

Abstract
Tissue-resident memory T cells (TRMs) can profoundly enhance mucosal immunity, but parameters governing TRM induction by vaccination remain poorly understood. Here we describe an approach exploiting natural albumin transport across the airway epithelium to enhance mucosal TRM generation by vaccination. Pulmonary immunization with albumin-binding amphiphile-conjugates of peptide antigens and CpG adjuvant (amph-vaccines) increased vaccine accumulation in the lung and mediastinal lymph nodes (MLNs). Amph-vaccines prolonged antigen presentation in MLNs over 2 weeks, leading to 25-fold increased lung-resident T-cell responses over traditional immunization and enhanced protection from viral or tumor challenge. Mimicking such prolonged exposure through repeated administration of soluble vaccine revealed that persistence of both antigen and adjuvant was critical for optimal TRM induction, mediated through T-cell priming in MLNs post prime, and directly in the lung tissue post boost. Thus, vaccine persistence strongly promotes TRM induction, and amph-conjugates may provide a practical approach to achieve such kinetics in mucosal vaccines.
One-sentence summary:
Pulmonary immunization with albumin-binding peptide vaccines promotes resident memory CD8+ T cell generation in the lung.
Introduction
Tissue-resident memory T cells (TRMs) provide immune surveillance at barrier tissues and provide a first line of defense against intracellular pathogens at mucosal surfaces (1–3). TRMs provide rapid protection in the tissue by detecting antigens presented by infected cells, triggering production of inflammatory cytokines/chemokines and mediating cytotoxicity (1, 4, 5). Studies profiling TRM specificity in different tissues have shown that the human TRM repertoire, while diverse, can be biased towards pathogens that infect a particular tissue, as evidenced by the presence of influenza-specific TRMs in lungs (6, 7) and hepatitis B-specific TRMs in the liver (8). In addition, TRMs play an important role in protection from tumors that develop from epithelial tissues (9–11). Based on these characteristics, the design of vaccines that efficiently generate TRM cells is of great interest for both infectious disease and cancer. Although much progress has been made in defining cues that promote TRM generation (12–15), it remains poorly understood how parameters of vaccination might be optimized to enhance the differentiation of long-lived tissue-resident lymphocyte populations.
Peptide-based vaccines are attractive for their safety, affordability, and compatibility with rapid manufacture for personalized cancer vaccines (16). While numerous ongoing clinical trials are exploring the use of peptide vaccines in cancer (17, 18) and infectious diseases (16, 19), their immunogenicity remains modest and there are currently no FDA-approved peptide vaccines. One major shortcoming of peptide-based vaccines is poor pharmacokinetics, a combination of inefficient accumulation in lymph nodes and rapid degradation in vivo (20, 21). To address this, we previously described a vaccine technology enabling massive expansion of antigen-specific T cells, based on coupling peptide antigens or molecular adjuvants to an amphiphilic phospholipid moiety, forming amphiphile vaccines (amph-vaccines) (20–22). On injection, the lipid tail of these amph-vaccine molecules binds to endogenous albumin in the interstitial fluid, and albumin then serves as an efficient chaperone delivering the antigen or adjuvant to draining lymph nodes. In lymph nodes, the lipid tail of the amph-vaccine transfers from albumin to insert in cell membranes, resulting in potent antigen-specific effector CD8+ T-cell responses (21, 22).
Albumin is the most abundant serum protein and like IgG, binds to the neonatal Fc receptor (FcRn), which delays its catabolism and increases its half-life (23). FcRn receptors expressed by pulmonary epithelial cells in the lungs, bi-directionally transport albumin and antibodies across the airway epithelium (23, 24). We hypothesized that amph-vaccines administered to the pulmonary mucosa could be transported from the airway to the lung parenchyma by albumin, and thereafter trafficked to lung-draining lymph nodes to prime tissue-resident T cells.
Here we show that vaccines comprised of peptide antigens and the Toll-like receptor-9 (TLR-9) agonist CpG DNA as a molecular adjuvant, show greatly enhanced uptake in lung tissue when administered as albumin-binding amphiphile conjugates, leading to potent T-cell priming in lung-draining MLNs. Pulmonary immunization with amph-vaccines elicited a long-lived TRM population in the lungs that protected against viral challenge and tumor lung metastasis. Mechanistically, amph-vaccines elicited sustained antigen presentation in MLNs. Mimicking sustained vaccine exposure by repeated administration of unmodified peptide/CpG revealed that optimal establishment of TRMs required prolonged availability of both antigen and adjuvant over ~2 weeks in MLNs and local lung tissue. This leads to efficient T-cell priming in MLNs during a primary immunization and expansion of cells locally in the lung following boosting. These findings provide insights into vaccine characteristics that can be optimized to amplify TRM induction for mucosal immunity.
Results
Albumin-binding vaccines accumulate in the lung parenchyma and draining MLNs
In agreement with previous reports (25), we measured ~200 μg/mL of albumin present in the bronchoalveolar lavage (BAL) fluid of naive mice, which rapidly increased to a few mg/mL following pulmonary administration of the TLR9 agonist CpG (Fig. 1A). Fluorescently-labeled albumin administered intratracheally (i.t.) was retained in the tissue after 24 hr at higher levels in wild type mice than FcRn−/− mice, suggesting uptake in the tissue is promoted by FcRn receptors (Fig. 1B–C).
Figure 1. Amphiphile peptides or CpG are retained in the lung parenchyma and draining MLNs.
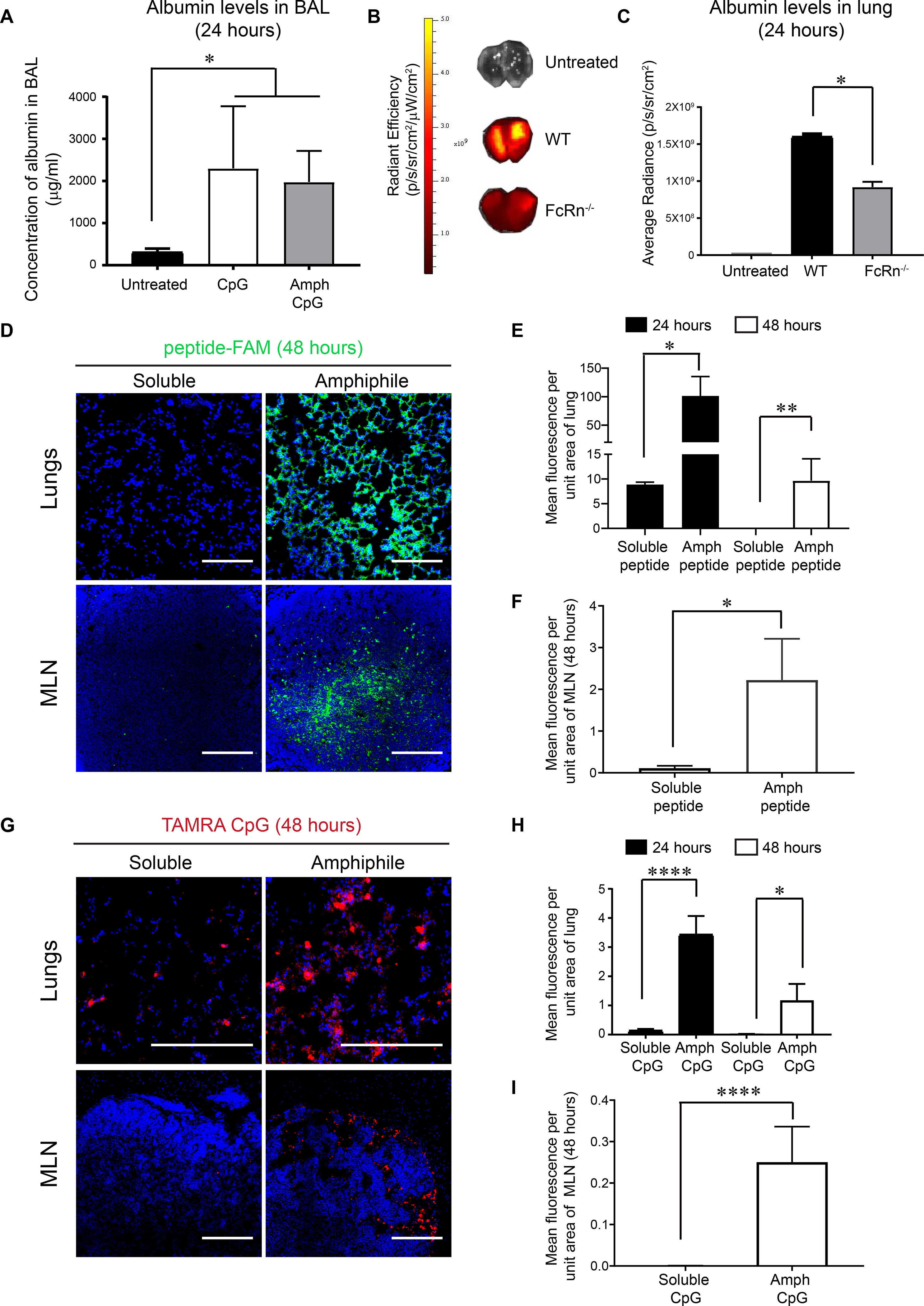
(A) Albumin levels measured by ELISA in bronchoalveolar lavage (BAL) from WT mice that were untreated or treated with inflammatory stimuli i.t. (B) Labeled mouse albumin was delivered i.t. into WT and FcRn−/− mice (n = 3/group) and fluorescence signal from lungs was measured by IVIS imaging 24 hours later. (C) Quantification of fluorescence signal from experiment described in (B). (D-I) C57BL/6 mice (n=3/group) were administered 20 μg FAM-labeled (green) gp100 peptide, an equimolar amount of labeled amph-gp100, 10 μg TAMRA-labeled (red) CpG, or an equimolar amount of labeled amph-CpG. Shown are representative immunofluorescence images of lungs (upper panel, scale bar = 100 μm in D and 200 μm in G) and MLNs (lower panel, scale bar = 100 μm) harvested from mice at 48 hours post immunization (D, G; DAPI (blue) was used to stain nuclei). Quantification of peptide or CpG signals from the lungs at 24 and 48 hours are shown in (E, H) and MLNs (F, I). Data shown are mean + SEM, *p<0.05, **p<0.001, ****p<0.0001 as determined by one-way ANOVA. Data are representative of at least two independent experiments.
To test whether an albumin-binding moiety would alter tissue distribution of peptide antigens or adjuvants in the lungs, we synthesized amphiphile-conjugate (amph-conjugate) vaccines comprised of a melanoma peptide antigen gp10020–39 linked to a DSPE phospholipid tail through a poly(ethylene glycol) spacer, and CpG DNA linked to a similar C18 lipid tail (fig. S1). We previously demonstrated that these amph-conjugate molecules bind to albumin and efficiently traffic to skin-draining lymph nodes following subcutaneous (s.c.) injection (20, 21). We first compared the tissue distribution of fluorescently-labeled free or amph-conjugate gp10020–39 and CpG following pulmonary administration in the lungs and lung-draining MLNs, using i.t. instillation as a surrogate for aerosol delivery in humans.
Twenty-four hours after i.t. administration, amph-gp100 conjugates were present in the tissue parenchyma at ~10-fold greater levels than the free peptide, and were still readily detectable 48 hours post immunization (Fig. 1D–E). Amph-gp100 further accumulated in MLNs, with significantly higher levels detected at 48 hours compared to soluble gp100 (Fig. 1D, F). Similarly, amph-CpG showed substantially greater accumulation in the lung tissue and MLNs compared to free CpG (Fig. 1G–I). Together, these results suggest that modification with an albumin-binding lipid tail enhanced the uptake and persistence of peptide antigens and CpG adjuvant in lung tissues.
Pulmonary vaccination with amph-vaccines elicits high levels of antigen-specific lung TRMs
We next evaluated the immunogenicity of amph-vaccines and their ability to prime lung parenchyma-homing T cells compared to the same vaccines administered parenterally. We tested both the gp100 antigen and a viral gag peptide termed AL11 (26), in anticipation of testing for protection against viral challenge. Groups of mice were vaccinated i.t. or s.c. on days 0 and 14 with either free AL11/CpG or amph-AL11/amph-CpG. One week later, tissues were collected and intravascular staining was used to differentiate T cells localized in the vasculature vs. the lung parenchyma (27). In accordance with our previous results (20), amph-conjugate vaccination elicited much greater AL11-specific T-cell expansion in the peripheral blood compared to soluble peptide/CpG immunization whether administered s.c. or i.t., but s.c. amph-vaccination elicited higher levels of circulating T cells than i.t. administration (Fig. 2A, B). By contrast, i.t. amph-vaccines elicited a large population of antigen-specific T cells in the lungs, 25-fold greater than soluble peptide/CpG given by the same route and 100-fold greater than amph-vaccines administered subcutaneously (Fig. 2A, C). We carried out similar experiments with soluble vs. amph-conjugate gp100/CpG vaccines, and evaluated the functionality of the resulting T-cell responses 1-week post boost via intracellular cytokine staining. As shown in fig. S2A–C, s.c.-delivered amph-vaccination elicited a strong IFN-γ/TNF-α-producing gp100-specific T-cell response in the lung vasculature, but essentially no cytokine-competent T cells were detected in the lung parenchyma. By contrast, pulmonary vaccination elicited a prominent population of cytokine-producing T cells in the lung tissue, with amph-conjugate immunization eliciting a 24-fold larger cytokine+ T-cell response compared to soluble vaccines (fig. S2C).
Figure 2: Mucosal amph-vaccines elicit robust expansion of antigen-specific CD8+ TRMs in the lungs.

C57BL/6 mice were immunized either s.c. or i.t. with 5 μg of free AL11 peptide and 8 μg CpG or equimolar doses of amph-AL11 combined with amph-CpG on days 0 and 14. (A) Representative flow cytometry plots of CD8+AL11-tetramer+ T cells in the blood (upper panel) and lung parenchyma (lower panel) at day 20. (B, C) Quantification of CD8+AL11-tetramer+ cells in the blood (B) and lung parenchyma (C). (D) Representative histograms of CD49a expression on CD8+ T cells and quantification of CD49a mean fluorescence intensity (MFI) following i.t. amph-vaccination. (E) Representative flow cytometry plots of CD69 and CD103 expression on CD8+AL11-tetramer+ T cells in the blood (left panel), lung vasculature (center panel) and lung parenchyma (right panel) following i.t. amph-vaccination. (F) Quantification of the percentage of lung CD8+AL11-tetramer+ T cells expressing CD69, CD103 or both. (G) WT and Batf3−/− mice were vaccinated with amph-gp100 and amph-CpG i.t. following the schedule in (A). Quantification of cytokine+ CD8+ lung T cells. (H) WT and FcRn−/− mice were vaccinated with amph-gp100 and amph-CpG i.t. following the schedule in (A) and lung antigen-specific CD8+ T cells were quantified. Data are shown are mean + SEM, **p<0.01, ***p<0.001, ****p<0.0001 as determined by one-way ANOVA. Data are representative of at least three independent experiments (n = 3 – 5 animals/group).
CD8+ T cells in the lung parenchyma following amph-conjugate vaccination expressed substantially higher levels of the collagen-binding mucosal α1β1 integrin CD49a compared to cells in the peripheral blood or lung vasculature (Fig. 2D) (28). The majority of lung antigen-specific CD8+ T cells following i.t. immunization also expressed the tissue-resident markers CD69, CD103, or both, while T cells primed by the parenteral immunizations expressed these markers at a much lower frequency (Fig. 2E–F) (29–31).
Similar to our previous findings with parenteral amph-peptide vaccination (21), T-cell priming by i.t. amph-vaccine immunization was mediated by Batf3-dependent dendritic cells (DCs), as indicated by the loss of responses in Batf3−/− animals (Fig. 2G). Additionally, antigen-specific T-cell responses trended lower in the lungs of FcRn−/− mice (Fig. 2H) and the generation of CD69+CD103+ cells was significantly reduced (fig. S3).
Since intranasal (i.n.) immunization is more easily implemented clinically than aerosol immunization, we also compared i.n. and i.t. vaccine delivery. We found that i.t. immunization elicited 4-fold higher T-cell responses in the lungs compared to i.n. immunization (fig. S4). Based on these data, we focused on i.t. administration to mimic aerosol immunization in humans. Collectively, these data suggest that i.t-delivered amph-conjugates greatly enhance the potency of lung mucosal peptide vaccination and that pulmonary amph-vaccines promote establishment of T RMs in the lung parenchyma.
Pulmonary vaccination with amph-conjugate vaccines is protective against viral and tumor challenge
To evaluate the persistence of lung TRMs, we primed and boosted mice with soluble or amphiphile-conjugate AL11 gag peptide vaccines, and analyzed lung tissues 4 months post boost (Fig. 3A). A significant population of antigen-specific lung T cells was only detectable in mice that received i.t. amph-vaccination (Fig. 3B–C), and ~25% of these T cells were CD103+ (Fig. 3D). To test the protective capacity of this TRM population, we challenged AL11-vaccinated mice i.t. with a lethal dose of vaccinia virus expressing the gag epitope five months after immunization. Animals that received the amph-vaccine delivered i.t. all survived viral challenge, while mice in all other groups continuously lost body weight and succumbed by day 7 (Fig. 3E, F). To determine whether protection was mediated by cells pre-positioned in the lungs versus recruited from the blood, we repeated this experiment but treated animals with FTY720 starting 5 days before challenge to block egress of T cells from lymph nodes (32) (fig. S5A–B). Protection from viral challenge in mice receiving the i.t. amph-vaccine was not significantly different in cohorts that received FTY720 or the vehicle alone control (Fig. 3G; p > 0.999), suggesting that de novo T-cell recruitment into the lung was not necessary for protection. Only i.t amph-vaccination significantly reduced viral burden in the lungs compared to non-vaccinated mice (Fig. 3H). FTY720 treated i.t.-vaccinated mice showed a trend toward higher viral loads in the lungs but this did not reach statistical significance, suggesting that protection was predominantly mediated by TRMs.
Figure 3: Antigen-specific CD8+ T cells elicited by i.t. amph-vaccination are long-lived and protect from viral challenge.

(A) C57BL/6 mice (n=5/group) were vaccinated with 5 μg AL11 peptide combined with 8 μg CpG or equimolar doses of amph-AL11 and amph-CpG administered i.t. or s.c. on days 0 and 28, and then challenged with a lethal dose of SIV-Gag vaccinia virus (106 pfu) i.t. or sacrificed to characterize the immune response in the lungs on day 150. (B) Representative flow cytometry plots of CD8+AL11-tetramer+ T cells in the lung parenchyma. (C) Enumeration of lung CD8+AL11-tetramer+ T cells at day 150. (D) Enumeration of CD44+CD103+CD8+AL11-tetramer+ T cells at day 150. (E, F) Animal weights over time (E) and survival (F) post vaccinia virus challenge. (G, H) C57BL/6 mice (n=5/group) were left naïve or vaccinated as in (A), then challenged with vaccinia virus at day 150 in the presence or absence of FTY720. Shown is overall survival (G) and number of viral plaques in the lungs at day 6 post challenge (H). (I) C57BL/6 mice (n=10/group) were left naïve or vaccinated with 10 μg gp100, 10 μg Trp1, and 10 μg Trp2 peptide antigens combined with 8 μg CpG, or vaccinated with equimolar doses of amph-peptides and amph-CpG on days 0 and 14. Thirty days post boost mice were challenged with 4×105 B16F10 melanoma cells i.v. and survival over time is plotted. In each cohort the number of mice surviving at 80 days is indicated. Data are represented as mean + SEM, from three independent experiments (A-H) and two independent experiments (I), *p<0.01, **p < 0.001, ***p<0.0001; ****p<0.00001; ns, not significant. Survival data were analyzed using a log rank test and comparisons between multiple groups were performed using one-way ANOVA.
To test the protective capacity of mucosal amph-conjugate vaccination against tumors, we prophylactically vaccinated mice with amph-conjugates of three melanoma peptides (Trp1455–463, Trp2180–188 and gp10020–39) and challenged animals 30 days later with an intravenous injection of B16F10 melanoma cells. A majority of animals were protected in the pulmonary-vaccinated group while only 40% were protected by s.c. vaccination, although these data did not reach statistical significance (Fig. 3I). To evaluate the potential of amph-vaccines to be useful in a therapeutic context, we performed a study treating B16F10 lung metastases with amph-vaccination in the presence of clinically-relevant checkpoint blockade. While anti-PD1 alone had no efficacy in this setting, 60% of mice treated with amph –vaccine + anti-PD1 showed complete tumor regression (fig. S6). Thus, amph-conjugate vaccination primed a long-lived pool of antigen-specific T cells in the lungs that was protective against viral or tumor challenge.
Sustained exposure to antigen and adjuvant in MLNs enhances TRM generation in lungs
We next sought to understand why amph-conjugate vaccination was so much more effective in generating TRM cells compared to soluble peptide immunization. We hypothesized that the duration of antigen presentation elicited by amph-conjugate vaccines might be an important factor contributing to the formation of TRMs. To define the kinetics of antigen presentation resulting from soluble vs. amph-conjugate vaccination, we immunized mice i.t. with either soluble or amph-conjugate gp100 vaccines, followed by adoptive transfer of CFSE-labeled Pmel-1 TCR-transgenic CD8+ T cells that recognize the gp100 epitope (33) as reporters of antigen presentation. Pmel-1 T cells transferred 5 days post vaccination proliferated to a greater extent in the MLNs following amph-vaccination compared to soluble peptide immunization (Fig. 4A, B). More strikingly, Pmel-1 T cells transferred as late as two weeks after amph-gp100 immunization were still induced to undergo several rounds of cell division, while no reporter cell proliferation was detected in the MLNs of soluble peptide/CpG-immunized mice (Fig. 4C).
Figure 4: Extended exposure to both antigen and adjuvant promotes TRM development.
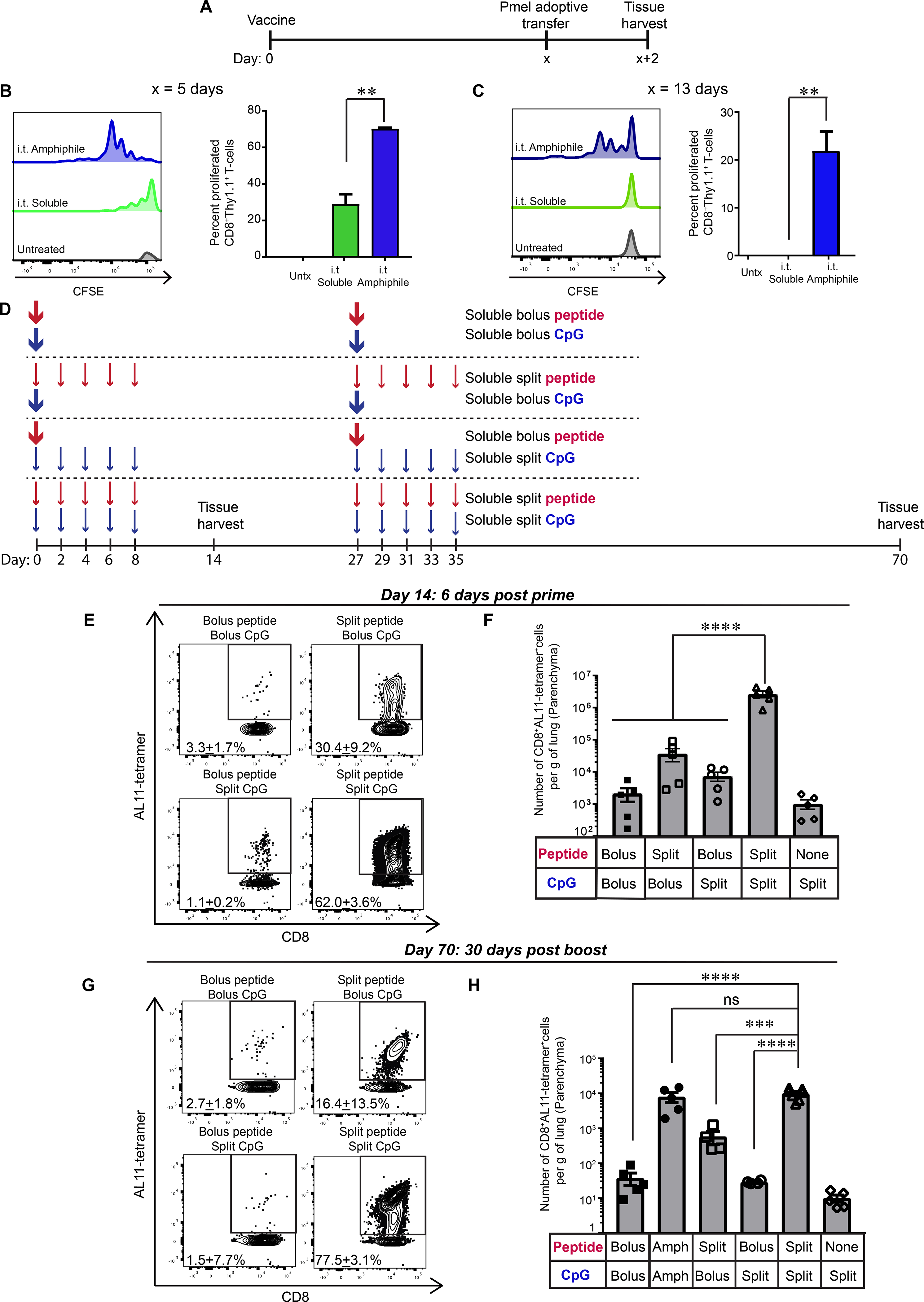
(A-C) C57BL/6 mice were vaccinated on day 0 with 10 μg gp100 and 8 μg CpG or equimolar doses of amph-gp100 and amph-CpG. At 5 or 13 days later, 5×105 CFSE-labeled Pmel-1 CD8+ T cells were adoptively transferred, and tissues were harvested 48 hours later for flow cytometry analysis. Shown are the experiment timeline (A), and representative histograms of CFSE dilution and percent of divided Pmel-1 T cells derived from MLNs (n = 4/group) of mice that received adoptive transfer on day 5 (B) or day 13 (C). (D-H) C57BL/6 mice (n = 5–7/group) were immunized with soluble AL11 peptide and CpG following the dosing patterns indicated. The total dose for each regimen was fixed at 10 μg AL11 and 8 μg CpG. Tissues were harvested on day 14 post prime or 35 days post boost on day 70. Shown are the dosing schedules (D), representative flow cytometry plots of lung CD8+AL11-tetramer+ T cells on day 14 post prime (E) or day 70 post boost (G), and enumeration of tetramer+ T cells on day 14 (F) and day 70 (H). Data are shown are mean + SEM, ***p<0.001, ****p<0.0001 as determined by one-way ANOVA. Data are representative of three independent experiments.
These data showed that amph-conjugates prolong antigen presentation in the MLN, but it was unclear if this was the dominant factor in responses to the conjugate vaccination, or if other potential effects (e.g., changes in which cells acquire antigen/adjuvant) could be important. To isolate the role of vaccine kinetics, we tested the impact of mimicking sustained antigen/adjuvant uptake in the amph-conjugate immunization by administering a simple soluble gag peptide/CpG vaccine as a bolus inoculation i.t. versus breaking up the dose into 5 injections administered every other day (“split” dosing, Fig. 4D). To differentiate the roles of peptide vs. adjuvant, we compared cohorts that received split peptide + bolus adjuvant or bolus peptide + split adjuvant dosing; the total dose of antigen and adjuvant received by each group was kept constant. As previously observed, bolus pulmonary peptide/CpG immunization elicited very low T-cell accumulation in the lungs (Fig. 4E, F). Split dosing of peptide combined with bolus CpG, or bolus peptide combined with split-dose CpG, were also ineffective. By contrast, split dosing of both antigen and adjuvant elicited a substantial antigen-specific T-cell population in lungs following a priming immunization, 70-fold greater than the split/bolus combinations (Fig. 4E, F).
To evaluate the impact of vaccine kinetics on the response to the booster immunization, we repeated these studies administering split or bolus dosing of vaccines at both prime and boost, and quantified TRM formation in the lungs 30 days after the last dose (day 70, Fig. 4D). Bolus amph-conjugate vaccination was included as a comparator group. Similar to the post prime experiments, split-dosing of both peptide and adjuvant was the most effective regimen, eliciting 17-fold greater T-cell responses than split-dosed antigen + bolus adjuvant and 28-fold greater than bolus antigen + split-dosed adjuvant (Fig. 4G, H). Notably, the sustained antigen presentation achieved by bolus immunization with amph-conjugates led to similar levels of antigen-specific TRMs as achieved by the optimal split-dosed vaccination. Similar to bolus amph-conjugate immunization, a majority of the lung-resident CD8+ T cells elicited by split-dosed vaccination expressed CD69 and ~25% were CD69+CD103+ (fig. S7). Thus, TRM establishment following mucosal vaccination is sensitive to both antigen and adjuvant kinetics, with prolonged exposure to both antigen and inflammatory cues promoting maximal resident memory establishment.
Amph-conjugates enhance T cell priming in MLNs at prime and directly in lung tissue at boost
Since amph-conjugates showed enhanced accumulation in both the lung parenchyma and MLNs relative to soluble vaccines, we next assessed whether i.t. vaccination primed T cells in the draining MLN, or alternatively, if T cells were expanded directly in the lung parenchyma. When mice were dosed with FTY720 daily starting 2 days before i.t. amph-conjugate vaccination, very few antigen-specific CD8+ T cells were detected in the lung parenchyma at day 7 (Fig. 5A, B). Administration of amph-CpG alone did not recruit antigen-specific T cells to the lung regardless of FTY720 treatment. This suggested that initial T-cell priming occurs in the MLN, as observed in prior studies (34). To assess the location of T-cell activation at boosting, we primed groups of mice, then boosted the animals in the presence of FTY720 two weeks later. In this case, FTY720 treatment had no impact on the numbers of CD8+ T cells accumulated in the lung parenchyma 7 days post boost (Fig. 5C, D), suggesting that the vaccine boost results in local antigen presentation and proliferation of CD8+ T cells within the lung parenchyma, without a significant contribution of T cells trafficking from the circulation. This result was not due to a lack of antigen presentation in draining MLNs during the boost, because antigen-specific T cells were expanded in the MLNs (Fig. 5E). Similar to the post prime results, local inflammation induced by amph-CpG alone had no impact on lung T-cell accumulation (Fig. 5D). Thus, amph-vaccines can expand T cells directly in the lung tissue following booster immunization.
Figure 5: Amph-conjugates elicit T-cell activation directly in lung tissue at boost.
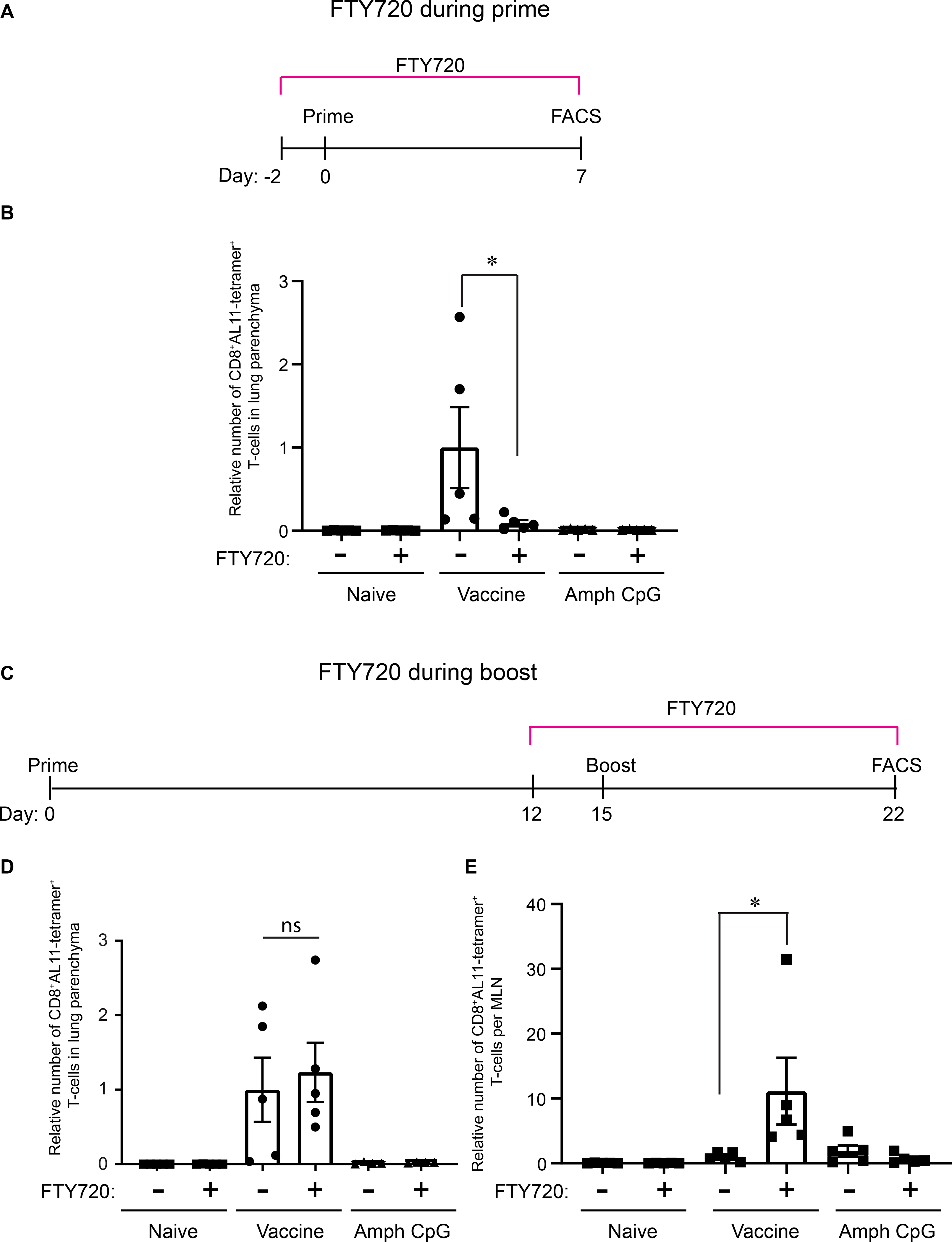
C57BL/6 mice were immunized with amph-conjugated AL11 peptide and CpG in the presence or absence of FTY720 treatment. (A, C) Schedule of amph-vaccine delivery in the presence of FTY720 during prime (A) and boost (C) immunizations. (B) Quantification of CD8+AL11-tetramer+ T cells in the lung parenchyma post prime. (D, E) Quantification of CD8+AL11-tetramer+ T cells in lungs (D) and MLN (E) 6 days post boost in the presence or absence of FTY720. Data are represented as mean + SEM, *p<0.05, as determined by one-way ANOVA, data are representative of two independent experiments (n = 5 animals/group).
Amph-conjugate vaccines promote TRM generation during booster immunization by recruitment and activation of cross-presenting DCs
Given the finding that trafficking of T cells from MLNs was not necessary for the expansion of TRMs during boosting, we finally examined the effects of amph-conjugate and soluble peptide immunization on the key antigen presenting DC populations in the lungs. CD103+ DCs and CD11b+DCs were gated as previously described (fig. S8) (35). We found no appreciable increase in either the number of CD11b+ or Batf3-dependent CD103+ DCs in the lung parenchyma 6 days following primary immunization with soluble or amph-conjugates (Fig. 6A, B). Phenotyping lung Batf3-dependent CD103+ DCs for surface expression of CD86 and MHC-II showed equivalent expression of activation markers in both soluble and amph-conjugate vaccinated mice (Fig. 6C, D). By contrast, at 6 days following administration of a booster immunization, we found that while soluble peptide vaccination again had no effect on DC populations in the lung tissue, amph-conjugate vaccination elicited a 3-fold increase in Batf3-dependent CD103+ DCs (Fig. 6E). Interestingly, the number of lung CD11b+ DCs in soluble- or amph-conjugate-vaccinated mice was significantly reduced compared to naïve mice at this time point (Fig. 6F). Approximately 30% of cross-presenting CD103+ DCs in the lung expressed high levels of MHC-II and CD86 (Fig. 6G,H). These data suggest that boosting of TRM expansion by pulmonary immunization with amph-conjugates is promoted by DC recruitment to the lung parenchyma.
Figure 6: Activated CD103+ DCs are expanded in the lung tissue following booster immunization with amph-vaccines.
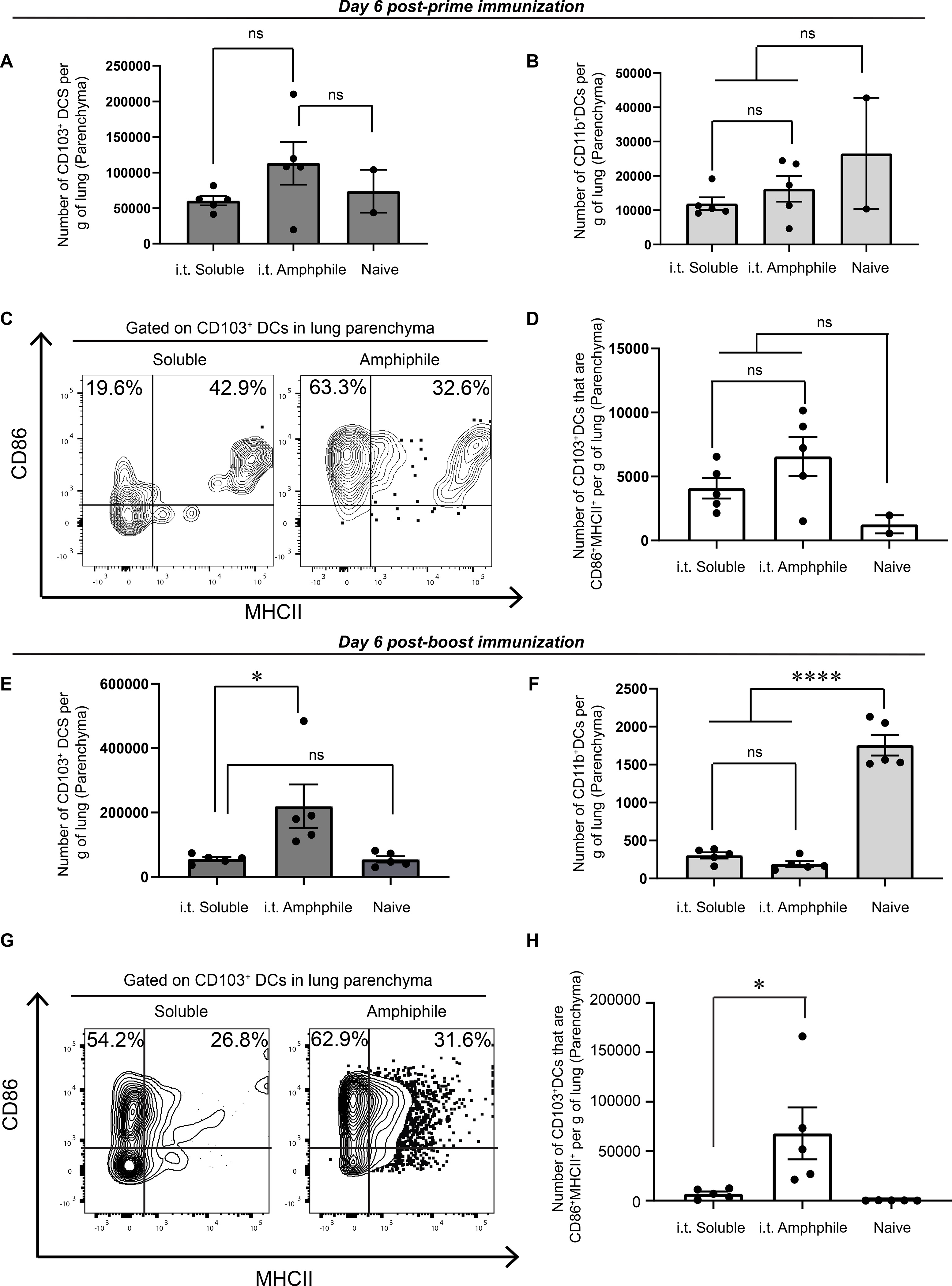
C57BL/6 mice were vaccinated on day 0 with 5 μg AL11 and 8 μg CpG or equimolar doses of amph-AL11 and amph-CpG. Tissues were harvested 6 days post prime immunization. (A, B) Enumeration of CD103+ (A) and CD11b+ (B) DCs in the lung. (C) Representative flow cytometry plots of CD86 and MHC-II expression on lung CD103+ DCs. (D) Quantification of lung MHC-II+CD86+CD103+DCs. (E-H) C57BL/6 mice were vaccinated on days 0 and 14 with 10 μg gp100 and 8 μg CpG or equimolar doses of amph-gp100 and amph-CpG. Tissues were harvested 6 days post boost immunization. (E, F) Quantification of lung CD103+ (E) and CD11b+ (F) DCs. (G) Representative flow cytometry plots of CD86 and MHC-II expression on lung CD103+ DCs. (H) Quantification of lung MHC-II+CD86+CD103+DCs. Data are represented as mean + SEM, *p<0.05, ****p<0.0001, ns: not significant, as determined by one-way ANOVA; data are representative of at least 2 independent experiments (n = 5 animals/ group).
Discussion
TRMs play an important role in protection from pathogens and tumors at barrier tissues both in pre-clinical animal models (10, 36) and in humans (11, 37). Thus, strategies to promote TRM generation at mucosal surfaces are of great interest for both cancer and infectious disease. Since vaccine efficacy is influenced by the anatomic location where immune responses are primed, one of the most effective strategies is to directly administer vaccines to mucosal surfaces (2, 9, 34). However, the natural barrier function of these tissues is an impediment to vaccination, e.g., in the lungs, where material is rapidly cleared to protect the airways. Here we exploited a natural transport pathway – bidirectional trafficking of albumin across the airway epithelium (23, 24) – as a strategy to enhance vaccination and induction of TRMs in the lung parenchyma. Albumin further serves to promote subsequent trafficking to MLNs, following its natural trafficking pathway (20). Peptide antigens and CpG adjuvant modified with a phospholipid tail to promote binding to albumin exhibited increased accumulation and persistence in the lung parenchyma. These changes in vaccine pharmacokinetics correlated with 25-fold greater lung antigen-specific T-cell populations, which expressed markers of tissue residence. Consistent with the expectations for TRMs in protective immunity, these responses correlated with enhanced protection from both pulmonary virus and tumor challenge.
Vaccines targeting respiratory immunity often employ intranasal (i.n.) or aerosol immunization routes (the latter typically modeled by i.t. administration in rodents). Although intranasal administration is easily implemented clinically, i.n. immunization preferentially primes upper respiratory tract immunity and is less effective for lower respiratory tract antibody and TRM responses (38). We confirmed these findings following rigorous i.n. vs. i.t. immunization with amph-vaccines. Small studies in human volunteers have also suggested that aerosol immunization reaching the lower respiratory tract is more immunogenic than i.n. immunization (39). Hence aerosol immunization may be desirable for more complete lung protection.
We previously showed that amph-conjugate peptide vaccines administered parenterally lead to much longer-lived antigen presentation compared to soluble peptide vaccines in draining lymph nodes (21). Similarly, here we found that amph-conjugate pulmonary immunization leads to antigen presentation persisting at least 13 days. Using simplified “split” dosing protocols with soluble peptide/adjuvant, we found that prolonged availability of both antigen and adjuvant is important for maximal TRM induction in the lungs, and that sustained peptide/adjuvant availability in the MLNs and/or lung tissue appears to be the major factor governing the potency of amph-conjugate immunization. These data are consistent with prior reports suggesting that inflammatory stimuli must be coupled with pulmonary antigen to induce TRM formation (40, 41). Additionally, several groups have demonstrated that continued viral antigen presentation influences the longevity and magnitude of both CD4+(42) and CD8+ (43) TRMs in the context of pulmonary infection. Recently, Braeckel-Brudimir et al. showed that repeated antigen encounter through multiple rounds of infection increases the magnitude and durability of lung CD8+ TRMs, through increased seeding and improved survival of T cells within the lung (13).
Analysis of the impact of FTY720 treatment on amph-conjugate vaccine responses revealed that the vaccine mechanisms of action differ during prime vs. booster immunizations. Post prime, T cell activation in MLNs and homing to the lung tissue was required for establishment of lung-resident responses. This finding is consistent with studies showing that DCs in the MLN can imprint responding T cells with lung homing receptor expression (44). By contrast, during boosting, T-cell expansion was not impacted by blocking T-cell egress from lymphoid organs. This is in accordance with data from other groups who have observed that proliferation of antigen-primed CD8+ T cells can occur in non-lymphoid tissues (34, 45).
Both soluble and amph-vaccination led to a reduction in CD11b+ DCs post boost (Figure 6F). Although it is unclear why CD11b+ DCs were reduced, the phenotype of myeloid cells in the lung is known to change with exposure to inflammatory stimuli (46, 47). Furthermore, the finding that amph-peptide immunization is entirely dependent on Batf3+ dendritic cells (Fig. 2H) suggests that these cells do not play a significant role in antigen presentation after pulmonary amphiphile vaccination. Interestingly, amph-conjugate immunization both substantially expanded Batf3-dependent CD103+ DCs in the lungs during boosting and induced greater DC activation. Thus, amph-conjugate immunization is able to enhance T-cell activation in draining lymph nodes and directly in the lung tissue, bolstering antigen presentation at both sites for effective responses during both the prime and boost.
In conclusion, we have shown that mucosal immunization with amph-vaccines efficiently generates functional TRMs in the lung, mediated by efficient uptake across the lung epithelium and prolonged antigen and adjuvant stimulation. Using amphiphile vaccines to generate robust mucosal immunity mediated by TRMs could be beneficial in the context of prophylaxis for infectious diseases, the need for which has been underscored by the recent global coronavirus pandemic for which peptide vaccine platforms are actively being explored (48).
Materials and Methods
Study Design
The aims of this study were to characterize the immune response to amph-vaccines delivered into the lungs, to assess whether this form of vaccination could protect from virus or tumor challenges, and define mechanisms of action for this vaccine. T cells in the lung were enumerated by flow cytometry following standard protocols. To study the functional efficacy of T cells generated in the lung, mice were infected intra-tracheally with lethal doses of vaccinia virus expressing a defined antigen. For tumor studies, melanoma cells were introduced into the lung by intravenous injection. All animal experiments were repeated at least twice with n = 2–10 animals/group being studied. No outliers were excluded from the data analyses and studies were not blinded.
Materials
Peptides with an N-terminal cysteine (AL11: CAAVKNWMTQTL, gp100: CAVGALEGPRNQDWLGVPRQL, Trp1: CTAPDNLGYM, Trp2: CSVYDFFVWL) (26, 49) were ordered from Genscript, NJ and conjugation to maleimide-PEG-DSPE amphiphiles was performed as previously described (20, 21). Amphiphile-conjugated CpG (Amph-CpG) was produced by solid-phase synthesis of Class B CpG 1826 oligonucleotide with a G2 spacer conjugated to an 18-carbon diacyl tail via the 5’ end as previously described (20).
Animals
All mice were purchased from The Jackson Laboratory. Vaccination experiments were performed in 6–8 week old WT C57BL/6 mice (Strain number: 000664), FcRn−/− (Strain number: 003982) and Batf3−/− mice (Strain number: 013755). Tumor studies were performed in albino C57BL/6 mice (Strain number: 000058). For T cell adoptive transfer experiments gp100-specific T cells were isolated from B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J Pmel-1 transgenic mice (Strain number: 005023). All experiments were performed in accordance with NIH ethical guidelines following an Institutional Animal Care and Use Committee-approved protocol.
Vaccinations
Mice were immunized with peptide and CpG either in their soluble or amph-conjugate forms at indicated doses. Intratracheal administration was performed using a platform and light source to visualize the tracheal opening of an anesthetized mouse and instill up to 50 μl of vaccine solution or 106 pfu of vaccinia virus as described (50). Subcutaneous vaccination was performed at the base of the tail.
Vaccinia Challenge
Four months after vaccine boosting, mice were infected intratracheally with vaccinia virus expressing the AL11 epitope from SIV gag (106 pfu per mouse) (50). Animal weights were recorded daily post challenge and animals were euthanized when their weight decreased by > 20%. To quantify viral titers, lung tissue homogenates were used to infect CV-1 cells in culture and resulting plaques were quantified (51). Mice were also treated with 1 mg/kg FTY720 (or vehicle control) delivered daily by intraperitoneal injection beginning 5 days prior to vaccinia infection.
Tumor Studies
100,000 or 400,000 B16F10 cells that were transduced with GFP and luciferase were injected intravenously to seed melanoma tumors in the lungs. When used, anti-PD1 was administered intra-peritoneally at 200 mg/kg every 3 days. Tumor growth was monitored by bioluminescence imaging using an IVIS Spectrum imager and mice were sacrificed when the average radiance or ROIs over the lungs of animals exceeded 5X106 photons/s/cm2/sr.
Adoptive Transfer
CD8+ T cells were isolated from spleens of Pmel-1 transgenic mice by negative selection using an EasySep mouse CD8+ T cell isolation kit (Stemcell Technologies, MA). T cells were labeled with CFSE and 500,000 cells were injected retro-orbitally followed by sacrifice 2 days later and recovery of mediastinal lymph nodes for analysis of CFSE dilution in Pmel-1 T cells by flow cytometry.
Flow Cytometry
Lungs, mediastinal lymph nodes and peripheral blood were collected from immunized and control animals. Lung lobes were separated and cut into small pieces in PBS containing 0.5% FBS and 0.05M HEPES buffer with 120U/ml Collagenase IV (Worthington Bio, NJ) and 40 U/m DNase I (bioWorld, OH). Single cell suspensions were made using the gentleMACS lung dissociation protocol (52). Mediastinal lymph nodes were passed through a 70 μm cell strainer to obtain a single cell suspension of lymphocytes. Red blood cells were lysed using ACK lysis buffer (Gibco, MD). Single cell suspensions were washed with PBS and re-suspended in flow cytometry staining buffer (PBS+2% FBS+0.1% sodium azide). Cells were stained with fluorescently labeled antibodies and tetramers and samples were analyzed on LSR Fortessa or Canto (Becton Dickinson, NJ) flow cytometry instruments. Data were analyzed using FlowJo (TreeStar). Antibodies used were the following: anti-CD8α (53–6.7), anti-CD69 (H1.2F3), anti-CD103 (2E7), anti-TNFα (MPX-XT22), anti-IFNγ (XMG1.2), anti-CD11a (H155–78), anti-CD49a (Hα31/8), anti-CD11c (N418), anti-CD11b (M1/70), anti-CD24 (M1/69), anti-CD64 (X54–5/7.1), anti-SiglecF (1RNM44N) and anti-CD45 (30-F11). CD103+ DCs and CD11b+ DCs in the lung were identified by using a previously established gating scheme (fig. S8) (53).
Immunofluorescence
Lungs from immunized or control mice were isolated, fixed in periodate-lysine-paraformaldehyde (PLP) buffer and inflated with 2 ml of a mixture of equal parts PBS and optimum cutting temperature (OCT) compound as described (52). Inflated lungs were embedded in OCT and frozen. Cryosections were cut and stained with DAPI. Images were acquired using a 30X objective on an Olympus FV1200 laser scanning confocal microscope. Image analysis was performed using ImageJ (NIH).
Statistical Analysis
Sample sizes were chosen based on known effect sizes of amph-conjugate immunization in prior work (20, 21) and pilot experiments. Statistical analyses were performed using GraphPad Prism version 8. Analysis of multiple groups was performed using one-way or two-way ANOVA using the Tukey post hoc test for multiple comparison within groups. Graphs indicate the mean and standard error of the mean (SEM) for the various groups. p<0.05 was considered to be statistically significant.
Supplementary Material
Table S1. Raw data file (Excel spreadsheet)
Fig. S1. Structure of amph-vaccines
Fig. S2. Intratracheal delivery of gp100 amph-vaccine results in functional antigen-specific T cells in the lung parenchyma
Fig. S3. Establishment of antigen-specific TRM cells in the lungs following amph-vaccine immunization is impaired in FcRn-deficient animals
Fig. S4. Intratracheal delivery of amph-vaccine results in increased antigen-specific T cells in the lung parenchyma compared to intra-nasal delivery
Fig. S5. FTY720 treatment significantly diminishes T cells in peripheral circulation
Fig. S6. Intratracheal therapeutic vaccination with amph-vaccine combined with checkpoint blockade causes tumor regression
Fig. S7. Antigen-specific T cells elicited by extended soluble dosing express markers of tissue residence
Fig. S8. Lung dendritic cell gating scheme
Acknowledgments
We thank the Koch Institute Swanson Biotechnology Center for technical support, specifically the flow cytometry and microscopy facilities. We thank Peter Choi for reviewing and editing this manuscript.
Funding:
This work was supported by the following grants: The Bridge Project, a partnership between the Koch Institute at MIT and the Dana-Farber/ Harvard Cancer Center, the Marble Center for Cancer Nanomedicine, the Ragon Institute of MGH, MIT, and Harvard, and the NIH (awards EB022433 and CA247632). KR was funded by a Koch Institute Quinquennial fellowship. DJI is an investigator of the Howard Hughes Medical Institute.
Footnotes
Competing interests: DJI and KDM are inventors on a patent application based on use of amphiphile vaccine technology. The patent has been licensed to Elicio Therapeutics, a company co-founded by DJI. The other authors declare that they have no competing interests.
Data and Materials Availability: All additional data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References
- 1.Wu T, Hu Y, Lee Y-TY-T, Bouchard KR, Benechet A, Khanna K, Cauley LS, Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J. Leukoc. Biol. 95, 215–24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perdomo C, Zedler U, Kühl AA, Lozza L, Saikali P, Sander LE, Vogelzang A, Kaufmann SHE, Kupz A, Mucosal BCG Vaccination Induces Protective Lung-Resident Memory T Cell Populations against Tuberculosis. MBio. 7, e01686–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan HX, Wheatley AK, Esterbauer R, Jegaskanda S, Glass JJ, Masopust D, De Rose R, Kent SJ, Induction of vaginal-resident HIV-specific CD8 T cells with mucosal prime-boost immunization. Mucosal Immunol. 11, 994–1007 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Schenkel JM, Fraser KA, Vezys V, Masopust D, Sensing and alarm function of resident memory CD8+ T cells. Nat. Immunol. 14, 509–513 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D, T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science. 346, 98–101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Purwar R, Campbell J, Murphy G, Richards WG, Clark RA, Kupper TS, Resident Memory T cells (TRM) are abundant in human lung: Diversity, function, and antigen specificity. PLoS One. 6, 16245 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turner DL, Bickham KL, Thome JJ, Kim CY, D’Ovidio F, Wherry EJ, Farber DL, Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol. 7, 501–10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pallett LJ, Davies J, Colbeck EJ, Robertson F, Hansi N, Easom NJW, Burton AR, Stegmann KA, Schurich A, Swadling L, Gill US, Male V, Luong TV, Gander A, Davidson BR, Kennedy PTF, Maini MK, IL-2high tissue-resident T cells in the human liver: Sentinels for hepatotropic infection. J. Exp. Med. 214, 1567–1580 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sandoval F, Terme M, Nizard M, Badoual C, Bureau M-F, Freyburger L, Clement O, Marcheteau E, Gey A, Fraisse G, Bouguin C, Merillon N, Dransart E, Tran T, Quintin-Colonna F, Autret G, Thiebaud M, Suleman M, Riffault S, Wu T-C, Launay O, Danel C, Taieb J, Richardson J, Zitvogel L, Fridman WH, Johannes L, Tartour E, Mucosal Imprinting of Vaccine-Induced CD8+ T Cells Is Crucial to Inhibit the Growth of Mucosal Tumors. Sci. Transl. Med. 5, 172ra20–172ra20 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nizard M, Roussel H, Diniz MO, Karaki S, Tran T, Voron T, Dransart E, Sandoval F, Riquet M, Rance B, Marcheteau E, Fabre E, Mandavit M, Terme M, Blanc C, Escudie J-B, Gibault L, Barthes FLP, Granier C, Ferreira LCS, Badoual C, Johannes L, Tartour E, Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun. 8, 15221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ganesan A-P, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, Samaniego-Castruita D, Singh D, Seumois G, Alzetani A, Woo E, Friedmann PS, V King E, Thomas GJ, Sanchez-Elsner T, Vijayanand P, Ottensmeier CH, Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat. Immunol. 18, 940–950 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergsbaken T, Bevan MJ, Fink PJ, Local Inflammatory Cues Regulate Differentiation and Persistence of CD8+ Tissue-Resident Memory T Cells. Cell Rep. 19, 114–124 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Braeckel-Budimir N, Varga SM, Badovinac VP, Harty Correspondence JT, Harty JT, Repeated Antigen Exposure Extends the Durability of Influenza-Specific Lung-Resident Memory CD8+ T Cells and Heterosubtypic Immunity. Cell Rep. 24, 3374–3382 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang N, Bevan MJ, Transforming growth factor-β signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity. 39, 687–696 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, Wang D, Getzler AJ, Nguyen T, Crotty S, Wang W, Pipkin ME, Goldrath AW, Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature. 552, 253–257 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li W, Joshi MD, Singhania S, Ramsey KH, Murthy AK, Peptide vaccine: Progress and challenges. Vaccines. 2 (2014), pp. 515–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bezu L, Kepp O, Cerrato G, Pol J, Fucikova J, Spisek R, Zitvogel L, Kroemer G, Galluzzi L, Trial watch: Peptide-based vaccines in anticancer therapy. Oncoimmunology. 7 (2018), doi:1 10.1080/2162402X.2018.1511506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Melief CJM, van Hall T, Arens R, Ossendorp F, van der Burg SH, Therapeutic cancer vaccines. J. Clin. Invest. 125, 1–12 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inoue N, Abe M, Kobayashi R, Yamada S, in Advances in Experimental Medicine and Biology (Springer New York LLC, 2018), vol. 1045, pp. 271–296. [DOI] [PubMed] [Google Scholar]
- 20.Liu H, Moynihan KD, Zheng Y, Szeto GL, V Li A, Huang B, Van Egeren DS, Park C, Irvine DJ, Structure-based programming of lymph-node targeting in molecular vaccines. Nature. 507, 519–22 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moynihan KD, Holden RL, Mehta NK, Wang C, Karver MR, Dinter J, Liang S, Abraham W, Melo MB, Zhang AQ, Li N, Le Gall S, Pentelute BL, Irvine DJ, Enhancement of Peptide Vaccine Immunogenicity by Increasing Lymphatic Drainage and Boosting Serum Stability. Cancer Immunol. Res. 6, 1025–1038 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma L, Dichwalkar T, Chang JYH, Cossette B, Garafola D, Zhang AQ, Fichter M, Wang C, Liang S, Silva M, Kumari S, Mehta NK, Abraham W, Thai N, Li N, Dane Wittrup K, Irvine DJ, Enhanced CAR–T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science (80-. ). 365, 162–168 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sand KMK, Bern M, Nilsen J, Noordzij HT, Sandlie I, Andersen JT, Unraveling the interaction between FcRn and albumin: opportunities for design of albumin-based therapeutics. Front. Immunol. 5 (2014), doi:10.3389/fimmu.2014.00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson CL, Chaudhury C, Kim J, Bronson CL, Wani MA, Mohanty S, Perspective-- FcRn transports albumin: relevance to immunology and medicine. Trends Immunol. 27, 343–8 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Kantrow SP, Shen Z, Jagneaux T, Zhang P, Nelson S, Neutrophil-mediated lung permeability and host defense proteins. Am. J. Physiol. Lung Cell. Mol. Physiol. 297, L738–45 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barouch DH, Pau MG, Custers JHHV, Koudstaal W, Kostense S, Havenga MJE, Truitt DM, Sumida SM, Kishko MG, Arthur JC, Korioth-Schmitz B, Newberg MH, Gorgone DA, Lifton MA, Panicali DL, Nabel GJ, Letvin NL, Goudsmit J, Immunogenicity of Recombinant Adenovirus Serotype 35 Vaccine in the Presence of Pre-Existing Anti-Ad5 Immunity. J. Immunol. 172, 6290–6297 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Anderson KG, Sung H, Skon CN, Lefrancois L, Deisinger A, Vezys V, Masopust D, Cutting edge: intravascular staining redefines lung CD8 T cell responses. J. Immunol. 189, 2702–6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ray SJ, Franki SN, Pierce RH, Dimitrova S, Koteliansky V, Sprague AG, Doherty PC, de Fougerolles AR, Topham DJ, The Collagen Binding α1β1 Integrin VLA-1 Regulates CD8 T Cell-Mediated Immune Protection against Heterologous Influenza Infection. Immunity. 20, 167–179 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Cyster JG, Schwab SR, Sphingosine-1-Phosphate and Lymphocyte Egress from Lymphoid Organs. Annu. Rev. Immunol. 30, 69–94 (2012). [DOI] [PubMed] [Google Scholar]
- 30.MacKay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, Tscharke DC, Heath WR, Inouye M, Carbone FR, Gebhardt T, The developmental pathway for CD103+ CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 14, 1294–1301 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, Braun A, Masson F, Kallies A, Belz GT, Carbone FR, T-box Transcription Factors Combine with the Cytokines TGF-β and IL-15 to Control Tissue-Resident Memory T Cell Fate. Immunity. 43, 1101–1111 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Adachi K, Chiba K, FTY720 story. Its discovery and the following accelerated development of sphingosine 1-phosphate receptor agonists as immunomodulators based on reverse pharmacology. Perspect. Medicin. Chem. 1, 11–23 (2007). [PMC free article] [PubMed] [Google Scholar]
- 33.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, Hwang LN, Feigenbaum L, Kruisbeek AM, Rosenberg SA, Restifo NP, Tumor Regression and Autoimmunity after Reversal of a Functionally Tolerant State of Self-reactive CD8 + T Cells. J. Exp. Med. 198, 569–580 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Çuburu N, Graham BS, Buck CB, Kines RC, Pang Y-YS, Day PM, Lowy DR, Schiller JT, Intravaginal immunization with HPV vectors induces tissue-resident CD8+ T cell responses. J. Clin. Invest. 122, 4606–20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GRS, Perlman H, Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol. 49, 503–510 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schenkel JM, Masopust D, Tissue-resident memory T cells. Immunity. 41, 886–97 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Piet B, de Bree GJ, Smids-Dierdorp BS, van der Loos CM, Remmerswaal EBM, von der Thüsen JH, van Haarst JMW, Eerenberg JP, ten Brinke A, van der Bij W, Timens W, van Lier RAW, Jonkers RE, CD8+ T cells with an intraepithelial phenotype upregulate cytotoxic function upon influenza infection in human lung. J. Clin. Invest. 121, 2254–2263 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanders MT, Deliyannis G, Pearse MJ, McNamara MK, Brown LE, Single dose intranasal immunization with ISCOMATRIX™ vaccines to elicit antibody-mediated clearance of influenza virus requires delivery to the lower respiratory tract. Vaccine. 27, 2475–2482 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Nardelli-Haefliger D, Lurati F, Wirthner D, Spertini F, Schiller JT, Lowy DR, Ponci F, De Grandi P, Immune responses induced by lower airway mucosal immunisation with a human papillomavirus type 16 virus-like particle vaccine. Vaccine. 23, 3634–3641 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Takamura S, Yagi H, Hakata Y, Motozono C, McMaster SR, Masumoto T, Fujisawa M, Chikaishi T, Komeda J, Itoh J, Umemura M, Kyusai A, Tomura M, Nakayama T, Woodland DL, Kohlmeier JE, Miyazawa M, Specific niches for lung-resident memory CD8+ T cells at the site of tissue regeneration enable CD69-independent maintenance. J. Exp. Med. 213, 3057–3073 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McMaster SR, Wein AN, Dunbar PR, Hayward SL, Cartwright EK, Denning TL, Kohlmeier JE, Pulmonary antigen encounter regulates the establishment of tissue-resident CD8 memory T cells in the lung airways and parenchyma article. Mucosal Immunol. 11, 1071–1078 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jelley-Gibbs DM, Brown DM, Dibble JP, Haynes L, Eaton SM, Swain SL, Unexpected prolonged presentation of influenza antigens promotes CD4 T cell memory generation. J. Exp. Med. 202, 697–706 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim TS, Hufford MM, Sun J, Fu YX, Braciale TJ, Antigen persistence and the control of local T cell memory by migrant respiratory dendritic cells after acute virus infection. J. Exp. Med. 207, 1161–1172 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mikhak Z, Strassner JP, Luster AD, Lung dendritic cells imprint T cell lung homing and promote lung immunity through the chemokine receptor CCR4. J. Exp. Med. 210, 1855–69 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stary G, Olive A, Radovic-Moreno AF, Gondek D, Alvarez D, Basto PA, Perro M, Vrbanac VD, Tager AM, Shi J, Yethon JA, Farokhzad OC, Langer R, Starnbach MN, von Andrian UH, VACCINES. A mucosal vaccine against Chlamydia trachomatis generates two waves of protective memory T cells. Science. 348, aaa8205 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ginhoux F, Schlitzer A, CD11b+ DCs rediscovered: Implications for vaccination. Expert Rev. Vaccines. 13 (2014), pp. 445–447. [DOI] [PubMed] [Google Scholar]
- 47.Desch AN, Henson PM, V Jakubzick C, Pulmonary dendritic cell development and antigen acquisition. Immunol. Res. 55, 178–86 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang N, Shang J, Jiang S, Du L, Subunit Vaccines Against Emerging Pathogenic Human Coronaviruses. Front. Microbiol. 11 (2020), doi:10.3389/fmicb.2020.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moynihan KDKD, Opel CFCF, Szeto GLGL, Tzeng A, Zhu EFEF, Engreitz JMJM, Williams RTRT, Rakhra K, Zhang MHMH, Rothschilds AMAM, Kumari S, Kelly RLRL, Kwan BHBH, Abraham W, Hu K, Mehta NKNK, Kauke MJMJ, Suh H, Cochran JRJR, Lauffenburger DADA, Wittrup KDD, Irvine DJDJ, Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med. 22, 1402–1410 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suh H, Elkhader J, Seidman MA, Abraham W, Im E-J, Irvine DJ, Barouch DH, Foley MH, Yen M, Moon JJ, Li AV, Generation of Effector Memory T Cell-Based Mucosal and Systemic Immunity with Pulmonary Nanoparticle Vaccination. Sci. Transl. Med. 5, 204ra130–204ra130 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakaya Y, Zheng H, García-Sastre A, Enhanced cellular immune responses to SIV Gag by immunization with influenza and vaccinia virus recombinants. Vaccine. 21, 2097–106 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Joshi NS, Akama-Garren EH, Lu Y, Lee D-Y, Chang GP, Li A, DuPage M, Tammela T, Kerper NR, Farago AF, Robbins R, Crowley DM, Bronson RT, Jacks T, Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-tumor T Cell Responses. Immunity. 43, 579–90 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.V Misharin A, Morales-Nebreda L, Mutlu GM, Budinger GRS, Perlman H, Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol. 49, 503–10 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Raw data file (Excel spreadsheet)
Fig. S1. Structure of amph-vaccines
Fig. S2. Intratracheal delivery of gp100 amph-vaccine results in functional antigen-specific T cells in the lung parenchyma
Fig. S3. Establishment of antigen-specific TRM cells in the lungs following amph-vaccine immunization is impaired in FcRn-deficient animals
Fig. S4. Intratracheal delivery of amph-vaccine results in increased antigen-specific T cells in the lung parenchyma compared to intra-nasal delivery
Fig. S5. FTY720 treatment significantly diminishes T cells in peripheral circulation
Fig. S6. Intratracheal therapeutic vaccination with amph-vaccine combined with checkpoint blockade causes tumor regression
Fig. S7. Antigen-specific T cells elicited by extended soluble dosing express markers of tissue residence
Fig. S8. Lung dendritic cell gating scheme