

Abstract
The aging process is represented by the time‐dependent decay in physiologic functions of living beings. Major interest has been focused in recent years on the determinants of this progressive condition due to its correlative relationship with the onset of diseases. Several hallmark features have been observed in aging, such as genetic alterations, mitochondrial impairment, and telomere shortening. At the cellular level, a senescent phenotype has been identified in response to aging that is characterized by a flat appearance, proliferative arrest, and production of specific molecules. The net effect of these cells in the course of diseases is an argument of debate. In fact, while the onset of a senescent phenotype may prevent tumor spreading, these cells appear to support pathological processes in some conditions. Several studies are now focused on clarifying the specific molecular pathways of aging/senescence in different cells, tissues, or organs. Biliary and vascular components, within the liver, have emerged as important determinants of some form of liver disease. In this review we summarize the most recent achievements on aging/senescence, focusing on the biliary and vascular liver system. Conclusion: Several findings, in both preclinical animal models and on human liver specimens, converge in supporting the presence of specific aging hallmarks in the diseases involving these hepatic compartments.
In this review we have summarized the most recent achievments on aging/ senescene, focusing, on the biliary and vacular liver system

Abbreviations
- Bcl‐xL
B‐cell lymphoma, extra‐large
- BDL
bile duct ligated
- CCL2
chemokine (C‐C motif) ligand 2
- ECM
extracellular matrix
- EMT
epithelial to mesenchymal transition
- FoxA2
forkhead box A2
- HCV
hepatitis C virus
- HGF
hepatocyte growth factor
- HSC
hepatic stellate cell
- IL
interleukin
- KC
Kupffer cell
- LSEC
liver sinusoidal endothelial cell
- Mdr2−/−
multidrug‐resistant knockout
- NK‐1R
neurokinin‐1 receptor
- PBC
primary biliary cholangitis
- PBP
peribiliary vascular plexus
- PSC
primary sclerosing cholangitis
- SASP
senescence‐associated secretory phenotype
- SA‐β‐gal
senescence‐associated‐β‐galactosidase
- SCF
stem cell factor
- Sec
secretin
- SP
substance P
- SR
secretin receptor
- TGF‐β1
transforming growth factor beta 1
- VEGF
vascular endothelial growth factor
- α‐CGRP
α‐calcitonin gene‐related peptide
Aging recapitulates the several processes impairing pathophysiological functions and homeostasis of a living organism over time. In aged organisms, including humans, several biochemical and cellular systems undergo a progressive time‐dependent impairment such as mitochondrial respiration, stem cell reserves, and genome repair function.( 1 ) The possibility to counteract the deleterious effects of aging requires an in‐depth knowledge of different intracellular mechanisms involved in this process. In fact, several studies have focused on this target during the past few decades.( 2 ) In 1939, the first observations of a prolonged life span in rodents maintained at a low caloric intake( 3 ) were made; and we are now using senolytic treatment in clinical trials.( 4 ) Moreover, possible individual aging patterns have been recently suggested in humans by multi‐omic analysis.( 5 ) Aging effects on the liver appear to be less relevant than those on heart, kidney or brain, possibly due to the specific enhanced regenerative properties of the liver.( 6 , 7 ) However, important hepatic deleterious changes occur with time, also demonstrated in the clinical setting observed by suboptimal outcomes of liver‐transplanted patients with hepatitis C virus (HCV), when older donors were used.( 8 ) In this review, we discuss the specific aging‐related biliary and vascular liver phenotypes, including those observed at the cellular/molecular level and with regard to liver diseases.
Liver Aging and Cellular Senescence
Liver aging is associated with impaired proliferative and metabolic functions. With regard to this latter point, evidence suggests that changes occurring in the liver over time may have a role in susceptibility to nonalcoholic fatty liver disease (NAFLD).( 9 ) Linked to the concept of tissue aging, cellular senescence has been recognized as the most important age‐associated phenotypic change.( 10 ) Cellular senescence is an irreversible halt of cell cycle progression; thus, the cell is no longer able to proliferate but can remain metabolically active.( 11 ) This process was first described in the early 1960s in diploid cell cultures, in which the exhaustion of replicative cell capacity over time led to a senescent phenotype characterized by growth arrest.( 12 ) Cellular senescence, together with autophagy and apoptosis, participates in the network of the possible cellular response to stress; in contrast, autophagy (a process removing damaged intracellular organelles or molecules) has been suggested to promote senescent transformation in some cases.( 13 ) Although the onset of senescence is postulated to be a protective mechanism against malignant transformation during aging, the secretion of several molecular factors, termed senescence‐associated secretory phenotype (SASP), is triggered by these cells.( 14 ) SASP contribute to several biological processes such as angiogenesis, tissue inflammation, and repair. At the same time, SASPs are able to maintain and to diffuse (in the neighboring cells) the senescence phenotype through autocrine and paracrine mechanisms.( 14 , 15 ) Although the correlation between tissue aging and cellular senescence may be, in part, supported by the possible accumulation of genomic injuries and/or mutational signals during time, the major role in this process appears to be played by telomere shortening.( 16 ) This time‐dependent reduction of telomeres, which is a loss of a maximum of 200 kD at the end of any replicative cycle, in the end determines apoptosis or the onset of the senescence phenotype.( 17 ) This process is generally regarded as the form of replicative senescence; however, a premature senescence may also arise in particular conditions of cellular stress and damage.( 14 ) Finally, telomere shortening and/or cellular senescence has been consistently reported in human liver diseases,( 18 , 19 ) suggesting their possible role in these maladies that present a worst outcome in aged livers.
Biliary and Vascular Compartment Interplay
The anatomical relationship between the biliary and the vascular component within the liver was already described by the identification, by means of scanning electron microscopy coupled with liver casting, of the peribiliary‐vascular‐plexus (PBP).( 10 , 20 ) A graphical representation of the relationship between PBP and biliary tract is depicted in Fig. 1. Originating from lateral branches of the hepatic artery, the PBP reaches the sinusoids with small direct anastomotic vessels or interconnecting with vessels of the portal vein, and becoming narrow when surrounding smaller ducts. A physiological cooperation occurs between the biliary tract and PBP, as the latter supports cholangiocyte nutrition and recirculation of biliary components back again to the liver.( 21 ) The strict relationship between the biliary compartment and PBP is further confirmed by the parallel proliferation of these two different systems during injury, as observed in bile duct ligated (BDL) rats.( 21 ) Moreover, cross‐talk occurs between the biliary and vascular compartment during normal, developmental, or pathological condition, primarily through vascular endothelial growth factor (VEGF) and angiopoietin 1 and 2.( 21 , 22 , 23 , 24 ) With regard to VEGF, this is produced by cholangiocytes and supports their growth with an autocrine mechanism. Moreover, in a rodent model, this mediator is capable of counteracting PBP vanishing induced by hepatic artery ligation.( 21 ) Taken together these data support the idea of a unique physiologic system in which biliary and vascular functions are integrated.
FIG. 1.
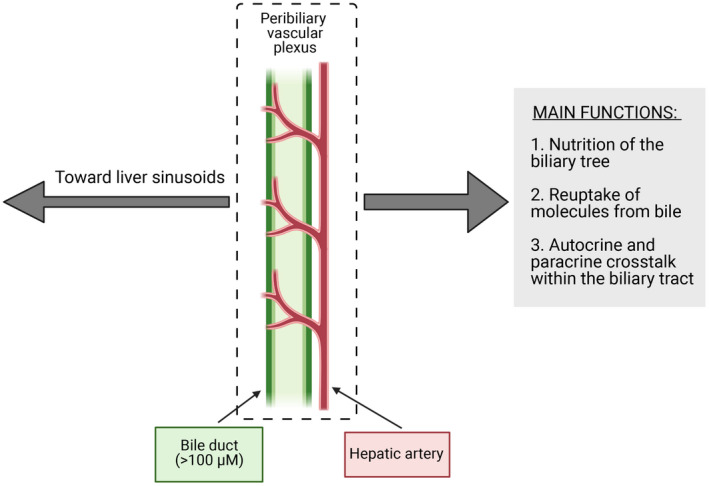
Schematic representation of biliary tract and peribiliary vascular plexus assembly. The main functions of peribiliary vascular plexus, in supporting biliary tract activities, are reported in the gray box on the right side. (This figure was made with BioRender.com under a purchased license agreement.)
Biliary Epithelium
The biliary epithelium is lined by cholangiocytes, which contribute to qualitative and quantitative changes in bile secretion/composition, before its release in the duodenum.( 25 ) Bile acid–independent ductal bile secretion is sustained primarily by the activity of secretin (Sec)/secretin receptor (SR) pathway within these cells.( 25 , 26 ) Following stimulation with its ligand, SR activates cyclic adenosine monophosphate/protein kinase A/cystic fibrosis transmembrane conductance regulator/Cl‐/HCO3 ‐ exchanger signaling, thus simulating a bicarbonate‐enriched choleresis.( 27 ) However, different from other liver cells, numerous hormones and hormone receptors are expressed by cholangiocytes, which regulate not only the secretive, but also the proliferative, response in normal as well as in pathological conditions. Moreover, several other organic molecules, such as bile acids, angiogenic factors or neuropeptides, modulate the activities of the biliary epithelium, suggesting cholangiocytes to be the main liver collectors of signals coming from circulating molecules.( 28 ) Finally, bile duct cells are composed of two different cellular types: small cholangiocytes and large cholangiocytes, lining small and large ducts, respectively.( 29 ) Large cholangiocytes are those promoting the main physiologic activities of biliary tract such as secretion, response to hormones, and mediators.( 29 ) In contrast, small cholangiocytes are considered a quiescent population that is able to proliferate and acquire the large cholangiocytes phenotype when these are damaged.( 29 )
Aging of the Biliary Epithelium
Age‐related impairment of cholangiocytes has emerged as an important field of research in the last decade. The evaluation of the aging processes may, in fact, shed light on the mechanisms of adult chronic cholestatic liver diseases. Among the latter, primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC) are the most represented in humans.( 30 ) PBC, the most frequent autoimmune liver disease with a prevalence of approximately 30/100,000, generally affects middle age women and is characterized by the presence of circulating anti‐mitochondrial antibodies.( 31 ) From a pathological point of view, the disease presents as a lymphocytic cholangitis that may evolve in ductopenia and fibrosis. PSC is less frequent, with a male‐to‐female ratio of 2:1. Abundant deposition of scar tissue, determining strictures of the biliary tract, is the main hallmark of the disease.( 32 ) Both PBC and PSC are conditions affected by a significant morbidity and mortality, whereas a definitive pharmacological treatment for severe cases has not been identified so far. This perspective, among the other molecular aspects also related to the aging process, deserves interest.
In the context of biliary damage, modulation of biliary senescence during cholestatic damage has become of increasing interest. Indeed, enhanced biliary senescence, specifically in ductular reactive cells, has been found in chronic liver diseases, particularly in PBC.( 11 ) Additionally, enhanced biliary senescence correlates with fibrosis progression in chronic liver diseases.( 11 ) Telomere shortening, which was stated previously as a key aspect of senescence, has been found in human PBC samples,( 33 ) and the effect of biliary senescence on PBC outcomes has been largely studied.( 34 , 35 , 36 ) Cholangiocytes isolated from patients with PSC show exacerbated senescence and SASP marker expression compared with other cholestatic diseases.( 19 ) Similarly, in a 3D organoid model using PSC cholangiocytes, these “cholangioids” displayed senescent and SASP features, which promoted macrophage recruitment and number.( 37 ) The role of senescence/SASP in biliary injury has been demonstrated; thus, evaluation of targeting this axis for therapeutic use is of great interest.
Data From Experimental Models
As previously discussed, the main cellular response to the aging process is represented by the onset of a senescent phenotype. Morphologically these cells are larger, with increased cytoplasm/nuclear ratio, and present an increased number of vacuoles( 14 ) The tumor suppressor retinoblastoma protein and p‐53 are considered the main molecular routes for the induction of senescent phenotypes.( 38 ) A cellular increase of cyclin‐dependent kinase inhibitors is found in senescent phenotype. Among these, p21 and p16 are usually considered as appropriate markers to establish the rate of senescence of a specific tissue in normal and pathological conditions. In addition, levels of senescence‐associated‐β‐galactosidase (SA‐β‐gal), an enzyme present in lysosomes, has been generally regarded as an important biochemical indicator of senescence and has been used widely in research.( 39 ) Senescent hallmarks have been studied in preclinical models of both PBC and PSC. One study focused on Forkhead box A2 (FoxA2) activity during experimental cholestasis in BDL and multidrug‐resistant knock‐out (Mdr2 −/−) mice.( 40 ) FoxA2 is regarded as an important regulator of cell differentiation, and the model of cholestasis is characterized by the decrease of this molecular mediator.( 41 ) In this study, an inverse relationship was observed between FoxA2 expression and hallmarks of senescence such as p16 expression or SA‐β‐gal staining in liver section. Interestingly, FoxA2 restoration obtained by small cholangiocyte cell therapy determined an important reduction of senescent phenotype.( 40 ) These data were extended by a study examining the role of substance P (SP), a tachykinin family neuropeptide, in the same preclinical models of murine cholestasis (BDL and Mdr2 −/−).( 42 ) The interest regarding SP and biliary tract came by observations demonstrating (1) decreased cholangiocarcinoma (CCA) cells growth and (2) BDL‐induced biliary proliferation, after inhibition or genetic suppression of the specific SP receptor neurokinin‐1 (NK‐1R).( 43 , 44 ) When senescent features were examined in both cholangiocytes and hepatic stellate cells (HSCs) coming from cholestatic rodent, these were increased in bile duct cells, while the opposite behavior (less senescence, more cellular activity) was observed in HSCs, thus resulting in increased collagen deposition and fibrosis. Moreover, in this study, the SP/NK‐1R pathway played a major role in differential modulation of cholangiocyte–HSC senescence balancing, as inhibition of NK‐1R increased biliary tract viability and reduced profibrotic processes. Although the specific mechanism linking SP/NK‐1R to differential cellular senescence remains undetermined, this research showed that possible regulation of senescent phenotype in different cells might be a possible target for therapy.( 45 ) The relationship of the main secretory and proliferative cholangiocyte pathway, the Sec/SR axis was also examined with regard to cellular senescence in preclinical models of cholestasis. In BDL mice, knockout of the SR gene reduces hyperplasia( 46 ) and inhibition of Sec/SR axis; in Mdr2 −/− mice, it ablates the transforming growth factor beta 1 (TGF‐β1)–mediated enhanced liver fibrosis.( 47 ) Changes in cellular senescence processes were evaluated in a study examining the double SR−/−/Mdr2 −/− knockout mice.( 48 ) The Sec/SR system exhibited effects similar to those observed with regard to SP. In fact, genetic ablation of SR greatly reduced fibrosis and proliferation, down‐regulating cholangiocyte senescence and up‐regulating the senescent phenotype of HSCs. This view was corroborated by data from the BDL model of Sec−/−/SR−/− mice.( 47 ) Again, the differential effect of Sec/SR on cholangiocytes–HSC senescence was confirmed; moreover, these findings were extended, suggesting that Sec‐related effects were achieved by TGF‐β1/TGF‐β1R stimulation.( 47 ) In the last few years, other biomolecules such as α‐Calcitonin gene‐related peptide (α‐CGRP), stem cell factor (SCF), or vimentin have been demonstrated to participate in experimental cholestatic injury, differentially regulating cholangiocyte and HSC senescence. In the α‐CGRP−/− mouse model, the typical senescence phenotypic distribution (cholangiocytes+; HSCs−) observed with BDL and in parallel with cholestatic damage was reverted.( 49 ) Accordingly, several senescence markers were decreases in cholangiocytes, such as p16, p21, chemokine (C‐C motif) ligand 2 (CCL2), and plasminogen activator inhibitor 1 (PAI‐1). SCF signaling also modulates experimental cholestatic biliary damage, as demonstrated by reduction of cholangiocytes injury/senescence when its axis is ablated.( 50 ) Similar results are also obtained by targeting vimentin. Its inhibition improves biliary damage and cellular senescence, possibly interfering with the epithelial to mesenchymal transition (EMT) process of bile duct cells, a mechanism linked to the onset of fibrosis.( 51 ) Finally, the direct targeting of senescent cellular machinery has been attempted, in experimental systems, to improve pathological features. Inhibition of the cyclin‐dependent kinase inhibitor p16 has been obtained in a model of cholestasis by the administration of p16 morpholino. This strategy decreased senescence, secretion of SASP, and ductular reaction in Mdr2 −/− mice.( 52 ) In another experimental approach, B‐cell lymphoma, extra‐large (Bcl‐xL) inhibition determined apoptosis of both senescent cholangiocytes and activated fibroblast, thus reducing liver damage in a Mdr2 −/− rodent model.( 53 ) In contrast, Bcl‐xL overexpression, in the course of cholestatic injury, was related to ETS proto‐oncogene factor 1 and p300 cooperation.( 54 ) In conclusion, the data coming from experimental studies support the hypothesis of a unifying model linking ductular reaction, EMT, fibrosis, and the differential cholangiocyte/HSC cellular senescence in an integrated machinery that determines chronic cholestatic liver diseases. This picture may be implemented in the future including also the role of mast cells. During liver injury, mast cells surround the biliary tract and play a critical role in the contribution by driving a senescent phenotype, thus promoting damage.( 55 ) The main molecular factors, supporting cholangiocyte senescence in experimental models of cholestasis, are summarized in Fig. 2. Correlative findings in human are reported in the following section.
FIG. 2.
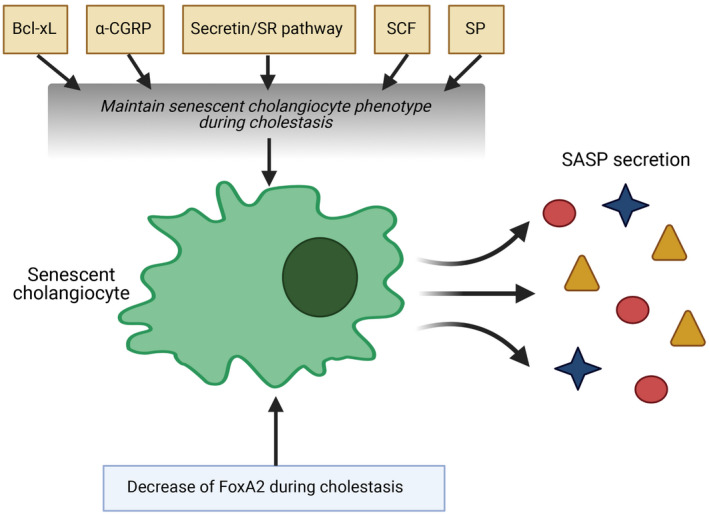
The molecular determinants of cholangiocyte senescence, identified in experimental models of cholestasis. (This figure was made with BioRender.com under a purchased license agreement.)
Human Studies
Several aspects observed in animal models related to aging, such as telomere shortening, cellular senescence, DNA alteration, mitochondrial impairment and others, have been also observed in human liver.( 56 ) These changes may impair the physiologic defense against liver injury or they could enhance pathological pathways with lipids accumulation, as observed in NAFLD.( 57 ) With regard to cholestatic diseases, for instance, more severe evolution of PBC are usually observed in middle‐aged women, with the postmenopausal drop of protective estrogens.( 58 ) However, in addition to unrelated liver aging factors that may worsen cholestatic injury, as in the previous case, specific aging hallmarks have been identified in human liver affected by disease of the biliary tract. Cholangiocytes freshly isolated from patients with PSC are enlarged, with tight junction defects, and stain positive for SA‐β‐gal (marker of senescence) in nearly 50% of cases.( 59 ) These cells also exhibit an increase production of SASP, such as interleukin (IL) 6 and IL‐8. In another study, comparison of liver tissue coming from healthy subjects and patients with PSC, PBC, and HCV demonstrated a maximum increase of markers of cellular senescence (p16) and SASP secretion (IL‐6, IL‐8, CCL‐2, and PAI‐1) in patients with PSC, followed by those with PBC.( 19 ) Patients with HCV exhibited cellular senescence features similar to the healthy control. Finally, in the same research, normal human cholangiocytes exposed to different damaging agents within 10 days evolved toward a senescent phenotype with production of SASP. Another study examined the hallmarks of senescence in human PBC liver tissue as a function of clinical outcome and response to therapy.( 60 ) This study demonstrated that (1) the number of senescent cells was proportional to the severity of the disease, and (2) increased expression of p16 was related to an inadequate response to ursodeoxycholic acid standard therapy.
Taken together, these findings suggest that cholangiocyte senescence might be a characteristic feature of biliary tract diseases, and that senescent phenotypes would rise as a standard response of these cells to damage. Other correlative studies matching rodent and human samples supported this view. In a previously mentioned study, FoxA2 activity was nearly undetectable in human PSC or PBC liver specimens, whereas SASP levels (TGF‐β1) were increased.( 41 ) The possible link between SP and cellular senescence in PSC was suggested by the finding of increased blood levels of this neuropeptide and its messenger RNA in liver of subjects affected by an advanced stage of this disease.( 42 ) Other correlative assessments regarding α‐CGRP, SCF, or vimentin levels were carried out in PSC human tissue to confirm the preclinical data obtained in rodents, as previously described.( 49 , 50 , 51 ) Finally, increased levels of twinfilin‐1 were detected in cholangiocytes of patients with PBC or PSC. This protein, in experimental cholestasis, appeared to respond to several microRNA signals and to play a major role in supporting the proliferative/senescent evolution of bile duct cells in this setting.( 61 )
With regard to other biliary diseases, cellular senesce was also examined in biliary atresia, which is a pediatric progressive cholestatic disease characterized by the preferential obliteration of the extrahepatic biliary duct.( 62 ) In a study, liver evaluation of cell cycle regulators p16 and p21 was carried out in 80 patients with biliary atresia. The results evidenced a constant occurrence of cellular senescence in this disease, similar to other forms of injury of the biliary tract. Moreover, a possible relationship with the progression of injury was suggested.( 63 ) In conclusion, preclinical animal and human data strongly support the role of aging, and in particular of cellular senescence, in the molecular machinery at the base of chronic cholestatic human diseases. Two aspects are particular intriguing at present: (1) how cellular senescence contributes in an integrated process with inflammation, ductular reaction, EMT, and fibrosis in the maintenance of the disease; and (2) how to attain a different regulation of cholangiocyte and HSC senescence, to reduce injury in the course of the diseases of the biliary tract. Further studies will hopefully answer these questions.
Liver Sinusoid
The hepatic vascular compartment has a unique structure that allows maintenance of liver homeostasis. Liver blood flow enters into the liver through two vessels, the portal vein and the hepatic artery, altogether supplying the hepatic tissue with oxygen, nutrients, hormones, and other substances including inflammatory factors and toxins. The segment of the hepatic microcirculation where the exchange of substances occurs is the hepatic sinusoid, a unique vascular bed consisting of a layer of liver sinusoidal endothelial cells (LSECs) surrounded by vascular perisinusoidal cells (HSCs), and flanked by plates of nonparenchymal cells or hepatocytes. The thin area where HSCs reside is called the space of Disse. In addition, the hepatic sinusoid houses an important part of the phagocytic system, Kupffer cells (KCs), the resident macrophages, and other immune cells like T lymphocytes and pit cells (also known as hepatic natural killer cells), altogether making the liver a key organ for innate immune system.( 64 ) Immune cells are anchored to the luminal side of the sinusoidal endothelium, and therefore are exposed to the bloodstream. The complex functions of the liver in molecule biosynthesis, metabolism, inflammation, and clearance of bloodstream are tightly dependent on an adequate microcirculation, guaranteed by a healthy phenotype and proper paracrine signaling within the liver sinusoid.( 65 )
Aging of Liver Sinusoidal Cells
Liver Sinusoidal Endothelial Cells
LSECs are specialized endothelial cells that form the vascular wall in the liver sinusoids. In normal conditions, LSECs form a permeable barrier due to fenestrae presence in their membrane and due to absence of basal membrane. These properties make the liver endothelium discontinuous, increasing crosstalk between hepatocytes and liver sinusoidal cells. LSECs also play a role in regulating hepatic vascular tone through the production of vasoactive molecules.( 66 )
Data from experimental models demonstrated that LSECs de‐differentiate during healthy aging, a process initially defined by partial loss in the number and diameter of fenestrae, also termed as pseudo‐capillarization.( 67 ) Subsequent studies improved the description of LSEC de‐differentiation in aging, clearly defining that such distinctiveness was not limited to morphologic changes but accompanied by significant deregulations in the LSEC phenotype.( 68 , 69 ) It has been described that alterations in key vasodilatory pathways, including the nitric oxide one, may contribute to increment the hepatic vascular resistance. The reduction in vasodilators is of relevance considering the roles of nitric oxide regulating the hepatic vascular tone,( 70 , 71 ) exerting anti‐inflammatory effects and maintaining other nonparenchymal cell phenotypes through paracrine interactions.( 72 , 73 ) Aged LSECs exhibit decreased expression of diverse angiocrine receptors (e.g., SE‐1, stabilin‐2 [Stab2], kdr) and angiocrine factors (including wingless‐type [wnt2] and hepatocyte growth factor [hgf]), which may affect hepatocyte regeneration.( 69 , 74 ) LSECs are in a moderate pro‐inflammatory state, which, together with down‐regulation of sirtuin 1 and up‐regulation of p16, suggest senescence of this cell type in aging.( 75 ) Finally, LSEC scavenger functions are also compromised in aging, as demonstrated by the reduction in endocytic capacity.( 76 ) Interestingly, a recent study suggested that scavenger capacity dynamics during a lifespan would be a key mechanism to promote LSEC senescence.( 75 ) Middle‐aged mice exhibit a significant increase in scavenger receptors expression, which may fuel the intake of toxic substances, ultimately leading to mitochondrial oxidative stress and senescence. Following senescence progression, heterochromatic silences, thus suppressing, the expression of several scavenger receptors and endocytosis genes to ultimately reduce LSEC endocytic capacity in aging.( 77 ) The mentioned deregulations due to aging have a clear effect on LSEC response in front of an injury, either acute or chronic. In fact, preclinical studies that aimed to analyze the effects of aging on the liver microcirculation demonstrated that LSECs exhibit profound changes in response to an injury, including further reduction in fenestrae, loss of angiocrine mediators, and decline in vasodilators synthesis.( 78 , 79 ) These alterations have an effect on vascular functionality, maintenance of other liver cells, and development of clinical complications of liver injury including portal hypertension.
Hepatic Stellate Cells
HSCs are the main collagen‐synthesis cells of the liver, and by producing extracellular matrix (ECM) and interacting with neighboring cells, they play a key role in liver architecture modification and function. HSCs’ first described function was to store fat in the capillary wall of the human liver (also known as fat‐storing cells or Ito cells). This property has led their identification in the liver tissue and their isolation using density gradient methods. However, during liver damage, these quiescent HSCs acquire an “activated” phenotype, losing their fat‐storing cell phenotype and becoming pro‐contractile, proliferative, and matrix‐secreting myofibroblast‐like cells.( 80 )
In healthy aging, HSCs exhibit a slight, but remarkable, activation state, as evidenced by increased proliferation, significant increments in the expression of specific markers including α‐smooth muscle actin, collagen I and collagen IV, together with increase in inflammatory, pro‐oxidant and senescence markers, with evidence of matrix deposition within the parenchyma or sinusoids.( 69 , 81 ) Interestingly, and opposite to what is observed in chronic liver disease, aged HSCs present increased intercellular accumulation of lipids, presumably due to alterations in retinoid metabolism and lipid breakdown like down‐regulation in cellular retinol binding protein 1 and overexpression of patatin‐like phospholipase domain containing 3.
In response to an injury, HSCs become overactivated and therefore actively contribute to the aggravation and perpetuation of liver diseases. Indeed, preclinical models of acute liver injury (due to ischemia and reperfusion injury) and chronic liver disease (due to chronic carbon tetrachloride [CCl4] administration) in aging demonstrated hyperresponse of HSCs, leading to hypercontraction, elevated production, and secretion of ECM components, which ultimately led to exaggerated architectural distortion and elevation in the hepatic vascular tone.( 78 )
Hepatic Macrophages
Liver resident macrophages, also known as KCs, are situated in the sinusoidal lumen. KCs are exposed to blood, as well as to antigens and bacterial endotoxins, therefore representing the first line of defense to maintain immune system in the liver. Current evidence indicates that macrophages perform a wide repertoire of functions in inflammation and repair. The factors determining the behavior of macrophages at sites of inflammation are complex, and dependent on several variables of the organ and model of tissue injury.( 82 )
During aging, and in addition to LSECs, KCs become pro‐inflammatory, displaying elevated levels of IL‐6, a cytokine with key roles in liver regeneration, infection defense, and metabolism regulation, together with alterations in other polarization makers.( 69 , 83 ) The pro‐inflammatory state of KCs, and LSECs may contribute to the recruitment of circulating inflammatory cells including neutrophils and monocyte‐derived macrophages, observed in aged livers. In situations of chronic liver disease, aged animals exhibit further deterioration in the hepatic inflammatory phenotype, exhibiting increased recruitment of inflammatory macrophages together with down‐regulation in anti‐inflammatory and pro‐resolutive cytokines.( 78 ) The polarization of resident macrophages and the increased myeloid content in the aged cirrhotic liver may indeed derive from the exaggerated activation of the hepatic endothelium (as described previously) and from increased gut‐derived bacterial products, ultimately aggravating and perpetuating CLD in aged individuals. In Table 1, we summarize the findings on the aging process of LSECs, HSCs, and macrophages.
Human Data
Little is known about the effect of aging in the human liver sinusoid. Epidemiological data evidenced higher prevalence of chronic liver diseases coursing with microcirculatory dysfunction in the elderly, together with faster progression and worse prognosis,( 57 , 84 , 85 ) altogether suggesting that the hepatic vascular bed might be unprotected in front of an injury also in the human being. In accordance with this hypothesis, morphological analysis of human livers suggested pseudo‐capillarization of LSECs and slight activation of HSCs,( 86 ) and recent data supported the concept of sinusoidal unprotection due to aging. In the context of healthy aging, characterization of the liver sinusoid in young and aged human liver tissues validated most of phenotypic deregulations observed in preclinical models.( 69 ) Human aged livers exhibit features of LSEC de‐differentiation, as suggested by reduced angiocrine and vasodilatory genes, including endothelial nitric oxide synthase, HGF, laminin subunit beta 1 and Stab2, together with moderate activation of HSCs. In the scenario of chronic liver disease, transcriptomic analysis of liver tissue from patients with cirrhosis identified substantial number of genes differentially expressed comparing young (<48 years) versus old (>58 years) individuals. Comprehensive characterization of these data revealed an aged cirrhotic signature that, interestingly, included up‐regulated pathways related to vascular pathobiology, thus confirming the deterioration of the sinusoidal vascular bed in aged liver disease.( 78 ) These findings, detected in well‐characterized patients, may have an important role in the discovery of new therapeutic approaches and treatment of liver diseases in aging. Indeed, these findings suggest that future preclinical studies and clinical trials should consider age in their design (Fig. 3).
FIG. 3.
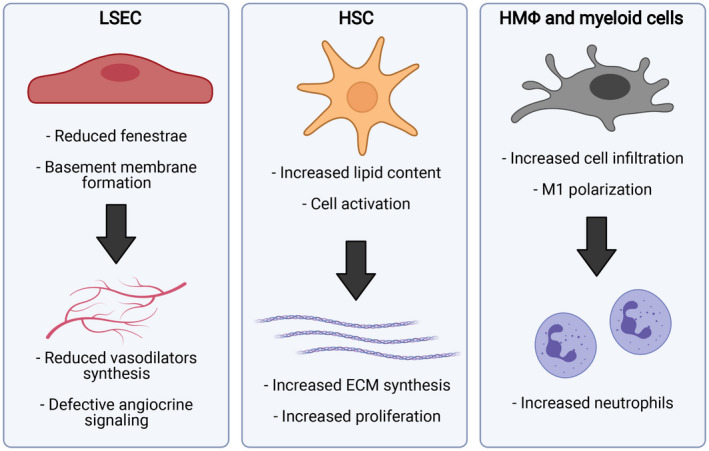
Major phenotypic modifications due to aging in liver sinusoidal cells. Abbreviation: HMΦ, hepatic macrophage. (This figure was made with BioRender.com under a purchased license agreement.)
Conclusions
In conclusion, while aging represents one of the most important risk factors for several neoplastic and nonneoplastic chronic diseases, important issues remain to be addressed. For example, the identification of appropriate markers of this time‐dependent process (as a function of disease onset and course) may possibly improve translational application of preclinical studies in human.( 2 ) Second, inhibition or modulation of cellular senescent phenotypes might be beneficial in pathological conditions. Finally, identification of senescence‐related pathways in different tissues would possibly allow us to design new treatment to organ‐specific disease. Preclinical animal studies have already shown that ablation of senescent cells (obtained by means of specific drugs or small interfering RNA treatment) was able to improve cardiac reserve in older mice or lifespan in progeroid Ercc1_/Δ rodents.( 87 ) Several drugs, including dasatinib, quercetin, fisetin and others, have been proposed to lead senescent cells toward deletion/apoptosis.( 88 ) In a pivotal study, intermittent administration of dasatinib and quercetin was conducted for 3 weeks in 14 patients affected by idiopathic pulmonary fibrosis. Physical function was improved by treatment, with an acceptable safety and tolerability.( 89 ) Further findings on senolytic treatment in humans were obtained in a study on patients with diabetes‐related renal disease.( 90 ) This research demonstrated, after a 3‐day short course of dasatinib + quercetin, a reduced number of adipose tissue senescent cells and decreased circulating levels of SASP. However, large trials on chronic administration of senolytic treatments would require an accurate evaluation of risk and benefit to be undertaken in the future. Moreover, a ubiquitous depletion of senescent cells might not be beneficial in different pathological settings. For instance, in this review we reported that the improvement in the animal model of cholestasis is not only associated with the ablation of senescent cholangiocytes, but also with the induction of senescence in HSCs. We also reported that different cells are present within the vascular liver compartment that may unevenly react and contribute to the aging/senescent process. This strongly suggests that a specific line of research focusing on aging of biliary and vascular compartments would be necessary to pursue therapeutic options for diseases afflicting these anatomical districts in humans.
TABLE 1.
Main Effects of Aging on the Cells of Liver Vasculature
| Cell Type | Pathophysiological Event | Specific Evidence | References |
|---|---|---|---|
| LSEC | Vasoconstriction | ⇩NO, ⇩eNOS, ⇩cGMP, ⇩HO‐1, ⇩KLF2 | ( 69, 78 ) |
| Inflammation | ⇧ICAM, ⇧IL‐6, ⇧CD6, SASP: ⇧(cytokines IL‐1α, IL‐1β, IL‐15, I‐L18), ⇧(chemokines Ccl2, Ccl6, Ccl8, Ccl24, Cxcl9, Cxcl12, Cxcl13, Cxcl16) | ( 68, 69 ) | |
| Scavenging dysfunction | ⇩SR(FSA), ⇩(Msr1, Marco, ScarB1, ScarB2, CD36r, CD68r, Mrc1, Stab1, Stab2), ⇩(Ox‐LDL, Ac‐LDL) | ( 75, 91 ) | |
| Angiocrine deregulation | ⇩Stab2, ⇩CB32b, ⇩VEGF‐R2, ⇩HGF, ⇩Wnt2, ⇩Hamp, ⇩Axin2 | ( 69, 78, 92 ) | |
| Capillarization | Pseudo‐capillarization (partial loss of fenestrae and basement membrane formation) | ( 86, 93 ) | |
| HSC | ECM synthesis | ⇑α‐SMA, ⇑collagen 1α1, ⇑collagen 1α2 | ( 56, 69, 78 ) |
| Proliferation | ⇑PDGF‐receptor β, ⇑desmin, ⇩telomere length, ⇑Ki67 | ( 69, 94 ) | |
| Hypercontraction | ⇑Rhok, ⇑ α‐SMA, ⇑LPA | ( 10, 95, 96, 97 ) | |
| Paracrine deregulation | ⇑SASP, ⇑SA‐β‐gal, ⇑p21, ⇑p53, ⇑p16, ⇑IL‐22, ⇑CCN1, ⇑retinoic acid, ⇑SP | ( 42, 98, 99 ) | |
| HMΦ | Inflammation | ⇧ICAM, ⇧IL‐6, ⇩TNFα, ⇩Mrc1, ⇩Arg1, ⇩IL‐10 | ( 68, 69, 78 ) |
Abbreviations: Ac‐LDL, acetylated low‐density lipoprotein; CCN1, cellular communication network factor 1; cGMP, cyclic guanine monophosphate; Cxcl, chemokine (C‐X‐C motif) ligand; eNOS, endothelial nitric oxide synthase; FSA, formaldehyde‐treated albumin; Hamp, hepcidin antimicrobial peptide; HMΦ, hepatic macrophage; HO‐1, heme oxygenase 1; ICAM, intercellular adhesion molecule; KLF2, Krüppel like factor 2; LPA, lysophosphatidic acid; Marco, macrophage receptor with collagenous structure; Mrc1, mannose receptor C‐type 1; Msr1, macrophage scavenger receptor 1; Ox‐LDL, oxidized low‐density lipoprotein; PDGF, platelet‐derived growth factor; Rhok, Rho‐associated protein kinase; ScarB, scavenger receptor class B; Stab2, stabilin‐2; TNF, tumor necrosis factor; VEGF‐R2, VEGF receptor 2; Wnt2, wingless‐type; α‐SMA, α‐smooth muscle actin.
Supported by PSC Partners Seeking a Cure, the Swiss National Science Foundation (SNF320030_189252/1), Indiana University Health (Hickam Endowed Chair, Gastroenterology, Medicine), National Institute of Diabetes and Digestive and Kidney Diseases (DK076898, DK107310, DK108959, DK110035, DK115184, and DK119421), United States Department of Veteran’s Affairs, Biomedical Laboratory Research and Development Service (1I01BX003031, 5I01BX000574, Research Career Scientist, Senior Research Career Scientist), and Instituto de Salud Carlos III/Spanish Ministry of Science (FIS PI20/00220).
Potential conflict of interest: Nothing to report.
Contributor Information
Gianfranco Alpini, Email: galpini@iu.edu, Email: jordi.gracia@idibaps.org.
Jordi Gracia‐Sancho, Email: jordi.gracia@idibaps.org.
References
Author names in bold designate shared co‐first authorship.
- 1. Lopez‐Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013;153:1194‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Campisi J, Kapahi P, Lithgow GJ, Melov S, Newman JC, Verdin E. From discoveries in ageing research to therapeutics for healthy ageing. Nature 2019;571:183‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McCay CM, Maynard LA, Sperling G, Barnes LL. Retarded growth, life span, ultimate body size and age changes in the albino rat after feeding diets restricted in calories. Nutr Rev 1975;33:241‐243. [DOI] [PubMed] [Google Scholar]
- 4. Ellison‐Hughes GM. First evidence that senolytics are effective at decreasing senescent cells in humans. EBioMedicine 2020;56:102473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ahadi S, Zhou W, Schüssler‐Fiorenza Rose SM, Sailani MR, Contrepois K, Avina M, et al. Personal aging markers and ageotypes revealed by deep longitudinal profiling. Nat Med 2020;26:83‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Durand F, Levitsky J, Cauchy F, Gilgenkrantz H, Soubrane O, Francoz C. Age and liver transplantation. J Hepatol 2019;70:745‐758. [DOI] [PubMed] [Google Scholar]
- 7. Isaev NK, Stelmashook EV, Genrikhs EE. Neurogenesis and brain aging. Rev Neurosci 2019;30:573‐580. [DOI] [PubMed] [Google Scholar]
- 8. Charlton M. The impact of advancing donor age on histologic recurrence of hepatitis C infection: the perils of ignored maternal advice. Liver Transpl 2003;9:535‐537. [DOI] [PubMed] [Google Scholar]
- 9. Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, et al. Cellular senescence drives age‐dependent hepatic steatosis. Nat Commun 2017;8:15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van Deursen JM. The role of senescent cells in ageing. Nature 2014;509:439‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sasaki M, Ikeda H, Yamaguchi J, Miyakoshi M, Sato Y, Nakanuma Y. Bile ductular cells undergoing cellular senescence increase in chronic liver diseases along with fibrous progression. Am J Clin Pathol 2010;133:212‐223. [DOI] [PubMed] [Google Scholar]
- 12. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 1961;25:585‐621. [DOI] [PubMed] [Google Scholar]
- 13. Nakanuma Y, Sasaki M, Harada K. Autophagy and senescence in fibrosing cholangiopathies. J Hepatol 2015;62:934‐945. [DOI] [PubMed] [Google Scholar]
- 14. Meng L, Quezada M, Levine P, Han Y, McDaniel K, Zhou T, et al. Functional role of cellular senescence in biliary injury. Am J Pathol 2015;185:602‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest 2018;128:1238‐1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hoare M, Das T, Alexander G. Ageing, telomeres, senescence, and liver injury. J Hepatol 2010;53:950‐961. [DOI] [PubMed] [Google Scholar]
- 17. Kong CM, Lee XW, Wang X. Telomere shortening in human diseases. FEBS J 2013;280:3180‐3193. [DOI] [PubMed] [Google Scholar]
- 18. Wiemann SU, Satyanarayana A, Tsahuridu M, Tillmann HL, Zender L, Klempnauer J, et al. Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. FASEB J 2002;16:935‐942. [DOI] [PubMed] [Google Scholar]
- 19. Tabibian JH, O'Hara SP, Splinter PL, Trussoni CE, LaRusso NF. Cholangiocyte senescence by way of N‐ras activation is a characteristic of primary sclerosing cholangitis. Hepatology 2014;59:2263‐2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kardon RH, Kessel RG. Three‐dimensional organization of the hepatic microcirculation in the rodent as observed by scanning electron microscopy of corrosion casts. Gastroenterology 1980;79:72‐81. [PubMed] [Google Scholar]
- 21. Gaudio E, Onori P, Pannarale L, Alvaro D. Hepatic microcirculation and peribiliary plexus in experimental biliary cirrhosis: a morphological study. Gastroenterology 1996;111:1118‐1124. [DOI] [PubMed] [Google Scholar]
- 22. Fabris L, Cadamuro M, Libbrecht L, Raynaud P, Spirlì C, Fiorotto R, et al. Epithelial expression of angiogenic growth factors modulate arterial vasculogenesis in human liver development. Hepatology 2008;47:719‐728. [DOI] [PubMed] [Google Scholar]
- 23. Gaudio E, Onori P, Franchitto A, Pannarale L, Alpini G, Alvaro D. Hepatic microcirculation and cholangiocyte physiopathology. Ital J Anat Embryol 2005;110:71‐75. [PubMed] [Google Scholar]
- 24. Gaudio E, Barbaro B, Alvaro D, Glaser S, Francis H, Franchitto A, et al. Administration of r‐VEGF‐A prevents hepatic artery ligation‐induced bile duct damage in bile duct ligated rats. Am J Physiol Gastrointest Liver Physiol 2006;291:G307‐G317. [DOI] [PubMed] [Google Scholar]
- 25. Kanno N, LeSage G, Glaser S, Alvaro D, Alpini G. Functional heterogeneity of the intrahepatic biliary epithelium. Hepatology 2000;31:555‐561. [DOI] [PubMed] [Google Scholar]
- 26. Alpini G, Lenzi R, Sarkozi L, Tavoloni N. Biliary physiology in rats with bile ductular cell hyperplasia. Evidence for a secretory function of proliferated bile ductules. J Clin Invest 1988;81:569‐578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu N, Baiocchi L, Zhou T, Kennedy L, Ceci L, Meng F, et al. Functional Role of the secretin/secretin receptor signaling during cholestatic liver injury. Hepatology 2020;72:2219‐2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hall C, Sato K, Wu N, Zhou T, Kyritsi K, Meng F, et al. Regulators of cholangiocyte proliferation. Gene Expr 2017;17:155‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Han Y, Glaser S, Meng F, Francis H, Marzioni M, McDaniel K, et al. Recent advances in the morphological and functional heterogeneity of the biliary epithelium. Exp Biol Med (Maywood) 2013;238:549‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. de Vries E, Beuers U. Management of cholestatic disease in 2017. Liver Int 2017;37(Suppl 1):123‐129. [DOI] [PubMed] [Google Scholar]
- 31. Lleo A, Wang GQ, Gershwin ME, Hirschfield GM. Primary biliary cholangitis. Lancet 2020;396:1915‐1926. [DOI] [PubMed] [Google Scholar]
- 32. Dyson JK, Beuers U, Jones DEJ, Lohse AW, Hudson M. Primary sclerosing cholangitis. Lancet 2018;391:2547‐2559. [DOI] [PubMed] [Google Scholar]
- 33. Sasaki M, Ikeda H, Yamaguchi J, Nakada S, Nakanuma Y. Telomere shortening in the damaged small bile ducts in primary biliary cirrhosis reflects ongoing cellular senescence. Hepatology 2008;48:186‐195. [DOI] [PubMed] [Google Scholar]
- 34. Sasaki M, Yoshimura‐Miyakoshi M, Sato Y, Nakanuma Y. A possible involvement of endoplasmic reticulum stress in biliary epithelial autophagy and senescence in primary biliary cirrhosis. J Gastroenterol 2015;50:984‐995. [DOI] [PubMed] [Google Scholar]
- 35. Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Chemokine‐chemokine receptor CCL2‐CCR2 and CX3CL1‐CX3CR1 axis may play a role in the aggravated inflammation in primary biliary cirrhosis. Dig Dis Sci 2014;59:358‐364. [DOI] [PubMed] [Google Scholar]
- 36. Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. A possible involvement of p62/sequestosome‐1 in the process of biliary epithelial autophagy and senescence in primary biliary cirrhosis. Liver Int 2012;32:487‐499. [DOI] [PubMed] [Google Scholar]
- 37. Loarca L, De Assuncao TM, Jalan‐Sakrikar N, Bronk S, Krishnan A, Huang B, et al. Development and characterization of cholangioids from normal and diseased human cholangiocytes as an in vitro model to study primary sclerosing cholangitis. Lab Invest 2017;97:1385‐1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature 2004;432:307‐315. [DOI] [PubMed] [Google Scholar]
- 39. Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol 2021;22:75‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McDaniel K, Meng F, Wu N, Sato K, Venter J, Bernuzzi F, et al. Forkhead box A2 regulates biliary heterogeneity and senescence during cholestatic liver injury in micedouble dagger. Hepatology 2017;65:544‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li Z, White P, Tuteja G, Rubins N, Sackett S, Kaestner KH. Foxa1 and Foxa2 regulate bile duct development in mice. J Clin Invest 2009;119:1537‐1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wan Y, Meng F, Wu N, Zhou T, Venter J, Francis H, et al. Substance P increases liver fibrosis by differential changes in senescence of cholangiocytes and hepatic stellate cells. Hepatology 2017;66:528‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Meng F, DeMorrow S, Venter J, Frampton G, Han Y, Francis H, et al. Overexpression of membrane metalloendopeptidase inhibits substance P stimulation of cholangiocarcinoma growth. Am J Physiol Gastrointest Liver Physiol 2014;306:G759‐G768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Glaser S, Gaudio E, Renzi A, Mancinelli R, Ueno Y, Venter J, et al. Knockout of the neurokinin‐1 receptor reduces cholangiocyte proliferation in bile duct‐ligated mice. Am J Physiol Gastrointest Liver Physiol 2011;301:G297‐G305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. O'Hara SP, La Russo NF. Cellular senescence, neuropeptides and hepatic fibrosis: additional insights into increasing complexity. Hepatology 2017;66:318‐320. [DOI] [PubMed] [Google Scholar]
- 46. Glaser S, Meng F, Han Y, Onori P, Chow BK, Francis H, et al. Secretin stimulates biliary cell proliferation by regulating expression of microRNA 125b and microRNA let7a in mice. Gastroenterology 2014;146:1795‐1808.e1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu N, Meng F, Invernizzi P, Bernuzzi F, Venter J, Standeford H, et al. The secretin/secretin receptor axis modulates liver fibrosis through changes in transforming growth factor‐beta1 biliary secretion in mice. Hepatology 2016;64:865‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhou T, Wu N, Meng F, Venter J, Giang TK, Francis H, et al. Knockout of secretin receptor reduces biliary damage and liver fibrosis in Mdr2−/− mice by diminishing senescence of cholangiocytes. Lab Invest 2018;98:1449‐1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wan Y, Ceci L, Wu N, Zhou T, Chen L, Venter J, et al. Knockout of alpha‐calcitonin gene‐related peptide attenuates cholestatic liver injury by differentially regulating cellular senescence of hepatic stellate cells and cholangiocytes. Lab Invest 2019;99:764‐776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Meadows V, Kennedy L, Hargrove L, Demieville J, Meng F, Virani S, et al. Downregulation of hepatic stem cell factor by Vivo‐Morpholino treatment inhibits mast cell migration and decreases biliary damage/senescence and liver fibrosis in Mdr2−/− mice. Biochim Biophys Acta Mol Basis Dis 2019;1865:165557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou T, Kyritsi K, Wu N, Francis H, Yang Z, Chen L, et al. Knockdown of vimentin reduces mesenchymal phenotype of cholangiocytes in the Mdr2−/− mouse model of primary sclerosing cholangitis (PSC). EBioMedicine 2019;48:130‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kyritsi K, Francis H, Zhou T, Ceci L, Wu N, Yang Z, et al. Downregulation of p16 decreases biliary damage and liver fibrosis in the Mdr2(/) mouse model of primary sclerosing cholangitis. Gene Exp 2020;20:89‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moncsek A, Al‐Suraih MS, Trussoni CE, O'Hara SP, Splinter PL, Zuber C, et al. Targeting senescent cholangiocytes and activated fibroblasts with B‐cell lymphoma‐extra large inhibitors ameliorates fibrosis in multidrug resistance 2 gene knockout (Mdr2−/−) mice. Hepatology 2018;67:247‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. O'Hara SP, Splinter PL, Trussoni CE, Guicciardi ME, Splinter NP, Al Suraih MS, et al. The transcription factor ETS1 promotes apoptosis resistance of senescent cholangiocytes by epigenetically up‐regulating the apoptosis suppressor BCL2L1. J Biol Chem 2019;294:18698‐18713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kundu D, Kennedy L, Meadows V, Baiocchi L, Alpini G, Francis H. The dynamic interplay between mast cells, aging/cellular senescence, and liver disease. Gene Expr 2020;20:77‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hunt NJ, Kang SWS, Lockwood GP, Le Couteur DG, Cogger VC. Hallmarks of aging in the liver. Comput Struct Biotechnol J 2019;17:1151‐1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sheedfar F, Di Biase S, Koonen D, Vinciguerra M. Liver diseases and aging: friends or foes? Aging Cell 2013;12:950‐954. [DOI] [PubMed] [Google Scholar]
- 58. Alvaro D, Mancino MG, Onori P, Franchitto A, Alpini G, Francis H, et al. Estrogens and the pathophysiology of the biliary tree. World J Gastroenterol 2006;12:3537‐3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tabibian JH, Trussoni CE, O'Hara SP, Splinter PL, Heimbach JK, LaRusso NF. Characterization of cultured cholangiocytes isolated from livers of patients with primary sclerosing cholangitis. Lab Invest 2014;94:1126‐1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sasaki M, Sato Y, Nakanuma Y. Increased p16(INK4a)‐expressing senescent bile ductular cells are associated with inadequate response to ursodeoxycholic acid in primary biliary cholangitis. J Autoimmun 2020;107:102377. [DOI] [PubMed] [Google Scholar]
- 61. Maroni L, Pinto C, Giordano DM, Saccomanno S, Banales JM, Spallacci D, et al. Aging‐related expression of twinfilin‐1 regulates cholangiocyte biological response to injury. Hepatology 2019;70:883‐898. [DOI] [PubMed] [Google Scholar]
- 62. Lakshminarayanan B, Davenport M. Biliary atresia: a comprehensive review. J Autoimmun 2016;73:1‐9. [DOI] [PubMed] [Google Scholar]
- 63. Sasaki M, Kuo FY, Huang CC, Swanson PE, Chen CL, Chuang JH, et al. Increased expression of senescence‐associated cell cycle regulators in the progression of biliary atresia: an immunohistochemical study. Histopathology 2018;72:1164‐1171. [DOI] [PubMed] [Google Scholar]
- 64. Shetty S, Lalor PF, Adams DH. Liver sinusoidal endothelial cells—gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol 2018;15:555‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Marrone G, Shah VH, Gracia‐Sancho J. Sinusoidal communication in liver fibrosis and regeneration. J Hepatol 2016;65:608‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. DeLeve LD, Maretti‐Mira AC. Liver sinusoidal endothelial cell: an update. Semin Liver Dis 2017;37:377‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cogger VC, Warren A, Fraser R, Ngu M, McLean AJ, Le Couteur DG. Hepatic sinusoidal pseudocapillarization with aging in the non‐human primate. Exp Gerontol 2003;38:1101‐1107. [DOI] [PubMed] [Google Scholar]
- 68. Ito Y, Sorensen KK, Bethea NW, Svistounov D, McCuskey MK, Smedsrod BH, et al. Age‐related changes in the hepatic microcirculation in mice. Exp Gerontol 2007;42:789‐797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Maeso‐Díaz R, Ortega‐Ribera M, Fernández‐Iglesias A, Hide D, Muñoz L, Hessheimer AJ, et al. Effects of aging on liver microcirculatory function and sinusoidal phenotype. Aging Cell 2018;17:e12829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gracia‐Sancho J, Marrone G, Fernandez‐Iglesias A. Hepatic microcirculation and mechanisms of portal hypertension. Nat Rev Gastroenterol Hepatol 2019;16:221‐234. [DOI] [PubMed] [Google Scholar]
- 71. Wiest R, Groszmann RJ. The paradox of nitric oxide in cirrhosis and portal hypertension: too much, not enough. Hepatology 2002;35:478‐491. [DOI] [PubMed] [Google Scholar]
- 72. Deleve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology 2008;48:920‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Marrone G, Russo L, Rosado E, Hide D, García‐Cardeña G, García‐Pagán JC, et al. The transcription factor KLF2 mediates hepatic endothelial protection and paracrine endothelial‐stellate cell deactivation induced by statins. J Hepatol 2013;58:98‐103. [DOI] [PubMed] [Google Scholar]
- 74. Ding B‐S, Nolan DJ, Butler JM, James D, Babazadeh AO, Rosenwaks Z, et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature 2010;468:310‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Grosse L, Wagner N, Emelyanov A, Molina C, Lacas‐Gervais S, Wagner KD, et al. Defined p16(High) senescent cell types are indispensable for mouse healthspan. Cell Metab 2020;32:87‐99.e86. [DOI] [PubMed] [Google Scholar]
- 76. Hilmer SN, Cogger VC, Fraser R, McLean AJ, Sullivan D, Le Couteur DG. Age‐related changes in the hepatic sinusoidal endothelium impede lipoprotein transfer in the rat. Hepatology 2005;42:1349‐1354. [DOI] [PubMed] [Google Scholar]
- 77. Grosse L, Bulavin DV. LSEC model of aging. Aging (Albany NY) 2020;12:11152‐11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Maeso‐Díaz R, Ortega‐Ribera M, Lafoz E, Lozano JJ, Baiges A, Francés R, et al. Aging influences hepatic microvascular biology and liver fibrosis in advanced chronic liver disease. Aging Dis 2019;10:684‐698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hide D, Warren A, Fernandez‐Iglesias A, Maeso‐Diaz R, Peralta C, Le Couteur DG, et al. Ischemia/reperfusion injury in the aged liver: the importance of the sinusoidal endothelium in developing therapeutic strategies for the elderly. J Gerontol A Biol Sci Med Sci 2020;75:268‐277. [DOI] [PubMed] [Google Scholar]
- 80. Pinzani M, Gentilini P. Biology of hepatic stellate cells and their possible relevance in the pathogenesis of portal hypertension in cirrhosis. Semin Liver Dis 1999;19:397‐410. [DOI] [PubMed] [Google Scholar]
- 81. Warren A, Cogger VC, Fraser R, Deleve LD, McCuskey RS, Le Couteur DG. The effects of old age on hepatic stellate cells. Curr Gerontol Geriatr Res 2011;2011:439835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases‐diversity, plasticity and therapeutic opportunities. Cell Mol Immunol 2021;18:45‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Stahl EC, Delgado ER, Alencastro F, LoPresti ST, Wilkinson PD, Roy N, et al. Inflammation and ectopic fat deposition in the aging murine liver is influenced by CCR2. Am J Pathol 2020;190:372‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tsochatzis EA, Bosch J, Burroughs AK. Liver cirrhosis. Lancet 2014;383:1749‐1761. [DOI] [PubMed] [Google Scholar]
- 85. Thabut D, Le Calvez S, Thibault V, Massard J, Munteanu M, Di Martino V, et al. Hepatitis C in 6,865 patients 65 yr or older: a severe and neglected curable disease? Am J Gastroenterol 2006;101:1260‐1267. [DOI] [PubMed] [Google Scholar]
- 86. McLean AJ, Cogger VC, Chong GC, Warren A, Markus AM, Dahlstrom JE, et al. Age‐related pseudocapillarization of the human liver. J Pathol 2003;200:112‐117. [DOI] [PubMed] [Google Scholar]
- 87. Zhu YI, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 2015;14:644‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kirkland JL, Tchkonia T. Senolytic drugs: from discovery to translation. J Intern Med 2020;288:518‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: results from a first‐in‐human, open‐label, pilot study. EBioMedicine 2019;40:554‐563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019;47:446‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Simon‐Santamaria J, Malovic I, Warren A, Oteiza A, Le Couteur D, Smedsrød B, et al. Age‐related changes in scavenger receptor‐mediated endocytosis in rat liver sinusoidal endothelial cells. J Gerontol A Biol Sci Med Sci 2010;65:951‐960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Cheluvappa R, Hilmer SN, Kwun SY, Jamieson HA, O’Reilly JN, Muller M, et al. The effect of old age on liver oxygenation and the hepatic expression of VEGF and VEGFR2. Exp Gerontol 2007;42:1012‐1019. [DOI] [PubMed] [Google Scholar]
- 93. Le Couteur DG, Cogger VC, Markus AM, Harvey PJ, Yin ZL, Ansselin AD, et al. Pseudocapillarization and associated energy limitation in the aged rat liver. Hepatology 2001;33:537‐543. [DOI] [PubMed] [Google Scholar]
- 94. Verma S, Tachtatzis P, Penrhyn‐Lowe S, Scarpini C, Jurk D, Von Zglinicki T, et al. Sustained telomere length in hepatocytes and cholangiocytes with increasing age in normal liver. Hepatology 2012;56:1510‐1520. [DOI] [PubMed] [Google Scholar]
- 95. Kaffe E, Katsifa A, Xylourgidis N, Ninou I, Zannikou M, Harokopos V, et al. Hepatocyte autotaxin expression promotes liver fibrosis and cancer. Hepatology 2017;65:1369‐1383. [DOI] [PubMed] [Google Scholar]
- 96. Yanase M, Ikeda H, Matsui A, Maekawa H, Noiri E, Tomiya T, et al. Lysophosphatidic acid enhances collagen gel contraction by hepatic stellate cells: association with rho‐kinase. Biochem Biophys Res Commun 2000;277:72‐78. [DOI] [PubMed] [Google Scholar]
- 97. Ikeda H, Yatomi Y, Yanase M, Satoh H, Nishihara A, Kawabata M, et al. Effects of lysophosphatidic acid on proliferation of stellate cells and hepatocytes in culture. Biochem Biophys Res Commun 1998;248:436‐440. [DOI] [PubMed] [Google Scholar]
- 98. Nishizawa H, Iguchi G, Fukuoka H, Takahashi M, Suda K, Bando H, et al. IGF‐I induces senescence of hepatic stellate cells and limits fibrosis in a p53‐dependent manner. Sci Rep 2016;6:34605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Schrader J, Fallowfield J, Iredale JP. Senescence of activated stellate cells: not just early retirement. Hepatology 2009;49:1045‐1047. [DOI] [PubMed] [Google Scholar]