

Abstract
(E/Z)-3-(4-((E)-1-(4-Hydroxyphenyl)-2-phenylbut-1-enyl)phenyl)acrylic acid (GW7604) as a derivative of (Z)-4-hydroxytamoxifen (4-OHT) was linked by diaminoalkane spacers to molecules that are known binders to the coactivator binding site (benzimidazole or thioxo-quinazolinone scaffolds). With this modification, an optimization of the pharmacological profile was achieved. The most active thioxo-quinazolinone derivative 16 showed extraordinarily high affinity to the estrogen receptor (ER) β (RBA = 110%), inhibited effectively the coactivator recruitment (IC50 = 20.88 nM (ERα) and 28.34 nM (ERβ)), acted as a pure estradiol (E2) antagonist in a transactivation assay (IC50 = 18.5 nM (ERα) and 7.5 nM (ERβ)), and downregulated the ERα content in MCF-7 cells with an efficacy of 60% at 1 μM. The cytotoxicity was restricted to hormone-dependent MCF-7 (IC50 = 4.2 nM) and tamoxifen-resistant MCF-7TamR cells (IC50 = 476.6 nM). The compounds bearing a thioxo-quinazolinone moiety can therefore be assigned as pure E2-antagonistic selective ER degraders/downregulators. By contrast, the benzimidazole derivatives acted solely as pure antagonists without degradation of the ER.
1. Introduction
Breast cancer is still the most common cancer in women worldwide, affecting one in eight women in high-income countries, and the incidence is further increasing.1 The majority of all types of mammary carcinomas (MCs) are, at least initially, hormone-dependent.2,3 Endocrine therapy, including aromatase inhibitors or selective estrogen receptor modulators (SERMs)/selective estrogen receptor downregulators (SERDs), consequently represents an indispensable treatment opportunity. Unfortunately, acquired endocrine resistance is an inevitable issue, which manifests after prolonged therapy.4,5
With regard to genomic alterations, endocrine-resistant breast tumors are divided into four groups: (i) tumors bearing estrogen receptor alpha (ERα) gene alterations (ESR1), (ii) tumors harboring lesions in the mitogen-activated protein kinase pathway, (iii) tumors with mutations in the transcriptional factors, and (iv) tumors with undiscovered resistance mechanisms.6,7 ESR1 alterations represent, with approximately 20%, the largest category.6,8 These ESR1 point mutations, for instance, are particularly frequently located in the ligand binding domain (LBD) and thus require the search for new antiestrogenic drugs to overcome this kind of resistance.9
One possibility to impede estrogen-mediated pathways, in general, is to induce specific conformations of the estrogen receptor (ER) upon drug binding, which prevents coactivator recruitment and ultimately gene transcription in hormone-dependent MC cells. The SERM tamoxifen (Figure 1), as the first targeted anti-breast-cancer therapeutic agent,10 acts via this mode of action. Upon attachment of an agonist at the ligand binding site (LBS), Helix 12 (H12) is oriented over the LBD. With parts from helices H3, H4, H5, and H12, and the turn between helices H3 and H4, a hydrophobic groove (activation function 2 (AF2)) that accommodates an LXXLL motif is formed. The AF2 is essential for coactivator binding. In the case of (Z)-4-hydroxytamoxifen (4-OHT), which is the active metabolite of tamoxifen, H12 is repositioned, AF2 is not formed, and interactions with coactivator peptides are blocked in hormone-dependent tumor cells, preventing their growth.11,12 Nevertheless, in some other target tissues (endometrium, bones), activation is possible due to an insufficient shielding of the charge at Asp351 (at ERα) by the basic side chain. It is noteworthy that in one-third of patients acquired resistance against tamoxifen occurs within 15 years.13 In this case, the use of aromatase inhibitors (e.g., anastrozol) or SERDs (fulvestrant (Figure 1)) is indicated.14
Figure 1.
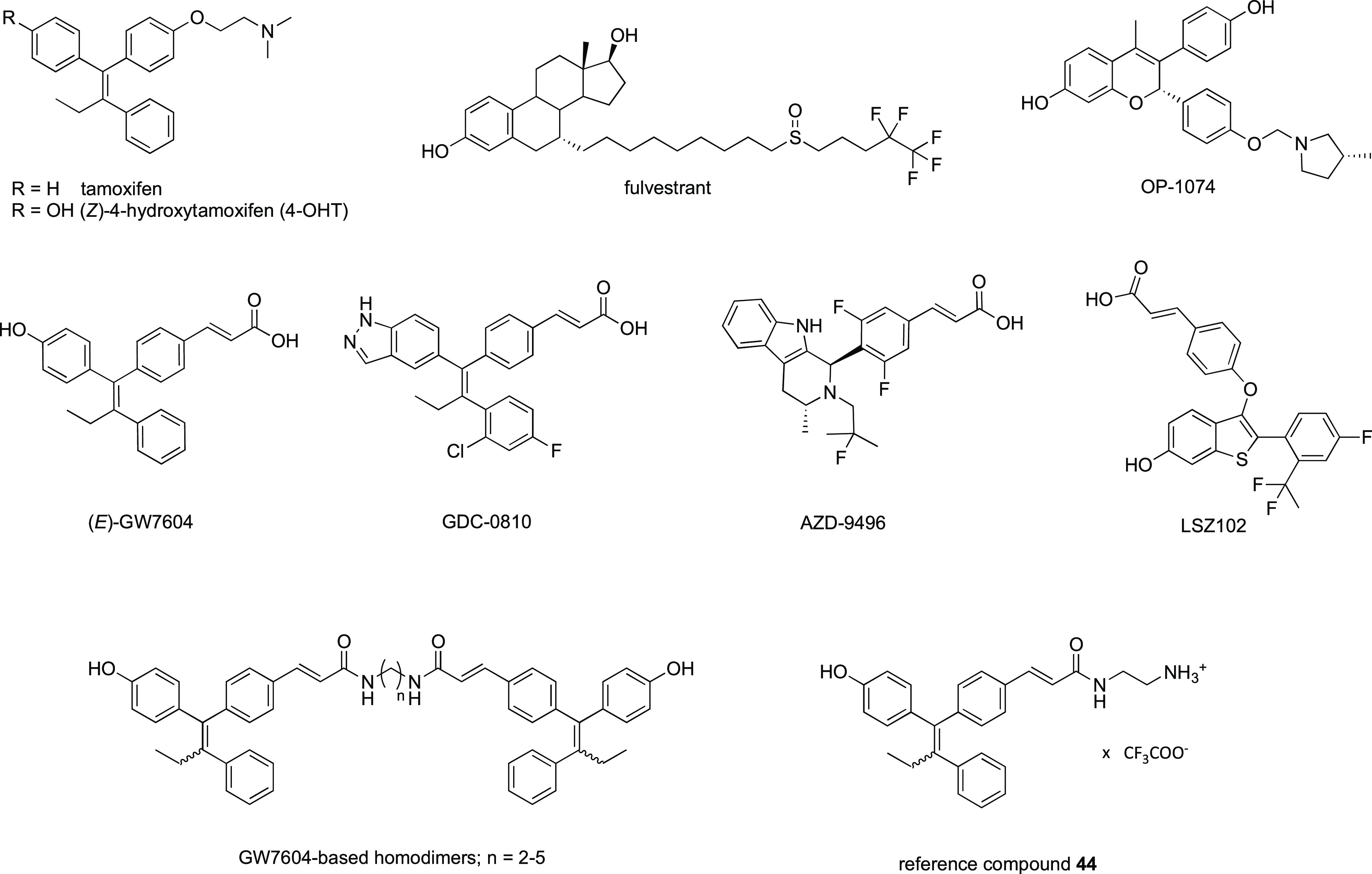
SERMs: tamoxifen and 4-OHT. SERDs: fulvestrant and OP-1074. SERM-SERDs: (E)-GW7604, GDC-0810, AZD-9496, and LSZ102. GW7604-based homodimers and reference compound 44.
Fulvestrant disrupts ER dimerization and nuclear localization. Furthermore, it accelerates receptor degradation. This mode of action is based on unusual conformational change of the ER upon drug binding. It sterically prevents H12 from forming any type of interaction with residues of the ER. Consequently, H12 is destabilized, hydrophobic surfaces at the ER are exposed, and the receptor is submitted to downregulation by the ubiquitin-proteasome pathway.12,15 Consequently, the ER-mediated transcriptional activity is completely blocked, leading to an effective growth reduction of hormone-dependent tumors. OP-1074 (Figure 1), declared as a pure antiestrogen and SERD, displays the same mechanism as fulvestrant.16 To derive an alternative after failure of tamoxifen treatment, 4-OHT was structurally modified in a way where the basic side chain was exchanged by a carboxylate bearing moiety. (E/Z)-3-(4-((E)-1-(4-Hydroxyphenyl)-2-phenylbut-1-enyl)phenyl)acrylic acid (GW7604) (Figure 1), the cinnamic acid analogue of 4-OHT, showed diminished hormonal effects, e.g., induction of ER expression and cell-growth-stimulating effects at low concentrations.17,18 This is ascribed to the repulsion of the carboxylate and the amino acid Asp351 (ERα). The conformational change disrupts the surface charge around this amino acid required for coactivator binding in the 4-OHT/ER complex.19 Unlike 4-OHT, the binding of GW7604 even causes downregulation of the receptor. Since H12 is not totally destabilized as found for the fulvestrant-bound receptor and less hydrophobic sites are exposed, degradation occurs, but to a low extent.18 Further modifications mainly include the change in the LBS-binding core. Examples are GDC-0810,20 AZD-9496,21 and LSZ10222 (Figure 1), referred to as SERM-SERDs16 or as SERM/SERD hybrids.23
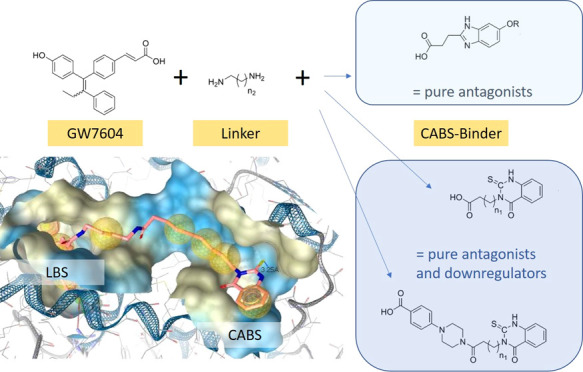
In our group, we used another approach and developed compounds in such a way that besides the LBS, the coactivator binding site (CABS) is targeted simultaneously. We evaluated the consequences on the receptor binding affinity and the intracellular responses.
In a first study, homodimers of GW7604 and of the related cyclofenilacrylic acid were designed, because an X-ray crystal structure revealed a hydrophobic groove at the CABS suitable to bind 1,1-diaryl- or 1,1,2-triarylalkenes (see chapter 2.1). Alkyl spacers of different lengths between the molecules should guarantee sufficient flexibility to reach two different pockets within this exposed surface, which emerge as potential binding areas.24 As proposed, it was possible to increase the binding affinity and to inhibit ER transactivation.
In continuation of this structure–activity relationship (SAR) study, we tried to optimize the CABS-binding properties. 1,1-Diarylalkene derivatives can bind at the ER surface, but steric repulsion might render accessibility to the proposed binding pockets. Therefore, GW7604 was linked to molecules, which were already described as CABS binders. We selected thioxo-quinazolinone derivatives (e.g., 4-(4-(4-(4-oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)butanoyl)piperazin-1-yl)benzoic acid or 4-(4-oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)butanoic acid25) and the 3-(5-methoxy-1H-benzo[d]imidazol-2-yl)propanoic acid (Chart 1) to be connected via the diaminoalkane spacer to GW7604. The 5-hydroxybenzimidazole scaffold was also investigated regarding their H-bond formation within the pockets at the ER surface.
Chart 1. Design of Heterodimeric ER Ligands.

The binding affinities of the compounds at the isolated LBD, their cellular responses such as inhibition of gene activation, ER downregulation, and antiproliferative effects in hormone-dependent/-independent as well as in tamoxifen-resistant tumor cells (resistance group (ii) as mentioned above) were evaluated. Furthermore, quantitative cellular uptake studies were performed to rationalize the influence of compound accumulation on the cellular activity.
2. Results and Discussion
2.1. Docking Studies
Previously, we described a theoretical model to evaluate the binding of bivalent molecules at the ER. It is based on the crystal structure of the ERβ-LBD (PDB entry 2FSZ)26 cocrystallized with two 4-OHT molecules.24 The first one is attached at the LBS and the second one at the CABS. Both can formally be connected by an alkyl spacer, enabling a view on possible binding modes of homodimeric compounds.
GW7604-based bivalent derivatives (Figure 1) were already synthesized and tested for ER interactions. It is postulated that the GW7604 moiety binds in the LBS of ERβ forming H-bonds to Arg346, Glu305, and one water molecule in a classic manner,27,28 while the terminal drug molecule interacts at the hydrophobic surface. These interactions were considered as a prerequisite for being a valid docking pose.
As a further development, GW7604 is linked to scaffolds of known CABS binders (formation of heterodimeric compounds). A suitable one represents the 4-(4-oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)butanic acid. The use of various diaminoalkyl spacers (n2 = 2, 3, Chart 1) allows binding to side pockets along the identified hydrophobic channel at the CABS. In addition, the influence of alkanoic acid (n1 = 2, 6, Chart 1) at the thioxo-quinazolinone core was studied.
The best docking results revealed the combination of an N-butanoic acid chain (n1 = 2) with a C3 spacer (n2 = 2: compound 15, Chart 1). In this case, the thioxo-quinazolinone core reached the hydrophobic groove 9 Å away from the nitrogen atom of the GW7604 amide (Figure 2A). The second binding region, which is about 18–20 Å apart, was targeted employing an octanoic acid residue (n1 = 6) and a 1,4-diaminobutane chain (n2 = 3: compound 16, Chart 1). In this pocket, cation−π interaction with Lys314 in addition to multiple hydrophobic contacts is possible. The distance to Gln327 is 3.25 Å, and there might be a possibility to render further interactions (Figure 2B).
Figure 2.

Ligand binding pocket of the ERβ-LBD (PDB entry 2FSZ)26 with (A) 15, (B) 16, and (C) 18 depicted in rose. Hydrophobic protein–ligand interactions are shown as yellow spheres and H-bonds by red and green arrows.
In a second attempt, we used 4-(4-(4-(4-oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)butanoyl)piperazin-1-yl)aryl derivatives initially discovered as CABS binders by Sun et al.25 Introduction of a 4-COOH group at the aryl ring makes the connection to GW7604 via a diaminoalkane spacer (1,3-diaminopropane (n2 = 2)) possible. The best results provided 17 (n1 = 2), allowing an attachment comparable to that postulated by Sun et al.25 To assess the influence of higher flexibility and to reach areas at the ER comparable to 16, the alkanoic acid chain at the thioxo-quinazolinone was elongated (n1: 2 → 6).
Compound 18 (n1 = 6) adapted indeed a similar position at the ER (compare Figure 2B,C). The position of the thioxo-quinazolinones differed from that described by Sun et al.,25 whereby the piperazinylbenzoate moiety was pulled away from the position close to charge clamp residues Glu493 and Met494 more toward the LBS, whereas the thioxo-quinazolinone ring remained in the area near Lys314 and Gln327. This orientation enabled an H-bond between the C=S group and Gln327 and diminished the interaction between the heteroaromatic amine group and Val320.
To further reduce the steric demand of the CABS binder, and to occupy the hydrophobic pocket closer to the LBS, the 3-(5-methoxy/hydroxy-1H-benzo[d]imidazol-2-yl)propanoic acid moiety was introduced. C-alkylated benzimidazole scaffolds have already been recognized as CABS binders at the ER,29 but were also used as essential cores for the design of partial peroxisome proliferator-activated receptor γ (PPARγ) agonists.30,31 The conjugation to GW7604 was again performed by diamide formation (n2 = 1–5). Interestingly, the 5-methoxybenzimidazole derivatives 31 and 32 with a short C2 or C3 linker (n1 = 1, 2) mainly targeted a hydrophobic pocket close to the LBS, while longer chains allowed the attachment as discussed above. Upon ether cleavage, the resulting 5-hydroxybenzimidazole derivatives with an extended alkyl chain (C5: 42; C6: 43) caused H-bond formation with Gln327, which is also in close vicinity (4–5 Å) to the charge clamp residue Lys314 (Figure 3A).
Figure 3.

Ligand binding pocket of the ERβ-LBD (PDB entry 2FSZ)26 with (A) 42 and (B) 38 in rose.
This interaction was prevented in 5-methoxybenzimidazoles. But surprisingly, an H-bond with Arg501 located in H12 was possible, causing reorientation in the binding cleft, inducing a similar position of both long-chained heterodimers in the LBD (Figure 3B).
In summary, the theoretical investigations document the accessibility of the proposed binding areas within the CABS using thioxo-quinazolinone and benzimidazole scaffolds. Hydrophobic contacts as well as H-bonding with either Lys314 or Gln327 depend on the spacer length and the used heteroaromatic scaffold. Based on the above-described theoretical investigations, a selection of compounds was synthesized to investigate the predicted interactions and to explore the assignability of those to the ERα.
2.2. Chemistry
2.2.1. Synthesis
The thioxo-quinazolinone-based CABS-binding motifs were obtained from building blocks 1 and 2, which differ in their alkyl chain (n1 = 2, 6). The carboxylate was derived in cases of 11 and 12 with a simple diaminoalkane chain (n2 = 2, 3), while in 13 and 14 a piperazinylbenzoate scaffold was used to combine the 1,3-diaminopropane spacer and the COOH group of 1 and 2via amide bonding (Scheme 1).
Scheme 1. Synthesis Pathway for the Thioxo-quinazolinone Trifluoroacetate Building Blocks 11–14.
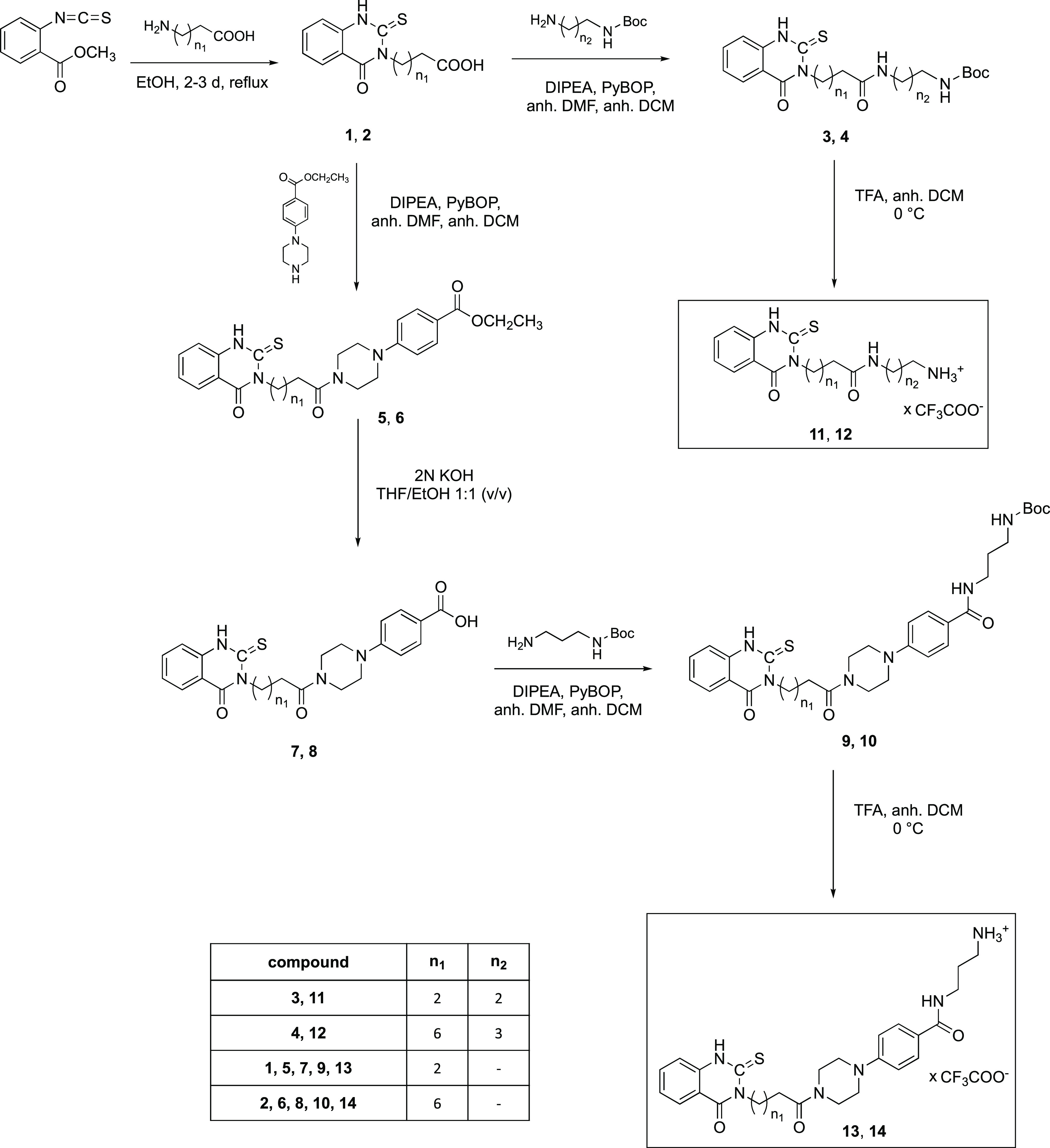
According to the published procedure by Sun et al.,25 the thioxo-quinazolinone ring closure to 1 and 2 occurred in ethanol (EtOH) under reflux, however, without the need for additional KOH. In fact, KOH led to a partial decomposition of the scaffold. 1H Nuclear magnetic resonance (NMR) spectroscopy and high-resolution mass spectrometry (HR-MS) confirmed the successful ring formation (see the Supporting Information).
The amide syntheses (3, 4, 5, 6, 9, 10) utilized the coupling reagent benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) and the auxiliary base diisopropylamine (DIPEA) in dry dichloromethane (DCM) and dimethylformamide (DMF).32−35 The workup under acidic conditions (pH 3–4) guaranteed removal of basic byproducts. Three out of the four piperazinylbenzoate containing compounds (5, 6, and 9) precipitated from the reaction mixture as a result of their poor solubility in DCM/DMF. Generally, the yields were good to excellent, ranging from 54 to 92%. Compound 10 was separated from unwanted side products by column chromatography.
The carboxylic acids 7 and 8 as educts for the syntheses of 9 and 10 were obtained from esters 5 and 6 by ester cleavage with KOH in tetrahydrofuran (THF) and ethanol 1:1 (v/v). The cleavage of the tert-butyloxycarbonyl (Boc) protecting group from 3, 4, 9, and 10 with trifluoroacetic acid (TFA) in dry DCM34,36 yielded the trifluoroacetate salts 11–14 in quantitative yield.
Finally, the reaction of GW7604 with amines 11–14 using PyBOP/DIPEA gave the heterodimeric products 15–18 (Scheme 2). In each case, the purification by column chromatography led to a loss of compound (yields: 15 (29%), 16 (26%), 17 (54%), and 18 (48%)).
Scheme 2. Reaction of Thioxo-quinazolinone Trifluoroacetate Building Blocks with GW7604.

The 3-(5-methoxy-1H-benzo[d]imidazol-2-yl)propanoic acid (20) acted as an educt for the syntheses of dimers 31, 32, 34, 36, and 38. To obtain this synthon (Scheme 3), 4-methoxyphenylenediamine was reacted with succinic anhydride to the benzimidazole, followed by esterification with EtOH (→19) as already described by Zeng et al.37 The last step was necessary for a better separation from side products. Subsequently, 19 was hydrolyzed under acidic conditions (2 N HCl in EtOH38), yielding 20 as a hydrochloride in quantitative yields.
Scheme 3. Synthetic Pathway for the 5-Methoxy-1H-benzo[d]imidazole Bis(trifluoroacetate) Building Blocks.
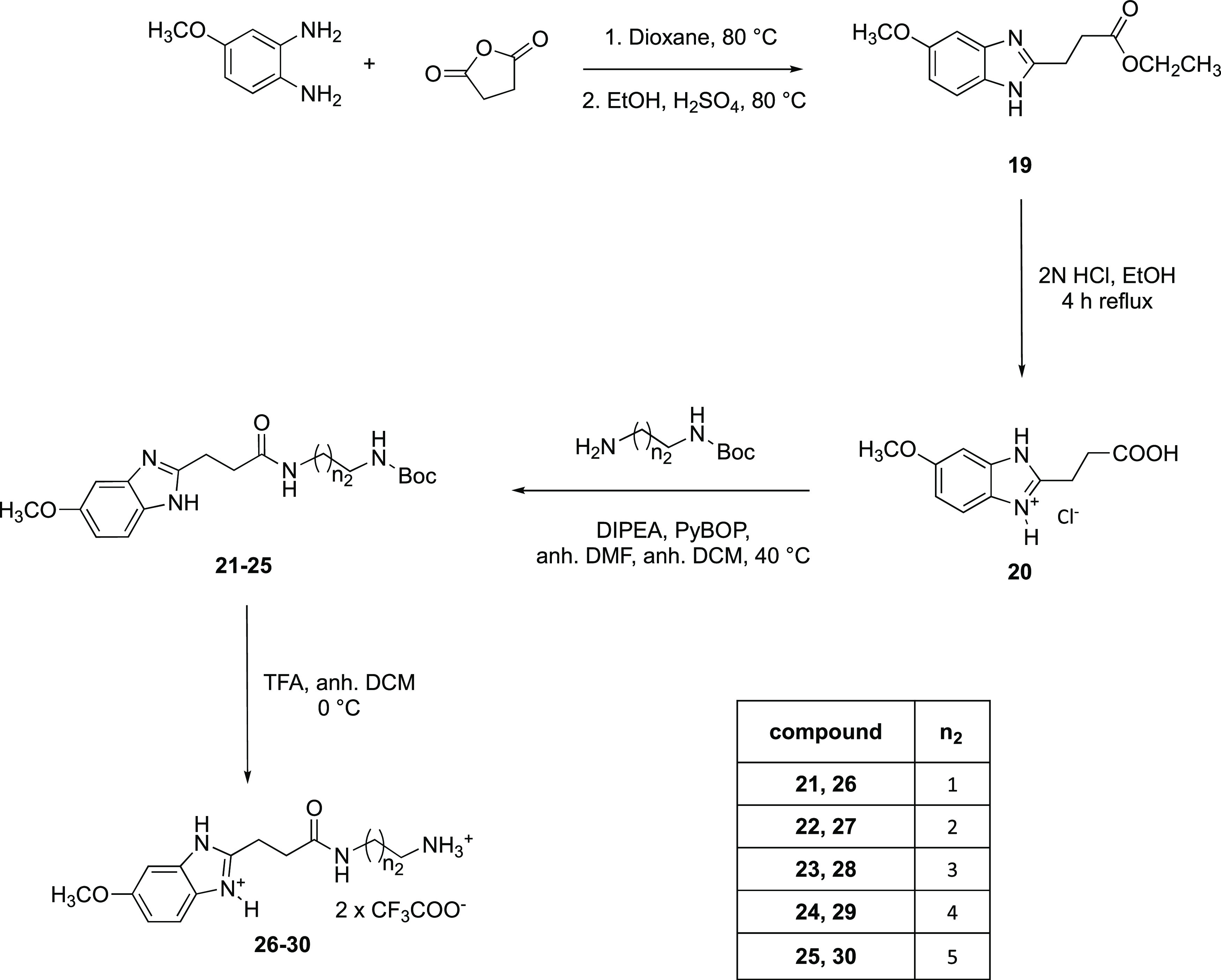
Amide coupling (PyBOP/DIPEA) of 20 with N-Boc-protected diaminoalkanes gained the respective amides 21–25 (yields: 43–87%), which were then deprotected (→26–30) with TFA in quantitative yields.
An analogous reaction of GW7604 with the 5-methoxybenzimidazoles 26–30 yielded 31, 32, 34, 36, and 38 (Scheme 4). Methoxy-GW7604 was used to obtain the methoxy series 33, 35, and 37. The yields for deprotection of compounds 31–38 were strongly dependent on their solubility in DCM or chlorobenzene and differed considerably among the homologues (12–81%).
Scheme 4. GW7604-Benzimidazole Formation.
Subsequent cleavage of the methoxy groups with BBr3 in dry DCM or chlorobenzene resulted in 39–43.
All final compounds 15–18, 31, 32, 34, 36, and 38–43 were characterized by 1H NMR and 13C NMR as well as HR-MS. Two-dimensional NMR spectra were recorded to assign the respective isomers. The purity was assessed by high-performance liquid chromatography (HPLC). The syntheses and the characterization of the intermediates as well as 2D NMR spectra of compound 17 as a representative can be found in the Supporting Information (Figures S4–S7).
2.2.2. Stability Studies
The heterodimeric compounds were synthesized from (E/Z)-GW7604 or (E/Z)-methoxy-GW7604. The use of isomerically pure educts is pointless, because fast isomerization of the double bond within the 1,1,2-triarylalkene core takes place in solution.39 Such a reaction was already observed in the case of GW7604-based homodimers.24 Among the new compounds, the benzimidazole derivatives 31, 32, 34, 36, 39–43 as well as 16 were isolated in a 50:50 ratio, while for the others, the Z isomer predominated: 15 (E/Z = 30:70), 17 (E/Z = 12:88), and 18 (E/Z = 20:80).
For the interpretation of the biological results, it is necessary to obtain information about the isomerization by simulating physiological conditions. Therefore, we incubated 17 and 36 as examples in a mixture of methanol (MeOH) and 2× phosphate-buffered saline (PBS) (75:25, v/v) and analyzed the E/Z isomerization by HPLC using an RP18 column and acetonitrile (ACN)/water (TFA, 0.1% or Na2SO4, 20 mM (pH 3), respectively) gradients.
The E/Z ratio of 17 (E/Z = 12:88) built during the reaction course was confirmed. Incubation at 37 °C for 72 h increased the amount of the E isomer to 25% (Figure 4A). Compound 36, incubated under the same conditions, held its E/Z distribution of 50:50 during the whole experiment (Figure 4B).
Figure 4.

Time-dependent determination of the isomer ratio of 17 and 36 (0.5 mM) in methanol and 2× PBS (75:25, v/v) at 37 °C by HPLC using an RP18 column as the stationary phase. (A) 17 (ACN/water (0.1% TFA) gradient; flow rate: 1.6 mL/min; oven temperature: 30 °C; 254 nm) and (B) 36 (ACN/water (Na2SO4, 20 mM (pH 3)) gradient; flow rate: 1.2 mL/min; oven temperature: 30 °C; 281 nm).
These results correspond to studies on the isomerization of (Z)-4-OHT to the less active (E)-4-OHT. In these experiments, 4-OHT of different isomeric ratios was incubated in various media for 72 h. In each case, a stable ratio of 30% of the E isomer was obtained.40 In long-time experiments of up to 6 months, 4-OHT isomerized to E/Z = 50:50, regardless of the applied conditions and solvents.41
2.3. Biological Evaluation
2.3.1. Ligand Binding Affinity
The relative binding affinity (RBA; compared to estradiol (E2): 100%) was determined with a time-resolved fluorescence resonance energy transfer (TR-FRET) competitive binding assay using the isolated LBDs of ERα and ERβ. GW7604, fulvestrant, and 4-OHT were applied as references.
The affinity of 4-OHT and GW7604 indicates the relevance of the side chain in the β-channel. The RBA values of 4-OHT (RBA(ERα) = 14.7%; RBA(ERβ) = 60.7%) decreased upon the exchange of the dimethylaminoethoxy group by an acrylic acid moiety (→GW7604) to 6.2% (ERα) and 27.1% (ERβ).
The binding affinities listed in Table 1 clearly document that the kind of CABS binder contributes to the affinity of the ERs. All compounds possess equal or higher RBA to ERα than GW7604. The results at ERβ are somewhat sophisticated.
Table 1. In Vitro Competitive Binding Assay.
| compound | TR-FRETa ERα | RBAb ERα | TR-FRETa ERβ | RBAb ERβ | |
|---|---|---|---|---|---|
| IC50 [nM] | [%] | IC50 [nM] | [%] | ||
| thioxo-quinazolinones | 15 | 4.01 ± 1.90 | 8.2 | 12.6 ± 4.8 | 5.9 |
| 16 | 1.41 ± 0.46 | 23.4 | 0.82 ± 0.07 | 110 | |
| 17 | 4.63 ± 1.74 | 7.1 | 18.5 ± 4.8 | 4.0 | |
| 18 | 4.23 ± 1.12 | 7.8 | 6.81 ±1.86 | 10.9 | |
| 5-methoxybenzimidazole | 31 | 2.75 ± 1.33 | 12.0 | 4.88 ± 2.65 | 15.2 |
| 32 | 4.11 ± 1.88 | 8.0 | 3.01 ± 4.00 | 24.7 | |
| 34 | 2.22 ± 0.76 | 14.9 | 5.26 ± 3.10 | 14.1 | |
| 36 | 1.56 ± 0.63 | 21.1 | 7.84 ± 0.25 | 9.5 | |
| 38 | 2.61 ± 2.25 | 12.6 | 5.22 ± 0.65 | 14.3 | |
| 5-hydroxybenzimidazole | 39 | 1.45 ± 0.37 | 22.8 | 1.99 ± 0.88 | 37.4 |
| 40 | 2.72 ± 1.12 | 12.1 | 3.78 ± 1.54 | 19.7 | |
| 41 | 1.62 ± 0.50 | 20.4 | 2.84 ± 1.31 | 26.2 | |
| 42 | 1.28 ± 0.58 | 25.7 | 3.81 ± 2.35 | 19.5 | |
| 43 | 3.41 ± 2.07 | 9.7 | 7.8 ± 2.30 | 9.5 | |
| references | E2 | 0.33 ± 0.19 | 100 | 1.02 ± 0.50 | 100 |
| GW7604 | 5.35 ± 1.39 | 6.2 | 3.77 ± 1.06 | 27.1 | |
| fulvestrant | 2.85 ± 0.83 | 11.6 | 10.2 ± 2.5 | 10.0 | |
| 4-OHT | 2.24 ± 0.89 | 14.7 | 1.68 ± 1.00 | 60.7 |
Displacement of fluorescent-labeled E2 from the LBD of ERα or ERβ by heterodimeric compounds and references.
Relative binding affinity (RBA) compared to E2 (100%).
The long and flexible spacer in 16 allows an effective attachment of the linked CABS binder at the proposed binding caves, especially at the surface of ERβ (Figure 2B). The cation−π contacts of the aromatic ring with the charge clamp residues Lys314 and Gln327 (Figure 2B) seem to be the key interaction responsible for the high binding affinity of 110%. Such a binding appears to be also relevant at ERα (RBA = 23.4%). The predicted attachment of 15 (Figure 2A) is less effective. The short alkyl chain mediates only nonspecific van der Waals interactions within the hydrophobic pocket closer to the LBS, resulting in distinctly lower RBA values (RBA(ERβ) = 5.9%; RBA(ERα) = 8.2%). Compound 17 bearing the 4-(4-(4-(4-oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)butanoyl)piperazin-1-yl)phenyl residue caused an RBA at ERβ of 4.0%, indicating that this moiety is not able to reach its essential binding cave.25 Elongation of the spacer between piperazine and 2-thioxo-1,4-dihydroquinazoline from C3 to C7 also failed to increase the ER affinity (18: RBA(ERβ) = 10.9%). A binding pose as depicted in Figure 2C seems not to be suitable to strengthen the attachment at the ER.
The RBA values of 7.1–8.2% determined for 15, 17, and 18 at ERα indicate comparable interactions at both subtypes. Only the RBA of 16 is lower at ERα (23.4%) compared to ERβ (110%). However, the binding affinity to ERα is still higher than that of 4-OHT and GW7604.
The terminal benzimidazole represents an efficient CABS binder. The 5-methoxy derivatives 31, 34, and 38 showed relatively high binding affinities without subtype selectivity (RBA(ERα) = 12.0–14.9%; RBA(ERβ) = 14.1–15.2%). In contrast, 32 more effectively bonded to ERβ (RBA = 24.7%) than to ERα (RBA = 8.0%). The effect of compound 36 is controversial. The RBA amounts to 21.1% (ERα) and 9.5% (ERβ). These findings contradict in part the results of the theoretical considerations, which documented a preference of the compounds with the C6 spacer (38) at ERβ.
The 5-hydroxybenzimidazoles 39 (RBA = 37.4%), 41 (RBA = 26.2%), and 42 (RBA = 19.5%) possessed higher receptor bindings to ERβ than their 5-methoxy derivatives. As predicted, the additional H-bond formation to Gln327 (Figure 3A) increased the affinity. 40 (RBA = 19.7%) and 43 (RBA = 9.5%) were slightly less potent after ether cleavage. The same trend was observed at ERα. The highest RBA values were observed for 39 (22.8%), 41 (20.4%), and 42 (25.7%).
In conclusion, the subtype selectivity regarding the interaction with the isolated receptor of the heterodimeric GW7604 derivatives was low. At ERα, only 36 (ERα/ERβ = 2.22) as well as 16 and 32 at ERβ (ERβ/ERα = 4.7) possessed higher than a 2-fold selectivity. It is noteworthy that the binding compared to GW7604 was improved for ERα, while it was the same (with the exception of 16) for ERβ.
2.3.2. Coactivator Recruitment
To assess the ability of the heterodimeric ligands to induce or inhibit coactivator binding, the interaction of the modified peroxisome proliferator-activated receptor-γ coactivator 1 (PGC-1) peptide with the LBD of ERα was studied by a TR-FRET assay with 16, 36, and 42 as examples, which possess the highest binding affinity.
E2 as a positive control caused maximum coactivator recruitment already at the concentration of 10 nM (for details, see the Supporting Information; Figure S33). In contrast, none of the compounds showed per se agonistic effects up to a concentration of 1 μM.
The ability to prevent the activating effects of E2 at ERα was evaluated in a competition experiment (E2: 4 nM, representing the concentration that activated the coactivator recruitment to 80% of the maximum; drug concentrations: 5 nM to 3 μM) after an incubation time of 10 or 30 min.
GW7604 (1 μM) completely reduced the PGC-1 binding at ERα after 10 min of incubation. This effect was strongly diminished after 30 min. Even at 5 μM, merely 45% coactivator binding was observed (Figure 5).
Figure 5.

Inhibition of coactivator recruitment by concomitant administration of compounds 16, 36, and 42 with E2 (ERα: 4 nM; ERβ: 34 nM). (A) ERα, incubation time: 10 min; (B) ERα, incubation time: 30 min; and (C) ERβ, incubation time: 30 min.
Compounds 16, 36, and 42 effectively prevented coactivator recruitment. The activity after 10 min at ERα increased in the series GW7604 (IC50 = 228.4 nM) < 36 (IC50 = 36.33 nM) < 42 (IC50 = 23.34 nM) < 16 (IC50 = 13.54 nM) (Figure 5A). The derivatives completely circumvented the attachment of PGC-1 even after a prolonged incubation of 30 min GW7604 (IC50 = 2433 nM) < 36 (IC50 = 153.1 nM) < 42 (IC50 = 125.3 nM) < 16 (IC50 = 20.88 nM) (Figure 5B). The thioxo- quinazolinone dimer 16 showed the strongest effects after 10 and 30 min. In contrast to the other compounds, its effect only marginally diminished with time. Furthermore, it was 100-fold more active than GW7604. This might be the consequence of the additional addressing of the CABS, which seems to be more effective in the case of the thioxo-quinazolinone than in the case of terminal benzimidazoles.
The inhibition of coactivator recruitment was also investigated at ERβ (E2-concentration: 34 nM) (Figure 5C). In contrast to the experiment with ERα, GW7604 caused a complete repression of PGC1 binding at 3 μM after an incubation time of 30 min (IC50 = 382.9 nM). 16 (IC50 = 28.34 nM) and 36 (IC50 = 154.4 nM) showed comparable effects at both subtypes, while the inhibitory potency at ERβ strongly increased to IC50 = 63.97 nM in the case of 42.
These findings point out that the dimers were capable of inhibiting the coactivator binding to the receptor not only by the blockage of the LBS but also, more importantly, by the spatial isolation of the CABS.
2.3.3. Solubility and Cellular Uptake
Prior to discussing the effects in cellular systems, the solubility of the compounds at relevant concentrations was estimated. The inherent fluorescence of the cinnamide scaffold is suitable for quantification not only in water but also in cellular systems by fluorometric measurements.
The solubility in aqueous solutions is, with the exception of 36 (∼10 μM), higher than 20 μM: thioxo-quinazolinones: 15: >40 μM; 16: >40 μM; 17: 22.3 μM; 18: >40 μM; 5-methoxybenzimidazoles: 31: 17.3 μM; 32: 25.2 μM; 34: 25.2 μM; 38: 25.9 μM; 5-hydroxybenzimidazoles: 39–43: >40 μM. It is worth mentioning that the solubility of fulvestrant is distinctly lower (11.1 μM).
Based on their fluorometric properties, 15, 34, and 41 were chosen as representatives for the cellular uptake studies.
The compounds were incubated at a concentration of 10 μM with either MCF-7 or COS-7 cells, and the intracellular amount, determined by fluorometry, was related to the protein content.42
The uptake in MCF-7 cells reached saturation already after 4 h (15 (2.8 nmol/mg), 34 (1.5 nmol/mg), and 41 (0.3 nmol/mg)), which remained constant for the duration of 48 h of incubation (Figure 6A). In COS-7 cells, a comparable kinetic was observed (Figure 6B). However, the uptake of 34 (0.4 nmol/mg) after 4 h was reduced, whereas 15 (1.6 nmol/mg) and 41 (0.4 nmol/mg) accumulated to the same extent as in MCF-7 cells. Furthermore, it is obvious that ether cleavage strongly reduced the intracellular amount, which can be attributed to the higher polarity of the CABS binder. The comparison with GW7604 is impossible because its relative fluorescence intensity is too weak to perform uptake studies.
Figure 6.
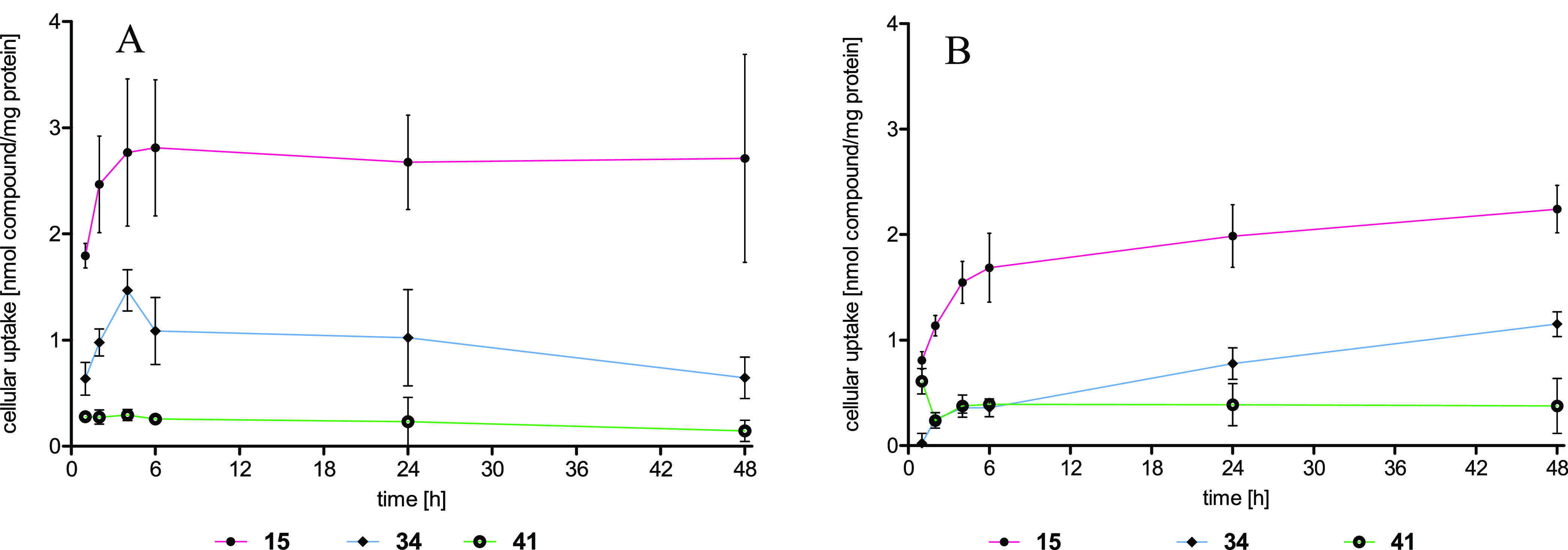
Cellular uptake expressed as nmol compound/mg protein into (A) MCF-7 cells and (B) COS-7 cells after 1, 2, 4, 6, 24, and 48 h of incubation. Data points represent the mean ± SD of ≥3 independent experiments.
2.3.4. Inhibition of Transactivation
The interaction of the heterodimeric compounds with the ERs in cellular systems was studied in a luciferase-based reporter gene assay. Thereto, U2OS osteosarcoma cells were transiently transfected with a receptor plasmid (pSG5-ERα or pSG5-ERβ), a firefly reporter plasmid (p(ERE)2-luc+), and a renilla plasmid (pRenilla-CMV) for standardization.24 Depending on the experimental setting, it is possible to define the agonistic and the antagonistic activity.
At concentrations of 0.1 and 1 μM, none of the compounds caused agonistic effects (Figure S36; Supporting Information). Applied at increasing concentrations (1 nM–10 μM) together with E2 (0.03 nM at ERα, 0.3 nM at ERβ), the antagonistic potency was quantified and expressed as IC50 values (Table 2).
Table 2. Inhibition of E2-Induced Transactivation Determined in a Luciferase Reporter Gene Assay (ERα and ERβ) Using U2OS Cells, Transiently Transfected with Plasmids pSG5-ERα or pSG5-ERβ and the Reporter Plasmid p(ERE)2-luc+.
| compound | transactivation ERα | transactivation ERβ | |
|---|---|---|---|
| IC50a [nM] | IC50a [nM] | ||
| thioxo-quinazolinones | 15 | 30.2 ± 14.3 | 4.1 ± 0.6 |
| 16 | 18.5± 1.8 | 7.5 ± 1.3 | |
| 17 | 104 ± 14 | 48.0 ± 12.0 | |
| 18 | 914 ± 50 | 157± 22 | |
| 5-methoxybenzimidazole | 31 | 9.7 ± 8.9 | 6.7 ± 2.7 |
| 32 | 21.5 ± 10.7 | 26.9 ± 5.7 | |
| 34 | 34.5 ± 3.5 | 11.6 ± 5.5 | |
| 36 | 9.2 ± 3.2 | 8.9 ± 4.0 | |
| 38 | 9.4 ± 2.7 | 9.0 ± 3.3 | |
| 5-hydroxybenzimidazole | 39 | 26.0 ± 6.6 | 20.2 ± 8.6 |
| 40 | 80.8 ± 16.4 | 18.7 ± 8.3 | |
| 41 | 107 ± 4 | 15.9 ± 1.7 | |
| 42 | 30.9 ± 8.7 | 46.5 ± 20.7 | |
| 43 | 20.0 ± 3.2 | 16.1 ± 7.0 | |
| references | GW7604 | 238 ± 74 | 154 ± 53 |
| fulvestrant | n.d.b | n.d.b | |
| 4-OHT | 2.3 ± 1.3 | 1.0 ± 1.3 | |
| 44 | 5.3 ± 3.2 | 3.6 ± 1.4 |
IC50 values represent the means ± SD of ≥3 independent experiments,.
n.d., not defined.
The suitability of the test was verified on the effects of 4-OHT. It reduced the E2-induced luciferase expression with IC50 = 2.3 nM (ERα) and 1.0 nM (ERβ), comparable to data from the literature.43 Fulvestrant completely inhibited the E2-stimulated luciferase expression even at the lowest concentration (0.05 nM) due to its extraordinarily high ER-downregulation potency (see below). Hence, no IC50 calculation was possible.
The reference GW7604 was distinctly less active than 4-OHT with IC50 = 238 nM (ERα) and 154 nM (ERβ). All heterodimers, except for 18, possessed a higher antagonistic activity at both subtypes than GW7604.
Within the thioxo-quinazolinone series, compound 16 was the most potent antagonist at ERα with IC50 = 18.5 nM. At ERβ, 15 and 16 showed IC50 values of 4.1 and 7.5 nM, respectively, which point to effects independent of the diaminoalkane spacer length (15: C3; 16: C7). On the other hand, the bulky phenylpiperazine moiety strongly reduced the transactivation activity (17: ERα: IC50 = 104 nM; ERβ: IC50 = 48.0 nM; 18: ERα: IC50 = 914 nM; ERβ: IC50 = 157 nM).
The most active compounds of the 5-methoxybenzimidazole series, and in general, were 31, 36, and 38 inhibiting the stimulating effects of E2 at both ER subtypes with IC50 of 6.7–9.7 nM. 32 (ERα: IC50 = 21.5 nM; ERβ: IC50 = 26.9 nM) and 34 (ERα: IC50 = 34.5 nM; ERβ: IC50 = 11.6 nM) were less active. Among these derivatives, only 34 possessed a slight subtype selectivity for ERβ.
The data revealed that the diaminoalkane spacer plays a subordinate role in the attachment of the 5-methoxybenzimidazole moiety at the ER. The contacts at ERα seem to be mainly of a hydrophobic nature, because ether cleavage reduced the antagonistic effects. IC50 values of 39, 42, and 43 increased to 20.0–30.9 nM. 40 and 41 were still less active with IC50 = 80.8 and 107 nM.
At ERβ, only 39 (IC50 = 20.2 nM) and 42 (IC50 = 46.5 nM) slightly lost their antagonistic activities. H-bridges to amino acids, e.g., Gln327 or Lys314 as proposed for 42 and 43 by theoretical studies (Figure 3A), appear to be part of the binding mode and strengthened the attachment to the CABS.
This interpretation has to be handled with care in view of the fact that hydroxy-substituted derivatives accumulated in cells to a lesser amount compared to the related methoxy-bearing compounds (as demonstrated with the examples of 34 and 41). This explains the weaker antitransactivation potency despite a similar inhibition of coactivator recruitment in the case of benzimidazoles. The thioxo-quinazolinones (e.g., 15) showed an even higher uptake than benzimidazoles (Figure 6). However, compound 16, which displayed the strongest inhibition of coactivator recruitment and a binding affinity in the low nanomolar range, did not reflect this inhibitory potency in the reporter gene assay.
2.3.5. Estrogen Receptor Downregulation
To evaluate the SERM-/SERD-like properties of the heterodimers, their impact on the ERα levels in MCF-7 cells after 24 h of incubation at 1 μM was quantified using an In-Cell Western immunoassay (Table 3 and Figure 7).
Table 3. ERα Levels in MCF-7 Cells Determined by the In-Cell Western Immunoassay.
| compound | % ERα remaininga [1 μM] | % efficacyb [1 μM] | |
|---|---|---|---|
| thioxo-quinazolinone | 15 | 39.9 ± 8.5 | 60 |
| 16 | 40.3 ± 8.6 | 60 | |
| 17 | 5.5 ± 5.0 | 95 | |
| 18 | 35.7 ± 11.3 | 64 | |
| 5-methoxybenzimidazole | 31 | 113.8 ± 4.3 | c |
| 32 | 120.3 ± 31.6 | c | |
| 34 | 132.9 ± 14.1 | c | |
| 36 | 130.0 ± 15.1 | c | |
| 38 | 101.8 ± 1.9 | c | |
| 5-hydroxybenzimidazole | 39 | 99.2 ± 3.5 | 1 |
| 40 | 90.0 ± 10.4 | 10 | |
| 41 | 83.8 ± 29.2 | 16 | |
| 42 | 92.0 ± 5.5 | 8 | |
| 43 | 85.2 ± 6.2 | 15 | |
| references | GW7604 | 44.3 ± 12.1 | 56 |
| fulvestrant | 0 | 100 | |
| 4-OHT | 263.0 ± 9.0 | c | |
| 44 | 165.0 ± 4.0 | c |
ERα levels compared to the solvent control (dimethyl sulfoxide (DMSO)). Values represent the mean ± SD of ≥3 independent experiments.
Efficacy, calculated as the downregulation of ERα compared to the efficacy of the reference compound fulvestrant.
Upregulation.
Figure 7.

Percent efficacy related to fulvestrant. Negative values indicate upregulation. Values represent the means ± SE of ≥3 independent experiments.
The SERD fulvestrant caused an almost complete ER destabilization/degradation (efficiency at 1 μM was set to 100%), while 4-OHT as mixed agonist/antagonist44,45 significantly upregulated the ERα content to 263% compared to the untreated control (100%). As already discussed in a previous paper,24 the exchange of the dimethylaminoethoxyphenyl group by a cinnamic acid moiety (GW7604) increased the downregulatory properties to an efficiency of 56% (Table 3).
This effect was attributed to the change of a positively charged side chain in the case of 4-OHT to a negative one in GW7604. The side chain of 4-OHT interacts with the surface amino acid Asp351 of ERα in an ionic attraction, allowing the binding of coactivators that are necessary for expression of the receptor. The carboxylate of GW7604 generates a strong repulsion of Asp351, which disrupts the surface charge around this amino acid required for coactivator binding in the 4-OHT/ER complex.19
The relevance of the positively charged side chain documented the derivation of the carboxylic group of GW7604 with a 1,2-diaminoethane chain. Compound 44 increased the ERα expression to about 165%, resembling the biological profile of the partial agonist 4-OHT.18,24,46,47
The binding of the long side chain at the steroidal core of fulvestrant to the surface of the receptor effectively destabilizes the protein, leading to ubiquitination and degradation. Accordingly, in the In-Cell Western immunoassay, a complete downregulation of ERα in MCF-7 cells (efficacy 100%) is visible.
Exceptional results provided the testing of the thioxo-quinazolinones. All of them were more active downregulators than the parent compound GW7604, although they do not bear a negative charge at the side chain. Furthermore, based on their structure, they cannot change the conformation of the ER as it is caused by the side chain of fulvestrant. Nevertheless, 15, 16, and 18 reduced the ERα content to about 40%, while 17 (efficacy: 95%) was almost as active as fulvestrant.
Compounds out of the benzimidazole series only marginally influenced the ERα content in the cells. Upon incubation with the 5-methoxy derivatives at a drug concentration of 1 μM, the level slightly increased (100–130%), while the hydroxy-substituted ones showed a trend toward downregulation (83–100%) (Figure 7).
The mode of downregulation with compounds bearing the thioxo-quinazolinone scaffold has not yet been completely clarified.
2.3.6. Antiproliferative Effects
Whether the ER interactions discussed above consequently influence the proliferation of hormone-dependent tumor cells was assessed via the crystal violet assay.48 The ER-positive MCF-7, the tamoxifen-resistant MCF-7TamR,49 as well as the hormone-independent MDA-MB-231 breast cancer cell lines were used to visualize these effects. Representative concentration–activity and time–activity curves are depicted in Figure 8. All calculated IC50 values are listed in Table 4.
Figure 8.


Antiproliferative effects against ER-positive MCF-7 (blue), tamoxifen-resistant MCF-7TamR (red), and ER-negative MDA-MB-231 (orange) breast cancer cells. Values represent the mean ± SD of ≥3 independent experiments. For further graphs, see the Supporting Information (Figures S34 and S35).
Table 4. Antiproliferative Effects: IC50 Values at ER-Positive MCF-7, Tamoxifen-Resistant MCF-7TamR, and ER-Negative MDA-MB-231 Breast Cancer Cells.
| compound | MCF-7 | IC50 [nM]a | MDA-MB-231 | |
|---|---|---|---|---|
| MCF-7TamR | ||||
| thioxo-quinazolinone | 15 | 1.9 | 2 296 | >1 μM |
| 16 | 4.2 | 476.6 | n.a. | |
| 17 | 27.8 | 550.8 | n.a. | |
| 18 | 12.6 | 1 208 | n.a. | |
| 5-methoxybenzimidazole | 31 | 2.8 | n.d. | n.a. |
| 32 | 2.4 | n.d. | n.a. | |
| 34 | 4.1 | 486.3 | n.a. | |
| 36 | 1.8 | 68.7 | n.a. | |
| 38 | 0.5 | n.d. | n.a. | |
| 5-hydroxybenzimidazole | 39 | 10.6 | 531.0 | n.a. |
| 40 | 4.5 | 543.7 | n.a. | |
| 41 | 9.8 | 1 711 | n.a. | |
| 42 | 11.5 | 235.7 | n.a. | |
| 43 | 5.6 | 325.1 | >1 μM | |
| references | GW7604 | 52.0 | 956.9 | n.a. |
| fulvestrant | 0.58 | 1.8 | n.d | |
| 4-OHT | n.d | n.d | n.d |
Values represent means of ≥3 independent experiments; n.d., not determined; n.a., not active.
The compounds were incubated at concentrations between 1 nM and 10 μM for 120 h, and the remaining adherent growing cells were stained by crystal violet.24 The photometric measurement after extraction of the dye from the chromatin with ethanol allows the quantification of the cell mass. Comparing the treated wells with the solvent-treated control wells results in % T/Ccorr values, from which conclusions can be drawn about the extent of the cytotoxicity. T/Ccorr > 80% is considered nonantiproliferative/nontoxic. T/Ccorr values between 80 and 20% are classified as antiproliferative. Below 20%, the compounds are cytostatic. A cytocidal effect is defined as a T/Ccorr value below 0%.
To obtain an insight into the kinetic of cell death, MCF-7 and MDA-MB-231 cells were further incubated at concentrations of 5, 10, and 20 μM for 12, 24, 36, 48, 72, and 144 h.
4-OHT did not influence the cells up to a concentration of 1 μM. At 10 μM, cytostatic and cytocidal effects occurred and reduced the T/Ccorr values of MDA-MB-231, MCF-7, and MCF-7TamR cells to 10, −55, and −57%, respectively. Time-dependent experiments at increased concentrations also indicated cytocidal effects against the MCF-7 cell line after 24 h at 20 μM and after 48 h at 10 μM. The growth of MDA-MB-231 cells was only reduced at 20 μM, reaching cytocidal effects after 24 h (Figure 8).
Fulvestrant was completely inactive in the MDA-MB-231 cell line, while it had strong effects in MCF-7 cells (IC50 = 0.58 nM). The time–activity curve (Figure 8) indicates no unspecific cytotoxicity but identical curves at concentrations of 5, 10, and 20 μM with a maximum of activity of T/Ccorr = 26% after 72 h. The proliferation of MCF-7TamR cells was reduced, too (IC50 = 1.8 nM).
It should be mentioned that the concentration–activity curves showed only antiproliferative effects. The maximum activity is in the range of T/Ccorr = 30–35%. The compounds displayed no cytostatic or cytocidal effects at the applied concentrations.
GW7604 did not influence MDA-MB-231 cells but reduced the growth of MCF-7 cells with IC50 = 52.0 nM. Against MCF-7TamR only at 10 μM, a marginal effect of T/Ccorr = 68% was observed. Treatment of MDA-MB-231 cells with the heterodimeric compounds did not affect the proliferation up to a used concentration of 1 μM. At higher concentrations (10 μM), only 15 and 43 reduced the cell growth unspecifically to T/Ccorr = 20 and 63%. This effect seems to be hormone/ER-independent, because the time-dependent experiment documented, e.g., for 15 at concentrations higher than 1 μM identical effects against MDA-MB-231 and MCF-7 cells (Figure 8).
The activity in the MCF-7 cell line depended on the used CABS binder and the kind of linkage to the GW7604 core. The thioxo-quinazolinone derivatives 15 and 16 caused IC50 values of 1.9 and 4.2 nM, while the piperazinylbenzoate bearing compounds 17 and 18 showed decreased cytotoxicity with IC50 = 27.8 and 12.6 nM.
The 5-methoxybenzimidazoles 31–38 reduced the cell growth of MCF-7 cells independent of the spacer length. The IC50 values were in the range of 0.5–4 nM, only marginally higher than the IC50 of fulvestrant (IC50 = 0.58 nM). 38 (IC50 = 0.50 nM) was even as active as fulvestrant. Ether cleavage diminished the cytotoxicity to IC50 = 4.5–11.5 nM. The most active compound was 40 with an IC50 of 4.5 nM.
It should be mentioned that none of the compounds stimulated the proliferation of hormone-dependent MCF-7 cells, indicating the absence of partial estrogenic properties.
All compounds were active against the MCF-7TamR cell line, showing a flat concentration–activity curve. The maximum effect at 10 μM was in most cases higher than T/Ccorr = 50%. Only 15, 17, 38, and 43 reached a T/Ccorr of about 40%. All compounds were more active than GW7604. Sigmoid curves allowed in some cases the calculation of IC50 values as a parameter of antiproliferative potency. From these data, it is obvious that the compounds were less potent than fulvestrant. The most active compound was 36 with an IC50 of 68.7 nM.
3. Conclusions
In this SAR study, GW7604 was linked to known CABS binders to evaluate the possibility of increasing ER binding and to inhibit coactivator recruitment. The biological activities were investigated at the isolated receptors and in cellular systems. The thioxo-quinazolinones mediated high binding affinity, with extraordinary RBA values of 23.4 and 110% measured for compound 16 at ERα and ERβ, respectively. The 5-methoxy/hydroxybenzimidazole increased the affinity, too. All derivatives showed higher affinity to ERα than GW7604, while only compound 16 and the 5-hydroxybenzimidazole derivative 39 exceeded the affinity of the reference to ERβ. The coactivator recruitment was also effectively inhibited at both receptor subtypes, as demonstrated for 16, 36, and 42, with 10- to 100-fold higher inhibitory potency compared to GW7604. Consequently, the heterodimeric compounds inhibited the E2-induced transactivation in the luciferase reporter gene assay more effectively. None of them stimulated the ER expression as 4-OHT.
ER downregulation showed the thioxo-quinazolinones derivatives. 17 caused nearly complete receptor degradation (94.5% efficacy). In contrast, the receptor content remained almost unchanged in the case of benzimidazole-bearing species.
The influence on cell growth is based on the interference in hormonal pathways. The compounds did not reduce the proliferation of hormone-independent MDA-MB-231 cells. Only compound 15 showed unspecific effects at concentrations higher than 1 μM. Against hormone-dependent MCF-7 cells, all heterodimers were more active than GW7604. Compound 38 (IC50 = 0.5 nM) reduced the proliferation as effectively as fulvestrant. Also, the growth of the tamoxifen-resistant subline MCF-7TamR decreased upon treatment, however, with a flat concentration−activity relation.
In conclusion, the attempt to modify the pharmacological profile of GW7604 by linking the cinnamic acid moiety by a diaminoalkane spacer to the CABS binder was successful. The compounds showed the profile either of pure antiestrogens (5-methoxy/hydroxybenzimidazoles) or of pure antiestrogens with ER-degradation potency (thioxo-quinazolinones).
4. Experimental Section
4.1. Chemistry
4.1.1. General
All reagents were purchased from Sigma-Aldrich, TCI, VWR, or Alfa Aesar and were used without further purification. All solvents were distilled before usage. Anhydrous solvents were obtained by distillation under argon over an appropriate drying agent. Reactions were performed under an inert argon atmosphere using oven-dried glassware, septa, and syringes for the addition of substances. Chromatography purification was performed employing either classic standard procedures or on a Biotage Isolera 1 Flash purification system. Silica gel 60 Å was used in both cases. 1H NMR and 13C NMR spectra were obtained on a Varian Gemini-200 (now Agilent), 400 MHz Avance 4 Neo (Bruker), 500 MHz Direct Drive 2 (Agilent), 600 MHz Avance II (Bruker), or 700 MHz Avance 4 Neo (Bruker) spectrometer. As solvents for NMR, deuterated chloroform (CDCl3), dimethyl sulfoxide (DMSO-d6), acetone ((CD3)2CO), and methanol (CD3OD) were used. The chemical shifts (δ) were referenced to tetramethylsilane (TMS) or the solvent peak and are given in parts per million (ppm). Coupling constants (J) are given in Hertz (Hz).
The purity of final compounds 15–18, 31, 32, 34, 36, 38, and 39–43 as well as the stability of compounds 17 and 36 was measured by HPLC (Shimadzu, LaChrom) on a 250 mm reversed-phase C18 column as the stationary phase (KNAUER) equipped with a diode-array detector (for HPLC spectra and methods, see the Supporting Information). HR-MS spectra were obtained from an Orbitrap Elite MS (Thermo Fisher Scientific). All final compounds were >95% pure.
4.1.2. Computational Design of the Heterodimers
The ligands were docked analogously to previous studies.24 The 2D and 3D chemical structures were generated with the ChemDraw14 package.50 GOLD suite v 5.251 was employed for docking experiments and the results were examined in LigandScout 4.1 alpha4.52
4.1.3. Synthesis of the Final Compounds
4.1.3.1. General Procedure for Amide Formation
First, DIPEA (6.0 equiv) and then PyBOP (1.1 equiv), dissolved in dry DCM, were added to a solution of GW7604 or methoxy-GW7604 (1.1 equiv) in dry DMF at 0 °C under an argon atmosphere. The solution was stirred for 5 min followed by the dropwise addition of the respective trifluoroacetate salt (1.0 equiv) in dry DMF. The reaction mixture was stirred initially at 0 °C for 30 min and then at 40 °C for 20 h. Subsequently, the solvents were concentrated and the residue was dissolved in ethyl acetate (EA). Water was added (pH = 8–9, adjusted with 2N NaOH if needed), and the aqueous phase was extracted twice with EA. The combined organic layers were washed with brine, dried over anhydrous Na2SO4, and filtered. Purification was achieved by column chromatography.32−35
4.1.3.2. Syntheses of the Thioxo-quinazolinones 15–18
Diamide coupling was carried out according to the general procedure for heterodimer formation, yet at rt instead of at 40 °C and performing an acidic workup with 1 N HCl at pH = 2–3.
4.1.3.2.1. N-[3-((E)-3-(4-((E/Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-enyl)phenyl)acrylamido)propyl]-4-[4-oxo-2-thioxodihydroquinazolin-3-yl]butanamide (15)
15 was synthesized following the general procedure described above, applying 55 mg of GW7604 (0.15 mmol) in 0.5 mL of dry DMF, 0.16 mL of DIPEA (0.94 mmol), and 85 mg of PyBOP (0.16 mmol) in 1 mL of dry DCM. The trifluoroacetate salt 11 (1.1 equiv, 71 mg, 0.17 mmol) in 1.0 mL of dry DMF was added dropwise. Purification was carried out by flash column chromatography with a petroleum ether (PE):EA:MeOH gradient (EA: 30 → 100% and then MeOH: 0 → 5%) followed by column chromatography with a DCM and MeOH gradient (98:2 → 95:5). 15 was obtained as a colorless powder (28 mg, 0.038 mmol, 26%). Purity: 98.2% (HPLC: 1.0 mM in MeOH, 254 nm). 1H NMR: (700 MHz, CD3OD, E/Z = 30:70): δ 0.92 (t, 3J = 7.4 Hz, 3H, CH2CH3), 1.71 (p, 3J = 6.8 Hz, 0.6H, NHCH2CH2CH2NH, E isomer), 1.76 (p, 3J = 6.8 Hz, 1.4H, NHCH2CH2CH2NH, Z isomer), 2.07–2.15 (m, 2H, NCH2CH2CH2CONH), 2.28–2.65 (m, 2H, NCH2CH2CH2CONH), 2.47 (q, 3J = 7.4 Hz, 1.4H, CH2CH3, Z isomer), 2.51 (q, 3J = 7.4 Hz, 0.6H, CH2CH3, E isomer), 3.20 (t, 3J = 6.7 Hz, 0.6H, NHCH2CH2CH2NH, E isomer), 3.24 (t, 3J = 6.7 Hz, 1.4H, NHCH2CH2CH2NH, Z isomer), 3.33–3.38 (2 × t, 2H, 2 × NHCH2CH2CH2NH), 4.54–4.61 (2 × t, 2H, 2 × NCH2CH2CH2CONH), 6.36–6.46 (m, 1.6H, ArH + CH=CHCONH), 6.60 (d, 3J = 15.8 Hz, 0.7H, CH=CHCONH, Z isomer), 6.66 (d, 3J = 8.6 Hz, 1.4H, ArH, Z isomer), 6.78 (d, 3J = 8.5 Hz, 0.6H, ArH, E isomer), 6.87 (d, 3J = 8.2 Hz, 0.6H, ArH, E isomer), 7.03 (d, 3J = 8.5 Hz, 0.6H, ArH, E isomer), 7.07–7.20 (m, 5.7H, ArH), 7.20–7.27 (m, 2.4H, ArH), 7.27–7.32 (m, 1H, ArH), 7.35 (d, 3J = 15.7 Hz, 0.3H, CH=CHCONH, E isomer), 7.50–7.58 (m, 2.1H, ArH + CH=CHCONH), 7.63–7.70 (m, 1H, ArH), 7.99–8.07 (m, 1H, ArH). 13C NMR: (176 MHz, CD3OD, E/Z = 30:70): δ 13.81, 13.87, 24.36, 29.86, 29.99, 30.18, 30.22, 34.57, 37.90, 37.94, 38.04, 38.11, 46.80, 115.28, 116.03, 116.13, 117.08, 121.07, 121.54, 125.49, 127.21, 127.37, 127.86, 128.68, 128.83, 128.93, 129.00, 130.84, 130.86, 131.10, 131.67, 132.44, 133.05, 133.56, 134.64, 135.29, 135.60, 136.56, 139.55, 139.69, 140.74, 141.47, 142.84, 143.64, 143.68, 143.90, 146.78, 147.03, 156.67, 157.56, 161.65, 168.83, 175.42, 177.40. HR-MS (m/z): calcd for C40H40N4O4S [M – H]−: 671.2698, found: 671.2720.
4.1.3.2.1.1. N-[4-((E)-3-(4-((E/Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-enyl)phenyl)acrylamido)butyl]-8-[4-oxo-2-thioxodihydroquinazolin-3-yl]octanamide (16)
16 was synthesized following the general procedure applying 56 mg of GW7604 (0.15 mmol) in 0.5 mL of dry DMF, 0.16 mL of DIPEA (0.94 mmol), and 87 mg of PyBOP (0.17 mmol) in 1 mL of dry DCM. 12 (1.1 equiv, 84 mg, 0.17 mmol) in 1.0 mL of dry DMF was added. Purification by flash column chromatography with a gradient of PE and EA (70 → 100% EA) led to 16 as a whitish-yellow powder (31 mg, 0.042 mmol, 29%). Purity: 97.3% (HPLC: 1.0 mM in MeOH, 254 nm). 1H NMR: (700 MHz, CD3OD, E/Z = 45:55): δ 0.88–0.93 (2 × t, 3H, 2 × CH2CH3), 1.34–1.41 (m, 6H, CH2), 1.53–1.65 (m, 6H, CH2), 1.71–1.78 (m, 2H, CH2), 2.13–2.22 (2 × t, 2H, 2 × CH2CON), 2.45 (q, 3J = 7.3 Hz, 1.1H, CH2CH3, Z isomer), 2.50 (q, 3J = 7.4 Hz, 0.9H, CH2CH3, E isomer), 3.18 (t, 3J = 6.3 Hz, 0.9H, NHCH2CHCH2CH2NH, E isomer), 3.21 (t, 3J = 6.4 Hz, 1.1H, NHCH2CH2CH2CH2NH, Z isomer), 3.28 (t, 3J = 6.3 Hz, 0.9H, NHCH2CH2CH2CH2NH, E isomer), 3.33 (t, 3J = 6.2 Hz, 1.1H, NHCH2CH2CH2CH2NH, Z isomer), 4.45–4.49 (2 × t, 2H, 2 × NCH2), 6.37–6.46 (m, 1.4H, ArH + CH=CHCONH), 6.60 (d, 3J = 15.8 Hz, 0.6H, CH=CHCONH, Z isomer), 6.64 (d, 3J = 8.5 Hz, 1.1H, ArH, Z isomer), 6.78 (d, 3J = 8.3 Hz, 0.9H, ArH, E isomer), 6.85 (d, 3J = 8.1 Hz, 0.9H, ArH, E isomer), 7.02 (d, 3J = 8.3 Hz, 0.9H, ArH, E isomer), 7.05–7.21 (m, 5.9H, ArH), 7.22 (d, 3J = 8.0 Hz, 1.1H, ArH, Z isomer), 7.24 (d, 3J = 8.3 Hz, 1H, ArH), 7.27–7.31 (m, 1H, ArH), 7.35 (d, 3J = 15.7 Hz, 0.4H, CH=CHCONH, E isomer), 7.50–7.57 (m, 1.7H, ArH + CH=CHCONH), 7.64–7.70 (m, 1H, ArH), 8.02 (d, 3J = 8.0 Hz, 1H, ArH). 13C NMR: (176 MHz, CD3OD, E/Z = 45:55): δ 13.81, 13.88, 27.01, 27.55, 27.77, 27.87, 27.91, 28.72, 29.86, 29.94, 29.99, 30.11, 37.13, 39.98, 40.18, 40.24, 47.45, 115.27, 116.02, 116.12, 117.06, 121.11, 121.57, 125.49, 127.20, 127.36, 127.84, 128.66, 128.76, 128.92, 129.00, 130.84, 130.86, 131.10, 131.68, 132.44, 133.05, 133.56, 134.64, 135.29, 135.60, 136.53, 139.53, 139.68, 140.70, 141.39, 142.82, 143.63, 143.68, 143.87, 146.73, 146.98, 156.65, 157.55, 161.52, 168.72, 176.34, 177.30. HR-MS (m/z): calcd for C45H50N4O4S [M – H]−: 741.3553, found: 741.3558.
4.1.3.2.1.2. N-[3-((E)-3-(4-((E/Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-enyl)phenyl)acrylamido)propyl]-4-[4-(4-(4-oxo-2-thioxodihydroquinazolin-3-yl)butanoyl)piperazin-1-yl]benzamide (17)
17 was synthesized following the general procedure applying 68 mg of GW7604 (0.18 mmol) in 1 mL of dry DMF, 0.2 mL of DIPEA (1.13 mmol), and 104 mg of PyBOP (0.20 mmol) in 2 mL of dry DCM. 13 (1.1 equiv, 125 mg, 0.20 mmol) in 1 mL of dry DMF was added dropwise. Purification was carried out by flash column chromatography with a PE:EA:MeOH gradient (EA: 30 → 100% and then MeOH: 0 → 5%). 17 was obtained as a whitish-yellow powder (85 mg, 0.10 mmol, 54%). Purity: 99.8% (HPLC: 0.5 mM in MeOH, 254 nm). 1H NMR: (600 MHz, DMSO-d6, E/Z = 12:88): δ 0.86 (t, 3J = 7.4 Hz, 3H, CH2CH3), 1.64–1.67 (m, 0.2H, CH2, E isomer), 1.70 (p, 3J = 6.8 Hz, 1.8H, CH2, Z isomer), 1.98 (p, 3J = 7.0 Hz, 2H, CH2), 2.40 (q, 3J = 7.2 Hz, 2H, CH2CH3), 2.46 (t, 3J = 7.3 Hz, NCH2CH2CH2CON), 3.22–3.26 (m, 4H, NHCH2CH2CH2NH), 3.27–3.31 (m, 4H CON(CH2CH2)2N), 3.51–3.61 (m, 4H, CON(CH2CH2)2N), 4.46 (t, 3J = 7.1 Hz, NCH2), 6.42 (d, 3J = 8.5 Hz, 1.8H, ArH, Z isomer), 6.46 (d, 3J = 15.8 Hz, 0.1H, CH=CHCONH, E isomer), 6.62–6.65 (m, 2.6H, ArH + CH=CHCONH, Z isomers), 6.77 (d, 3J = 8.4 Hz, 0.2H, ArH, E isomer), 6.84 (d, 3J = 8.2 Hz, 0.2H, ArH, E isomer), 6.95–6.98 (m, 2.1H, ArH), 7.01 (d, 3J = 8.3 Hz, 0.2H, ArH, E isomer), 7.10–7.27 (m, 7H, ArH + CH=CHCONH, E isomer), 7.31–7.33 (m, 1H, ArH), 7.38 (d, 3J = 8.2 Hz, 1H, ArH), 7.44 (d, 3J = 15.7 Hz, 0.9H, CH=CHCONH, Z isomer), 7.56 (d, 3J = 7.8 Hz, 1.8H, ArH, Z isomer), 7.72–7.76 (m, 3H, ArH), 7.94 (d, 3J = 7.8 Hz, 1H, ArH), 8.06 (brt, 3J = 5.4 Hz, 0.1H, NH, E isomer), 8.16 (brt, 3J = 5.5 Hz, 0.9H, NH, Z isomer), 8.20 (brt, 0.1H, NH, E isomer), 8.24 (brt, 3J = 5.5 Hz, 0.9H, NH, Z isomer), 9.21 (s, 0.9H, OH, Z isomer), 9.46 (s, 0.1H, OH, E isomer), 12.90 (s, 1H, NH). 13C NMR: (151 MHz, DMSO-d6, E/Z = 12:88): δ 13.27, 22.13, 28.36, 29.38, 29.80, 36.56, 36.76, 40.54, 44.31, 45.19, 46.93, 47.21, 113.75, 114.33 Z isomer, 114.99 E isomer, 115.47, 121.88, 123.98, 124.02, 124.30, 126,03, 126.54 E isomer, 127.18, 127.34 Z isomer, 127.78 Z isomer, 127.85 E isomer, 128.30, 129.24, 129.49, 130.11 E isomer, 130.69 E isomer, 131.29 Z isomer, 132.90, 133.16, 135.26, 137.64, 138.09, 138.98, 140.59, 141.73, 144.52, 152.32, 155.31, 159.31, 164.94, 165.72, 169.89, 175.04. HR-MS (m/z): calcd for C51H52N6O5S [M – H]−: 859.3647, found: 859.3679.
4.1.3.2.1.3. N-[3-((E)-3-(4-((E/Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-enyl)phenyl)acrylamido)propyl]-4-[4-(8-(4-oxo-2-thioxodihydroquinazolin-3-yl)octanoyl)piperazin-1-yl]benzamide (18)
18 was synthesized according to the general procedure applying 68 mg of GW7604 (0.18 mmol) in 1 mL of dry DMF, 0.20 mL of DIPEA (1.13 mmol), and 104 mg of PyBOP (0.20 mmol) in 2 mL of dry DCM. 14 (1.1 equiv, 136 mg, 0.20 mmol) in 1 mL of dry DMF was added slowly. The crude product was purified by column chromatography with a DCM and MeOH gradient (96:4 → 95:5). 18 was obtained as a whitish-yellow powder (80 mg, 0.10 mmol, 48%). Purity: 99.7% (HPLC: 0.5 mM in MeOH, 254 nm). 1H NMR: (700 MHz, DMSO-d6, E/Z = 20:80): δ 0.86 (t, 3J = 7.3 Hz, 3H, CH2CH3), 1.30–1.36 (m, 6H, CH2), 1.52 (p, 3J = 6.7 Hz, 2H, CH2), 1.65–1.73 (m, 4H, CH2), 2.34 (t, 3J = 7.4 Hz, 2H, CH2CON(CH2CH2)2N), 2.39 (q, 3J = 7.2 Hz, 1.6H, CH2CH3, Z isomer), 2.44 (q, 3J = 7.4 Hz, 0.4H, CH2CH3, E isomer), 3.18–3.25 (m, 4H, NHCH2CH2CH2NH), 3.25–3.28 (m, 4H, CON(CH2CH2)2N), 3.56–3.62 (m, 4H, CON(CH2CH2)2N), 4.39 (t, 3J = 7.7 Hz, 2H, NCH2), 6.42 (d, 3J = 8.5 Hz, 1.6H, ArH, Z isomer), 6.46 (d, 3J = 15.8 Hz, 0.2H, CH=CHCONH, E isomer), 6.59–6.66 (m, 2.4H, ArH, Z isomer + CH=CHCONH, Z isomer), 6.77 (d, 3J = 8.3 Hz, 0.4H, ArH, E isomer), 6.84 (d, 3J = 8.1 Hz, 0.4H, ArH, E isomer), 6.94–6.99 (m, 2.2H, ArH), 7.01 (d, 3J = 8.3 Hz, 0.4H, ArH, E isomer), 7.04–7.29 (m, 7H, ArH + CH=CHCONH, E isomer), 7.30–7.36 (m, 1H, ArH), 7.39 (d, 3J = 8.2 Hz, 1H, ArH), 7.44 (d, 3J = 15.7 Hz, 0.8H, CH=CHCONH, Z isomer), 7.56 (d, 3J = 8.0 Hz, 1.6H, ArH, Z isomer), 7.72–7.76 (m, 3H, ArH), 7.95 (d, 3J = 7.8 Hz, 1H, ArH), 8.05 (brt, 3J = 5.5 Hz, 0.2H, NH, E isomer), 8.15 (brt, 3J = 5.4 Hz, 0.8H, NH, Z isomer), 8.20 (brt, 3J = 5.4 Hz, 0.2H, NH, E isomer), 8.23 (brt, 3J = 5.4 Hz, 0.8H, NH, Z isomer), 9.20 (s, 0.8H, OH, Z isomer), 9.45 (s, 0.2H, OH, E isomer), 12.91 (s, 1H, NH). 13C NMR: (176 MHz, DMSO-d6, E/Z = 20:80): δ 13.36, 24.68, 26.16, 26.26, 28.47, 28.63, 29.48, 32.22, 36.66, 36.87, 40.57, 44.46, 45.66, 47.14, 47.52, 113.86, 114.43 Z isomer, 115.09 E isomer, 115.48, 115.62, 121.99, 124.17, 124.43, 126.12, 126.63 E isomer, 127.24, 127.43 Z isomer, 127.87 Z isomer, 128.40, 129.33, 129.58, 130.20 E isomer, 130.78 E isomer, 131.38 Z isomer, 133.00, 133.26, 135.39, 137.75, 138.18, 139.09, 140.69, 141.83, 144.62, 152.40, 155.41, 159.21, 165.04, 165.81, 170.74, 174.98. HR-MS (m/z): calcd for C55H60N6O5S [M – H]−: 915.4273, found: 915.4283.
4.1.3.2.2. Syntheses of the 5-Methoxybenzimidazoles 31, 32, 34, 36, 38
4.1.3.2.2.1. (E)-3-[4-((E/Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-enyl)phenyl]-N-[2-(3-(5-methoxy-1H-benzo[d]imidazol-2-yl)propanamido)ethyl]acrylamide (31)
31 was synthesized according to the general procedure for heterodimer formation using 140 mg of GW7604 (0.38 mmol) in 2 mL of dry DMF, 0.4 mL of DIPEA (2.27 mmol), and 216 mg of PyBOP (0.42 mmol) in 2 mL of dry DCM. 26 (186 mg, 0.38 mmol) in 1 mL of dry DMF was added dropwise to this mixture. Column chromatography with DCM and MeOH (95:5 → 93:7) afforded 31 as orange crystals (116 mg, 0.19 mmol, 50%). Purity: 98.5% (HPLC: 0.5 mM in ACN + water (Na2SO4, 20 mM (pH 3)), 254 nm). 1H NMR: (700 MHz, CD3OD, E/Z = 55:45): δ 0.94 (t, 3J = 7.4 Hz, 3H, CH2CH3), 2.49 (q, 3J = 7.4 Hz, 0.9H, CH2CH3, Z isomer), 2.54 (q, 3J = 7.4 Hz, 1.1H, CH2CH3, E isomer), 2.73 (t, 3J = 7.5 Hz, 1.1H, CH2CH2CONH, E isomer), 2.76 (t, 3J = 7.5 Hz, 0.9H, CH2CH2CONH, Z isomer), 3.15 (t, 3J = 7.5 Hz, 1.1H, CH2CH2CONH, E isomer), 3.18 (t, 3J = 7.5 Hz, 0.9H, CH2CH2CONH, Z isomer), 3.34 (t, 1.1H, GW7604NHCH2CH2, E isomer), 3.36–3.41 (m, 2H, GW7604NHCH2CH2), 3.43 (t, 3J = 5.8 Hz, 0.9H, GW7604NHCH2CH2, Z isomer), 3.79 (s, 1.6H, OCH3, E isomer), 3.80 (s, 1.4H, OCH3, Z isomer), 6.36 (d, 3J = 15.7 Hz, 0.5H, CH=CHCONH, E isomer), 6.44 (d, 3J = 8.6 Hz, 0.9H, ArH, Z isomer), 6.53 (d, 3J = 15.8 Hz, 0.5H, CH=CHCONH, Z isomer), 6.68 (d, 3J = 8.6 Hz, 0.9H, ArH, Z isomer), 6.81 (d, 3J = 8.4 Hz, 1.1H, ArH, E isomer), 6.82–6.86 (m, 1H, ArH), 6.89 (d, 3J = 8.3 Hz, 1.1H, ArH, E isomer), 6.98–7.04 (m, 1H, ArH), 7.06 (d, 3J = 8.5 Hz, 1.1H, ArH, E isomer), 7.08–7.23 (m, 6.1H, ArH), 7.25 (d, 3J = 8.1 Hz, 0.9H, ArH, Z isomer), 7.30–7.43 (m, 1.6H, ArH + CH=CHCONH, E isomer), 7.51 (d, 3J = 8.1 Hz, 0.9H, ArH, Z isomer), 7.54 (d, 3J = 15.8 Hz, 0.5H, CH=CHCONH, Z isomer). 13C NMR: (700 MHz, CD3OD, E/Z = 55:45): δ 13.80, 13.86, 25.50, 29.85, 29.99, 34.64, 40.16, 40.20, 40.26, 56.19, 98.08, 112.87, 115.28, 116.03, 116.21, 120.96, 121.43, 127.22, 137.37, 127.87, 128.68, 128.93, 129.00, 130.83, 130.86, 131.09, 131.67, 132.42, 133.04, 133.48, 134.55, 135.29, 135.59, 139.54, 139.68, 141.57, 141.59, 142.86, 143.64, 143.67, 143.92, 146.82, 147.06, 155.04, 155.06, 156.67, 157.56, 157.90, 169.04, 174.52, 174.56. HR-MS (m/z): calcd for C38H38N4O4 [M + H]+: 615.2966, found: 615.2964.
4.1.3.2.2.2. (E)-3-[4-((E/Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-enyl)phenyl]-N-[3-(3-(5-methoxy-1H-benzo[d]imidazol-2-yl)propanamido)propyl]acrylamide (32)
32 was synthesized according to the general procedure for heterodimer formation using 44 mg of GW7604 (0.12 mmol) in 1.3 mL of dry DMF, 0.13 mL of DIPEA (0.71 mmol), and 68 mg of PyBOP (0.13 mmol) in 1 mL of dry DCM. 27 (60 mg, 0.12 mmol) in 1 mL of dry DMF was added dropwise. Purification by column chromatography with a gradient using DCM and MeOH (95:5 → 90:10) led to 32 as a light-yellow powder (44 mg, 0.07 mmol, 60%). Purity: 98.7% (HPLC: 1.0 mM in ACN + water (Na2SO4, 20 mM (pH 3)), 254 nm). 1H NMR: (700 MHz, CD3OD, E/Z = 55:45): δ 0.92 (t, 3J = 7.4 Hz, 3H, CH2CH3), 1.66 (p, 3J = 6.8 Hz, 1.1H, NHCH2CH2CH2NH, E isomer), 1.71 (p, 3J = 6.8 Hz, 0.9H, NHCH2CH2CH2NH, Z isomer), 2.48 (q, 3J = 7.3 Hz, 0.9H, CH2CH3, Z isomer), 2.52 (q, 3J = 7.4 Hz, 1.1H, CH2CH3, E isomer), 2.69–2.78 (2 × t, 3J = 7.5 Hz, 2H, 2 × CH2CH2CONH), 3.14–3.19 (2 × t, 3J = 7.5 Hz, 2H, 2 × CH2CH2CONH), 3.19–3.23 (m, 2H, GW7604NHCH2CH2CH2NH), 3.25 (t, 3J = 6.7 Hz, 1.1H, GW7604NHCH2, E isomer), 3.27 (t, 3J = 6.7 Hz, 0.9H, GW7604NHCH2, Z isomer), 3.79 (s, 1.6H, OCH3, E isomer), 3.80 (s, 1.4H, OCH3, Z isomer), 6.38–6.45 (m, 1.5H, ArH, Z isomer + CH=CHCONH, E isomer), 6.58 (d, 3J = 15.8 Hz, 0.5H, CH=CHCONH, Z isomer), 6.66 (d, 3J = 8.4 Hz, 0.9H, ArH, Z isomer), 6.78 (d, 3J = 8.3 Hz, 1.1H, ArH, E isomer), 6.81–6.86 (m, 1H, ArH), 6.87 (d, 3J = 8.1 Hz, 1.1H, ArH, E isomer), 6.98–7.02 (m, 1H, ArH), 7.04 (d, 3J = 8.3 Hz, 1.1H, ArH, E isomer), 7.09–7.19 (m, 6.1H, ArH), 7.24 (d, 3J = 7.9 Hz, 0.9H, ArH, Z isomer), 7.32–7.40 (m, 1.6H, ArH + CH=CHCONH, E isomer), 7.51–7.57 (m, 1.4H, ArH + CH=CHCONH, Z isomer). 13C NMR: (700 MHz, CD3OD, E/Z = 55:45): δ 13.81 E isomer, 13.86 Z isomer, 25.54 E isomer, 29.85 Z isomer, 29.99 E isomer, 30.16 E isomer, 30.20 Z isomer, 34.71, 37.81, 37.86 E isomer, 37.89 Z isomer, 56.21, 97.96, 113.10, 115.28 Z isomer, 116.03 E isomer, 116.21, 121.04 E isomer, 121.50 Z isomer, 127.22 Z isomer, 127.37 E isomer, 127.85 E isomer, 128.67 Z isomer, 128.93 Z isomer, 129.00 E isomer, 130.84 Z isomer, 130.86 E isomer, 131.11 Z isomer, 131.67 E isomer, 132.44 E isomer, 133.04 Z isomer, 133.54 E isomer, 134.62 Z isomer, 135.29 Z isomer, 135.60 E isomer, 139.54 Z isomer, 139.68 E isomer, 141.47, 142.86 Z isomer, 143.65 E isomer, 143.67 Z isomer, 143.92 E isomer, 146.80 E isomer, 147.05 Z isomer, 154.95 E isomer, 154.97 Z isomer, 156.67 Z isomer, 157.57 E isomer, 158.03, 168.80, 174.05 Z isomer, 174.08 E isomer. HR-MS (m/z): calcd for C39H40N4O4 [M + H]+: 629.3122, found: 629.3115.
4.1.3.2.2.3. (E)-3-[4-((E/Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-enyl)phenyl]-N-[4-(3-(5-methoxy-1H-benzo[d]imidazol-2-yl)propanamido)butyl]acrylamide (34)
34 was synthesized according to the general procedure using 50 mg of GW7604 (0.14 mmol) in 1.3 mL of dry DMF, 0.14 mL of DIPEA (0.81 mmol), and 84 mg of PyBOP (1.2 equiv, 0.13 mmol) in 1 mL of dry DCM. 28 (70 mg, 0.14 mmol) in 1 mL of dry DMF was added dropwise. Purification by column chromatography with DCM and MeOH (95:5) and subsequent crystallization from MeOH/water provided 34 as a peach-colored powder (43 mg, 0.07 mmol, 51.8%). Purity: 96.4% (HPLC: 1.0 mM in ACN + water (Na2SO4, 20 mM (pH 3)), 254 nm). 1H NMR: (600 MHz, CD3OD, E/Z = 55:45): δ 0.92 (t, 3J = 7.3 Hz, 3H, CH2CH3), 1.44–1.49 (m, 2H, CH2), 1.49–1.55 (m, 2H, CH2), 2.47 (q, 3J = 7.4 Hz, 0.9H, CH2CH3, Z isomer), 2.52 (q, 3J = 7.4 Hz, 1.1H, CH2CH3, E isomer), 2.63–2.76 (2 × t, 2H, 2 × CH2CH2CONH), 3.10–3.16 (2 × t, 3J = 7.5 Hz, 2H, 2 × CH2CH2CONH), 3.18 (t, 3J = 6.2 Hz, 1.1H, GW7604NHCH2CH2CH2CH2NH, E isomer) 3.19–3.23 (m, 2H, CH2), 3.26 (t, 3J = 6.3 Hz, 0.9H GW7604NHCH2, Z isomer), 3.78 (s, 1.6H, OCH3, E isomer), 3.80 (s, 1.4H, OCH3, Z isomer), 6.38–6.45 (m, 1.5H, ArH + CH=CHCONH), 6.58 (d, 3J = 15.8 Hz, 0.5H, CH=CHCONH, Z isomer), 6.66 (d, 3J = 8.7 Hz, 0.9H, ArH, Z isomer), 6.78 (d, 3J = 8.6 Hz, 1.1H, ArH, E isomer), 6.80–6.84 (2 × dd, 1H, ArH), 6.87 (d, 3J = 8.3 Hz, 1.1H, ArH, E isomer), 6.97–7.02 (m, 1H, ArH), 7.04 (d, 3J = 8.6 Hz, 1.1H, ArH, E isomer), 7.07–7.20 (m, 6.1H, ArH), 7.24 (d, 3J = 8.2 Hz, 0.9H, ArH, Z isomer), 7.31–7.39 (m, 1.6H, ArH + CH=CHCONH), 7.50–7.57 (m, 1.4H, ArH + CH=CHCONH). 13C NMR: (151 MHz, CD3OD, E/Z = 55:45): δ 13.81, 13.87, 25.77, 27.68, 27.72, 27.75, 27.80, 29.85, 29.99, 34.86, 40.00, 40.03, 40.13, 40.19, 56.20, 98.04, 112.75, 115.29, 116.04, 116.41, 121.11, 121.58, 127.22, 127.36, 127.83, 128.66, 128.93, 129.00, 129.87, 130.84, 130.86, 131.11, 131.67, 132.44, 133.04, 133.58, 134.66, 135.30, 135.61, 139.54, 139.69, 141.37, 142.85, 143.65, 143.68, 143.90, 146.76, 147.01, 155.04, 156.67, 157.56, 157.83, 168.70, 168.72, 174.05, 174.07. HR-MS (m/z): calcd for C40H42N4O4 [M + H]+: 643.3279, found: 643.3271.
4.1.3.2.2.4. (E)-3-[4-((E/Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-enyl)phenyl]-N-[5-(3-(5-methoxy-1H-benzo[d]imidazol-2-yl)propanamido)pentyl]acrylamide (36)
36 was synthesized according to the general procedure for heterodimer formation using 77 mg of GW7604 (0.21 mmol) in 2 mL of dry DMF, 0.22 mL of DIPEA (1.24 mmol), and 118 mg of PyBOP (0.23 mmol) in 1.5 mL of dry DCM. 29 (110 mg, 0.21 mmol) in 1 mL of dry DMF was added dropwise. Purification by column chromatography with a DCM and MeOH gradient (95:5 → 93:7) led to 36 as orange, sparkling crystals (70 mg, 0.11 mmol, 52%). Purity: 98.5% (HPLC: 1.0 mM in ACN + water (Na2SO4, 20 mM (pH 3)), 254 nm and 281 nm). 1H NMR: (500 MHz, CD3OD, E/Z = 50:50): δ 0.83 – 0.99 (2 × t, 3H, 2 × CH2CH3), 1.26–1.37 (m, 2H, NHCH2CH2CH2CH2CH2NH), 1.45–1.55 (m, 4H, NHCH2CH2CH2CH2CH2NH), 2.44–2.48 (m, 1H, CH2CH3, Z isomer), 2.52 (d, 3J = 7.4 Hz, 1H, CH2CH3, E isomer), 2.66–2.75 (2 × t, 2H, 2 × CH2CH2CONH), 3.10–3.23 (m, 5H, CH2), 3.26 (t, 3J = 7.0 Hz, 1H, GW7604NHCH2, Z isomer), 3.74–3.88 (2 × s, 3H, 2 × OCH3), 6.37–6.47 (m, 1.5H, ArH + CH=CHCONH), 6.60 (d, 3J = 15.8 Hz, 0.5H, CH=CHCONH, Z isomer), 6.62–6.71 (m, 1H, ArH, Z isomer), 6.79 (d, 3J = 8.5 Hz, 1H, ArH, E isomer), 6.82–6.89 (m, 2H, ArH), 6.97–7.02 (m, 1H, ArH), 7.03 (d, 3J = 8.4 Hz, 1H, ArH, E isomer), 7.08–7.18 (m, 6H, ArH), 7.20–7.25 (m, 1H, ArH), 7.32–7.40 (m, 1.5H, ArH + CH=CHCONH), 7.49–7.57 (m, 1.5H, ArH + CH=CHCONH). 13C NMR: (126 MHz, CD3OD, E/Z = 50:50): δ 13.81, 13.86, 25.10, 25.13, 25.75, 25.78, 29.85, 29.93, 29.95, 30.01, 30.05, 34.85, 34.88, 40.22, 40.30, 40.36, 56.21, 98.06, 112.87, 115.27, 116.02, 116.18, 121.12, 121.58, 127.21, 127.35, 127.80, 128.62, 128.92, 128.98, 130.83, 130.85, 131.09, 131.67, 132.43, 133.04, 133.55, 134.63, 135.28, 135.60, 139.53, 139.67, 141.33, 142.82, 143.63, 143.67, 143.88, 146.72, 146.97, 154.99, 156.66, 157.55, 157.90, 168.70, 173.96. HR-MS (m/z): calcd for C41H44N4O4 [M + H]+: 657.3435, found: 657.3462.
4.1.3.2.2.5. (E)-3-[4-((E/Z)-1-(4-Hydroxyphenyl)-2-phenylbut-1-enyl)phenyl]-N-[6-(3-(5-methoxy-1H-benzo[d]imidazol-2-yl)propanamido)hexyl]acrylamide (38)
38 was synthesized according to the general procedure for heterodimer formation using 139 mg of GW7604 (0.38 mmol) in 2 mL of dry DMF, 0.39 mL of DIPEA (2.26 mmol), and 215 mg of PyBOP (0.41 mmol) in 2 mL of dry DCM. 30 (200 mg, 0.38 mmol) in 1 mL of dry DMF was added dropwise. Purification by column chromatography with a gradient of DCM and MeOH (96:4 → 95:5) afforded 38 as a light-yellow powder (139 mg, 0.21 mmol, 55%). Purity: 99.0% (HPLC: 1.0 mM in ACN + water (Na2SO4, 20 mM (pH 3)), 254 nm). 1H NMR: (500 MHz, CD3OD, E/Z = 50:50): δ 0.93 (t, 3J = 7.4 Hz, 3H, CH2CH3), 1.20–1.39 (m, 4H, CH2), 1.39–1.56 (m, 4H, CH2), 2.48 (q, 3J = 7.3 Hz, 1H, CH2CH3, Z isomer), 2.52 (q, 3J = 7.5 Hz, 1H, CH2CH3, E isomer), 2.71 (2 × t, 2H, 2 × CH2CH2CONH), 3.09–3.19 (m, 4H, CH2NH + CH2CH2CONH), 3.22 (t, 3J = 7.0 Hz, 1H, GW7604NHCH2, E isomer), 3.27 (t, 3J = 7.1 Hz, 1H, GW7604NHCH2, Z isomer), 3.81–3.85 (2 × s, 3H, 2 × OCH3), 6.41–6.45 (m, 1.5H, ArH + CH=CHCONH), 6.61 (d, 3J = 15.8 Hz, 0.5H, CH=CHCONH, Z isomer), 6.67 (d, 3J = 8.7 Hz, 1H, ArH, Z isomer), 6.79 (d, 3J = 8.6 Hz, 1H, ArH, E isomer), 6.82–6.85 (2 × dd, 1H, ArH), 6.88 (d, 3J = 8.2 Hz, 1H, ArH, E isomer), 6.98–7.02 (m, 1H, ArH), 7.04 (d, 3J = 8.6 Hz, 1H, ArH, E isomer), 7.07–7.20 (m, 6H, ArH), 7.25 (d, 3J = 8.1 Hz, 1H, ArH, Z isomer), 7.33–7.38 (m, 1.5H, ArH + CH=CHCONH), 7.52–7.55 (m, 1.5H, ArH + CH=CHCONH). 13C NMR: (126 MHz, CD3OD, E/Z = 50:50): δ 13.81, 13.87, 25.78, 27.43, 27.46, 27.57, 27.60, 29.86, 30.00, 30.26, 30.31, 34.86, 40.24, 40.39, 40.45, 56.21, 98.02, 112.79, 115.27, 116.02, 121.16, 121.62, 127.21, 127.35, 127.81, 128.63, 128.92, 128.99, 130.83, 130.85, 131.09, 131.66, 132.43, 133.03, 133.59, 134.67, 135.28, 135.60, 139.54, 139.68, 141.29, 142.83, 143.63, 143.66, 143.88, 146.72, 146.97, 154.99, 156.66, 157.55, 157.86, 168.65, 173.94. HR-MS (m/z): calcd for C42H46N4O4 [M + H]+: 671.3592, found: 671.3588.
4.1.3.2.3. Syntheses of the 5-Hydroxybenzimidazoles 39–43
General Procedure for Methyl Ether Cleavage:
The respective methoxy-protected GW7604-benzimidazole derivative (1 equiv) was dissolved in dry DCM or dry chlorobenzene. The suspension was cooled to −78 °C, and a BBr3 solution in DCM (1 M, 3.5 equiv for one methoxy group, 6–7 equiv for two methoxy groups) was added dropwise. The solution was further stirred at 0–25 °C for 3–24 h. MeOH (10 mL) was added to stop the reaction, stirred for 30 min, and concentrated under reduced pressure. The residue was neutralized with 6 N NaOH (pH = 8–9), and EA was added. The aqueous phase was extracted three times, and the combined organic phases were dried over anhydrous Na2SO4, evaporated to dryness, and further purified by column chromatography.
4.1.3.2.3.1. (E)-N-[2-(3-(5-Hydroxy-1H-benzo[d]imidazol-2-yl)propanamido)ethyl]-3-[4-((E/Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-enyl)phenyl]acrylamide (39)
39 was synthesized according to the general procedure applying 93 mg of 31 (0.15 mmol) in 5 mL of dry chlorobenzene and 0.52 mL of a BBr3 solution (0.52 mmol) at ambient temperature. The reaction time was 24 h. Purification was performed by column chromatography with DCM and MeOH (9:1), affording 39 as a white-yellowish powder (30 mg, 0.05 mmol, 42%). Purity: 95.0% (HPLC: 1.0 mM in MeOH, 254 nm). 1H NMR: (500 MHz, CD3OD, E/Z = 50:50): δ 0.92 (t, 3J = 7.2 Hz, 3H, CH2CH3), 2.47 (q, 3J = 7.3 Hz, 1H, CH2CH3, Z isomer), 2.51 (q, 3J = 7.3 Hz, 1H, CH2CH3, E isomer), 2.65–2.76 (2 × t, 2H, 2 × CH2CH2CONH), 3.07–3.16 (2 × t, 2H, 2 × CH2CH2CONH), 3.33–3.46 (m, 4H, NHCH2CH2NH), 6.37 (d, 3J = 15.7 Hz, 0.5H, CH=CHCONH, E isomer), 6.42 (d, 3J = 8.0 Hz, 1H, ArH, Z isomer), 6.53 (d, 3J = 15.7 Hz, 0.5H, CH=CHCONH, Z isomer), 6.66 (d, 3J = 8.1 Hz, 1H, ArH, Z isomer), 6.69–6.75 (m, 1H, ArH), 6.78 (d, 3J = 7.9 Hz, 1H, ArH, E isomer), 6.82–6.92 (m, 2H, ArH), 7.04 (d, 3J = 8.1 Hz, 1H, ArH, E isomer), 7.05–7.22 (m, 6H, ArH), 7.24 (d, 3J = 7.7 Hz, 1H, ArH, Z isomer), 7.26–7.32 (m, 1H, ArH), 7.34 (d, 3J = 15.7 Hz, 0.5H, CH=CHCONH, E isomer), 7.46–7.57 (m, 1.5H, ArH + CH=CHCONH). 13C NMR: (126 MHz, CD3OD, E/Z = 50:50): δ 13.85, 13.90, 25.54, 29.85, 29.99, 34.70, 40.16, 99.98, 112.77, 115.26, 116.01, 116.22, 120.88, 121.35, 127.19, 127.33, 127.85, 128.67, 128.92, 128.97, 130.81, 131.08, 131.66, 132.42, 133.04, 133.40, 133.95, 134.47, 135.24, 135.54, 139.45, 139.60, 141.61, 142.79, 143.56, 143.60, 143.85, 146.77, 147.01, 154.68, 156.60, 157.49, 168.98, 169.00, 174.49, 174.53. HR-MS (m/z): calcd for C37H36N4O4 [M + H]+: 601.2809, found: 601.2809.
4.1.3.2.3.2. (E)-N-[3-(3-(5-Hydroxy-1H-benzo[d]imidazol-2-yl)propanamido)propyl]-3-[4-((E/Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-enyl)phenyl]acrylamide (40)
Synthesis of 40 was carried out according to the general procedure applying 68 mg of 33 (0.11 mmol) dissolved in 3.6 mL of dry DCM and 0.71 mL of BBr3 solution (0.71 mmol) at 0 °C. The reaction time was 2 h. Purification was performed by column chromatography with DCM and MeOH (9:1), affording 40 as a white-yellowish powder (58 mg, 0.09 mmol, 81%). Purity: 99.7% (HPLC: 1.0 mM in ACN + water (Na2SO4, 20 mM (pH 3)), 254 nm). 1H NMR: (700 MHz, CD3OD, E/Z = 50:50): δ 0.92 (t, 3J = 7.4 Hz, 3H, CH2CH3), 1.66 (p, 3J = 6.8 Hz, 1H, NHCH2CH2CH2NH, E isomer), 1.71 (p, 3J = 6.8 Hz, 1H, NHCH2CH2CH2NH, Z isomer), 2.47 (q, 3J = 7.4 Hz, 1H, CH2CH3, Z isomer), 2.52 (q, 3J = 7.4 Hz, 1H, CH2CH3, E isomer), 2.68–2.74 (2 × t, 2H, 2 × CH2CH2CONH), 3.11 (t, 3J = 7.5 Hz, 1H, CH2CH2CONH, E isomer), 3.13 (t, 3J = 7.5 Hz, 1H, CH2CH2CONH, Z isomer), 3.19–3.23 (2 × t, 2H, 2 × GW7604NHCH2CH2CH2NH), 3.24 (t, 3J = 6.9 Hz, 1H, GW7604NHCH2, E isomer), 3.27 (t, 3J = 6.9 Hz, 1H, GW7604NHCH2, Z isomer), 6.40–6.43 (m, 1.5H, ArH + CH=CHCONH), 6.59 (d, 3J = 15.8 Hz, 0.5H, CH=CHCONH, Z isomer), 6.66 (d, 3J = 8.7 Hz, 1H, ArH, Z isomer), 6.69–6.74 (2 × dd, 1H, ArH), 6.78 (d, 3J = 8.5 Hz, 1H, ArH, E isomer), 6.83–6.91 (m, 2H, ArH), 7.04 (d, 3J = 8.5 Hz, 1.1H, ArH, E isomer), 7.07–7.21 (m, 6H, ArH), 7.24 (d, 3J = 8.1 Hz, 1H, ArH, Z isomer), 7.27–7.32 (m, 1H, ArH), 7.35 (d, 3J = 15.7 Hz, 0.5H, CH=CHCONH, E isomer), 7.52–7.58 (m, 1.5H, ArH + CH=CHCONH). 13C NMR: (176 MHz, CD3OD, E/Z = 50:50): δ 13.81, 13.86, 25.72, 29.85, 30.00, 30.18, 30.22, 34.91, 37.83, 37.85, 37.87, 37.92, 112.71, 115.29, 116.04, 121.06, 121.52, 127.22, 127.37, 127.86, 128.68, 128.93, 129.00, 130.85, 130.87, 131.11, 131.68, 132.45, 133.05, 133.56, 134.64, 135.30, 135.61, 139.55, 139.70, 141.48, 142.86, 143.65, 143.69, 143.91, 146.79, 147.04, 154.67, 154.75, 156.68, 157.57, 168.83, 174.26. HR-MS (m/z): calcd for C38H38N4O4 [M + H]+: 615.2966, found: 615.2960.
4.1.3.2.3.3. (E)-N-[4-(3-(5-Hydroxy-1H-benzo[d]imidazol-2-yl)propanamido)butyl]-3-[4-((E/Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-enyl)phenyl]acrylamide (41)
41 was synthesized according to the general procedure with 112 mg of 35 (0.17 mmol) dissolved in 5.5 mL of dry DCM and 1.02 mL of BBr3 solution (1.02 mmol) at 0 °C. The reaction time was 4 h. Purification was achieved by column chromatography with DCM and MeOH (9:1), affording 41 as a white-yellowish powder (48 mg, 0.076 mmol, 45%). Purity: 98.3% (HPLC: 1.0 mM in ACN + water (Na2SO4, 20 mM (pH 3)), 283 nm). 1H NMR: (600 MHz, CD3OD, E/Z = 50:50): δ 0.92 (t, 3J = 7.4 Hz, 3H, CH2CH3), 1.44–1.50 (m, 2H, CH2), 1.44–1.50 (m, 2H, CH2), 1.50–1.53 (m, 2H, CH2), 2.47 (q, 3J = 7.4 Hz, 1H, CH2CH3, Z isomer), 2.51 (q, 3J = 7.4 Hz, 1H, CH2CH3, E isomer), 2.68 (t, 3J = 7.6 Hz, 1H, CH2CH2CONH), 2.70 (t, 3J = 7.6 Hz, 1H, CH2CH2CONH), 3.08–3.14 (2 × t, 2H, 2 × CH2CH2CONH), 3.17 (t, 3J = 6.4 Hz, 1H, GW7604NHCH2CH2CH2CH2NH, E isomer), 3.19–3.23 (m, 2H, CH2), 3.26 (t, 3J = 6.4 Hz, 1H, GW7604NHCH2, Z isomer), 6.37–6.46 (m, 1.5H, ArH + CH=CHCONH), 6.59 (d, 3J = 15.8 Hz, 0.5H, CH=CHCONH, Z isomer), 6.66 (d, 3J = 8.8 Hz, 1H, ArH, Z isomer), 6.70–6.76 (2 × dd, 1H, ArH), 6.78 (d, 3J = 8.6 Hz, 1H, ArH, E isomer), 6.84–6.91 (m, 2H, ArH), 7.03 (d, 3J = 8.6 Hz, 1H, ArH, E isomer), 7.06–7.21 (m, 6H, ArH), 7.24 (d, 3J = 8.2 Hz, 1H, ArH, Z isomer), 7.26–7.32 (2 × d, 1H, ArH), 7.35 (d, 3J = 15.7 Hz, 0.5H, CH=CHCONH, E isomer), 7.52–7.57 (m, 1.5H, ArH + CH=CHCONH). 13C NMR: (151 MHz, CD3OD, E/Z = 50:50): δ 13.81, 13.87, 25.70, 25.71, 27.69, 27.73, 27.78, 29.85, 29.99, 34.84, 34.85, 40.00, 40.02, 40.11, 40.17, 99.97, 112.83, 115.27, 116.02, 116.23, 121.08, 121.54, 127.21, 127.36, 127.84, 128.66, 128.93, 129.00, 130.83, 130.86, 131.10, 131.67, 132.44, 133.04, 133.56, 134.64, 135.28, 135.59, 139.53, 139.53, 139.67, 141.40, 142.84, 143.63, 143.67, 143.89, 146.76, 147.01, 154.69, 154.76, 156.66, 157.56, 168.71, 168.73, 174.03, 174.05. HR–zMS (m/z): calcd for C39H40N4O4 [M + H]+: 629.3050, found: 629.3098.
4.1.3.2.3.4. (E)-N-[5-(3-(5-Hydroxy-1H-benzo[d]imidazol-2-yl)propanamido)pentyl]-3-[4-((E/Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-enyl)phenyl]acrylamide (42)
42 was synthesized according to the general procedure with 61 mg of 37 (0.091 mmol) dissolved in 3.5 mL of dry DCM and 0.64 mL of BBr3 solution (0.64 mmol) at 0 °C. The reaction time was 4.5 h. Purification was achieved by column chromatography with DCM and MeOH (9:1) followed by crystallization from MeOH/water. 42 was obtained as a white powder (12 mg, 0.019 mmol, 21%). Purity: 98.6% (HPLC: 1.0 mM in ACN + water (Na2SO4, 20 mM (pH 3)), 283 nm). 1H NMR: (500 MHz, CD3OD, E/Z = 50:50): δ 0.83–0.99 (2 × t, 3J = 7.4 Hz, 3H, 2 × CH2CH3), 1.23–1.36 (m, 2H, NHCH2CH2CH2CH2CH2NH), 1.40–1.59 (m, 4H, NHCH2CH2CH2CH2CH2NH), 2.46 (q, 3J = 7.4 Hz, 1H, CH2CH3, Z isomer), 2.51 (q, 3J = 7.4 Hz, 1H, CH2CH3, E isomer), 2.62–2.79 (2 × t, 2H, 2 × CH2CH2CONH), 3.07–3.19 (m, 4H, CH2CH2CONH + GW7604NHCH2CH2CH2CH2CH2NH), 3.21 (t, 3J = 7.1 Hz, 1H, GW7604NHCH2, E isomer), 3.26 (t, 3J = 7.0 Hz, GW7604NHCH2, Z isomer), 6.37–6.47 (m, 1.5H, ArH + CH=CHCONH), 6.60 (d, 3J = 15.7 Hz, 0.5H, CH=CHCONH, Z isomer), 6.65 (d, 3J = 8.6 Hz, 1H, ArH, Z isomer), 6.72–6.82 (m, 2H, ArH), 6.85 (d, 3J = 8.3 Hz, 1H, ArH, E isomer), 6.87–6.92 (m, 1H, ArH), 7.03 (d, 3J = 8.5 Hz, 1H, ArH, E isomer), 7.05–7.19 (m, 6H, ArH), 7.22 (d, 3J = 8.2 Hz, 1H, ArH, Z isomer), 7.27–7.39 (m, 1.5H, ArH + CH=CHCONH), 7.47–7.58 (m, 1.5H, ArH + CH=CHCONH). 13C NMR: (126 MHz, CD3OD, E/Z = 50:50): δ 13.87, 13.92, 25.20, 25.57, 29.90, 30.04, 30.07, 34.72, 40.29, 40.37, 99.95, 113.37, 115.32, 116.07, 116.14, 121.15, 121.62, 127.26, 127.40, 127.86, 128.68, 128.97, 129.03, 130.89, 131.13, 131.71, 132.47, 133.08, 133.58, 134.66, 135.32, 135.64, 139.70, 141.39, 142.87, 143.66, 143,70, 143.92, 146.77, 147.03, 154.61, 155.17, 156.68, 157.58, 168.76, 173.86. HR-MS (m/z): calcd for C40H42N4O4 [M + H]+: 643.3279, found: 643.3252.
4.1.3.2.3.5. (E)-N-[6-(3-(5-Hydroxy-1H-benzo[d]imidazol-2-yl)propanamido)hexyl]-3-[4-((E/Z)-1-(4-hydroxyphenyl)-2-phenylbut-1-enyl)phenyl]acrylamide (43)
43 was synthesized according to the general procedure with 130 mg of 38 (0.19 mmol) dissolved in 3.9 mL of dry DCM and 0.68 mL of BBr3 solution (0.68 mmol) at 10 °C. The reaction time was 4.5 h. Purification was performed by column chromatography first with EA (100%) and then with a gradient of DCM and MeOH (98:2 → 95:5). 43 was obtained as an off-white powder (15 mg, 0.023 mmol, 12%). Purity: 95.0% (HPLC: 1.0 mM in ACN + water (Na2SO4, 20 mM (pH 3)), 283 nm). 1H NMR: (700 MHz, CD3OD, E/Z = 50:50): δ 0.92 (2 × t, 3J = 7.4 Hz, 3H, 2 × CH2CH3), 1.22–1.34 (m, 4H, CH2), 1.40–1.52 (m, 3H, CH2), 1.50 (p, 3J = 7.7 Hz, 1H, CH2, Z isomer), 2.47 (q, 3J = 7.4 Hz, 1H, CH2CH3, Z isomer), 2.52 (q, 3J = 7.5 Hz, 1H, CH2CH3, E isomer), 2.65–2.72 (2 × t, 2H, 2 × CH2CH2CONH), 3.09–3.17 (m, 4H, CH2NH + CH2CH2CONH), 3.22 (t, 3J = 7.0 Hz, 1H, GW7604NHCH2, E isomer), 3.27 (t, 3J = 7.1 Hz, 1H, GW7604NHCH2, Z isomer), 6.41–6.45 (m, 1.5H, ArH, Z isomer + CH=CHCONH, E isomer), 6.61 (d, 3J = 15.8 Hz, 0.5H, CH=CHCONH, Z isomer), 6.66 (d, 3J = 8.7 Hz, 1H, ArH, Z isomer), 6.72–6.74 (2 × dd, 4J = 2.3 Hz, 1H, ArH), 6.78 (d, 3J = 8.5 Hz, 1H, ArH, E isomer), 6.84–6.90 (m, 2H, ArH), 7.03 (d, 3J = 8.6 Hz, 1H, ArH, E isomer), 7.07–7.20 (m, 6H, ArH), 7.24 (d, 3J = 8.2 Hz, 1H, ArH, Z isomer), 7.29–7.31 (m, 1H, ArH), 7.35 (d, 3J = 15.7 Hz, 0.5H, CH=CHCONH, E isomer), 7.53–7.56 (m, 1.5H, ArH + CH=CHCONH, Z isomer). 13C NMR: (176 MHz, CD3OD, E/Z = 1:1): δ 13.81 E isomer, 13.86 Z isomer, 25.75, 27.46 E isomer, 27.48 Z isomer, 27.59 E isomer, 27.62 Z isomer, 29.85 Z isomer, 30.00 E isomer, 30.24 E isomer, 30.27 Z isomer, 30.29 E isomer, 30.33 Z isomer, 34.89, 40.25 E isomer, 40.26 Z isomer, 40.42 Z isomer, 40.48 E isomer, 100.05, 112.85, 115.28 Z isomer, 116.03 E isomer, 116.14, 121.16 E isomer, 121.63 Z isomer, 127.21 Z isomer, 127.37 E isomer, 127.83 E isomer, 128.65 Z isomer, 128.93 Z isomer, 129.00 E isomer, 130.84 Z isomer, 130.86 E isomer, 131.10 Z isomer, 131.67 E isomer, 132.44 E isomer, 133.04 Z isomer, 133.60, 134.68 Z isomer, 135.30 Z isomer, 135.61 E isomer, 139.55 Z isomer, 139.69 E isomer, 141.32, 142.84 Z isomer, 143.65 Z isomer, 143.68 E isomer, 143.89 E isomer, 146.73 E isomer, 146.99 Z isomer, 147.93, 154.68, 154.75, 156.67 Z isomer, 157.56 E isomer, 168.70, 173.98. HR-MS (m/z): calcd for C41H44N4O4 [M + H]+: 657.3435, found: 657.3438.
4.2. Cell Culture Experiments
4.2.1. General
The human osteosarcoma cell line U2OS, the hormone-dependent breast cancer cell line MCF-7, the ER-negative breast cancer cell line MDA-MB-231, and the African green monkey kidney cell line COS-7 were obtained from the cell line service (CLS, Eppelheim, Germany). The MCF-7TamR cell line was generated and kindly provided by Cardiff University (Great Britain). The cells were maintained as monolayer cultures. McCoy’s 5A medium supplemented with 10% fetal bovine serum (FBS) (both from Biochrome GmbH, Berlin, Germany) was used for the osteosarcoma cell line, and Dulbecco’s modified Eagle’s medium (DMEM) without phenol red, with glucose (4.5 g/L) (GE Healthcare, Pasching, Austria), supplemented with 10% FBS and 1% pyruvate (GE Healthcare) was used for the breast cancer cell lines. The COS-7 cell line was maintained with DMEM supplemented with 10% FBS. MCF-7TamR were cultured in RPMI medium supplemented with 5% FBS, L-glutamine (4 mM), and 4-OHT (10–7 M). All cell lines were cultivated in a humidified atmosphere (5% CO2/95% air) at 37 °C and passaged twice a week. DMSO was used as the solvent for the investigated compounds. The final concentration of DMSO never exceeded 0.1% in cell-based assays. Vehicle-treated controls were always included.
4.2.2. Binding Assays
LanthaScreen TR-FRET ER alpha/beta competitive binding assays (Invitrogen, Carlsbad) were used to investigate the binding affinity to the isolated receptors according to the manufacturer′s protocols. Measurement was performed with an Enspire multimode plate reader (PerkinElmer Life Sciences, Waltham). Calculations were performed with Excel (Microsoft, Redmond) and GraphPad Prism (GraphPad Software, San Diego).
4.2.3. Cellular Uptake
The determination of the cellular uptake in MCF-7 and COS-7 cells was performed as already described.24 Measurement was conducted with the Enspire multimode plate reader. The values represent the mean ± SD of ≥3 independent experiments.
4.2.4. Luciferase Reporter Gene Assay
Transactivation evaluation with respect to ERα/β was performed essentially as previously described.24 The transfection reagent (TansIT-LT1, MoBiTec) and the dual-luciferase reporter assay (Promega) were applied according to the manufacturer’s protocol. Renilla luciferase activity was used as the internal control and for normalization. The values represent the mean ± SD of ≥3 independent experiments.
4.2.5. In-Cell Western Immunoassay
Degradation was evaluated with the CellTagTM 700 In-Cell Western kit (LI-COR, Lincoln) according to the manufacturer’s protocol. MCF-7 cells were treated, and calculation was performed as previously described.24
4.2.6. Crystal Violet Assay
The cytotoxicity evaluation was performed using MCF-7, MCF-7TamR49 (2 × 103 cells per well), and MDA-MB-231 cell lines (1.5 × 103 cells per well) according to a modified protocol previously described.48 Cells were seeded in 96-well microtiter plates in DMEM supplemented with 10% charcoal dextran-treated FBS and 1% pyruvate. Twenty-four hours after seeding, the selected compounds, controls, and the vehicle (DMSO) were added at indicated concentrations in quadruples. After an incubation time of 120 h in a humidified atmosphere (5% CO2/95% air) at 37 °C, the medium was aspirated, and cells were washed with PBS (GE Healthcare) and fixed with a solution of 1% (v/v) glutaric dialdehyde in PBS. The cell biomass was determined via staining of adherent cells with crystal violet, extraction of the stain with ethanol (70%, v/v), and measurement of absorbance at 590 nm. Cell viability is expressed as the percentage of cell viability of the vehicle-treated control, which was set to 100%. Results are the mean ± SD of ≥3 independent experiments.
Acknowledgments
The authors gratefully acknowledge the Cardiff University School of Pharmacy and Pharmaceutical Sciences for providing the tamoxifen-resistant MCF-7TamR cells. We thank Christoph Kreutz (Institute of Organic Chemistry, University of Innsbruck) and Peter Schneider (Institute of Pharmacognosy, University of Innsbruck) for recording the 1H and 13C NMR spectra. We also thank Inte:Ligand GmbH for providing an academic license of LigandScout. D.S. is an Ingeborg Hochmair professor of the University of Innsbruck. The Austrian Research Promotion Agency FFG [West Austrian BioNMR 858017] is also kindly acknowledged.
Glossary
Abbreviations
- 4-OHT
4-hydroxytamoxifen
- ACN
acetonitrile
- AF2
activation function 2
- anh.
anhydrous
- Boc
tert-butyloxycarbonyl
- CABS
coactivator binding site
- DCM
dichloromethane
- DIPEA
diisopropylamine
- DMEM
Dulbecco′s modified Eagle’s medium
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- E2
estradiol
- EA
ethyl acetate
- equiv
equivalent
- ER
estrogen receptor
- ESR1
ERα gene alterations
- EtOH
ethanol
- FBS
fetal bovine serum
- GW7604
(E/Z)-3-(4-((E)-1-(4-hydroxyphenyl)-2-phenylbut-1-enyl)phenyl)acrylic acid
- H12
Helix 12
- HPLC
high-performance liquid chromatography
- HR-MS
high-resolution mass spectrometry
- LBD
ligand binding domain
- LBS
ligand binding site
- MeOH
methanol
- NMR
nuclear magnetic resonance spectra
- PBS
phosphate-buffered saline
- PE
petroleum ether
- PGC-1
peroxisome proliferator-activated receptor-γ coactivator 1
- PPARγ
peroxisome proliferator-activated receptor γ
- PyBOP
benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate
- RBA
relative binding affinity
- SAR
structure–activity relationship
- SD
standard deviation
- SE
standard error
- SERDs
selective ER degraders/downregulators
- SERMs
selective ER modulators
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TMS
tetramethylsilane
- TR-FRET
time-resolved fluorescence resonance energy transfer
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c02230.
Author Contributions
A.K.: conceptualization, data curation, formal analysis, investigation, methodology, writing of original draft, writing the review. C.K.: data curation, formal analysis, investigation, methodology, writing of original draft, writing, including review and editing. D.S.: resources, software, investigation. F.G.: investigation. R.G.: conceptualization, supervision, writing, including review and editing.
The authors declare no competing financial interest.
Supplementary Material
References
- Fitzmaurice C.; Abate D.; Abbasi N.; et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 1990 to 2017: a systematic analysis for the global burden of disease study. JAMA Oncol. 2019, 5, 1749–1768. 10.1001/jamaoncol.2019.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson B. E.; Ross R. K.; Pike M. C.; Casagrande J. T. Endogenous hormones as a major factor in human cancer. Cancer Res. 1982, 42, 3232–3239. [PubMed] [Google Scholar]
- Bai Z.; Gust R. Breast cancer, estrogen receptor and ligands. Arch. Pharm. Chem. Life Sci. 2009, 342, 133–149. 10.1002/ardp.200800174. [DOI] [PubMed] [Google Scholar]
- Jordan V. C.; O’Malley B. W. Selective estrogen-receptor modulators and antihormonal resistance in breast cancer. J. Clin. Oncol. 2007, 25, 5815–5824. 10.1200/JCO.2007.11.3886. [DOI] [PubMed] [Google Scholar]
- Musgrove E. A.; Sutherland R. L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- Razavi P.; Chang M. T.; Xu G.; Bandlamudi C.; Ross D. S.; Vasan N.; Cai Y.; Bielski C. M.; Donoghue M. T. A.; Jonsson P.; Penson A.; Shen R.; Pareja F.; Kundra R.; Middha S.; Cheng M. L.; Zehir A.; Kandoth C.; Patel R.; Huberman K.; Smyth L. M.; Jhaveri K.; Modi S.; Traina T. A.; Dang C.; Zhang W.; Weigelt B.; Li B. T.; Ladanyi M.; Hyman D. M.; Schultz N.; Robson M. E.; Hudis C.; Brogi E.; Viale A.; Norton L.; Dickler M. N.; Berger M. F.; Iacobuzio-Donahue C. A.; Chandarlapaty S.; Scaltriti M.; Reis-Filho J. S.; Solit D. B.; Taylor B. S.; Baselga J. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell 2018, 34, 427–438. 10.1016/j.ccell.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Wang Q.; Wang Q.; Cao J.; Sun J.; Zhu Z. Mechanisms of resistance to estrogen receptor modulators in ER+/HER2- advanced breast cancer. Cell. Mol. Life Sci. 2020, 77, 559–572. 10.1007/s00018-019-03281-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeselsohn R.; Yelensky R.; Buchwalter G.; Frampton G.; Meric-Bernstam F.; Gonzalez-Angulo A. M.; Ferrer-Lozano J.; Perez-Fidalgo J. A.; Cristofanilli M.; Gomez H.; Arteaga C. L.; Giltnane J.; Balko J. M.; Cronin M. T.; Jarosz M.; Sun J.; Hawryluk M.; Lipson D.; Otto G.; Ross J. S.; Dvir A.; Soussan-Gutman L.; Wolf I.; Rubinek T.; Gilmore L.; Schnitt S.; Come S. E.; Pusztai L.; Stephens P.; Brown M.; Miller V. A. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res. 2014, 20, 1757–1767. 10.1158/1078-0432.CCR-13-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan P.; Jordan V. C. New insights into acquired endocrine resistance of breast cancer. Cancer Drug Res. 2019, 2, 198–209. 10.20517/cdr.2019.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan V. C. Tamoxifen: a most unlikely pioneering medicine. Nat. Rev. Drug Discovery 2003, 2, 205–213. 10.1038/nrd1031. [DOI] [PubMed] [Google Scholar]
- Shiau A. K.; Barstad D.; Loria P. M.; Cheng L.; Kushner P. J.; Agard D. A.; Greene G. L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. 10.1016/S0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- Pike A. C. W.; Brzozowski A. M.; Walton J.; Hubbard R. E.; Thorsell A.-G.; Li Y.-L.; Gustafsson J.-Å.; Carlquist M. Structural insights into the mode of action of a pure antiestrogen. Structure 2001, 9, 145–153. 10.1016/S0969-2126(01)00568-8. [DOI] [PubMed] [Google Scholar]
- Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 2005, 365, 1687–1717. 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- Adamo V.; Iorfida M.; Montalto E.; Festa V.; Garipoli C.; Scimone A.; Zanghi M.; Caristi N. Overview and new strategies in metastatic breast cancer (MBC) for treatment of tamoxifen-resistant patients. Ann. Oncol. 2007, 18, 53–57. 10.1093/annonc/mdm225. [DOI] [PubMed] [Google Scholar]
- Long X.; Nephew K. P. Fulvestrant (ICI 182,780)-dependent interacting proteins mediate immobilization and degradation of estrogen receptor-α. J. Biol. Chem. 2006, 281, 9607–9615. 10.1074/jbc.M510809200. [DOI] [PubMed] [Google Scholar]
- Fanning S. W.; Hodges-Gallagher L.; Myles D. C.; Sun R.; Fowler C. E.; Plant I. N.; Green B. D.; Harmon C. L.; Greene G. L.; Kushner P. J. Specific stereochemistry of OP-1074 disrupts estrogen receptor alpha helix 12 and confers pure antiestrogenic activity. Nat. Commun. 2018, 9, 2368. 10.1038/s41467-018-04413-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willson T. M.; Henke B. R.; Momtahen T. M.; Charifson P. S.; Batchelor K. W.; Lubahn D. B.; Moore L. B.; Oliver B. B.; Sauls H. R.; et al. 3-[4-(1,2-Diphenylbut-1-enyl)phenyl]acrylic acid: a non-steroidal estrogen with functional selectivity for bone over uterus in rats. J. Med. Chem. 1994, 37, 1550–1552. 10.1021/jm00037a002. [DOI] [PubMed] [Google Scholar]
- Wu Y.-L.; Yang X.; Ren Z.; McDonnell D. P.; Norris J. D.; Willson T. M.; Greene G. L. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol. Cell 2005, 18, 413–424. 10.1016/j.molcel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Bentrem D.; Dardes R.; Liu H.; MacGregor-Schafer J.; Zapf J.; Jordan V. Molecular mechanism of action at estrogen receptor alpha of a new clinically relevant antiestrogen (GW7604) related to tamoxifen. Endocrinology 2001, 142, 838–846. 10.1210/endo.142.2.7932. [DOI] [PubMed] [Google Scholar]
- Lai A.; Kahraman M.; Govek S.; Nagasawa J.; Bonnefous C.; Julien J.; Douglas K.; Sensintaffar J.; Lu N.; Lee K.-J.; Aparicio A.; Kaufman J.; Qian J.; Shao G.; Prudente R.; Moon M. J.; Joseph J. D.; Darimont B.; Brigham D.; Grillot K.; Heyman R.; Rix P. J.; Hager J. H.; Smith N. D. Identification of GDC-0810 (ARN-810), an orally bioavailable selective estrogen receptor degrader (SERD) that demonstrates robust activity in tamoxifen-resistant breast cancer xenografts. J. Med. Chem. 2015, 58, 4888–4904. 10.1021/acs.jmedchem.5b00054. [DOI] [PubMed] [Google Scholar]
- De Savi C.; Bradbury R. H.; Rabow A. A.; Norman R. A.; de Almeida C.; Andrews D. M.; Ballard P.; Buttar D.; Callis R. J.; Currie G. S.; Curwen J. O.; Davies C. D.; Donald C. S.; Feron L. J. L.; Gingell H.; Glossop S. C.; Hayter B. R.; Hussain S.; Karoutchi G.; Lamont S. G.; MacFaul P.; Moss T. A.; Pearson S. E.; Tonge M.; Walker G. E.; Weir H. M.; Wilson Z. Optimization of a novel binding motif to (E)-3-(3,5-difluoro-4-((1R,3R)-2-(2-fluoro-2-methylpropyl)-3-methyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)phenyl)acrylic acid (AZD9496), a potent and orally bioavailable selective estrogen receptor downregulator and antagonist. J. Med. Chem. 2015, 58, 8128–8140. 10.1021/acs.jmedchem.5b00984. [DOI] [PubMed] [Google Scholar]
- Tria G. S.; Abrams T.; Baird J.; Burks H. E.; Firestone B.; Gaither L. A.; Hamann L. G.; He G.; Kirby C. A.; Kim S.; Lombardo F.; Macchi K. J.; McDonnell D. P.; Mishina Y.; Norris J. D.; Nunez J.; Springer C.; Sun Y.; Thomsen N. M.; Wang C.; Wang J.; Yu B.; Tiong-Yip C. L.; Peukert S. Discovery of LSZ102, a potent, orally bioavailable selective estrogen receptor degrader (SERD) for the treatment of estrogen receptor positive breast cancer. J. Med. Chem. 2018, 61, 2837–2864. 10.1021/acs.jmedchem.7b01682. [DOI] [PubMed] [Google Scholar]
- Wardell S. E.; Nelson E. R.; Chao C. A.; Alley H. M.; McDonnell D. P. Evaluation of the pharmacological activities of RAD1901, a selective estrogen receptor degrader. Endocr.-Relat. Cancer 2015, 22, 713–724. 10.1530/ERC-15-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox A.; Kalchschmid C.; Schuster D.; Gaggia F.; Manzl C.; Baecker D.; Gust R. Development of bivalent triarylalkene- and cyclofenil-derived dual estrogen receptor antagonists and downregulators. Eur. J. Med. Chem. 2020, 192, 112191. 10.1016/j.ejmech.2020.112191. [DOI] [PubMed] [Google Scholar]
- Sun A.; Moore T. W.; Gunther J. R.; Kim M.-S.; Rhoden E.; Du Y.; Fu H.; Snyder J. P.; Katzenellenbogen J. A. Discovering small-molecule estrogen receptor α/coactivator binding inhibitors: high-throughput screening, ligand development, and models for enhanced potency. ChemMedChem 2011, 6, 654–666. 10.1002/cmdc.201000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Chirgadze N. Y.; Briggs S. L.; Khan S.; Jensen E. V.; Burris T. P. A second binding site for hydroxytamoxifen within the coactivator-binding groove of estrogen receptor beta. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 9908–9911. 10.1073/pnas.0510596103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egner U.; Heinrich N.; Ruff M.; Gangloff M.; Mueller-Fahrnow A.; Wurtz J.-M. Different ligands–different receptor conformations: modeling of the hERα LBD in complex with agonists and antagonists. Med. Res. Rev. 2001, 21, 523–539. 10.1002/med.1024. [DOI] [PubMed] [Google Scholar]
- Katzenellenbogen J. A. The 2010 Philip S. portoghese medicinal chemistry lectureship: addressing the “core issue” in the design of estrogen receptor ligands. J. Med. Chem. 2011, 54, 5271–5282. 10.1021/jm200801h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibitz N. E.Synthese und Analytik von Benzimidazolderivaten als potentielle ″Nuclear receptor alternate-site modulators″ für den Estrogenrezeptor α. Diploma Thesis, Universität Innsbruck: Innsbruck, 2014. [Google Scholar]
- Obermoser V.; Urban M. E.; Murgueitio M. S.; Wolber G.; Kintscher U.; Gust R. New telmisartan-derived PPARγ agonists: impact of the 3D-binding mode on the pharmacological profile. Eur. J. Med. Chem. 2016, 124, 138–152. 10.1016/j.ejmech.2016.08.027. [DOI] [PubMed] [Google Scholar]
- Obermoser V.; Mauersberger R.; Schuster D.; Czifersky M.; Lipova M.; Siegl M.; Kintscher U.; Gust R. Importance of 5/6-aryl substitution on the pharmacological profile of 4′-((2-propyl-1H-benzo[d]imidazol-1-yl)methyl)-[1,1′-biphenyl]-2-carboxylic acid derived PPARγ agonists. Eur. J. Med. Chem. 2017, 126, 590–603. 10.1016/j.ejmech.2016.11.051. [DOI] [PubMed] [Google Scholar]
- Shan M.Design, synthesis, and evaluation of bivalent estrogen ligands. PhD-Thesis, Freie Universität Berlin: Berlin, 2011. [Google Scholar]
- Lin L.-H.; Lee L.-W.; Sheu S.-Y.; Lin P.-Y. Study on the stevioside analogues of steviolbioside, steviol, and isosteviol 19-alkyl amide dimers: synthesis and cytotoxic and antibacterial activity. Chem. Pharm. Bull. 2004, 52, 1117–1122. 10.1248/cpb.52.1117. [DOI] [PubMed] [Google Scholar]
- Coste J.; Le-Nguyen D.; Castro B. PyBOP: a new peptide coupling reagent devoid of toxic by-product. Tetrahedron Lett. 1990, 31, 205–208. 10.1016/S0040-4039(00)94371-5. [DOI] [Google Scholar]
- Rivero-Buceta E.; Carrero P.; Doyagüez E. G.; Madrona A.; Quesada E.; Camarasa M. J.; Peréz-Pérez M. J.; Leyssen P.; Paeshuyse J.; Balzarini J.; Neyts J.; San-Félix A. Linear and branched alkyl-esters and amides of gallic acid and other (mono-, di- and tri-) hydroxy benzoyl derivatives as promising anti-HCV inhibitors. Eur. J. Med. Chem. 2015, 92, 656–671. 10.1016/j.ejmech.2015.01.033. [DOI] [PubMed] [Google Scholar]
- Vu C. B.; Bemis J. E.; Jirousek M. R.; Milne J. C.; Smith J. J.. Preparation of fatty acid acylated salicylates for the treatment of inflammatory disorders. US20100184730A1, 2010.
- Zeng Q.; Toro A.; Patterson J. B.; Wade W. S.; Zubovics Z.; Yang Y.; Wu Z.. Benzoheterocyclecarboxaldehyde derivatives as IRE-1α inhibitors and their preparation and use for the treatment of diseases. WO2011127070A2, 2011.
- Hack S.; Wörlein B.; Höfner G.; Pabel J.; Wanner K. T. Development of imidazole alkanoic acids as mGAT3 selective GABA uptake inhibitors. Eur. J. Med. Chem. 2011, 46, 1483–1498. 10.1016/j.ejmech.2011.01.042. [DOI] [PubMed] [Google Scholar]
- Katzenellenbogen J. A.; Carlson K. E.; Katzenellenbogen B. S. Facile geometric isomerization of phenolic non-steroidal estrogens and antiestrogens: limitations to the interpretation of experiments characterizing the activity of individual isomers. J. Steroid Biochem. 1985, 22, 589–596. 10.1016/0022-4731(85)90210-9. [DOI] [PubMed] [Google Scholar]
- Malet C.; Spritzer P.; Cumins C.; Guillaumin D.; Mauvais-Jarvis P.; Kuttenn F. Effect of 4-hydroxytamoxifen isomers on growth and ultrastructural aspects of normal human breast epithelial (HBE) cells in culture. J. Steroid Biochem. Mol. Biol. 2002, 82, 289–296. 10.1016/S0960-0760(02)00226-1. [DOI] [PubMed] [Google Scholar]
- Hilt D. C.; Masini-Eteve V.; Fedynec R.; Taravella B.. Chemically stable compositions of 4-hydroxy tamoxifen. WO2005/092310, 2005.
- Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Singh K.; Munuganti R. S.; Leblanc E.; Lin Y. L.; Leung E.; Lallous N.; Butler M.; Cherkasov A.; Rennie P. S. In silico discovery and validation of potent small-molecule inhibitors targeting the activation function 2 site of human oestrogen receptor alpha. Breast Cancer Res. 2015, 17, 27 10.1186/s13058-015-0529-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan V. C. Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. 2. Clinical considerations and new agents. J. Med. Chem. 2003, 46, 1081–1111. 10.1021/jm020450x. [DOI] [PubMed] [Google Scholar]
- Allen K. E.; Clark E. R.; Jordan V. C. Evidence for the metabolic activation of non-steroidal antioestrogens: a study of structure-activity relationships. Br. J. Pharmacol. 1980, 71, 83–91. 10.1111/j.1476-5381.1980.tb10912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J.; Guillen V. S.; Sharma A.; Zhao Y.; Ziegler Y.; Gong P.; Mayne C. G.; Srinivasan S.; Kim S. H.; Carlson K. E.; Nettles K. W.; Katzenellenbogen B. S.; Katzenellenbogen J. A. Adamantyl antiestrogens with novel side chains reveal a spectrum of activities in suppressing estrogen receptor mediated activities in breast cancer cells. J. Med. Chem. 2017, 60, 6321–6336. 10.1021/acs.jmedchem.7b00585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan M.; Rickert E. L.; Chen L.; Aftab S. A.; Nephew K. P.; Weatherman R. V. Characterization of molecular and structural determinants of selective estrogen receptor downregulators. Breast Cancer Res. Treat. 2007, 103, 37–44. 10.1007/s10549-006-9353-2. [DOI] [PubMed] [Google Scholar]
- Obermoser V.; Baecker D.; Schuster C.; Braun V.; Kircher B.; Gust R. Chlorinated cobalt alkyne complexes derived from acetylsalicylic acid as new specific antitumor agents. Dalton Trans. 2018, 47, 4341–4351. 10.1039/C7DT04790H. [DOI] [PubMed] [Google Scholar]
- Knowlden J. M.; Hutcheson I. R.; Jones H. E.; Madden T.; Gee J. M.; Harper M. E.; Barrow D.; Wakeling A. E.; Nicholson R. I. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology 2003, 144, 1032–1044. 10.1210/en.2002-220620. [DOI] [PubMed] [Google Scholar]
- PerkinElmer Informatics, 1998-2014.
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Wolber G.; Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.