

Abstract
Hepatocellular carcinoma (CC) is a common and deadly cancer with complex molecular pathogenesis. Little is known about dual‐specificity phosphatases (DUSPs) in HCC. We investigated DUSP9 expression in human HCC, associations between DUSP9 and patient outcomes, and effects of altered DUSP9 expression on HCC biology. We studied public data sets as well as 196 patients at our institution who had HCC resections. Quantitative real‐time reverse transcription polymerase chain reaction and western blot demonstrated that DUSP9 expression was increased >10‐fold in HCC compared to adjacent liver and healthy controls (P = 0.005). Kaplan‐Meier and multivariable regression analyses revealed that higher DUSP9 expression was associated with shorter disease‐free survival (high DUSP9, 1.6; 95% confidence interval, 0.9‐2.3 vs. low DUSP9, 3.4; 95% confidence interval, 1.8‐5.0 years; P = 0.04) and increased risk of recurrence (hazard ratio 1.55; 95% confidence interval, 1.01‐2.67; P = 0.05) after resection. DUSP9 complementary DNA (cDNA) was cloned using rapid amplification of cDNA ends, revealing two DUSP9 isoforms in human HCC cells. Studies of transcriptional regulation using promoter‐luciferase reporter constructs suggested that DUSP9 transcription is regulated by E26 transformation‐specific transcription factors. Proliferation of hepatic cells in vitro was enhanced by lentiviral‐mediated overexpression of DUSP9. In contrast, DUSP9 knockout HCC cells generated using clustered regularly interspaced short palindromic repeats (CRISPR) demonstrated decreased HCC proliferation and doxorubicin resistance in vitro and impaired xenograft growth in vivo. RNA sequencing, gene set enrichment, and network/pathway analysis revealed that DUSP9 knockout is associated with activation of protein kinase activity and apoptosis. Conclusion: DUSP9 regulates cell proliferation and predicts recurrence after surgery in HCC. DUSP9 may represent a novel prognostic candidate and therapeutic target. Additional studies are warranted to further explore the role and regulation of DUSP9 in HCC.
Abbreviations
- AFP
alpha‐fetoprotein
- Bp
base pair
- Cas9n
clustered regularly interspaced short palindromic repeats‐associated protein 9
nickase
- cDNA
complementary DNA
- CDS
coding sequence
- CI
confidence interval
- CRISPR
clustered regularly interspaced short palindromic repeats
- DEG
differentially expressed gene
- DFS
disease‐free survival
- DUSP
dual‐specificity phosphatase
- ECM
extracellular matrix
- ERK
extracellular signal‐regulated kinase
- ETS
E26 transformation specific
- Fer
ferritin
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- GEO
Gene Expression Omnibus
- GEP
gene expression profiling
- GFP
green fluorescent protein
- GO
gene ontology
- GPC3
glypican 3
- h
human
- HCC
hepatocellular carcinoma
- HR
hazard ratio
- IQR
interquartile range
- KO
knockout
- LV
lentiviral
- MAPK
mitogen‐activated protein kinase
- mEF1
mitochondrial elongation factor 1
- MKP
mitogen‐activated protein kinase phosphatase
- mRNA
messenger RNA
- MW
molecular weight
- OS
overall survival
- p
plasmid
- PCR
polymerase chain reaction
- PP5
PLC/PRF/5 cell line
- qPCR
quantitative polymerase chain reaction
- RACE
rapid amplification of complementary DNA ends
- RNA‐Seq
RNA sequencing
- sgRNA
single‐guide RNA
- TCGA
The Cancer Genome Atlas
- TF
transcription factor
- UHN
University Health Network
- WST
water‐soluble tetrazolium salt
- WT
wild type
Hepatocellular carcinoma (HCC) accounts for 75%‐85% of primary liver cancers.( 1 ) Curative surgery or ablation is available for early stage HCC, although recurrence is common.( 2 , 3 , 4 ) For advanced disease, median survival is less than 1 year.( 5 )
Identification of mechanisms contributing to HCC is necessary for new therapeutic strategies. HCC has complex pathogenesis,( 6 ) and recent work suggests that processes important in stem/progenitor cells may play a role.( 7 ) Mitogen‐activated protein kinase (MAPK) signaling is a major oncogenic pathway active in HCC as well as normal liver development.( 8 , 9 ) Dual‐specificity phosphatases (DUSPs), also known as MAPK phosphatases (MKPs), regulate MAPKs by dephosphorylating phosphotyrosine and phosphoserine/phosphothreonine residues on extracellular signal‐regulated kinase (ERK), c‐Jun N‐terminal kinase, and p38.( 10 ) DUSPs are increasingly recognized to participate in many cellular processes.( 11 , 12 ) While dysregulated MAPK signaling is a driver of HCC against which therapies are being targeted,( 8 ) the role of DUSPs remains poorly described.
In this study, we focused on DUSP9 (MKP4), the only DUSP with increased expression in both fetal liver and HCC. We characterized DUSP9 expression in human HCC, determined whether DUSP9 was associated with clinical outcomes, and investigated the effects of altered DUSP9 expression on HCC biology.
Materials and Methods
Analysis of Public Data Sets
Microarray data on human fetal liver (FLiver) versus normal adult liver (GSE1133) and HCC tumors versus adjacent normal tissues (GSE25097) were downloaded from Gene Expression Omnibus (GEO) DataSets (ncbi.nlm.nih.gov/gds) using the R package GEOquery. Human NCI60 cell lines data (NCI60_U133A_CEL) were obtained from BioGPS (biogps.org/downloads/). Data were analyzed using limma version 3.42.2. The Cancer Genome Atlas (TCGA) liver cancer HCC IlluminaHiSeq RNA sequencing (RNA‐Seq) data were acquired from the University of California Santa Cruz Xena Browser (xenabrowser.net/datapages/) for correlation expression analysis by running the ggscatterstats function of the R package ggstatsplot (rdocumentation.org/packages/ggstatsplot). DUSP9 pan‐cancer analysis was conducted using Gene Expression Profiling (GEP) Interactive Analysis 2 (GEPIA2) web tools (gepia2.cancer‐pku.cn/#index). Expression analysis of DUSPs was conducted using GraphPad Prism 8 software (San Diego, CA) based on TCGA data. To assess whether DUSP9 was associated with recurrence after resection for HCC, we used TCGA clinical data( 13 ) and grouped patients based on median DUSP9 expression (i.e., fiftieth percentile).
Analysis of Patients in the University Health Network
Research Ethics Board approval was obtained, and written informed consent was provided by patients, who were managed by international guidelines.( 14 ) All patients ≥18 years of age who underwent liver resection for HCC between October 1, 2009, and November 30, 2017, were included. Specimens were sectioned within 30 minutes of removal, and viable tumor tissue and adjacent nontumorous liver tissue were collected. Demographics and tumor characteristics were extracted from a prospectively maintained database and analyzed retrospectively. Diagnosis of recurrence was based on standard radiologic criteria or biopsy.
Cell Culture
Cell lines were incubated at 37°C in 5% CO2 and confirmed negative for mycoplasma. Human normal liver cell line THLE‐2 and HCC cell lines HepG2 and PLC/PRF/5 cell line (PP5) were from the American Type Culture Collection (CRL‐2706, HB‐8065, and CRL‐8024); HepaRG bipotential hepatic progenitor cells were from MilliporeSigma (MMHPR116); Hep3B HCC cells were from Dr. Burton Yang (Sunnybrook Research Institute, Toronto, Canada); and Huh7 HCC cells were from Dr. Paolo Parini (Karolinska Institute, Sweden). Cells were cultured according to vendors’ instructions. HCC cells were cultured in Low Glucose 1X modified Eagle’s medium (10370021; Gibco) plus 1X GlutaMAX, 1 mM sodium pyruvate (11360070; Gibco), and 10% fetal bovine serum. Cells were counted with a Countess II FL cell counter (AMQAF1000; Thermo Fisher). Cell lines were authenticated by short tandem repeat (STR) genotyping using the AmpFlSTR Identifiler Polymerase Chain Reaction (PCR) Amplification Kit (4322288; Thermo Fisher) on an ABI 3100 genetic analyzer and analyzed with GeneMapper.
RNA Isolation and Quantitative PCR
Total RNA was isolated using the PureLink RNA Minikit (Invitrogen) followed by DNA elimination using the TURBO DNA‐free Kit (AM1907; Ambion). Major vault protein human liver/fetal liver total RNA pool (540017/540173; Agilent) was used as control. Complementary DNA (cDNA) was synthesized by SuperScript IV reverse transcriptase (18090050; Thermo Fisher) using random hexamers (SO142; Thermo Scientific) from 5 µg total RNA. Quantitative PCR (qPCR) was performed with a LightCycler 480 SYBR Green I Master (04707516001; Roche) on a LightCycler 480 real‐time PCR platform (Roche). Primers are listed in Supporting Tables S1 and S3. Messenger RNA (mRNA) expression of relevant genes was normalized against glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). Quantitative analysis was performed according to the 2−ΔΔCt method. PCR products were recovered from 384‐well plates, purified using GenepHlow Gel/PCR isolation kit (DFH300; Geneaid Biotech), ligated into plasmid (p)GEM‐T Vector (A3600; Promega), and sequenced.
Rapid Amplification of cDNA Ends PCR
Total RNA of HepG2 and Huh7 cell lines and HCC‐13 tumor tissues were pooled (1 μg per sample) for cDNA synthesis using the SMARTer rapid amplification of cDNA ends (RACE) 5’/3’ Kit (634858; Takara Bio) with a nested PCR strategy. Universal primer mix and DUSP9‐specific primer D9‐r were introduced in the first round of amplification. We used 1:100 times diluted PCR products for the secondary nested PCR with forward primer NUPin and BC034936‐specific reverse primer Bc‐r (Supporting Table S1). PCR products were sequenced.
Luciferase Reporter Assay
The pBME vector inserted with the DUSP9 promoter (−2,000 base pair [bp] to +489 bp of NM_001395) was synthesized by Biomatik (Kitchener, Canada) as a PCR template. DUSP9 promoter plasmids were constructed by amplifying promoter regions from template DNA and cloning into pGL3‐basic (firefly luciferase, E1751; Promega) between Nhe I and Hind III. E26 transformation‐specific (ETS) binding site mutations were created using the QuikChange Site‐Directed Mutagenesis Kit (200523; Agilent). Primers are presented in Supporting Table S1. The pGL3 reporter was cotransfected with the Renilla luciferase‐thymidine kinase plasmid (phRL‐TK) control reporter (Renilla luciferase, E2241; Promega) into Hep3B, PP5, or Huh7 cells at a 15:1 ratio using Lipofectamine 2000 transfection (11668027; Thermo Fisher). Firefly luciferase activity was normalized to Renilla luciferase activity using the dual‐luciferase reporter assay (E1910; Promega) on a GloMax 20/20 luminometer (E5311; Promega).
Lentiviral Studies
Lentiviral (LV) vector LV.Fer‐DUSP9 was generated by inserting the human DUSP9 coding region (1,155 bp coding sequence [CDS] of NM_001395.2, synthesized by VectorBuilder, Chicago, IL) into pLV vector at the BamH I site. Human (h) ferritin (Fer) composite promoter (hFerH‐mitochondrial elongation factor 1 [mEF1]; 1,476 bp) was amplified from pVITRO2‐neo‐mcs plasmid (pvitro2‐nmcs; InvivoGen, San Diego, CA) using primers Fer‐f/Fer‐r (Supporting Table S1), then digested with Nhe I and BamH I. Fragments of hFerH‐mEF1 (Nhe I, BamH I cleaved) and DUSP9 CDS (BamH I cleaved) were cloned into Nhe I and BamH I digested LV vector LeGO‐iG2 (27341; Addgene, from Boris Fehse).( 15 ) For vehicle control vector LV.Fer‐mock, LeGO‐iG2 spleen focus forming virus promoter was replaced with an hFerH‐mEF1 fragment using the Nhe I–BamH I double‐digestion strategy (Supporting Fig. S2A). High‐titer LV.Fer‐DUSP9 and LV.Fer‐mock LV were produced with Lenti‐X Single Shots packaging system (631275; Clontech). LV‐transduced cells were sorted 4‐5 days posttransduction using a FACSAria TMIII Sorter (BD Biosciences) based on green fluorescent protein (GFP).
Clustered Regularly Interspaced Short Palindromic Repeats‐Associated Protein 9 Nickase Studies
DUSP9 knockout (KO) was accomplished using the clustered regularly interspaced short palindromic repeats (CRISPR)‐associated protein 9 nickase (Cas9n) strategy, as described.( 16 ) Separate pairs of single‐guide RNA (sgRNA) duplexes were targeted after first or second initiation codons of human DUSP9 using the sgRNA design tool (http://crispr.mit.edu). sgRNA duplex sgRNA‐1a, sgRNA‐1b, sgRNA‐2a, and sgRNA‐2b were cloned into Bbs I‐cleaved pSpCas9n(BB)‐2A‐GFP (pX461) (48140; Addgene) individually. The T2A‐GFP encoding DNA fragments in sgRNA‐1b‐PX461 and sgRNA‐2b‐PX461 were replaced with P2A‐mCherry (from plasmid 60954; Addgene) between two EcoR I sites to generate sgRNA‐1b‐PX461‐mCherry and sgRNA‐2b‐PX461‐mCherry. Hep3B or PP5 cells were cotransfected with sgRNA‐1a‐pX461 and sgRNA‐1b‐pX461‐mCherry constructs and HepG2 or Huh7 cells were cotransfected with sgRNA‐2a‐pX461 and sgRNA‐2b‐pX461‐mCherry constructs using Lipofectamine 3000 Reagent (L3000008; Thermo Fisher). At 48 hours after transfection, GFP+/mCherry+ cells were individually sorted into 96‐well plates (10 plates for PP5 and Huh7, 20 plates for HepG2 and Hep3B) using the FACSAria III Sorter (BD Biosciences). Colonies were amplified through 12‐and 6‐well plates, and individual clones were screened by genomic DNA sequencing and western blotting. Colonies in which DUSP9 nonsense mutations occurred (based on nonhomologous end joining) at or immediately downstream of CRISPR‐Cas9n cleavage sites were selected as DUSP9 KO colonies. Colonies in which the DUSP9 mutation was not detected were selected as DUSP9 wild‐type (WT) controls.
Western Blotting
Total protein was isolated using PRO‐PREP Protein Extraction Kit (17081; iNtRON), and concentrations were determined using Pierce Bicinchoninic Acid Protein Assay Kit (23227; Thermo Scientific). We separated 10‐50 μg protein extracts by sodium dodecyl sulfate–polyacrylamide gel electrophoresis using 12% TGX FastCast acrylamide gel (1610175; Bio‐Rad), transferred these onto Immobilon‐P 0.45‐μm polyvinylidene fluoride membrane (IPVH00010; Millipore), probed this with antibodies (Supporting Table S2), and visualized the results with a Pierce ECL Plus (32132; Thermo Scientific).
Histopathology and Immunofluorescence
Tissues were fixed in 10% neutral‐buffered formalin, processed, and paraffin‐embedded. Hematoxylin and eosin staining was performed on 5‐μm sections. Paraffin sections were labeled with polyclonal anti‐human DUSP9 overnight, followed by 1‐hour incubation with Cy3‐conjugated secondary antibody (Supporting Table S2). Images were obtained using a Quorum Wave FX Spinning Disc Confocal System and analyzed using Olympus DP2‐BSW software.
Cell Proliferation and Viability Assay
HCC cells were cultured in 96‐well plates with 5,000 cells/well (nontreatment) or 10,000 cells/well (doxorubicin treated) for up to 8 days. Cell viability was measured using Vybrant 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) Cell Proliferation Assay Kit (M6494; Invitrogen) or water‐soluble tetrazolium 1 (WST‐1) reagent (MK400; Takara). Formazan absorbance was measured by a Multiskan FC microplate photometer (51119000; Thermo Fisher) at 570 nm (MTT) or 450 nm/620 nm (absorbance/reference, WST‐1).
Mouse Experiments
Approval was obtained from the University Health Network (UHN) Animal Care Committee. Cultured DUSP9 KO or WT HCC cells were resuspended in Matrigel (354234; BD Biosciences). We subcutaneously injected 1 × 106 cells/70 µL (PP5) or 7.5 × 105 cells/70 μL (Huh7) into 8‐12‐week‐old female nonobese diabetic severe combined immunodeficiency gamma (NSG) mice, as described,( 17 ) with eight replicates per sample. Mice injected with KO and WT cells were analyzed concurrently and monitored until the largest xenograft in the cohort measured 2 cm in diameter; they were then euthanized. Xenografts were removed, weighed, photographed, and stored.
Statistics
Clinical data were analyzed using STATA 15.0 (Stata Corp., College Station, TX). Continuous variables were expressed as median and interquartile range (IQR) and compared using the Mann‐Whitney U test. Categorical data were expressed as proportions and compared by the chi‐square test with Fisher’s correction. Survival probabilities were assessed by the Kaplan‐Meier method and compared by the log‐rank test. Multivariable Cox regression was applied to identify predictors for disease‐free survival (DFS) and overall survival (OS). DFS was the time between resection and tumor relapse, death, or end of follow‐up. OS was the time between resection and date of death or end of study. We performed multivariable regression to investigate whether DUSP9 was associated with DFS/OS after adjustment for confounders. Variables for multivariable regression were sex, tumor differentiation, resection R status, tumor staging, microvascular invasion, macrovascular invasion, and serum alpha‐fetoprotein (AFP). DUSP9 expression was adjusted to pathologic characteristics. The final model was chosen by stepwise backward regression applying P < 0.1 to removal and P < 0.05 to inclusion. Results of multivariable analysis were expressed as hazard ratios (HRs) with 95% confidence intervals (CIs). Proportional hazard assumption for Cox modeling was not violated as assessed through Schoenfeld residuals and related global tests. Expression correlation between DUSP9 and other genes was assessed by Pearson’s r analysis with 95% CIs. Statistical analysis for laboratory data was performed using GraphPad Prism version 8. For comparison between groups, unpaired two‐tailed t test was used. Results are expressed as mean ± SD unless otherwise indicated. P < 0.05 was considered significant.
RNA‐Seq
For each of three HCC cell lines (Hep3B, PP5, and Huh7), total RNA samples were isolated separately from three distinct colonies of DUSP KO and WT cells, and 200 ng was submitted for library preparation using TruSeq Stranded Total RNA Library Prep Gold (20020598; Illumina). Libraries were sequenced on an Illumina NextSeq500 System with 75‐bp length paired‐end mode aiming for 40 million reads per sample. Raw reads were trimmed using Trimmomatic,( 18 ) aligned to human genome (hg38) using HISAT2 version 2.1.0( 19 ) and sorted/indexed using SAMTOOLS version 1.3.1.( 20 ) Transcript assembly was done using StringTie version 1.3.4d.( 21 ) Differential expression analysis between DUSP9 KO and WT colonies was performed separately with R packages DESeq2 version 1.26.0, edgeR version 3.28.0, and limma‐voom version 3.42.2( 22 , 23 , 24 ); results were visualized using pheatmap.( 25 ) Differentially expressed genes (DEGs) defined individually by three different packages were visualized using R package VennDiagram.( 26 ) Gene ontology (GO) analysis was conducted using Metascape( 27 ) on overlapped DEG lists from the three pipelines. A list of significant GO terms based on DUSP9 KO‐associated up‐regulated DEGs was clustered (K‐Means) into multiple groups using R package simplifyEnrichment( 28 ) based on semantic similarity. Pathway analysis was done using GAGE and Pathview.( 29 , 30 )
Results
DUSP9 is Increased in HCC and Predicts Recurrence After Surgery in Public Data Sets
We interrogated GEO DataSets and BioGPS to identify genes that demonstrated differential expression in fetal liver (FLiver GSE1133: 12,402 genes in two fetal livers vs. two healthy livers), HCC tumor tissues (GSE25097: 18,076 genes in 268 HCC tumors vs. 243 normal adjacent liver tissues), and liver cancer cell lines (NCI60_U133A_CEL: HepG2 and Huh7 vs. 95 other non‐HCC cancer cell lines) (Fig. 1A).
FIG. 1.
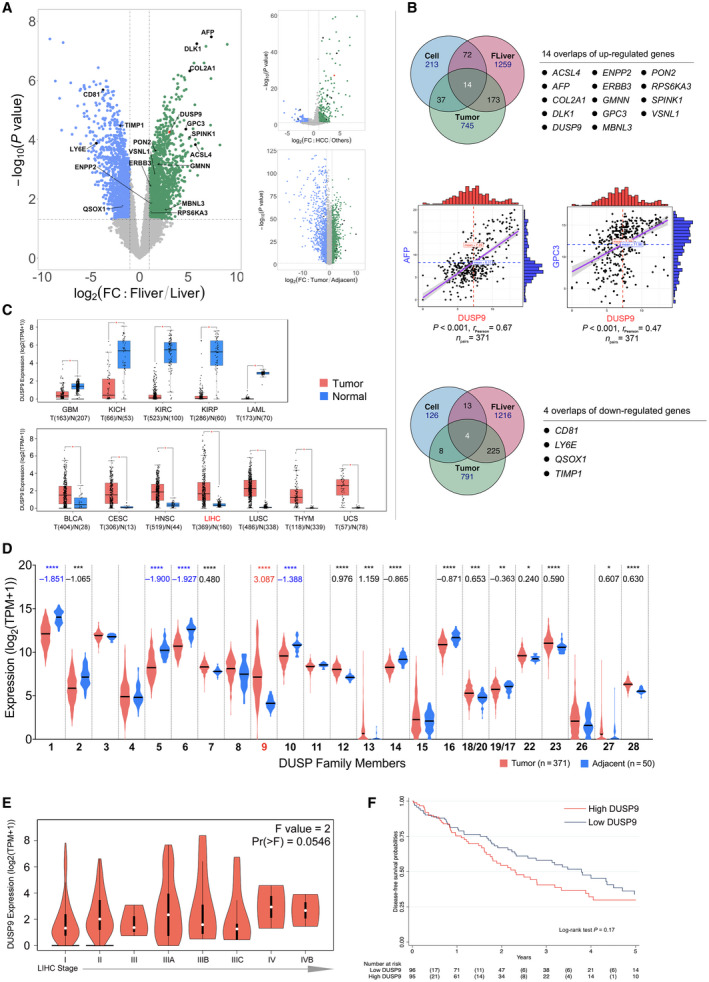
DUSP9 expression in HCC in public data sets. (A) Volcano plot of gene expression data from GSE1133 (left), NCI60_U133A_CEL (right top), and GSE25097. (B) Venn diagrams showing up‐regulated (top) and down‐regulated (bottom) genes shared by all three comparisons in (A). Correlation plots (middle) of DUSP9, AFP, and GPC3 expression in TCGA data. (C) Box plot of DUSP9 expression in different cancers; horizontal bars indicate the median, box indicates interquartile range from Q1 to Q3 with jitter size of 0.4; *P < 0.05. (D) Violin plot showing expression of DUSPs in HCC; violin plots indicate probability density and horizontal bars indicate the median; significant mean fold changes of gene expression in tumor versus adjacent are indicated below asterisks; ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05. (E) Violin plot of DUSP9 expression in progressive stages of HCC; white dots indicate median, vertical black boxes indicate interquartile range from Q1 to Q3. (F) Kaplan‐Meier analysis of DFS following HCC resection in TCGA cohort according to DUSP9 expression. Abbreviations: BLCA, bladder cancer; CD81, cluster of differentiation 81; CESC, cervical cancer; COL2A1, collagen type 2 α1; DLK1, delta like non‐canonical Notch ligand 1; ERBB3, erb‐b2 receptor tyrosine kinase 3; FC, fold change; FLiver, fetal liver; GBM, glioblastoma; HNSC, head‐neck squamous cell carcinoma; KICH/KIRC/KIRP, kidney cancers: chromophobe renal cell carcinoma/clear cell renal cell carcinoma/papillary renal cell carcinoma; LAML, acute myeloid leukemia; LIHC, liver hepatocellular carcinoma; LUSC, lung squamous cell carcinoma; RPS6KA3, ribosomal protein S6 kinase A3; THYM, thymoma; TIMP1, tissue inhibitor of metalloproteinase 1; TPM, transcripts per million; UCS, uterine cancer.
We identified 14 genes with increased expression and four genes with decreased expression in fetal liver, HCC tumors, and HCC cell lines (Fig. 1B). DUSP9 was the only DUSP with increased expression, along with other genes, including the HCC markers AFP and glypican 3 (GPC3). Analysis of gene expression data from TCGA further confirmed a correlation between increased expression of DUSP9 and increased expression of both AFP and GPC3.
We visualized HCC RNA‐Seq data from TCGA and Genotype‐Tissue Expression using GEPIA2 and observed that DUSP9 expression varied across different cancers (Fig. 1C). Further analysis of TCGA data on 371 HCCs revealed that DUSP9 was the only DUSP that demonstrated significantly increased expression in HCC (Fig. 1D). In addition, DUSP9 expression appeared to increase with HCC American Joint Committee on Cancer stage (Fig. 1E).
Finally, we analyzed clinical data associated with HCC samples in TCGA. Of 196 patients who underwent liver resection for HCC, DUSP9 expression levels were available for 191. We divided patients into two groups based on median DUSP9 expression level (i.e., fiftieth percentile). Patients with higher DUSP9 expression had higher serum AFP, more advanced tumor stage, and higher frequency of microvascular invasion (Supporting Table S4). The median DFS was 2.3 (95% CI, 1.5‐3.0) years in patients with higher DUSP9 levels and 3.9 (95% CI, 2.7‐4.8) years in patients with lower DUSP9 levels (P = 0.17) (Fig. 1F). In a multivariable Cox model, we identified that higher DUSP9 level was associated with increased risk of recurrence after liver resection, with an HR of 1.02 (95% CI, 1.00‐1.04; P = 0.02) (Supporting Table S5).
DUSP9 is Increased in HCC and Predicts Recurrence After Surgery in Patients in the UHN
To validate the findings in public data sets, we studied 196 patients undergoing surgery for HCC at our institution. qPCR revealed significantly increased DUSP9 expression in tumors compared with adjacent liver tissue and normal healthy human liver controls (Fig. 2A). Compared with normal controls, DUSP9 expression was significantly increased by a mean ± SD of 11.35 ± 3.78‐fold in tumors compared with 0.64 ± 0.07‐fold in adjacent liver tissues (P = 0.0051).
FIG. 2.
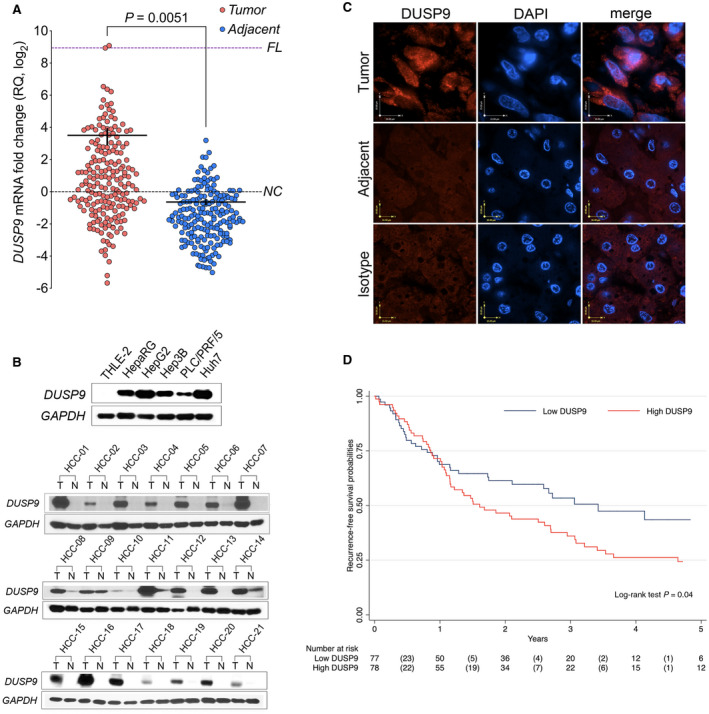
DUSP9 expression in HCC in patients in the UHN. (A) Dot plot of qPCR (using primer pair fp2/rp2) for DUSP9 expression in HCC tumors (red) and adjacent liver tissues (blue). Horizontal black bar indicates mean, vertical black bar indicates SEM. (B) Western blot of DUSP9 in hepatic cell lines, HCC tumors, and adjacent liver. (C) Immunofluorescence staining of human HCC tumor and adjacent liver. White scale bars, 15 μm; yellow scale bar, 10 μm. (D) Kaplan‐Meier analysis of DFS following HCC resection in UHN cohort according to DUSP9 expression. Abbreviations: DAPI, 4´,6‐diamidino‐2‐phenylindole; FL, fetal liver; N, adjacent liver; NC, normal control; RQ, relative quantification; T, hepatocellular carcinoma tumors.
We confirmed this observation with western blots for DUSP9 expression in human liver‐derived cell lines and tissues. DUSP9 was detected in four human HCC cell lines (HepG2, Hep3B, PP5, and Huh7) as well as the human hepatic progenitor cell line HepaRG but could not be detected in the normal human liver cell line THLE‐2 (Fig. 2B, top). Representative results from paired tumor and adjacent liver tissues from 21 patients, revealing increased DUSP9 in HCC tissues compared with matched adjacent liver, are shown in Fig. 2B (bottom). Immunofluorescence revealed robust cytosolic expression of DUSP9 in human HCC tissues but not in matched adjacent liver tissue (Fig. 2C).
We analyzed clinical and pathologic data from 155 of the 196 patients for whom DUSP9 expression data were available in order to investigate the relationship between DUSP9 and clinically relevant outcomes. The median follow‐up was 1.6 (IQR, 0.7‐3.0) years. The median increase in DUSP9 expression was 5.0‐fold (IQR, 1.7‐17.4). Therefore, we compared patients with HCC tissues that demonstrated a >5‐fold increase in DUSP9 (high DUSP9) to those with ≤5‐fold increase in DUSP9 (low DUSP9). Baseline characteristics of both groups are presented in Table 1. The median DFS was 1.6 (95% CI, 0.9‐2.3) years in patients with higher DUSP9 and 3.4 (95% CI, 1.8‐5.0) years in the group with lower DUSP9 (P = 0.04) (Fig. 2D). There was no statistical difference in OS between groups (P = 0.19). We performed a multivariable Cox regression analysis to identify predictors of recurrence after liver resection in our cohort. Well‐established predictors of poor clinical outcomes, including tumor size, tumor number, and vascular invasion, were validated in our cohort of patients (Table 2). In addition to these, a >5‐fold increase in DUSP9 expression also emerged as a significant predictor of recurrence, with an HR of 1.55 (95% CI, 1.01‐2.67; P = 0.05). DUSP9 expression was not associated with OS.
Table 1.
Characteristics of Patients in the Study
| Variable | Overall n = 155 | Study Groups | ||
|---|---|---|---|---|
| Low DUSP‐9 n = 77 | High DUSP‐9 n = 78 | P Value | ||
| Preoperative | ||||
| Sex, male (%) | 123 (79.4) | 66 (85.7) | 57 (73.1) | 0.05 |
| Age, years (IQR) | 65.0 (58.4‐71.9) | 63.5 (56.2‐70.1) | 66.3 (58.8‐74.4) | 0.14 |
| BMI, kg/m2 (IQR) | 25.2 (21.9‐28.7) | 25.2 (22.4‐28.1) | 25.8 (21.8‐29.4) | 0.77 |
| Etiology liver disease (%) | 0.08 | |||
| Hepatitis C | 41 (26.5) | 17 (22.1) | 24 (30.8) | |
| Hepatitis B | 69 (44.5) | 41 (53.2) | 28 (35.9) | |
| Alcohol | 6 (3.9) | 4 (5.2) | 2 (2.6) | |
| NASH | 6 (3.9) | 3 (3.9) | 3 (3.8) | |
| Other | 5 (3.2) | 0 (0) | 5 (6.4) | |
| None | 28 (18.1) | 12 (15.6) | 16 (20.5) | |
| MELD (IQR) | 7 (6‐8) | 7.0 (6.4‐8.1) | 7.0 (6.4‐8.3) | 0.82 |
| Child‐Pugh (%) | ||||
| A | 155 (100) | 77 (100) | 78 (100) | ‐ |
| B | 0 (0) | 0 (0) | 0 (0) | |
| Tumor biggest size, cm (IQR) | 4.5 (3.3‐6.9) | 4.5 (3.3‐6.8) | 4.4 (3.3‐7.0) | 0.88 |
| Tumor number (IQR) | 1 (1‐4) | 1 (1‐1) | 1 (1‐1) | 0.85 |
| AFP, ng/dL (IQR) | 13 (3‐333) | 18 (3‐368) | 8 (4‐335) | 0.81 |
| DUSP9 (fold change) | 5.0 (1.7‐17.4) | 1.7 (0.5‐2.5) | 17.0 (8.4‐44.1) | <0.001 |
| Pathology | ||||
| Tumor biggest size, cm (IQR) | 5 (3.5‐7.5) | 4.8 (3.3‐7.0) | 5.2 (3.9‐8.4) | 0.16 |
| Tumor number (IQR) | 1 (1‐22) | 1 (1‐1) | 1 (1‐1) | 0.59 |
| R0, yes (%) | 145 (93.5) | 73 (94.8) | 72 (92.3) | 0.53 |
| Microvascular invasion, yes (%) | 90 (58.1) | 43 (55.8) | 47 (60.3) | 0.58 |
| Macrovascular invasion, yes (%) | 14 (9.0) | 7 (9.1) | 7 (9.0) | 0.98 |
| Tumoral differentiation (%) | 0.72 | |||
| Well | 6 (3.9) | 2 (2.6) | 4 (5.1) | |
| Moderate | 127 (81.9) | 64 (83.1) | 63 (80.8) | |
| Poor | 22 (14.2) | 11 (14.3) | 11 (14.1) | |
Abbreviations: BMI, body mass index; MELD, Model for End‐Stage Liver Disease; NASH, nonalcoholic steatohepatitis; R0, negative resection margin.
Table 2.
Univariable and Multivariable Regression For Predictors of Recurrence in the UHN Cohort
| Variable | Univariable | Multivariable | ||
|---|---|---|---|---|
| HR (95% CI) | P Value | HR (95% CI) | P Value | |
| Sex (ref., male) | 0.62 (0.39‐0.99) | 0.05 | ‐ | ‐ |
| Age (per year) | 1.03 (1.00‐1.05) | 0.01 | 1.03 (1.01‐1.05) | 0.01 |
| Etiology (ref., HCV) | ||||
| HBV | 0.43 (0.25‐0.72) | 0.001 | ‐ | ‐ |
| Alcohol | 0.64 (0.19‐2.11) | 0.46 | ‐ | ‐ |
| NASH | 1.46 (0.60‐3.54) | 0.40 | ‐ | ‐ |
| Other | 1.09 (0.42‐2.84) | 0.85 | ‐ | ‐ |
| None | 0.99 (0.55‐1.77) | 0.97 | ‐ | ‐ |
| BMI (per kg/m2) | 1.00 (0.99‐1.01) | 0.69 | ‐ | |
| MELD (per point) | 1.03 (0.91‐1.17) | 0.61 | ‐ | |
| Serum AFP (ref., <20 ng/mL) | ||||
| 20‐100 ng/mL | 1.05 (0.54‐2.02) | 0.89 | ‐ | ‐ |
| 100‐1,000 ng/mL | 0.85 (0.50‐1.49) | 0.57 | ‐ | ‐ |
| >1,000 ng/mL | 0.99 (0.53‐1.87) | 0.99 | ‐ | ‐ |
| Specimen tumor size, cm | 1.08 (1.04‐1.14) | <0.001 | ‐ | ‐ |
| Specimen tumor number | 1.11 (1.03‐1.20) | 0.01 | 1.10 (1.07‐2.67) | 0.01 |
| DUSP9 level (ref., low) | 1.53 (1.01‐2.43) | 0.05 | 1.55 (1.01‐2.37) | 0.05 |
| R0 status (ref., yes) | 0.91 (0.37‐2.25) | 0.84 | ‐ | ‐ |
| Microvascular invasion (ref., no) | 2.16 (1.39‐3.35) | 0.01 | 1.69 (1.07‐2.67) | 0.02 |
| Macrovascular invasion (ref., no) | 5.69 (3.01‐10.75) | <0.001 | 5.05 (2.59‐9.85) | <0.001 |
| Tumoral differentiation (ref., well) | ||||
| Moderate | 2.35 (0.58‐9.57) | 0.23 | ‐ | ‐ |
| Poorly | 1.66 (0.36‐7.57) | 0.51 | ‐ | ‐ |
Abbreviations: BMI, body mass index; HBV, chronic hepatitis B; HCV, chronic hepatitis C; MELD, Model for End‐Stage Liver Disease; NASH, nonalcoholic steatohepatitis; R0, negative resection margin.
Cloning of Full‐Length DUSP9 cDNA Sequences From Human HCC and Identification of a Novel DUSP9 Isoform
In order to further investigate DUSP9 in human HCC, we identified full‐length cDNA sequences corresponding to DUSP9 from human HCC cell lines and tissues. The human DUSP9 gene, exon assembly, and transcripts based on the experiments described below are schematically illustrated in Fig. 3A.
FIG. 3.
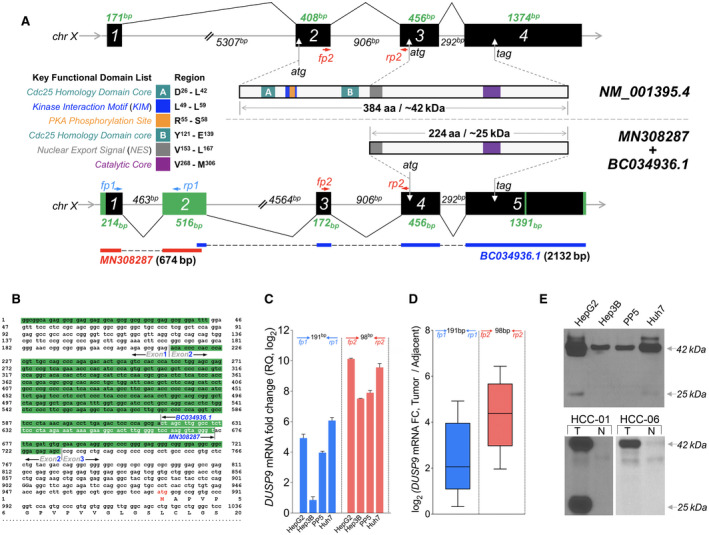
Identification of HCC‐specific DUSP9 isoform. (A) Schematic illustration comparing WT DUSP9 (NM_001395.4 at top) and N‐terminal truncated DUSP9 isoform (bottom, combination of MN308287 and BC034936.1). Black/green boxes indicate exons. Coding sequences are shown with key functional domains. Green segments in truncated isoform represent sequence differences compared to WT DUSP9. Primer pairs (fp1/rp1, fp2/rp2) are also indicated. (B) Combination of 5’RACE‐PCR amplified fragment (MN) with published partial sequence BC034936.1. MN308287 consists of g1 to t674, including 57 nucleotides of overlap with BC034936.1 (white letters), revealing 617 nucleotides of novel 5’ sequence. Red “atg” represents the predicted translation initiation site of the HCC‐specific DUSP9 isoform. (C) qPCR using primers selective for the truncated DUSP9 isoform (fp1/rp1, blue bars) or all DUSP9 isoforms (fp2/rp2, red bars) in HCC cell lines (RQ normalized to DUSP9 mRNA level in THLE‐2). Graph shows mean + SEM. (D) qPCR using primers selective for the truncated DUSP9 isoform (fp1/rp1, blue box) or all DUSP9 isoforms (fp2/rp2, red box) in 10 HCC tumors and adjacent liver tissues. Graph shows IQR (box), median (horizontal line), and outliers (whiskers). (E) Western blot of DUSP9 in four HCC cell lines, human HCC tumors, and adjacent liver. Abbreviations: aa, amino acid; Cdc25, cell division cycle 25; chr, chromosome; FC, fold change; N, adjacent liver; RQ, relative quantification; T, HCC tumors.
The National Center for Biotechnology Information reported two DUSP9 reference mRNA sequences (GenBank NM_001318503 and NM_001395) and a partial CDS of HCC‐specific DUSP9 cDNA (GenBank BC034936) that is missing the 5’ end. Using 5’ RACE‐PCR, we amplified a novel 674‐bp fragment (Fig. 3B) unique from the two published transcript variants, providing 617 bp of novel sequence upstream of BC034936. This sequence has been deposited in GenBank as MN308287.
Our experiments identified 43 bp of novel sequence extending exon 1 to the 5’ end, a novel second exon (Fig. 3A, green bar), absence of a fragment within exon 3 that aligns to the first ATG start codon in exon 2 of NM_001395, and two insertions of 19 and 6 bp in exon 5. When combined with BC034936, this predicted a novel DUSP9 cDNA present in HCC that contains an ATG start codon in exon 4. This would generate an N‐terminal truncated protein product of 224 amino acids with a predicted molecular weight (MW) of ~25 kDa, compared with the 384‐amino acid (42 kDa) product generated by the full‐length reference sequence in NM_001395. Both proteins contain the catalytic core (purple bar) as well as the nuclear export signal (gray bar), but the truncated protein lacks the kinase interaction motif (blue bar).
In order to characterize the relative expression of the two mRNAs in human HCC, we performed qPCR using primers selective for the novel cDNA (Fig. 3A, fp1/rp1 in blue) as well as primers that would amplify both the reference and novel cDNAs (Fig. 3B, fp2/rp2 in red). Both transcripts were detected in human tissues and human HCC cell lines, with approximately half of the increase in DUSP9 mRNA levels attributable to the novel transcript, although there was variability between cell lines (Fig. 3C,D). To validate this observation, we performed western blot analysis on HCC cell lines and human liver tissues using a C‐terminal targeted monoclonal antibody against a shared epitope encoded by both transcripts. This showed strong expression of the full‐length human DUSP9 protein product encoded by the reference cDNA in all tested HCC tissues and cell lines with a predicted MW of ~42 kDa (Fig. 3E). However, bands corresponding to the shorter protein product, with a predicted MW of ~25 kDa, were only visible in approximately 40% of human tumors and in HepG2 and Huh7 cell lines.
DUSP9 Expression in HCC Cells is Regulated by ETS Transcription Factors
To investigate DUSP9 transcriptional regulation in HCC, HCC cells were transfected with luciferase reporter constructs under the transcriptional control of serial truncations of the DUSP9 promoter. Truncation of the region between −938 bp and −707 bp significantly reduced luciferase activity (Supporting Fig. S1A, top). Analysis of serial truncations of this region revealed a 64‐bp segment (−771 bp to −707 bp) that was critical for luciferase activity. Interrogation of the JASPAR database with this 64‐bp fragment revealed a palindromic TTCCGGAA sequence containing two ETS transcription factor (TF) binding core motifs (GGA(A/T)). Single‐silencing mutations of either the GGAA (or TTCC) motif resulted in modest reductions in luciferase activity, while double‐silencing mutations of TTCC and GGAA resulted in significant luciferase activity reduction similar to truncation of this entire segment of the promoter (Supporting Fig. S1A, bottom).
We performed quantitative real‐time reverse transcription PCR to determine the expression profiles of 27 ETS family members among HepG2, Hep3B, PP5, and Huh7 HCC cell lines. mRNA of 16 ETS TFs could be detected in at least two of the HCC cell lines tested (Supporting Fig. S1B). Using HCC gene expression data from TCGA, we assessed co‐expression of ETS TFs with DUSP9 and observed that four ETS TFs (ETV4, ELF3, ERF, and ETV5) demonstrated a significant correlation with DUSP9 (Supporting Fig. S1C).
DUSP9 Overexpression Enhances Proliferation of Liver Cells In Vitro
To investigate the effects of further increasing DUSP9 expression in HCC, we transduced cells with an LV vector encoding the full‐length human DUSP9 coding region (Supporting Fig. S2A). Greater increases in DUSP9 mRNA were observed in the HepaRG, Hep3B, and PP5 cell lines that are known to have lower constitutive DUSP9 expression (Fig. 2B; Supporting Fig. S2B; additional data not shown). HepG2 and Huh7 cells, which have much higher constitutive DUSP9 expression, showed only a modest increase in DUSP9 mRNA. Significant increases in cell proliferation were observed in the cell lines in which the greatest increase in DUSP9 mRNA was achieved (Supporting Fig. S2C‐G).
DUSP9 KO Decreases HCC Cell Proliferation and Chemotherapy Resistance
In order to investigate the effects of decreased DUSP9 expression on HCC, we used CRISPR to generate DUSP9 KO human HCC cell lines devoid of both DUSP9 transcripts and protein products. Cas9n‐mediated DNA cleavage sites were designed after both the first and second start codons, as illustrated in Fig. 4A, and cellular clones incorporating the intended mutations were screened and confirmed by DNA sequencing. We successfully generated multiple DUSP9 KO lines in Hep3B, PP5, and Huh7 HCC cell lines. Western blot revealed no DUSP9 expression in KO cells compared with WT cells (Fig. 4B).
FIG. 4.
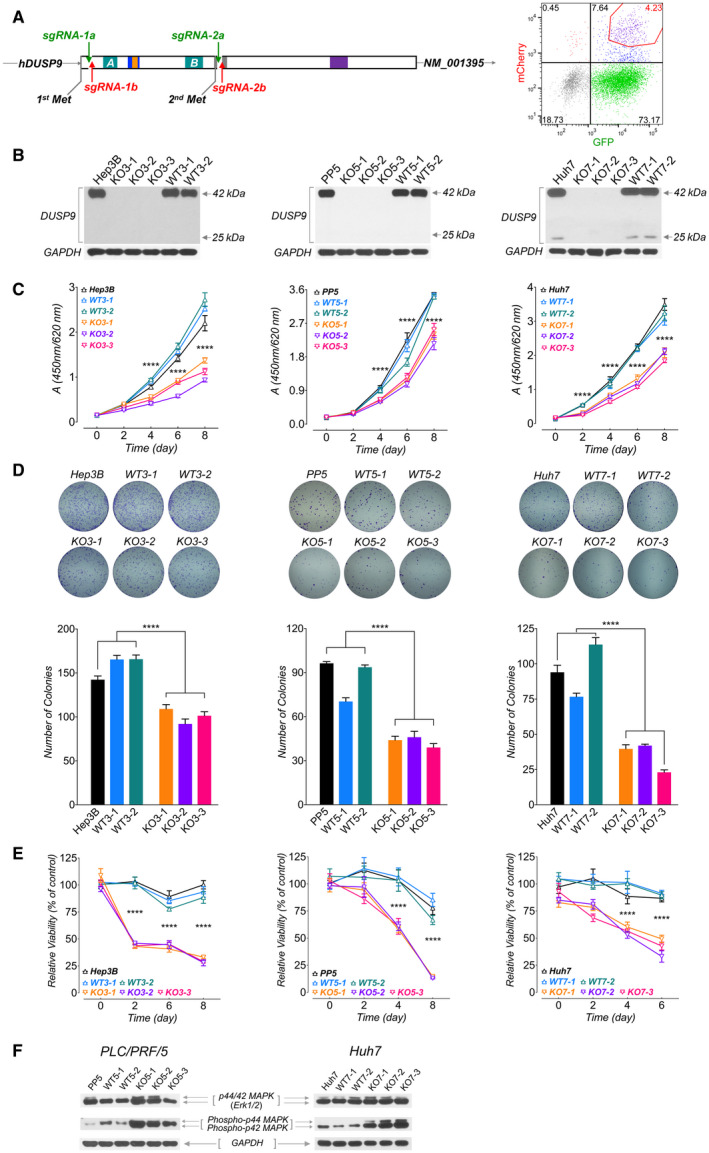
Characterization of DUSP9 KO HCC cells. (A) Targeting strategy for CRISPR‐mediated DUSP9 KO. Cas9n‐mediated DNA cleavage sites were designed after the first start codon (Hep3B and PP5 cells, using sgRNA‐1a/1b) or after the second start codon (HepG2 and Huh7 cells, using sgRNA‐2a/2b). FACS plot (right) shows gating of transfected cells with dual fluorescent tags (GFP for sgRNA‐1a or 2a, mCherry for sgRNA‐1b or 2b) for further screening. (B) Western blots for DUSP9 in WT and KO cell lines. (C) WST‐1 assay on DUSP9 WT and KO cells. Data represent mean of six replicates per group; ****P < 0.0001. (D) Colony formation assays on DUSP9 KO and WT cells. Data show mean + SEM of three replicates per group; ****P < 0.0001. (E) Relative viability assays of doxorubicin‐treated DUSP9 KO versus WT Hep3B (200 ng/mL), PP5 (400 ng/mL), and Huh7 (1 µg/mL) cells. Data show mean + SEM of six replicates per group; ****P < 0.0001. (F) Western blot of DUSP9 KO and WT cells for Erk1/2 (p44/42 MAPK) and phosphorylated Erk1/2 (phospho‐p44/42 MAPK). Abbreviation: FACS, fluorescence‐activated cell sorting.
To assess the functional impact of DUSP9 KO, we measured HCC cell proliferation using WST‐1 assays and observed that DUSP9 KO cells had decreased proliferation in vitro (Fig. 4C). Furthermore, we examined colony formation in soft agar and observed that DUSP9 KO cells showed reduced colony‐forming capacity (Fig. 4D). We also investigated whether DUSP9 KO affected HCC cell viability in response to doxorubicin treatment and found that DUSP9 KO cells demonstrated significantly reduced resistance to chemotherapy treatment (Fig. 4E).
To determine whether DUSP9 KO had any effect on MAPK activity, we used western blot to assay for ERK phosphorylation. ERK phosphorylation was significantly increased in DUSP9 KO PP5 and Huh7 cells (Fig. 4F).
DUSP9 KO Decreases HCC Xenograft Growth
To investigate the effect of DUSP9 KO on human HCC growth in vivo, we generated subcutaneous xenografts in immunodeficient mice. DUSP9 KO PP5 (left) and Huh7 (right) cells generated smaller xenografts with a reduced growth rate (Fig. 5A,B). Xenografts generated by both DUSP9 KO and WT cells had similar histopathologic features of HCC, including hepatocyte‐like cells with nuclear atypia, high nuclear‐to‐cytoplasmic ratio, frequent mitotic figures, absence of portal tracts, and increased thickness of hepatocellular plates (Fig. 5C). Although Hep3B WT cells generated xenografts similar to WT PP5 and Huh7 cells (data not shown), DUSP9 KO Hep3B cells failed to generate xenografts, preventing quantitative comparisons.
FIG. 5.
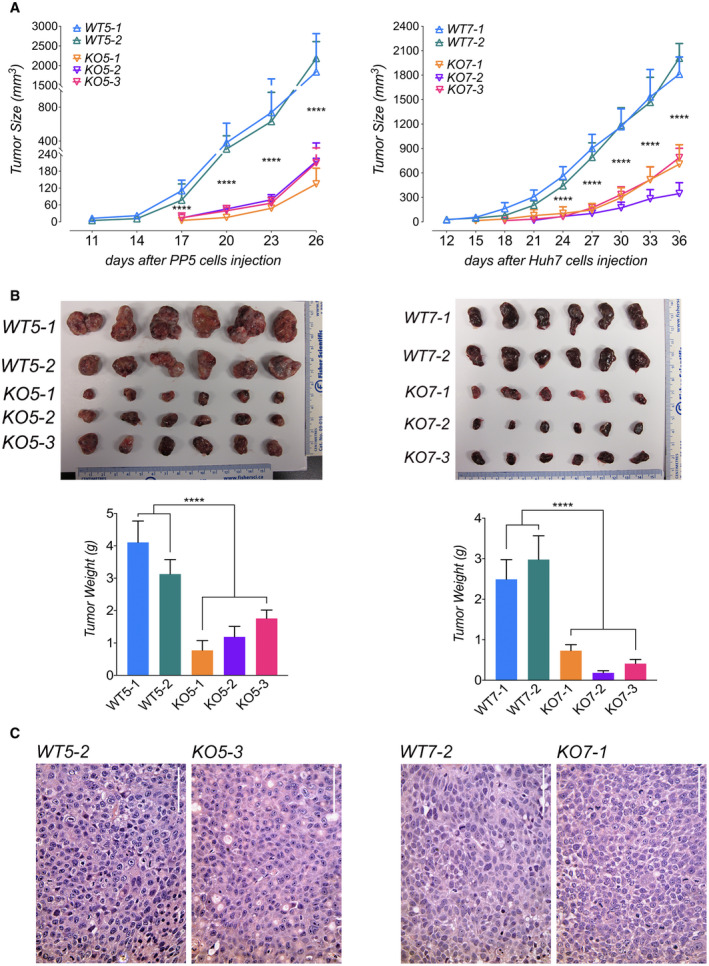
DUSP9 KO decreases human HCC xenograft growth in vivo. (A) Xenograft tumor growth curves generated by DUSP9 WT and KO HCC cells. (B) Xenografts generated by DUSP9 WT and KO HCC cells and corresponding bar graphs of tumor weights. Data in (A,B) show mean + SEM of eight replicates per group; ****P < 0.0001. (C) HCC xenografts generated by DUSP9 WT and KO HCC cells. Scale bar, 100 μm.
RNA‐Seq Reveals Multiple Pathways Affected by DUSP9 KO in HCC
In order to identify pathways influenced by DUSP9 in HCC, we performed RNA‐Seq on multiple DUSP9 KO and WT colonies derived from Hep3B, PP5, and Huh7 cells. Expression cluster analysis clearly differentiated between DUSP9 KO and WT cells across cell lines (Fig. 6A). Pooled analysis of KO versus WT colonies with DESeq2, edgeR, and limma allowed for the generation of a consensus list of DEGs, with 343 up‐regulated genes and 109 down‐regulated genes (Fig. 6B).
FIG. 6.
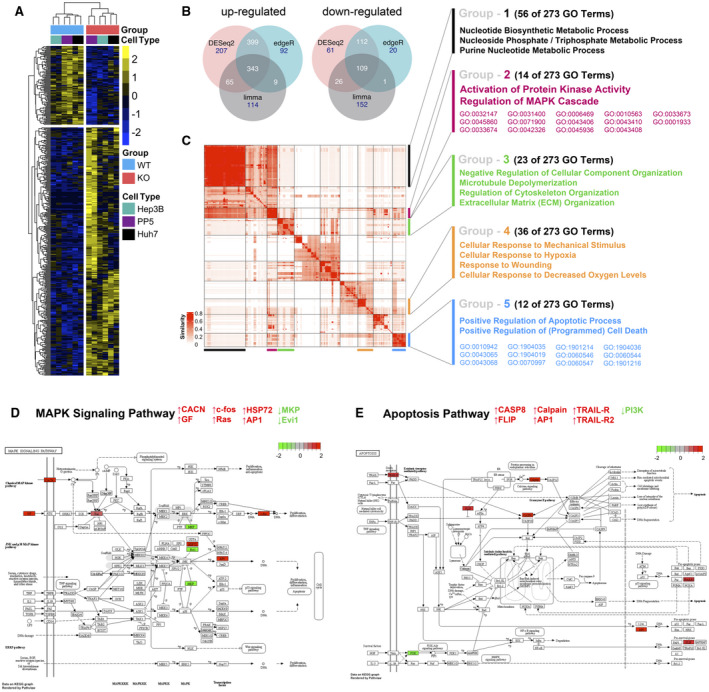
RNA‐Seq analysis of DUSP9 WT and KO HCC cells. (A) Representative heatmap of expression clustering differentiates between DUSP9 KO and WT colonies across cell lines. DUSP9 KO is associated with decreased expression of 109 genes (top) and increased expression of 343 genes (bottom) (yellow, increased expression; blue, decreased expression). (B) Venn diagrams illustrating 343 up‐regulated and 109 down‐regulated genes consistently identified by pooled analysis of RNA‐Seq data from DUSP9 KO versus WT HCC cells using the R packages DESeq2, edgeR, and limma‐voom. (C) Clustering of 273 GO terms returned by Metascape functional enrichment analysis into five major groups. (D,E) Schematic illustrations of MAPK signaling and apoptosis pathways generated by pathview, highlighting specific genes with altered expression in association with DUSP9 KO (red, up‐regulated; green, down‐regulated). Abbreviations: AP1, activator protein 1; CACN, calcium voltage‐gated channel gene group; CASP8, caspase 8; Evi1, ecotropic viral integration site‐1; FLIP, FLICE‐inhibitory protein; GF, growth factor; HSP72, heat shock protein 72; PI3K, phosphoinositide 3‐kinase; TRAIL‐R, tumor necrosis factor–related apoptosis‐inducing ligand receptor.
To characterize the functional relevance of DEGs associated with DUSP9 KO, we conducted GO functional enrichment analysis using Metascape. Clustering of 273 GO terms into five major groups is illustrated in Fig. 6C and Supporting Table S6. In addition to common cellular metabolic processes, DUSP9 KO was clearly associated with activation of protein kinase activity and MAPK regulation as well as positive regulation of apoptotic processes. Individual genes in the MAPK signaling pathway and apoptosis pathway are more specifically highlighted in Fig. 6D,E, demonstrating altered expression in association with DUSP9 KO.
Discussion
In this report, we have shown that DUSP9 expression is increased in human HCC and that higher DUSP9 expression is associated with shorter disease‐free survival and a higher risk of disease recurrence after liver resection. We cloned HCC‐specific full‐length DUSP9 cDNA sequences and identified a novel DUSP9 isoform expressed in HCC. We demonstrated that DUSP9 KO impairs HCC cellular proliferation and chemotherapy resistance in vitro as well as tumor growth and progression in vivo. RNA‐Seq revealed that DUSP9 influences many processes in HCC cells, including protein kinase activity and apoptosis as well as regulation of the cytoskeleton and extracellular matrix (ECM).
We identified DUSP9 as a molecule of interest in HCC by screening public data sets for genes differentially expressed in both HCC and fetal liver. This screen also identified AFP, GPC3, and delta like non‐canonical Notch ligand 1 (DLK1), which have well‐characterized roles in HCC,( 31 , 32 , 33 ) supporting the validity of our approach. Our screen also identified erb‐b2 receptor tyrosine kinase 3 (ErbB3), which plays a role in many cancers,( 34 ) as well as ribosomal protein S6 kinase A3 (RPS6KA3) (RSK2), a negative regulator of MAPK signaling.( 35 )
Despite increased expression of DUSP9 in HCC, analysis of TCGA data revealed that DUSP9 shows variable expression across different cancers, suggesting that the biology of DUSP9 is tissue specific. This likely accounts for the heterogeneity reported with regards to both tumor suppressor and oncogene roles of DUSP9 in breast, gastric, colon, and renal cancer.( 36 , 37 , 38 , 39 )
Only two groups have reported on DUSP9 in HCC, with disparate findings. Using bioinformatics, Petrochilos et al.( 40 ) found that DUSP9 expression was increased in human HCC samples, while Liu et al.( 41 ) found that DUSP9 (MKP4) expression was decreased in HCC. We reproduced the latter group’s methods but found that PCR products generated by the published primers did not correspond to DUSP9, suggesting confounding of the data by off‐target amplification artifacts.
Higher DUSP9 expression was a significant predictor of HCC recurrence after resection. For patients in the UHN, higher DUSP9 was also associated with a significantly shorter DFS after surgery. A significant relationship between DUSP9 expression and OS was not detected and may require a longer period of follow‐up than was possible in this study. While higher DUSP9 expression was associated with features of more advanced HCC in the TCGA cohort, this relationship was not observed in patients in the UHN. potentially exposing the independent impact of DUSP9 on clinical outcomes more clearly. Future studies of a wider spectrum of patients with HCC will help to better characterize DUSP9 in HCC.
Our experiments revealed two mRNA transcripts corresponding to DUSP9 in human HCC cells. Further research is required to determine the specific role and regulation of the shorter protein. Our experiments suggest that DUSP9 is transcriptionally regulated by ETS TFs. As ETS TFs are downstream effectors of MAPKs, this finding is consistent with theories that DUSPs function as part of a negative feedback loop controlling MAPK activity and are transcriptionally regulated by MAPK signaling themselves.( 42 , 43 ) Increased DUSP9 expression in HCC may thus reflect aberrant activation of ETS TFs, as described in many solid cancers.( 44 )
On a background of high constitutive DUSP9 expression in HCC cells, LV overexpression of human DUSP9 drove modest increases in cell proliferation in cells with lower constitutive DUSP9 expression. We therefore focused on the effects of reducing DUSP9 by generating DUSP9 KO human HCC cell lines using CRISPR. DUSP9 KO cells demonstrated sustained reduction in proliferation and chemotherapy resistance. This observation contrasts with the failure of MAPK pathway inhibitors in clinical trials due to the activation of compensatory mechanisms in tumor cells that circumvent targeted inhibition of single molecules.( 8 , 45 ) DUSP9 may thus represent a vulnerable therapeutic target in cancer cells.
As DUSP9 is an ERK‐specific phosphatase,( 12 , 42 ) we observed a predicted increase in ERK phosphorylation with DUSP9 KO. Despite this, HCC cell proliferation and tumor growth decreased. These observations may be explained by the interaction of DUSP9 with other pathways in HCC cells. As negative regulators of MAPKs, DUSPs are predicted to function as tumor suppressors, but studies have revealed both loss and gain of DUSP expression in association with cancer progression, treatment resistance, and poor patient outcomes,( 46 ) suggesting as yet undefined roles in other cellular processes.
We used RNA‐Seq to broadly investigate molecular pathways and networks affected by DUSP9 in HCC. Consistent with the known function of DUSP9 as a negative regulator of MAPKs, DUSP9 KO was associated with activation of protein kinase activity and regulation of the MAPK cascade. Interestingly, DUSP9 KO was also associated with positive regulation of apoptotic processes, providing a potential explanation for our observations in this study. The specific mechanism by which DUSP9 may regulate apoptosis in HCC cells requires further investigation. Although it is widely recognized that MAPK signaling can both promote and inhibit apoptosis in different cell types and in response to different stimuli,( 47 ) there are no published reports describing a role for DUSP9 in apoptosis of cancer cells.
Our analysis also revealed that DUSP9 KO was associated with changes in metabolism, ECM and cytoskeletal organization, and stress response. These processes are implicated in regulation of the tumor microenvironment, which plays a critical role in tumor biology.( 48 ) Perturbed ECM deposition and remodeling are hallmarks of cirrhosis, the major risk factor for HCC.( 49 ) Similarly, cytoskeletal dysregulation contributes to HCC by driving epithelial–mesenchymal transition, tumor invasion, and metastasis.( 50 ) Because DUSP9 has not previously been implicated in these processes, further studies are required to validate these observations.
In conclusion, we demonstrate that DUSP9 contributes to human HCC pathobiology and may represent a novel candidate for prognostic use and therapeutic intervention. Additional studies are warranted to investigate mechanisms regulating DUSP9 expression in HCC as well as mechanisms through which DUSP9 affects HCC biology and clinical outcomes.
Supporting information
Fig S1
Fig S2
Table S1‐5
Supported by the Canadian Liver Foundation (2017‐2019 Operating Grant awarded to A. Ghanekar).
Potential conflict of interest: Nothing to report.
References
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Villanueva A. Hepatocellular carcinoma. N Engl J Med 2019;380:1450‐1462. [DOI] [PubMed] [Google Scholar]
- 3. Roayaie S, Jibara G, Tabrizian P, Park J‐W, Yang J, Yan L, et al. The role of hepatic resection in the treatment of hepatocellular cancer. Hepatology 2015;62:440‐451. [DOI] [PubMed] [Google Scholar]
- 4. Sapisochin G, Goldaracena N, Laurence JM, Dib M, Barbas A, Ghanekar A, et al. The extended Toronto criteria for liver transplantation in patients with hepatocellular carcinoma: a prospective validation study. Hepatology 2016;64:2077‐2088. [DOI] [PubMed] [Google Scholar]
- 5. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc J‐F, et al.; SHARP Investigators Study Group . Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378‐390. [DOI] [PubMed] [Google Scholar]
- 6. Craig AJ, von Felden J, Garcia‐Lezana T, Sarcognato S, Villanueva A. Tumour evolution in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2020;17:139‐152. [DOI] [PubMed] [Google Scholar]
- 7. Sia D, Villanueva A, Friedman SL, Llovet JM. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 2017;152:745‐761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Braicu C, Buse M, Busuioc C, Drula R, Gulei D, Raduly L, et al. A Comprehensive review on MAPK: a promising therapeutic target in cancer. Cancers (Basel) 2019;11:1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gordillo M, Evans T, Gouon‐Evans V. Orchestrating liver development. Development 2015;142:2094‐2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Patterson KI, Brummer T, O'Brien PM, Daly RJ. Dual‐specificity phosphatases: critical regulators with diverse cellular targets. Biochem J 2009;418:475‐489. [DOI] [PubMed] [Google Scholar]
- 11. Kidger AM, Keyse SM. The regulation of oncogenic Ras/ERK signalling by dual‐specificity mitogen activated protein kinase phosphatases (MKPs). Semin Cell Dev Biol 2016;50:125‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seternes OM, Kidger AM, Keyse SM. Dual‐specificity MAP kinase phosphatases in health and disease. Biochim Biophys Acta Mol Cell Res 2019;1866:124‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cancer Genome Atlas Research Network . Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017;169:1327‐1341.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marrero JA, Kulik LM, Sirlin CB, Zhu AX, Finn RS, Abecassis MM, et al. Diagnosis, staging, and management of hepatocellular carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2018;68:723‐750. [DOI] [PubMed] [Google Scholar]
- 15. Weber K, Bartsch U, Stocking C, Fehse B. A multicolor panel of novel lentiviral "gene ontology" (LeGO) vectors for functional gene analysis. Mol Ther 2008;16:698‐706. [DOI] [PubMed] [Google Scholar]
- 16. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 2013;8:2281‐2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ahmed SU, Zair M, Chen K, Iu M, He F, Adeyi O, et al. Generation of subcutaneous and intrahepatic human hepatocellular carcinoma xenografts in immunodeficient mice. J Vis Exp 2013;79:e50544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114‐2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nat Biotechnol 2019;37:907‐915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al.; 1000 Genome Project Data Processing Subgroup . The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25:2078‐2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript‐level expression analysis of RNA‐seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 2016;11:1650‐1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ritchie ME, Phipson B, Wu DI, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kolde R. Pheatmap: pretty heatmaps. R package version 2012;61:1‐7. [Google Scholar]
- 26. Chen H, Boutros PC. VennDiagram: a package for the generation of highly‐customizable Venn and Euler diagrams in R. BMC Bioinformatics 2011;12:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist‐oriented resource for the analysis of systems‐level datasets. Nat Commun 2019;10:1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gu ZG, Hübschmann D. SimplifyEnrichment: an R/Bioconductor package for Clustering and Visualizing Functional Enrichment Results. bioRxiv 2020;312116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luo W, Friedman MS, Shedden K, Hankenson KD, Woolf PJ. GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 2009;10:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luo W, Brouwer C. Pathview: an R/Bioconductor package for pathway‐based data integration and visualization. Bioinformatics 2013;29:1830‐1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Galle PR, Foerster F, Kudo M, Chan SL, Llovet JM, Qin S, et al. Biology and significance of alpha‐fetoprotein in hepatocellular carcinoma. Liver Int 2019;39:2214‐2229. [DOI] [PubMed] [Google Scholar]
- 32. Haruyama Y, Kataoka H. Glypican‐3 is a prognostic factor and an immunotherapeutic target in hepatocellular carcinoma. World J Gastroenterol 2016;22:275‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu X, Liu R‐F, Zhang X, Huang L‐Y, Chen F, Fei Q‐L, et al. DLK1 as a potential target against cancer stem/progenitor cells of hepatocellular carcinoma. Mol Cancer Ther 2012;11:629‐638. [DOI] [PubMed] [Google Scholar]
- 34. Kiavue N, Cabel L, Melaabi S, Bataillon G, Callens C, Lerebours F, et al. ERBB3 mutations in cancer: biological aspects, prevalence and therapeutics. Oncogene 2020;39:487‐502. [DOI] [PubMed] [Google Scholar]
- 35. Saha M, Carriere A, Cheerathodi M, Zhang X, Lavoie G, Rush J, et al. RSK phosphorylates SOS1 creating 14‐3‐3‐docking sites and negatively regulating MAPK activation. Biochem J 2012;447:159‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu S, Wang Y, Sun L, Zhang Z, Jiang Z, Qin Z, et al. Decreased expression of dual‐specificity phosphatase 9 is associated with poor prognosis in clear cell renal cell carcinoma. BMC Cancer 2011;11:413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu F, Lv T, Chen G, Ye H, Wu W, Li G, et al. Epigenetic silencing of DUSP9 induces the proliferation of human gastric cancer by activating JNK signaling. Oncol Rep 2015;34:121‐128. [DOI] [PubMed] [Google Scholar]
- 38. Imajo M, Kondoh K, Yamamoto T, Nakayama K, Nakajima‐Koyama M, Nishida E. Antagonistic interactions between extracellular signal‐regulated kinase mitogen‐activated protein kinase and retinoic acid receptor signaling in colorectal cancer cells. Mol Cell Biol 2017;37:e00012‐e00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lu H, Tran L, Park Y, Chen I, Lan J, Xie Y, et al. Reciprocal regulation of DUSP9 and DUSP16 expression by HIF1 controls ERK and p38 MAP kinase activity and mediates chemotherapy‐induced breast cancer stem cell enrichment. Cancer Res 2018;78:4191‐4202. [DOI] [PubMed] [Google Scholar]
- 40. Petrochilos D, Shojaie A, Gennari J, Abernethy N. Using random walks to identify cancer‐associated modules in expression data. BioData Min 2013;6:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu J, Ni W, Xiao M, Jiang F, Ni R. Decreased expression and prognostic role of mitogen‐activated protein kinase phosphatase 4 in hepatocellular carcinoma. J Gastrointest Surg 2013;17:756‐765. [DOI] [PubMed] [Google Scholar]
- 42. Jeffrey KL, Camps M, Rommel C, Mackay CR. Targeting dual‐specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat Rev Drug Discov 2007;6:391‐403. [DOI] [PubMed] [Google Scholar]
- 43. Tetsu O, McCormick F. ETS‐targeted therapy: can it substitute for MEK inhibitors? Clin Transl Med 2017;6:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sizemore GM, Pitarresi JR, Balakrishnan S, Ostrowski MC. The ETS family of oncogenic transcription factors in solid tumours. Nat Rev Cancer 2017;17:337‐351. [DOI] [PubMed] [Google Scholar]
- 45. von Manstein V, Yang CM, Richter D, Delis N, Vafaizadeh V, Groner B. Resistance of cancer cells to targeted therapies through the activation of compensating signaling loops. Curr Signal Transduct Ther 2013;8:193‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Keyse SM. Dual‐specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev 2008;27:253‐261. [DOI] [PubMed] [Google Scholar]
- 47. Yue J, Lopez JM. Understanding MAPK signaling pathways in apoptosis. Int J Mol Sci 2020;21:2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hernandez‐Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology 2013;144:512‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu XZ, Chen D, Xie GR. Extracellular matrix remodeling in hepatocellular carcinoma: effects of soil on seed? Med Hypotheses 2006;66:1115‐1120. [DOI] [PubMed] [Google Scholar]
- 50. Peng J‐M, Bera R, Chiou C‐Y, Yu M‐C, Chen T‐C, Chen C‐W, et al. Actin cytoskeleton remodeling drives epithelial‐mesenchymal transition for hepatoma invasion and metastasis in mice. Hepatology 2018;67:2226‐2243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Table S1‐5