

Abstract
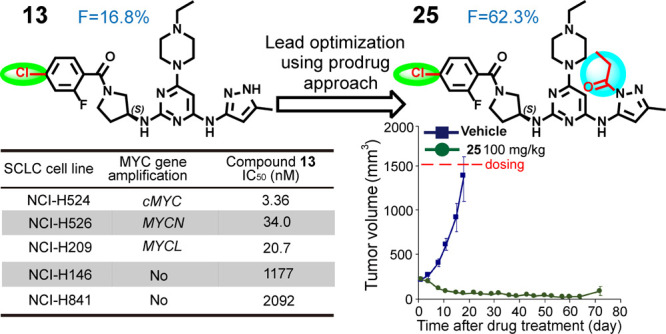
The A-type Aurora kinase is upregulated in many human cancers, and it stabilizes MYC-family oncoproteins, which have long been considered an undruggable target. Here, we describe the design and synthesis of a series of pyrimidine-based derivatives able to inhibit Aurora A kinase activity and reduce levels of cMYC and MYCN. Through structure-based drug design of a small molecule that induces the DFG-out conformation of Aurora A kinase, lead compound 13 was identified, which potently (IC50 < 200 nM) inhibited the proliferation of high-MYC expressing small-cell lung cancer (SCLC) cell lines. Pharmacokinetic optimization of 13 by prodrug strategies resulted in orally bioavailable 25, which demonstrated an 8-fold higher oral AUC (F = 62.3%). Pharmacodynamic studies of 25 showed it to effectively reduce cMYC protein levels, leading to >80% tumor regression of NCI-H446 SCLC xenograft tumors in mice. These results support the potential of 25 for the treatment of MYC-amplified cancers including SCLC.
Introduction
Deregulation of MYC-family oncogenes (i.e., cMYC, MYCN, and MYCL) is associated with a poor prognosis and unfavorable survival of cancer patients.1 Amplification of MYC-family oncogenes has been observed in 28% cancers in The Cancer Genome Atlas (TCGA).2 Sustained MYC-family protein levels can initiate tumor formation, accelerate tumor progression, and help in tumor maintenance. Many MYC-driven metabolic changes such as glycolysis and glutaminolysis support the increased need of nucleic acids, proteins, and lipids during rapid cell proliferation.3,4 Despite the pivotal role of MYC in normal tissue regeneration,5,6 several murine-based studies have supported MYC as a potential therapeutic target for cancers. For example, a conditional transgenic mouse model for MYC-induced tumorigenesis demonstrated that brief inactivation of cMYC was sufficient to elicit sustained regression of transplanted osteogenic sarcoma cells,7 and knockdown of cMYC in glioma cancer stem cells reduced proliferation with concomitant cell cycle arrest and increased apoptosis, whereas nonstem glioma cells displayed limited dependence on MYC expression for survival and proliferation.8
MYC has long been considered “undruggable” by small-molecule inhibitors due to its lack of enzymatic activity and an accessible affinity pocket. Nevertheless, alternative approaches toward the modulation of MYC oncogenic functions via indirect strategies have been extensively investigated.9 For example, use of Omomyc, a 90 amino acid dominant-negative form of MYC, which competes with cellular MYC, prevented formation of MYC/MAX heterodimers and thus, transcriptional activation of a specific set of genes.10 The demonstrated antitumor efficacy of the Omomyc miniprotein in several preclinical mouse models has paved the way to clinical trials.11,12 MYC transcription was downregulated by the small-molecule bromodomain inhibitor JQ1, which showed potent anticancer effects both in vitro and in vivo in multiple hematopoietic cancers and pancreatic ductal adenocarcinoma exhibiting high cMYC.9,13 CD532, a kinase inhibitor that induces allosteric conformation change of Aurora A kinase, was found to weaken MYCN–Aurora A interaction, thereby releasing MYCN protein for proteasome degradation, resulting in tumor growth inhibition.14−18 Although CD532 lacks drug-like properties due to its short half-life and poor oral bioavailability,14 this evidence validates an effective strategy for the targeting of MYC-family oncoproteins.
Several small-molecule inhibitors targeting Aurora kinases have been developed in the last two decades,19−25 including inhibitors which bind to the protein at the adenosine triphosphate (ATP) binding site which contains a DFG (Asp–Phe–Gly) motif that could adopt two different conformations, the active DFG-in and the inactive DFG-out states. In the DFG-out conformation, the Asp and Phe of the DFG motif of the activation loop swap positions, resulting in the formation of a new allosteric pocket and better enzymatic selectivity.26 Using a typical DFG-in inhibitor scaffold of Aurora A, Martin et al. discovered that induced-dipole forces along the Ala273 side chain alter the charge distribution of the DFG backbone, leading to DFG-out inhibitors that are highly potent for Aurora A.27 We applied this concept to structure-based drug design (SBDD) guided with enzymatic assays and western blot analyses and identified a novel class of 6-methyl-N4-(5-methyl-1H-pyrazol-3-yl)-N2-(pyrrolidin-3-yl) pyrimidine-2,4-diamine derivatives (A), which potentially induce a flip in the DFG activation loop of Aurora A, resulting in reduced levels of cMYC and MYCN. Pharmacokinetic (PK) optimization using prodrug strategies for the lead compound 13 resulted in 25, which demonstrated sufficient oral bioavailability and led to >80% regression of a cMYC-amplified small-cell lung cancer (SCLC) in a xenograft mouse model.
Results and Discussion
Chemistry
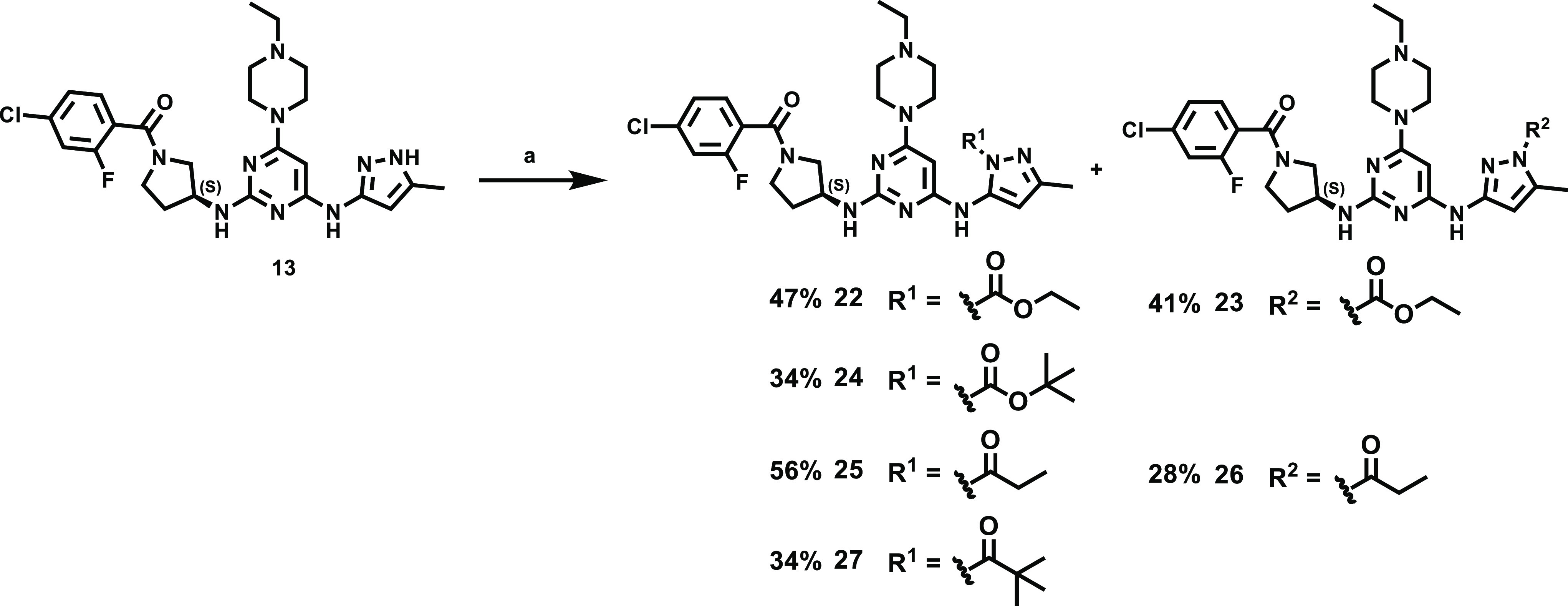
Compounds 1–21 (Table 1) were prepared according to the general synthetic method depicted in Schemes 1 and 2. A substitution reaction of compound 28 with 3-amino-5-methylpyrazole in THF gave the desired compound 29, which was converted to the corresponding compound 30 by the reaction with (S)-(3-aminopyrrolidin-1-yl) (3-chloro-2-fluorophenyl)methanone in 1-pentanol at 120 °C. Finally, compound 30 was reacted with 1-ethyl piperazine in the presence of trimethylamine to give the desired target compound 1 in 58% yield. Likewise, SNAr (nucleophilic aromatic substitution) reactions of compound 30 with a variety of amines gave the corresponding compounds 2–5 in moderate yields (26–57%) (Scheme 1). Treatment of 4,6-dichloro-2-(methylsulfonyl)pyrimidine (31) with tert-butyl (S)-3-aminopyrrolidine-1-carboxylate in the presence of triethylamine gave rise to 32 (56%), which was coupled with 5-methyl-1H-pyrazol-3-amine in the presence of NaI and triethylamine in DMSO to provide ether 33 in 85% yield (Scheme 2). Coupling of 33 with 1-ethylpiperazine proceeded at 140 °C to afford 34 in 84% yield. Acidification of 34 gave the corresponding hydrochloride amine salt, which in turn was treated with 4-chloro-2-fluorobenzoic acid and propanephosphonic acid anhydride (T3P) in dichloromethane to yield amide 13 in 56% yield over two steps. Similarly, coupling of the hydrochloride amine salt (from 34) with a selection of differentially substituted benzoic and pyridinecarboxylic acids and sulfonyl chlorides gave the corresponding amide and sulfonamide derivatives 6–21 in moderate to good yields (44–85%). In addition, the regioisomers 25 and 26 (2:1) were obtained at 140 °C in the presence of propionic anhydride in good yield (84%) (Scheme 3). By the same token, compound 13 directly underwent acylation with selected anhydrides or dicarbonates to afford amide prodrug compound 27 and carbamate prodrug compounds 22–24 in moderate to good yields (34–47%, Scheme 3). The purity of all synthesized compounds was established to be at least 95% by HPLC (see the Experimental Section) prior to their utilization in biological assays and animal studies.
Table 1. Biological Evaluation of Pyrimidines Derivatives on the Enzymatic Activity of Aurora A and Levels of MYC in MYC-Amplified Cancer Cells.

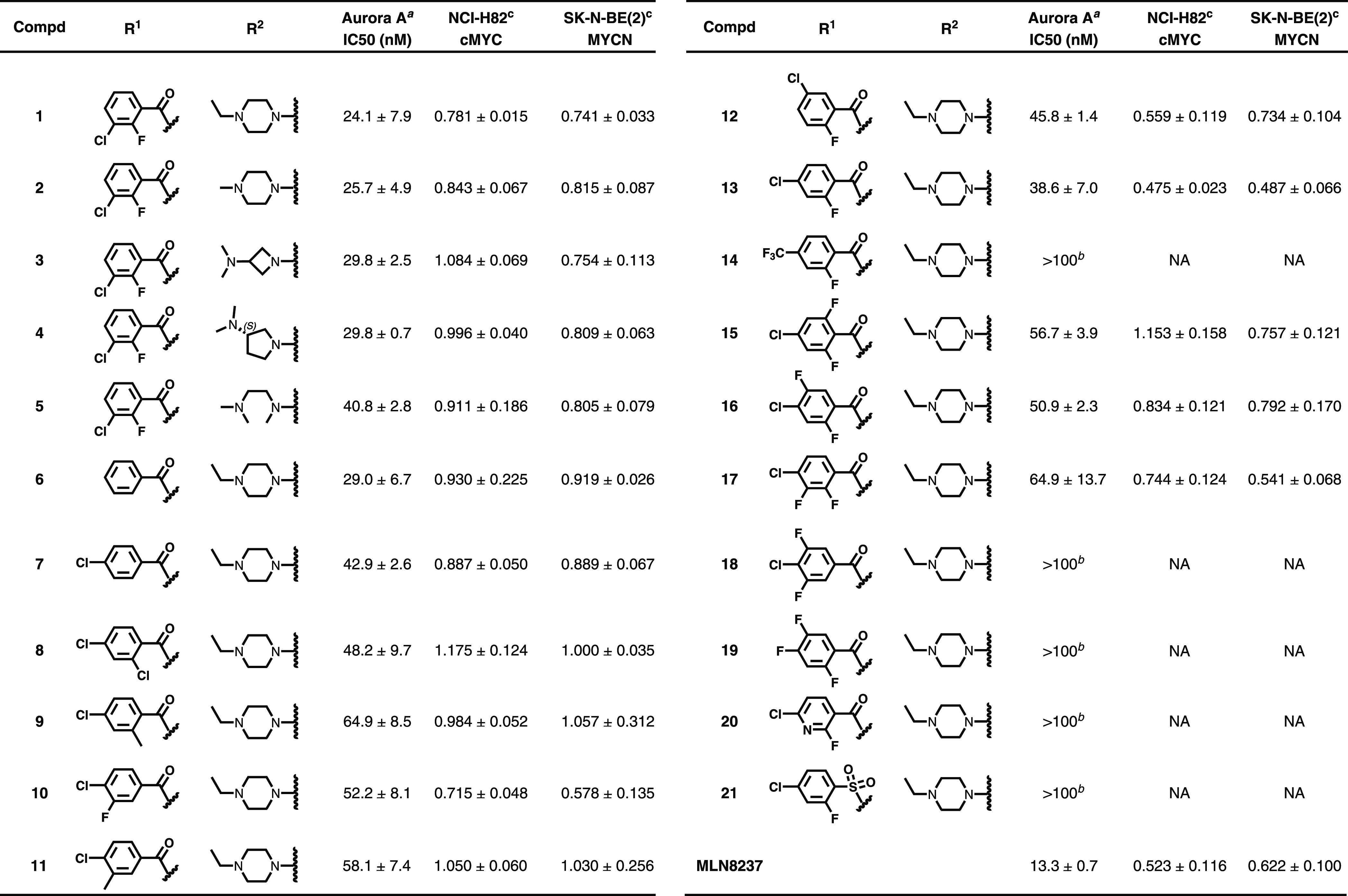
Values are the mean of 2–3 independent determinations ± SD (standard deviation).
Inhibition < 50% at 100 nM.
Relative protein level (mean ± SD) of cMYC in NCI-H82 and MYCN in SK-N-BE(2) under 1.0 μM of compound treatment. Experiments were undertaken in triplicate.
Scheme 1. Synthesis of Pyrimidine Derivatives with a Variety of Amines.
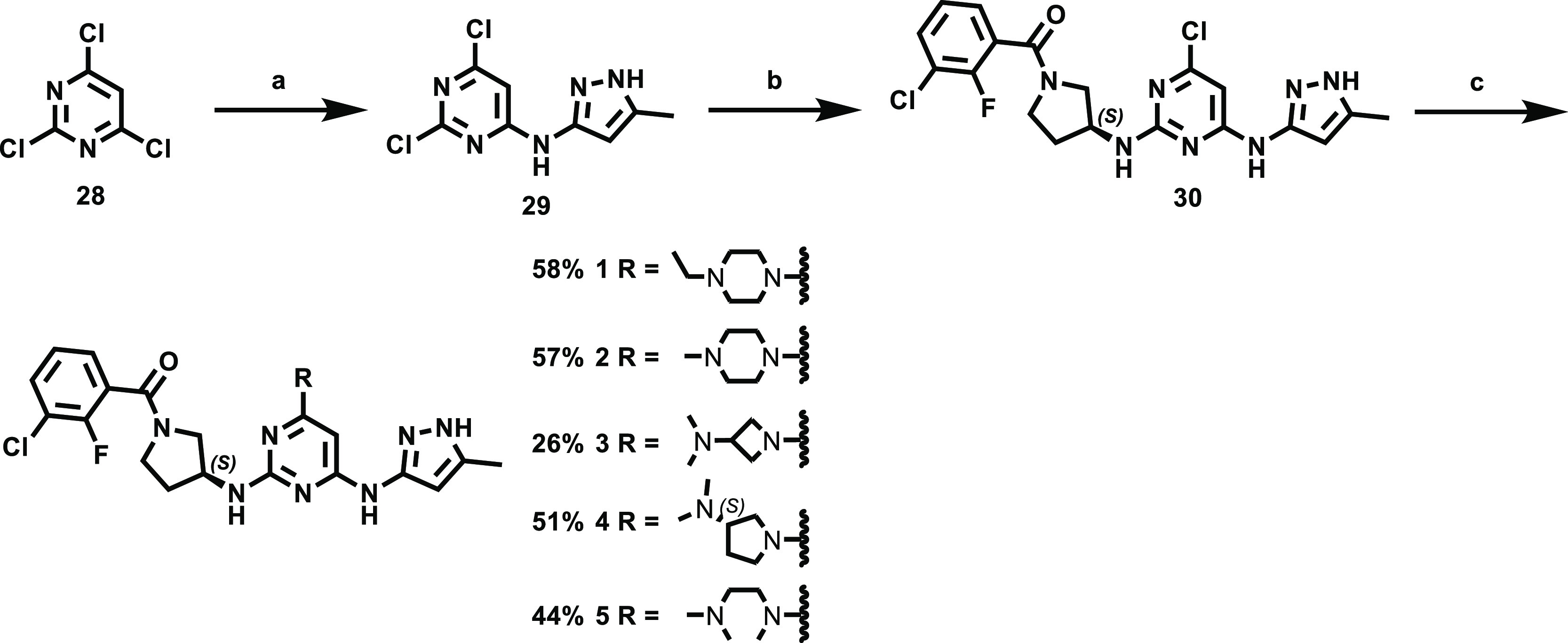
Reagents and conditions: (a) 3-amino-5-methylpyrazole, triethylamine, THF, 50 °C, 16 h, 75%; (b) (S)-(3-aminopyrrolidin-1-yl) (3-chloro-2-fluorophenyl)methanone, triethylamine, 1-pentanol, 120 °C, 6 h, 51%; (c) various amine, triethylamine, 1-pentanol, 140 °C, 2 h, 26–58%.
Scheme 2. Synthesis of Pyrimidine Derivatives.
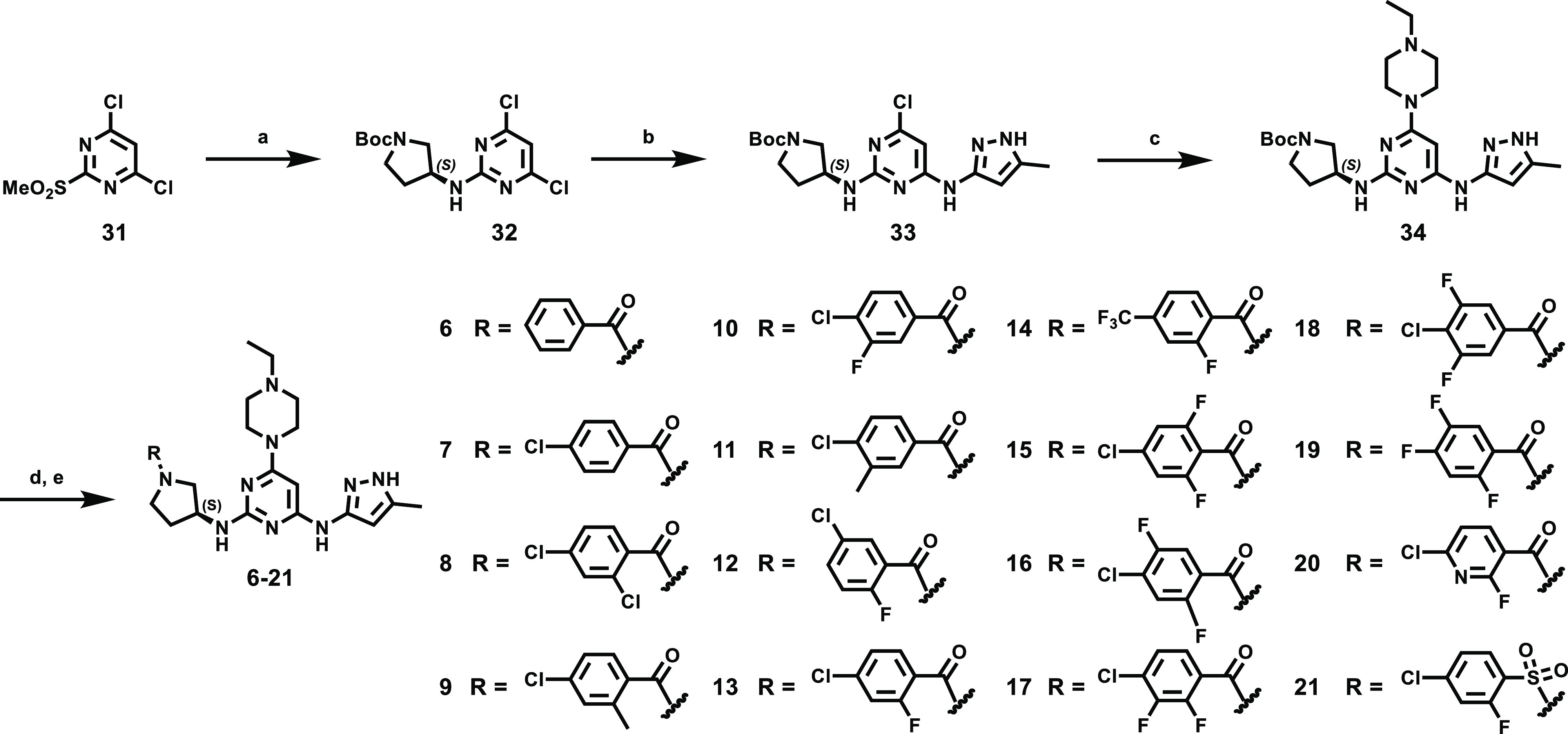
Reagents and conditions: (a) (S)-(−)-1-boc-3-aminopyrrolidine, triethylamine, THF, −70 °C, 6 h, 56%; (b) 3-amino-5-methylpyrazole, NaI, triethylamine, DMSO, 90 °C, 16 h, 85%; (c) 1-ethylpiperazine, triethylamine, 1-pentanol, 140 °C, 2 h, 84%; (d) 2 N HCl in ether, methanol, dichloromethane, 4 h, 99%; (e) various benzoic acid, T3P, triethylamine, DMF, dichloromethane, 16 h, 44–68% or 6-chloro-2-fluoropyridine-3-carboxylic acid, T3P, triethylamine, DMF, dichloromethane, 16 h, 46% (20) or 4-chloro-2-fluorobenzenesulfonyl chloride, triethylamine, dichloromethane, rt, 4 h, 85% (21).
Scheme 3. Synthesis of Compound 13 Prodrugs.
Reagents and conditions: (a) various anhydride or dicarbonate, 1,4-dioxane, 140 °C, 30 min.
SBDD, Biological Evaluation, and Structure–Activity Relationship
Informed by structure modeling, we developed a novel class of 6-methyl-N4-(5-methyl-1H-pyrazol-3-yl)-N2-(pyrrolidin-3-yl) pyrimidine-2,4-diamine derivatives (structure A, Figure 1) as a versatile scaffold for the development of Aurora A kinase inhibitors adopting the DFG-out conformation. Compound 1 was designed through structure modeling of compound B, wherein the halogen group establishes an electrostatic dipole–dipole interaction with the methyl group of Ala273, causing the DFG activation loop to flip.27 Structural modeling revealed that the chloride substituent of compound 1 aligned well with compound B in the same binding pocket of Aurora A, and the distance between the chlorine atom to carbon atom of Ala273 in 1 is similar to B (i.e., 3.4 Å) (Figure 1).
Figure 1.

Molecular docking model of structure A-derived compound 1 (cyan) aligned with compound B (magenta) in complex with Aurora A (PDBID: 3UO6, backbone in gray, DFG-loop in magenta) and compared with an X-ray structure of Aurora A in complex with ATP (PDBID: 5DNR, DFG-loop in yellow). SBDD, structure-based drug design.
The inhibitory potency of the newly synthesized small molecules against Aurora A was first measured in ATP consumption assays using purified Aurora A protein. Aurora A kinase inhibitors capable of inducing a DFG flip have been previously demonstrated to cause degradation of MYC-family oncoproteins in neuroblastoma and SCLC cells.14,16 Therefore, compounds that exhibited IC50 < 100 nM enzymatic efficacy were subjected to western blot analysis for protein levels of cMYC and MYCN, respectively, in NCI-H82 [a SCLC cell line with cMYC amplification] and SK-N-BE(2) [a neuroblastoma cell line with MYCN amplification] cells (Table 1 and Supporting Information Figure S1A–C). Western blot analyses showed compound 1 marginally reduced MYCN level at a concentration of 1.0 μM after 24 h (Table 1). Compounds 2–5 bearing modifications to the R2 water-solubilizing group, such as the acyclic polar group on the 4-position of the pyrimidine ring showed limited ability to reduce levels of the cMYC/MYCN oncoproteins. Derivatization of the 4-position of the ethyl piperazine moiety presumably changed the physiological properties (including solubility, absorption, and metabolic rate) of the compounds but did not affect cMYC/MYCN levels. Compound 6 (lacking substitution on the benzene ring) showed no effect on cMYC or MYCN levels, although its enzymatic Aurora A inhibition activity was maintained.
Based on the above results and the availability of different halogenated benzoic acid derivatives, we investigated the contribution of such substituents to cMYC/MYCN levels. Compound 13, bearing a 4-chloro-2-fluorophenyl pyrrolidinyl methanone group, exhibited a slight reduction in enzymatic activity (Aurora A IC50 = 38.6 ± 7.0 nM) compared to compound 1 (Aurora A IC50 = 24.1 ± 7.9 nM) but was associated with the highest degree of cMYC and MYCN level reduction among compounds 1–21. Compared with compound 13, compound 7 lacking a fluorine atom showed a slight reduction in enzyme activity to Aurora A but lost the capability to reduce cMYC/MYCN levels. The 4-chloro-3-fluorophenyl compound 10 and 4-chloro-2,3-difluorophenyl compound 17 were less potent enzymatic inhibitors (Aurora A IC50 = 52.2 ± 8.1 and 64.9 ± 13.7 nM) than 13 and were moderately capable of decreasing cMYC/MYCN levels. On the other hand, introduction of fluoride at the ortho or meta positions of the phenyl ring (i.e., compounds 15, 16, and 18) was associated with a smaller decrease in cMYC/MYCN levels. The position of chlorine on the benzene ring was found to influence cMYC/MYCN levels; relocation of the chlorine atom of compound 13 from the para to the meta position (to give compounds 1 and 12) diminished the reduction in cMYC/MYCN levels compared to compound 13, as did replacement of the fluorine of 13 with chlorine (to give compound 8) or trifluoromethyl (to give 14) or its isostere methyl group (to give 9 and 11). In addition, replacement of the chlorine atom on the benzene ring of compound 16 with a fluorine atom (i.e., to give compound 19) decreased Aurora A inhibition activity (IC50 > 100 nM). These results indicate that the positions of the halogen substituents on the benzene are crucial for Aurora A kinase activity and cMYC/MYCN level reduction. To improve the physicochemical properties of compound 13, extensive structure–activity relationship studies were carried out by replacing its benzene moiety with a pyridine to give 20 and replacing the amide moiety of 13 with a sulfonamide to give the amide isostere 21. However, the Aurora A inhibitory potency of these two compounds was reduced by more than 2.5-fold (IC50 > 100 nM).
Single treatment of 13 in NCI-H82 and SK-N-BE(2) showed dose-dependent loss of cMYC and MYCN proteins. The phase II investigational drug MLN8237, although showed better activity than 13 in reducing cMYC/MYCN levels at lower concentrations (i.e., 50 and 200 nM), was not able to induce further cMYC/MYCN level reduction at higher concentrations (i.e., 500 and 1000 nM, Figure 2A,B and Supporting Information Figure S1C). In addition, whereas compound 13 is less promising in reducing the MYCN level compared to CD532 at 1.0 μM, it showed a similar trend in reducing the cMYC level in NCI-H82 cells (Supporting Information Figure S1D). The differences in the dose-dependent efficacy of 13, CD532, and MLN8237 on cMYC/MYCN destabilization could be derived from the various conformation-specific interacting modes between Aurora A, the inhibitors, and cMYC/MYCN, which would require biochemical assays, as reported by Gilburt et al. for further verification.28
Figure 2.

(A) Western blot analysis for the expression levels of cMYC and MYCN, phosphorylation of Aurora kinase A/B/C and cell cycle markers Cyclin B1 and pH3(Ser10) in NCI-H82 and SK-N-BE(2) cells, respectively, treated with 13 and MLN8237 for 24 h. Immunoblotting of β-ACTIN was used as loading controls. (B) Quantification of the relative protein level of cMYC and MYCN in cells treated as shown in (A). The values were obtained from triplicate experiments. (C) Percent survival of the indicated SCLC cell lines treated with 13 for 72 h. Amp, amplification. (D) Western blotting analysis for the cMYC protein expression in NCI-H82, NCI-H211, NCI-H446, NCI-H524, NCI-H2171, NCI-H146, and NCI-H841. Immunoblotting of GAPDH is shown as a loading control. Relative cMYC protein level is quantified in (E). (E) Summary of half-proliferation inhibition concentration (IC50) of 13 and cMYC protein levels and AURKA and AURKB mRNA levels (retrieved from the Cancer Cell Line Encyclopedia database) in SCLC cell lines shown in (C,D) and their genomic features. (F) Tumor growth curve of the xenografted NCI-H446 cells in nude mice. When the tumor size reached 200 mm3, mice were intravenously (IV) injected with vehicle (5 mL/kg) or 13 (50 mg/kg) using a 5-on-2-off cycle for two consecutive weeks. P < 0.05 from day 8 compares the 13-treated group and vehicle. (G) Body weight of the mice treated as described in (F) was monitored twice a week for 24 days.
Proliferation Inhibition of Cancer Cells
SCLC is an aggressive malignancy that accounts for around 10–15% of all lung cancers, for which no targeted therapy is currently available.29,30 SCLC cells are characterized by rapid proliferation, universal RB1 and TP53 inactivation, and high rates of MYC-family amplification.31 A recent study of MYC amplification in 77 formalin-fixed paraffin-embedded SCLC tumor samples using chromogenic in situ hybridization identified amplification of the MYC-family oncogenes in 20% of the tumors.32,33 Thus, we evaluated the growth-inhibitory effects of 13 on nine different SCLC cell lines: five with cMYC amplification, one with MYCN amplification, one with MYCL amplification, and two with no MYC-family amplification. As shown in Figure 2C, the sensitivity of these SCLC cells to compound 13 is significantly correlated (R2 = 0.7473, Supporting Information Figure S2A) with the protein level of cMYC and is not correlated with the mRNA levels of AURKA or AURKB (Figure 2D,E).34 NCI-H524 (which expressed with the highest level of cMYC) was the most sensitive to 13, with a growth inhibition IC50 = 3.36 nM. The other four cMYC-amplified SCLC cells also demonstrated IC50 < 200 nM, whereas the MYC-unamplified SCLC cell lines (i.e., NCI-H146 and NCI-H841) were less sensitive to 13 with IC50 > 1000 nM (Figure 2E). NCI-H526 and NCI-H209 which acquired MYCN and MYCL amplification, respectively, also showed high sensitivity to 13 (IC50 < 100 nM).
The antiproliferative effect of Aurora kinase inhibitors on SCLCs with cMYC-amplification has been reported.35,36 Sos et al. suggested that the viability of cMYC-amplified SCLC cells depends on cMYC and Aurora B protein expression, though knocking down Aurora B by RNA silencing did not alter the protein level of cMYC.36 The western blot result (Figure 2A) indicated that 13 and MLN8237 inhibited phosphorylation of both Aurora A and Aurora B in a dose-dependent manner, and compound 13 had a preferential in inhibiting Aurora B [evidenced with reduced pAurora B (Thr232) along with reduced pH3 (Ser10) and Cyclin B1] over Aurora A [evidenced with reduced pAurora A (Thr288)]. We noted that the trend of cMYC/MYCN reduction coincides better with the level of pAurora A (Thr288) than pAurora B (Thr232) in cells treated with various concentrations of 13 (Figure 2A). To elucidate the antiproliferation activity of 13 is attributed to cMYC/MYCN reduction or cell-cycle arrest induced by Aurora A and/or Aurora B kinase inhibition, we compared percent cell survival of NCI-H82 and SK-N-BE(2) with short-term (i.e., 24 h) treatment of an Aurora A-selective inhibitor (i.e., Aurora A inhibitor I, reported cell-free IC50 = 3.4 nM), an Aurora B-selective inhibitor (i.e., AZD1152, reported cell-free IC50 = 0.37 nM), 13, or MLN8237. Treatment of NCI-H82 with Aurora A inhibitor I or AZD1152 had a limited effect on cMYC levels (Supporting Information Figure S2B). Compound 13 and MLN8237 had better dose-dependent efficacy than Aurora A inhibitor I or AZD1152 in reducing cell viability. AZD1152 showed similar potency to 13 at lower concentrations (i.e., between 1 and 15.6 nM) but failed to reduce cell viability further at higher concentrations (Supporting Information Figure S2C). The 13-induced reduction in cMYC level is not due to reduced gene transcription (Supporting Information Figure S2D), and the cMYC protein level could be recovered by treatment with a proteasomal inhibitor MG132 (Supporting Information Figure S2E), suggesting that 13 had a minimal effect on the transcription or translation of cMYC. In addition, whereas 13 induced cell cycle arrest similar to AZD1152 (Supporting Information Figure S2F) through the inhibition on Aurora B, AZD1152 treatment had little effect on the cMYC level in NCI-H82 (Supporting Information Figure S2B). This result indicated that the reduced cMYC level was unlikely to be due to Aurora B inhibition-induced cell cycle perturbation, a conclusion similar to that of Sos et al.(36) On the other hand, while the hypothesis that MYC levels are impacted through a destabilization/degradation mechanism is intriguing, it is at present unclear whether the reduced cellular levels of cMYC/MYCN induced by 13 are caused by cell-cycle effects resulting from direct inhibition of Aurora kinases or Aurora A-dependent protein destabilization due to the intrinsic dependence of cMYC/MYCN levels on the cell cycle.
Xenograft Tumor Growth Inhibition in Mice
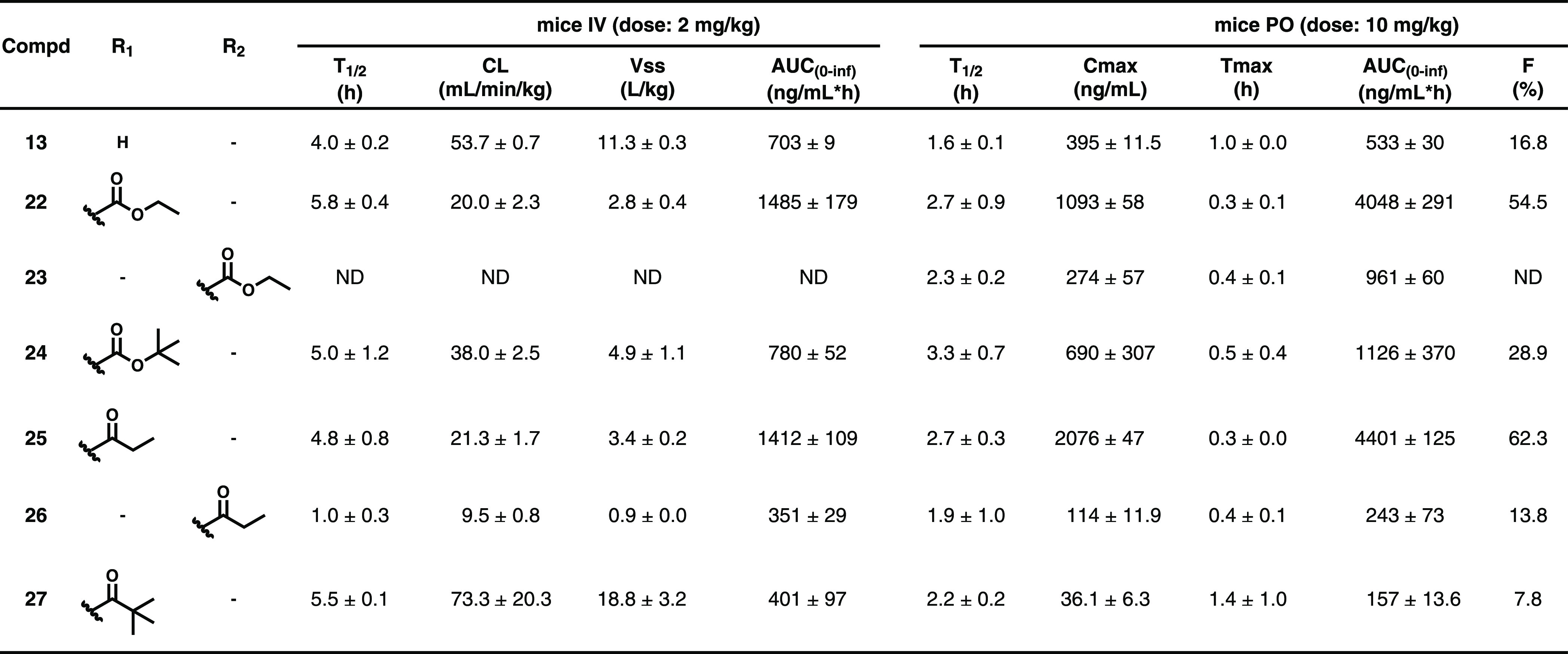
PK studies (Table 2) of 13 undertaken in mice revealed a moderate plasma half-life (4.0 ± 0.2 h), providing an area under the curve (AUC) of 703 ± 9 ng/mL·h when delivered at 2 mg/kg intravenous (IV). Although the bioavailability of 13 is suboptimal, its cellular activity suggested potential efficacy in vivo. A cMYC-amplified SCLC cell line NCI-H446 was selected for in vivo efficacy study due to its stem cell-like properties and consistently high growth rate when transplanted in mice.37,38 Intravenous administration of 13 at 50 mg/kg led to >90% tumor growth inhibition on a 5-on-2-off dosing schedule (Figure 2F), and less than 5% body weight change was observed during the treatment (Figure 2G).
Table 2. Lead Optimization of Compound 13 Using Prodrug Approachesa.

Values indicate the mean and standard deviation (SD) of three mice (n = 3). ND, not detected.
PK Improvements to the Lead Using Prodrug Approaches
The PK study in mice established 13 to have a high clearance rate (53.7 ± 0.7 mL/min/kg) in blood. Because 13 exhibited high in vivo SCLC tumor inhibition efficacy with intravenous administration (Figure 2F), we attempted to improve its oral bioavailability using prodrug approaches, which balance membrane permeation, P-glycoprotein efflux, hydrolysis in the gastrointestinal lumen and intestinal cells, and nonesterase metabolism in the liver.39 Several classes of nitrogen-containing prodrugs have been reported, wherein polar functional groups such as carbamates, carboxamides, and sulfamides40−42 are used to increase permeability. Here, two types of prodrug derivatives were designed: N-carboxylate derivatives (22–24) and N-acyl derivatives (25–27). The PK properties of 13 were greatly improved by its derivatization as N-carboxylate derivative 22 and N-acyl derivative 25; the released 13 showed AUC = 4048 ± 291 and 4401 ± 125 ng/mL·h with oral bioavailability = 54.5 and 62.3%, respectively (Table 2). On the other hand, the regioisomers 23 and 26 showed much lower AUC and had poor oral bioavailability. The reduced steric hindrance of the prodrug derivatives 23 and 26 may result in higher in vivo metabolic rate compared to 22 and 25. In addition, prodrugs bearing larger substituents such as 24 (N-carboxylate derivative) and 27 (N-acyl derivative) were also designed; however, the AUC revealed no further enhancement (Table 2). The bulky substituents may increase lipophilicity, thereby reducing tissue absorption.
We then examined the in vivo tumor growth inhibition efficacy of the N-carboxylate derivative 22 and N-acyl derivative 25, both of which showed superior oral bioavailability to 13 (Table 2). In the NCI-H446 xenograft tumor model, oral administration of 22 and 25 in mice on a 5-on-2-off regimen demonstrated dose-dependent anticancer activity (Figure 3A,B). Increasing the dose of 25 from 50 to 100 mg/kg in the fourth week further reduced the tumor volume (Figure 3B). In addition, oral administration of 25 at 100 mg/kg for four consecutive weeks induced >80% volume regression of the tumors, an observation similar to the reference compound MLN8237. Furthermore, the xenografted tumors treated with 25 remained stable for 44 days after the last dosage, whereas the MLN8237-treated group started to grow at day 22 after the last dosage. Body weight change in mice treated with 25 was less than 5% during the 4-week dosing period, suggesting its safety in mice at the 100 mg/kg dosage (Figure 3B). We noted that the body weight loss in mice administered with 25 was less severe compared to animals administered with 22 at the same dosage, although they both potentially induce neutropenia via the inhibition on the B-type Aurora kinase.43 Because 25 and 22 are both prodrugs of 13, this result suggested that the prodrug design may alter the PK properties of the compound and thus, the overall therapeutic index. In addition to shrinking of the NCI-H446 xenograft tumors with volume ≅200 mm3, 25 also showed great potency to shrink tumors with volume >750 mm3 (Figure 3C). These results demonstrate the in vivo activity of 22 and 25 on cMYC-amplified SCLC.
Figure 3.

(A,B) Growth curve of xenografted NCI-H446 in mice orally (PO) administered with vehicle and the indicated dosages of 22, 25, and MLN8237 using a 5-on-2-off cycle, respectively. Body weight of the mice during the course is shown at right. P < 0.05 for tumor volume from day 4 or day 8 compares the drug-treated groups (22, 25, and MLN8237) and the vehicle. (C) Mice bearing with >750 mm3 NCI-H446 xenograft tumors were orally treated with vehicle or 25 at 100 mg/kg from week 3 using a 5-on-2-off cycle for 2 weeks. Wk, week.
In Vivo MYC Protein Level and Cancer Cell Apoptosis
We next evaluated the influence of 25 and MLN8237 on the cMYC level in vivo. Mice bearing xenograft NCI-H446 tumors >500 mm3 in volume were orally administered a single dose of 25 or MLN8237 at 100 mg/kg. Tumors were harvested after 2, 4, 8, and 24 h of the treatment, respectively (Figure 4A). Whole tumor tissues were collected for western blotting and immunohistochemistry (IHC) analyses. The phosphorylation level of histone H3 at Ser10 [i.e., pH3(Ser10)], a substrate of Aurora B kinase, was reduced by >90% in the tumors treated with either 25 or MLN8237 at 2 h after drug administration, suggesting the effectiveness of the compounds on Aurora kinase inhibition and cell-cycle arrest at this time point. The western blot result of phosphorylated Aurora A/B/C was not provided due to the inability of detecting the signal in the tumor tissues. The protein level of cMYC gradually decreased in the tumors treated with 25 and was around 37% (p = 0.0037, t-test) of that in the vehicle-treated tumors (1.0 ± 0.230 in vehicle vs 0.374 ± 0.204 in tumors at 24 h post-treatment, Figure 4B,C) 24 h after the dosing of 25. On the other hand, treatment of MLN8237 had marginal effect on the cMYC level, an observation similar to the report of Mollaoglu et al.(29) This observation is perhaps due to the less significant dose-dependent efficacy of MLN8237 on cMYC level reduction (Figure 2A) or the overall PK properties of the compound.
Figure 4.

(A) Schematic illustration of the NCI-H446 tumor xenograft and drug treatment protocol in nude mice. (B) Western blot analysis for the expression of cMYC, cleaved PARP-1 (cPARP-1), a mitotic marker pH3(Ser10), and a histone H3 loading control in xenografted NCI-H446 tumors in mice oral (PO) administered with the vehicle (10 mL/kg), 25 at 100 mg/kg, or MLN8237 at 100 mg/kg. Tumors were harvested after 2, 4, 8, and 24 h of drug treatment, respectively. Relative protein levels of cMYC and cleaved cPARP-1 in tumors were quantified in (C,D). (E) Immunohistochemical analysis for the expression of cleaved caspase-3 (i.e., CASP3, brown color) to mark apoptotic cells in NCI-H446 xenograft tumors harvested from mice with vehicle treatment or orally administered with 25. The images were recorded at 400× magnification. (F) Quantification of the number of cleaved CASP3-positive cells in the IHC profile shown in (E). Cell numbers in tissues from four to six mice were quantified for each set of experimental conditions.
Apoptosis was assessed using levels of PARP-1, one of several known cellular substrates of caspases; cleavage of PARP-1 by caspases is considered a hallmark of apoptosis.44 Cleaved PARP-1 (cPARP-1) levels were significantly upregulated in NCI-H446 tumor tissues isolated from mice administered with 25 or MLN8237 after 24 h (p = 0.0013 for 25 and p = 0.0083 for MLN8237, t-test), although the level of increment was less significant in MLN8237- than in 25-treated tumors (Figure 4B,D). The IHC results showed that the incidence of cleaved Caspase-3 (CASP3) was also greatly increased (Figure 4E,F). These data suggested that 25 induced cMYC level reduction, resulting in CASP3-associated programmed cell death, and tumor regression.
Conclusions
Novel small molecules that promote degradation of MYC-family oncoproteins would be a great advancement in cancer treatment. By the rational design of an Aurora A kinase inhibitor able to induce the DFG-out conformation using computer modeling, we successfully identified an initial hit compound 1, which marginally reduced levels of cMYC and MYCN. Screening of different halogen substituents on the phenyl group resulted in compound 13 (S)-(4-chloro-2-fluorophenyl) (3-((4-(4-ethylpiperazin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone), which reduced cMYC and MYCN levels by >50% at 1.0 μM. The reduced cMYC/MYCN levels could be attributed to the inhibitor’s effects on cell cycle progression and/or protein stability. Compound 13 showed significantly enhanced inhibition potency against MYC-amplified SCLCs than MYC-unamplified SCLCs and tumor growth inhibition activity in a cMYC-amplified SCLC cell line NCI-H446 xenograft mouse model when administered intravenously. Lead optimization focusing on oral bioavailability led to the discovery of compound 25, a N-acyl prodrug of 13, which demonstrated improved oral availability and excellent NCI-H446 SCLC xenografted tumor regression in mice. These results support the potential of 25 for further development as an anticancer drug targeting MYC-amplified malignancies such as SCLC.
Experimental Section
General Chemistry
Unless otherwise stated, all reagents used were commercially available with suppliers and used as supplied. Reactions requiring anhydrous conditions were performed in flame-dried glassware and cooled under an argon or nitrogen atmosphere. All reactions were carried out under argon or nitrogen and monitored by analytical thin-layer chromatography using glass-backed plates (5 × 10 cm) precoated with silica gel 60 F254 as supplied by Merck; zones were detected visually under UV irradiation (254 nm) or by spraying with phosphomolybdic acid reagent (Sigma-Aldrich, USA), followed by heating to 80 °C. Flash column chromatography using silica gel 60 of 230–400 mesh size (Merck) was used routinely for purification and separation of product mixtures. 1H and 13C NMR spectra were recorded on Varian Mercury-300, Varian Mercury-400, Bruker NMR DMX-600, or Varian VNMRS-700 spectrometers. Chloroform-d or dimethyl sulfoxide-d6 or methanol-d4 was used as the solvent and tetramethylsilane (TMS) (δ 0.00 ppm) as an internal standard. Chemical shift values are reported in parts per million relative to the TMS in delta (δ) units. Multiplicities are recorded as s (singlet), br s (broad singlet), d (doublet), t (triplet), q (quartet), quint (quintet), dd (doublet of doublets), dt (doublet of triplets), ddd (doublet of doublets of doublets), and m (multiplet). Coupling constants (J) are expressed in hertz. Electrospray mass spectroscopy (ESMS) spectra were recorded as m/z values using an Agilent 1100 MSD mass spectrometer. All test compounds displayed more than 95% purity, as determined by a Hitachi 2000 series HPLC system using a C-18 column (Agilent Eclipse XDB-C18 5 μm, 4.6 mm × 150 mm, USA). Mobile phase A: acetonitrile; mobile phase B: 2 mM ammonium acetate aqueous solution containing 0.1% formic acid. The gradient system started from A/B (10%:90%) to A/B (90%:10%) with a flow rate of 0.5 mL/min, and the injection volume was 20 μL. The system was operated at 25 °C. Peaks were detected at 254 nm. The IUPAC nomenclature of compounds was determined with ACD/Name Pro software. Detailed characterization data for each compound are reported in the Supporting Information.
2,6-Dichloro-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (29)
3-Amino-5-methylpyrazole (7.96 g, 81.9 mmol) was added to a solution of 2,4,6-trichloropyrimidine 28 (10 g, 54.6 mmol), triethylamine (11.4 mL, 81.9 mmol) in THF (200 mL). The reaction mixture was heated at 50 °C, stirred for 16 h, and then quenched with brine (100 mL). The aqueous phase was extracted with ethyl acetate (3 × 200 mL). The combined organic extracts were washed with water and brine, dried over magnesium sulfate, and filtered. The filtrate was concentrated to get crude residue. The residue was purified by flash column chromatography over silica gel with n-hexane/ethyl acetate (1:1) to afford compound 29 (10 g, 41.2 mmol, 75% yield) as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.75 (s, 1H), 5.99 (s, 1H), 2.35 (s, 3H); LCMS (ESI) m/z: 244 [M + H]+.
(S)-(3-Chloro-2-fluorophenyl) (3-((4-chloro-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone (30)
A solution of compound 29 (5 g, 20.5 mmol), (S)-(3-aminopyrrolidin-1-yl) (3-chloro-2-fluorophenyl)methanone (6.5 g, 26.7 mmol), triethylamine (4.3 mL, 30.8 mmol) in 1-pentanol (5 mL) was heated at 120 °C for 6 h and then poured into water (300 mL). A precipitate formed, which was collected and purified by flash column chromatography over silica gel with dichloromethane/methanol (94:6) to give compound 30 (4.7 g, 10.5 mmol, 51% yield) as a yellow solid. 1H NMR (300 MHz, CDCl3): δ 10.65 (br s, 1H), 7.45 and 7.45 (t, J = 7.2 Hz, 1H), 7.22–7.08 (m, 1H), 6.35 and 6.31 (s, 1H), 5.98 and 5.92 (s, 1H), 4.85 and 4.64 (br s, 1H), 4.28–4.10 (m, 1H), 3.87–3.65 (m, 2H), 3.48–3.25 (m, 2H), 2.34 and 2.33 (s, 3H), 2.28–2.43 (m, 2H); LCMS (ESI) m/z: 450.1 [M + H]+.
General Procedure for the Synthesis of Compounds 1–6
The general procedure is illustrated below with compound 1 as a specific example.
(S)-(3-Chloro-2-fluorophenyl) (3-((4-(4-ethylpiperazin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone (1)
A solution of compound 30 (200 mg, 0.45 mmol) and 1-ethylpiperazine (103 mg, 0.9 mmol) in 1-pentanol (1 mL) was heated at 140 °C for 2 h and then diluted with brine (100 mL). The aqueous phase was extracted with ethyl acetate (3 × 200 mL). The combined organic extracts were washed with water and brine, dried over magnesium sulfate, and filtered. The filtrate was concentrated to give the crude residue, which was purified by flash column chromatography over silica gel with dichloromethane/methanol (90:10) to afford compound 1 (137 mg, 0.26 mmol, 58% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.43 and 7.43 (t, J = 7.5 Hz, 1H), 7.21 and 7.20 (q, J = 7.5 Hz, 1H), 7.09 (t, J = 7.5 Hz, 1H), 5.93 and 5.87 (s, 1H), 5.62 and 5.59 (s, 1H), 4.72 and 4.58 (br s, 1H), 4.13 (m, 1H), 3.82–3.66 (m, 2H), 3.62–3.42 (m, 5H), 3.41–3.25 (m, 1H), 2.51–2.40 (m, 6H), 2.31 and 2.29 (s, 3H), 2.28–2.21 (m, 1H), 2.16–2.05 (m, 1H), 1.12 and 1.12 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 163.51, 163.26, 162.82, 161.00 × 2, 160.81, 153.13 (d, JC–F = 244.0 Hz), 131.60, 127.52, 127.02 and 126.92 (d, JC–F = 17.9 Hz), 125.97, 120.25 and 120.18 (d, JC–F = 17.9 Hz), 75.55, 53.39 and 51.62, 52.14, 52.02, 51.73, 50.80 and 49.70, 46.11 and 44.06, 43.72, 31.21 and 29.54, 11.92 × 2; LCMS (ESI) m/z: 528.3 [M + H]+; HRMS (ESI) calcd for C25H31ClFN9O [M + H]+m/z: 528.2402; found, 528.2402; HPLC purity = 100%, tR = 10.60 min.
(S)-(3-Chloro-2-fluorophenyl) (3-((4-((5-methyl-1H-pyrazol-3-yl)amino)-6-(4-methylpiperazin-1-yl)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone (2)
Similar to the reaction procedures for 1, a solution of compound 30 (200 mg, 0.45 mmol) and 1-methylpiperazine (90 mg, 0.9 mmol) in 1-pentanol (0.9 mL) was heated at 140 °C for 2 h. After a usual workup, the crude residue was purified by silica gel column chromatography with dichloromethane/methanol (90:10) to afford compound 2 (131 mg, 0.26 mmol, 57% yield) as a yellow solid. 1H NMR (300 MHz, CDCl3): δ 7.43 (t, J = 6.6 Hz, 1H), 7.24–7.18 (m, 1H), 7.09 (t, J = 7.8 Hz, 1H), 5.93 and 5.87 (s, 1H), 5.63 and 5.60 (s, 1H), 4.72 and 4.78 (br s, 1H), 4.17–4.07 (m, 1H), 3.82–3.24 (m, 8H), 2.46–2.37 (m, 4H), 2.35–2.28 (m, 6H), 2.57–2.04 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 163.49, 163.23, 162.80, 160.97 × 2, 160.81, 153.09 (d, JC–F = 247.0 Hz), 131.59 (d, JC–F = 5.3 Hz), 127.50, 127.00 and 126.91 (d, JC–F = 18.0 Hz), 125.98, 120.24 and 120.17 (d, JC–F = 17.5 Hz), 75.57, 54.35, 54.20, 53.34 and 51.59, 50.76 and 49.67, 46.09 and 44.05, 45.82, 43.62, 31.20 and 29.52, 11.47; LCMS (ESI) m/z: 514.2 [M + H]+; HRMS (ESI) calcd for C24H29ClFN9O [M + H]+m/z: 514.2246; found, 514.2249; HPLC purity = 100%, tR = 11.01 min.
(S)-(3-Chloro-2-fluorophenyl) (3-((4-(3-(dimethylamino)azetidin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone (3)
Similar to the reaction procedures for 1, a solution of compound 30 (200 mg, 0.45 mmol) and N,N-dimethylazetidin-3-amine (90 mg, 0.9 mmol) in 1-pentanol (0.9 mL) was heated at 140 °C for 2 h. After a usual workup, the crude residue was purified by silica gel column chromatography with dichloromethane/methanol (90:10) to afford compound 3 (60 mg, 0.12 mmol, 26% yield) as a yellow solid. 1H NMR (300 MHz, CDCl3): δ 7.42 and 7.42 (t, J = 7.2 Hz, 1H), 7.24–7.17 (m, 1H), 7.09 (t, J = 7.8 Hz, 1H), 5.91 and 5.85 (s, 1H), 5.29 and 5.26 (s, 1H), 4.71 and 4.58 (br s, 1H), 4.09–3.90 (m, 3H), 3.81–3.65 (m, 4H), 3.51–3.10 (m, 3H), 2.29 and 2.28 (s, 3H), 2.23–2.03 (m, 8H); 13C NMR (100 MHz, DMSO-d6): δ 165.09, 164.93, 162.76, 161.16, 161.11, 159.83, 153.09 (d, JC–F = 246.3 Hz), 131.58 (d, JC–F = 3.8 Hz), 127.52 (d, JC–F = 3.8 Hz), 126.99 and 126.91 (d, JC–F = 18.3 Hz), 125.98, 120.19 and 120.14 (d, JC–F = 17.6 Hz), 74.46, 55.94, 55.90, 53.69, 53.56, 53.36 and 51.56, 50.71 and 49.58, 46.07 and 44.03, 41.49, 41.45, 31.13 and 29.56; LCMS (ESI) m/z: 514.2 [M + H]+; HRMS (ESI) calcd for C24H29ClFN9O [M + H]+m/z: 514.2246; found, 514.2238; HPLC purity = 100%, tR = 10.95 min.
(3-Chloro-2-fluorophenyl) ((S)-3-((4-((S)-3-(dimethylamino)pyrrolidin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone (4)
Similar to the reaction procedures for 1, a solution of compound 30 (200 mg, 0.45 mmol) and (S)–N,N-dimethylpyrrolidin-3-amine (102 mg, 0.9 mmol) in 1-pentanol (0.9 mL) was heated at 140 °C for 2 h. After workup, the crude residue was purified by silica gel column chromatography with dichloromethane/methanol (90:10) to afford compound 4 (121 mg, 0.23 mmol, 51% yield) as a yellow solid. 1H NMR (300 MHz, CDCl3): δ 7.47–7.40 (m, 1H), 7.33–7.27 (m, 1H), 7.16–7.08 (m, 1H), 5.90 and 5.85 (s, 1H), 5.46–5.36 (m, 2H), 4.59 (br s, 1H), 4.10–3.60 (m, 3H), 3.60–3.10 (m, 5H), 2.88–2.75 (m, 1H), 2.35–2.24 (m, 11H), 1.96–1.80 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 162.84, 161.38, 161.22, 161.02 and 160.98, 159.96, 159.93, 153.14 (d, JC–F = 247.1 Hz), 131.62 (d, JC–F = 5.4 Hz), 127.52 (d, JC–F = 3.0 Hz), 127.02 and 126.95 (d, JC–F = 17.6 Hz), 126.03, 120.25 and 120.22 (d, JC–F = 17.6 Hz), 75.32, 64.65 and 64.57, 53.50 and 51.69, 50.86 and 49.75, 50.22, 49.96, 46.14 and 44.12, 45.14, 44.99, 43.81, 43.75, 31.20 and 29.25, 29.62; LCMS (ESI) m/z: 528.2 [M + H]+; HRMS (ESI) calcd for C25H31ClFN9O [M + H]+m/z: 528.2402; found, 528.2389; HPLC purity = 98.47%, tR = 11.19 min.
(S)-(3-Chloro-2-fluorophenyl) (3-((4-((2-(dimethylamino)ethyl) (methyl)amino)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone (5)
Similar to the reaction procedures for 1, a solution of compound 30 (200 mg, 0.45 mmol) and N1,N1,N2-trimethylethane-1,2-diamine (92 mg, 0.9 mmol) in 1-pentanol (0.9 mL) was heated at 140 °C for 2 h. After workup, the crude residue was purified by silica gel column chromatography with dichloromethane/methanol (90:10) to afford compound 5 (102 mg, 0.2 mmol, 44% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.43 (t, J = 7.6 Hz, 1H), 7.25–7.20 (m, 1H), 7.10 (t, J = 7.8 Hz, 1H), 5.93 and 5.88 (s, 1H), 5.51 and 5.49 (s, 1H), 4.69 and 4.60 (br s, 1H), 4.15–4.04 (m, 1H), 3.83–3.44 (m, 5H), 3.42–3.25 (m, 1H), 2.98 and 2.94 (s, 3H), 2.41–2.35 (m, 2H), 2.32–2.25 (m, 7H), 2.23 (s, 3H), 2.20–2.05 (m, 1H); 13C NMR (100 MHz, DMSO-d6): δ 162.81, 162.67, 160.79 × 2, 160.42 × 2, 153.09 (d, JC–F = 247.0 Hz), 131.61 (d, JC–F = 6.9 Hz), 127.49, 126.98 and 126.91 (d, JC–F = 17.9 Hz), 126.03, 120.22 (d, JC–F = 17.5 Hz), 74.63, 56.39, 53.61 and 51.63, 50.81 and 49.73, 46.53, 46.38, 46.07 and 44.15, 45.50, 45.40, 35.38, 31.20 and 29.59; LCMS (ESI) m/z: 516.2 [M + H]+; HRMS (ESI) calcd for C24H31ClFN9O [M + H]+m/z: 516.2402; found, 516.2402; HPLC purity = 99.91%, tR = 11.16 min.
tert-Butyl (S)-3-((4,6-dichloropyrimidin-2-yl)amino)pyrrolidine-1-carboxylate (32)
tert-Butyl (S)-3-aminopyrrolidine-1-carboxylate (20 g, 105.7 mmol) was added dropwise at −70 °C over 1 h to a solution of the starting material 4,6-dichloro-2-(methylsulfonyl)pyrimidine 31 (20 g, 88.1 mmol) and triethylamine (25.5 mL, 176.2 mmol) in THF (200 mL). The reaction mixture was warmed up to room temperature, stirred for 6 h, and then quenched with brine (100 mL). The aqueous phase was extracted with ethyl acetate (3 × 200 mL). The combined organic extracts were washed with water and brine, dried over magnesium sulfate, and filtered. The filtrate was concentrated to afford the crude residue. The residue was purified by flash column chromatography over silica gel with n-hexane/ethyl acetate (4:1) to afford compound 32 (16.4 g, 49.3 mmol, 56% yield) as a white solid. 1H NMR (400 MHz, CDCl3): δ 6.65 (s, 1H), 5.42 (br s, 1H), 4.60–4.47 (m, 1H), 3.69 (dd, J = 11.2, 6.0 Hz, 1H), 3.52–3.40 (m, 3H), 3.35–3.15 (m, 1H), 2.23 (m, 1H), 1.85–1.75 (m, 1H), 1.47 (s, 9H); LCMS (ESI) m/z: 355.1 [M + Na]+.
tert-Butyl (S)-3-((4-chloro-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidine-1-carboxylate (33)
A solution of compound 32 (7 g, 21.0 mmol), 3-amino-5-methylpyrazole (8.1 g, 84.0 mmol), triethylamine (3.5 mL, 25.2 mmol), and NaI (4.7 g, 31.5 mmol) in DMSO (70 mL) was stirred at 90 °C for 16 h. The solution was cooled down to room temperature and poured into water. A precipitate formed, which was collected and purified by flash column chromatography over silica gel with n-hexane/ethyl acetate (1:1) to give compound 33 (7 g, 17.9 mmol, 85% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 6.32 (s, 1H), 5.93 (s, 1H), 4.48 (br s, 1H), 3.85–3.60 (m, 1H), 3.60–3.40 (m, 4H), 2.31 (s, 3H), 1.46 (s, 9H); LCMS (ESI) m/z: 394.1 [M + H]+.
tert-Butyl (S)-3-((4-(4-ethylpiperazin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidine-1-carboxylate (34)
A solution of compound 33 (7 g, 17.8 mmol) and 1-ethylpiperazine (4.1 g, 35.6 mmol) in 1-pentanol (14 mL) was heated at 140 °C for 2 h and then quenched with brine (100 mL). The aqueous phase was extracted with ethyl acetate (3 × 200 mL). The combined organic extracts were washed with water and brine, dried over magnesium sulfate, and filtered. The filtrate was concentrated to give the crude residue, which was purified by flash column chromatography over silica gel with ethyl acetate/methanol (90:10) to afford compound 34 (7 g, 14.9 mmol, 84% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 5.87 (s, 1H), 5.62 (s, 1H), 4.70–4.39 (m, 1H), 3.80–3.30 (m, 7H), 2.52–2.40 (m, 6H), 2.27 (s, 3H), 2.24–2.04 (m, 2H), 1.46 (s, 9H), 1.11 (t, J = 7.2 Hz, 3H); LCMS (ESI) m/z: 472.1 [M + H]+.
General Procedure for the Synthesis of Compounds 6–21
The general procedure is illustrated below with compound 13 as a specific example.
(S)-(4-Chloro-2-fluorophenyl) (3-((4-(4-ethylpiperazin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone (13)
A solution of 2 N hydrochloric acid in ether (1.06 mL, 2.1 mmol) was added to a solution of compound 34 (200 mg, 0.42 mmol) in dichloromethane/methanol (2:1, 1 mL) at room temperature with stirring. The resulting mixture was stirred at room temperature for 4 h and then concentrated in vacuo to give the crude amine salt, which was used without further purification.
Triethylamine (0.35 mL, 2.52 mmol), 4-chloro-2-fluorobenzoic acid (88 mg, 0.5 mmol), and propanephosphonic acid anhydride (T3P) ≥50 wt % in ethyl acetate (400 mg, 0.63 mmol) were added to a solution of amine salt in DMF/dichloromethane (1:3, 4 mL) at room temperature. The resulting mixture was stirred at room temperature for 16 h and then quenched with brine (20 mL). The aqueous phase was extracted with ethyl acetate (3 × 30 mL). The combined organic extracts were washed with water and brine, dried over magnesium sulfate, and filtered. The filtrate was concentrated to afford the crude residue, which was purified by flash column chromatography over silica gel with ethyl acetate/methanol (85:15) to give compound 13 (128 mg, 0.24 mmol, 58% yield) as a white solid. 1H NMR (400 MHz, CD3OD): δ 7.46–7.25 (m, 3H), 5.95–5.75 (m, 1H), 5.62–5.45 (m, 1H), 4.53 and 4.36 (br s, 1H), 3.98–3.64 (m, 3H), 3.63–3.56 (m, 2H), 3.54–3.40 (m, 4H), 2.60–2.40 (m, 6H), 2.36–2.29 (m, 1H), 2.23 and 2.22 (s, 3H), 2.10–1.90 (m, 1H), 1.15 and 1.14 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CD3OD): δ 166.60, 165.34, 165.17, 162.61 × 3, 159.78 (d, JC–F = 249.3 Hz), 137.99 (d, JC–F = 9.9 Hz), 131.19 (d, JC–F = 4.6 Hz), 126.52 (d, JC–F = 3.1 Hz), 125.05 (t, JC–F = 16.8 Hz), 118.01 and 117.92 (d, JC–F = 25.1 Hz), 77.43, 55.30 and 53.37, 53.57, 53.51, 52.49 and 51.48, 47.82 and 45.63, 44.88, 44.85, 32.73 and 31.11, 11.86, 11.82; LCMS (ESI) m/z: 528.2 [M + H]+; HRMS (ESI) calcd for C25H31ClFN9O [M + H]+m/z: 528.2402; found, 528.2408; HPLC purity = 99.57%, tR = 10.68 min.
(S)-(3-((4-(4-Ethylpiperazin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl) (phenyl)methanone (6)
Similar to the reaction procedures for 13, a solution of 2 N hydrochloric acid in ether (1.06 mL, 2.1 mmol) was added to a solution of compound 34 (200 mg, 0.42 mmol) in dichloromethane/methanol (2:1, 1 mL), and the resulting mixture was stirred at room temperature for 4 h. After concentration in vacuo, the residue was dissolved in DMF/dichloromethane (1:3, 4 mL), and then, triethylamine (0.35 mL, 2.52 mmol), benzoic acid (61 mg, 0.5 mmol), and propanephosphonic acid anhydride (T3P) ≥50 wt % in ethyl acetate (400 mg, 0.63 mmol) were added. The solution was stirred at room temperature for 16 h. After workup, the crude residue was purified by silica gel column chromatography with ethyl acetate/methanol (85:15) to afford compound 6 (132 mg, 0.28 mmol, 66% yield) as a yellow solid. 1H NMR (300 MHz, CDCl3): δ 7.49–7.44 (m, 2H), 7.42–7.34 (m, 3H), 5.91 and 5.84 (s, 1H), 5.63 and 5.59 (s, 1H), 4.68 and 4.55 (br s, 1H), 4.15–4.05 (m, 1H), 3.82–3.40 (m, 8H), 2.54–2.42 (m, 6H), 2.26 and 2.29 (s, 3H), 2.25–2.03 (m, 2H), 1.14 and 1.13 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 168.49, 163.44, 163.21, 160.94 × 2, 160.74, 137.01 and 136.94, 129.77, 128.26 and 128.19, 127.10, 75.50, 54.81 and 51.66, 52.01, 51.90, 51.12 and 49.49, 47.31 and 44.34, 43.55 × 2, 31.76 and 29.40, 11.78 × 2; LCMS (ESI) m/z: 476.2 [M + H]+; HRMS (ESI) calcd for C25H33N9O [M + H]+m/z: 476.2887; found, 476.2883; HPLC purity = 96.41%, tR = 9.63 min.
(S)-(4-Chlorophenyl) (3-((4-(4-ethylpiperazin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone (7)
Similar to the reaction procedures for 13, a solution of 2 N hydrochloric acid in ether (1.06 mL, 2.1 mmol) was added to a solution of compound 34 (200 mg, 0.42 mmol) in dichloromethane/methanol (2:1, 1 mL), and the resulting mixture was stirred at room temperature for 4 h. After concentration in vacuo, the residue was dissolved in DMF/dichloromethane (1:3, 4 mL), and then, triethylamine (0.35 mL, 2.52 mmol), 4-chlorobenzoic acid (78 mg, 0.5 mmol), and propanephosphonic acid anhydride (T3P) ≥50 wt % in ethyl acetate (400 mg, 0.63 mmol) were added. The solution was stirred at room temperature for 16 h. After workup, the crude residue was purified by silica gel column chromatography with ethyl acetate/methanol (85:15) to afford compound 7 (144 mg, 0.28 mmol, 67% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.42 and 7.42 (d, J = 8.4 Hz, 2H), 7.36 and 7.34 (d, J = 8.4 Hz, 2H), 5.91 and 5.85 (s, 1H), 5.62 and 5.57 (s, 1H), 4.69 and 4.56 (br s, 1H), 4.20–4.14 (m, 1H), 3.77–3.66 (m, 2H), 3.64–3.40 (m, 8H), 2.50–2.40 (m, 6H), 2.29 and 2.28 (s, 3H), 2.26–2.17 (m, 1H), 2.14–2.05 (m, 1H), 1.12 and 1.11 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 167.46, 163.52, 163.25, 161.03 × 2, 160.84, 135.68 and 135.64, 134.57, 129.19, 128.37 and 128.31, 75.52, 54.84 and 52.16, 52.02 × 2, 51.78, 51.21 and 49.53, 47.31 and 44.56, 43.76, 31.81 and 29.42, 11.94 × 2; LCMS (ESI) m/z: 510.2 [M + H]+; HRMS (ESI) calcd for C25H32ClN9O [M + H]+m/z: 510.2497; found, 510.2498; HPLC purity = 99.63%, tR = 10.60 min.
(S)-(2,4-Dichlorophenyl) (3-((4-(4-ethylpiperazin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone (8)
Similar to the reaction procedures for 13, a solution of 2 N hydrochloric acid in ether (1.06 mL, 2.1 mmol) was added to a solution of compound 34 (200 mg, 0.42 mmol) in dichloromethane/methanol (2:1, 1 mL), and the resulting mixture was stirred at room temperature for 4 h. After concentration in vacuo, the residue was dissolved in DMF/dichloromethane (1:3, 4 mL), and then, triethylamine (0.35 mL, 2.52 mmol), 2,4-dichlorobenzoic acid (96 mg, 0.5 mmol), and propanephosphonic acid anhydride (T3P) ≥50 wt % in ethyl acetate (400 mg, 0.63 mmol) were added. The solution was stirred at room temperature for 16 h. After workup, the crude residue was purified by silica gel column chromatography with ethyl acetate/methanol (85:15) to afford compound 8 (101 mg, 0.18 mmol, 44% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.42 and 7.41 (d, J = 11.8 Hz, 1H), 7.25–7.23 (m, 1H), 7.22 and 7.20 (t, J = 7.6 Hz, 1H), 5.91 and 5.86 (s, 1H), 5.65 and 5.64 (s, 1H), 4.57 (br s, 1H), 4.11–4.00 (m, 1H), 3.80–3.68 (m, 1H), 3.63–3.47 (m, 5H), 3.45–3.14 (m, 2H), 2.51–2.40 (m, 6H), 2.29 and 2.28 (s, 3H), 2.27–2.02 (m, 2H), 1.12 (t, J = 7.6 Hz, 3H); 13C NMR (175 MHz, DMSO-d6): δ 164.60, 163.41, 163.14, 160.92 × 2, 160.70, 136.04 and 135.88, 134.16 and 136.12, 130.15 and 130.04, 129.26 and 129.21, 129.09 and 129.00, 127.87, 75.47, 53.16 and 51.17, 52.12, 51.99, 51.69, 50.71 and 49.62, 45.85 and 43.72, 43.67, 31.19 and 29.41, 11.95 × 2; LCMS (ESI) m/z: 544.2 [M + H]+; HRMS (ESI) calcd for C25H31Cl2N9O [M + H]+m/z: 544.2107; found, 544.2094; HPLC purity = 99.97%, tR = 12.04 min.
(S)-(4-Chloro-2-methylphenyl) (3-((4-(4-ethylpiperazin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone (9)
Similar to the reaction procedures for 13, a solution of 2 N hydrochloric acid in ether (1.06 mL, 2.1 mmol) was added to a solution of compound 34 (200 mg, 0.42 mmol) in dichloromethane/methanol (2:1, 1 mL), and the resulting mixture was stirred at room temperature for 4 h. After concentration in vacuo, the residue was dissolved in DMF/dichloromethane (1:3, 4 mL), and then, triethylamine (0.35 mL, 2.52 mmol), 4-chloro-2-methylbenzoic acid (85 mg, 0.5 mmol), and propanephosphonic acid anhydride (T3P) ≥50 wt % in ethyl acetate (400 mg, 0.63 mmol) were added. The solution was stirred at room temperature for 16 h. After workup, the crude residue was purified by silica gel column chromatography with ethyl acetate/methanol (90:10) to afford compound 9 (160 mg, 0.31 mmol, 68% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.22–7.07 (m, 3H), 5.91 and 5.86 (s, 1H), 5.66 (s, 1H), 4.55 (br s, 1H), 4.15–4.05 (m, 1H), 3.79–3.32 (m, 7H), 3.20–3.13 (m, 1H), 2.54–2.40 (m, 6H), 2.30–2.25 (m, 6H), 2.15–2.05 (m, 2H), 1.13 and 1.12 (t, J = 7.0 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 168.04, 163.90, 163.65, 161.42 × 2, 161.19, 137.19 and 137.00, 136.80 and 136.60, 133.38 and 133.33, 130.32 and 130.25, 127.93 and 127.84, 126.17, 75.95, 54.00 and 51.50, 52.52, 52.39, 52.09, 51.25 and 50.12, 46.61 and 43.82, 44.18, 44.14, 40.58, 31.79 and 29.99, 18.83, 18.68, 12.32; LCMS (ESI) m/z: 524.3 [M + H]+; HRMS (ESI) calcd for C26H34ClN9O [M + H]+m/z: 524.2653; found, 524.2655; HPLC purity = 99.65%, tR = 11.29 min.
(S)-(4-Chloro-3-fluorophenyl) (3-((4-(4-ethylpiperazin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone (10)
Similar to the reaction procedures for 13, a solution of 2 N hydrochloric acid in ether (1.06 mL, 2.1 mmol) was added to a solution of compound 34 (200 mg, 0.42 mmol) in dichloromethane/methanol (2:1, 1 mL), and the resulting mixture was stirred at room temperature for 4 h. After concentration in vacuo, the residue was dissolved in DMF/dichloromethane (1:3, 4 mL), and then, triethylamine (0.35 mL, 2.52 mmol), 4-chloro-3-fluorobenzoic acid (87 mg, 0.5 mmol), and propanephosphonic acid anhydride (T3P) ≥50 wt % in ethyl acetate (400 mg, 0.63 mmol) were added. The solution was stirred at room temperature for 16 h. After workup, the crude residue was purified by silica gel column chromatography with ethyl acetate/methanol (85:15) to afford compound 10 (122 mg, 0.23 mmol, 55% yield) as a yellow solid. 1H NMR (700 MHz, DMSO-d6): δ 8.70 (br s, 1H), 7.68 and 7.64 (t, J = 8.1 Hz, 1H), 7.60 and 7.57 (dd, J = 9.8, 1.4 Hz, 1H), 6.51 (br s, 1H), 6.06 (br s, 1H), 5.80 (br s, 1H), 4.41 and 4.20 (br s, 1H), 3.85–3.73 (m, 1H), 3.67–3.62 (m, 1H), 3.61–3.50 (m, 1H), 3.50–3.40 (m, 3H), 2.49–2.22 (m, 6H), 2.16 and 2.13 (s, 3H), 2.11–2.09 (m, 1H), 2.03–1.86 (m, 1H), 1.07–0.99 (m, 3H); 13C NMR (175 MHz, DMSO-d6): δ 166.00, 165.96, 163.35, 163.08, 160.89 × 2, 156.80 and 156.76 (d, JC–F = 245.9 Hz), 137.83 and 137.80 (d, JC–F = 12.3 Hz), 130.69 (d, JC–F = 10.5 Hz), 124.53, 120.95 and 120.92 (d, JC–F = 17.3 Hz), 115.78 (d, JC–F = 22.0 Hz), 75.56, 54.54 and 51.11, 51.96, 51.84, 51.63, 49.37 and 49.09, 44.39 and 40.00, 43.53, 31.69 and 29.37, 11.76 × 2; LCMS (ESI) m/z: 528.2 [M + H]+; HRMS (ESI) calcd for C25H31ClFN9O [M + H]+m/z: 528.2402; found, 528.2400; HPLC purity = 98.79%, tR = 11.67 min.
(S)-(4-Chloro-3-methylphenyl) (3-((4-(4-ethylpiperazin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone (11)
Similar to the reaction procedures for 13, a solution of 2 N hydrochloric acid in ether (1.06 mL, 2.1 mmol) was added to a solution of compound 34 (200 mg, 0.42 mmol) in dichloromethane/methanol (2:1, 1 mL), and the resulting mixture was stirred at room temperature for 4 h. After concentration in vacuo, the residue was dissolved in DMF/dichloromethane (1:3, 4 mL), and then, triethylamine (0.35 mL, 2.52 mmol), 4-chloro-3-methylbenzoic acid (85 mg, 0.5 mmol), and propanephosphonic acid anhydride (T3P) ≥50 wt % in ethyl acetate (400 mg, 0.63 mmol) were added. The solution was stirred at room temperature for 16 h. After workup, the crude residue was purified by silica gel column chromatography with ethyl acetate/methanol (90:10) to afford compound 11 (170 mg, 0.32 mmol, 60% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.36–7.30 (m, 2H), 7.23 and 7.23 (d, J = 8.0 Hz, 1H), 5.91 and 5.84 (s, 1H), 5.641 and 5.61 (s, 1H), 4.65 and 4.56 (br s, 1H), 4.12–4.00 (m, 1H), 3.80–3.70 (m, 2H), 3.65–3.40 (m, 6H), 2.53–2.40 (m, 6H), 2.36 (s, 3H), 2.29 and 2.28 (s, 3H), 2.25–2.16 (m, 1H), 2.15–2.03 (m, 1H), 1.12 and 1.11 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 167.39 × 2, 163.38, 163.13, 160.87, 160.68, 135.77, 135.46 and 135.39, 134.47, 129.83, 128.59 and 128.53, 126.23, 75.40, 54.60 and 52.01, 51.87, 51.81, 51.58, 51.05 and 49.38, 47.12 and 44.17, 43.65, 31.64 and 29.30, 19.34, 11.80; LCMS (ESI) m/z: 524.3 [M + H]+; HRMS (ESI) calcd for C26H34ClN9O [M + H]+m/z: 524.2653; found, 524.2653; HPLC purity = 97.33%, tR = 11.61 min.
(S)-(5-Chloro-2-fluorophenyl) (3-((4-(4-ethylpiperazin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone (12)
Similar to the reaction procedures for 13, a solution of 2 N hydrochloric acid in ether (1.06 mL, 2.1 mmol) was added to a solution of compound 34 (200 mg, 0.42 mmol) in dichloromethane/methanol (2:1, 1 mL), and the resulting mixture was stirred at room temperature for 4 h. After concentration in vacuo, the residue was dissolved in DMF/dichloromethane (1:3, 4 mL), and then, triethylamine (0.35 mL, 2.52 mmol), 5-chloro-2-fluorobenzoic acid (87 mg, 0.5 mmol), and propanephosphonic acid anhydride (T3P) ≥50 wt % in ethyl acetate (400 mg, 0.63 mmol) were added. The solution was stirred at room temperature for 16 h. After workup, the crude residue was purified by silica gel column chromatography with ethyl acetate/methanol (90:10) to afford compound 12 (162 mg, 0.31 mmol, 68% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.39–7.30 (m, 2H), 7.06 and 7.03 (t, J = 8.8 Hz, 1H), 5.89 and 5.85 (s, 1H), 5.73 and 5.71 (s, 1H), 4.56 (br s, 1H), 3.99–3.80 (m, 1H), 3.80–3.50 (m, 6H), 3.46–3.24 (m, 2H), 2.62–2.45 (m, 6H), 2.31 and 2.29 (s, 3H), 2.23–2.15 (m, 2H), 1.17 and 1.15 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 163.67, 163.49, 162.81, 161.33 × 2, 161.25, 156.88 and 156.83 (d, JC–F = 245.6 Hz), 131.57, 129.04, 128.70, 127.60 and 127.51 (d, JC–F = 20.1 Hz), 118.47 and 118.40 (d, JC–F = 23.6 Hz), 76.11, 53.56 and 51.90, 51.76 × 3, 51.15 and 50.03, 46.39 and 44.38, 43.26, 31.61 and 29.98, 11.37 × 2; LCMS (ESI) m/z: 528.2 [M + H]+; HRMS (ESI) calcd for C25H31ClFN9O [M + H]+m/z: 528.2402; found, 528.2410; HPLC purity = 97.89%, tR = 10.86 min.
(S)-(3-((4-(4-Ethylpiperazin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl) (2-fluoro-4-(trifluoromethyl)phenyl)methanone (14)
Similar to the reaction procedures for 13, a solution of 2 N hydrochloric acid in ether (1.06 mL, 2.1 mmol) was added to a solution of compound 34 (200 mg, 0.42 mmol) in dichloromethane/methanol (2:1, 1 mL), and the resulting mixture was stirred at room temperature for 4 h. After concentration in vacuo, the residue was dissolved in DMF/dichloromethane (1:3, 4 mL), and then, triethylamine (0.35 mL, 2.52 mmol), 2-fluoro-4-(trifluoromethyl)benzoic acid (104 mg, 0.5 mmol), and propanephosphonic acid anhydride (T3P) ≥50 wt % in ethyl acetate (400 mg, 0.63 mmol) were added. The solution was stirred at room temperature for 16 h. After workup, the crude residue was purified by silica gel column chromatography with ethyl acetate/methanol (85:15) to afford compound 14 (129 mg, 0.23 mmol, 55% yield) as a yellow solid. 1H NMR (300 MHz, CD3OD): δ 7.74–7.55 (m, 3H), 5.87 and 5.82 (s, 1H), 4.56 and 4.42 (br s, 1H), 4.01–3.77 (m, 1H), 3.77–3.62 (m, 4H), 3.62–3.57 (m, 3H), 3.57–3.40 (m, 1H), 2.83–2.57 (m, 6H), 2.40–2.30 (m, 1H), 2.24 and 2.22 (s, 3H), 2.15–2.02 (m, 1H), 1.27–1.14 (m, 3H); 13C NMR (100 MHz, DMSO-d6): δ 163.24, 163.05, 162.70, 169.94 × 2, 160.82, 157.54 (d, JC–F = 247.0 Hz), 131.56 (q, JC–F = 31.2 Hz), 130.21, 129.45 (d, JC–F = 17.6 Hz), 123.20 (q, JC–F = 247.8 Hz), 121.97, 113.72 (d, JC–F = 25.9 Hz), 75.66, 53.23 and 51.56, 51.37 × 3, 50.79 and 49.66, 46.03 and 44.09, 42.80, 31.18 and 29.54, 10.95 × 2; LCMS (ESI) m/z: 562.3 [M + H]+; HRMS (ESI) calcd for C26H31F4N9O [M + H]+m/z: 562.2666; found, 562.2659; HPLC purity = 98.61%, tR = 11.59 min.
(S)-(4-Chloro-2,6-difluorophenyl) (3-((4-(4-ethylpiperazin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone (15)
Similar to the reaction procedures for 13, a solution of 2 N hydrochloric acid in ether (1.06 mL, 2.1 mmol) was added to a solution of compound 34 (200 mg, 0.42 mmol) in dichloromethane/methanol (2:1, 1 mL), and the resulting mixture was stirred at room temperature for 4 h. After concentration in vacuo, the residue was dissolved in DMF/dichloromethane (1:3, 4 mL), and then, triethylamine (0.35 mL, 2.52 mmol), 4-chloro-2,6-difluorobenzoic acid (96 mg, 0.5 mmol), and propanephosphonic acid anhydride (T3P) ≥50 wt % in ethyl acetate (400 mg, 0.63 mmol) were added. The solution was stirred at room temperature for 16 h. After workup, the crude residue was purified by silica gel column chromatography with ethyl acetate/methanol (85:15) to afford compound 15 (140 mg, 0.26 mmol, 61% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.10–6.95 (m, 2H), 5.88 and 5.85 (s, 1H), 5.76 and 5.74 (s, 1H), 4.58 (br s, 1H), 4.21–3.87 (m, 2H), 3.80–3.68 (m, 1H), 3.67–3.47 (m, 5H), 3.43–3.17 (m, 1H), 2.57–2.43 (m, 6H), 2.38–2.30 (m, 1H), 2.29 and 2.28 (s, 3H), 2.24–2.25 (m, 1H), 1.14 and 1.14 (t, J = 7.2 Hz, 3H); 13C NMR (175 MHz, DMSO-d6): δ 163.57, 163.35, 161.23, 161.02, 158.65, 158.56, 158.27 (t, JC–F = 247.4 Hz), 135.72 (t, JC–F = 12.8 Hz), 114.12 (q, JC–F = 23.1 Hz), 113.72 (d, JC–F = 23.3 Hz), 75.89, 53.12 and 50.84, 51.84 × 3, 50.01 and 45.86, 44.34 and 40.00, 43.39, 31.01 and 29.66, 11.57, 11.49; LCMS (ESI) m/z: 546.2 [M + H]+; HRMS (ESI) calcd for C25H30ClF2N9O [M + H]+m/z: 546.2308; found, 546.2314; HPLC purity = 99.75%, tR = 11.23 min.
(S)-(4-Chloro-2,5-difluorophenyl) (3-((4-(4-ethylpiperazin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone (16)
Similar to the reaction procedures for 13, a solution of 2 N hydrochloric acid in ether (1.06 mL, 2.1 mmol) was added to a solution of compound 34 (200 mg, 0.42 mmol) in dichloromethane/methanol (2:1, 1 mL), and the resulting mixture was stirred at room temperature for 4 h. After concentration in vacuo, the residue was dissolved in DMF/dichloromethane (1:3, 4 mL), and then, triethylamine (0.35 mL, 2.52 mmol), 4-chloro-2,5-difluorobenzoic acid (96 mg, 0.5 mmol), and propanephosphonic acid anhydride (T3P) ≥50 wt % in ethyl acetate (400 mg, 0.63 mmol) were added. The solution was stirred at room temperature for 16 h. After workup, the crude residue was purified by silica gel column chromatography with ethyl acetate/methanol (85:15) to afford compound 16 (119 mg, 0.22 mmol, 52% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.23–7.10 (m, 2H), 5.91 and 5.86 (s, 1H), 5.70 and 5.68 (s, 1H), 4.63 and 4.58 (br s, 1H), 4.02–3.95 (m, 1H), 3.80–3.70 (m, 2H), 3.65–3.47 (m, 5H), 3.44–3.25 (m, 1H), 2.55–2.42 (m, 6H), 2.30 and 2.29 (s, 3H), 2.22–2.03 (m, 2H), 1.14 and 1.13 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 163.37, 163.12, 161.78, 160.93, 160.79 × 2, 153.81 (d, JC–F = 242.5 Hz), 153.48 (d, JC–F = 244.8 Hz), 125.72, 121.46, 118.63 and 118.57 (d, JC–F = 28.2 Hz), 116.46 and 116.26, 76.50, 53.24 and 51.81, 51.74, 51.59 × 2, 50.75 and 49.61, 46.00 and 44.05, 43.38, 31.17 and 29.20, 11.57 × 2; LCMS (ESI) m/z: 546.2 [M + H]+; HRMS (ESI) calcd for C25H30ClF2N9O [M + H]+m/z: 546.2308; found, 546.2312; HPLC purity = 97.98%, tR = 11.07 min.
(S)-(4-Chloro-2,3-difluorophenyl) (3-((4-(4-ethylpiperazin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone (17)
Similar to the reaction procedures for 13, a solution of 2 N hydrochloric acid in ether (1.06 mL, 2.1 mmol) was added to a solution of compound 34 (200 mg, 0.42 mmol) in dichloromethane/methanol (2:1, 1 mL), and the resulting mixture was stirred at room temperature for 4 h. After concentration in vacuo, the residue was dissolved in DMF/dichloromethane (1:3, 4 mL), and then, triethylamine (0.35 mL, 2.52 mmol), 4-chloro-2,3-difluorobenzoic acid (96 mg, 0.5 mmol), and propanephosphonic acid anhydride (T3P) ≥50 wt % in ethyl acetate (400 mg, 0.63 mmol) were added. The solution was stirred at room temperature for 16 h. After workup, the crude residue was purified by silica gel column chromatography with ethyl acetate/methanol (85:15) to afford compound 17 (101 mg, 0.18 mmol, 44% yield) as a yellow solid. 1H NMR (700 MHz, CDCl3): δ 7.18 (t, J = 7.0 Hz, 1H), 7.04 (s, 1H), 5.93 and 5.87 (s, 1H), 5.63 and 5.60 (s, 1H), 4.70 and 4.58 (br s, 1H), 4.11 (m, 1H), 3.79–3.68 (m, 2H), 3.60–3.57 (m, 1H), 3.56–3.46 (m, 4H), 3.41–3.27 (m, 1H), 2.50–2.41 (m, 6H), 2.30 and 2.29 (s, 3H), 2.27–2.22 (m, 1H), 2.16–2.08 (m, 1H), 1.12 and 1.11 (t, J = 7.0 Hz, 3H); 13C NMR (175 MHz, DMSO-d6): δ 164.82 × 2, 164.20, 164.17, 161.03, 147.78 and 147.33 (dd, JC–F = 237.0, 15.2 Hz), 147.42 (dd, JC–F = 237.6, 13.8 Hz), 126.09, 125.79 and 125.71 (dd, JC–F = 48.6, 14.4 Hz), 124.41 and 124.33 (dd, JC–F = 60.9, 14.3 Hz), 123.75, 123.04, 75.03, 55.23 and 51.13, 52.73, 52.67, 52.63, 51.79 and 50.64, 46.35 and 45.27, 44.36, 32.62 and 30.73, 12.12, 11.73; LCMS (ESI) m/z: 546.2 [M + H]+; HRMS (ESI) calcd for C25H30ClF2N9O [M + H]+m/z: 546.2308; found, 546.2300; HPLC purity = 99.16%, tR = 11.82 min.
(S)-(4-Chloro-3,5-difluorophenyl) (3-((4-(4-ethylpiperazin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone (18)
Similar to the reaction procedures for 13, a solution of 2 N hydrochloric acid in ether (1.06 mL, 2.1 mmol) was added to a solution of compound 34 (200 mg, 0.42 mmol) in dichloromethane/methanol (2:1, 1 mL), and the resulting mixture was stirred at room temperature for 4 h. After concentration in vacuo, the residue was dissolved in DMF/dichloromethane (1:3, 4 mL), and then, triethylamine (0.35 mL, 2.52 mmol), 4-chloro-3,5-difluorobenzoic acid (96 mg, 0.5 mmol), and propanephosphonic acid anhydride (T3P) ≥50 wt % in ethyl acetate (400 mg, 0.63 mmol) were added. The solution was stirred at room temperature for 16 h. After workup, the crude residue was purified by silica gel column chromatography with ethyl acetate/methanol (90:10) to afford compound 18 (138 mg, 0.25 mmol, 56% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.18 and 7.15 (d, J = 7.0 Hz, 2H), 5.88 and 5.84 (s, 1H), 5.76 and 5.73 (s, 1H), 4.57 (br s, 1H), 4.01–3.85 (m, 1H), 3.82–3.53 (m, 6H), 3.52–3.40 (m, 2H), 2.58–2.45 (m, 6H), 2.30 and 2.29 (s, 3H), 2.20–2.15 (m, 2H), 1.20–1.10 (m, 3H); 13C NMR (150 MHz, DMSO-d6): δ 165.44, 163.66, 163.45, 161.29 × 3, 158.18 and 158.14 (d, JC–F = 248.9 Hz), 138.18 (t, JC–F = 6.8 Hz), 112.06 (d, JC–F = 23.7 Hz), 110.11 (t, JC–F = 20.2 Hz), 76.06, 54.71, 52.40 and 51.55, 51.74 × 2, 49.84 and 47.43, 44.88 and 43.30, 43.30, 32.08 and 29.91, 11.84, 11.33; LCMS (ESI) m/z: 546.2 [M + H]+; HRMS (ESI) calcd for C25H30ClF2N9O [M + H]+m/z: 546.2308; found, 546.2306; HPLC purity = 98.78%, tR = 12.07 min.
(S)-(3-((4-(4-Ethylpiperazin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl) (2,4,5-trifluorophenyl)methanone (19)
Similar to the reaction procedures for 13, a solution of 2 N hydrochloric acid in ether (1.06 mL, 2.1 mmol) was added to a solution of compound 34 (200 mg, 0.42 mmol) in dichloromethane/methanol (2:1, 1 mL), and the resulting mixture was stirred at room temperature for 4 h. After concentration in vacuo, the residue was dissolved in DMF/dichloromethane (1:3, 4 mL), and then, triethylamine (0.35 mL, 2.52 mmol), 2,4,5-trifluorobenzoic acid (88 mg, 0.5 mmol), and propanephosphonic acid anhydride (T3P) ≥50 wt % in ethyl acetate (400 mg, 0.63 mmol) were added. The solution was stirred at room temperature for 16 h. After workup, the crude residue was purified by silica gel column chromatography with ethyl acetate/methanol (85:15) to afford compound 19 (100 mg, 0.19 mmol, 45% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.33–7.23 (m, 1H), 7.01–6.92 (m, 1H), 5.87 and 5.84 (s, 1H), 5.74 and 5.72 (s, 1H), 4.54 (br s, 1H), 3.96–3.80 (m, 2H), 3.78–3.50 (m, 5H), 3.49–3.26 (m, 2H), 2.60–2.50 (m, 6H), 2.31 and 2.30 (s, 3H), 2.22–2.03 (m, 2H), 1.17 and 1.16 (t, J = 7.0 Hz, 3H); 13C NMR (175 MHz, DMSO-d6): δ 172.74, 163.37, 163.19, 162.41, 161.17, 161.07, 153.62 (dt, JC–F = 244.9, 8.6 Hz), 150.14 (dt, JC–F = 250.0, 13.4 Hz), 146.46 (dt, JC–F = 244.3, 12.7 Hz), 122.14 (t, J = 21.8 Hz), 117.20 (d, J = 20.3 Hz), 107.08 (ddd, J = 29.2, 21.4, 11.1 Hz), 75.97, 53.55, 51.92 and 50.98, 51.61, 51.25, 49.91 and 46.37, 44.45 and 42.60, 42.51, 31.36 and 29.76, 10.66, 10.55; LCMS (ESI) m/z: 530.3 [M + H]+; HRMS (ESI) calcd for C25H30F3N9O [M + H]+m/z: 530.2604; found, 530.2617; HPLC purity = 96.24%, tR = 10.37 min.
(S)-(6-Chloro-2-fluoropyridin-3-yl) (3-((4-(4-ethylpiperazin-1-yl)-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)amino)pyrrolidin-1-yl)methanone (20)
Similar to the reaction procedures for 13, a solution of 2 N hydrochloric acid in ether (1.06 mL, 2.1 mmol) was added to a solution of compound 34 (200 mg, 0.42 mmol) in dichloromethane/methanol (2:1, 1 mL), and the resulting mixture was stirred at room temperature for 4 h. After concentration in vacuo, the residue was dissolved in DMF/dichloromethane (1:3, 4 mL), and then, triethylamine (0.35 mL, 2.52 mmol), 6-chloro-2-fluoropyridine-3-carboxylic acid (87 mg, 0.5 mmol), and propanephosphonic acid anhydride (T3P) ≥50 wt % in ethyl acetate (400 mg, 0.63 mmol) were added. The solution was stirred at room temperature for 16 h. After workup, the crude residue was purified by silica gel column chromatography with ethyl acetate/methanol (85:15) to afford compound 20 (102 mg, 0.19 mmol, 46% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.82 and 7.82 (t, J = 8.2 Hz, 1H), 7.29–7.24 (m, 1H), 5.90 and 5.85 (s, 1H), 5.68 and 5.66 (s, 1H), 4.63 and 4.59 (br s, 1H), 4.04–3.93 (m, 1H), 3.83–3.72 (m, 2H), 3.63–3.26 (m, 6H), 2.53–2.42 (m, 6H), 2.31 and 2.30 (s, 3H), 2.22–2.04 (m, 2H), 1.13 and 1.13 (t, J = 7.2 Hz, 3H); 13C NMR (175 MHz, DMSO-d6): δ 163.69, 163.45, 162.03, 162.01, 161.23, 161.03, 157.25 and 157.13 (d, JC–F = 242.2 Hz), 148.26 (t, JC–F = 12.1 Hz), 143.65 (d, JC–F = 12.8 Hz), 123.02, 118.76 and 118.67 (d, JC–F = 31.0 Hz), 75.79, 53.59 and 52.05, 52.21, 52.12, 51.96, 51.09 and 49.89, 46.35 and 44.52, 43.75, 31.43 and 29.73, 11.90 × 2; LCMS (ESI) m/z: 529.2 [M + H]+; HRMS (ESI) calcd for C24H30ClFN10O [M + H]+m/z: 529.2355; found, 529.2353; HPLC purity = 97.47%, tR = 9.99 min.
(S)-N2-(1-((4-Chloro-2-fluorophenyl)sulfonyl)pyrrolidin-3-yl)-6-(4-ethylpiperazin-1-yl)-N4-(5-methyl-1H-pyrazol-3-yl)pyrimidine-2,4-diamine (21)
A solution of 2 N hydrochloric acid in ether (1.06 mL, 2.1 mmol) was added to a solution of compound 34 (200 mg, 0.42 mmol) in dichloromethane/methanol (2:1, 1 mL) at room temperature. The resulting mixture was stirred at room temperature for 4 h and then concentrated to give crude amine salt, which was used without further purification.
Triethylamine (0.35 mL, 2.52 mmol) and 4-chloro-2-fluorobenzenesulfonyl chloride (114 mg, 0.5 mmol) were added to a solution of amine salt in dichloromethane (4 mL) at room temperature. The resulting mixture was stirred at room temperature for 4 h and then quenched with brine (20 mL). The aqueous phase was extracted with ethyl acetate (3 × 30 mL). The combined organic extracts were washed with water and brine, dried over magnesium sulfate, and filtered. The filtrate was concentrated to get a crude residue, which was purified by flash column chromatography over silica gel with ethyl acetate/methanol (85:15) to afford compound 21 (201 mg, 0.36 mmol, 85% yield) as a light-purple solid. 1H NMR (300 MHz, CD3OD): δ 7.81 and 7.80 (t, J = 7.5 Hz, 1H), 7.34–7.28 (m, 2H), 5.82 (s, 1H), 5.57 (s, 1H), 4.27 (br s, 1H), 3.80–3.66 (m, 4H), 3.63–3.45 (m, 5H), 3.02–2.80 (m, 6H), 2.26 (s, 3H), 2.23–2.15 (m, 1H), 2.04–1.93 (m, 1H), 1.28 and 1.27 (t, J = 7.1 Hz, 3H); 13C NMR (175 MHz, DMSO-d6): δ 162.84 × 2, 160.96 × 3, 158.37 (d, JC–F = 255.5 Hz), 139.33 (d, JC–F = 9.6 Hz), 132.27, 125.45 (d, JC–F = 2.6 Hz), 124.09 (d, JC–F = 15.3 Hz), 118.36 (d, JC–F = 25.9 Hz), 76.00, 64.32, 61.58, 53.24, 50.76, 46.36, 38.57, 38.53, 30.64, 7.17 × 2; LCMS (ESI) m/z: 564.2 [M + H]+; HRMS (ESI) calcd for C24H31ClFN9O2S [M + H]+m/z: 564.2072; found, 564.2063; HPLC purity = 96.34%, tR = 11.11 min.
General Procedure for the Synthesis of Compounds 22–27
The general procedure is illustrated below with compounds 25 as a specific example.
(S)-1-(5-((2-((1-(4-Chloro-2-fluorobenzoyl)pyrrolidin-3-yl)amino)-6-(4-ethylpiperazin-1-yl)pyrimidin-4-yl)amino)-3-methyl-1H-pyrazol-1-yl)propan-1-one (25)
A solution of propionic anhydride (60 mg, 0.46 mmol) in 1,4-dioxane (0.5 mL) was added to a solution of compound 13 (300 mg, 0.42 mmol) in 1,4-dioxane (6 mL) at 140 °C. The resulting mixture was stirred at 140 °C for 30 min, cooled to room temperature, and then concentrated in vacuo. The residue was purified by flash column chromatography over silica gel with n-hexane/ethyl acetate/triethylamine (60:35:5) to afford compound 25 (137 mg, 0.24 mmol, 56% yield) and 26 (69 mg, 0.12 mmol, 28% yield) as a pale-yellow solid. 1H NMR (300 MHz, CDCl3): δ 9.88 and 9.84 (br s, 1H), 7.34 and 7.32 (t, J = 7.7 Hz, 1H), 7.12 (q, J = 8.3 Hz, 1H), 7.05 (d, J = 9.3 Hz, 1H), 6.51 and 6.42 (s, 1H), 5.30 and 5.25 (s, 1H), 4.72 (br s, 1H), 4.60–4.40 (m, 1H), 4.00–3.60 (m, 3H), 3.60–3.15 (m, 5H), 3.06 and 3.05 (q, J = 8.3 Hz, 2H), 2.65–2.37 (m, 6H), 2.35–2.23 (m, 1H), 2.19 and 2.17 (s, 3H), 2.03–1.92 (m, 1H), 1.23–1.07 (m, 6H); 13C NMR (100 MHz, CDCl3): δ 178.03, 164.45, 163.88 and 163.77, 161.40 and 161.32, 159.13, 158.31 and 158.28 (d, JC–F = 250.1 Hz), 153.91 and 153.85, 144.18 and 144.12, 136.62 (d, JC–F = 9.9 Hz), 130.13 and 130.08 (d, JC–F = 10.0 Hz), 125.18 and 125.15, 123.84 and 123.80 (d, JC–F = 18.0 Hz), 116.82 and 116.74 (d, JC–F = 25.2 Hz), 95.80 and 95.69, 78.39, 53.70 and 52.24, 52.47, 52.41, 51.20 and 50.28, 46.01 and 44.35, 44.05, 32.21 and 30.55, 29.00, 14.45, 11.95, 8.32; LCMS (ESI) m/z: 584.2 [M + H]+; HRMS (ESI) calcd for C28H35ClFN9O2 [M + H]+m/z: 584.2665; found, 584.2645; HPLC purity = 96.81%, tR = 16.39 min.
(S)-1-(3-((2-((1-(4-Chloro-2-fluorobenzoyl)pyrrolidin-3-yl)amino)-6-(4-ethylpiperazin-1-yl)pyrimidin-4-yl)amino)-5-methyl-1H-pyrazol-1-yl)propan-1-one (26)
1H NMR (400 MHz, CDCl3): δ 7.35 (q, J = 6.8 Hz, 1H), 7.15 (q, J = 9.7 Hz, 1H), 7.09 (d, J = 9.6 Hz, 1H), 6.35 and 6.30 (s, 1H), 6.04 and 6.00 (s, 1H), 5.03 and 4.93 (br s, 1H), 4.55 and 4.44 (d, J = 5.7 Hz, 1H), 4.03–3.34 (m, 7H), 3.30–3.16 (m, 1H), 3.04 (t, J = 7.8 Hz, 2H), 2.60–2.36 (m, 5H), 2.47–2.36 (m, 4H), 2.35–2.18 (m, 2H), 1.29–1.19 (m, 3H), 1.11 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 174.26 and 174.24, 164.55, 164.36 and 164.28, 160.78, 159.64 and 159.56, 158.29 (d, JC–F = 230.3 Hz), 150.78 and 150.71, 144.61, 136.72 (d, JC–F = 9.9 Hz), 130.23 and 130.19 (d, JC–F = 9.1 Hz), 125.27, 123.94 and 123.85 (d, JC–F = 18.0 Hz), 116.91 and 116.83 (d, JC–F = 25.2 Hz), 103.03 and 102.95, 78.22, 53.78 and 52.32, 52.59, 52.53, 51.15 and 50.24, 46.11 and 44.42, 44.18, 32.25 and 30.64, 28.90, 14.88, 12.02, 8.85; LCMS (ESI) m/z: 584.3 [M + H]+; HRMS (ESI) calcd for C28H35ClFN9O2 [M + H]+m/z: 584.2655; found, 584.2665; HPLC purity = 97.32%, tR = 12.99 min.
Ethyl (S)-5-((2-((1-(4-chloro-2-fluorobenzoyl)pyrrolidin-3-yl)amino)-6-(4-ethylpiperazin-1-yl)pyrimidin-4-yl)amino)-3-methyl-1H-pyrazole-1-carboxylate (22)
Similar to the reaction procedures for 25, diethyl dicarbonate (375 mg, 2.3 mmol) was added to a solution of compound 13 (300 mg, 0.42 mmol) in 1,4-dioxane (6 mL). The resulting mixture was stirred at 140 °C for 30 min. After workup, the crude residue was purified by silica gel column chromatography with dichloromethane/methanol (97:3) to give compound 22 (118 mg, 0.2 mmol, 47% yield) and compound 23 (103 mg, 0.17 mmol, 41% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 9.35 and 9.31 (br s, 1H), 7.38 (q, J = 7.3 Hz, 1H), 7.22–7.07 (m, 2H), 6.56 and 6.48 (s, 1H), 5.37 and 5.33 (s, 1H), 4.90 and 4.84 (br s, 1H), 4.62–4.44 (m, 3H), 4.04–3.68 (m, 2H), 3.66–3.45 (m, 4H), 3.43–3.21 (m, 2H), 2.57–2.40 (m, 6H), 2.38–2.29 (m, 1H), 2.27 and 2.26 (s, 3H), 2.06–1.95 (m, 1H), 1.47 and 1.46 (t, J = 7.2 Hz, 3H), 1.13 and 1.12 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 164.61, 164.01 and 163.90, 161.49 and 161.40, 158.45 and 158.41 (d, JC–F = 7.2 Hz), 159.17 and 159.14, 154.36 and 154.31, 152.59, 144.31 and 144.25, 136.80 (d, JC–F = 9.9 Hz), 130.26 and 130.21 (d, JC–F = 9.6 Hz), 125.33 and 125.31, 123.95 and 123.91 (d, JC–F = 17.9 Hz), 116.97 and 116.88 (d, JC–F = 25.2 Hz), 95.73 and 95.64, 78.43, 64.60, 53.87 and 52.38, 52.56, 52.50, 51.33 and 50.41, 46.14 and 44.45, 44.13, 32.39 and 30.75, 14.65, 14.50, 12.01 and 11.98; LCMS (ESI) m/z: 600.3 [M + H]+; HRMS (ESI) calcd for C28H35ClFN9O3 [M + H]+m/z: 600.2614; found, 600.2612; HPLC purity = 97.55%, tR = 16.35 min.
Ethyl (S)-3-((2-((1-(4-chloro-2-fluorobenzoyl)pyrrolidin-3-yl)amino)-6-(4-ethylpiperazin-1-yl)pyrimidin-4-yl)amino)-5-methyl-1H-pyrazole-1-carboxylate (23)
1H NMR (400 MHz, CDCl3): δ 7.37 (q, J = 7.5 Hz, 1H), 7.17 (q, J = 10.3 Hz, 1H), 7.10 (d, J = 9.6 Hz, 1H), 6.31 and 6.26 (s, 1H), 5.97 and 5.89 (s, 1H), 4.98 and 4.88 (br s, 1H), 4.58–4.39 (m, 3H), 4.04–3.67 (m, 2H), 3.65–3.51 (m, 4H), 3.50–3.19 (m, 2H), 2.53 and 2.52 (s, 3H), 2.50–2.42 (m, 6H), 2.35–2.17 (m, 2H), 1.43 and 1.42 (t, J = 7.0 Hz, 3H), 1.13 and 1.12 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 164.61, 164.15 and 164.11, 161.02, 160.01 and 159.88, 158.44 (d, JC–F = 250.8 Hz), 151.35 and 151.28, 150.51 and 150.48, 145.18, 136.78 (d, JC–F = 10.7 Hz), 130.28 and 130.23 (d, JC–F = 9.5 Hz), 125.32 (d, JC–F = 2.2 Hz), 124.00 and 123.94 (d, JC–F = 17.5 Hz), 116.97 and 116.90 (d, JC–F = 25.2 Hz), 103.05 and 102.96, 77.81 and 77.73, 63.94, 53.88 and 52.38, 52.63, 52.57, 51.25 and 50.35, 46.18 and 44.47, 44.09, 32.34 and 30.73, 14.68, 14.45, 12.01 and 11.98; LCMS (ESI) m/z: 600.3 [M + H]+; HRMS (ESI) calcd for C28H35ClFN9O3 [M + H]+m/z: 600.2614; found, 600.2616; HPLC purity = 97.55%, tR = 16.35 min.
tert-Butyl (S)-5-((2-((1-(4-chloro-2-fluorobenzoyl)pyrrolidin-3-yl)amino)-6-(4-ethylpiperazin-1-yl)pyrimidin-4-yl)amino)-3-methyl-1H-pyrazole-1-carboxylate (24)
Similar to the reaction procedures for 25, di-tert-butyl dicarbonate (100 mg, 0.46 mmol) was added to a solution of compound 13 (300 mg, 0.42 mmol) in 1,4-dioxane (6 mL). The resulting mixture was stirred at 140 °C for 30 min. After workup, the crude residue was purified by silica gel column chromatography with dichloromethane/methanol (97:3) to give compound 24 (90 mg, 0.14 mmol, 34% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 9.44 and 9.42 (br s, 1H), 7.39 (q, J = 7.7 Hz, 1H), 7.24–7.09 (m, 2H), 6.55 and 6.47 (s, 1H), 5.39 and 5.35 (s, 1H), 4.83 and 4.78 (br s, 1H), 4.63–4.45 (m, 2H), 4.23–4.00 (m, 2H), 3.92–3.72 (m, 2H), 3.65–3.48 (m, 2H), 3.47–3.23 (m, 1H), 2.55–2.39 (m, 4H), 2.39–2.32 (m, 2H), 2.28 and 2.27 (s, 3H), 2.07–1.99 (m, 2H), 1.56 (s, 9H), 1.14 and 1.12 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 164.66, 164.09 and 163.96, 161.52 and 161.43, 159.26, 158.46 (d, JC–F = 253.2 Hz), 153.63, 151.51, 144.34 and 144.28, 130.28 (d, JC–F = 10.7 Hz), 130.28 and 130.23 (d, JC–F = 9.6 Hz), 125.35, 123.97 (d, JC–F = 17.6 Hz), 116.99 and 116.91 (d, JC–F = 25.2 Hz), 95.56 and 95.49, 86.14, 78.40, 53.88 and 52.43, 52.66, 52.59, 51.34 and 50.43, 46.17 and 44.49, 44.25, 32.43 and 30.77, 28.26, 14.82, 12.11; LCMS (ESI) m/z: 628.3 [M + H]+; HRMS (ESI) calcd for C30H39ClFN9O3 [M + H]+m/z: 628.2927; found, 628.2922; HPLC purity = 97.10%, tR = 16.70 min.
(S)-1-(5-((2-((1-(4-Chloro-2-fluorobenzoyl)pyrrolidin-3-yl)amino)-6-(4-ethylpiperazin-1-yl)pyrimidin-4-yl)amino)-3-methyl-1H-pyrazol-1-yl)-2,2-dimethylpropan-1-one (27)
Similar to the reaction procedures for 25, pivalic anhydride (86 mg, 0.46 mmol) was added to a solution of compound 13 (300 mg, 0.42 mmol) in 1,4-dioxane (6 mL). The resulting mixture was stirred at 140 °C for 30 min. After workup, the crude residue was purified by silica gel column chromatography with dichloromethane/methanol (97:3) to give compound 27 (87 mg, 0.14 mmol, 34% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.42–7.35 (m, 1H), 7.23–7.09 (m, 2H), 6.55 and 6.50 (s, 1H), 5.87 and 5.84 (s, 1H), 4.92 and 4.79 (br s, 1H), 4.56 and 4.47 (q, J = 5.7 Hz, 1H), 4.06–3.79 (m, 1H), 3.77–3.55 (m, 5H), 3.54–3.19 (m, 2H), 2.56–2.40 (m, 9H), 2.36–2.22 (m, 1H), 2.03–1.94 (m, 1H), 1.53 and 1.51 (s, 9H), 1.16–1.12 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 177.66 and 177.63, 164.58, 164.45 and 164.35, 160.66, 159.25 and 159.17, 158.36 (d, JC–F = 250.9 Hz), 149.93 and 149.83, 145.61, 136.69 (d, JC–F = 9.9 Hz), 130.19 and 130.14 (d, JC–F = 9.6 Hz), 125.24, 123.88 and 123.78 (d, JC–F = 17.6 Hz), 116.88 and 116.80 (d, JC–F = 24.8 Hz), 101.95 and 101.85, 78.60, 53.81 and 52.27, 52.55, 52.49, 51.10 and 50.19, 46.15 and 42.07, 44.01, 44.31, 44.27, 44.14, 32.21 and 30.57, 24.79, 27.76, 27.71, 15.60, 12.00; LCMS (ESI) m/z: 612.3 [M + H]+; HRMS (ESI) calcd for C30H39ClFN9O2 [M + H]+m/z: 612.2978; found, 612.2974; HPLC purity = 97.45%, tR = 16.89 min.
In Vitro Inhibition of Aurora A Kinase Activity
The efficacy of the compounds as inhibitors of Aurora A kinase was assessed in vitro using the Kinase-Glo Plus Luminescent Kinase assay (Promega, USA), as described previously.45 In brief, recombinant glutathione S-transferase (GST)-tagged N-terminal truncated human Aurora A (amino acids 123–401) was expressed in Sf9 insect cells and purified by glutathione affinity chromatography to obtain recombinant Aurora A. Samples of purified recombinant Aurora A (150 ng) were reacted with each test compound in 50 μL of 50 mM Tris-HCl pH 7.4, 10 mM NaCl, 10 mM MgCl2, 0.01% bovine serum albumin, 5.0 μM ATP, 1 mM dithiothreitol, 15 μM tetra(-LRRASLG) peptide at 37 °C for 120 min, followed by addition of 50 μL Kinase-Glo Plus Reagent. The resulting mixture was incubated at 25 °C for 20 min. A 70 μL aliquot of the mixture was transferred to a black microtiter plate. Luminescence was measured using a Wallac Vector 1420 multilabel counter (PerkinElmer, USA).
Cell Culture
The SCLC cell lines NCI-H82, NCI-H446, NCI-H211, NCI-H524, NCI-H526, NCI-H146, NCI-H841, and NCI-H209, and the neuroblastoma cell line SK-N-BE(2) were obtained from American Type Culture Collection (ATCC, USA). All SCLC cell lines were maintained in RPMI1640 medium (ThermoFisher Scientific, USA) supplemented with 10% fetal bovine serum (FBS, Hyclone, USA) and antibiotics. SK-N-BE(2) was maintained in Minimum Essential Medium (MEM, ThermoFisher Scientific, USA) supplemented with 10% FBS (HyClone, USA) and antibiotics.
Western Blot Analysis
Cancer cells were treated with each of the compounds at different compound concentrations. After 24 h, cells were collected, washed with 1× phosphate buffered saline (PBS), lysed in 1× Laemmli protein sample buffer, and boiled at 100 °C for 10 min. Each lysate was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene fluoride (PVDF, Millipore, USA) membrane, and blotted with antibodies. Primary antibodies used for western blotting were cMYC (Cell Signaling, 5605S), MYCN (Cell Signaling, 9405S), PARP-1(Abcam, ab32378), GAPDH (Genetex, GTX100118), pAurora A/B/C (Cell Signaling, #2914), Aurora A (Abcam, ab52973), pHistone H3 Ser10 (Epitomics, 1173–1), Histone H3 (Millipore, 07-690), Cyclin B1 (Santa Cruz, sc-245), and β-ACTIN (Sigma-Aldrich, A1978). After the blotting (0.2% casein, 1% BSA, or 5% nonfat milk), membranes were washed with blotting buffer (0.2% casein in 1× PBS or 1× TBST), and corresponding alkaline phosphatase (AP)- or horseradish peroxidase (HRP)-conjugated secondary antibodies (Sigma-Aldrich) were added. The blots were developed by chemiluminescence (PerkinElmer, USA).
Cell Proliferation Inhibition Assay
The efficacy of the compounds as inhibitors of cancer cell proliferation was assessed using PrestoBlue Cell Viability Reagent (ThermoFisher Scientific, USA). Cells were seeded at a density of 5000–10,000 cells per well in 96-well plates. After 24 h, cells were treated with each compound at various concentrations (0–10 μM). IC50 values were computed based on a triplicate, eight-point titration. After 72 h of drug treatment, 10 μL of PrestoBlue Cell Viability Reagent was added to each well, and fluorescence signals were collected using a Wallac Vector 1420 multilabel counter (PerkinElmer, USA).
Real-Time Quantitative PCR (qRT-PCR)
Total mRNAs were isolated from cells using RNeasy mini kit (Qiagen). Complementary DNAs were produced using SuperScript III Reverse Transcriptase (Thermo Fisher Scientific) and oligo dT (Invitrogen) and random hexamer (Fermentas) as primers. Quantitative PCR was carried out using Power SYBR Green master mix (Thermo Fisher Scientific) using primers for cMYC (5′-AAACACAAACTTGAACAGCTAC-3′ and 5′-ATTTGAGGCAGTTTACATTATGG-3′) and GAPDH (5′-GGAAGGTGAAGGTCGGAGTCA-3′ and 5′-GTCATTGATGGCAACAATATCCACT-3′).
Pharmacokinetics
Procedures and use of animals were approved by the Institutional Animal Care and Use Committee (IACUC) of the National Health Research Institutes (NHRI, Zhunan, Miaoli, Taiwan). The facility where this research was conducted is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council. Six-week-old male ICR mice obtained from BioLASCO, Taiwan, were used for the PK studies. A single 2 mg/kg intravenous and 10 mg/kg oral dose were separately administered to two groups of three male mice. Serial blood samples were collected from each mouse at times of 0.03, 0.08 (IV only), 0.25, 0.5, 1, 2, 4, 6, 8, and 24 h after dosing. Plasma was separated from blood by centrifugation and analyzed for compounds by LC–MS/MS. The chromatographic system consisted of an Agilent 1200 series LC system and an Agilent ZORBAX Eclipse XDB-C8 column (5 μm, 3.0 × 150 mm) interfaced to an MDS Sciex API3000 tandem mass spectrometer, equipped with an ESI in the positive scanning mode. A gradient HPLC method was employed for separation. PK parameters were calculated by the noncompartmental model using the Kinetica program.
In Vivo Xenograft Tumor Growth Inhibition
Male athymic nu/nu nude mice (BioLASCO, Taiwan) 6 weeks old were housed in sterile cages maintained under 12 h light/dark cycles with controlled temperature and humidity. Mice were inoculated subcutaneously with 1 × 106 NCI–H446 cells (ATCC@ HTB-171) resuspended in saline mixed with 50% Matrigel matrix (Corning, USA). The sizes of the xenografted tumors were measured by a digital caliper (GMC-190; Goldsun Electronics Co.) and calculated using the formula: tumor volume (mm3) = length × (width)2/2. Body weight and tumor size were measured at least twice a week. When the xenograft tumor reached ≥200 mm3 in size, mice were intravenous or orally administered with the vehicle (5% dimethylacetamide/95% PEG400) or the compounds on a 5-on-2-off dosing regimen for 2–4 weeks. Tumor growth was analyzed for a statistically significant difference using ANOVA, followed by the Student–Newman–Keuls test. P < 0.05 was considered a significant difference between groups.
In Vivo Target Validation
The efficacy of a compound in reducing the MYC protein level and in inducing cell apoptosis was assessed using an NCI-H446 xenograft tumorigenicity mouse model. Six-week-old male athymic nu/nu nude mice (BioLASCO, Taiwan) were inoculated subcutaneously with 1 × 106 NCI-H446 cells resuspended in saline mixed with 50% Matrigel matrix (Corning, USA). When the xenograft tumor reached a size ≥500 mm3, mice were orally administered with one dosage of the vehicle (5% dimethylacetamide/95% PEG400, 10 mL/kg) or 25 or MLN8237 at 100 mg/kg. Tumors were harvested after 2, 4, 8, and 24 h of drug treatment, respectively. Half of the tumor was cryopreserved and then subjected to western analysis; the other half was fixed in 10% formalin and then embedded in paraffin. For immunohistochemistry, tissue paraffin sections were deparaffinized using xylene and ethanol. After rehydration, antigen retrieval was achieved by placing the slides in 100 °C citrate buffer, pH 6.0 for 1 h. Endogenous peroxides were quenched by 0.5% H2O2 for 10 min. Slides were blocked with 3% BSA for 30 min and then incubated with a rabbit anticleaved Caspase-3 antibody (Cell signaling, 9661S) at room temperature for 1.5 h. After three washes with 1× PBS, a goat antirabbit HRP-polymer (Biocare Medical, USA) was added, followed by development of the color using 3,3′-diaminobenzidine substrate chromogen (Biocare Medical, USA). The nucleus was stained with hematoxylin.
Acknowledgments
The authors would like to thank K. S. Shia, J. C. Lee, and L. K. Tsou for helpful discussions and their review of the paper. The authors also acknowledge the assistance of the staff members at the Animal Center of the National Health Research Institutes (NHRI), Taiwan.
Glossary
Abbreviations
- ATP
adenosine triphosphate
- AUC
area under the curve
- BSA
bovine serum albumin
- CASP3
caspase-3
- cCASP3
cleaved caspase-3
- cPARP-1
cleaved PARP-1
- IHC
immunohistochemistry
- IV
intravenous
- PK
pharmacokinetic
- PO
per os
- SBDD
structure-based drug design
- SCLC
small-cell lung cancer
- TBST
Tris-buffered saline and 0.1% Tween-20
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01806.
A docking model of compound 1 (PDB) aligned with compound B (PDB) in complex with Aurora A (PDBID: 3UO6) (PDB)
Molecular formula strings and Aurora A enzymatic inhibition activities for 1−27 (CSV)
Western blot profile of cMYC and MYCN in cells treated with various Aurora kinase inhibitors, cell viability, cell cycle analysis and expression profile of the target proteins in cells treated with various Aurora kinase inhibitors, 1H, 13C NMR, and HPLC spectra of compounds 1–27, and 1H spectra of compounds 29, 30, and 32–34 (PDF)
Author Contributions
∥ Y.-H.C. and T.-K.Y. contributed equally to this manuscript. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by NHRI intramural grants 104-0324-01-09-02, 105-0324-01-13-06, 106-0324-01-13-06, and 107-0324-01-19-10 and a grant from the Ministry of Science and Technology, Taiwan (MOST109-2622-B-400-004).
The authors declare no competing financial interest.
Supplementary Material
References
- McKeown M. R.; Bradner J. E. Therapeutic strategies to inhibit MYC. Cold Spring Harbor Perspect. Med. 2014, 4, a014266. 10.1101/cshperspect.a014266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub F. X.; Dhankani V.; Berger A. C.; Trivedi M.; Richardson A. B.; Shaw R.; Zhao W.; Zhang X.; Ventura A.; Liu Y.; Ayer D. E.; Hurlin P. J.; Cherniack A. D.; Eisenman R. N.; Bernard B.; Grandori C.; Pan-cancer alterations of the MYC oncogene and its proximal network across the cancer genome atlas. Cell Syst. 2018, 6, 282–300 e2. 10.1016/j.cels.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang C. V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harbor Perspect. Med. 2013, 3, a014217. 10.1101/cshperspect.a014217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejure F. R.; Eilers M. MYC and tumor metabolism: chicken and egg. EMBO J. 2017, 36, 3409–3420. 10.15252/embj.201796438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis A. C.; Wims M.; Spotts G. D.; Hann S. R.; Bradley A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993, 7, 671–682. 10.1101/gad.7.4.671. [DOI] [PubMed] [Google Scholar]
- Sawai S.; Shimono A.; Wakamatsu Y.; Palmes C.; Hanaoka K.; Kondoh H. Defects of embryonic organogenesis resulting from targeted disruption of the N-myc gene in the mouse. Development 1993, 117, 1445–1455. 10.1242/dev.117.4.1445. [DOI] [PubMed] [Google Scholar]
- Jain M.; Arvanitis C.; Chu K.; Dewey W.; Leonhardt E.; Trinh M.; Sundberg C. D.; Bishop J. M.; Felsher D. W. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 2002, 297, 102–104. 10.1126/science.1071489. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wang H.; Li Z.; Wu Q.; Lathia J. D.; McLendon R. E.; Hjelmeland A. B.; Rich J. N. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One 2008, 3, e3769 10.1371/journal.pone.0003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Liu H.; Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther. 2018, 3, 5. 10.1038/s41392-018-0008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers M.; Eisenman R. N. Myc’s broad reach. Genes Dev. 2008, 22, 2755–2766. 10.1101/gad.1712408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu M.-E.; Jauset T.; Massó-Vallés D.; Martínez-Martín S.; Rahl P.; Maltais L.; Zacarias-Fluck M. F.; Casacuberta-Serra S.; Serrano Del Pozo E.; Fiore C.; Foradada L.; Cano V. C.; Sánchez-Hervás M.; Guenther M.; Romero Sanz E.; Oteo M.; Tremblay C.; Martín G.; Letourneau D.; Montagne M.; Morcillo Alonso M. Á.; Whitfield J. R.; Lavigne P.; Soucek L. Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci. Transl. Med. 2019, 11, eaar5012 10.1126/scitranslmed.aar5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massó-Vallés D.; Soucek L. Blocking Myc to treat cancer: reflecting on two decades of omomyc. Cells 2020, 9, 883. 10.3390/cells9040883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore J. E.; Issa G. C.; Lemieux M. E.; Rahl P. B.; Shi J.; Jacobs H. M.; Kastritis E.; Gilpatrick T.; Paranal R. M.; Qi J.; Chesi M.; Schinzel A. C.; McKeown M. R.; Heffernan T. P.; Vakoc C. R.; Bergsagel P. L.; Ghobrial I. M.; Richardson P. G.; Young R. A.; Hahn W. C.; Anderson K. C.; Kung A. L.; Bradner J. E.; Mitsiades C. S. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson W. C.; Meyerowitz J. G.; Nekritz E. A.; Chen J.; Benes C.; Charron E.; Simonds E. F.; Seeger R.; Matthay K. K.; Hertz N. T.; Eilers M.; Shokat K. M.; Weiss W. A. Drugging MYCN through an allosteric transition in Aurora kinase A. Cancer Cell 2014, 26, 414–427. 10.1016/j.ccr.2014.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockmann M.; Poon E.; Berry T.; Carstensen A.; Deubzer H. E.; Rycak L.; Jamin Y.; Thway K.; Robinson S. P.; Roels F.; Witt O.; Fischer M.; Chesler L.; Eilers M. Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell 2013, 24, 75–89. 10.1016/j.ccr.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauch D.; Rudalska R.; Cossa G.; Nault J.-C.; Kang T.-W.; Wuestefeld T.; Hohmeyer A.; Imbeaud S.; Yevsa T.; Hoenicke L.; Pantsar T.; Bozko P.; Malek N. P.; Longerich T.; Laufer S.; Poso A.; Zucman-Rossi J.; Eilers M.; Zender L. A MYC-aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med. 2016, 22, 744–753. 10.1038/nm.4107. [DOI] [PubMed] [Google Scholar]
- Lee J. K.; Phillips J. W.; Smith B. A.; Park J. W.; Stoyanova T.; McCaffrey E. F.; Baertsch R.; Sokolov A.; Meyerowitz J. G.; Mathis C.; Cheng D.; Stuart J. M.; Shokat K. M.; Gustafson W. C.; Huang J.; Witte O. N. N-Myc drives neuroendocrine prostate cancer initiated from human prostate epithelial cells. Cancer Cell 2016, 29, 536–547. 10.1016/j.ccell.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto T.; Horn S.; Brockmann M.; Eilers U.; Schüttrumpf L.; Popov N.; Kenney A. M.; Schulte J. H.; Beijersbergen R.; Christiansen H.; Berwanger B.; Eilers M. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 2009, 15, 67–78. 10.1016/j.ccr.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Carmena M.; Earnshaw W. C. The cellular geography of aurora kinases. Nat. Rev. Mol. Cell Biol. 2003, 4, 842–854. 10.1038/nrm1245. [DOI] [PubMed] [Google Scholar]
- Lens S. M. A.; Voest E. E.; Medema R. H. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat. Rev. Cancer 2010, 10, 825–841. 10.1038/nrc2964. [DOI] [PubMed] [Google Scholar]
- Fu J.; Bian M.; Jiang Q.; Zhang C. Roles of Aurora kinases in mitosis and tumorigenesis. Mol. Cancer Res. 2007, 5, 1–10. 10.1158/1541-7786.mcr-06-0208. [DOI] [PubMed] [Google Scholar]
- Agnese V.; Bazan V.; Fiorentino F. P.; Fanale D.; Badalamenti G.; Colucci G.; Adamo V.; Santini D.; Russo A. The role of Aurora-A inhibitors in cancer therapy. Ann. Oncol. 2007, 18, vi47–vi52. 10.1093/annonc/mdm224. [DOI] [PubMed] [Google Scholar]
- Tatsuka M.; Katayama H.; Ota T.; Tanaka T.; Odashima S.; Suzuki F.; Terada Y. Multinuclearity and increased ploidy caused by overexpression of the aurora- and Ipl1-like midbody-associated protein mitotic kinase in human cancer cells. Cancer Res. 1998, 58, 4811–4816. [PubMed] [Google Scholar]
- Kollareddy M.; Zheleva D.; Dzubak P.; Brahmkshatriya P. S.; Lepsik M.; Hajduch M. Aurora kinase inhibitors: progress towards the clinic. Invest. New Drugs 2012, 30, 2411–2432. 10.1007/s10637-012-9798-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand S.; Penrhyn-Lowe S.; Venkitaraman A. R. Aurora-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell 2003, 3, 51–62. 10.1016/s1535-6108(02)00235-0. [DOI] [PubMed] [Google Scholar]
- Vijayan R. S. K.; He P.; Modi V.; Duong-Ly K. C.; Ma H.; Peterson J. R.; Dunbrack R. L. Jr; Levy R. M. Conformational analysis of the DFG-out kinase motif and biochemical profiling of structurally validated type II inhibitors. J. Med. Chem. 2015, 58, 466–479. 10.1021/jm501603h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. P.; Zhu J.-Y.; Lawrence H. R.; Pireddu R.; Luo Y.; Alam R.; Ozcan S.; Sebti S. M.; Lawrence N. J.; Schönbrunn E. A novel mechanism by which small molecule inhibitors induce the DFG flip in Aurora A. ACS Chem. Biol. 2012, 7, 698–706. 10.1021/cb200508b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilburt J. A. H.; Sarkar H.; Sheldrake P.; Blagg J.; Ying L.; Dodson C. A. Dynamic equilibrium of the Aurora A kinase activation loop revealed by single-molecule spectroscopy. Angew Chem. Int. Ed. Engl. 2017, 56, 11409–11414. 10.1002/anie.201704654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollaoglu G.; Guthrie M. R.; Böhm S.; Brägelmann J.; Can I.; Ballieu P. M.; Marx A.; George J.; Heinen C.; Chalishazar M. D.; Cheng H.; Ireland A. S.; Denning K. E.; Mukhopadhyay A.; Vahrenkamp J. M.; Berrett K. C.; Mosbruger T. L.; Wang J.; Kohan J. L.; Salama M. E.; Witt B. L.; Peifer M.; Thomas R. K.; Gertz J.; Johnson J. E.; Gazdar A. F.; Wechsler-Reya R. J.; Sos M. L.; Oliver T. G. MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to Aurora kinase inhibition. Cancer Cell 2017, 31, 270–285. 10.1016/j.ccell.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oronsky B.; Reid T. R.; Oronsky A.; Carter C. A. What’s new in SCLC? A review. Neoplasia 2017, 19, 842–847. 10.1016/j.neo.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakawa R.; Takenaka M.; Kohno T.; Shimada Y.; Totoki Y.; Shibata T.; Tsuta K.; Nishikawa R.; Noguchi M.; Sato-Otsubo A.; Ogawa S.; Yokota J. Genome-wide identification of genes with amplification and/or fusion in small cell lung cancer. Gene Chromosome Canc. 2013, 52, 802–816. 10.1002/gcc.22076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rita de Cássia S. A.; Meurer R. T.; Roehe A. V. MYC amplification is associated with poor survival in small cell lung cancer: a chromogenic in situ hybridization study. J. Canc. Res. Clin. Oncol. 2014, 140, 2021–2025. 10.1007/s00432-014-1769-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfrich B. A.; Kim J.; Gao D.; Chan D. C.; Zhang Z.; Tan A.-C.; Bunn P. A. Jr Barasertib (AZD1152), a small molecule Aurora B inhibitor, inhibits the growth of SCLC cell lines in vitro and in vivo. Mol. Cancer Ther. 2016, 15, 2314–2322. 10.1158/1535-7163.mct-16-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J.; Caponigro G.; Stransky N.; Venkatesan K.; Margolin A. A.; Kim S.; Wilson C. J.; Lehár J.; Kryukov G. V.; Sonkin D.; Reddy A.; Liu M.; Murray L.; Berger M. F.; Monahan J. E.; Morais P.; Meltzer J.; Korejwa A.; Jané-Valbuena J.; Mapa F. A.; Thibault J.; Bric-Furlong E.; Raman P.; Shipway A.; Engels I. H.; Cheng J.; Yu G. K.; Yu J.; Aspesi P. Jr; de Silva M.; Jagtap K.; Jones M. D.; Wang L.; Hatton C.; Palescandolo E.; Gupta S.; Mahan S.; Sougnez C.; Onofrio R. C.; Liefeld T.; MacConaill L.; Winckler W.; Reich M.; Li N.; Mesirov J. P.; Gabriel S. B.; Getz G.; Ardlie K.; Chan V.; Myer V. E.; Weber B. L.; Porter J.; Warmuth M.; Finan P.; Harris J. L.; Meyerson M.; Golub T. R.; Morrissey M. P.; Sellers W. R.; Schlegel R.; Garraway L. A. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook K. E.; Garza S. J.; Lira M. E.; Ching K. A.; Lee N. V.; Cao J.; Yuan J.; Ye J.; Ozeck M.; Shi S. T.; Zheng X.; Rejto P. A.; Kan J. L. C.; Christensen J. G.; Pavlicek A. An integrated genomic approach to identify predictive biomarkers of response to the aurora kinase inhibitor PF-03814735. Mol. Cancer Ther. 2012, 11, 710–719. 10.1158/1535-7163.mct-11-0184. [DOI] [PubMed] [Google Scholar]
- Sos M. L.; Dietlein F.; Peifer M.; Schottle J.; Balke-Want H.; Muller C.; Koker M.; Richters A.; Heynck S.; Malchers F.; Heuckmann J. M.; Seidel D.; Eyers P. A.; Ullrich R. T.; Antonchick A. P.; Vintonyak V. V.; Schneider P. M.; Ninomiya T.; Waldmann H.; Buttner R.; Rauh D.; Heukamp L. C.; Thomas R. K. A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 17034–17039. 10.1073/pnas.1207310109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Zhou Y.; Qian H.; Shao G.; Lu X.; Chen Q.; Sun X.; Chen D.; Yin R.; Zhu H.; Shao Q.; Xu W. Stemness and inducing differentiation of small cell lung cancer NCI-H446 cells. Cell Death Dis. 2013, 4, e633 10.1038/cddis.2013.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J.; Yan L.; Torres R.; Gong X.; Bian H.; Marugán C.; Boehnke K.; Baquero C.; Hui Y.-H.; Chapman S. C.; Yang Y.; Zeng Y.; Bogner S. M.; Foreman R. T.; Capen A.; Donoho G. P.; Van Horn R. D.; Barnard D. S.; Dempsey J. A.; Beckmann R. P.; Marshall M. S.; Chio L.-C.; Qian Y.; Webster Y. W.; Aggarwal A.; Chu S.; Bhattachar S.; Stancato L. F.; Dowless M. S.; Iversen P. W.; Manro J. R.; Walgren J. L.; Halstead B. W.; Dieter M. Z.; Martinez R.; Bhagwat S. V.; Kreklau E. L.; Lallena M. J.; Ye X. S.; Patel B. K. R.; Reinhard C.; Plowman G. D.; Barda D. A.; Henry J. R.; Buchanan S. G.; Campbell R. M. Aurora A-selective inhibitor LY3295668 leads to dominant mitotic arrest, apoptosis in cancer cells, and shows potent preclinical antitumor efficacy. Mol. Cancer Ther. 2019, 18, 2207–2219. 10.1158/1535-7163.mct-18-0529. [DOI] [PubMed] [Google Scholar]
- Beaumont K.; Webster R.; Gardner I.; Dack K. Design of ester prodrugs to enhance oral absorption of poorly permeable compounds: challenges to the discovery scientist. Curr. Drug Metab. 2003, 4, 461–485. 10.2174/1389200033489253. [DOI] [PubMed] [Google Scholar]
- Ettmayer P.; Amidon G. L.; Clement B.; Testa B. Lessons learned from marketed and investigational prodrugs. J. Med. Chem. 2004, 47, 2393–2404. 10.1021/jm0303812. [DOI] [PubMed] [Google Scholar]
- Galasiti Kankanamalage A. C.; Kim Y.; Rathnayake A. D.; Alliston K. R.; Butler M. M.; Cardinale S. C.; Bowlin T. L.; Groutas W. C.; Chang K.-O. Design, synthesis, and evaluation of novel prodrugs of transition state inhibitors of norovirus 3CL protease. J. Med. Chem. 2017, 60, 6239–6248. 10.1021/acs.jmedchem.7b00497. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Brindisi M. Organic carbamates in drug design and medicinal chemistry. J. Med. Chem. 2015, 58, 2895–2940. 10.1021/jm501371s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung C. H. A.; Sarvagalla S.; Lee J. Y.-C.; Huang Y.-C.; Coumar M. S. Aurora kinase inhibitor patents and agents in clinical testing: an update (2011 - 2013). Expert Opin. Ther. Pat. 2014, 24, 1021–1038. 10.1517/13543776.2014.931374. [DOI] [PubMed] [Google Scholar]
- Chaitanya G.; Alexander J. S.; Babu P. PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. 10.1186/1478-811X-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y.-Y.; Chang C.-P.; Lin W.-H.; Tsai C.-H.; Chiu I.-C.; Wang W.-P.; Wang P.-C.; Chen P.-Y.; Lin W.-H.; Chang C.-F.; Kuo P.-C.; Song J.-S.; Shih C.; Hsieh H.-P.; Chi Y.-H. Design and synthesis of BPR1K653 derivatives targeting the back pocket of Aurora kinases for selective isoform inhibition. Eur. J. Med. Chem. 2018, 151, 533–545. 10.1016/j.ejmech.2018.03.064. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.