yang XR, et al. Germline mutations in protection of telomeres 1 in two families with Hodgkin lymphoma. Br J Haematol 2018; 181:372–377. Doi: 10.1111/bjh.15203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hussin J, Sinnett D, Casals F, Idaghdour Y, Bruat V, Saillour V, et al. Rare allelic forms of PRDM9 associated with childhood leukemogenesis. Genome Res 2013; 23:419–430. Doi: 10.1101/gr.144188.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Powell BC, Jiang L, Mazny DM, Treviño LR, Dreyer ZE, Strong LC, et al. Identification of TP53 as an acute lymphocytic leukemia susceptibility gene through exome sequencing. Pediatr Blood Cancer 2013; 60:E1–E3. Doi: 10.1002/pbc.24417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waanders E, Scheijen B, Jongmans MC, Venselaar H, van Reijmersdal SV, van Dijk AH, et al. Germline activating TYK2 mutations in pediatric patients with two primary acute lymphoblastic leukemia occurrences. Leukemia 2017; 31:821–828. Doi: 10.1038/leu.2016.277 [DOI] [PubMed] [Google Scholar]
- 52.Roccaro AM, Sacco A, Shi J, Chiarini M, Perilla-Glen A, Manier S, et al. Exome sequencing reveals recurrent germ line variants in patients with familial Waldenström macroglobulinemia. Blood 2016; 127:2598–2606. Doi: 10.1182/blood-2015-11-680199 [DOI] [PubMed] [Google Scholar]
- 53.Lawrie A, Han S, Sud A, Hosking F, Cezard T, Turner D, et al. Combined linkage and association analysis of classical Hodgkin lymphoma. Oncotarget 2018; 9:20377–20385. Doi: 10.18632/oncotarget.24872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Speedy HE, Kinnersley B, Chubb D, Broderick P, Law PJ, Litchfield K, et al. Germ line mutations in shelterin complex genes are associated with familial chronic lymphocytic leukemia. Blood 2016; 128:2319–2326. Doi: 10.1182/blood-2016-01-695692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hirvonen EAM, Pitkänen E, Hemminki K, Aaltonen LA, Kilpivaara O. Whole-exome sequencing identifies novel candidate predisposition genes for familial polycythemia vera. Hum Genomics 2017; 11:6. Doi: 10.1186/s40246-017-0102-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spinella JF, Healy J, Saillour V, Richer C, Cassart P, Ouimet M, et al. Whole-exome sequencing of a rare case of familial childhood acute lymphoblastic leukemia reveals putative predisposing mutations in Fanconi anemia genes. BMC Cancer 2015; 15:539. Doi: 10.1186/s12885-015-1549-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Donner I, Katainen R, Kaasinen E, Aavikko M, Sipilä LJ, Pukkala E, et al. Candidate susceptibility variants in angioimmunoblastic T-cell lymphoma. Fam Cancer 2019; 18:113–119. Doi: 10.1107/s10689-018-0099-x [DOI] [PubMed] [Google Scholar]
- 58.Valentine MC, Linabery AM, Chasnoff S, Hughes AE, Mallaney C, Sanchez N, et al. Excess congenital non-synonymous variation in leukemia-associated genes in MLL-infant leukemia: a Children’s Oncology Group report. Leukemia 2014; 28:1235–1241. Doi: 10.1038/leu.2013.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Al-Dewik N, Ben-Omran T, Zayed H, Trujillano D, Kishore S, Rolfs A, et al. Clinical exome sequencing unravels new disease-causing mutations in the myeloproliferative neoplasms: A pilot study in patients from the state of Qatar. Gene 2019; 689:34–42. Doi: 10.1016/j.gene.2018/12.009 [DOI] [PubMed] [Google Scholar]
- 60.Andreeva TV, Tyazhelova TV, Rykalina VN, Gusev FE, Goltsov AY, Zolotareva OI, et al. Whole exome sequencing links dental tumor to an autosomal-dominant mutation in ANO5 gene associated with gnathodiaphyseal dysplasia and muscle dystrophies. Sci Rep 2016; 6:26440. Doi: 10.1038/srep26440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heddar A, Fermey P, Coutant S, Angot E, Sabourin JC, Michelin P, et al. Familial solitary chondrosarcoma resulting from germline EXT2 mutation. Genes Chromosomes Cancer 2017; 56:1280134. Doi: 10.1002/gcc.22419 [DOI] [PubMed] [Google Scholar]
- 62.Pillay N, Plagnol V, Tarpey PS, Lobo SB, Presneau N, Szuhai K, et al. A common single-nucleotide variant in T is strongly associated with chordoma. Nat Genet 2012; 44:1185–1187. Doi: 10.1038/ng.2419 [DOI] [PubMed] [Google Scholar]
- 63.Ronellenfitsch MW, Oh JE, Satomi K, Sumi K, Harter PN, Steinbach JP, et al. CASP9 germline mutation in a family with multiple brain tumors. Brain Pathol 2018; 28:94–102. Doi: 10.1111/bpa.12471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Mariano M, Gallesio R, Chierici M, Furlanello C, Conte M, Garaventa A, et al. Identification of GALNT14 as a novel neuroblastoma predisposition gene. Oncotarget 2015; 6:26335–26346. Doi: 10.18632/oncotarget.4501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang L, Yamaguchi S, Burstein MD, Terashima K, Chang K, Ng HK, et al. Novel somatic and germline mutations in intracranial germ cell tumours. Nature 2014; 511:241–245. Doi: 10.1038/nature13296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Andrianova MA, Chetan GK, Sibin MK, McKee T, Merkler D, Narasinga RK, et al. Germline PMS2 and somatic POLE exonuclease mutations cause hypermutability of the leading DNA strand in biallelic mismatch repair deficiency syndrome brain tumours. J Pathol 2017; 243:331–341. Doi: 10.1002/path.4957 [DOI] [PubMed] [Google Scholar]
- 67.Bainbridge MN, Armstrong GN, Gramatges MM, Bertuch AA, Jhangiani SN, Doddapaneni H, et al. Germline mutations in shelterin complex genes are associated with familial glioma. J Natl Cancer Inst 2014; 107:384. Doi: 10.1093/jnci/dju384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Smith MJ, O’Sullivan J, Bhaskar SS, Hadfield KD, Poke G, Caird J, et al. Loss-of-function mutations in SMARCE1 cause an inherited disorder of multiple spinal meningiomas. Nat Genet 2013; 45:295–298. Doi: 10.1038/ng.2552 [DOI] [PubMed] [Google Scholar]
- 69.Aavikko M, Li SP, Saarinen S, Alhopuro P, Kaasinen E, Morgunova E, et al. Loss of SUFU function in familial multiple meningioma. Am J Hum Genet 2012; 91:520–526. Doi: 10.1016/j.ajhg.2012.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nordfors K, Haapasalo J, Afyounian E, Tuominen J, Annala M, Häyrynen S, et al. Whole-exome sequencing identifies germline mutation in TP53 and ATRX in a child with genomically aberrant AT/RT and her mother with anaplastic astrocytoma. Cold Spring Harb Mol Case Stud. 2018; 4(2). Doi: 10.1101/mcs.a002246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim YH, Ohta T, Oh JE, Le Calvez-Kelm F, McKay J, Voegele C, et al. TP53, MSH4, and LATS1 germline mutations in a family with clustering of nervous system tumors. Am J Pathol 2014; 184:2374–2381. Doi: 10.1016/j.ajpath.2014.05.017 [DOI] [PubMed] [Google Scholar]
- 72.Backes C, Harz C, Fischer U, Schmitt J, Ludwing N, Petersen BS, et al. New insights into the genetics of glioblastoma multiforme by familial exome sequencing. Oncotarget 2015; 6:5918–5931. Doi: 10.18632/oncotarget.2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Park DJ, Odefrey FA, Hammet F, Giles GG, Baglietto L, Hopper JL, et al. FAN1 variants identified in multiple-case early-onset breast cancer families via exome sequencing: no evidence for association with risk for breast cancer. Breast Cancer Res Treat 2011; 130:1043–1049. Doi: 10.1107/s10549-011-1704-y [DOI] [PubMed] [Google Scholar]
- 74.Hilbers FS, Meijers CM, Laros JF, van Galen M, Hoogerbrugge N, Vasen HF, et al. Exome sequencing of germline DNA from non-BRCA1/2 familial breast cancer cases selected on basis of aCGH tumor profiling. PLoS One 2013; 8:e55732. Doi: 10.1371/journal.pone.0055734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Radmanesh H, Spethmann T, Enben J, Schümann P, Bhuju S, Geffers R, et al. Assessment of an APOBEC3B truncating mutation, c.783del G, in patients with breast cancer. Breast Cancer Res Treat 2017; 162:31–37. Doi: 10.1007/s10549-016-4100-9 [DOI] [PubMed] [Google Scholar]
- 76.Tavera-Tapia A, Pérez-Cabornero L, Macías JA, Ceballos MI, Roncador G, de la Hoya M, et al. Almost 2% of Spanish breast cancer families are associated to germline pathogenic mutations in the ATM gene. Breast Cancer Res Treat 2017; 161: 597–604. Doi: 10.1007/s10549-016-40580-7. [DOI] [PubMed] [Google Scholar]
- 77.Vijai J, Topka S, Villano D, Ravichandran V, Maxwell KN, Maria A, et al. A recurrent ERCC3 truncating mutation confers moderate risk for breast cancer. Cancer Discov 2016; 6:1267–1275. Doi: 10.1158/2159-8290.CD-16-0487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kiiski JI, Pelttari LM, Khan S, Frysteinsdottir ES, Reynisdottir I, Hart SN, et al. Exome sequencing identified FANCM as a susceptibility gene for triple-negative breast cancer. Proc Natl Acad Sci USA 2014; 111:15172–15177. Doi: 10.1073/pnas.1407909111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sokolenko AP, Bulanova DR, Iyevleva AG, Aleksakhina SN, Preobrazhenskaya EV, Ivanstov AO, et al. High prevalence of GPRC5A germline mutations in BRCA1-mutant breast cancer patients. Int J Cancer 2014; 134:2352–2358. Doi: 10.1002/ijc.28569 [DOI] [PubMed] [Google Scholar]
- 80.Lynch H, Wen H, Kim YC, Snyder C, Kinarsky Y, Chen PX, et al. Can unknown predisposition in familial breast cancer be family-specific? Breast J 2013; 19:520–528. Doi: 10.1111/tbj.12145 [DOI] [PubMed] [Google Scholar]
- 81.Silvestri V, Zelli V, Valentini V, Rizzolo P, Navazio AS, Coppa A, et al. Whole-exome sequencing and targeted gene sequencing provide insights into the role of PALB2 as a male breast cancer susceptibility gene. Cancer 2017; 123: 210–218. Doi: 10.1002/cncr.30337 [DOI] [PubMed] [Google Scholar]
- 82.Riahi A, Radmanesh H, Schümann P, Bogdanova N, Geffers R, Meddeb R, et al. Exome sequencing and case-control analyses identify RCC1 as a candidate breast cancer susceptibility gene. Int J Cancer 2018; 142:2512–2517. Doi: 10.1002/ijc.31273 [DOI] [PubMed] [Google Scholar]
- 83.Cybulski C, Carrot-Zhang J, Kluźniak W, Rivera B, Kashyap A, Wokolorczyk D, et al. Germline RECQL mutations are associated with breast cancer susceptibility. Nat Genet 2015; 47:643–646. Doi: 10.1038/ng.3284 [DOI] [PubMed] [Google Scholar]
- 84.Sun J, Wang Y, Xia Y, Xu Y, Ouyang T, Li J, et al. Mutations in RECQL gene are associated with predisposition to breast cancer. PLoS Genet 2015; 11:e1005228. Doi: 10.1372/journal.pgen.1005228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Park DJ, Tao K, Le Calvez-Kelm F, Nguyen-Dumont T, Robinot N, Hammet F, et al. Rare mutations in RINT1 predispose carriers to breast and Lynch syndrome-spectrum cancers. Cancer Discov 2014; 4:804–815. Doi: 10.1158/2159-8290.CD-14-0212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nguyen-Dumont T, Teo ZL, Hammet F, Roberge A, Mahmoodi M, Tsimiklis H, et al. Is RNASEL:p.Glu265* a modifier of early-onset breast cancer risk for carriers of high-risk mutations? BMC Cancer 2018; 18:165. Doi: 10.1186/s12885-018-4028-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Park DJ, Lesueur F, Nguyen-Dumont T, Pertesi M, Odefrey F, Hammet F, et al. Rare mutations in XRCC2 increase the risk of breast cancer. Am J Hum Genet 2012; 90:734–739. Doi: 10.1016/j.ajhg.2012.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ataei-Kachouei M, Nadaf J, Akbari MT, Atri M, Majewski J, Riazalhosseini Y, et al. Double heterozygosity of BRCA2 and STK11 in familial breast cancer detected by exome sequencing. Iran J Public Health 2015; 44:1348–1352. [PMC free article] [PubMed] [Google Scholar]
- 89.Thompson ER, Doyle MA, Ryland GL, Rowley SM, Choong DY, Tothill RW, et al. Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet 2012; 8:31002894. Doi: 10.1371/journal.pgen.1002894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Merdad A, Gari MA, Hussein S, Al-Khayat S, Tashkandi H, Al-Maghrabi J, et al. Characterization of familial breast cancer in Saudi Arabia. BMC Genomics 2015; 16 Suppl 1:S3. Doi: 10.1186/1471-2164-16-S1-S3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guo X, Shi J, Cai Q, Shu XO, He J, Wen W, et al. Use of deep whole-genome sequencing data to identify structure risk variants in breast cancer susceptibility genes. Hum Mol Genet 2018; 27:853–859. Doi: 10.1093/hmg/ddy005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Noh JM, Kim J, Cho DY, Choi DH, Park W, Huh SJ. Exome sequencing in a breast cancer family without BRCA mutation. Radiat Oncol J 2015; 33:149–154. Doi: 10.3857/roj/2015.33.2.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cybulski C, Lubiński J, Wokolorczyk D, Kuźniak W, Kashyap A, Sopik V, et al. Mutations predisposing to breast cancer in 12 candidate genes in breast cancer patients from Poland. Clin Genet 2015; 88:366–370. Doi: 10.1111/cge.12524 [DOI] [PubMed] [Google Scholar]
- 94.Garcia-Aznarez FJ, Fernandez V, Pita G, Peterlongo P, Dominguez O, de la Hoya M, et al. Whole exome sequencing suggests much of non-BRCA1/BRCA2 familial breast cancer is due to moderate and low penetrance susceptibility alleles. PLoS One 2013; 8:e55681. Doi: 10.1371/journal.pone.0055681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hamdi Y, Boujemaa M, Ben Rekaya M, Ben Hamda C, Mighri N, El Benna H, et al. Family specific genetic predisposition to breast cancer: results from Tunisian whole exome sequenced breast cancer cases. J Transl Med 2018; 16:158. Doi: 10.1186/s12967-018-1504-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Torrezan GT, de Almeida FGDSR, Figueiredo MCP, Barros BDF, de Paula CAA, Valieris R, et al. Complex landscape of germline variants in Brazilian patients with hereditary and early onset breast cancer. Front Genet 2018; 9:161. Doi: 10.3389/fgene.2018.00161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jalkh N, Chouery E, Haidar Z, Khater C, Atallah D, Ali H, et al. Next-generation sequencing in familial breast cancer patients from Lebanon. BMC Med Genomics 2017; 10:8. Doi: 10.1186/s12920-017-0244-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kim YC, Soliman AS, Cui J, Ramadan M, Hablas A, Abouelhoda M, et al. Unique features of germline variation in five Egyptian familial breast cancer families revealed by exome sequencing. PLoS One 2017; 12:E0167581. Doi: 10.1371/journal.pone.0167581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wen H, Kim YC, Snyder C, Xiao F, Fleissner EA, Becirovic D, et al. Family-specific, novel, deleterious germline variants provide a rich resource to identify genetic predispositions for BRCAx familial breast cancer. BMC Cancer 2014; 14:470. Doi. 10.1186/1471-2407-14-470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Snape K, Rurak E, Tarpey P, Renwick A, Turnbull C, Seal S, et al. Predisposition gene identification in common cancers by exome sequencing: insights from familial breast cancer. Breast Cancer Res Treat 2012; 134:429–433. Doi: 10.1007/s10549-012-2057-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Piccolo SR, Hoffman LM, Conner T, Shrestha G, Cohen AL, Marks JR, et al. Integrative analyses reveal signaling pathways underlying familial breast cancer susceptibility. Mol Syst Biol 2016; 12:860. Doi: 10.15252/msb.20156506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bellido F, Sowada N, Mur P, Lázaro C, Pons T, Valdés-Mas R, et al. Association between germline mutations in BRF1, a subunit of the RNA polymerase III transcription complex, and hereditary colorectal cancer. Gastroenterology 2018; 154: 181–194. Doi: 10.1053/j.gastro.2017.09.005 [DOI] [PubMed] [Google Scholar]
- 103.Seguí N, Mina LB, Lázaro C, Sanz-Pamplona R, Pons T, Navarro M, et al. Germline mutations in FAN1 cause hereditary colorectal cancer by impairing DNA repair. Gastroenterology 2015; 149:563–566. Doi: 10.1053/j.gastro.2015.05.056 [DOI] [PubMed] [Google Scholar]
- 104.Seguí N, Navarro M, Pineda M, Köger N, Bellido F, González S, et al. Exome sequencing identified MUTYH mutations in a family with colorectal cancer and an atypical phenotype. Gut 2015. 64:355–356. Doi: 10.1136/gutjnl-2014-307084 [DOI] [PubMed] [Google Scholar]
- 105.Nieminen TT, O’Donohue MF, Wu Y, Lohi H, Scherer SW, Paterson AD, et al. Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology 2014; 147:595–598. Doi: 10.1053/j.gastro.2014.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Martín-Morales L, Feldman M, Vershinin Z, Garre P, Caldés T, Levy D. SETD6 dominant negative mutation in familial colorectal cancer type X. Hum Mol Genet 2017; 26:4481–4493. Doi: 10.1093/hmg/ddx336 [DOI] [PubMed] [Google Scholar]
- 107.DeRycke MS, Gunawardena SR, Middha S, Asmann YW, Schaid DJ, McDonnell SK, et al. Identification of novel variants in colorectal cancer families by high-throughput exome sequencing. Cancer Epidemiol Biomarkers Prev 2013; 22:1239–1251. Doi: 10.1158/1055-9965.EPI-12-1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Palles C, Cazier JG, Howarth KM, Domingo E, Jones AM, Broderick P, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 2013; 45:136–144. Doi: 10.1038/ng.2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.109Arora S, Yan H, Cho I, Fan HY, Luo B, Gai X, et al. Genetic variants that predispose to DNA double-strand breaks in lymphocytes from a subset of patients with familial colorectal carcinomas. Gastroenterology 2015; 149:1872–1883. Doi: 10.1053/j.gastro.2015.08.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.de Voer RM, Hahn MM, Weren RD, Mensenkamp AR, Gillissen C, van Zelst-Stams WA, et al. Identification of novel candidate genes for early-onset colorectal cancer susceptibility. PLoS Genet 2016; 12:31005880. Doi: 10.1371/journal.pgen.1005880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Franch-Expósito S, Esteban-Jurado C, Garre P, Quintanilla I, Duran-Sanchon S, Díaz-Gay M, et al. Rare germline copy number variants in colorectal cancer predisposition characterized by exome sequencing analysis. J Genet Genomics 2018; 45:41–45. Doi: 10.1016/j.jgg.2017.12.001 [DOI] [PubMed] [Google Scholar]
- 112.Esteban-Jurado C, Franch-Expósito S, Muñoz J, Ocaña T, Carballal S, López-Cerón M, et al. The Fanconi anemia DNA damage repair pathway in the spotlight for germline predisposition to colorectal cancer. Eur J Hum Genet 2016; 24:1501–1505. Doi: 10.1038/ejhg.2016.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Smith CG, Naven M, Harris R, Colley J, West H, Li N, et al. Exome resequencing identifies potential tumor-suppressor genes that predispose to colorectal cancer. Hum Mutat 2013; 34: 1026–1034. Doi: 10.1002/humu.22333 [DOI] [PubMed] [Google Scholar]
- 114.Chubb D, Broderick P, Dobbins SE, Frampton M, Kinnersley B, Penegar S, et al. Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat Commun 2016; 7:11883. Doi: 10.1038/ncomms11883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Esteban-Jurado C, Vila-Casadesús M, Garre P, Lozano JJ, Pristoupilova A, Beltran S, et al. Whole-exome sequencing identified rare pathogenic variants in new predisposition genes for familial colorectal cancer. Genet Med 2015; 17: 131–42. Doi: 10.1038/gim.2014.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tanskanen T, Gylfe AE, Katainen R, Taipale M, Renkonen-Sinisalo L, Järvinen H, et al. Systematic search for rare variants in Finnish early-onset colorectal cancer patients. Cancer Genet 2015. 208:35–40. Doi: 10.1016/j.cancergen.2014.12.004 [DOI] [PubMed] [Google Scholar]
- 117.Gylfe AE, Katainen R, Kondelin J, Tanskanen T, Cajuso T, Hänninen U, et al. Eleven candidate susceptibility genes for common familial colorectal cancer. PLoS Genet 2013; 9:e1003876. Doi: 10.1371/journal.pgen.1003876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yu L, Yin B, Qu K, Li J, Jin Q, Liu L, et al. Screening for susceptibility genes in hereditary non-polyposis colorectal cancer. Oncol Lett 2018; 15:9413–9419. Doi: 10.3892/ol.2018.8504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang JX, Fu L, de Voer RM, Hahn MM, Jin P, Lv CX, et al. Candidate colorectal cancer predisposing gene variants in Chinese early-onset and familial cases. World J Gastroenterol 2015; 21:4136–4149. Doi: 10.3748/wjg.v21.i14.4136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Khalilipour N, Baranova A, Jebelli A, Heravi-Moussavi A, Bruskin S, Abbaszadegan MR. Familial esophageal squamous cell carcinoma with damaging rare/germline mutations in KCNJ12/KCNJ18 and GPRIN2 genes. Cancer Genet 2018; 221:46–52. Doi: 10.1016/j.cancergen.2017.11.011 [DOI] [PubMed] [Google Scholar]
- 121.Sherman SK, Maxwell JE, Qian Q, Bellizzi AM, Braun TA, Iannettoni MD, et al. Esophageal cancer in a family with hamartomatous tumors and germline PTEN frameshift and SMAD7 missense mutations. Cancer Genet 2015; 208: 41–46. Doi: 10.1016/j.cancergen.2014.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Donner I, Katainen R, Tanskanen T, Kaasinen E, Aavikko M, Ovaska K, et al. Candidate susceptibility variants for esophageal squamous cell carcinoma. Genes Chromosomes Cancer. 2017; 56:453–459. Doi: 10.1002/gcc.22448 [DOI] [PubMed] [Google Scholar]
- 123.Forouzanfar N, Baranova A, Milanizadeh S, Heravi-Moussavi A, Jebelli A, Abbaszadegan MR. Novel candidate genes may be possible predisposing factors revealed by whole exome sequencing in familial esophageal squamous cell carcinoma. Tumour Biol 2017; 39. Doi: 10.1177/1010428317699115 [DOI] [PubMed] [Google Scholar]
- 124.Vogelaar IP, van der Post RS, van Krieken JHJ, Spruijt L, van Zelst-Stams WA, Kets CM, et al. Unraveling genetic predisposition to familial or early onset gastric cancer using germline whole-exome sequencing. Eur J Hum Genet 2017; 25:1246–1252. Doi: 10.1038/ejhg.2017.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Calvete O, Reyes J, Zuñiga S, Paumard-Hernández B, Fernández V, Bujanda L, et al. Exome sequencing identifies ATP4A gene as responsible of an atypical familial type I gastric neuroendocrine tumour. Hum Mol Genet 2015; 24:2914–2922. Doi: 10.1093/hmg/ddv054 [DOI] [PubMed] [Google Scholar]
- 126.Majewski IJ, Kluijt I, Cats A, Scerri TS, de Jong D, Kluin RJ, et al. An α-E-catenin (CTNNA1) mutation in hereditary diffuse gastric cancer. J Pathol 2013; 229:621–629. Doi: 10.1002/path.4152 [DOI] [PubMed] [Google Scholar]
- 127.Donner I, Kiviluoto T, Ristimäki A, Aaltonen LA, Vahteristo P. Exome sequencing reveals three novel candidate predisposition genes for diffuse gastric cancer. Fam Cancer 2015; 14:241–246. Doi: 10.1007/s10689-015-9778-z [DOI] [PubMed] [Google Scholar]
- 128.Sahasrabudhe R, Lott P, Bohorquez M, Toal T, Estrada AP, Suarez JJ, et al. Germline mutations in PALB2, BRCA1, and RAD1C, which regulate DNA recombination repair, in patients with gastric cancer. Gastroenterology 2017; 152:983–986.e6. doi: 10.1053/j.gastro.2016.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fewings E, Larionov A, Redman J, Goldgraben MA, Scarth J, Richardson S, et al. Germline pathogenic variants in PALB2 and other cancer-predisposing genes in families with hereditary diffuse gastric cancer without CHD1 mutation: a whole-exome sequencing study. Lancet Gastrolenterol Hepatol 2018; 3:489–498. Doi: 10.1016//s2468-1253(18)30079-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Thutkawkorapin J, Picelli S, Kontham V, Liu T, Nilsson D, Lindblom A. Exome sequencing in one family with gastric and rectal cancer. BMC Genet 2017; 17:41. Doi: 10.1186/s12863-016-0351-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Comino-Méndez I, Garcia-Aznárez FJ, Schiavi F, Landa I, Leandro-Garcia LJ, Letón R, et al. Exome sequencing identified MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet 2011; 43:663–667. Doi: 10.1038/ng.861 [DOI] [PubMed] [Google Scholar]
- 132.Cao M, Sun F, Huang X, Dai J, Cui B, Ning G. Analysis of the inheritance pattern of a Chinese family with phaeochromocytomas through whole exome sequencing. Gene 2013; 526:164–169. Doi: 10.1016/j.gene.2013.04.081 [DOI] [PubMed] [Google Scholar]
- 133.Channir HI, van Overeem Hansen T, Andreasen S, Yde CW, Kiss K, Charabi BW. Genetic characterization of adenoid cystic carcinoma of the minor salivary glands: A potential familial occurrence in first-degree relatives. Head Neck Pathol 2017; 11:546–551. Doi: 10.1007/s12105-017-0801-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sasaki MM, Slol AD, Bao R, Rhodes LV, Chambers R, Vokes EE, et al. Integrated genomic analysis suggests MLL3 is a novel candidate susceptibility gene for familial nasopharyngeal carcinoma. Cancer Epidemiol Biomarkers Prev 24:1222–1228. Doi: 10.1158/1055-9965.EPI-15-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dai W, Zheng H, Cheung AK, Tang CS, Ko JM, Wong BW, et al. Whole-exome sequencing identified MST1R as a genetic susceptibility gene in nasopharyngeal carcinoma. Proc Natl Acad Sci USA 2016; 113:3317–3322. Doi: 10.1073/pnas.1523436113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Das R, Kundu S, Laskar S, Choudhury Y, Ghosh SK. Assessment of DNA repair susceptibility genes identified by whole exome sequencing in head and neck cancer. DNA Repair (Amst) 2018; 66-67:50–63. Doi: 10.1016/j.dnarep.2018.04.005 [DOI] [PubMed] [Google Scholar]
- 137.Sei Y, Zhao X, Forbes J, Szymczak S, Li Q, Trivedi A, et al. A hereditary form of small intestinal carcinoid associated with a germline mutation in inositol polyphosphate multikinase. Gastroenterology 2015; 149:67–78. Doi: 10.1053/j.gastro.2015.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dumanski JP, Rasi C, Björklund P, Davies H, Ali AS, Grönberg M, et al. A MUTYH germline mutation is associated with small intestinal neuroendocrine tumors. Endocr Relat Cancer 2017; 24:427–445. Doi: 10.1530/ERC-17-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Popova T, Hebert L, Jacquemin V, Gad S, Caux-Moncoutier V, Dubois-d’Enghien C, et al. Germline BAP1 mutations predispose to renal cell carcinomas. Am J Hum Genet 2013; 92:974–980. Doi: 10.1016/j.ajhg.2013.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hanks S, Perdeaux ER, Seal S, Ruark E, Mahamdallie SS, Murray A, et al. Germline mutations in the PAF1 complex gene CTR9 predispose to Wilms tumour. Nat Commun 2014; 5:4396. Doi: 10.1038/ncomms5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mahamdallie SS, Hanks S, Karlin KL, Zachariou A, Perdeaux ER, Ruark E, et al. Mutations in the transcriptional repressor REST predispose to Wilms tumor. Nat Genet 2015; 47:1471–1474. Doi: 10.1038/ng.3440 [DOI] [PubMed] [Google Scholar]
- 142.Palculict TB, Ruteshouser EC, Fan Y, Wang W, Strong L, Huff V. Identification of germline DICER1 mutations and loss of heterozygosity in familial Wilms tumour. J Med Genet 2016; 53:385–388. Doi: 10.1136/jmedgenet-2015-103311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Caruso S, Calderaro J, Letouzé E, Nault JC, Couchy G, Goulai A, et al. Germline and somatic DICER1 mutations in familial and sporadic liver tumors. J Hepatol 2017; 66:734–742. Doi: 10.1016/j.jhep.2106.12.010 [DOI] [PubMed] [Google Scholar]
- 144.Kukita Y, Okami J, Yoneda-Kato N, Nakamae I, Kawabata T, Higashiyama M, et al. Homozygous inactivation of CHEK2 is linked to a familial case of multiple primary lung cancer with accompanying cancers in other organs. Cold Spring Harb Mol Case Study 2016; 2:a001032. Doi: 10.1101/mcs/a001032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yamamoto H, Higasa K, Sakaguchi M, Shien K, Soh J, Ichimura K, et al. Novel germline mutation in the transmembrane domain of HER2 in familial lung adenocarcinomas. J Natl Cancer Inst 2014; 106:djt338. Doi: 10.1093/jnci/djt338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lusk CM, Wenzlaff AS, Dyson G, Purrington KS, Watza D, Land S. Whole-exome sequencing reveals genetic variability among lung cancer cases subphenotyped for emphysema. Carcinogenesis 2016; 37:139–144. Doi: 10.1093/carcin/bgv248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tode N, Kikuchi T, Sakakibara T, Hirano T, Inoue A, Ohkouchi S, et al. Exome sequencing deciphers a germline MET mutation in familial epidermal growth factor receptor-mutant lung cancer. Cancer Sci 2017; 108:1263–1270. Doi: 10.111/cas.13233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Xiong D, Wang Y, Kupert E, Simpson C, Pinney SM, Gaba CR, et al. A recurrent mutation in PARK2 is associated with familial lung cancer. Am J Hum Genet 2015; 96:301–308. Doi: 10.1016/j.ajhg.2014.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chen HY, Yu SL, Ho BC, Su KY, Hsu YC, Chang CS, et al. R331W missense mutation of oncogene YAP1 is a germline risk allele for lung adenocarcinoma with medical actionability. J Clin Oncol 2015; 33:2303–2310. Doi: 10.1200/JCO.2014.59.3590 [DOI] [PubMed] [Google Scholar]
- 150.He Y, Li Y, Qui Z, Zhou B, Shi S, Zhang K, et al. Identification and validation of PROM1 and CRTC2 mutations in lung cancer patients. Mol Cancer 2014; 13:19. Doi: 10.1186/1476-4598-13-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tomoshige K, Matsumoto K, Tsuchiya T, Oikawa M, Miyazaki T, Yamasaki N, et al. Germline mutations causing familial lung cancer. J Hum Genet 2015; 69:597–603. Doi: 10.1038/jhg.2015.75 [DOI] [PubMed] [Google Scholar]
- 152.Kanwal M, Ding XJ, Ma ZH, Li LW, Wang P, Chen Y, et al. Characterization of germline mutations in familial lung cancer from the Chinese population. Gene 2018; 641:94–104. doi: 10.1016/j.gene.2017.10.020 [DOI] [PubMed] [Google Scholar]
- 153.Luo W, Tian P, Wang Y, Xu H, Chen L, Tang C, et al. Characteristics of genomic alterations of lung adenocarcinoma in young never-smokers. Int J Cancer 2018; 143:1696–1705. Doi: 10.1002/ijc.31542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Esai Selvan M, Klein RJ, Gümüş ZH. Rare, pathogenic germline variants in Fanconi Anemia genes increase risk for squamous lung cancer. Clin Cancer Res 2019; 25:1517–1525. Doi: 10.1158/1078-0432.CCR-18-2660 [DOI] [PubMed] [Google Scholar]
- 155.Renieri A, Mencarelli MA, Cetta F, Baldassarri M, Mari F, Furini S, et al. Oligogenic germline mutations identified in early non-smokers lung adenocarcinoma patients. Lung Cancer 2014; 85:168–174. Doi: 10.1016/j.lungcan.2014.05.020 [DOI] [PubMed] [Google Scholar]
- 156.Donner I, Katainen R, Sipilä LJ, Aavikko M, Pukkala E, Aaltonen LA. Germline mutations in young non-smoking women with lung adenocarcinoma. Lung Cancer 2018; 122:76–82. Doi: 10.1016/j.lungcan.2018.05.027 [DOI] [PubMed] [Google Scholar]
- 157.Dębniak T, Scott RJ, Lea RA, Görski B, Masojć B, Cybulski C, et al. Founder mutations for early onset melanoma as revealed by whole exome sequencing suggests that this is not associated with the increasing incidence of melanoma in Poland. Cancer Res Treat 2019; 51:337–344. Doi: 10.4143/crt.2018.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Aoude LG, Wadt K, Bojesen A, Crüger D, Borg A, Trent JM, et al. A BAP1 mutation in a Danish family predisposes to uveal melanoma and other cancers. PLoS One. 2013; 8:e72144. Doi: 10.1371/journal.pone.0072144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Teerlink CC, Huff C, Stevens J, Yu Y, Holmen SL, Silvis MR, et al. A nonsynonymous variant in the GOLM1 gene in cutaneous malignant melanoma. J Natl Cancer Inst 2018; 110:1380–1385. Doi: 10.1093/jnci/djy058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Aoude LG, Heitzer E, Johansson P, Gartside M, Wadt K, Pritchard AL, et al. POLE mutations in families predisposed to cutaneous melanoma. Fam Cancer 2015; 14:621–628. Doi: 10.1007/s10689-015-9826-8 [DOI] [PubMed] [Google Scholar]
- 161.Shi J, Yang XR, Ballew B, Rotunno M, Calista D, Fargnoli MC, et al. Rare missense variants in POT1 predisposes to familial cutaneous malignant melanoma. Nat Genet 2014; 46:482–486. Doi: 10.1038/ng.2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Robles-Espinoza CD, Harland M, Ramsay AJ, Aoude LG, Quesada V, Ding Z, et al. POT1 loss-of-function variants predispose to familial melanoma. Nat Genet 2014; 46:478–481. Doi: 10.1038/ng.2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wadt KA, Aoude LG, Golmard L, Hansen TV, Sastre-Garau X, Hayward NK, et al. Germline RAD51B truncating mutation in a family with cutaneous melanoma. Fam Cancer 2015; 14:337–340. Doi: 10.1007/s10689-015-9781-4 [DOI] [PubMed] [Google Scholar]
- 164.Benfodda M, Gazal S, Descamps V, Basset-Seguin N, Deschamps L, Thomas L, et al. Truncating mutations of TP53AIP1 gene predispose to cutaneous melanoma. Genes Chromosomes Cancer 2018; 57:294–303. Doi: 10.1002/gcc.22528 [DOI] [PubMed] [Google Scholar]
- 165.Ibarrola-Villava M, Kumar R, Nagore E, Benfodda M, Guedj M, Gazal S, et al. Genes involved in the WNT and vesicular trafficking pathways are associated with melanoma predisposition. Int J Cancer 2015; 136:2109–2119. Doi: 10.1002/ijc.29257 [DOI] [PubMed] [Google Scholar]
- 166.Artomov M, Stratigos AJ, Kim I, Kumar R, Lauss M, Reddy BY, et al. Rare variant, gene-based associations study of hereditary melanoma using whole-exome sequencing. J Natl Cancer Inst 2017; 109(12). Doi: 10.1093/jnci/djx083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yu Y, Hu H, Chen JS, Hu F, Fowler J, Scheet P, et al. Integrated case-control and somatic-germline interaction analyses of melanoma susceptibility genes. Biochim Biophys Acta Mol Basis Dis 2018; 1864:2247–2254. Doi: 10.1016/j.bbadis.2018.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Franceschi S, Spugnesi L, Aretini P, Lessi F, Scarpitta R, Galli A, et al. Whole-exome analysis of a Li-Fraumeni family trio with a novel TP53 PRD mutation and anticipation profile. Carcinogenesis 2017; 38:938–943. Doi: 10.10093/carcin/bgx069 [DOI] [PubMed] [Google Scholar]
- 169.Wadt K, Choi J, Chung JY, Kiilgaard J, Heedgaard S, Drzewiecki KT, et al. A cryptic BAP1 splice mutation in a family with uveal and cutaneous melanom, and paraganglioma. Pigment Cell Melanoma Res 2012; 25:815–818. Doi: 10.1111/pcmr.12006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wilson TL, Hattangady N, Lerario AM, Williams C, Koeppe E, Quinonez S, et al. A new POT1 germline mutation-expanding the spectrum of POT1-associated cancers. Fam Cancer 2017; 16:561–566. Doi: 10.1007/s10689-017-9984-y [DOI] [PubMed] [Google Scholar]
- 171.Li WD, Li QR, Xu SN, Wei FJ, Ye ZJ, Cheng JK, et al. Exome sequencing identifies an MLL3 gene germline mutation in a pedigree of colorectal cancer and acute myeloid leukemia. Blood 2013; 121:1478–1479. Doi: 10.1182/blood-2012-12-470559 [DOI] [PubMed] [Google Scholar]
- 172.Rohlin A, Zagoras T, Nilsson S, Lundstam U, Wahlström J, Hultén L, et al. A mutation in POLE predisposing to a multi-tumour phenotype. Int J Oncol 2014; 45:77–81. Doi: 10.3892/ijo.2014.2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hansen MF, Johansen J, Bjornevoll I, Sylvander AE, Steinsbekk KS, Sӕtrom P, et al. A novel POLE mutation associated with cancers of colon, pancreas, ovaries and small intestine. Fam Cancer 2015; 14:437–448. Doi: 10.1007/s10689-015-9803-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wei C, Peng B, Han Y, Chen WV, Rother J, Tomlinson GE, et al. Mutations of HNRNPA0 and WIF1 predispose members of a large family to multiple cancers. Fam Cancer 2015; 14:297–306. Doi: 10.1007/s10689-014-9738-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lu C, Xie M, Wendl MC, Wang J, McLellan MD, Leiserson MD, et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nat Commun 2015; 6:10086. Doi: 10.1038/ncomms10086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Huang KL, Mashl RJ, Wu Y, Ritter DI, Wang J, Oh C, et al. Pathogenic germline variants in 10,389 adult cancers. Cell 2018; 173:355–370.e14. doi: 10.1016/j.cell.2018.03.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Diets IJ, Waanders E, Ligtenberg MJ, van Bladel DAG, Kamping EJ, Hoogerbrugge PM, et al. High yield of pathogenic germline mutations causative or likely causative of the cancer phenotype in selected children with cancer. Clin Cancer Res 2018; 24:1594–1603. Doi: 10.1158/1078-0432.CCR-17-1725 [DOI] [PubMed] [Google Scholar]
- 178.Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med 2015; 373:2336–2346. Doi: 10.1056/NEJMoa1508054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Dicks E, Song H, Ramus SJ, Oudenhove EV, Tyrer JP, Intermaggio MP, et al. Germline whole exome sequencing and large-scale replication identified FANCM as a likely high grade serous ovarian cancer susceptibility gene. Oncotarget 2017; 8:50930–50940. Doi: 10.18632/oncotarget.15871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Witkowski L, Carrot-Zhang J, Albrecht S, Fahiminiya S, Hamel N, Tomiak E, et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet 2014; 46:438–443. Doi: 10.1038/ng.2931 [DOI] [PubMed] [Google Scholar]
- 181.Stafford JL, Dyson G, Levin NK, Chaudhry S, Rosati R, Kalpage H, et al. Reanalysis of BRCA1/2 negative high-risk ovarian cancer patients reveals novel germline risk loci and insights into missing heritability. PLoS One 2017; 12:e0178450. Doi: 10.1371/journal.pone.0178450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kanchi KL, Johnson KJ, Lu C, McLellan MD, Leiserson MD, Wendl MC, et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat Commun 2014; 5:3156. Doi: 10.1038/ncomms4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Määttä K, Rantapero T, Lindström A, Nykter M, Kankuri-Tammilehto M, Laasanen SL, et al. Whole-exome sequencing of Finnish hereditary breast cancer families. Eur J Hum Genet 2016; 25:85–93. Doi: 10.1038/ejhg.2016.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lu HM, Li S, Black MH, Lee S, Hoiness R, Wu S, et al. Association of breast and ovarian cancers with predisposition genes identified by large-scale sequencing. JAMA Oncol 2019; 5:51–57. Doi: 10.1001/jamaoncol.2018.2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy ML, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov 2012; 2:41–46. Doi: 10.1158/2159-8290.CD-11-0194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Grant RC, Denroche RE, Borgida A, Virtanen C, Cook N, Smith AL, et al. Exome-wide association study of pancreatic cancer risk. Gastroenterology 2018; 154:719–722. Doi: 10.1053/j.gastro.2017.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Grant RC, Al-Sukhni W, Borgida AE, Holter S, Kanji ZS, McPherson T, et al. Exome sequencing identifies nonsegregating nonsense ATM and PALB2 variants in familial pancreatic cancer. Hum Genomics 2013; 7:11. Doi: 10.1186/1479-7364-7-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Smith AL, Alirezaie N, Connor A, Chan-Seng-Yue M, Grant R, Selander I, et al. Candidate DNA repair susceptibility genes identified by exome sequencing in high-risk pancreatic cancer. Cancer Lett 2016; 370:302–312. Doi: 10.1016/j.canlet.2015.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Roberts NJ, Norris AL, Petersen GM, Bondy ML, Brand R, Gallinger S, et al. Whole genome sequencing defines the genetic heterogeneity of familial pancreatic cancer. Cancer Discov 2016; 6:166–175. Doi: 10.1158/2159-8290.CD-15-0402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Yang XR, Rotunno M, Xiao Y, Ingvar C, Helgadottir H, Pastorino L, et al. Multiple rare variants in high-risk pancreatic cancer-related genes may increase risk for pancreatic cancer in a subset of patients with and without germline CDKN2A mutations. Hum Genet 2016; 135:1241–12. Doi: 10.1007/s00439-016-1715-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Rand KA, Rohland N, Tandon A, Stram A, Sheng X, Do R, et al. Whole-exome sequencing of over 4100 men of African ancestry and prostate cancer risk. Hum Mol Genet 2016; 25:371–381. Doi: 10.1093/hmg/ddv462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Fitzgerald LM, Kumar A, Boyle EA, Zhang Y, McIntosh LM, Kolb S, et al. Germline missense variants in the BTNL2 gene are associated with prostate cancer susceptibility. Cancer Epidemiol Biomarkers Prev 2013; 22:1520–1528. Doi: 10.1158/1055-9965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Mijuskovic M, Saunders EJ, Leongamornlert DA, Wakerell S, Whitmore I, Dadaev T, et al. Rare germline variants in DNA repair genes and the angiogenesis pathway predispose prostate cancer patients to develop metastatic disease. Br J Cancer 2018; 119:96–104. Doi: 10.1038/s41416-018-014-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hayano T, Matsui H, Nakaoka H, Ohtake N, Hosomichi K, Suzuki K, et al. Germline variants of prostate cancer in Japanese families. PLoS One 2016; 11:e0164233. Doi: 10.1371/journal.pone.0164233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Karyadi DM, Geybels MS, Karlins E, Decker B, McInstosh L, Hutchinson A, et al. Whole exome sequencing in 75 high-risk families with validation and replication in independent case-control studies identifies TANGO2, OR5H14, and CHAD as new prostate cancer susceptibility genes. Oncotarget 2017; 8:1495–1507. Doi: 10.18632/oncotarget.13646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Johnson AM, Zuhlke KA, Plotts C, McDonnell SK, Middha S, Riska SM, et al. Mutational landscape of candidate genes in familial prostate cancer. Prostate 2014; 74:1371–1378. Doi: 10.1002/pros.22849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hart SN, Ellingson MS, Schahl K, Vedell PT, Carlson RE, Sinnwell JP, et al. Determining the frequency of pathogenic germline variants from exome sequencing in patients with castrate-resistant prostate cancer. BMJ Open 2016; 6:e010332. Doi: 10.1136/bmjopen-2015-010332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hunter SM, Rowley SM, Clouston D, KCon Fab Investigators, Li J, Lupat R, et al. Searching for candidate genes in familial BRCAX mutation carriers with prostate cancer. Urol Oncol 2016; 34:120.e9–16. Doi: 10.1016/j.urolonc.2015.10.009 [DOI] [PubMed] [Google Scholar]
- 199.Koboldt DC, Kanchi KL, Gui B, Larson DE, Fulton RS, Isaacs WB, et al. Rare variation in TET2 is associated with clinically relevant prostate carcinoma in African Americans. Cancer Epidemiol Biomarkers Prev 2016; 25:1456–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Nicolas E, Arora S, Zhou Y, Serebriiskii IG, Andrake MD, Handorf ED, et al. Systematic evaluation of underlying defects in DNA repair as an approach to case-only assessment of familial prostate cancer. Oncotarget 2015; 6:39614–39633. Doi: 10.18632/oncotarget.5554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Calvete O, Martinez P, Garcia-Pavia P, Benitez-Buelga C, Paumard-Hernández B, Fernandez V, et al. A mutation in the POT1 gene is responsible for cardiac angiosarcoma in TP53-negative Li-Fraumeni-like families. Nat Commun 2015; 6:8383. Doi: 10.1038/ncomms9383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Witkowski L, Lalonde E, Zhang J, Albrecht S, Hamel N, Cavallone L, et al. Familial rhabdoid tumor ‘avant la lettre’—from pathology review to exome sequencing and back again. J Pathol 2013; 231:35–43. Doi: 10.1002/path.4225 [DOI] [PubMed] [Google Scholar]
- 203.Aavikko M, Kaasinen E, Nieminen JK, Byun M, Donner I, Mancuso R, et al. Whole-genome sequencing identifies STAT4 as a putative susceptibility gene in classic Kaposi sarcoma. J Infect Dis 2015; 211:1842–1851. Doi: 10.1093/infdis/jiu667 [DOI] [PubMed] [Google Scholar]
- 204.Jouenne F, Chauvot de Beauchene I, Bollaert E, Avril MF, Caron O, Ingster O, et al. Germline CDKN2A/P16INK4A mutations contribute to genetic determinism of sarcoma. J Med Genet 2017; 54:607–612. Doi: 10.1136/jmedgenet-2016-014402 [DOI] [PubMed] [Google Scholar]
- 205.Brohl AS, Patidar R, Turner CE, Wen X, Song YK, Wei JS, et al. Frequent inactivating germline mutations in DNA repair genes in patients with Ewing sarcoma. Genet Med 2017; 19:955–958. Doi: 10.1038/gim.2016.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Litchfield K, Loveday C, Levy M, Dudakia D, Rapley E, Nsengimana J, et al. Large-scale sequencing of testicular germ cell tumour (TGCT) cases excludes major TGCT predisposition gene. Eur Urol 2018; 73:828–831. Doi: 10.1016/j.eururo.2018.01.021 [DOI] [PubMed] [Google Scholar]
- 207.Truscott L, Gell J, Chang VY, Lee H, Strom SP, Pillai R, et al. Novel association of familial testicular germ cell tumor and autosomal dominant polycystic kidney disease with PKD1 mutation. Pediatr Blood Cancer 2017; 64:100–102. Doi: 10.1002/pfc.26197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Paumard-Hernández B, Calvete O, Inglada Pérez L, Tejero H, Al-Hahrour F, Pita G, et al. Whole exome sequencing identified PLEC, EXO5 and DNAH7 as novel susceptibility genes in testicular cancer. Int J Cancer 2018; 143:1954–1962. Doi: 10.1002/ijc.31604 [DOI] [PubMed] [Google Scholar]
- 209.Litchfield K, Levy M, Dudakia D, Proszek P, Shipley C, Basten S, et al. Rare disruptive mutations in ciliary function genes contribute to testicular cancer susceptibility. Nat Commun 2016; 7:13840. Doi: 10.1038/ncomms13840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Liu C, Yu Y, Yin G, Zhang J, Wen W, Ruan X, Li D, et al. C14orf93 (RTFC) is identified as a novel susceptibility gene for familial nonmedullary thyroid cancer. Biochem Biophys Res Commun 2017; 482:590–596. Doi: 10.1016/j.bbrc.2016.11.078 [DOI] [PubMed] [Google Scholar]
- 211.Gara SK, Jia L, Merino MJ, Agarwal SK, Zhang L, Cam M, et al. Germline HABP2 mutation causing familial nonmedullary thyroid cancer. N Engl J Med 2015; 373:448–455. Doi: 10.1056/NEJMoa1502449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ye F, Gao H, Xiao L, Zuo Z, Liu Y, Zhao Q, et al. Whole exome and target sequencing identified MAP2K5 as novel susceptibility gene for familial non-medullary thyroid carcinoma. Int J Cancer 2019; 144:1321–1330. Doi: 10.1002/ijc.31825 [DOI] [PubMed] [Google Scholar]
- 213.Sponziello M, Benvenuti S, Gentile A, Pecce V, Rosignolo F, Virzi AR, et al. Whole exome sequencing identifies a germline MET mutation in two siblings with hereditary wild-type RET medullary thyroid cancer. Hum Mutat 2018; 39:371–377. Doi: 10.1002/humu.23378 [DOI] [PubMed] [Google Scholar]
- 214.Ikeda Y, Kiyotani K, Yew PY, Kato T, Tamura K, Yap KL, et al. Germline PARP4 mutations in patients with primary thyroid and breast cancers. Endocr Relat Cancer 2016; 23:171–179. Doi: 10.1530/ERC-15-0359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Tomsic J, He H, Akagi K, Liyanarachchi S, Pan Q, Bertani B, et al. A germline mutation in SRRM2, a splicing factor gene, is implicated in papillary thyroid carcinoma predisposition. Sci Rep 2015; 5:10566. Doi: 10.1038/srep10566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Ni Y, Seballos S, Fletcher B, Romigh T, Yehia L, Mester J, et al. Germline compound heterozygous poly-glutamine deletion in USF3 may be involved in predisposition to heritable and sporadic epithelial thyroid carcinoma. Hum Mol Genet 2017; 262:243–257. Doi: 10.1093/hmg/ddw382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Pinheiro M, Drigo SA, Tonhosolo R, Andrade SCS, Marchi FA, Jurisica I, et al. HABP2 p.G534E variant in patients with family history of thyroid and breast cancer. Oncotarget 2017; 8:40896–40905. Doi: 10.18632/oncotarget.16639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ernst C, Hahnen E, Engel C, Nothnagel M, Weber J, Schmutzler RK, et al. Performance of in silico prediction tools for the classification of rare BRCA1/2 missense variants in clinical diagnostics. BMC Med Genomics 2018; 11:35. Doi: 10.1186/s12920-018-0353-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Leong IU, Stuckey A, Lai D, Skinner JR, Love DR. Assessment of the predictive accuracy of five in silico prediction tools, alone or in combination, and two metaservers to classify long QT syndrome gene mutations. BMC Med Genet 2015. 16:34. Doi: 10.1186/s12881-015-0176-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.McCarthy DJ, Humburg P, Kanapin A, Rivas MA, Gaulton K, Cazier JB, et al. Choice of transcripts and software has a large effect on variant annotation. Genome Med. 2014. March 31;6(3):26. doi: 10.1186/gm543. eCollection 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Rehm HL, Bale SJ, Bayrak-Toydemir P, Berg JS, Brown KK, Deignan JL, et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med 2013; 15(9): 733–747. Doi: 10.1038/gim.2013.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Landrum MJ, Kattman BL. ClinVar at five years: Delivering on the promise. Hum Mutation 2018; 39(11):1623–1630. Doi:10.1002.humu.23641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Melchor L and Benítez J. The complex genetic landscape of familial breast cancer. Hum Genet. 2013; 132(8):845–63. Doi: 10.1007/s00439-013-1299-y. [DOI] [PubMed] [Google Scholar]
- 224.Kiezun A, Garimella K, Do R, Stitziel NO, Neale BM, McLaren PJ, et al. Exome sequencing and the genetic basis of complex traits. Nat Genet 2012; 44:623–630. Doi: 10.1038/ng.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011. September 27;12(11):745–55. doi: 10.1038/nrg3031. Review. [DOI] [PubMed] [Google Scholar]
- 226.Lappalainen T, Scott AJ, Brandt M, and Hallet IM. Genomic analysis in the age of human genome sequencing. Cell 2019; 177:70. Doi: 10.1016/j.cell.2019.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, et al. Exome sequencing identifies PALB2 as a pancreatic cancer susceptibility bene. Science 2009; 324:217. Doi: 10.1126/science.1171202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Saarinen S, Aavikko M, Aittomaki K, Launonen V, Lehtonen R, Franssila K, et al. Exome sequencing reveals germline NPAT mutation as a candidate risk factor for Hodgkin lymphoma. Blood 2011; 118:493–498. Doi: 10.1182/blood-2011-03-341560 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.