

Abstract
Nonalcoholic fatty liver disease (NAFLD) has emerged as a leading cause of chronic liver disease worldwide, primarily because of the massive global increase in obesity. Despite intense research efforts in this field, the factors that govern the initiation and subsequent progression of NAFLD are poorly understood, which hampers the development of diagnostic tools and effective therapies in this area of high unmet medical need. Here we describe a regulator in molecular pathogenesis of NAFLD: STE20‐type protein kinase MST4. We found that MST4 expression in human liver biopsies was positively correlated with the key features of NAFLD (i.e., hepatic steatosis, lobular inflammation, and hepatocellular ballooning). Furthermore, the silencing of MST4 attenuated lipid accumulation in human hepatocytes by stimulating β‐oxidation and triacylglycerol secretion, while inhibiting fatty acid influx and lipid synthesis. Conversely, overexpression of MST4 in human hepatocytes exacerbated fat deposition by suppressing mitochondrial fatty acid oxidation and triacylglycerol efflux, while enhancing lipogenesis. In parallel to these reciprocal alterations in lipid storage, we detected substantially decreased or aggravated oxidative/endoplasmic reticulum stress in human hepatocytes with reduced or increased MST4 levels, respectively. Interestingly, MST4 protein was predominantly associated with intracellular lipid droplets in both human and rodent hepatocytes. Conclusion: Together, our results suggest that hepatic lipid droplet–decorating protein MST4 is a critical regulatory node governing susceptibility to NAFLD and warrant future investigations to address the therapeutic potential of MST4 antagonism as a strategy to prevent or mitigate the development and aggravation of this disease.
Abbreviations
- 1H‐MRS
single‐proton magnetic resonance spectroscopy
- 8‐oxoG
8‐oxoguanine
- ADRP
adipose differentiation‐related protein
- BMI
body mass index
- CHOP
C/EBP‐homologous protein
- Ctrl
control
- DHE
dihydroethidium
- ER
endoplasmic reticulum
- FFA
free fatty acid
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- GCK
germinal center kinase
- HCC
hepatocellular carcinoma
- HNE
4‐hydroxynonenal
- IHHs
immortalized human hepatocytes
- mRNA
messenger RNA
- NAFL
nonalcoholic fatty liver
- NAFLD
nonalcoholic fatty liver disease
- NAS
NAFLD activity score
- NASH
nonalcoholic steatohepatitis
- NTC
nontargeting control
- OA
oleic acid
- PEX5
peroxisomal biogenesis factor 5
- PLIN5
perilipin 5
- PMP70
peroxisomal membrane protein 70 kDa
- RT‐PCR
real‐time polymerase chain reaction
- siRNA
small interfering RNA
- T2D
type 2 diabetes
- TAG
triacylglycerol
The human kinome encodes a large group of STE20 kinases, named after their yeast homologue Sterile20 involved in the mating pathway.( 1 ) Based on phylogenetic relationships, mammalian STE20 kinases can be divided into germinal center kinase (GCK) and p21‐activated kinase (PAK) families. The GCKs share a conserved N‐terminal catalytic domain and possess a variable noncatalytic C‐terminal region that mediates protein–protein interactions.( 2 ) The PAKs all contain a C‐terminal kinase domain and an N‐terminal regulatory GTPase‐binding domain.( 2 ) STE20‐type kinases are implicated in a broad range of cellular functions, such as regulation of proliferation, differentiation, apoptosis, polarity, migration, and cytoskeleton rearrangements.( 2 , 3 , 4 ) Notably, several STE20 kinases have recently been shown to aggravate ectopic lipid storage within nonadipose tissue in the context of nutritional stress, exacerbating the development of systemic insulin resistance in obesity.( 5 )
MST4 (also known as STK26 or MASK) is a member of the GCKIII subfamily of STE20 kinases and has been demonstrated to regulate diverse biological processes through phosphorylation of multiple substrates. To date, most of the studies have focused on the role of MST4 in cancer. MST4 is known to activate ERK and ezrin pathways to induce proliferation and epithelial–mesenchymal transition in several types of tumor cells.( 6 , 7 , 8 ) Recently, MST4 phosphorylation of ATG4B was also shown to promote autophagy, contributing to glioblastoma malignancy.( 9 ) The interaction of MST4 with the multi‐subunit STRIPAK complex has been associated with breast cancer metastasis.( 10 ) In line with the suggested role of MST4 in tumorigenesis, the expression of MST4 has been found to be up‐regulated in several different tumor types including gastric, breast, pancreatic, and prostate cancer, glioblastoma, and hepatocellular carcinoma (HCC), significantly correlating with poor patient prognosis.( 6 , 8 , 9 , 11 , 12 , 13 ) Recently, MST4 function has been associated with inflammatory responses through phosphorylation of TRAF6( 14 ) and gastric acid secretion by phosphorylating ACAP4 and ezrin.( 15 ) MST4 was previously also reported to act via activation of ezrin downstream of the LKB1/STRAD/MO25 polarization complex in brush border formation.( 16 )
Our recent investigations have revealed that several STE20‐type kinases contribute to the molecular pathogenesis of nonalcoholic fatty liver disease (NAFLD) by critically controlling hepatic lipid deposition as well as oxidative and endoplasmic reticulum (ER) stress in the liver.( 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 ) We now hypothesize that MST4 is also involved in the regulation of liver lipotoxicity. Here, we provide evidence that MST4 expression in human liver biopsies is positively correlated with the key features of NAFLD (i.e., hepatic steatosis, lobular inflammation, and hepatocellular ballooning). By modifying the expression levels of MST4 in cultured human hepatocytes, we also demonstrate a cell‐autonomous role of MST4 in the regulation of liver lipid partitioning, and oxidative and ER stress. Interestingly, we found that MST4 protein is predominantly associated with intracellular lipid droplets in both human and rodent hepatocytes.
NAFLD refers to a spectrum of histological abnormalities, ranging from nonalcoholic fatty liver (NAFL) defined as fatty infiltration in >5% of hepatocytes (steatosis) to nonalcoholic steatohepatitis (NASH), which in addition to hepatic steatosis is characterized by local inflammation and hepatocyte ballooning.( 25 ) Although NAFL is usually asymptomatic, patients with NASH have a high risk of liver‐related morbidity and mortality due to progression into cirrhosis, liver failure, and HCC.( 26 ) Despite intensive research in this field, no pharmacological therapy is currently approved to treat NASH, which is projected to become the most common indication for liver transplantation in the near future.( 26 ) Hence, deciphering the molecular mediators that control the initiation and aggravation of NAFLD is of high importance to develop improved methods for disease prediction, prevention, and treatment in this area of significant unmet medical need.
Materials and Methods
Cell Culture, RNA Interference, and Transient Overexpression
Immortalized human hepatocytes (IHHs; a gift from B. Staels, the Pasteur Institute of Lille, University of Lille Nord de France, Lille, France) were maintained in William’s E Medium (GlutaMAX supplemented; Gibco, Paisley, United Kingdom) supplemented with human insulin (20 U/L; Actarapid Penfill; Novo Nordisk, Bagsværd, Denmark) and dexamethasone (50 nmol/L; Sigma‐Aldrich, St. Louis, MO). IHHs, originally obtained from healthy liver tissue removed surgically from a 59‐year‐old man and immortalized by stable transfection with SV40 large T antigen–expressing plasmid, have been shown to retain several differentiated features of primary human hepatocytes.( 27 , 28 ) HepG2 cells (HCC, human; American Type Culture Collection, Manassas, VA) were maintained in Dulbecco’s modified Eagle’s medium (GlutaMAX supplemented; Gibco). Both culture media were supplemented with 10% (vol/vol) fetal bovine serum and 1% (vol/vol) penicillin/streptomycin (Gibco). Cells were demonstrated to be free of mycoplasma infection by MycoAlert Mycoplasma Detection Kit (Lonza, Basel, Switzerland).
IHHs and HepG2 cells were transfected with MST4 small interfering RNA (siRNA; HS01_00030410; Sigma‐Aldrich) or scrambled siRNA (SIC001; Sigma‐Aldrich) using Lipofectamine RNAiMax (Thermo Fisher Scientific, Waltham, MA). Cells were also transfected with human MYC‐tagged MST4 expression plasmid (EX‐W0097‐M43; GeneCopoeia; Labomics, Nivelles, Belgium) or an empty control plasmid (EX‐NEG‐M43; GeneCopoeia) using Lipofectamine 2000 (Thermo Fisher Scientific).
Assessment of Lipotoxicity in Cultured Human Hepatocytes
In all experiments described subsequently, transfected cells were exposed to 50 µmol/L oleic acid (OA; Sigma‐Aldrich) for 48 hours, which is known to efficiently induce steatosis in vitro (see Figs. 3A and 4A for schematic presentation of the experimental design).
Fig. 3.
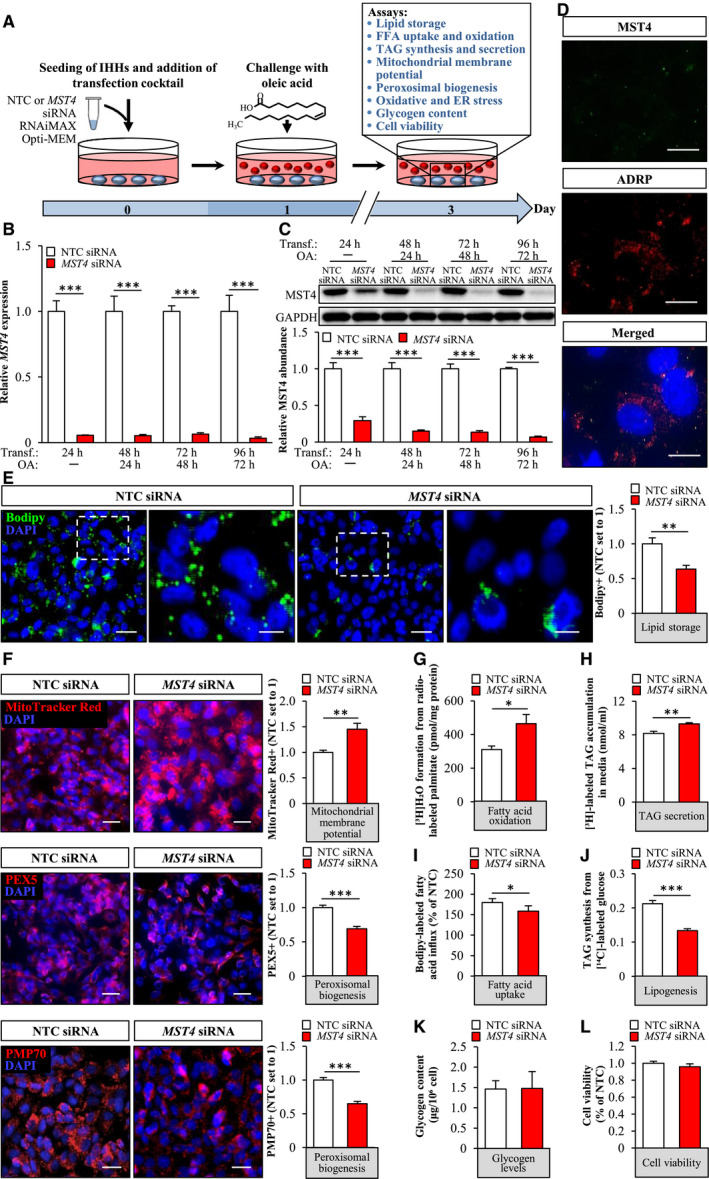
Silencing of MST4 decreases lipid accumulation in IHHs. IHHs were transfected with MST4 siRNA or NTC siRNA and challenged with OA for 48 hours. (A) Schematic illustration of the study design. (B,C) MST4 mRNA (B) and protein (C) abundance. Protein levels were analyzed by densitometry; representative western blots are shown with GAPDH used as a loading control. (D) Representative immunofluorescence images of cells double‐stained with antibodies for MST4 (green) and ADRP (red); nuclei stained with DAPI (blue). (E,F) Representative images of cells stained with Bodipy (green) or MitoTracker Red (red), or processed for immunofluorescence with anti‐PEX5 (red) or anti‐PMP70 (red) antibodies; nuclei stained with DAPI (blue). Quantification of the staining. (G) Oxidation of radiolabeled palmitate. (H) Secretion of [3H]TAG into the media. (I) Fatty acid uptake rate. (J) TAG synthesis from [14C]‐labeled glucose. (K) Glycogen levels. (L) Cell viability was assessed using resazurin. In (D) and (F), the scale bars represent 10 µm and 25 µm, respectively; in (E), the scale bars at the left and right represent 25 µm and 10 µm, respectively. Data are presented as mean ± SEM from 4‐10 wells per group. Abbreviation: Transf, transfection. *P < 0.05, **P < 0.01, ***P < 0.001.
Fig. 4.
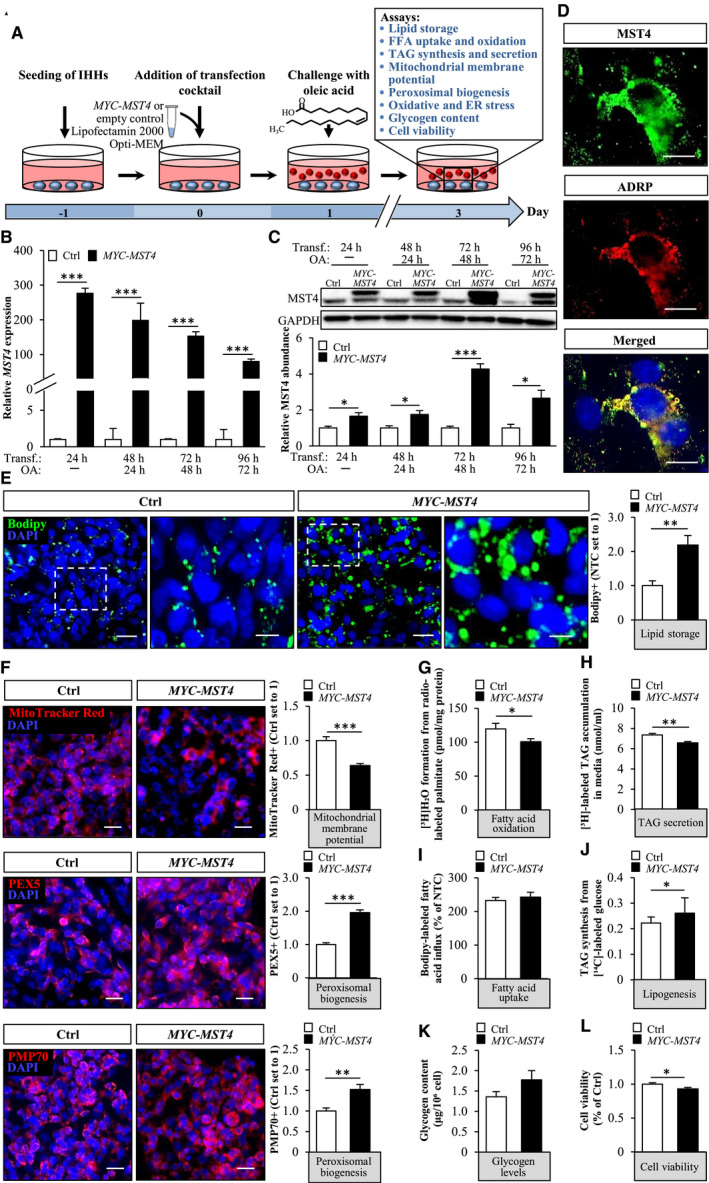
Overexpression of MST4 aggravates lipid accumulation in IHHs. IHHs were transfected with MYC‐tagged MST4 expression plasmid or an empty‐control plasmid and challenged with OA for 48 hours. (A) Schematic illustration of the study design. (B,C) MST4 mRNA (B) and protein (C) abundance. Protein levels were analyzed by densitometry; representative western blots are shown with GAPDH used as a loading control. (D) Representative immunofluorescence images of cells double‐stained with antibodies for MST4 (green) and ADRP (red); nuclei stained with DAPI (blue). (E,F) Representative images of cells stained with Bodipy (green) or MitoTracker Red (red), or processed for immunofluorescence with anti‐PEX5 (red) or anti‐PMP70 (red) antibodies; nuclei stained with DAPI (blue). Quantification of the staining. (G) Oxidation of radiolabeled palmitate. (H) Secretion of [3H]TAG into the media. (I) Fatty acid uptake rate. (J) TAG synthesis from [14C]‐labeled glucose. (K) Glycogen levels. (L) Cell viability was assessed using resazurin. In (D) and (F), the scale bars represent 10 µm and 25 µm, respectively; in (E), the scale bars at the left and right represent 25 µm and 10 µm, respectively. Data are presented as mean ± SEM from 4‐12 wells per group. Abbreviations: Ctrl, control; Transf, transfection. *P < 0.05, **P < 0.01, ***P < 0.001.
Cells were stained with Bodipy 493/503 (Invitrogen, Carlsbad, CA) for neutral lipids, MitoTracker Red (Thermo Fisher Scientific) for mitochondrial membrane potential,( 29 ) or dihydroethidium (DHE; Life Technologies, Grand Island, NY) for superoxide radicals, as previously described.( 22 ) In parallel, cells were processed for immunofluorescence with anti‐MST4, anti‐adipose differentiation‐related protein (ADRP), anti‐peroxisomal biogenesis factor 5 (PEX5), anti‐peroxisomal membrane protein 70 kDa (PMP70), anti‐MYC, anti‐8‐oxoguanine (8‐oxoG), anti‐4‐hydroxynonenal (HNE), anti‐E06, anti‐KDEL, or anti‐C/EBP‐homologous protein (CHOP) antibodies (see Supporting Table S1 for antibody information). The labeled area was quantified in 6‐8 randomly selected microscopic fields (×20) per well using the ImageJ software (1.47v; National Institutes of Health, Bethesda, MD).
To measure β‐oxidation, cells were incubated in the presence of (9,10‐3H[N])palmitic acid (Perkin‐Elmer, Waltham, MA), and [3H]‐labeled water was quantified as the product of free fatty acid (FFA) oxidation (see Supporting Information Materials for details).( 20 ) Mitochondrial oxygen consumption rate (OCR) was measured by the Seahorse XFe96 Analyzer and the SeaHorse XF Cell Mito Stress Test Kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s recommendations, using Seahorse XF base media containing 10 mmol/L glucose, 2 mmol/L glutamine, and 1 mmol/L pyruvate (Agilent Technologies). Triacylglycerol (TAG) secretion and incorporation of [14C]glucose (Perkin‐Elmer) into TAGs were measured as previously described (see Supporting Information Materials for details).( 21 ) Fatty acid uptake was quantified using the QBT (Quencher‐Based Technology) Fatty Acid Uptake Assay Kit (Molecular Devices, San Jose, CA), glycogen levels were measured using the Glycogen Assay Kit (Sigma‐Aldrich), and cell viability was assessed using the CellTiter‐Blue Cell Viability Assay (Promega, Stockholm, Sweden) (see Supporting Information Materials for details). The TAG hydrolase activity was determined in total cell lysates using [3H]triolein (PerkinElmer) as the substrate in an assay buffer containing 0.25 mmol/L triolein, 0.8 mmol/L phosphatidylcholine, 20 mmol/L Tris, 150 mmol/L NaCl, and 1 mmol/L ethylene diamine tetraacetic acid, pH 8.0 (Sigma‐Aldrich). To measure autophagic flux, cells were treated with 50 nmol/L or 100 nmol/L bafilomycin A1 (Sigma‐Aldrich) for 2 hours.
Quantitative Real‐Time Polymerase Chain Reaction and Western Blot
The methods used for the isolation of RNA, the synthesis of complementary DNA, and the quantitative real‐time polymerase chain reaction (RT‐PCR) assay have been described previously.( 21 ) Western blot was carried out as previously described( 30 ) (see Supporting Table S1 for antibody information).
Analysis of MST4 Expression in Human Liver Biopsies
The levels of MST4 messenger RNA (mRNA) were quantified in liver biopsy material collected from Caucasian individuals (men, n = 35; women, n = 27) who underwent laparoscopic abdominal surgery for Roux‐en‐Y bypass (n = 12), sleeve gastrectomy (n = 9), or elective cholecystectomy (n = 41). The participants fulfilled the following inclusion criteria: (1) men and women, age >18 years; (2) indication for elective laparoscopic or open abdominal surgery; (3) body mass index (BMI) between 18 and 50 kg/m2; (4) feasible for abdominal MRI; and (5) signed written informed consent. The exclusion criteria for liver biopsy donors were (1) significant acute or chronic inflammatory disease or clinical signs of infection; (2) C‐reactive protein >10 mg/dL; (3) type 1 diabetes and/or antibodies against glutamic acid decarboxylase and islet cell antibodies; (4) systolic blood pressure >140 mm Hg and diastolic blood pressure >95 mm Hg; (5) clinical evidence of cardiovascular or peripheral artery disease; (6) thyroid dysfunction; (7) alcohol or drug abuse; and (8) pregnancy. Type 2 diabetes (T2D) was defined by a fasting plasma glucose >7.0 mmol/L and/or a 120‐minute oral glucose tolerance test glucose >11.1 mmol/L. For participant characteristics, see Cansby et al.( 21 )
Total body fat was measured by dual X‐ray absorptiometry, and liver fat was measured by single‐proton magnetic resonance spectroscopy (1H‐MRS) as previously described.( 31 ) A small liver biopsy was obtained during the surgery (between 8 and 10 hours after an overnight fast), immediately snap‐frozen in liquid nitrogen, and stored at −80°C. NAFLD activity score (NAS) was assessed on liver sections by a certified pathologist.( 32 ) Quantitative RT‐PCR was carried out in liver biopsies as described previously using the probes for MST4 (Hs01550830_m1; Life Technologies) and 18S rRNA (Hs99999901_s1; Life Technologies), which span exon–exon boundaries to improve the specificity.
All participants gave their written informed consent before taking part in the study. All investigations were approved by the Ethics Committee of the University of Leipzig, Germany (159‐12‐21052012 and 017‐12‐23012012) and were performed in accordance with the Declaration of Helsinki.
Statistical Analysis
Statistical significance between the groups was evaluated using the two‐sample Student t test with a value of P < 0.05 considered statistically significant. Correlation between MST4 expression in human liver biopsies and hepatic lipid content, inflammation, and ballooning was investigated by Spearman’s rank correlation analysis after the Kolmogorov–Smirnov test was performed to assess normality of data. All statistical analyses were conducted using SPSS statistics (v24; IBM Corporation, Armonk, NY).
Results
Hepatic MST4 Expression Is Positively Correlated With the Severity of NAFLD in Humans
The hallmark of NAFLD is the excessive accumulation of fat in the liver.( 25 ) Thus, we first analyzed the expression of MST4 mRNA in liver biopsies in relation to the hepatic fat levels measured by 1H‐MRS in a cohort of 62 subjects (BMI: 22.7‐45.6 kg/m2; body fat: 19.5%‐57.9%; and liver fat: 1.1%‐50.0%). We found that hepatic MST4 expression was positively correlated with liver fat content (Fig. 1A). Notably, there was no correlation between liver MST4 mRNA and the BMI, body fat, or waist‐to‐hip ratio of the participants (Supporting Fig. S1).
Fig. 1.
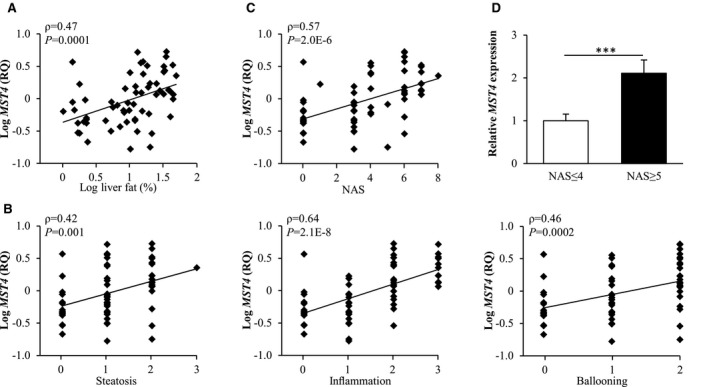
Expression of MST4 mRNA in human liver is significantly and positively correlated with the severity of NAFLD. (A) Correlation between hepatic fat content measured by 1H‐MRS and MST4 mRNA expression assessed in human liver biopsies by quantitative RT‐PCR. (B,C) Correlation between MST4 mRNA expression in human liver biopsies and three individual histologic lesions of NAS (i.e., steatosis, lobular inflammation, and hepatocellular ballooning (B) as well as the total NAS (C). (D) MST4 mRNA expression in livers of individuals with low (NAS ≤ 4) versus high (NAS ≥ 5) NAS. Results are presented as mean ± SEM. Abbreviation: RQ, relative quantification.
We further examined the correlation between hepatic MST4 transcript levels and severity of NAFLD by applying the widely used semiquantitative histological scoring NAS, which is used in today’s clinical diagnosis.( 32 ) We found that MST4 mRNA expression was positively correlated with all three individual features of NAS (liver steatosis, lobular inflammation, and hepatocellular ballooning) as well as total NAS (Fig. 1B,C). Moreover, the subjects with NAS ≥ 5, which defines definite NASH (n = 24), displayed about 2‐fold higher MST4 mRNA levels compared to subjects with NAS ≤ 4, which defines simple steatosis or borderline NASH (n = 38) (Fig. 1D).
In the past few years, several epidemiological studies have reported a convincing association between NAFLD and an increased risk of developing T2D; once developed, T2D also has the potential to promote the progression to NASH.( 33 ) Based on this complex and bidirectional relationship between NAFLD and T2D, we decided to perform additional correlation analyses in a subset of patients who had been diagnosed with T2D (n = 24). We found that the liver fat assessed by MRS as well as all three individual histological components of NAS correlated significantly and positively with MST4 mRNA expression even in this cohort (Supporting Fig. S2).
MST4 Protein Is Associated With Intrahepatocellular Lipid Droplets
We determined the subcellular localization of MST4 protein in mouse liver sections and in two human cell lines of hepatic origin (IHHs and HepG2) using immunofluorescence microscopy. We found that MST4 fully colocalized with ADRP (also known as adipophilin or perilipin‐2), the main hepatic lipid droplet coating protein( 34 ) (Fig. 2A). Notably, ADRP has been shown to only exist in cells associated with lipid droplets, as, in the absence of lipid droplet binding, ADRP is rapidly degraded through the ubiquitin‐proteasome system.( 35 ) The full overlap of the immunostainings for MST4 and ADRP therefore suggests that the localization of MST4 protein in hepatocytes is essentially restricted to lipid droplets.
Fig. 2.
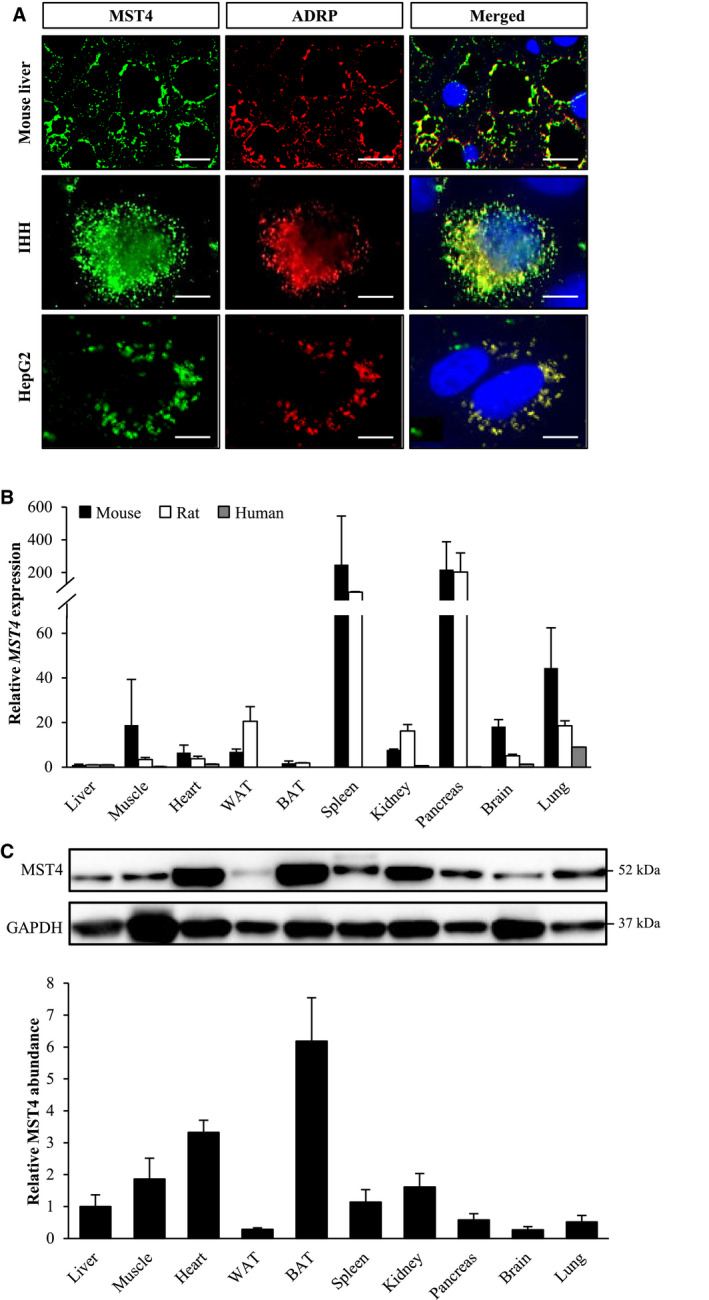
MST4, expressed in a broad range of tissues, associates with intracellular lipid droplets in hepatocytes. (A) Representative immunofluorescence images of liver sections from high fat–fed mice, and IHHs and HepG2 cells, double‐stained with antibodies for MST4 (green) and ADRP (red); merged image shows colocalization in yellow with nuclei stained with DAPI (blue). The scale bars at the top and two bottom represent 20 µm and 10 µm, respectively. (B) Relative MST4 mRNA expression was assessed by quantitative RT‐PCR in mouse, rat, and human tissues. The transcript level of MST4 in the liver of each species is set to 1. Mouse and rat data are presented as mean ± SEM from three to six animals. Human MTC Panel I (Takara, Kyoto, Japan) was analyzed for human expression data. (C) Mouse tissue lysates were analyzed by western blot using antibodies specific for MST4; MST4 protein levels were quantified by densitometry with the protein abundance in the liver set to 1; representative western blots are shown with glyceraldehyde 3‐phosphate dehydrogenase used as a loading control. Results are presented as mean ± SEM from five mice. Abbreviations: BAT, brown adipose tissue; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; WAT, white adipose tissue.
We also quantified the relative abundance of MST4 mRNA and protein across a broad panel of tissues. Similarly to previous reports describing a broad distribution of MST4 transcript in human body,( 36 ) we detected MST4 mRNA in all tissues examined in human, mouse, and rat (Fig. 2B). Western blot analysis performed in mice also identified MST4 protein in all organs studied (Fig. 2C). The relative abundance of MST4 protein in mouse liver was lower compared with several other tissues such as brown adipose tissue, heart, muscle, and kidney (Fig. 2C). Notably, immunostainings with anti‐MST4 antibody demonstrated homogenous protein expression in mouse liver, adipose tissue, heart, and skeletal muscle, with most of the hepatocytes, adipocytes, cardiomyocytes, and muscle fibers expressing MST4; the presence of MST4 was restricted to the tubulointerstitial region in kidney sections (Fig. 2A and Supporting Fig. S3).
The differences in relative quantities of mouse Mst4 mRNA (highest levels in spleen and pancreas) and protein (highest abundance in brown adipose tissue and heart) suggest the involvement of post‐transcriptional mechanisms in regulating the Mst4 gene function.
Silencing of MST4 Suppresses Lipid Accumulation in Human Hepatocytes
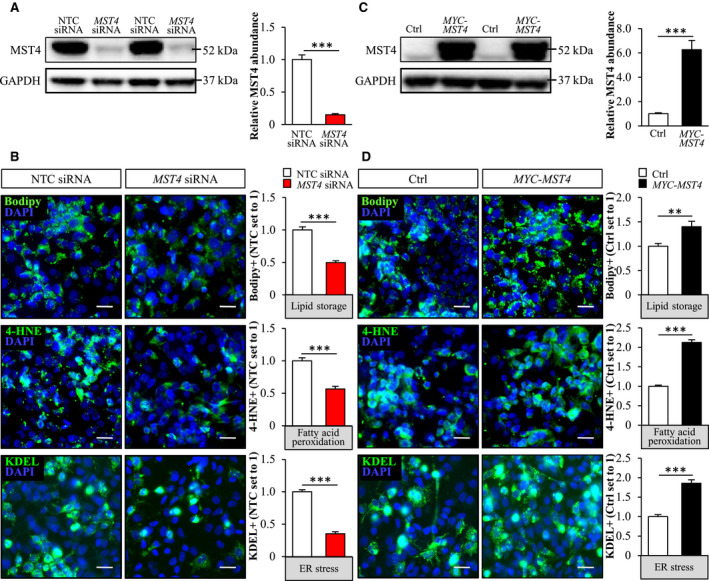
To explore the impact of MST4 knockdown on intrahepatocellular lipid storage, we transfected IHHs with MST4‐specific siRNA or with a nontargeting control (NTC) siRNA (Fig. 3A). In all experiments, we subsequently exposed the IHHs to OA, to mimic the metabolic environment in high‐risk individuals (Fig. 3A). Cells transfected with MST4 siRNA displayed substantially lower target mRNA and protein expression assessed by quantitative RT‐PCR and western blot, respectively (Fig. 3B,C). Consistently, we detected no immunostaining using MST4 antibodies in cells transfected with MST4 siRNA (Fig. 3D).
First, we stained IHHs with Bodipy 493/503, which detects neutral lipids within lipid droplets. We found that the Bodipy‐positive area was 1.6 ± 0.1‐fold lower in cells transfected with MST4 siRNA compared with NTC siRNA (Fig. 3E). Next, we examined the mechanisms underlying the reduced lipid storage observed in IHHs by MST4 silencing. We found that the staining with MitoTracker Red, a fluorescent dye that monitors the membrane potential of mitochondria and is an indirect indicator of the mitochondrial activity,( 29 ) was 1.4 ± 0.1‐fold higher in MST4‐deficent cells (Fig. 3F). Consistently, the depletion of MST4 resulted in 1.5 ± 0.2‐fold increase in β‐oxidation, assessed by quantification of [3H]‐labeled water as the product of [9,10‐3H(N)]palmitic acid oxidation (Fig. 3G). To further study the mitochondrial function, we measured OCR in live cells by the SeaHorse technology using glucose as substrate. We did not detect any significant differences in basal or maximal respiration, whereas proton leak across mitochondrial membrane was lower, and mitochondrial coupling efficiency was higher, in MST4‐deficient cells (Supporting Fig. S4). We also revealed slightly enhanced secretion of de novo synthesized TAG into the media in IHHs, where MST4 was knocked down (Fig. 3H). Reciprocally, we found that fatty acid influx and TAG synthesis were significantly lower in IHHs transfected with MST4 siRNA compared with NTC siRNA (Fig. 3I,J).
Notably, in contrast to enhanced mitochondrial function, the peroxisomal activity was decreased in MST4‐deficient cells, as evidenced by diminished immunostaining for peroxisome biogenesis marker PEX5 and peroxisomal membrane protein PMP70 (Fig. 3F).
Glycogen content was similar in cells transfected with MST4 siRNA versus NTC siRNA (Fig. 3K), and cell viability was unaffected (Fig. 3L).
Overexpression of MST4 Aggravates Lipid Storage in Human Hepatocytes
To examine whether an increase in MST4 abundance would lead to an opposite effect on hepatocyte lipid metabolism compared with MST4 knockdown, we transfected IHHs with human MYC‐tagged MST4 expression plasmid or an empty control plasmid (Fig. 4A). Similarly to the experimental setup used in IHHs transfected with MST4 siRNA, we also challenged cells with OA before assessments (Fig. 4A). Immunostainings with anti‐MYC antibody demonstrated a high transfection efficacy of about 90% in IHHs transfected with MYC‐MST4 expression plasmid (Supporting Fig. S5A). IHHs transfected with MYC‐MST4 displayed substantially higher MST4 mRNA and protein abundance (152.7 ± 12.8 and 4.3 ± 0.3‐fold increase in mRNA and protein levels, respectively, at 72 hours following transfection when assays were performed (Fig. 4B,C). Nonsense‐mediated mRNA decay, triggered by the termination codon of the ORF situated upstream of the most 3’ splice site in the expression plasmid, is likely the reason for the discrepancy observed between the very high increase in MST4 transcript levels versus the relatively modest raise in protein abundance.( 37 ) Immunofluorescence microscopy also demonstrated that, similarly to the endogenous protein, MYC‐tagged MST4 was targeted to the lipid droplets visualized by ADRP staining (Fig. 4D).
We found that, reciprocally to the effect of MST4 knockdown, the accumulation of intracellular lipids was higher in MST4‐overexpressing IHHs, as evidenced by 2.2 ± 0.3‐fold increase in Bodipy‐positive area (Fig. 4E). Mechanistically, we observed 1.6 ± 0.1‐fold lower staining with MitoTracker Red in IHHs transfected with MST4 expression plasmid compared with vector control (Fig. 4F), in parallel with significant reduction in β‐oxidation rate (Fig. 4G). Furthermore, the secretion of TAG into the media was slightly suppressed in MST4‐overexpressing cells (Fig. 4H). In contrary, de novo lipogenesis was augmented in IHHs transfected with MST4 expression plasmid, without any change in fatty acid uptake rate (Fig. 4I,J). Notably, overexpression of MST4 in IHHs substantially enhanced immunostaining for PEX5 and PMP70 (Fig. 4F), suggesting higher peroxisomal activity.
Glycogen accumulation was not affected by MST4 overexpression (Fig. 4K). Interestingly, cell viability was slightly lower in IHHs transfected with MYC‐MST4 versus vector control (Fig. 4L).
MST4 Controls Oxidative and ER Stress in Hepatocytes
Excessive lipid storage in hepatocytes is considered a primary event, which causes oxidative and ER stress, initiating the activation of inflammatory and apoptotic pathways and pro‐fibrotic responses.( 38 ) To this end, we found that lower intracellular lipid deposition in MST4‐deficient IHHs was accompanied by reduced oxidative stress, as evidenced by repressed levels of superoxide radicals (O•−) quantified by DHE staining, abrogated oxidative DNA damage detected by immunostaining for 8‐oxoG, and protection against deposition of lipid peroxidation products and oxidized phospholipids measured by immunostaining for 4‐HNE and E06, respectively (Fig. 5A). Reciprocally, overexpression of MST4 resulted in substantially aggravated oxidative damage in IHHs, as evidenced by elevated levels of DHE, 8‐oxoG, 4‐HNE, and E06 (Fig. 5B). In parallel with alterations in oxidative stress markers, we detected reduced or enhanced immunostaining for KDEL (a signal motif for ER retrieval) and CHOP (a marker for ER stress‐induced cell death) in IHHs where MST4 was depleted or overexpressed, respectively (Fig. 5A,B). Western blot analysis also revealed a lower abundance of ER stress markers ATF4 and XBP1s, but not BiP, in MST4‐deficient hepatocytes (Supporting Fig. S6).
Fig. 5.
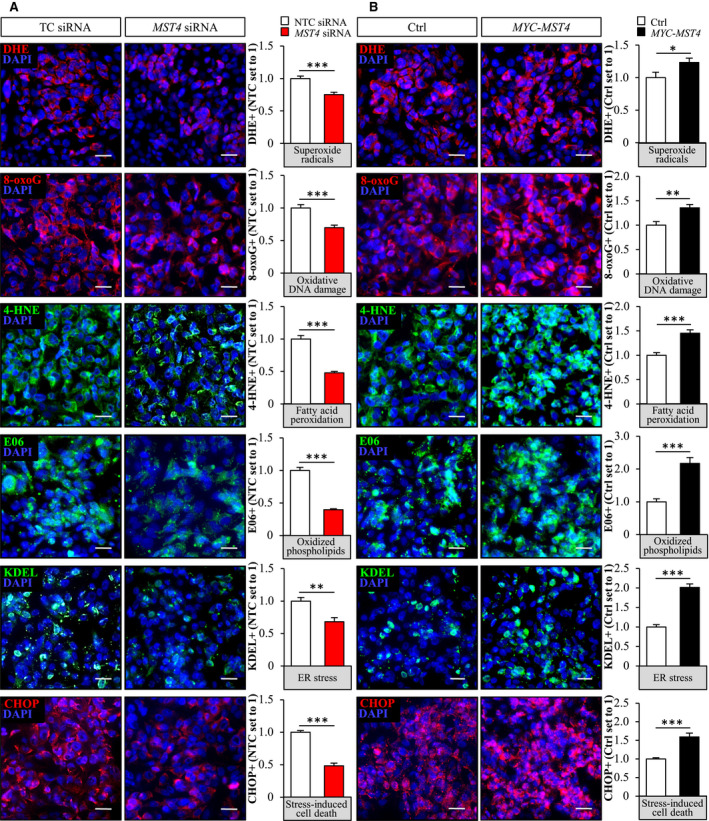
MST4 regulates oxidative and ER stress in IHHs. IHHs were transfected with MST4 siRNA or NTC siRNA (A), or with MYC‐tagged MST4 expression plasmid or an empty control plasmid (B), and challenged with OA for 48 hours. Representative images of cells stained with DHE (red) or processed for immunofluorescence with anti‐8‐oxoG (red), anti‐4‐HNE (green), anti‐E06 (green), anti‐KDEL (green), or anti‐CHOP (red) antibodies; nuclei stained with DAPI (blue). Quantification of the staining. The scale bars represent 25 µm. Data are presented as mean ± SEM from eight wells per group. *P < 0.05, **P < 0.01, ***P < 0.001.
In line with our observations in IHHs, we found that, along with lower lipid storage, the oxidative and ER stress were significantly decreased in human hepatoma cell line HepG2 transfected with MST4 siRNA compared with NTC siRNA (Fig. 6A,B). Reciprocally, lipid accumulation and oxidative and ER stress were markedly exacerbated in HepG2 cells transfected with MST4 expression plasmid versus an empty control plasmid (Fig. 6C,D). Notably, similarly to IHHs, a high transfection efficacy of about 90% was reached in HepG2 transfected with MYC‐MST4 expression plasmid (Supporting Fig. S5B).
Fig. 6.
Silencing of MST4 suppresses lipid storage as well as oxidative and ER stress in HepG2 cells, and reciprocal effect is seen with MST4 overexpression. HepG2 cells were transfected with MST4 siRNA or NTC siRNA (A,B) or with MYC‐tagged MST4 expression plasmid or an empty control plasmid (C,D), and challenged with OA for 48 hours. (A,C) MST4 protein levels were analyzed by densitometry; representative western blots are shown with GAPDH used as a loading control. (B,D) Representative images of cells stained with Bodipy (green) or processed for immunofluorescence with anti‐4‐HNE (green) or anti‐KDEL (green) antibodies; nuclei stained with DAPI (blue). Quantification of the staining. In (B) and (D), the scale bars represent 25 µm. Data are presented as mean ± SEM from four to eight wells per group. **P < 0.01, ***P < 0.001.
Silencing of MST4 Has No Impact on Lipolytic Activity or Autophagic Flux in Hepatocytes
Based on the association of MST4 with intrahepatocellular lipid droplets, we hypothesized that MST4 regulates lipid mobilization from the droplets locally by increasing the rate of canonical lipolysis, where a series of lipases acting on the lipid droplet surface [i.e., adipose triglyceride lipase (ATGL), hormone‐sensitive lipase (HSL), and monoglyceride lipase (MGL)] sequentially reduce TAG into FFAs, which are then directed to mitochondria for β‐oxidation or forwarded to the ER and Golgi for synthesis and secretion of very low‐density lipoprotein (VLDL)‐TAG.( 39 ) However, the analysis of TAG hydrolase activity using triolein as the substrate (assay that measures the rate of canonical lipolysis( 40 )) did not detect any differences in IHHs transfected with MST4 siRNA compared with NTC siRNA (Supporting Fig. S7).
Notably, we found that the abundance of lipid droplet–associated protein perilipin 5 (PLIN5) was higher in IHHs transfected with MST4 siRNA compared with NTC siRNA (Fig. 7A). This is of interest, because PLIN5 has recently emerged as a critical node that regulates hepatic lipid droplet dynamics by promoting autophagy but also by enhancing mitochondrial biogenesis and preventing fatty acid–induced inflammation.( 41 ) To evaluate the possible role of increased autophagy in the enhanced lipid catabolism observed in MST4‐deficient hepatocytes, we next measured the abundance of the key marker of autophagic flux LC3‐II in IHHs transfected with MST4 siRNA versus NTC siRNA. However, we did not find any evidence that the silencing of MST4 in IHHs would alter hepatocyte autophagy, when assessed both under basal conditions or after treatment with lysosomal inhibitor bafilomycin A1 (Fig. 7B).
Fig. 7.
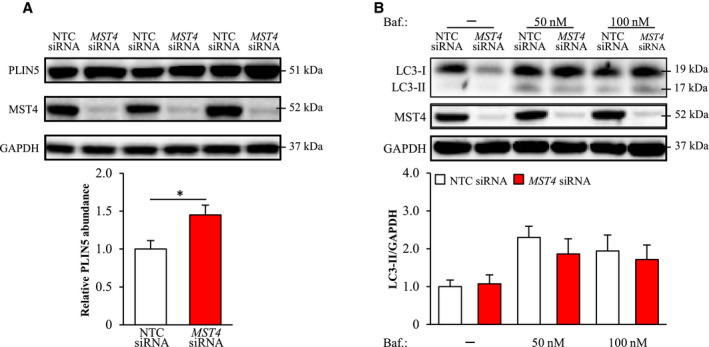
Knockdown of MST4 in IHHs increases PLIN5 levels but has no impact on autophagic flux. IHHs were transfected with MST4 siRNA or NTC siRNA and challenged with OA for 48 hours. In (B), bafilomycin A1 (inhibitor of autophagosome‐lysosome fusion) was added 2 hours before harvesting cells. Cell lysates were analyzed by western blot using antibodies specific for PLIN5, LC3, or MST4. Protein levels were analyzed by densitometry; representative western blots are shown with GAPDH used as a loading control. Results are presented as mean ± SEM from 6‐11 wells per group. Abbreviation: Baf., bafilomycin A1. *P < 0.05.
Consistent with these findings in IHHs, we detected significantly increased levels of PLIN5 without any alterations in LC3‐II abundance in HepG2 transfected with MST4 siRNA versus NTC siRNA (Supporting Fig. S8).
Discussion
NAFLD is the most common chronic liver disease with an estimated global prevalence of about 25%.( 25 ) Despite this high medical importance, the mechanisms governing hepatic lipid accumulation, and the reasons whereby a subgroup of patients with NAFLD with simple liver steatosis (i.e., NAFL) progress into hepatic inflammation and cellular damage (i.e., NASH), remain incompletely understood.( 25 ) This study provides several lines of evidence for a hitherto unknown role of hepatic lipid droplet–associated protein kinase MST4 in molecular pathogenesis of NAFLD.
Excessive intrahepatocellular lipid accumulation is considered the primary insult in NAFLD.( 25 ) Here we found that MST4 transcript levels in human liver biopsies were positively correlated with hepatic steatosis measured both by MRS and histological scoring. Furthermore, we observed attenuated lipid deposition within lipid droplets in MST4‐deficient human hepatocytes, and, reciprocally, aggravated lipid storage was detected in MST4‐overexpressing hepatocytes. Conceptually, imbalance in liver lipid homeostasis caused either by an increased substrate availability for lipid droplet formation (elevated FFA uptake and de novo lipogenesis) or impaired lipid use (decreased β‐oxidation and VLDL‐TAG secretion) can lead to hepatic steatosis. Indeed, we found that MST4 cell autonomously regulates both hepatocellular lipid anabolism and catabolism: de novo lipogenesis was significantly suppressed, and β‐oxidation and lipid export were markedly enhanced, in MST4‐deficient hepatocytes, with an opposite effect seen in cells overexpressing MST4 (Fig. 8).
Fig. 8.
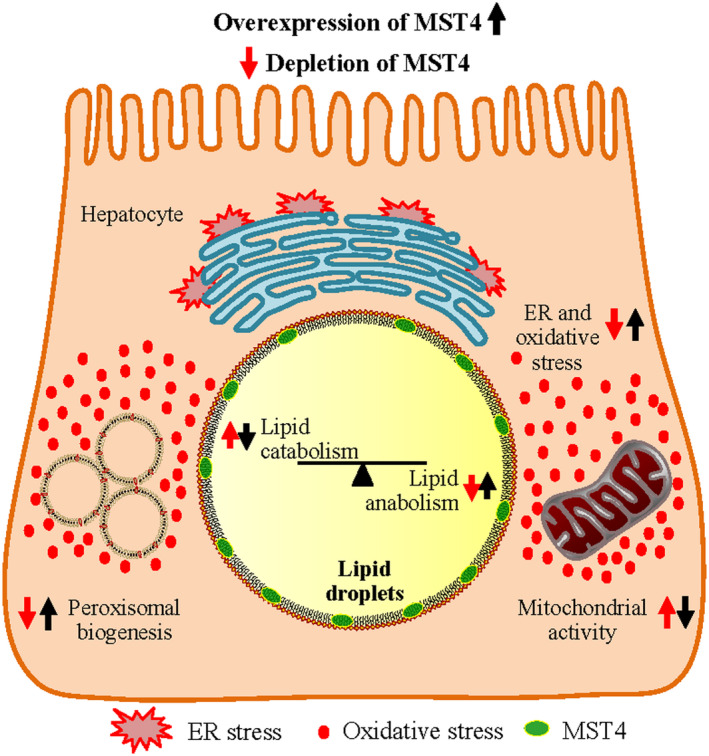
Schematic presentation of the function of MST4 in regulating intrahepatocellular lipid storage. Silencing of MST4 stimulates lipid droplet catabolism through enhanced β‐oxidation and TAG secretion, and inhibits lipid droplet anabolism through suppressed FFA influx and TAG synthesis. Furthermore, knockdown of MST4 protects hepatocytes from oxidative and ER stress. The opposite effects on lipid storage and metabolic stress are seen when MST4 is overexpressed.
Ectopic lipid storage within hepatocytes in NAFL triggers oxidative and ER stress in the liver, which in turn fuels local inflammation, fibrosis, and cell damage, ultimately leading to the disease progression toward NASH.( 38 ) Remarkably, in parallel with reciprocal alterations in lipid accumulation, we found substantially attenuated or aggravated oxidative/ER stress in human hepatocytes with reduced or increased MST4 levels, respectively. Furthermore, MST4 mRNA expression in human liver biopsies was positively correlated not only with hepatic steatosis but also with lobular inflammation and hepatocellular ballooning (i.e., the key histological lesions characterizing NASH). Together, these results suggest that MST4, in addition to exacerbating liver lipid storage, also triggers disease progression from NAFL to NASH.
As a central feature in patients with obesity and NAFLD, liver mitochondrial dysfunction driving hepatic ROS production has been suggested to play a pivotal role during the transition from simple steatosis to NASH.( 25 ) Ultrastructural mitochondrial abnormalities have been documented in patients and experimental models of NASH.( 42 ) Furthermore, rodents and patients with NASH display decreased mitochondrial coupling efficiency and, consequently, increased proton leak in liver.( 43 ) In light of this evidence, it is likely that lower mitochondrial proton leak and more efficient oxidative flux detected in MST4‐deficient hepatocytes in this study contributed to the protection against oxidative damage and inflammatory insult. Remarkably, an improved mitochondrial function detected in MST4‐deficent hepatocytes was paralleled by diminished peroxisomal biogenesis. This is in line with previous evidence showing that compromised mitochondrial activity in NAFLD triggers peroxisomal fatty acid oxidation, which further escalates the oxidative stress and NASH progression.( 44 )
Interestingly, we found that MST4 has a very distinct subcellular localization pattern in both human and mouse hepatocytes, coating intracellular lipid droplets. Hepatic lipid droplets were initially assumed to be inert sites of fat sequestration; however, during the past decade, new insights have emerged with respect to liver lipid droplet biology, and in particular, the complexity of their proteome composition.( 39 ) Today, hepatic lipid droplets are recognized as highly dynamic organelles that orchestrate not only liver lipid metabolism but also cell signaling, protein quality control and storage, mitochondrial function, and membrane trafficking as examples; however, the molecular mechanisms of these complex functions still remain largely elusive. Notably, the most well‐characterized genetic risk factors that confer susceptibility to the full spectrum of NAFLD are PNPLA3 and HSD17B13, which both encode hepatic lipid droplet proteins,( 45 , 46 ) thus highlighting a critical role of lipid droplets in the molecular pathogenesis of NAFLD. In contrast to our experiments using immunostainings with anti‐MST4 antibody, several recent studies characterizing the liver lipid droplet proteome by liquid chromatography–tandem mass spectrometry (LC/MS‐MS) analysis have failed to identify MST4.( 22 , 47 , 48 , 49 ) Because MST4 lacks a classical membrane‐spanning domain consisting of hydrophobic amino acids (search performed using the TM Finder tool), it is likely to be targeted to lipid droplets through protein–protein interactions. These types of interactions may be disrupted during the purification of the lipid droplet fraction before LC/MS‐MS runs, providing one possible explanation for this apparent discrepancy.
In addition to MST4, the GCKIII subfamily of STE20‐type kinases includes STK25 (also known as YSK1 or SOK1) and MST3 (also known as STK24), which are almost 90% identical in the amino acid sequence with MST4 in the kinase domain, but have less than 20% alignment in the noncatalytic C‐terminal domain. Similarly to MST4, both STK25 and MST3 proteins associate with intrahepatocellular lipid droplets( 18 , 20 , 21 ) and regulate liver lipotoxicity in the context of obesity. First, we found that obese mice overexpressing STK25 accumulate excessive fat in the liver, which is accompanied by exacerbated influx of macrophages and nutritional fibrosis.( 18 , 19 , 30 ) Reciprocally, diet‐induced liver steatosis, inflammation, and fibrosis are efficiently prevented in mice with reduced STK25 activity by genetic ablation or antisense oligonucleotide treatment.( 17 , 19 , 23 ) Furthermore, STK25 mRNA and protein levels in human liver biopsies correlate with the severity of NAFLD,( 19 , 20 ) and several common nonlinked SNPs in the human STK25 gene are associated with altered liver fat content.( 23 ) Similarly, our recent studies have revealed that antagonizing the activity of MST3 in cultured human hepatocytes protects against intrahepatocellular lipid storage and associated damage.( 21 ) Interestingly, the silencing of MST4 in IHHs in this study resulted in a significant compensatory increase in the levels of MST3 and STK25 proteins (Supporting Fig. S9). Importantly, our results clearly demonstrate that, although these three closely related kinases are all present on the surface of intrahepatocellular lipid droplets and they carry out similar functions, the enhanced abundance of MST3 and STK25 is not able to compensate for the impact of loss of MST4.
Today, no targeted pharmacological therapy is approved against NAFLD/NASH, and the treatment options are restricted to lifestyle changes and liver transplantation.( 26 ) This study highlights MST4 as a regulator of hepatic lipid storage and associated liver damage, and provides evidence that antagonizing MST4 activity may provide a strategy to prevent or mitigate the development and aggravation of NAFLD.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Fig S9
Table S1
Supplementary Material
Supported by grants from the Swedish Research Council, European Foundation for the Study of Diabetes, West Sweden Avtal om Läkarutbildning och Forskning Program, Novo Nordisk Foundation, Swedish Heart‐Lung Foundation, Swedish Diabetes Foundation, Å. Wiberg Foundation, Adlerbert Research Foundation, I. Hultman Foundation, F. Neubergh Foundation, Prof. N. Svartz Foundation, L. and J. Grönberg Foundation, W. and M. Lundgren Foundation, and I.‐B. and A. Lundberg Research Foundation.
Potential conflict of interest: Dr. Hanns‐Ulrich consults for Mirum. He received grants from Intercept. Dr. Bluher consults, advises, and is on the speakers’ bureau for Boehringer Ingelheim, Lilly, and Novo Nordisk.
References
- 1. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science 2002;298:1912‐1934. [DOI] [PubMed] [Google Scholar]
- 2. Strange K, Denton J, Nehrke K. Ste20‐type kinases: evolutionarily conserved regulators of ion transport and cell volume. Physiology (Bethesda) 2006;21:61‐68. [DOI] [PubMed] [Google Scholar]
- 3. Thompson BJ, Sahai E. MST kinases in development and disease. J Cell Biol 2015;210:871‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Radu M, Semenova G, Kosoff R, Chernoff J. PAK signalling during the development and progression of cancer. Nat Rev Cancer 2014;14:13‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pombo CM, Iglesias C, Sartages M, Zalvide JB. MST kinases and Metabolism. Endocrinology 2019;160:1111‐1118. [DOI] [PubMed] [Google Scholar]
- 6. Li T, Deng L, He X, Jiang G, Hu F, Ye S, et al. MST4 predicts poor prognosis and promotes metastasis by facilitating epithelial‐mesenchymal transition in gastric cancer. Cancer Manag Res 2019;11:9353‐9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lin ZH, Wang L, Zhang JB, Liu Y, Li XQ, Guo L, et al. MST4 promotes hepatocellular carcinoma epithelial‐mesenchymal transition and metastasis via activation of the p‐ERK pathway. Int J Oncol 2014;45:629‐640. [DOI] [PubMed] [Google Scholar]
- 8. Schmitt DC, Madeira da Silva L, Zhang W, Liu Z, Arora R, Lim S, et al. ErbB2‐intronic microRNA‐4728: a novel tumor suppressor and antagonist of oncogenic MAPK signaling. Cell Death Dis 2015;6:e1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang T, Kim CK, Alvarez AA, Pangeni RP, Wan X, Song X, et al. MST4 phosphorylation of ATG4B regulates autophagic activity, tumorigenicity, and radioresistance in glioblastoma. Cancer Cell 2017;32:840‐855, e848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Madsen CD, Hooper S, Tozluoglu M, Bruckbauer A, Fletcher G, Erler JT, et al. STRIPAK components determine mode of cancer cell migration and metastasis. Nat Cell Biol 2015;17:68‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen M, Zhang H, Shi Z, Li Y, Zhang X, Gao Z, et al. The MST4‐MOB4 complex disrupts the MST1‐MOB1 complex in the Hippo‐YAP pathway and plays a pro‐oncogenic role in pancreatic cancer. J Biol Chem 2018;293:14455‐14469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang H, Ma X, Peng S, Nan X, Zhao H. Differential expression of MST4, STK25 and PDCD10 between benign prostatic hyperplasia and prostate cancer. Int J Clin Exp Pathol 2014;7:8105‐8111. [PMC free article] [PubMed] [Google Scholar]
- 13. Ye Q‐H, Qin L‐X, Forgues M, He P, Kim JW, Peng AC, et al. Predicting hepatitis B virus‐positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med 2003;9:416‐423. [DOI] [PubMed] [Google Scholar]
- 14. Jiao S, Zhang Z, Li C, Huang M, Shi Z, Wang Y, et al. The kinase MST4 limits inflammatory responses through direct phosphorylation of the adaptor TRAF6. Nat Immunol 2015;16:246‐257. [DOI] [PubMed] [Google Scholar]
- 15. Yuan X, Yao PY, Jiang J, Zhang Y, Su Z, Yao W, et al. MST4 kinase phosphorylates ACAP4 protein to orchestrate apical membrane remodeling during gastric acid secretion. J Biol Chem 2017;292:16174‐16187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. ten Klooster JP, Jansen M, Yuan J, Oorschot V, Begthel H, Di Giacomo V, et al. Mst4 and Ezrin induce brush borders downstream of the Lkb1/Strad/Mo25 polarization complex. Dev Cell 2009;16:551‐562. [DOI] [PubMed] [Google Scholar]
- 17. Amrutkar M, Cansby E, Chursa U, Nuñez‐Durán E, Chanclón B, Ståhlman M, et al. Genetic disruption of protein kinase STK25 ameliorates metabolic defects in a diet‐induced type 2 diabetes model. Diabetes 2015;64:2791‐2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Amrutkar M, Cansby E, Nuñez‐Durán E, Pirazzi C, Ståhlman M, Stenfeldt E, et al. Protein kinase STK25 regulates hepatic lipid partitioning and progression of liver steatosis and NASH. FASEB J 2015;29:1564‐1576. [DOI] [PubMed] [Google Scholar]
- 19. Amrutkar M, Chursa U, Kern M, Nuñez‐Durán E, Ståhlman M, Sütt S, et al. STK25 is a critical determinant in nonalcoholic steatohepatitis. FASEB J 2016;30:3628‐3643. [DOI] [PubMed] [Google Scholar]
- 20. Amrutkar M, Kern M, Nuñez‐Durán E, Ståhlman M, Cansby E, Chursa U, et al. Protein kinase STK25 controls lipid partitioning in hepatocytes and correlates with liver fat content in humans. Diabetologia 2016;59:341‐353. [DOI] [PubMed] [Google Scholar]
- 21. Cansby E, Kulkarni NM, Magnusson E, Kurhe Y, Amrutkar M, Nerstedt A, et al. Protein kinase MST3 modulates lipid homeostasis in hepatocytes and correlates with nonalcoholic steatohepatitis in humans. FASEB J 2019;33:9974‐9989. [DOI] [PubMed] [Google Scholar]
- 22. Nerstedt A, Kurhe Y, Cansby E, Caputo M, Gao L, Vorontsov E, et al. Lipid droplet‐associated kinase STK25 regulates peroxisomal activity and metabolic stress response in steatotic liver. J Lipid Res 2020;61:178‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nuñez‐Durán E, Aghajan M, Amrutkar M, Sütt S, Cansby E, Booten SL, et al. Serine/threonine protein kinase 25 antisense oligonucleotide treatment reverses glucose intolerance, insulin resistance, and nonalcoholic fatty liver disease in mice. Hepatol Commun 2018;2:69‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Caputo M, Kurhe Y, Kumari S, Cansby E, Amrutkar M, Scandalis E, Booten SL, et al. Silencing of STE20‐type kinase MST3 in mice with antisense oligonucleotide treatment ameliorates diet‐induced nonalcoholic fatty liver disease. Under the review at Mol Ther Nucleic Acids 2020. [DOI] [PubMed]
- 25. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol 2018;15:349‐364. [DOI] [PubMed] [Google Scholar]
- 26. Younossi ZM, Loomba R, Rinella ME, Bugianesi E, Marchesini G, Neuschwander‐Tetri BA, et al. Current and future therapeutic regimens for nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2018;68:361‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Samanez CH, Caron S, Briand O, Dehondt H, Duplan I, Kuipers F, et al. The human hepatocyte cell lines IHH and HepaRG: models to study glucose, lipid and lipoprotein metabolism. Arch Physiol Biochem 2012;118:102‐111. [DOI] [PubMed] [Google Scholar]
- 28. Schippers IJ, Moshage H, Roelofsen H, Muller M, Heymans HSA, Ruiters M, et al. Immortalized human hepatocytes as a tool for the study of hepatocytic (de‐)differentiation. Cell Biol Toxicol 1997;13:375‐386. [DOI] [PubMed] [Google Scholar]
- 29. Li HY, Ham A, Ma TC, Kuo SH, Kanter E, Kim D, et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy 2019;15:113‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cansby E, Amrutkar M, Holm LM, Nerstedt A, Reyahi A, Stenfeldt E, et al. Increased expression of STK25 leads to impaired glucose utilization and insulin sensitivity in mice challenged with a high‐fat diet. FASEB J 2013;27:3660‐3671. [DOI] [PubMed] [Google Scholar]
- 31. Kannt A, Pfenninger A, Teichert L, Tönjes A, Dietrich A, Schön MR, et al. Association of nicotinamide‐N‐methyltransferase mRNA expression in human adipose tissue and the plasma concentration of its product, 1‐methylnicotinamide, with insulin resistance. Diabetologia 2015;58:799‐808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander‐Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol 1999;94:2467‐2474. [DOI] [PubMed] [Google Scholar]
- 33. Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol 2013;10:330‐344. [DOI] [PubMed] [Google Scholar]
- 34. McManaman JL, Bales ES, Orlicky DJ, Jackman M, MacLean PS, Cain S, et al. Perilipin‐2‐null mice are protected against diet‐induced obesity, adipose inflammation, and fatty liver disease. J Lipid Res 2013;54:1346‐1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takahashi Y, Shinoda A, Kamada H, Shimizu M, Inoue J, Sato R. Perilipin2 plays a positive role in adipocytes during lipolysis by escaping proteasomal degradation. Sci Rep 2016;6:20975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Qian Z, Lin C, Espinosa R, LeBeau M, Rosner MR. Cloning and characterization of MST4, a novel Ste20‐like kinase. J Biol Chem 2001;276:22439‐22445. [DOI] [PubMed] [Google Scholar]
- 37. Lykke‐Andersen S, Jensen TH. Nonsense‐mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol 2015;16:665‐677. [DOI] [PubMed] [Google Scholar]
- 38. Friedman SL, Neuschwander‐Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 2018;24:908‐922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gluchowski NL, Becuwe M, Walther TC, Farese RV Jr. Lipid droplets and liver disease: from basic biology to clinical implications. Nat Rev Gastroenterol Hepatol 2017;14:343‐355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang X, Lu X, Lombès M, Rha GB, Chi Y‐I, Guerin TM, et al. The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab 2010;11:194‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang E, Cui W, Lopresti M, Mashek MT, Najt CP, Hu H, et al. Hepatic PLIN5 signals via SIRT1 to promote autophagy and prevent inflammation during fasting. J Lipid Res 2020;61:338‐350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Verbeek J, Lannoo M, Pirinen E, Ryu D, Spincemaille P, Vander Elst I, et al. Roux‐en‐y gastric bypass attenuates hepatic mitochondrial dysfunction in mice with non‐alcoholic steatohepatitis. Gut 2015;64:673‐683. [DOI] [PubMed] [Google Scholar]
- 43. Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, et al. Adaptation of hepatic mitochondrial function in humans with non‐alcoholic fatty liver is lost in steatohepatitis. Cell Metab 2015;21:739‐746. [DOI] [PubMed] [Google Scholar]
- 44. Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 2004;114:147‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Abul‐Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P, et al. A protein‐truncating HSD17B13 variant and protection from chronic liver disease. N Engl J Med 2018;378:1096‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Khan SA, Wollaston‐Hayden EE, Markowski TW, Higgins L, Mashek DG. Quantitative analysis of the murine lipid droplet‐associated proteome during diet‐induced hepatic steatosis. J Lipid Res 2015;56:2260‐2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Crunk AE, Monks J, Murakami A, Jackman M, MacLean PS, Ladinsky M, et al. Dynamic regulation of hepatic lipid droplet properties by diet. PLoS One 2013;8:e67631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Su W, Wang Y, Jia X, Wu W, Li L, Tian X, et al. Comparative proteomic study reveals 17beta‐HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A 2014;111:11437‐11442. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Fig S9
Table S1
Supplementary Material