

Abstract
The sarcomere is the basic contractile unit of muscle, composed of repeated sets of actin thin filaments and myosin thick filaments. During muscle development, sarcomeres grow in size to accommodate the growth and function of muscle fibers. Failure in regulating sarcomere size results in muscle dysfunction; yet, it is unclear how the size and uniformity of sarcomeres are controlled. Here we show that the formin Diaphanous is critical for the growth and maintenance of sarcomere size: Dia sets sarcomere length and width through regulation of the number and length of the actin thin filaments in the Drosophila flight muscle. To regulate thin filament length and sarcomere size, Dia interacts with the Gelsolin superfamily member Flightless I (FliI). We suggest that, through controlling actin dynamics and turnover, that these actin regulators generate uniformly sized sarcomeres tuned for the muscle contractions required for flight.
Keywords: Sarcomere, actin filaments, Diaphanous, Formins, Actin polymerization, Flightless I, Gelsolin, Actin severing, Drosophila, Flight muscle, Muscle maintenance
Introduction:
Sarcomeres are the basic contractile units of striated muscles. Sarcomeres arrange themselves sequentially to form a myofibril, and multiple myofibrils are found within each muscle fiber. The proper size, uniformity, and arrangement of sarcomeres are prerequisites for muscle function. Over the past several years, significant progress has been made in identifying proteins that are involved in sarcomere assembly (Burkart et al., 2007; Molnár et al., 2014; Orfanos et al., 2015; Pappas et al., 2010). However, it is still unclear how the size and uniformity of sarcomeres are determined and maintained, particularly during periods of muscle growth.
The Drosophila indirect flight muscles (IFMs) provide an ideal system to study sarcomere size and function: firstly, the components and structure of sarcomeres are conserved from Drosophila to humans. Secondly, IFMs are a sensitive system with an easily accessible readout: minor changes in muscle structure can lead to deficits in flight ability (Reedy et al., 2000). Thirdly, IFMs most resemble human cardiac muscle both structurally (Spletter et al., 2015) and functionally (Tu and Daniel, 2004). Hence, identifying genes that regulate sarcomeric structure and function in this system will have important ramifications for human skeletal and cardiac muscle development and homeostasis. Lastly, this system allows the use of the powerful genetics and imaging approaches available in the Drosophila model system.
In both Drosophila and humans, sarcomeres are primarily composed of thick filaments and thin filaments and their associated proteins (Figure 1A). Thick filaments are composed of myosin bundles that are crosslinked at the M-line. Thin filaments contain parallel actin filaments of uniform length. The barbed ends of the actin filaments are anchored to the Z-disc through α-actinin, and their pointed ends are oriented to the edge of the M-line. Mutations in genes that encode thin filament proteins such as Actin (ACTA1), Troponin I (TNNI1, TNNI2, TNNI3), Tropomyosin (TPM2, TPM3), and Nebulin (NEB) are known to cause congenital myopathies in human, such as actin myopathy, intranuclear rod myopathy, and Nemaline Myopathy (Clarkson et al., 2004; Malfatti and Romero, 2016; Ochala, 2008; Sewry et al., 2019).
Figure 1: Knockdown of Diaphanous impairs flight ability and sarcomere size in IFMs.
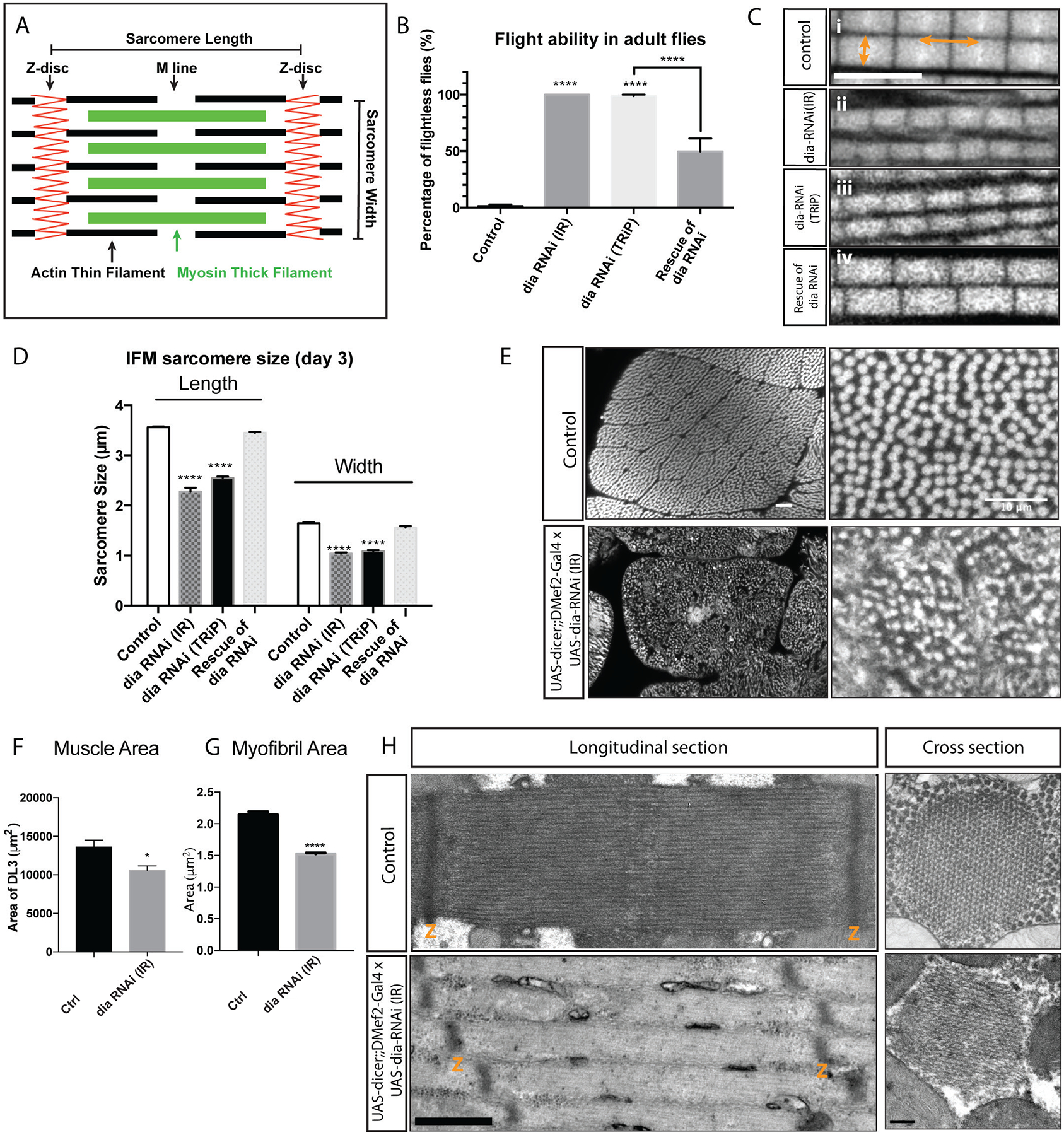
A. Schematic diagram of the structure of the sarcomere, noting Z disc, M line, thin and thick filaments. Length and width of the sarcomere are indicated. B. Flight assay: 25 flies per replicate, 3 replicates per genotype, n= 75 flies. The flight ability of individual flies was tested 3 days after eclosion. dia RNAi constructs were expressed in muscles with UAS-dicer2;;DMef2-Gal4. Reduced Dia activity resulted in impaired flight ability (p<0.0001). Rescue experiments were performed by expressing Dia::GFP in muscles along with dia RNAi (TRiP). The ability to fly was partially rescued by expressing Dia::GFP (p<0.0001). C. Myofibril and Sarcomere organization in flight muscle. Sarcomere length is indicated by the horizontal arrows, and sarcomere width is marked by the vertical arrows. Scale bar: 5μm; D. Quantification of sarcomere length and width: 5 flies per genotype, n = 5 average sarcomere length or width measurements per fly. In muscles with reduced Dia activity, both sarcomere length and width are significantly reduced (p<0.0001). Expressing Dia::GFP in the muscles restores flight capability, sarcomere length and width. E. Muscle cross sections indicating muscle size and myofibril organization. Scale bar: 10μm. F. Quantification of DLM3 cross-sectional areas: DLM3 from 5 flies per genotype, control n = 6 and dia-RNAi = 7 DLM3 muscles). In muscles with reduced Dia activity, muscle area is smaller than that of the controls, *p<0.05). G. Quantification of flight muscle myofibril area: 5 flies per genotype, 5 average myofibril area measurements per fly, n = 25 average myofibril area measurements per genotype. In muscles with reduced Dia activity, myofibril area is significantly smaller than that of the controls (****p<0.0001). H. Sarcomere morphology of newly eclosed adults using Transmission Electron Microscopy (TEM). Dia knockdown results in shorter and thinner sarcomeres. The numbers of thin and thick filaments are reduced. Scale bar: longitudinal sections: 600nm, cross sections: 200nm.
Despite its highly ordered, uniform, and nearly crystalline-like appearance, the proteins that constitute the sarcomere are subject to turnover: during sarcomere assembly and growth, actin incorporates into thin filaments at both the barbed and the pointed ends of the actin filaments, with the majority of the actin incorporation occurring at the pointed ends (Littlefield et al., 2001; Perkins and Tanentzapf, 2014). After sarcomere growth is completed, the dissociation and incorporation of actin at both ends of thin filaments still occur, without disrupting the overall sarcomere organization and function (Ono, 2010; Perkins and Tanentzapf, 2014). The continuous turnover of actin suggests that actin dynamics are regulated to preserve uniform sarcomere size and thus, optimal contraction. Previous studies have identified actin regulatory factors that influence sarcomere size by controlling actin filament elongation, stabilization, or severing. These factors include the actin elongating protein Sarcomere Length Short (SALS) (Bai et al., 2007), capping proteins: CapZ (Cpa and Cpb) (Pappas et al., 2008; Schafer et al., 1995) and Tropomodulin (Tmod) (Littlefield et al., 2001; Littlefield and Fowler, 2008; Mardahl-Dumesnil and Fowler, 2001; Sussman et al., 1998), the scaffolding proteins Titin (Sallimus) (Orfanos et al., 2015) and Nebulin (Lasp) (Fernandes and Schöck, 2014), and the severing protein Cofilin-2 (Twinstar/DmCofilin) (Balakrishnan et al., 2020; Kremneva et al., 2014). However, how these different types of proteins coordinate to ensure the uniform length of thin filaments remain largely unclear.
Formins are a family of proteins that control actin nucleation and polymerization in a number of cell contexts, including muscle. In C. elegans body wall muscles, the formins FHOD-1 and CYK-1 are involved in determining and maintaining sarcomere size and in anchoring thin filaments to Z-discs (Mi-Mi and Pruyne, 2015). In mammalian cardiac muscles, thirteen formins are expressed and play critical roles in myofibril development and maintenance (Arimura et al., 2013; Iskratsch et al., 2013; Kan et al., 2012; Rosado et al., 2014; Taniguchi et al., 2009; Wooten et al., 2013). In Drosophila, the formins DAAM and Fhos are required for thin filament assembly (Molnár et al., 2014; Shwartz et al., 2016). Despite our understanding that formins are involved in thin filament assembly and maintenance, much remains unknown: it is unclear how formins coordinate with other actin regulators to control actin turnover, the types of proteins with which they interact at the sarcomere, and whether there are other targets of formin activity in sarcomeres.
Diaphanous is the first identified member of the Drosophila formin family of proteins (Afshar et al., 2000). Dia has been shown to regulate actin polymerization in various developmental events in Drosophila, such as myoblast fusion (Deng et al., 2015), wing hair morphogenesis (Lu and Adler, 2015), dorsal closure (Bilancia et al., 2014; Nowotarski et al., 2014), wound healing (Matsubayashi et al., 2015), apical secretion of epithelial tubes (Rousso et al., 2013), synaptic growth (Pawson et al., 2008), and cytokinesis (Afshar et al., 2000). Dia is also known to stabilize microtubules (Daou et al., 2014) by interacting with the microtubule plus end tracking proteins, End binding 1 (EB1), and Adenomatous polyposis coli (APC) (Palazzo et al., 2001; Wen et al., 2004). Drosophila embryos homozygous for Dia null alleles die during embryogenesis (Afshar et al., 2000). Dia function has been extensively studied in migrating and dividing cells; these studies have provided insights to the mechanisms by which Dia, and potentially other formins, regulate different subcellular behaviors. Although the function of Dia in early Drosophila muscle development has been reported (Deng et al., 2015), whether Dia plays a role in muscle function and maintenance has not been investigated.
In this study, we show that the Diaphanous is required during sarcomere growth in the IFMs. Dia localizes to the M-line during sarcomere growth, determining the length and width of each sarcomere. Subsequently Dia is relocalized to the Z-disc where it is involved in thin filament maintenance. Taking advantage of the highly sensitive IFM system, we identified Flightless I (FliI), the Gesolin ortholog and an actin severing protein, as a Dia interacting partner. We propose that these proteins together regulate actin structure and turnover during sarcomere growth and maintenance; this, in turn, generates sarcomeres with optimal thin filament number and length and provides the conditions necessary for the muscle contraction required for flight.
Materials and Methods :
Fly stocks:
UAS-dia-RNAi (IR) (VDRC #103914), UAS-dia-RNAi (TRiP) (Bloomington #28541), UAS-DiaDN::GFP (Deng et al., 2015), UAS-dia::GFP (M. Peifer) (Homem and Peifer, 2008); UAS-dicer-2, Dmef2-Gal4, MHC-Gal4, tubP-Gal80ts/FM7 (Bloomington #7016), UAS-Act88F-GFP (Bloomington #9253), UAS-fliI-RNAi (VDRC #39528), UAS-fliI::HA (this study), UAS-chic-RNAi (VDRC #102759), UAS-Jupiter::GFP (Karpova et al., 2006). Fly stocks used in screen are listed in Supplementary Table 2. The GAL4-UAS system was used for expression studies (Brand and Perrimon, 1993).
Immunohistochemistry:
Adult flies were dissected, fixed in 4% PFA, and stained according to protocol described below. Antibodies were used at the following concentrations: α-Dia (1:1000) (S. Wasserman), α-Alpha-actinin (1:200) (Abcam #ab50599), α-GFP (1:400)(Abcam #ab6556), α-FliI (1:50) (Santa Cruz Biotechnology SC-30046), α-HA (1:100) (Roche #11867423001), α-Alpha-tubulin (1:400) (Sigma Aldric #T9026), Hoechst (Invitrogen H1399), α-Beta PS integrin (1: 50) (DSHB #CF.6G11), α-Tmod, α-Obscurin (1:100)(gift of B. Bullard, Burkart et al., 2007), Alexa Fluor 555-Phalloidin (1:100) (Invitrogen A34055), α-Chickadee/Profilin (1:100) (DSHB #chi 1J). For secondary antibodies, Alexa Fluor 488-, Alexa Fluor 555-, and Alexa Fluor 647-conjugated fluorescent secondary antibodies at 1:200 dilution (Invitrogen) were used. Fluorescent images were acquired on a Leica SP5 laser scanning confocal microscope equipped with a 63X 1.4 NA HCX PL Apochromat oil objective and LAS AF 2.2 software. Maximum intensity projections of confocal Z-stacks were rendered using Volocity.
Transmission electron microscopy:
IFM muscles were dissected into half thoraces as described above and were fixed in a solution composed of 2% Glutaraldehyde, 4% Paraformaldehyde, 2mM CaCl2, 0.5% Tannic acid in 0.1M Sodium Cacodylate buffer, pH 7.4. The samples were then fixed for 3 minutes using the Pelco Biowave (Ted Pella), followed by overnight fixation at 4°C. Samples were post-fixed with 1% osmium tetroxide containing 1.5% Potassium Ferrocyanide in Cacodylate buffer for 1 hour on ice. After fixation, samples were stained with 1% Uranyl Acetate aqueous solution at room temperature for 30 minutes. The samples were then dehydrated in a graded series of ethanol using the Pelco Biowave, followed by Acetone dehydration at room temperature for 10 minutes. After dehydration, each half thorax was infiltrated and embedded with Eponate 12™ (Ted Pella) and was subsequently sectioned into 60–70nm ultrathin sections. For each genotype, 3 samples were sectioned using a Reichert Jung Ultracut E microtome. After sectioning, samples were stained with 2% Uranyl Acetate and Sato’s lead stain. Images were taken under a JEOL 100CX Transmission Electron Microscope at 80kV equipped with an XR41-C camera and AmtV600 software (Advanced Microscopy Technology Corp., Woburn, MA).
Signal intensity measurement:
ImageJ (Fiji) was used to measure and plot signal intensities of the sarcomeres. The area to be measured was defined by dropping a box that covers the length of a sarcomere (box spanned from the end of M-line to the end of M-line of the following sarcomere). The size of box is adjusted according to the size of sarcomere. Fluorescent intensity is measured along the long axis of box and plotting into intensity curve.
To measure the relative ratio of fluorescence signal at the M-line and Z-disc, three boxes with the same size were dropped at Z-disc (Z), M-line (M) and between Z-disc and M-line (B). The relative ratio of signal at the Z-disc was calculated as (Z-B)/[(Z-B)+(M-B)]. The relative ratio of signal at the M-line was calculated as (M-B)/[(Z-B)+(M-B)]. Quantifications were made using 5 flies per genotype, 3 sarcomeres per fly, n = 15 sarcomeres signal measurements.
Fusion index:
The Fusion index was quantified by counting the number of nuclei in a 100μm × 5μm area of the IFM. For each genotype, 5 flies were dissected; the number of nuclei in 2 sample areas was counted in each fly (n = 10 nuclear counts per genotype).
Molecular cloning:
Drosophila fliI was amplified from cDNA (BDGP DGC clones: LD21753) with primer : forward: 5’CACCATGAGCGTGCTGCCGTT3’, reverse:5’ TAAATACACCTTGAAGGC3’. The gene was cloned into pUAST vectors with C-terminal HA tag and sequenced. The UAS-fliI::HA construct was validated by sequencing before being sent out for injection.
Flight assay:
Individual flies were dropped into a vial, and its flight ability was recorded (Banerjee et al., 2004). For each genotype, 25 flies were collected each day and tested for their flight ability on the 3rd day after eclosion. Three repeats were done for each genotype (n=75). Flies were separated by their flight ability and dissected for IFM staining.
Flight performance assay:
Flight performance was measured as described by (Babcock and Ganetzky, 2014). Vials containing ~20 flies were dropped down a tube suspended over a 90cm graduated cylinder. The cylinder was lined with a removable acrylic sheet coated with adhesive. A tray of oil was placed on the bottom of the cylinder. Once the flies were dropped into the tube, attempted to fly and were stuck on the adhesive, the acrylic sheet was removed and imaged using a digital camera. Fiji software was used to calculate the landing heights of flies. The number of flies that fell into the oil at the bottom were also counted and landing height recorded as 0 cm. The experiment was repeated 3–5 times, until the landing heights were recorded for a minimum of n=50 flies for each genotype (Control Day 0: 140 flies; Control Day 4: 51; Control Day 11: 103; dia-RNAi Day 0: 67; dia-RNAi Day 4: 54; dia-RNAi Day 11:85).
Flight muscle dissection:
Flies were collected immediately after eclosion and were kept at room temperature. Thoraces from 1–7 day old adult flies were dissected using published protocols (Hunt and Demontis, 2013; Schnorrer et al., 2010). For 1 day old adults, newly eclosed flies with white cuticles were used. After anaesthetizing the flies with CO2, the head, abdomen, legs and wings from thorax were removed with scissors. Thoraces were kept in Relaxing buffer before fixation (20mM PBS, pH=7.0, 5mM MgCl2, 5mM EGTA). Thoraces were then pre-fixed in fresh 4% paraformaldehyde (PFA) in relaxing buffer for 10min. After pre-fixation, thoraces were dissected into hemi-thoraces by cutting longitudinally with scissors and transferred into 24 well plates. Samples were washed twice with PBT (PBS+0.3% Triton X-100), and incubated in relaxing buffer for 15min, followed by fixation in 4% PFA for 20 minutes. After fixation, samples were washed in PBT 2×10min, and incubated in primary antibody overnight at 4°C. On the second day, thoraces were removed from the primary antibody, and washed in PBT 3×20min. After washing, samples were incubated in secondary antibody for 2h at room temperature. After rinsing in PBT 3×20min, hemi-thoraces were mounted in Prolong gold (Invitrogen P36930) and analyzed using Leica SP5 confocal microscope. For each experiment, unless further specified, the average myofibril length or width was calculated from each fly, using 5 flies per genotype, n =5 average myofibril length or widths.
For transverse muscle cross-sections, day 5–7 old flies were frozen in liquid nitrogen. Thoraces were removed and cut at the transverse suture. Cross sections were fixed in 4% paraformaldehyde for 1.5 hours, washed in PBST (PBS + 3% Triton X-100) three times for 10 minutes, and stained with Hoescht (1:100, Invitrogen H1399) and Phalloidin (1:200, Invitrogen A34055) for 1 hour at room temperature. Following staining, cross-sections were washed in PBST and mounted in Prolong gold.
Co-IP:
Drosophila S2 cells were co-transfected with Ubiquitin-Gal4, UAS- Dia::GFP (or UAS-GFP alone) and UAS-3xHA::FliI constructs. Cells were lysed at day3 in Lysis buffer (150mM NaCl, 2mM MgOAc, 20mM Tris, pH 7.5, 5% Glycerol, 0.5% NP40) for 30 minutes. Protein levels in the lysates were quantified using a Bradford assay (Bio-Rad 500–0006). Cell lysates were then incubated with Protein G-Agarose beads overnight at 4°C using rabbit anti-GFP (Torrey Pines #TP401) or anti-rabbit IgG (Santa Cruz # SC-7392) for mock immunoprecipitation controls. The beads were washed four times with Lysis buffer and boiled in 4x Laemmli Buffer. GFP-purified samples were then run on SDS-PAGE and blotted with rat anti-HA (Roche # 1867423).
Muscle Area Measurements
Images of muscle cross sections were taken using Zeiss LSM 880 scope. The area of the 3rd dorsal longitudinal muscle (DLM3) was measured using ImageJ (Fiji) polygon tracing tool. Area measurements of DLM3 muscles were made from 5 flies in control (n =6 DLM3 muscles) and Dia-RNAi (n = 7 DLM3 muscles).
Myofibril Area Measurements
An approximately 36 × 36 μM portion of DLM3 transverse muscle image was selected and converted into a binary image for averaged myofibril area analysis using Fiji. Objects outside the range of 0.4 – 3μM were omitted from myofibril area averages. Five sections were measured per fly, using 5 flies per genotype, n = 25 average myofibril area measurements.
Viability Assay
Flies were monitored for viability on the indicated days following transgene induction using control and dia-RNAi flies. Viability experiments were repeated three times using 17–20 flies per experiment, n = 3.
Statistical analyses:
Statistic tests were done in GraphPad Prism. Means of two groups were compared with Student’s t-test. Means of three or more groups were compared with one-way ANOVA test. Means of 2 groups over time were compared using 2-way ANOVA test. The difference between groups is considered significant when p<0.05.
Results:
Diaphanous knockdown impairs flight ability and sarcomere size in IFMs
To investigate the role of Dia in the indirect flight muscles, we reduced Dia levels specifically in muscles and examined the behavioral and anatomical phenotypes in adult flies. When we knocked down Dia using RNAi, we found that the flight ability was significantly impaired (Supplemental movie 1–2, Figure 1B; Percentage of flightless flies: control: 1.3%±2.31%, dia RNAi (IR): 100%±0%, dia RNAi (TRiP): 98.7%±2.31%, p<0.0001). The flightless phenotype associated with Dia knockdown was specific for abrogation of Dia function: expression of Dia::GFP in muscles rescued the flight phenotype associated with dia RNAi (Figure 1B; Percentage of flightless flies: dia RNAi (TRiP): 98.7%±2.31%, rescue: 49.7%±19.86%, p<0.0001). Expression of a DiaDN construct that abrogates Dia activity (Deng et al., 2015) also increased the percentage of flightless flies (Supplemental Figure 1A), reinforcing a role for Dia in flight ability.
As Dia is known to play a role in myoblast fusion and cell adhesion (Deng et al., 2015), we examined whether aberrations in these cellular functions contributed to the loss in flight ability seen upon Dia knockdown. We first assessed whether myoblast fusion was affected by counting the number of myonuclei in the IFMs (Supplemental Figure 1B). Expression of dia RNAi in the developing muscle did not significantly change the myonuclear number, as compared to control flies (Supplemental Figure 1B and C). As Dia is required for cell adhesion, we also investigated whether the loss of flight ability was due to detachment of IFMs from their tendon cells. No detached muscles or myospheres were detected in Dia knockdown thoraces. Moreover, we assessed the integrity of the myotendinous junctions (MTJ) by immunostaining with antibodies to β-PS integrin, a critical component of MTJ (Fernandes et al., 1996). We found that β-PS integrin was present at the myotendinous junctions similar to that seen in controls. No obvious changes in distribution or levels were noted in dia RNAi conditions (Supplemental Figure 1D). Taken together, our data suggest that Dia plays a role in flight muscle development after fusion and attachment.
As an additional control for these experiments, we confirmed the dia-RNAi –mediated reduction in Dia levels by immunostaining for Dia in the flight muscles. We found that dia-RNAi reduced Dia levels in the knockdown muscles compared to the controls (Supplemental Figure 1E). However, when we adjusted the brightness and over saturated the image (e.g. increasing the signal 3-fold using Volocity software), we found that residual Dia was detected in muscles expressing dia-RNAi. These data indicated that Dia knockdown was not 100% efficient and may explain why muscles with Dia knockdown can bypass earlier events in myogenesis, such as myoblast fusion.
We next investigated how reduction in Dia impaired flight ability. Dia is known to regulate actin filament nucleation and polymerization (Chesarone et al., 2010; Evangelista et al., 2002; Pruyne et al., 2002). As actin filaments are the major components of the sarcomere thin filaments (Figure 1A), we examined the morphology of myofibrils and sarcomeres in IFMs by immunostaining for F-actin. We first observed that the myofibrils in the Dia knockdown muscles are disorganized (Supplemental Figure 1F). The myofibrils at the surface of the muscle fiber and around the nuclei contain fraying actin filaments (Supplemental movie 3,4). In addition, the sarcomeres that make up these myofibrils appeared to be smaller (Figure 1C, Supplemental Figure 1F). We confirmed our observations through quantification of sarcomere length and width (Figure 1D) and found that, in muscles with reduced Dia function, the sarcomeres were shorter (length) and thinner (width) compared to controls (Figure 1C and D) (Sarcomere length: control: 3.6±0.15μm, dia-RNAi (IR): 2.3±0.39μm, dia-RNAi (TRiP): 2.6±0.28μm, p<0.0001, 20%−40% reduction in length. Sarcomere width: control: 1.6±0.13μm, dia-RNAi (IR): 1.0±0.10μm, dia-RNAi (TRiP): 1.1±0.11μm, p<0.0001, 30%−40% reduction in width). Sarcomeres with reduced lengths were also seen in muscles expressing DiaDN (Supplemental Figure 1G and H). When we examined Dia knockdown flies in which the flight ability was rescued by expression of Dia::GFP, we found that the length and width of the thin filaments were restored (Figure 1C and D) (length: 3.5±0.14μm, width: 1.6±0.14μm, p<0.0001).
To further investigate muscle morphology and myofibril size as well as organization, we examined cross sections of the Drosophila flight muscle. Our analysis showed that muscles with Dia knockdown were reduced in size compared to control (3rd dorsal longitudinal muscle (DLM3), control: 13603.7μM ± 898.93μM, dia-RNAi (IR): 10584.9μM ± 572.65μM, p<0.05) (Figure 1E and F). Closer examination of these muscles revealed that reduced Dia levels lead to disorganized myofibrils as well as reduced myofibril cross-sectional areas compared to controls (control: 2.1μM ± 0.05μM; dia-RNAi (IR), 1.5μM ± 0.02μM, p<0.0001) (Figure 1E, G).
We further investigated the morphological modifications to the sarcomere in Dia knockdown muscles using Transmission Electron Microscopy (TEM). Similar to what we observed with immunofluorescence, the sarcomere phenotype included highly irregular myofibrils as well as myofibrils consisting of sarcomeres with reduced size (Figure 1H, Supplemental Figure 1I and J). TEM of flight muscles from newly eclosed flies showed that reduced Dia levels resulted in a reduced number of thin and thick filaments, which led to sarcomeres with reduced widths (Figure 1H). In addition to reduced sarcomere size, both longitudinal and cross section showed that the organization of the actin filaments is disrupted in Dia knockdown muscles (Figure 1H, Supplemental Figure 1I and J). From these data, we conclude that Dia is important for IFM function by regulating different aspects of muscle structure: Dia controls myofibril arrangement and alignment, and it also regulates sarcomere size by controlling the length, number, and organization of the actin thin filaments.
Dia regulates sarcomere growth and maintenance
The sarcomeres in Drosophila IFM assemble at the early pupae stage (36hr APF) (Orfanos et al., 2015; Reedy and Beall, 1993). After the initiation of assembly, sarcomeres continue to grow in both length and width for the next, approximately 64 hours of pupal development (Mardahl-Dumesnil and Fowler, 2001; Spletter et al., 2015) (Supplemental Figure 2A). In newly eclosed flies, we find that the sarcomeres also continue to grow for several hours. Subsequently, sarcomere length and width are maintained over the remainder of adulthood (Figure 2A–B). To investigate Dia’s role in sarcomere growth, we measured sarcomere size in IFMs with Dia knockdown on the first day after eclosion. While the sarcomeres are correctly established, we found that they are smaller in size compared to control, suggesting impaired sarcomere growth during the pupal stage. We also examined sarcomere size in IFMs with Dia knockdown on days 1, 3, and 7 after eclosion. We found that, unlike wild-type controls in which sarcomere size continued to grow during early adulthood (Figure 2A–B), the length and width of the sarcomere in Dia knockdown did not increase (Figure 2C–D). Together, these data suggest Dia is important for sarcomere growth in Drosophila IFMs.
Figure 2: Knock down of Diaphanous impairs thin filament growth in IFMs.
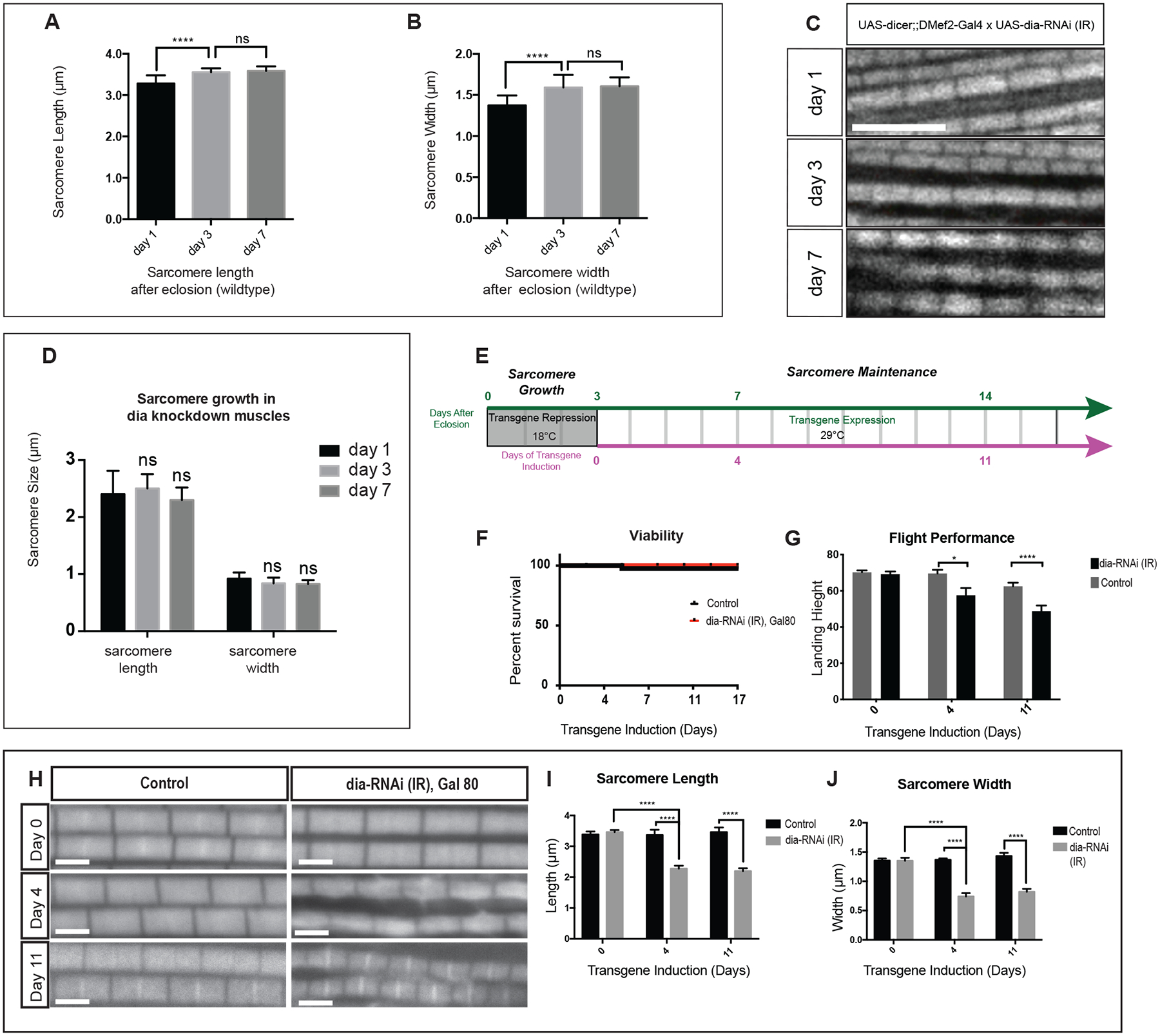
A-B. Changes in IFM sarcomere size in wild-type adult flies. Wild-type flies were dissected at days 1 (newly eclosed), 3, and 7 after eclosion. The length and width of the individual sarcomeres was measured and recorded (5 flies per genotype, n = 5 average sarcomere length or width measurements/fly). Sarcomeres in newly eclosed flies are significantly shorter (p<0.0001) and thinner (p<0.0001) than the sarcomeres of flies 3 days after eclosion. There is no significant difference in sarcomere size between day 3 and day 7 flies. These data are consistent with continued growth of the sarcomeres in newly eclosed flies (day 1), followed by sarcomere maintenance (thereafter). Scale bar: 5μm C-D. Sarcomere phenotypes upon Dia knockdown in the muscle. Sarcomere size was measured on Day 1, 3 and 7 after eclosion (5 flies per genotype, n = 5 average sarcomere length or width measurements/fly). There is no significant difference in sarcomere size between each group. E. Diagram of experimental design. dia-RNAi was induced in adult muscle 3 days after eclosion. F. Viability of adult fly was examined after induction of dia-RNAi. No significant difference in viability was observed between control (n = 56 flies) and Dia knockdown (n = 52 flies) groups at the indicated time points. G. Quantification of Flight Performance following Dia knockdown. Flight performance was measured after 0, 4 and 11 days of induced dia-RNAi (3, 7, and 14 days after eclosion). Difference of flight performance was observed between control (Day 0; n = 140 flies, Day 4; n =51 flies, Day 7;n = 103 flies) and Dia knockdown (Day 0; n = 67 flies, Day 4; n = 54 flies, Day 7;n = 85 flies) groups following 4 (p<0.05) and 11 (p<0.0001) days of induction. H-J. Sarcomere morphology and size was examined after dia-RNAi induction. Average myofibril measurements were made at the indicated days of knockdown induction. Four days after induced Dia knockdown, sarcomere length and width are significantly reduced compare to control (5 flies per genotype, n = 5 average sarcomere length or width measurements/fly, ****p<0.0001). Scale bar: 2μm.
To investigate Dia’s role in sarcomere maintenance, we used the temperature sensitive Gal80 system (Caygill and Brand, 2016) to knock down Dia in the IFMs 3 days after eclosion when the final adult sarcomere length and width are achieved (Figure 2E). Using this experimental design, we first determined that expressing dia-RNAi in IFM after eclosion did not change viability (Figure 2F). Flight performance, however, was significantly altered under these conditions (Figure 2G). Flight performance was measured using landing height as an indicator of flight muscle function (Babcock and Ganetzky, 2014). After 4 days of transgene induction, flies with dia-RNAi showed decreased flight performance with a 17 % reduction in landing height relative to control (control: 69.55cm±1.0cm; dia-RNAi: 57.49cm±3.99cm, p<0.05). There was a similar performance difference between control and dia-RNAi flies after 11 days of transgene induction (22% reduction, control: 62.47cm±1.98cm; dia-RNAi: 48.69cm±4.69cm, p<0.0001).
Given the defects in flight performance, we next examined the IFM sarcomere phenotype in flies subjected to this experimental design (Figure 2F). At day 0 of dia-RNAi induction, the sarcomeres display no morphological differences between control and Dia knockdown group (Figure 2H–J). We examined sarcomere phenotype at 4 days and 7 days after induced dia-RNAi expression. While control sarcomere size remains constant over time during the maintenance phase, there is a reduction in length (control: 3.36μm±0.17μm; dia-RNA: 2.28μm±0.09μm, p<0.0001) and width (control: 1.37μm±0.02μm; dia-RNAi: 0.74μm±0.06μm, p <0.0001) after 4 days of dia-RNAi induction. After 11 days of transgene induction, similar differences are observed in dia-RNAi flies compared to control in length (control: 3.461 μm±0.15 μm; dia-RNAi: 2.195 μm±0.09 μm, p <0.0001) and width (control: 1.43 μm±0.05 μm; dia-RNAi: 0.82 μm±0.05 μm, p-value<0.0001). This sarcomere phenotype is consistent with the defects in flight performance that we detect. Based on these data, we conclude that Dia contributes to IFM function through regulating sarcomere growth and maintenance.
Diaphanous knockdown changes the localization of actin capping proteins
Thus far, our data suggest that Dia regulates sarcomere size through controlling the growth of sarcomeres via its actin polymerization activity, namely the addition of actin to the preexisting actin thin filaments as well as through the addition of new actin thin filaments to the Z disc. Actin dynamics at either end of the thin filament is regulated by two different actin capping proteins, Cap Z and Tropomodulin (Tmod). CapZ stabilizes the barbed end of the actin thin filament, which is anchored to the Z-disc, and promotes actin filament organization (Schafer et al., 1995). Tmod caps the pointed end, which localizes towards the center of the sarcomere near the M-line, and prevents actin filament elongation (Littlefield and Fowler, 2008). A two-segment model has been proposed to explain how thin filament length is regulated: the actin thin filament is composed of two parts, the proximal segment that starts at the Z-disc with constant length and the distal segment close to the M-line with variable length. During sarcomere growth, actin monomers (Gokhin and Fowler, 2013) or short actin filaments (Molnár et al., 2014) compete with Tmod capping at the pointed end to associate with the thin filament and, therefore, elongate the thin filament. After growth and during muscle homeostasis, a higher level of actin turnover is seen at the barbed ends at the Z-discs (Perkins and Tanentzapf, 2014). To gain additional insight to the impact of Dia on sarcomere size, as well thin filament length and organization during growth, we examined the effect of Dia knockdown on the localization of actin capping proteins CapZ and Tmod.
CapZ, the barbed end actin capping protein, localizes mainly at the Z-disc in wild-type muscles (Figure 3A). Using antibodies against the CapZ subunit Cpa, we found that the localization of CapZ is disrupted with Dia knockdown. Although CapZ was still observed at the Z-disc, an increased CapZ signal was detected at the M line as well as in the region between the myofibrils (Figure 3A). This change in the localization pattern of CapZ suggested that fewer barbed ends are available at the Z-disc for CapZ to bind, which is consistent with the thinner sarcomeres found in Dia knockdown muscles. However, more barbed ends of the actin filaments are also available near the M-line to associate with CapZ in the Dia knockdown.
Figure 3: Knock down of Diaphanous results in changes in the localization of thin filament capping proteins.
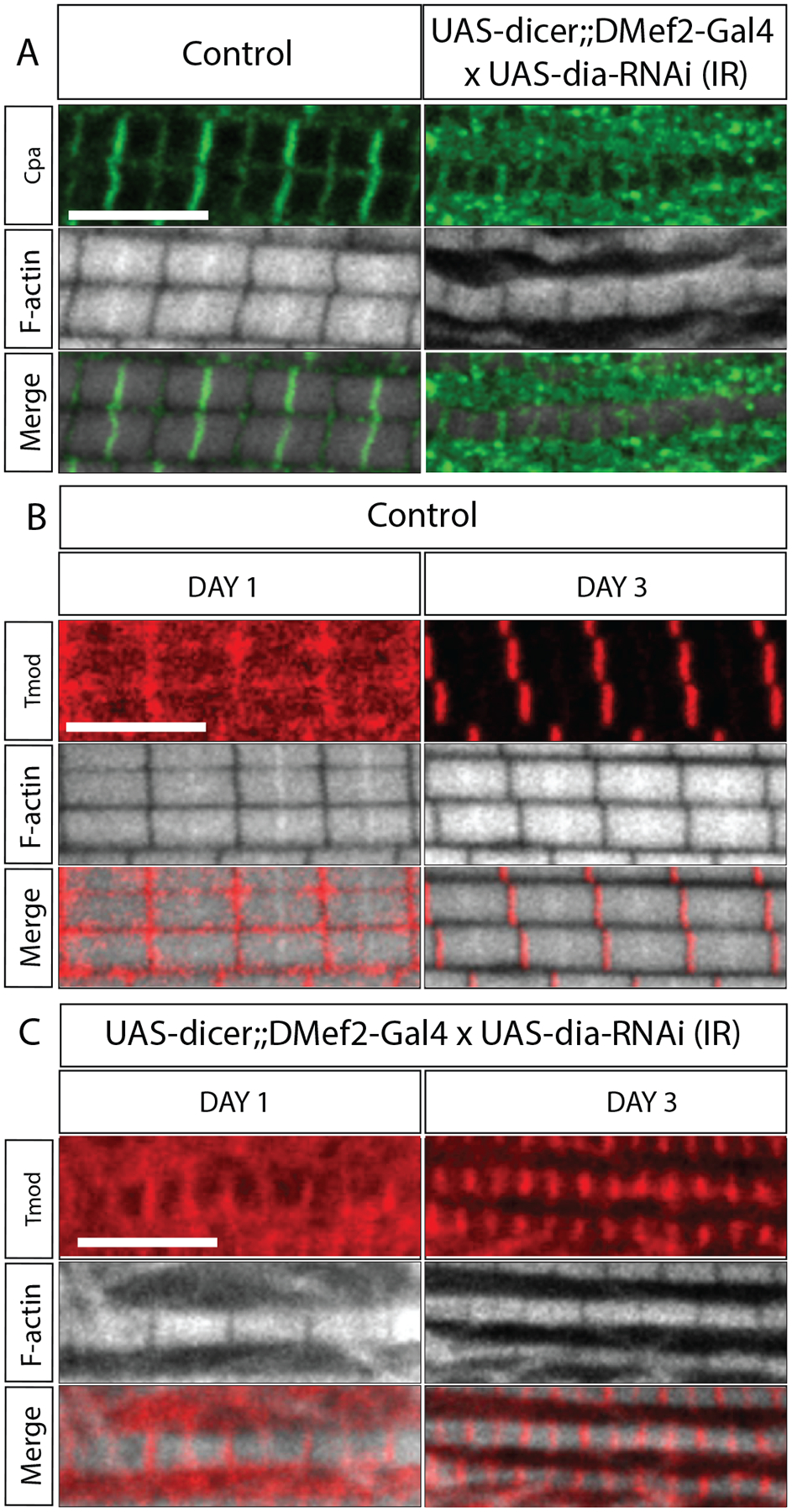
A. CapZ localization upon Dia knockdown in the IFMs. Flies were dissected 3 days after eclosion. 5 flies per genotype were dissected. 4 myofibrils/fly were imaged for analysis. Muscles are labeled with Phalloidin (white) and with an antibody against Cpa, a subunit of CapZ. B-C. Tmod localization in the IFMs upon dia knockdown. Flies were dissected 1 and 3 days after eclosion. 5 flies per genotype were dissected. 4 myofibrils/fly were imaged for analysis. Muscles are labeled with Phalloidin (white) and with an antibody against Tmod (red). Scale bar: 5 μm.
Tmod, the pointed end actin capping protein, is reported to localize to the M-line in wild-type muscles (Bai et al., 2007) (Figure 3B–C). Immunostaining of Tmod revealed that, in control IFMs, Tmod mainly localized to the M-line during the period of sarcomere growth, with diffused signal throughout the sarcomere (Figure 3B). After sarcomere growth is completed (day 3 and later), Tmod is restricted to the M-line, consistent with it labeling the pointed ends of actin thin filaments (Figure 3B). In IFMs with Dia knockdown, however, Tmod is present at both Z-disc and M-line (Figure 3C). The localization pattern of Tmod in the Dia knockdown condition suggested the presence of actin pointed ends, even at the proximal segments of thin filaments. We suggest that these filaments fail to elongate due to Dia knockdown.
Altogether, these data indicated that there is a mislocalization of actin capping proteins during sarcomere growth in the Dia knockdown IFMs. We suggest that the specific patterns of these proteins found in the Dia knockdown reflect alterations to actin dynamics that occur in the sarcomere during growth. Specifically, these data would suggest a function for Dia in regulating thin filament elongation at the Z-disc as well as near the M-line during growth.
The localization of Dia in sarcomeres changes during IFM growth
To better understand how Dia regulates thin filament growth and organization in sarcomere, we investigated the localization of Dia in the sarcomere. We used antibodies generated against Dia to examine Dia localization in Drosophila IFMs 1–7 days after eclosion. In flies which have just eclosed (day 1), Dia localizes mainly to the M-line (Figure 4Ai, arrows). As the flies age, sarcomere growth ceases, and homeostasis occurs (days 3 and 7). At these timepoints, higher Dia levels were found at the Z-discs compared to day 1 (Figure 4Aiii–v). Specifically, at day 3, similar Dia levels were observed at M-line and Z-disc, while at day 7, higher levels of Dia were found at Z-disc compared to the M-line (Figure 4A, arrowheads). This change in Dia’s localization was confirmed by measuring the fluorescence intensity across the sarcomere at the different time points, as well as by determining the relative ratio of Dia at the Z-disc and M-line (Figure 4A and B). Since studies have shown that actin incorporation occurs at both ends of the thin filaments, but preferably at the pointed ends near the M-line during growth (Littlefield et al., 2001; Perkins and Tanentzapf, 2014), the localization of Dia at the M line suggested that Dia regulates the elongation of thin filaments during growth; it subsequently relocates to the Z-discs when the sarcomere size stabilizes and is then maintained.
Figure 4: Diaphanous localization in IFM sarcomeres.
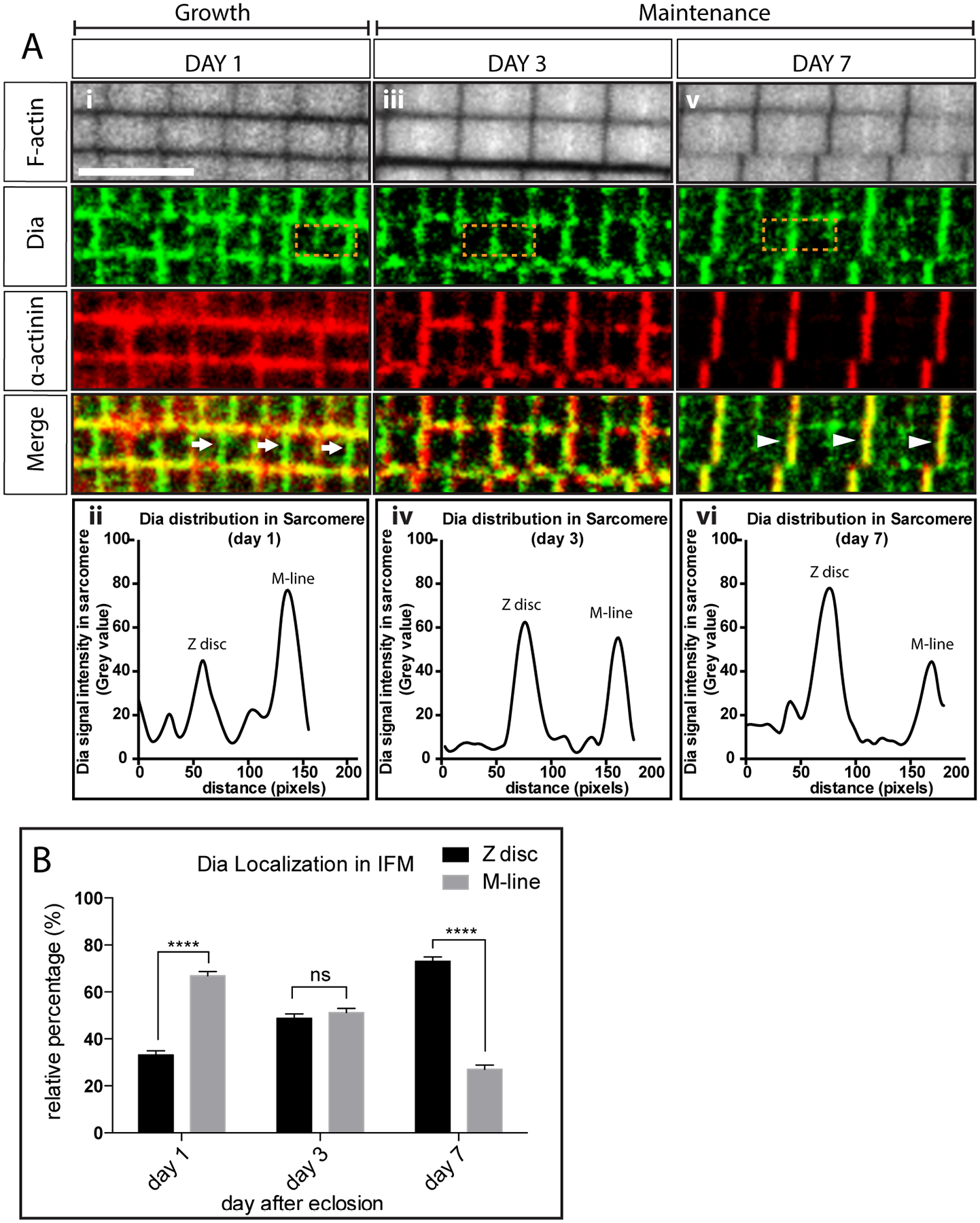
A. Dia localization in flies from day 1 (i), 3 (iii) and 7(v) after eclosion. Sarcomere thin filaments are labeled with Phalloidin (white). Z-discs are labeled with an antibody against α-actinin (red). Dia localization is visualized by staining with an antibody against Dia (green). Dia localizes mainly to the M-lines (arrow) 1 day after eclosion, and then shifts to the Z-discs as the sarcomeres cease growing (arrow head). The shift in Dia localization is verified by plotting the fluorescent signals in the boxed area: day 1 (ii), day 3 (iv) and day 7 (vi). B. Quantification of Dia localization in adult flies 1–7 days after eclosion. Fluorescence intensities of Dia were measured at the Z-disc, M-line, and at the area between the two regions from immunostained images. The relative percentage of Dia signal at the Z-disc and M-line was calculated and plotted (5 flies per genotype, 3 sarcomere signal measurements per fly, n = 15 sarcomere signal measurements, **** p<0.0001).
Dia interacts with FliI to regulate sarcomere assembly
To further understand how Dia controls sarcomere growth, we performed a limited genetic screen to identify Dia-interacting proteins (Supplemental Table 1). We selected our candidate genes based on their known involvement in sarcomere formation, actin polymerization activity, or actin depolymerization activity (Supplemental Table 2). We examined whether knock down of these genes would enhance the Dia knockdown phenotypes, leading to a change in viability rates. Using this assay, we isolated two genes that genetically interacted with dia during adult myogenesis: chickadee (chic) and flightless I (fliI). Flies expressing chic RNAi constructs alone in the muscles are viable and able to fly. Flies expressing fliI RNAi constructs in the muscles are viable but flightless. Double knockdowns of dia and chic, or dia and fliI, in the developing musculature resulted in pupal lethality, an earlier defect than seen with knockdown of any of the three genes alone (Supplemental Table 2).
Chickadee (Chic), the sole Drosophila Profilin, is a known Dia-interacting protein that promotes actin filament elongation (Geisbrecht and Montell, 2004; Webb et al., 2009). Isolation of Chic thus validated our screen. Knockdown of dia did not change Chic localization in muscles (Supplemental Figure 3A). Examination of the developing IFMs at 100h APF revealed that reducing both Dia and Chic blocked myoblast fusion and impaired the formation of both myotendinous junctions and myofibrils (Supplemental Figure 3B and C), consistent with previously published data (Deng et al., 2015). As the focus of this paper is the sarcomere, we no longer pursued the interaction between Chic and Dia in this context.
The other Dia-interacting protein that we identified in the screen was Flightless I (FliI). FliI is a member of the Gelsolin superfamily of proteins that severs actin filaments (Janmey et al., 1985; Kinosian et al., 1998). In Drosophila, FliI is known to play roles in actin distribution during cellularization (Straub et al., 1996), and it was uncovered in a large scale RNAi screen that focused on adult myogenesis (Schnorrer et al., 2010). Consistent with its flightless phenotype (Supplemental Table 2) and previous reports (Schnorrer et al., 2010), knockdown of FliI in the IFM resulted viable flies with increased filamentous actin and disorganized myofibrils (Figure 5A and B). We then examined, in more detail, the actin structures in FliI knockdown muscles. We categorized these actin structures within individual IFMs into two groups. The first group was composed of bundles of filamentous actin that exhibited longer actin filaments in comparison to the wild-type controls (Figure 5A, arrow). The second group consisted of myofibrils that were composed of, unexpectedly, shorter and thinner sarcomeres (Figure 5A, arrowhead and Figure 5 C and D) and that also labeled with antibodies directed to other sarcomeric proteins, such as α-actinin (Figure 5E). The presence of these two types of actin structures in the FliI knockdown muscles lead us to hypothesize: 1- depletion of FliI leads to a reduction of actin severing activities in the muscle cell and unbridled actin polymerization as seen in group 1 actin structures, and 2- the smaller sarcomeres found in the group 2 actin structures result from insufficient FliI actin severing and, as a result, reduced actin recycling in the cell (Bearer et al., 2002). We investigated these hypotheses and FliI’s relationship with Dia in more detail below.
Figure 5: Dia and FliI genetically interact to regulate sarcomere size.
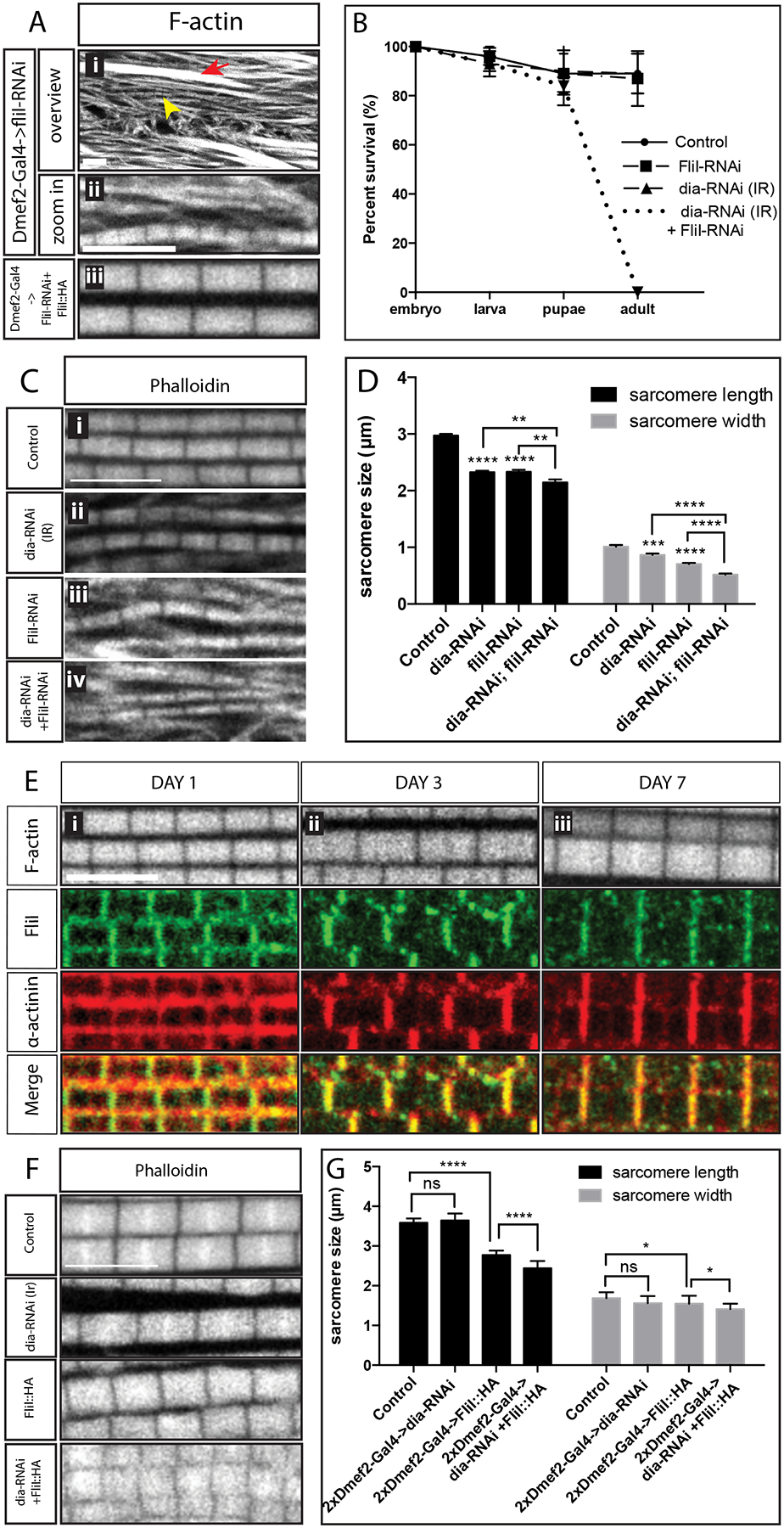
A. Sarcomere phenotype in muscles upon FliI knockdown. Flies were dissected 3 days after eclosion. Myofibrils are stained with Phalloidin (white), myofibrils without M-lines are marked with an arrow. Short sarcomeres are marked with an arrowhead and shown in the enlarged images. The bottom panel shows the sarcomere phenotype in flies that are rescued with FliI::HA. B. Survival assay. 25 embryos/genotype/day are selected for 3 days. Survival rates were calculated at the larval, pupal and adult stages. Knockdown of both Dia and FliI results in 100% lethality at the pupal stage. C. Sarcomere phenotypes in the pupae. 100 APF pupae were dissected. The morphology of myofibrils and sarcomeres are visualized with Phalloidin (white) Scale bar: 5 μm D. Quantification of sarcomere size. Knockdown of both FliI and Dia significantly reduces sarcomere length (p<0.01) and width (p<0.0001) compared to individually depleting either of the 2 proteins (5 flies per genotype, n = 5 average sarcomere length or width measurements/fly). E. Localization of FliI in an IFM. Control flies were dissected 1 to 7 days after eclosion. Muscles are labeled with Phalloidin (white) and with an antibody against α-actinin (red). The localization of FliI is visualized with a FliI antibody (green), which co-localizes with α-actinin at the Z-discs. Scale bars: 5 μm. F. Sarcomere phenotype in IFMs with FliI overexpression and Dia knockdown. Morphology of myofibrils and sarcomeres are visualized by labeling with Phalloidin (white). Scale bar: 5μm. G. Quantification of sarcomere size (5 flies per genotype, n = 5 average sarcomere length or width measurements/fly). Expressing both dia RNAi and FliI::HA in the muscles with two copies of DMef2-Gal4 significantly reduces sarcomere length (p<0.001) and width (p<0.1) in comparison to dia RNAi or FliI::HA alone.
To first confirm that the fliI knockdown phenotype was not due to off-target effects, we created an fliI::HA rescue construct. When we expressed FliI::HA in muscles in a fliI knockdown background, we found that the sarcomere phenotype were 100% rescued (Figure 5A). Consistent with a function for the actin severing activity of FliI in sarcomerogenesis, overexpression of FliI::HA in otherwise wild-type muscles resulted in shorter (control: 3.6±0.11; fliI-HA: 2.8±0.11; p<0.01, 22% reduction in length on average) and slightly thinner (control: 1.7±0.15; fliI-HA: 1.5±0.20, p<0.1, 12% reduction in width on average) sarcomeres (Figure 5C and D). Despite the sarcomere size change, the orientation and the parallel alignment of the myofibrils were not disrupted with FliI expression (Figure 5C), suggesting that sarcomere size and myofibril alignment are regulated through independent mechanisms. In addition to the fliI rescue experiment, we also examined the flight muscle phenotype in fliI3 flies. fliI3 is a viable mutant allele with a specific point mutation (G601A) in the conserved gelsolin-like domain, which is thought to abrogate FliI’s severing ability (de Couet et al., 1995; Miklos and De Couet, 1990). Similar to fliI-RNAi flies, analysis of fliI3 mutant IFMs revealed bundles of actin filaments as well as shorter and thinner sarcomeres (Supplemental 4A–C). Altogether, our data indicate that the actin severing activity of FliI is required for proper sarcomere size.
Our screen revealed that knockdown of both Dia and FliI in muscle led to lethality (Figure 5B). Given that knockdown of either Dia or FliI alone resulted in smaller sarcomeres, we next investigated the actin phenotypes in IFMs that were knocked down for both Dia and FliI (Figure 5C and D). As double knockdown of FliI and Dia resulted in lethality prior to eclosion, we examined the IFMs in 100h APF pupae. We found that a double knockdown of Dia and FliI resulted in shorter and thinner sarcomeres compared to wild-type controls (length 2.1±0.05μm, 30% reduction; width 0.5±0.02μm, 50% reduction) or compared to a knockdown of either Dia (length: 2.3±0.03μm, p<0.01, ~10% reduction, width: 0.9±0.02μm, p<0.0001, ~40% reduction), or FliI alone (length: 2.3±0.04μm, p<0.01, ~10% reduction, width: 0.7±0.02μm, p<0.0001, ~30% reduction) (Figure 5C and D). Similar as fliI-RNAi, the fliI3 mutant allele enhances Dia knockdown phenotype by further reducing sarcomere width (0.7±0.07μm, p<0.05), but unlike fliI-RNAi, it does not further reduce sarcomere length in Dia knockdown flies (Supplemental Figure 4A–C). The long actin filament bundles that do not stain with Z disc markers and are characteristic of the FliI knockdown were still found in the FliI and Dia double knockdown (Figure 5C iv).
Both the viability and the sarcomere data suggested Dia and FliI work together to promote sarcomere growth. These observations are consistent with an earlier report from mammalian cell culture and in vitro experiments that found that FliI promotes Dia activity (Higashi et al., 2010) This work suggested that FliI directly binds to the Dia C-terminal DAD domain: there, it competitively accelerates the dissociation between DID and DAD domain and thus enhances the actin assembly activity of Dia (Higashi et al., 2010). To investigate whether such a direct interaction between Dia and FliI exists in Drosophila, we examined physical interactions between FliI and Dia in Drosophila S2 cells. We expressed FliI::HA and Dia::GFP in Drosophila S2 cells and tested whether FliI-Dia physically interacted by co-IP. Despite the strong genetic interaction in the developing adult musculature (Supplemental Table 2), we found no evidence of physical interactions between FliI and Dia in S2 cells (Supplemental Figure 4D). Moreover, the additive nature of the double knockdown sarcomere phenotype (Figure 5C and D) suggest that FliI and Dia act in parallel pathways that act together to regulate sarcomere size.
We also examined the localization of FliI in wild-type muscles during sarcomere growth and maintenance. Using antibody staining, we found that FliI localizes to the sarcomere Z-discs at day 1, day 3, and day 7 (Figure 5E). Unlike Dia (Figure 4A), which mainly localizes at the M-line during growth and relocates to Z-disc during maintenance, FliI is localized to the Z-disc and remains at the Z-disc throughout the growth and homeostasis phases (Figure 5E, Supplemental Figure 4E). The different localization patterns supported our data that FliI does not physically interact with Dia, at least during the period of IFM growth. Together, these data suggest that FliI does not work directly with Dia to regulate sarcomere size during growth.
Based on the phenotype of the sarcomeres, myofibrils, and actin bundles in Dia and FliI knockdown muscles, we generated an alternative hypothesis to explain how Dia and FliI could indirectly act together to regulate sarcomere size. We hypothesized that both the polymerization activity of Dia and the severing activity of FliI are needed to regulate thin filament size and, thus, sarcomere size. However, both activities are also needed to control the level of available G-actin: actin severed by FliI from a thin filament’s barbed end re-enters the G-actin pool, while Dia incorporates G-actin from this pool into thin filaments to regulate sarcomere length and width. This hypothesis would explain the sarcomere phenotypes that we observed in the Dia and FliI knockdown muscles: when both Dia and FliI levels are reduced in muscles, long actin filament bundles characteristic of the FliI knockdown were observed due to the lack of actin severing activity, and the pool of available G-actin is reduced. Due to the reduced G-actin pool, as well as reduced actin polymerization activity resulting from Dia knockdown, the sarcomeres are shorter and thinner (Figure 6).
Figure 6: Model: Dia and FliI collaborate to regulate sarcomere size and uniformity.
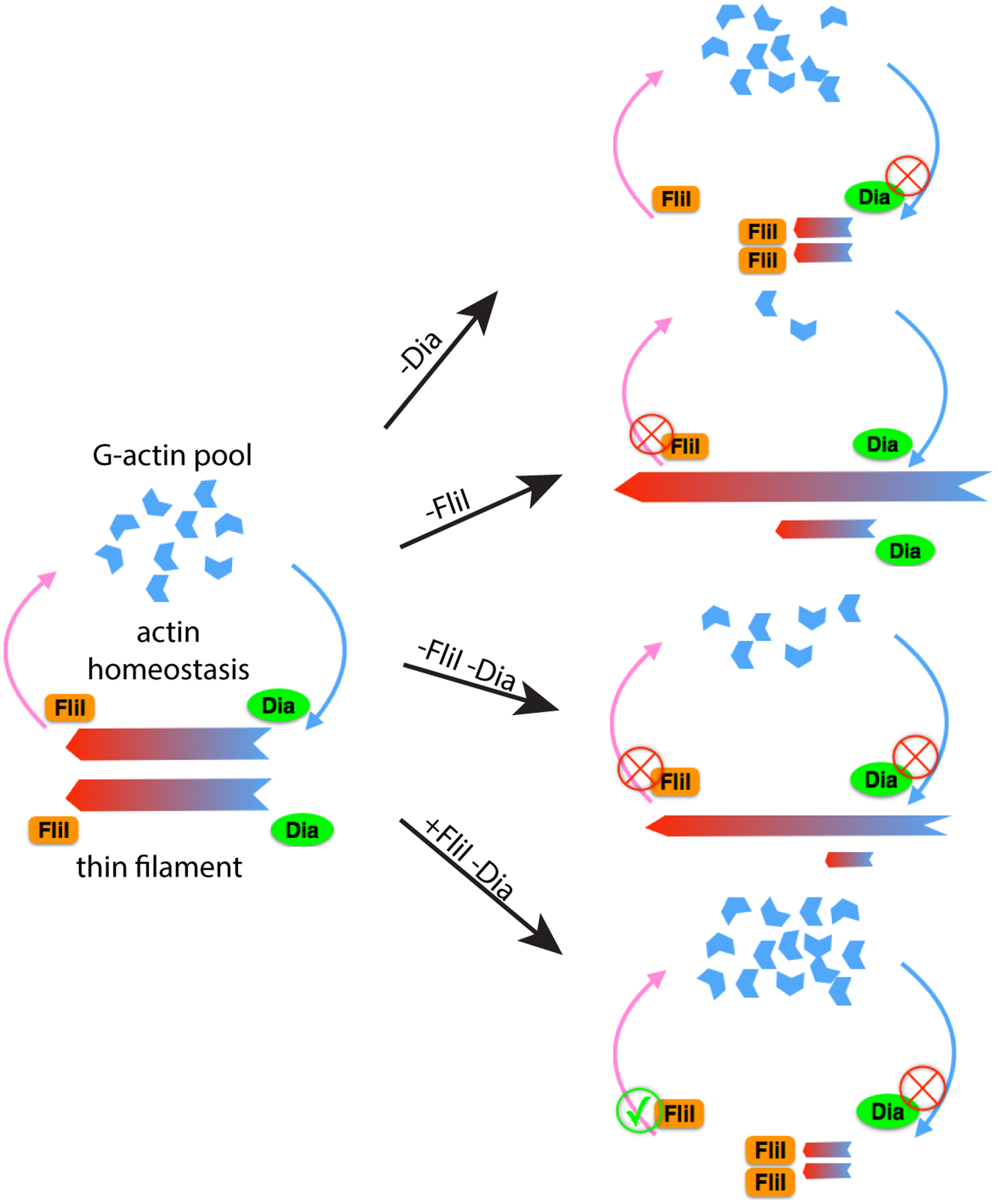
Under wild-type conditions, Dia and FliI act upon actin thin filaments during sarcomere growth to determine sarcomere size. Dia nucleates and polymerizes actin filaments, while FliI severs actin filaments. Together, Dia and FliI regulate the dynamics of actin thin filaments and maintain the balance between the F-actin and G-actin pools. Without Dia activity, actin filament elongation is impaired and the sarcomeres are shorter and thinner when compared to the control. Without FliI activity, long F-actin bundles form as a result of impaired actin-severing activity. Reduced actin severing also decreases the G-actin pool that limits actin polymerization by Diaphanous into the thin filament. As a result, the sarcomeres are shorter and thinner in comparison to the wild-type control. Knockdown of both FliI and Dia results in long F-actin bundles that form due to reduced actin severing activity and polymerization by residual Dia or other formins; however, the sarcomeres are even smaller than the single knock downs, due to both reduced G-actin pools and reduced actin elongation. When FliI is overexpressed in a background where Dia is knocked down, excessive actin severing and insufficient actin elongation results in sarcomeres that are smaller than just by manipulating either of the proteins individually.
As a test of our model, we predicted that we would see further shortening of sarcomeres when we express dia RNAi in a FliI overexpression background. In this context, we would have excessive actin filament severing caused by FliI overexpression but insufficient actin polymerization due to Dia knockdown. Consistent with our hypothesis, expressing dia RNAi and FliI::HA in muscles led to thin filaments that were both shorter (2.4±0.04μm, 33% reduction) and thinner (1.4±0.03μm, 18% reduction) compared to control as well as compared to expression of dia RNAi alone (length: 3.6±0.04μm, p<0.0001, width: 1.5±0.04μm, p<0.1), or FliI::HA alone (length: 2.8±0.02μm, p<0.0001, width: 1.6±0.04μm, p<0.1) (Figure 5F–G). Due to the excess severing capabilities with FliI overexpression, we did not observe the long actin bundles that were seen in Fli knockdown, which is also consistent with our hypothesis that these actin bundles are formed through insufficient severing activity (Figure 6).
Together, our data show that the function of Dia is required for sarcomere growth and maintenance. Moreover, the collaboration between Dia and FliI contributes to the regulation of sarcomere size. Dia and FliI localize at different ends of the thin filaments during growth. The actin polymerization activity of Dia and the F-actin severing activity of FliI coordinate to control actin dynamics and turnover in flight muscles.
Discussion:
In this study, we identified Diaphanous as a novel regulator of sarcomere size. We found that reduction of Dia function in the indirect flight muscles of Drosophila resulted in disorganized myofibrils that contained fraying actin filaments and smaller sarcomeres. As a functional readout of the sarcomeric defects that we have uncovered, the flies are flightless. Immunostaining of actin, TEM images, and localization of actin capping proteins suggest that the nucleation and elongation of actin thin filaments are impaired in the Dia knockdown muscles. We found that Dia localizes primarily to the M-lines during sarcomere growth but relocates to Z-discs as sarcomere growth ceases; these data suggest that Dia functions at the M-line during sarcomere growth to determine the length and width of sarcomere and at the Z-disc to maintain sarcomere size. Importantly, we also find that Dia genetically interacts with the actin severing protein Flightless I (FliI). Together we propose that these key actin regulators control sarcomere size through their actin polymerization and severing abilities and through their regulation of the balance between F-actin and G-actin pools (Figure 6).
Dia determines sarcomere size by regulating thin filament growth
Despite their nearly crystalline-like structures, sarcomeres are dynamic structures, with G-actin incorporating into, and depolymerizing from, thin filaments during sarcomere assembly, growth, and homeostasis (Littlefield et al., 2001; Mardahl-Dumesnil and Fowler, 2001; Perkins and Tanentzapf, 2014). How thin filaments are organized in sarcomeres has been under extensive investigation, and studies have reported numerous proteins that regulate thin filament length, such as Sallimus (Titin), Tropomodulin (Tmod), Lasp (Nebulin), Fhos, and DAAM. It has been proposed that the thin filament is composed of two segments: the proximal segment that is capped by CapZ at the Z-disc and its length is controlled by Nebulin, and the distal segment that is capped by Tmod and may undergo faster actin turnover (Gokhin and Fowler, 2013). Our observations fit with this two-segment model of thin filament growth and are consistent with Dia’s activity in polymerizing actin filaments: the localization of Dia, as well as the short actin thin filaments found in the Dia knockdown suggest that Dia elongates the distal segment of the thin filament during the sarcomere growth phase. The localization of Dia during the maintenance phase suggests that Dia regulates G-actin polymerization mainly at the barbed ends of the proximal segment after muscle maturation. Based on the thinner sarcomere phenotype in the Dia knockdown muscles, Dia may also function to nucleate and elongate additional actin filaments. Further analysis of how actin thin filaments are added to the Z disc to increase the width of the sarcomere remains a goal of future studies.
It is notable that in our Dia knockdown studies, sarcomeres and myofibrils still form. This is likely due to incomplete Dia knockdown as well as the partial redundant effects of other formins, including DAAM and Fhos (Molnár et al., 2014; Shwartz et al., 2016). We have shown that residual Dia could be detected in the muscles with Dia knockdown (Supplemental Figure 1E). This incomplete Dia knockdown would explain why the earlier steps in myogenesis, such as myoblast fusion and myotendinous junction formation, are unaffected, despite Dia’s known roles in these processes (Deng et al., 2015). Nevertheless, the muscle specific knock down allowed us to investigate Dia’s role in sarcomerogenesis. By using temporally-restricted induction of UAS-dia-RNAi after eclosion, we were able to separate the role of Dia in sarcomere formation and sarcomere maintenance. We found that reducing Dia levels after the sarcomere is fully formed results in smaller sarcomeres, indicating that Dia is required to maintain the structure and function of sarcomere.
In addition to Dia, DAAM, another formin family member, is known to be expressed in Drosophila IFM and to play a critical role in the initial assembly of thin filament (Molnár et al., 2014). Likewise, other formins, such as Fhos, have also been reported to determine and maintain sarcomere size in C. elegans body wall muscles (Mi-Mi and Pruyne, 2015). A recent study in Drosophila showed that Fhos plays an important role in the assembly and growth of the sarcomere thin-filament during adult myogenesis (Shwartz et al., 2016). Thus, redundant roles with these other formins could explain why sarcomeres still form and actin polymerization is still detected upon Dia knockdown. However, it is notable that the other formins do not completely complement Dia’s function, as the sarcomeres are smaller and the myofibrils are disorganized, in Dia knockdown muscles.
Dia works with the severing protein FliI to regulate sarcomere size
We identified FliI as a functional partner of Dia in sarcomere formation, growth, and homeostasis. In vitro experiments have reported that FliI enhances the activity of Dia by directly binding to the C-terminal of Dia (Higashi et al., 2010). However, the different localization patterns of Dia and FliI in the sarcomeres during growth suggest that it is unlikely that FliI and Dia directly interact with each other during this period. With the caveat that we are not getting complete removal of Dia or FliI, the additive effect of the double knockdown on sarcomere size would also suggest the two proteins working in parallel pathways. These observations were further supported by Co-IP experiments with FliI::HA and Dia::GFP from S2 cells, which indicated that FliI and Dia did not physically interact under our IP conditions. We note that our S2 cell experiment cannot completely rule out the possibility that Dia and FliI physically interact in the adult Drosophila muscles. Therefore, it remains possible that Dia and FliI directly interact to maintain sarcomere structure in mature muscle.
Staining for F-actin in muscles with FliI knockdown revealed two groups of actin structures: group one had multiple long actin filaments without Z-disc proteins and group two were myofibrils with shorter and thinner sarcomeres with M-line and Z-disc proteins. Based on these two groups of actin structures, as well as FliI’s known role in actin severing (Burger et al., 2016; Nag et al., 2013), we propose that the severing activity of FliI and the polymerization activity of Dia regulate thin filament length and that these actin regulators collaborate to control actin pools required for these activities (Figure 6). Our model suggests that during sarcomere formation, FliI localizes to the barbed ends of thin filaments, severing actin filaments. The severed and depolymerized G-actin then enters into the muscle G-actin pool and is subsequently incorporated via Diaphanous (and other formins) into the thin filament’s pointed ends. Our model (Figure 6) explains the actin/sarcomere phenotypes that we observed in the FliI-Dia genetic experiments. In FliI and Dia double knockdown muscles, reduced FliI activity leads to reduced severing of thin filaments and reduced G-actin pools; reduced Dia activity leads to impaired actin polymerization at the thin filaments. Therefore, the sarcomeres are both shorter and thinner than in each single knockdown. In muscles with FliI overexpression and Dia knockdown, increased FliI activity leads to enhance severing and increased G-actin levels. Reduction of Dia in this background, reduces polymerization and use of the available G-actin. Hence, the sarcomeres are still shorter and thinner than sarcomeres in muscles that only express dia RNAi or FliI::HA. To further support our hypothesis about G-actin pool, we attempted to rescue the FliI knockdown phenotype by overexpressing an IFM specific actin, Actin88F, in the muscles. However, over-expression of Actin88F alone resulted in shorter, yet wider, sarcomeres. Future experiments using different actin reagents will provide further tests of our model.
The Drosophila fliI gene encodes a protein that shares a 56% identity with its human homologue. The human FLII gene is located at chromosome 17p11.2. This region of the chromosome is deleted in patients with Smith-Magenis Syndrome. With the 29 genes located on chromosome 17p11.2, RAI1 is the gene located at this region of chromosome that is believed to be the cause of Smith-Magenis Syndrome. However, there are clinical features that are seen in patients carrying the chromosome deletion and not seen in patients with RAI1 mutations alone; these include cardiac abnormality (45% vs 20%), renal anomaly (19% vs 0%), and hearing loss (67% vs 20%) (Bi et al., 2004), suggesting in addition to RAI1, other genes on the region of the chromosome might contribute to the syndrome. Interestingly, these clinical features are all related to mutations in Dia or Dia-related formins (Lynch et al., 1997; Rosado et al., 2014; Sun et al., 2014; Thelen et al., 2015). Hence, the interactions that we have uncovered between Dia and FliI provide a potential explanation for the expressivity and penetrance of Smith-Magenis Syndrome.
Thick filaments are disrupted in dia knockdown muscles
In addition to thin filaments, the assembly and stability of thick filaments also play a role in controlling sarcomere size (Contompasis et al., 2010; Reedy et al., 2000). Thick filaments are myosin-based structures with myosin bundles cross-linked at the M-line. We found that with Dia knockdown, the thick filaments were shorter and each sarcomere has fewer thick filaments (Figure 1F, Supplemental Figure 1F–G). The integrity of M-line was not disrupted in muscles in Dia knockdown condition (Supplemental Figure 5A). Thus our data suggest that Dia also plays a role in thick filaments organization. Interestingly, it has been reported that microtubules are required to transport muscle myosin to the M line to form the thick filaments in mouse muscles(Pizon et al., 2005; Pizon et al., 2002), and more recently, it has been shown that microtubules are required for myofibril and sarcomere assembly in the Drosophila IFMs (Dhanyasi et al., 2020). Given that Dia regulates microtubule formation and stabilization (Palazzo et al., 2001), we examined microtubule organization in the muscle and found that the microtubule network was disrupted (Supplemental Figure 5). Whether Dia regulates thick filament organization and sarcomere size through microtubule organization remains to be investigated.
In conclusion, we report that Diaphanous is a novel actin regulator that controls sarcomere assembly and maintenance. Dia regulates the formation of actin networks in myofibrils, and therefore controls thin filament formation. We also identified FliI as the interacting partner of Dia. We propose a model that describes FliI and Dia working together to generate sarcomere size via their severing and polymerization activities on thin filaments, respectively, and via regulation of G-actin levels. These data identify new proteins involved in sarcomere formation and homeostasis that may be likely targets in human muscle disease.
Supplementary Material
Acknowledgements:
We thank S. Windner for technical support and Iliona Wolfowicz for the Drosophila adult image in the graphical abstract. We also thank J. Zallen, J. Dittman for discussion and comments. This study was supported by the National Institute of Health (NIH) [GM078318, AR108981, AR067361] to MKB and NCI [P30 CA 008748] core grant to MSKCC.
Footnotes
Competing interests: None
References:
- Afshar K, Stuart B, Wasserman SA, 2000. Functional analysis of the Drosophila diaphanous FH protein in early embryonic development. Development 127, 1887–1897. [DOI] [PubMed] [Google Scholar]
- Arimura T, Takeya R, Ishikawa T, Yamano T, Matsuo A, Tatsumi T, Nomura T, Sumimoto H, Kimura A, 2013. Dilated cardiomyopathy-associated FHOD3 variant impairs the ability to induce activation of transcription factor serum response factor. Circ J 77, 2990–2996. [DOI] [PubMed] [Google Scholar]
- Babcock DT, Ganetzky B, 2014. An improved method for accurate and rapid measurement of flight performance in Drosophila. J Vis Exp, e51223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Hartwig JH, Perrimon N, 2007. SALS, a WH2-domain-containing protein, promotes sarcomeric actin filament elongation from pointed ends during Drosophila muscle growth. Dev Cell 13, 828–842. [DOI] [PubMed] [Google Scholar]
- Balakrishnan M, Yu SF, Chin SM, Soffar DB, Windner SE, Goode BL, Baylies MK, 2020. Cofilin Loss in Drosophila Muscles Contributes to Muscle Weakness through Defective Sarcomerogenesis during Muscle Growth. Cell Rep 32, 107893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Lee J, Venkatesh K, Wu CF, Hasan G, 2004. Loss of flight and associated neuronal rhythmicity in inositol 1,4,5-trisphosphate receptor mutants of Drosophila. J Neurosci 24, 7869–7878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearer EL, Prakash JM, Li Z, 2002. Actin dynamics in platelets. Int Rev Cytol 217, 137–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi W, Saifi GM, Shaw CJ, Walz K, Fonseca P, Wilson M, Potocki L, Lupski JR, 2004. Mutations of RAI1, a PHD-containing protein, in nondeletion patients with Smith-Magenis syndrome. Hum Genet 115, 515–524. [DOI] [PubMed] [Google Scholar]
- Bilancia CG, Winkelman JD, Tsygankov D, Nowotarski SH, Sees JA, Comber K, Evans I, Lakhani V, Wood W, Elston TC, Kovar DR, Peifer M, 2014. Enabled negatively regulates diaphanous-driven actin dynamics in vitro and in vivo. Dev Cell 28, 394–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N, 1993. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415. [DOI] [PubMed] [Google Scholar]
- Burger D, Fickentscher C, de Moerloose P, Brandt KJ, 2016. F-actin dampens NLRP3 inflammasome activity via Flightless-I and LRRFIP2. Sci Rep 6, 29834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkart C, Qiu F, Brendel S, Benes V, Hååg P, Labeit S, Leonard K, Bullard B, 2007. Modular proteins from the Drosophila sallimus (sls) gene and their expression in muscles with different extensibility. J Mol Biol 367, 953–969. [DOI] [PubMed] [Google Scholar]
- Caygill EE, Brand AH, 2016. The GAL4 System: A Versatile System for the Manipulation and Analysis of Gene Expression. Methods Mol Biol 1478, 33–52. [DOI] [PubMed] [Google Scholar]
- Chesarone MA, DuPage AG, Goode BL, 2010. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat Rev Mol Cell Biol 11, 62–74. [DOI] [PubMed] [Google Scholar]
- Clarkson E, Costa CF, Machesky LM, 2004. Congenital myopathies: diseases of the actin cytoskeleton. J Pathol 204, 407–417. [DOI] [PubMed] [Google Scholar]
- Contompasis JL, Nyland LR, Maughan DW, Vigoreaux JO, 2010. Flightin is necessary for length determination, structural integrity, and large bending stiffness of insect flight muscle thick filaments. J Mol Biol 395, 340–348. [DOI] [PubMed] [Google Scholar]
- Daou P, Hasan S, Breitsprecher D, Baudelet E, Camoin L, Audebert S, Goode BL, Badache A, 2014. Essential and nonredundant roles for Diaphanous formins in cortical microtubule capture and directed cell migration. Mol Biol Cell 25, 658–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Couet HG, Fong KS, Weeds AG, McLaughlin PJ, Miklos GL, 1995. Molecular and mutational analysis of a gelsolin-family member encoded by the flightless I gene of Drosophila melanogaster. Genetics 141, 1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng S, Bothe I, Baylies MK, 2015. The Formin Diaphanous Regulates Myoblast Fusion through Actin Polymerization and Arp2/3 Regulation. PLoS Genet 11, e1005381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanyasi N, VijayRaghavan K, Shilo BZ, Schejter ED, 2020. Microtubules provide guidance cues for myofibril and sarcomere assembly and growth. Dev Dyn. [DOI] [PubMed] [Google Scholar]
- Evangelista M, Pruyne D, Amberg DC, Boone C, Bretscher A, 2002. Formins direct Arp2/3-independent actin filament assembly to polarize cell growth in yeast. Nat Cell Biol 4, 260–269. [DOI] [PubMed] [Google Scholar]
- Fernandes I, Schöck F, 2014. The nebulin repeat protein Lasp regulates I-band architecture and filament spacing in myofibrils. J Cell Biol 206, 559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes JJ, Celniker SE, VijayRaghavan K, 1996. Development of the indirect flight muscle attachment sites in Drosophila: role of the PS integrins and the stripe gene. Dev Biol 176, 166–184. [DOI] [PubMed] [Google Scholar]
- Geisbrecht ER, Montell DJ, 2004. A role for Drosophila IAP1-mediated caspase inhibition in Rac-dependent cell migration. Cell 118, 111–125. [DOI] [PubMed] [Google Scholar]
- Gokhin DS, Fowler VM, 2013. A two-segment model for thin filament architecture in skeletal muscle. Nat Rev Mol Cell Biol 14, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi T, Ikeda T, Murakami T, Shirakawa R, Kawato M, Okawa K, Furuse M, Kimura T, Kita T, Horiuchi H, 2010. Flightless-I (Fli-I) regulates the actin assembly activity of diaphanous-related formins (DRFs) Daam1 and mDia1 in cooperation with active Rho GTPase. J Biol Chem 285, 16231–16238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homem CC, Peifer M, 2008. Diaphanous regulates myosin and adherens junctions to control cell contractility and protrusive behavior during morphogenesis. Development 135, 1005–1018. [DOI] [PubMed] [Google Scholar]
- Hunt LC, Demontis F, 2013. Whole-mount immunostaining of Drosophila skeletal muscle. Nat Protoc 8, 2496–2501. [DOI] [PubMed] [Google Scholar]
- Iskratsch T, Reijntjes S, Dwyer J, Toselli P, Dégano IR, Dominguez I, Ehler E, 2013. Two distinct phosphorylation events govern the function of muscle FHOD3. Cell Mol Life Sci 70, 893–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janmey PA, Chaponnier C, Lind SE, Zaner KS, Stossel TP, Yin HL, 1985. Interactions of gelsolin and gelsolin-actin complexes with actin. Effects of calcium on actin nucleation, filament severing, and end blocking. Biochemistry 24, 3714–3723. [DOI] [PubMed] [Google Scholar]
- Kan OM, Takeya R, Abe T, Kitajima N, Nishida M, Tominaga R, Kurose H, Sumimoto H, 2012. Mammalian formin Fhod3 plays an essential role in cardiogenesis by organizing myofibrillogenesis. Biol Open 1, 889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova N, Bobinnec Y, Fouix S, Huitorel P, Debec A, 2006. Jupiter, a new Drosophila protein associated with microtubules. Cell Motil Cytoskeleton 63, 301–312. [DOI] [PubMed] [Google Scholar]
- Kinosian HJ, Newman J, Lincoln B, Selden LA, Gershman LC, Estes JE, 1998. Ca2+ regulation of gelsolin activity: binding and severing of F-actin. Biophys J 75, 3101–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremneva E, Makkonen MH, Skwarek-Maruszewska A, Gateva G, Michelot A, Dominguez R, Lappalainen P, 2014. Cofilin-2 controls actin filament length in muscle sarcomeres. Dev Cell 31, 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlefield R, Almenar-Queralt A, Fowler VM, 2001. Actin dynamics at pointed ends regulates thin filament length in striated muscle. Nat Cell Biol 3, 544–551. [DOI] [PubMed] [Google Scholar]
- Littlefield RS, Fowler VM, 2008. Thin filament length regulation in striated muscle sarcomeres: pointed-end dynamics go beyond a nebulin ruler. Semin Cell Dev Biol 19, 511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Adler PN, 2015. The diaphanous gene of Drosophila interacts antagonistically with multiple wing hairs and plays a key role in wing hair morphogenesis. PLoS One 10, e0115623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch ED, Lee MK, Morrow JE, Welcsh PL, León PE, King MC, 1997. Nonsyndromic deafness DFNA1 associated with mutation of a human homolog of the Drosophila gene diaphanous. Science 278, 1315–1318. [PubMed] [Google Scholar]
- Malfatti E, Romero NB, 2016. Nemaline myopathies: State of the art. Rev Neurol (Paris) 172, 614–619. [DOI] [PubMed] [Google Scholar]
- Mardahl-Dumesnil M, Fowler VM, 2001. Thin filaments elongate from their pointed ends during myofibril assembly in Drosophila indirect flight muscle. J Cell Biol 155, 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubayashi Y, Coulson-Gilmer C, Millard TH, 2015. Endocytosis-dependent coordination of multiple actin regulators is required for wound healing. J Cell Biol 210, 419–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi-Mi L, Pruyne D, 2015. Loss of Sarcomere-associated Formins Disrupts Z-line Organization, but does not Prevent Thin Filament Assembly in Caenorhabditis elegans Muscle. J Cytol Histol 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklos GL, De Couet HG, 1990. The mutations previously designated as flightless-I3, flightless-O2 and standby are members of the W-2 lethal complementation group at the base of the X-chromosome of Drosophila melanogaster. J Neurogenet 6, 133–151. [DOI] [PubMed] [Google Scholar]
- Molnár I, Migh E, Szikora S, Kalmár T, Végh AG, Deák F, Barkó S, Bugyi B, Orfanos Z, Kovács J, Juhász G, Váró G, Nyitrai M, Sparrow J, Mihály J, 2014. DAAM is required for thin filament formation and Sarcomerogenesis during muscle development in Drosophila. PLoS Genet 10, e1004166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag S, Larsson M, Robinson RC, Burtnick LD, 2013. Gelsolin: the tail of a molecular gymnast. Cytoskeleton (Hoboken) 70, 360–384. [DOI] [PubMed] [Google Scholar]
- Nowotarski SH, McKeon N, Moser RJ, Peifer M, 2014. The actin regulators Enabled and Diaphanous direct distinct protrusive behaviors in different tissues during Drosophila development. Mol Biol Cell 25, 3147–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochala J, 2008. Thin filament proteins mutations associated with skeletal myopathies: defective regulation of muscle contraction. J Mol Med (Berl) 86, 1197–1204. [DOI] [PubMed] [Google Scholar]
- Ono S, 2010. Dynamic regulation of sarcomeric actin filaments in striated muscle. Cytoskeleton (Hoboken) 67, 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orfanos Z, Leonard K, Elliott C, Katzemich A, Bullard B, Sparrow J, 2015. Sallimus and the dynamics of sarcomere assembly in Drosophila flight muscles. J Mol Biol 427, 2151–2158. [DOI] [PubMed] [Google Scholar]
- Palazzo AF, Cook TA, Alberts AS, Gundersen GG, 2001. mDia mediates Rho-regulated formation and orientation of stable microtubules. Nat Cell Biol 3, 723–729. [DOI] [PubMed] [Google Scholar]
- Pappas CT, Bhattacharya N, Cooper JA, Gregorio CC, 2008. Nebulin interacts with CapZ and regulates thin filament architecture within the Z-disc. Mol Biol Cell 19, 1837–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas CT, Krieg PA, Gregorio CC, 2010. Nebulin regulates actin filament lengths by a stabilization mechanism. J Cell Biol 189, 859–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson C, Eaton BA, Davis GW, 2008. Formin-dependent synaptic growth: evidence that Dlar signals via Diaphanous to modulate synaptic actin and dynamic pioneer microtubules. J Neurosci 28, 11111–11123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins AD, Tanentzapf G, 2014. An ongoing role for structural sarcomeric components in maintaining Drosophila melanogaster muscle function and structure. PLoS One 9, e99362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizon V, Gerbal F, Diaz CC, Karsenti E, 2005. Microtubule-dependent transport and organization of sarcomeric myosin during skeletal muscle differentiation. Embo j 24, 3781–3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizon V, Iakovenko A, Van Der Ven PF, Kelly R, Fatu C, Fürst DO, Karsenti E, Gautel M, 2002. Transient association of titin and myosin with microtubules in nascent myofibrils directed by the MURF2 RING-finger protein. J Cell Sci 115, 4469–4482. [DOI] [PubMed] [Google Scholar]
- Pruyne D, Evangelista M, Yang C, Bi E, Zigmond S, Bretscher A, Boone C, 2002. Role of formins in actin assembly: nucleation and barbed-end association. Science 297, 612–615. [DOI] [PubMed] [Google Scholar]
- Reedy MC, Beall C, 1993. Ultrastructure of developing flight muscle in Drosophila. I. Assembly of myofibrils. Dev Biol 160, 443–465. [DOI] [PubMed] [Google Scholar]
- Reedy MC, Bullard B, Vigoreaux JO, 2000. Flightin is essential for thick filament assembly and sarcomere stability in Drosophila flight muscles. J Cell Biol 151, 1483–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado M, Barber CF, Berciu C, Feldman S, Birren SJ, Nicastro D, Goode BL, 2014. Critical roles for multiple formins during cardiac myofibril development and repair. Mol Biol Cell 25, 811–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousso T, Shewan AM, Mostov KE, Schejter ED, Shilo BZ, 2013. Apical targeting of the formin Diaphanous in Drosophila tubular epithelia. Elife 2, e00666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DA, Hug C, Cooper JA, 1995. Inhibition of CapZ during myofibrillogenesis alters assembly of actin filaments. J Cell Biol 128, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnorrer F, Schönbauer C, Langer CC, Dietzl G, Novatchkova M, Schernhuber K, Fellner M, Azaryan A, Radolf M, Stark A, Keleman K, Dickson BJ, 2010. Systematic genetic analysis of muscle morphogenesis and function in Drosophila. Nature 464, 287–291. [DOI] [PubMed] [Google Scholar]
- Sewry CA, Laitila JM, Wallgren-Pettersson C, 2019. Nemaline myopathies: a current view. J Muscle Res Cell Motil 40, 111–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shwartz A, Dhanyasi N, Schejter ED, Shilo BZ, 2016. The Drosophila formin Fhos is a primary mediator of sarcomeric thin-filament array assembly. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spletter ML, Barz C, Yeroslaviz A, Schönbauer C, Ferreira IR, Sarov M, Gerlach D, Stark A, Habermann BH, Schnorrer F, 2015. The RNA-binding protein Arrest (Bruno) regulates alternative splicing to enable myofibril maturation in Drosophila flight muscle. EMBO Rep 16, 178–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub KL, Stella MC, Leptin M, 1996. The gelsolin-related flightless I protein is required for actin distribution during cellularisation in Drosophila. J Cell Sci 109 (Pt 1), 263–270. [DOI] [PubMed] [Google Scholar]
- Sun H, Al-Romaih KI, MacRae CA, Pollak MR, 2014. Human Kidney Disease-causing INF2 Mutations Perturb Rho/Dia Signaling in the Glomerulus. EBioMedicine 1, 107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman MA, Baqué S, Uhm CS, Daniels MP, Price RL, Simpson D, Terracio L, Kedes L, 1998. Altered expression of tropomodulin in cardiomyocytes disrupts the sarcomeric structure of myofibrils. Circ Res 82, 94–105. [DOI] [PubMed] [Google Scholar]
- Taniguchi K, Takeya R, Suetsugu S, Kan OM, Narusawa M, Shiose A, Tominaga R, Sumimoto H, 2009. Mammalian formin fhod3 regulates actin assembly and sarcomere organization in striated muscles. J Biol Chem 284, 29873–29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelen S, Abouhamed M, Ciarimboli G, Edemir B, Bähler M, 2015. Rho GAP myosin IXa is a regulator of kidney tubule function. Am J Physiol Renal Physiol 309, F501–513. [DOI] [PubMed] [Google Scholar]
- Tu MS, Daniel TL, 2004. Cardiac-like behavior of an insect flight muscle. J Exp Biol 207, 2455–2464. [DOI] [PubMed] [Google Scholar]
- Webb RL, Zhou MN, McCartney BM, 2009. A novel role for an APC2-Diaphanous complex in regulating actin organization in Drosophila. Development 136, 1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Eng CH, Schmoranzer J, Cabrera-Poch N, Morris EJ, Chen M, Wallar BJ, Alberts AS, Gundersen GG, 2004. EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration. Nat Cell Biol 6, 820–830. [DOI] [PubMed] [Google Scholar]
- Wooten EC, Hebl VB, Wolf MJ, Greytak SR, Orr NM, Draper I, Calvino JE, Kapur NK, Maron MS, Kullo IJ, Ommen SR, Bos JM, Ackerman MJ, Huggins GS, 2013. Formin homology 2 domain containing 3 variants associated with hypertrophic cardiomyopathy. Circ Cardiovasc Genet 6, 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.