

Abstract
Currently, the hepatocellular trafficking pathways that are used by the hepatitis B virus (HBV) during viral infection and shedding are poorly defined. It is known that the HBV uses late endosomal and multivesicular body (MVB) compartments for assembly and release. The intraluminal vesicles (ILVs) generated within MVBs have also been implicated in the late synthesis stages of a variety of pathogenic viruses. We recently observed that the HBV within infected hepatocytes appears to associate with the tetraspanin protein CD63, known to be a prominent and essential component of ILVs. Immunofluorescence microscopy of HBV‐expressing cells showed that CD63 colocalized with HBV proteins (large hepatitis B surface antigens [LHBs] and hepatitis B core) and labeled an exceptionally large number of secreted extracellular vesicles of uniform size. Small interfering RNA (siRNA)–mediated depletion of CD63 induced a substantial accumulation of intracellular LHBs protein but did not alter the levels of either intracellular or extracellular HBV DNA, nor pregenomic RNA. Consistent with these findings, we found that markedly less LHBs protein was associated with the released HBV particles from CD63 siRNA‐treated cells. Importantly, the HBV viral particles that were shed from CD63‐depleted cells were substantially less infective than those collected from control cells with normal CD63 levels. Conclusion: These findings implicate the tetraspanin protein CD63 as a marker and an important component in the formation and release of infectious HBV particles.
This study describes novel findings in regard to the host cell components important for appropriate packaging, trafficking and shedding of nascent HBVs. Specifically, a significant role for the tetraspanin, endosomal coat protein CD63, was observed in this process.

Abbreviations
- cDNA
complementary DNA
- DAPI
4′,6‐diamidino‐2‐phenylindole
- EGFP
enhanced green fluorescent protein
- ESCRT
endosomal sorting complex required for transport
- EV
extracellular vesicle
- GFP
green fluorescent protein
- HBc
hepatitis B core
- HBeAg
hepatitis B e antigen
- HBsAg
hepatitis B surface antigen
- HBV
hepatitis B virus
- ILV
intraluminal vesicle
- IP
immunoprecipitation
- LHBs
large hepatitis B surface antigen
- MHBs
middle hepatitis B surface antigen
- mRNA
messenger RNA
- MVB
multivesicular body
- NT
nontargeting
- PBS
phosphate‐buffered saline
- RT‐PCR
real‐time polymerase chain reaction
- SHBs
small hepatitis B surface antigen
- siNT
nontargeting control siRNA
- siRNA
small interfering RNA
- SVP
subviral particle
- TSG101
tumor susceptibility gene 101
Hepatitis B virus (HBV) remains an important health challenge in both Western and Eastern cultures with almost 292 million people chronically infected worldwide,( 1 ) with a substantial percentage of those infected with HBV eventually developing chronic hepatitis, cirrhosis, and hepatocellular carcinoma.( 2 ) HBV has a circular genome of partially double‐stranded DNA within a spherical particle 42 nm in diameter. It includes an icosahedral nucleocapsid, which consists of hepatitis B core (HBc) protein, and is surrounded by an envelope containing hepatitis B surface antigen (HBsAg). This envelope consists of cellular lipids and three surface proteins, small (SHBs), middle (MHBs) and large (LHBs), that are believed to have distinct functions including viral entry, morphogenesis, and infectivity, respectively.( 3 ) Importantly, while the envelopes of infectious HBV virions contain all three envelope proteins, the noninfectious empty subviral particles (SVPs) are assembled with just SHBs and MHBs, and are missing the LHBs protein.( 4 , 5 )
Although the LHBs protein is required for nucleocapsid envelopment,( 6 ) several reports have shown that LHBs is also essential for infectivity.( 7 ) The N‐terminal myristoylated amino acids 3‐77 in the preS1 region of LHBs are believed to be crucial for viral infectivity by mediating attachment to specific receptors.( 8 , 9 ) Because only several aspects of the HBV life cycle have been characterized, the mechanisms that contribute to the important process of viral assembly and release are poorly defined. Several studies have implicated the multivesicular body (MVB) as an essential sorting and packaging station in the maturation of several infectious viruses including the HBV. These findings also suggest that HBV maturation may require the formation of intraluminal vesicles (ILVs) in late endosomes/MVBs through AIP1/ALIX, the endosomal sorting complex required for transport (ESCRT)‐III complex, charged MVB proteins, and the AAA ATPase Vps4.( 10 , 11 ) Several reports have also suggested that the ESCRT pathway is used for hepatitis C virus (HCV) production.( 12 ) Many of these findings suggest that nascent viral particles bud into ILVs of the MVB and are transferred to the plasma membrane for release through a putative “exosome”‐like compartment.( 13 ) The release of these exosomes has been implicated in the transmission and infectivity of HCV,( 14 ) while the relationship of exosomes in HBV infection is still unknown.
We have recently reported that HBV infection induces a dramatic mobilization of the endo‐lysosomal and autophagic pathways via activation of the small regulatory GTPase Rab7. The consequences of this activation appear, in part, as the formation of numerous tubules extending from viral‐laden MVBs that contribute to subsequent HBV secretion.( 15 ) In an attempt to understand how this activation drives viral shedding and subsequent infection, we have observed that HBV‐infected cells, when grown on a charged substrate of poly‐l‐lysine, become surrounded by a substantial number of small vesicles of a uniform diameter (40‐50 nm) that stain positive for an exosome marker: the tetraspanin protein CD63. Importantly, small interfering RNA (siRNA)–mediated depletion of CD63 led to an accumulation of LHBs, while the levels of both intracellular and released extracellular HBV DNA or pregenomic RNA remained unchanged. Subsequent characterization of HBV particles derived from CD63‐depleted cells revealed markedly reduced levels of LHBs and were in turn substantially less infectious. These findings suggest that the exosome membrane protein CD63 may play an essential role in HBV release and infection. The implications of CD63 as a target to attenuate viral transmission are discussed.
Materials and Methods
Plasmids and siRNA
The CD63‐pEGFP N1 construct (enhanced green fluorescent protein [EGFP]–CD63) was cloned as follows: Specific oligonucleotide primers (synthesized by Integrated DNA Technologies, Coralville, IA) for human CD63 isoform A were designed (MacVector, New Haven, CT) using human CD63 isoform A complementary DNA (cDNA) sequences from GenBank (Accession number NM_001257389). Full‐length cDNAs encoding the CD63 isoform A were amplified by real‐time polymerase chain reaction (RT‐PCR). The primers used for green fluorescent protein (GFP)–CD63 were 5′ primer (5′‐GAA TTC ATG GCG GTG GAA GGA GGA A‐3′) and 3′ primer (5′‐CT C GAG AGA CCC CTA CAT CAC CTC G ‐3′). Human CD63 was cloned into pEGFP N1 vector using EcoRI (5′) and XhoI (3′) enzymes. GFP‐Rab7 wild type was previously described.( 16 ) Small interfering RNA targeting human CD63 (target sequence: 5′‐ATG TGT GAA GTT CTT GCT CTA‐3′) and nontargeting (NT) control siRNA (siNT) were purchased from Qiagen (Hilden, Germany) and Dharmacon (Thermo Fisher Scientific, Lafayette, CO), respectively.
Antibodies
Anti‐CD63 antibody was purchased from Developmental Studies Hybridoma Bank (Iowa City, IA), anti‐preS1 from Santa Cruz Biotechnology (Dallas, TX), anti‐actin from Sigma‐Aldrich (St. Louis, MO), and antitumor susceptibility gene 101 (TSG101) from GeneTex (Irvine, CA). Anti‐HBc antibodies for western blotting and immunofluorescence were purchased from DAKO (Carpinteria, CA) and Santa Cruz Biotechnology, respectively. An anti‐HBs horse polyclonal antibody was a kind gift from the Institute of Immunology (Tokyo, Japan).
Cell Culture and Transfection
The HBV‐expressing stable cell line HepG2.2.15 was kindly provided by Dr. Andrea Cuconati (Institute of Hepatitis and Virus Research, Doylestown, PA), and the parental human hepatoma cell line HepG2 was incubated in Roswell Park Memorial Institute 1640 medium with l‐glutamine (Corning Cellgro, Manassas, VA) supplemented with 10% fetal bovine serum, 50 U/mL penicillin, and 50 μg/mL streptomycin. The HBV‐expressing stable cell line HepAD38 (a gift from Christoph Seeger at Fox Chase Cancer Center) was incubated as described previously.( 17 ) HepG2.2.15 and HepG2 cells were seeded onto coverslips or plates coated with poly‐l‐lysine (Sigma‐Aldrich), as described previously.( 18 ) Cells were transiently transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. HepG2.2.15 cells were transfected twice in a 24‐hour interval to increase the transfection efficiency. Transfection of HepG2.2.15 cells with siRNA was performed using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. The re‐expression was performed as done previously, 24 hours after the siRNA transfection. An immortalized human primary hepatocyte cell line HuS‐E/2, which can be infected with live HBV, was kindly provided by Dr. Makoto Hijikata (Kyoto University, Japan) and was grown as described previously.( 19 ) HBV infection experiments with HuS‐E/2 cells were performed as previously described.( 20 ) Briefly, culture supernatant collected from CD63‐depleted or siNT (NT negative control) treated HepG2.2.15 cells was concentrated with an Amicon Ultra‐15 100K device (Merck Millipore, Billerica, MA). The concentrated supernatant containing HBV particles was used to infect HuS‐E/2 cells for 20 hours followed by several washes with fresh media. To assess the infectivity, we serially collected the culture supernatant after 6, 24, and 48 hours of infection and quantified the HBV‐DNA titer by RT‐PCR, and the HBV‐infected HuS‐E/2 cells were subjected to western blotting for LHBs.
Western Blotting
For western blotting, cells were lysed in NP buffer (137 mM NaCl, 1% NP‐40, 10% glycerol, 2 mM EDTA, 20 mM Tris‐base [pH 8.0] and complete protease inhibitors [Roche Diagnostics, Indianapolis, IN]). Soluble proteins (40 μg) was resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to a PVDF membrane. After blocking with 5% milk in phosphate‐buffered saline (PBS), the membrane was incubated with primary antibodies at room temperature for 2 hours, washed, and incubated with secondary antibodies for 1 hour. The signals were detected with SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific).
Real‐Time PCR and Chemiluminescence Immunoassay Using Culture Supernatant
Total DNA was extracted with a QIAamp DNA Blood Mini Kit (Qiagen) from 100 μL of the culture supernatant or 1.0 × 107 cells, and 5 μL of the extracted DNA solution was subjected to RT‐PCR using a LightCycler 480 system (Roche Diagnostics) with SYBR Green I Master (Roche Diagnostics), primers HBXF11 (5′‐ATG GCT GCT AGG CTG TGC TG‐3′) and HBXR4 (5′‐GTC CGC GTA AAG AGA GGT GC‐3′). Total RNA was extracted from 1.0 × 107 cells using Trizol (Invitrogen) and subjected to cDNA synthesis by SuperScript III (Invitrogen). RT‐PCR for quantifying total amount of pregenomic RNA and precore messenger RNA (mRNA) was conducted with SYBR Green I Master, primers HBCF7 (5′‐GCT ACC TGG GTG GGT GGT AAT TTG G‐3′) and HBCR5 (5′‐AGG GGA CCT GCC TCG TCG TCT AAC‐3′). The HBsAg and hepatitis B e antigen (HBeAg) levels in the culture supernatant were quantified with a chemiluminescence immunoassay (CLIA) using the ARCHITECT HBsAg reagent kit and ARCHITECT HBeAg reagent kit (Abbott Laboratories, Chicago, IL), respectively.
Immunoprecipitation of Enveloped Viral Particles
Immunoprecipitation (IP) with anti‐HBs antibody to quantify viral particles with envelope was performed as described previously.( 21 ) Briefly, the culture supernatant was incubated with the polyclonal anti‐HBs antibody at 4°C for 20 hours, and after the addition of protein G Plus‐agarose (Santa Cruz), it was incubated for 2 hours. After washing with PBS, 0.1 M glycine‐HCl (pH 2.2) was added to remove the antibody. The supernatant was neutralized with NaOH and subjected to RT‐PCR for HBV DNA.
Immunofluorescence Labeling and Microscopy
Cells were prepared for immunofluorescence as described previously.( 22 ) Briefly, cells were incubated with primary antibodies for 2 hours at 37°C, washed with D‐PBS, and incubated with labeled secondary antibodies (Life Technologies, Carlsbad, CA) for 1 hour at 37°C. Actin was stained using TRITC‐Phalloidin, and the nucleus was stained with 4′,6‐diamidino‐2‐phenylindole (DAPI; Sigma‐Aldrich). The coverslips were mounted on glass slides using ProLong mounting medium (Life Technologies), and cells were imaged with an AxioObserver D.1 epifluorescence microscope (Carl Zeiss, Thornwood, NY) equipped with a 100‐W mercury lamp using a × 63, 1.4N.A objective lens. Contrast and intensity for each image were manipulated uniformly using Adobe Photoshop software (Adobe Systems Inc., San Jose, CA). Images were analyzed with the IVision software (BioVision, Mountain View, CA).
Isolation of Extracellular Vesicles Containing HBV Particles
After treatment with siRNAs, HepG2.2.15 cells were cultured in serum‐free medium for 24 hours. Dead cells and cell debris were eliminated by successive centrifugations at 2,000g for 10 minutes and 10,000g for 30 minutes. The pellet was thrown away after each centrifugation, and supernatant was used for the following step. The supernatant was then centrifuged three times at 200,000g for 90 minutes. The pellet was washed with PBS and centrifuged one last time at the same high speed. The final pellet of extracellular vesicles (EVs) was resuspended in PBS or lysis buffers to use for subsequent experiments or stored at −80°C.
Nanoparticle Tracking Analysis
The Nanosight NTA NS300 (Malvern Instruments, Malvern, United Kingdom) was used to count EV particles.( 23 ) The instrument was operated according to the manufacturer’s protocol.
Sucrose Density Gradient Centrifugation
Sucrose density gradient centrifugation was performed to determine the buoyant density of viral particles in the culture supernatants as described previously.( 21 ) The centrifugation was done using himac CP80WX (HITACHI, Tokyo, Japan) with 5‐mL tubes. The 100‐μL fractions were collected from the top, and DNA was extracted from 10 of 100 μL and then subjected to RT‐PCR for HBV DNA. Two of the 100‐μL fractions were subjected to dot blot using anti‐HBs antibody, and the signal levels were quantified as HBsAg.
Statistical Analysis
Quantitative data are expressed as means ± SD. Statistical comparisons were made using Student t test unless otherwise indicated, and P < 0.05 was considered significant.
Results
HBV Proteins Colocalize With CD63 in HBV‐Expressing Cells
Previous reports by us and others have indicated that MVBs and autophagic compartments play an important role in the maturation and trafficking of HBV.( 10 , 11 , 15 , 24 , 25 , 26 ) In an attempt to better define the specific membranous compartments of the hepatocyte through which nascent virions are transported, we stained HepG2.2.15 cells, known to stably express HBV,( 27 ) with antibodies to distinct internal membrane compartment marker proteins. Although CD63 is a tetraspanin family member and a prominent component of both late endosomes and exosomes, its function at these compartments is largely undefined.( 28 ) HepG2.2.15 were cultured on poly‐l‐lysine‐coated coverslips and either transfected to express GFP‐CD63 or fixed and immunostained for anti‐CD63 and antibodies to preS1 of LHBs or HBc (Fig. 1A,B). Viral antigens can be seen colocalizing with the GFP‐CD63 compartments (arrows). Of note, the cells stained with the CD63 antibody were observed to be surrounded by elaborate “lawns” consisting of vast numbers of CD63 puncta that often appeared as dotted lines left behind as the cells migrated along the coverslip (Fig. 1C‐E). The number of CD63 puncta observed surrounding HepG2.2.15 cells were 1.69‐fold more prevalent than that observed for the parental HepG2 cells (Supporting Fig. S1). Higher‐resolution scanning electron micrographs of both control and infected cells (Fig. 1F,G) indicated that the CD63 puncta represent small membranous vesicles of a remarkably conserved size (40‐50 nm). Such images suggested that CD63‐positive EVs might be released from the cells and attach to the coated coverslips, and that they have diameters that are similar to the known size of HBV particles.
FIG. 1.
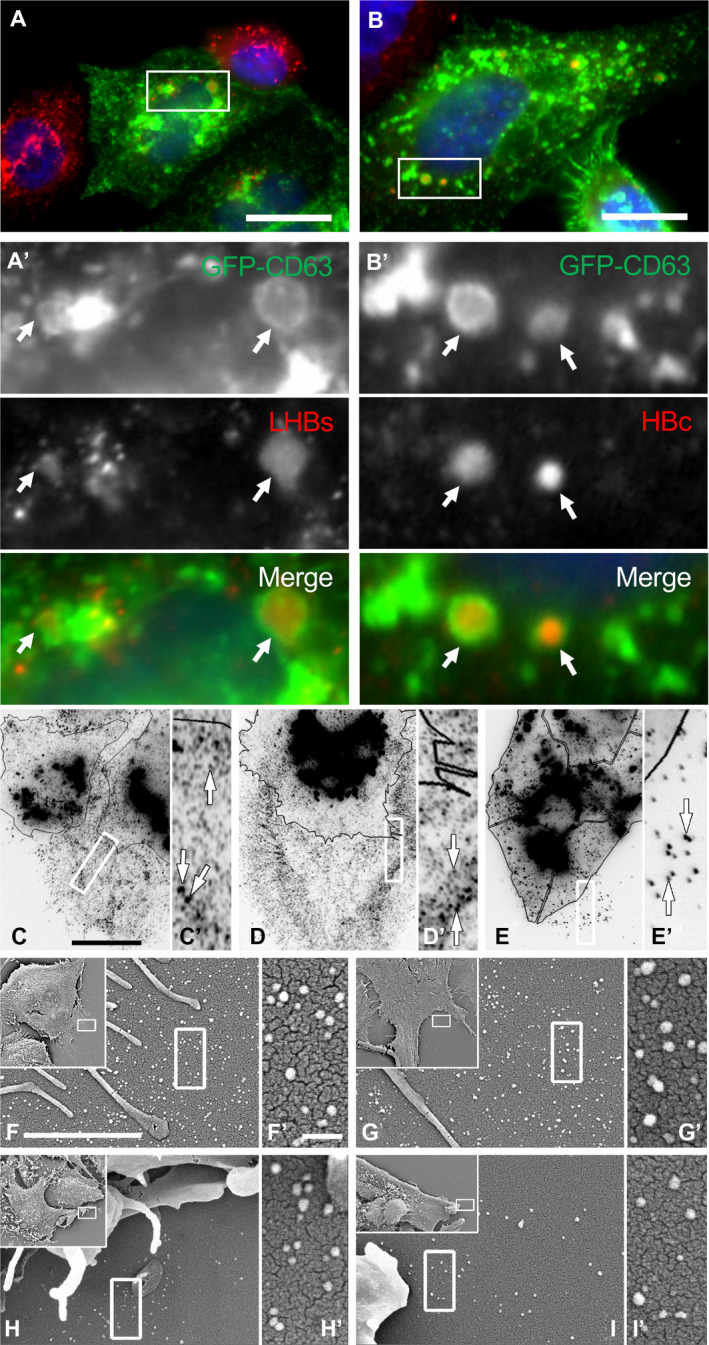
Association between intracellular CD63‐positive compartments and HBV proteins in HBV‐expressing cells that display CD63‐positive vesicular structures around cells. (A,A′,B,B′) HepG2.2.15 cells were transfected with GFP‐tagged CD63 (green) as a late endosome/MVB marker, then subsequently fixed and immunostained with antibodies to HBV components (red), including the LHBs protein (A′) or the HBc protein (B′). Arrows point to vesicles that are positive for both endosomal and viral antigens. DAPI (blue) for nuclear staining. (C‐E) Immunofluorescence images of HepG2.2.15 cells (C,D) or parental HepG2 cells (E) fixed and stained with antibodies to CD63. Images are presented as inverted gray scale to better contrast the substantial number of uniformly sized CD63‐positive vesicles surrounding each cell. Cells are outlined with black lines for clarity. Higher magnifications of boxed regions are provided in C′‐E′. (F,F′,G,G′,H,H′,I,I′) Scanning electron micrographs of cell borders showing the uniformity in size (40‐50 nm) of the HepG2.2.15 (F,F′,G,G′) and control HepG2 (H,H′,I,I′) generated vesicles. Scale bars: A‐C, 10 μm; F, 2 μm; F′, 200 nm.
HBV‐Infected Cells, Depleted of CD63, Accumulate Specific HBV Protein Components
Based on the substantial numbers of CD63‐positive vesicles released from HBV‐expressing cells (Fig. 1), we tested the role of this tetraspanin protein in the assembly and release of HBV. HepG2.2.15 cells were treated with dual, staggered, CD63‐specific siRNAs to reduce the endogenous levels of this tetraspanin protein, as confirmed by both immunostaining and western blot analysis (Fig. 2A,B). Subsequently, expression levels of the LHBs protein were assessed from these CD63 siRNA‐treated HepG2.2.15 cells by western blot analysis using an anti‐PreS1 antibody. Importantly, HepG2.2.15 cells with reduced CD63 protein levels retained a remarkable 7‐fold increase in LHBs levels compared with control‐treated cells, suggesting a significant retention of this viral coat protein (Fig. 2C,C′). The increase in MHBs and SHBs, as well as LHBs, was confirmed from HepAD38 cell lysates using an anti‐HBs antibody (Supporting Fig. S2A,B). In contrast to the LHBs protein, no accumulation of the HBc protein was observed in these siRNA‐treated cells (Fig. 2C). These biochemical findings were supported by immunofluorescence staining of knockdown cells that revealed a significant morphological accumulation of the LHBs viral protein (Fig. 2D,D′) but not the HBc protein (Fig. 2E,E′). These observations provide strong evidence for a central role of the CD63 tetraspanin protein in the packaged export of select HBV components. We confirmed these findings with HepAD38 cells using an anti‐HBs antibody that detects LHBs, MHBs, and SHBs. All of these envelope proteins, but not HBc, were increased after the CD63 depletion (Supporting Fig. S2).
FIG. 2.
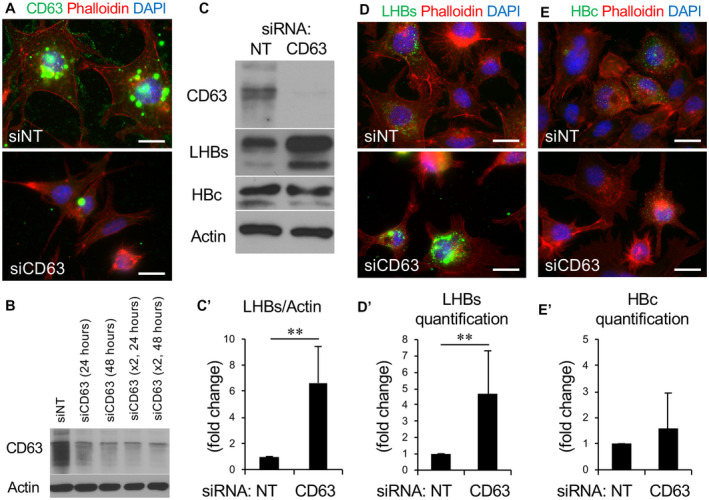
CD63‐depleted HBV‐expressing cells accumulate LHBs but not HBc protein. (A) Immunofluorescence images of HepG2.2.15 cells stained for phalloidin (red) and CD63 (green) transfected either with siNT or CD63 siRNA. (B) Western blot analysis of cells exposed to either single or double transfections of siRNA to CD63 revealing different knockdown efficiencies. (C) Western blot showing expression levels of LHBs and HBc in HepG2.2.15 cells with or without CD63 knockdown. (C′) CD63 depletion substantially increased the retention of intracellular LHBs but not HBc. Quantification of LHBs in five independent experiments shows a 6‐fold to 8‐fold increase in the retention of this viral protein in the CD63‐depleted cells. (D,E) Immunofluorescence microscopy images and corresponding quantification (graphs) of LHBs (D, green) and HBc (E, green) protein expression in HepG2.2.15 cells following siRNA knockdown of CD63. A 3‐fold to 7‐fold increase in the retention of LHBs protein is observed in the CD63‐depleted cells, while HBc protein levels appear unchanged. The y axis is expressed as fold changes in fluorescence intensity. **P < 0.01.
As CD63 localizes predominantly to late endosomes, MVBs, and lysosomes, with some association with endoplasmic reticulum and Golgi,( 28 ) we speculated that CD63 did not affect the synthesis of the HBV genome and nucleocapsid. To measure the effects of CD63 expression on the secretion of the HBV genome, the levels of both intracellular and extracellular HBV DNA were measured with RT‐PCR in control and CD63 siRNA‐treated cells. Intracellular DNA was extracted from 1.0 × 107 cells, and extracellular DNA was obtained from 100 μL of the culture supernatant. We found no significant differences in either intracellular or extracellular HBV DNA (Fig. 3A,B). Next, we tested whether the amount of HBV DNA that is contained in the enveloped viral particles is altered by the CD63 depletion. The amounts of HBV DNA after IP with anti‐HBs antibody were similar with, or without, CD63 siRNA (Fig. 3C). In contrast to the results of HBV DNA, the CD63 depletion significantly increased the HBsAg levels as assayed with CLIA using an anti‐HBs antibody (Fig. 3D). Western blot analysis of culture supernatant showed LHBs, MHBs, and SHBs were equally increased after CD63 depletion (Supporting Fig. S2C). To separate SVPs, enveloped HBV particles, and naked nucleocapsid particles, sucrose density gradient centrifugation was performed and showed that CD63 depletion did not alter the proportion of each particle in each fraction, while the total amount of HBsAg was increased (Supporting Fig. S3). These findings suggested that CD63 regulates the secretion of SVPs, but not the amount of enveloped viral particles. Because intracellular increase of LHBs/MHBs/SHBs led to the increase of these proteins in the supernatant, it was speculated that hepatocytes might have a high capacity to secret SVPs. Additionally, the secreted levels of another HBV secretory protein (HBeAg) were assessed from culture supernatant (Supporting Fig. S2D), and showed only modest changes in release of this protein into the media, suggesting that the increase of HBsAg may not be due to a general alteration of the viral secretory process.
FIG. 3.
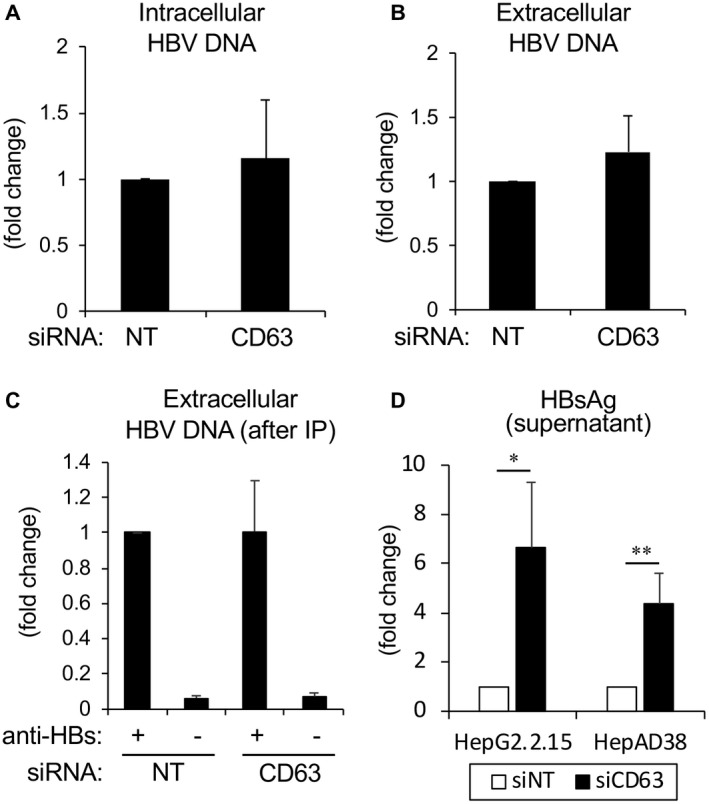
Replication and extracellular release of HBV DNA is independent of CD63 function. (A,B) To test and measure any potential role of CD63 in the release of the HBV genome from CD63‐depleted cells, total DNA was collected from both cell lysates (A) and supernatants (B) from HepG2.2.15 cells with or without CD63 depletion followed by RT‐PCR. No significant differences were observed. The findings in (A) and (B) represent the combined data of five independent experiments. (C) Using supernatant from HepG2.2.15 cells with or without CD63 depletion, IP with anti‐HBs antibody was performed and total DNA was extracted from the precipitates. The result of RT‐PCR shows no significant differences, indicating that the amount of HBV particles with an envelope was not altered after CD63 depletion. (D) HBsAg quantification using supernatant from HepG2.2.15 and HepAD38 cells with or without CD63 depletion shows that HBsAg was significantly increased after CD63 depletion. The findings in (C) and (D) represent the combined data of three independent experiments. **P < 0.01.
Because the observed alterations in HBV DNA, and that of the envelope proteins, were not consistent, we measured total viral RNA by northern blotting using HepAD38 cells (Fig. 4A). Interestingly, whereas the levels of 3.5 kb mRNA containing pregenomic RNA and precore mRNA remained the same, those of 2.4/2.1 kb mRNA coding for envelope proteins were increased after the CD63 depletion by 1.84‐fold. To pursue this finding further, the levels of 3.5 kb mRNA were assayed by RT‐PCR. Again, no difference was observed between HepG2.2.15 cells with and without CD63 depletion (Fig. 4B). Therefore, it was determined that CD63 regulates the transcription of 2.4/2.1 kb mRNA in a specific way.
FIG. 4.
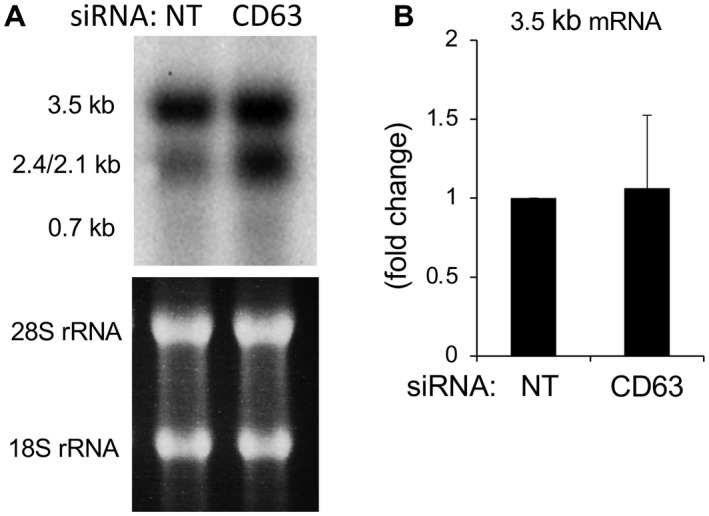
CD63 depletion increases mRNAs of HBV envelope proteins. (A) HepAD38 cells were transfected with NT control or CD63 siRNA followed by extraction of total RNA from cells and northern blotting with a full‐genome probe. Whereas the amount of 3.5 kb mRNA was similar, that of 2.4/2.1 kb mRNA was increased in CD63‐depleted cells. (B) Total RNA was extracted from HepG2.2.15 cells transfected with NT control or CD63 siRNA, and reverse‐transcription reactions and subsequent RT‐PCR were performed to detect 3.5 kb mRNA including both pregenomic RNA and precore mRNA. The results from five independent experiments showed no significant difference. Abbreviation: rRNA, ribosomal RNA.
EVs Collected From CD63‐Depleted HBV‐Producing Cells Exhibit an Altered Protein Composition and Are Less Infective
The findings described previously implicate CD63 in mediating normal HBV protein content in secreted virions and prompted us to further examine the role of this tetraspanin protein in EV secretion. Although many exosome preparations are traditionally collected by ultracentrifugation at 100,000g,( 29 , 30 ) several studies have performed ultracentrifugation at 200,000g,( 31 ) and we found that 200,000g was needed to pellet all HBV particles containing HBV DNA from the supernatant (Supporting Fig. S4). Subsequently we used this method to collect and compare EVs from HepG2.2.15 cells treated with control siRNA or siRNA against CD63. Interestingly, EVs collected from CD63‐depleted cells possessed substantially lower levels of LHBs and higher levels of a traditional MVB‐EV protein marker TSG101 (Fig. 5A,B), even though the cell lysates indicated that the CD63‐depleted cells had more internal LHBs. The finding that total amount of LHBs in the culture supernatant from the CD63‐depleted cells was higher than that from the control cells suggested that SVPs containing LHBs might not be precipitated in a meaningful way by the centrifugation. Also, because CD63 depletion did not change the relative proportion of HBsAg in SVP fractions and in HBV particle fractions (Supporting Fig. S3), SHBs ± MHBs might be maintained on the HBV particles. To isolate EVs, all culture media from siNT‐treated or siCD63‐treated HepG2.2.15 cells were collected and subjected to ultracentrifugation. As the levels of TSG101 appeared to be altered after CD63 depletion, thus making equal EV loading difficult, we used the NanoSight NTA NS300 technology to isolate cellular particles by size using a combination of light scattering and Brownian motion properties. The numbers of EVs isolated from siNT‐treated and siCD63‐treated cells appeared similar (mean numbers, 9.13 × 1010 vs. 7.17 × 1010/mL) (Fig. 5C). Using this method, equal numbers of EVs between siNT‐treated and siCD63‐treated cells were collected and subjected to RT‐PCR. CD63 depletion did not change HBV‐DNA levels in EVs (Fig. 5D), suggesting that, first, the CD63 positive EVs collected from the supernatants of infected cells represent intact HBV, and second, CD63‐depleted and siNT‐treated cells release the same number of EVs and HBV particles but with an altered protein composition with regard to TSG101 and LHBs proteins.
FIG. 5.
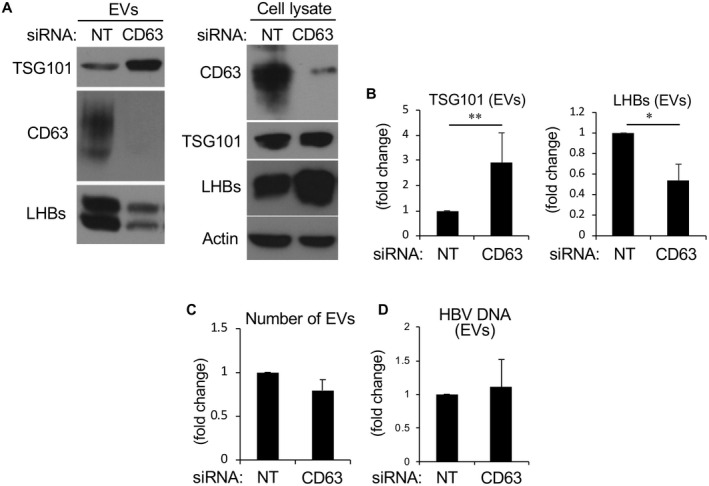
EVs collected from CD63‐depleted, HBV‐producing cells possess reduced LHBs protein. (A) Western blot analysis of lysates from HepG2.2.15 cells that were transfected with NT control siRNA or CD63 siRNA. Cells were lysed and EVs were purified by ultracentrifugation at 200,000g for 90 minutes and dissolved in lysis buffer. The expression levels of TSG101, CD63, and LHBs in EVs were determined by western blotting. CD63 depletion resulted in EVs with more TSG101, and less CD63 and LHBs. (B) Corresponding bar graphs quantifying TSG101 and LHBs protein expression from EVs. (C) EVs were isolated and counted by NanoSight with the vesicles ranging in size from 40 nm to 120 nm in diameter. (The y axis shows fold increase.) The numbers of EVs showed no significant difference between EVs with and without CD63 depletion. (D) The DNA titer contained within the EV preparation was determined by RT‐PCR and showed no significant difference between EVs isolated from control cells or cells depleted of CD63 protein. *P < 0.05 and **P < 0.01.
Several studies have reported that preS1 in the LHBs protein is essential for infectivity,( 32 ) as the acylation of glycine 2 of preS1 with myristic acid or N‐terminal myristoylation is important.( 33 , 34 , 35 ) However, the role of preS1 during infection of HBV is still unclear. We speculated that HBV with less LHBs derived from CD63‐depleted cells would be less infective, which was tested by collecting virus from HepG2.2.15 cells in which CD63 levels had been depleted, and infecting the immortalized human hepatocyte cell line HuS‐E/2. Culture supernatants were collected from the chronically infected HepG2.2.15 that were first treated with siNT control or CD63 siRNA. HBV DNA from cell supernatants were quantified using RT‐PCR and then added to HuS‐E/2 cells using a viral titer of 1.5 × 108 copies/mL. Cells were washed 20 hours later and returned to the incubator to allow the infection process to continue for different time periods (Fig. 6A). Subsequent to this incubation, the supernatant was collected and HBV‐DNA titer was quantified by RT‐PCR. As shown in Fig. 6B, the HBV particles contained within the culture media derived from CD63‐depleted cells were significantly less infective (as measured by HBV DNA) compared with HBV particles from control‐treated cells (Fig. 6B). These results suggest that CD63 is required for the formation of a mature LHBs containing viral envelope, which is important for efficient infection.
FIG. 6.
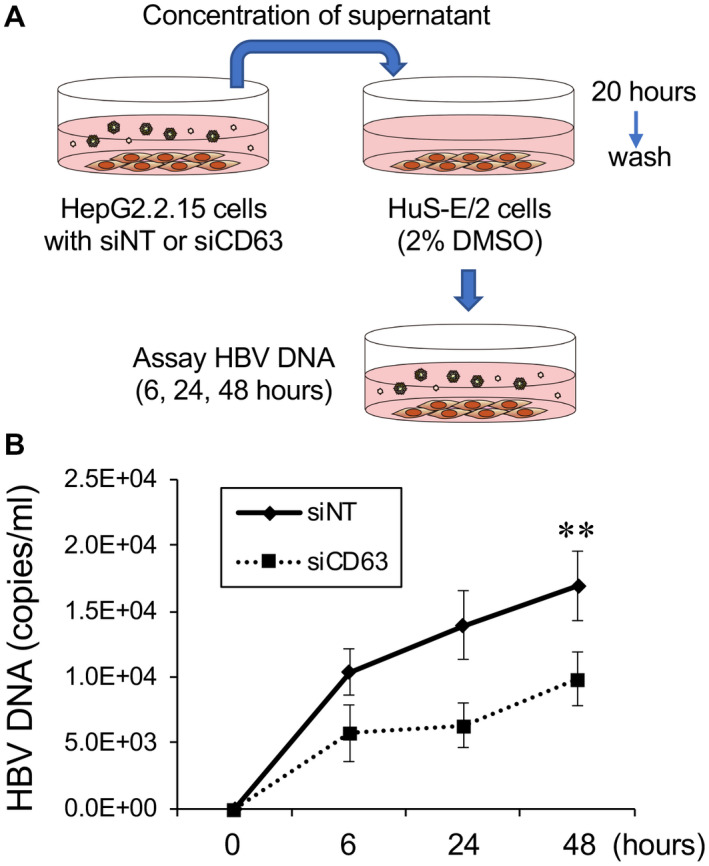
HBV particles isolated from infected cells with CD63 depletion exhibit less infectivity. (A) Illustration depicting the HBV infectivity assay, as described in the “Materials and Methods” section. Briefly, HepG2.2.15 cells were transfected with NT control or CD63 siRNA, and culture supernatants were collected from these cells, concentrated, and used to infect HuS‐E/2 cells for 20 hours. After infection and washing, the HBV‐DNA titer of culture supernatant collected 6, 24, and 48 hours after the infection was measured with RT‐PCR. (B) Results of RT‐PCR for HBV‐DNA quantification from three independent experiments. The HBV‐DNA level in the supernatant of HuS‐E/2 cells that were infected with HBV from CD63‐depleted cells was significantly lower than control, suggesting that HBV particles from CD63‐depleted cells were markedly less infective. **P < 0.01.
Discussion
In this study, we report new findings with regard to the host cell components that are important for appropriate packaging, trafficking, and shedding of nascent HBVs. Specifically, a significant role for the tetraspanin, endosomal coat protein CD63, was observed in this process. HBV protein components were found within CD63 vesicles in infected HepG2.2.15 cells that secrete “lawns” of CD63‐positive vesicles of a remarkably uniform size (Fig. 1). Importantly, reducing CD63 protein levels in these cells, via siRNA treatment, results in a substantial intracellular accumulation (5‐8 fold) of select viral proteins such as LHBs, but not HBc (Fig. 2) or the viral genome (Fig. 3). The transcription of mRNA for the envelope proteins was regulated by CD63 (Fig. 4). Interestingly, CD63 depletion results in an increase of TSG101 levels, a key member of the ESCRT complex, on late endosomes (Fig. 5A,B). This increase may compensate for the loss of the CD63 protein, as these cells secrete similar numbers of small EVs with a preserved HBV genome copy number (Fig. 5C,D), but, importantly, are less infective (Fig. 6). Based on these findings, CD63 could have two potential roles in the formation of HBV particles: first, inclusion of LHBs in the viral envelope; and second, translational regulation of the 2.4/2.1 kb mRNA. Figure 7 shows a model depicting the proposed roles of CD63 in the production of HBV particles.
FIG. 7.
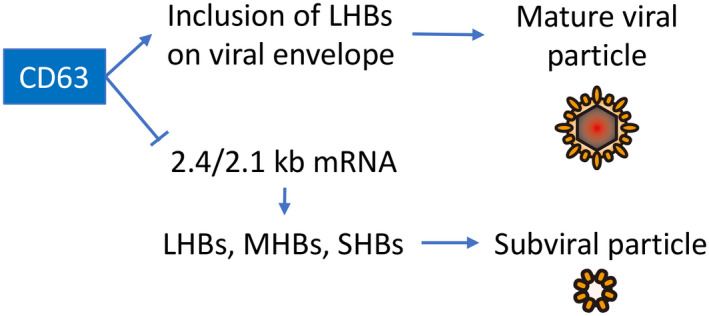
Model depicting the roles of CD63 in the production of mature HBV particles and subviral particles. CD63 might enhance the incorporation of LHBs to the viral envelope, and simultaneously, it might suppress the transcription of 2.4/2.1 kb mRNA, leading to reduction of envelope proteins LHBs, MHBs, and SHBs. Overall, CD63 promotes the efficient production and release of infectious HBV particles, and its suppression induces secretion of subviral particles and viral particles that are less infective.
CD63 belongs to the tetraspanin family and is mostly present in MVBs, including ILVs, lysosomes, and exosomes.( 28 ) Several studies have reported that CD63 is endocytosed through AP‐2 and clathrin‐coated pits from the cell surface,( 36 ) but may also be internalized through caveolae.( 28 ) Its prominent localization appears to be on late endocytic compartments in which the adaptor protein AP‐3 plays a role in its trafficking.( 37 , 38 ) Although little is known about its precise function, CD63 has been shown to play an important role in the trafficking of cargo to melanosomes that are considered a lysosome‐related organelle.( 39 ) Our findings are supportive of this study, as CD63 depletion leads to the decrease of LHBs in HBV particles that were collected by the EV ultracentrifugation assay (Fig. 5). These results suggest that CD63 might mediate the formation of the mature HBV envelope. Interestingly, a recent report regarding Epstein‐Barr virus (EBV) showed that the exosomal packaging of the EBV latent membrane protein 1 is regulated by CD63.( 40 ) A similar mechanism could be used for the incorporation of LHBs into the exosomal membrane.
It has been suggested that MVBs participate in HBV maturation and are released through a pathway that resembles exosome secretion.( 15 , 41 ) A previous study from our laboratory showed a colocalization between the HBV antigen and the MVB markers HRS (hepatorenal syndrome) and CD63 by confocal microscopy, with numerous HBV‐like particles residing within MVB compartments as viewed by transmission electron microscopy.( 15 ) It is predicted that the ILVs that reside within MVBs contain HBV particles and are released during viral secretion. Interestingly, it has been suggested that ILV formation may be supported by the CD63 protein.( 42 ) Therefore, we were somewhat surprised to find no significant difference in the levels of extracellular HBV DNA, even after IP with anti‐HBs antibody, between control and CD63 siRNA‐treated HepG2.2.15 cells (Fig. 3). This suggests that there are compensatory mechanisms such as the increased expression of the TSG101 protein (Fig. 5), or that a parallel pathway is used.
Previous studies have shown that the supernatants of HBV‐producing cell lines contain HBV‐enveloped virions, enveloped subviral particles, and nonenveloped naked nucleocapsid particles. Moreover, these are secreted from the cells by distinct pathways.( 11 , 43 ) HBV‐enveloped virions appear to use the conventional ESCRT machinery for budding,( 10 , 11 , 44 ) whereas the pathway of HBV‐naked nucleocapsid release is unconventional and requires select ESCRT components, such as ALIX and Bro1, without requiring the complete ESCRT machinery.( 45 ) In the present study, it was revealed that CD63 depletion reduced mature enveloped HBV and increased immature enveloped nucleocapsids, which may be secreted through unconventional pathways, although the transcription of envelope proteins was enhanced. We speculate that this may explain why the total levels of extracellular HBV DNA do not show significant differences between cells with and without CD63 depletion. Despite an alternative pathway, virion release appears to be defective, as reflected by reduced LHBs protein and attenuated infectivity. It is still unclear how CD63 might regulate the transcription of 2.4/2.1 kb mRNA, and the detailed mechanisms should be clarified in the future.
How might assembled virions in CD63‐deficient cells prove less infective? It is well established that HBV particles with a DNA‐containing nucleocapsid are enclosed within an envelope composed of LHBs, MHBs, and SHBs proteins.( 5 ) The LHBs protein has been reported to provide some functions toward binding of the nucleocapsid during envelopment as well as receptor binding during cell entry.( 46 ) The receptor for HBV infection has recently been identified as the sodium taurocholate co‐transporting polypeptide (NTCP),( 47 ) which requires epidermal growth factor receptor for the internalization of HBV.( 48 ) The preS1 sequence (residues 2‐48) of LHBs interacts with the NTCP at residues 9‐15, which provide a highly conserved motif that is crucial for their binding.( 49 ) Furthermore, myristoylation within the N‐terminus of LHBs is presumed to increase receptor binding.( 50 ) Thus, HBV particles lacking the LHBs protein coat would be expected to be markedly less interactive with the hepatocyte surface, and hence less infective. Current studies are focused on defining the hepatocellular compartment in which the LHBs protein resides when CD63 levels are reduced, and the alternative, parallel pathways that are used to drive release of the compromised HBV in these altered cells. Most importantly, these findings suggest that manipulation of CD63 protein expression or function could provide an attractive approach for antiviral therapy.
Supporting information
Fig S1‐S4
Acknowledgment
The authors thank Ms. Mary Schulz for reading the manuscript, Prof. Yasuhito Tanaka at Kumamoto University for the technical advice, and Biomedical Research Unit of Tohoku University Hospital for the technical support.
Supported by Uehara Memorial Foundation, Eisenberg Foundation, National Institute of Diabetes and Digestive and Kidney Diseases (DK44650 and P30DK084567), Japan Agency for Medical Research and Development (JP18fk0310101h0002), and Japan Society for the Promotion of Science (19K08385).
Potential conflict of interest: Nothing to report.
Contributor Information
Jun Inoue, Email: jinoue-drgn@umin.net.
Mark A. McNiven, Email: mcniven.mark@mayo.edu.
References
Author names in bold designate shared co‐first authorship.
- 1. Polaris Observatory Collaborators . Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: a modelling study. Lancet Gastroenterol Hepatol 2018;3:383‐403. [DOI] [PubMed] [Google Scholar]
- 2. Terrault NA, Lok ASF, McMahon BJ, Chang K‐M, Hwang JP, Jonas MM, et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology 2018;67:1560‐1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Inoue J, Ninomiya M, Shimosegawa T, McNiven MA. Cellular membrane trafficking machineries used by the hepatitis viruses. Hepatology 2018;68:751‐762. [DOI] [PubMed] [Google Scholar]
- 4. Heermann KH, Goldmann U, Schwartz W, Seyffarth T, Baumgarten H, Gerlich WH. Large surface proteins of hepatitis B virus containing the pre‐s sequence. J Virol 1984;52:396‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bruss V. A short linear sequence in the pre‐S domain of the large hepatitis B virus envelope protein required for virion formation. J Virol 1997;71:9350‐9357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Prange R, Streeck RE. Novel transmembrane topology of the hepatitis B virus envelope proteins. EMBO J 1995;14:247‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Urban S, Bartenschlager R, Kubitz R, Zoulim F. Strategies to inhibit entry of HBV and HDV into hepatocytes. Gastroenterology 2014;147:48‐64. [DOI] [PubMed] [Google Scholar]
- 8. Gripon P, Leseyec J, Rumin S, Guguenguillouzo C. Myristylation of the hepatitis‐B virus large surface protein is essential for viral infectivity. Virology 1995;213:292‐299. [DOI] [PubMed] [Google Scholar]
- 9. Schulze A, Schieck A, Ni Y, Mier W, Urban S. Fine mapping of pre‐S sequence requirements for hepatitis B virus large envelope protein‐mediated receptor interaction. J Virol 2010;84:1989‐2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lambert C, Doring T, Prange R. Hepatitis B virus maturation is sensitive to functional inhibition of ESCRT‐III, Vps4, and gamma 2‐adaptin. J Virol 2007;81:9050‐9060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Watanabe T, Sorensen EM, Naito A, Schott M, Kim S, Ahlquist P. Involvement of host cellular multivesicular body functions in hepatitis B virus budding. Proc Natl Acad Sci U S A 2007;104:10205‐10210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ariumi Y, Kuroki M, Maki M, Ikeda M, Dansako H, Wakita T, et al. The ESCRT system is required for hepatitis C virus production. PLoS One 2011;6:e14517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alenquer M, Amorim MJ. Exosome biogenesis, regulation, and function in viral infection. Viruses 2015;7:5066‐5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ramakrishnaiah V, Thumann C, Fofana I, Habersetzer F, Pan Q, de Ruiter PE, et al. Exosome‐mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc Natl Acad Sci U S A 2013;110:13109‐13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Inoue J, Krueger EW, Chen J, Cao H, Ninomiya M, McNiven MA. HBV secretion is regulated through the activation of endocytic and autophagic compartments mediated by Rab7 stimulation. J Cell Sci 2015;128:1696‐1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schroeder B, Srivatsan S, Shaw A, Billadeau D, McNiven MA. CIN85 phosphorylation is essential for EGFR ubiquitination and sorting into multivesicular bodies. Mol Biol Cell 2012;23:3602‐3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Watashi K, Liang G, Iwamoto M, Marusawa H, Uchida N, Daito T, et al. Interleukin‐1 and tumor necrosis factor‐alpha trigger restriction of hepatitis B virus infection via a cytidine deaminase activation‐induced cytidine deaminase (AID). J Biol Chem 2013;288:31715‐31727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Abdulkarim AS, Cao H, Huang B, McNiven MA. The large GTPase dynamin is required for hepatitis B virus protein secretion from hepatocytes. J Hepatol 2003;38:76‐83. [DOI] [PubMed] [Google Scholar]
- 19. Aly HH, Watashi K, Hijikata M, Kaneko H, Takada Y, Egawa H, et al. Serum‐derived hepatitis C virus infectivity in interferon regulatory factor‐7‐suppressed human primary hepatocytes. J Hepatol 2007;46:26‐36. [DOI] [PubMed] [Google Scholar]
- 20. Huang HC, Chen CC, Chang WC, Tao MH, Huang C. Entry of hepatitis B virus into immortalized human primary hepatocytes by clathrin‐dependent endocytosis. J Virol 2012;86:9443‐9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Inoue J, Ninomiya M, Umetsu T, Nakamura T, Kogure T, Kakazu E, et al. Small interfering RNA screening for the small GTPase Rab proteins identifies Rab5B as a major regulator of hepatitis B virus production. J Virol 2019;93:e00621‐e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cao H, Garcia F, McNiven MA. Differential distribution of dynamin isoforms in mammalian cells. Mol Biol Cell 1998;9:2595‐2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van der Pol E, Coumans FAW, Grootemaat AE, Gardiner C, Sargent IL, Harrison P, et al. Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nanoparticle tracking analysis, and resistive pulse sensing. J Thromb Haemost 2014;12:1182‐1192. [DOI] [PubMed] [Google Scholar]
- 24. Sir D, Tian Y, Chen WL, Ann DK, Yen TS, Ou JH. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc Natl Acad Sci U S A 2010;107:4383‐4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li J, Liu Y, Wang Z, Liu K, Wang Y, Liu J, et al. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J Virol 2011;85:6319‐6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tian Y, Sir D, Kuo CF, Ann DK, Ou JH. Autophagy required for hepatitis B virus replication in transgenic mice. J Virol 2011;85:13453‐13456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sells MA, Chen ML, Acs G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci U S A 1987;84:1005‐1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pols MS, Klumperman J. Trafficking and function of the tetraspanin CD63. Exp Cell Res 2009;315:1584‐1592. [DOI] [PubMed] [Google Scholar]
- 29. Lobb RJ, Becker M, Wen Wen S, Wong CSF, Wiegmans AP, Leimgruber A, et al. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J Extracell Vesicles 2015;4:27031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol 2006;Chapter 3:Unit 3.22. [DOI] [PubMed] [Google Scholar]
- 31. Pisitkun T, Shen RF, Knepper MA. Identification and proteomic profiling of exosomes in human urine. Proc Natl Acad Sci U S A 2004;101:13368‐13373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Glebe D, Urban S. Viral and cellular determinants involved in hepadnaviral entry. World J Gastroenterol 2007;13:22‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Le Seyec J, Chouteau P, Cannie I, Guguen‐Guillouzo C, Gripon P. Infection process of the hepatitis B virus depends on the presence of a defined sequence in the pre‐S1 domain. J Virol 1999;73:2052‐2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bruss V, Hagelsten J, Gerhardt E, Galle PR. Myristylation of the large surface protein is required for hepatitis B virus in vitro infectivity. Virology 1996;218:396‐399. [DOI] [PubMed] [Google Scholar]
- 35. Gripon P, Cannie I, Urban S. Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface protein. J Virol 2005;79:1613‐1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem 2003;72:395‐447. [DOI] [PubMed] [Google Scholar]
- 37. Peden AA, Oorschot V, Hesser BA, Austin CD, Scheller RH, Klumperman J. Localization of the AP‐3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J Cell Biol 2004;164:1065‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rous BA, Reaves BJ, Ihrke G, Briggs JAG, Gray SR, Stephens DJ, et al. Role of adaptor complex AP‐3 in targeting wild‐type and mutated CD63 to lysosomes. Mol Biol Cell 2002;13:1071‐1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van Niel G, Charrin S, Simoes S, Romao M, Rochin L, Saftig P, et al. The tetraspanin CD63 regulates ESCRT‐independent and ‐dependent endosomal sorting during melanogenesis. Dev Cell 2011;21:708‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hurwitz SN, Nkosi D, Conlon MM, York SB, Liu X, Tremblay DC, et al. CD63 regulates epstein‐barr virus LMP1 exosomal packaging, enhancement of vesicle production, and noncanonical NF‐kappaB signaling. J Virol 2017;91:e02251‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hanson PI, Cashikar A. Multivesicular body morphogenesis. Annu Rev Cell Dev Biol 2012;28:337‐362. [DOI] [PubMed] [Google Scholar]
- 42. Edgar JR, Eden ER, Futter CE. Hrs‐ and CD63‐dependent competing mechanisms make different sized endosomal intraluminal vesicles. Traffic 2014;15:197‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wittkop L, Schwarz A, Cassany A, Grün‐Bernhard S, Delaleau M, Rabe B, et al. Inhibition of protein kinase C phosphorylation of hepatitis B virus capsids inhibits virion formation and causes intracellular capsid accumulation. Cell Microbiol 2010;12:962‐975. [DOI] [PubMed] [Google Scholar]
- 44. Kian Chua P, Lin MH, Shih C. Potent inhibition of human hepatitis B virus replication by a host factor Vps4. Virology 2006;354:1‐6. [DOI] [PubMed] [Google Scholar]
- 45. Bardens A, Doring T, Stieler J, Prange R. Alix regulates egress of hepatitis B virus naked capsid particles in an ESCRT‐independent manner. Cell Microbiol 2011;13:602‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Engelke M, Mills K, Seitz S, Simon P, Gripon P, Schnölzer M, et al. Characterization of a hepatitis B and hepatitis delta virus receptor binding site. Hepatology 2006;43:750‐760. [DOI] [PubMed] [Google Scholar]
- 47. Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012;1:e00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Iwamoto M, Saso W, Sugiyama R, Ishii K, Ohki M, Nagamori S, et al. Epidermal growth factor receptor is a host‐entry cofactor triggering hepatitis B virus internalization. Proc Natl Acad Sci U S A 2019;116:8487‐8492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Glebe D, Urban S, Knoop EV, Çaǧ N, Krass P, Grün S, et al. Mapping of the hepatitis B virus attachment site by use of infection‐inhibiting preS1 lipopeptides and tupaia hepatocytes. Gastroenterology 2005;129:234‐245. [DOI] [PubMed] [Google Scholar]
- 50. Meier A, Mehrle S, Weiss TS, Mier W, Urban S. Myristoylated PreS1‐domain of the hepatitis B virus L‐protein mediates specific binding to differentiated hepatocytes. Hepatology 2013;58:31‐42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S4