

Abstract
Cellular senescence is a biological process triggered in response to time-accumulated DNA damage, which prioritizes cell survival over cell function. Particularly, senescent T lymphocytes can be generated prematurely during chronic inflammatory diseases regardless of chronological aging. These senescent T lymphocytes are characterized by the loss of CD28 expression, a co-stimulatory receptor that mediates antigen presentation and effective T-cell activation. An increased number of premature senescent CD4+CD28- T lymphocytes has been frequently observed in osteolytic diseases, including rheumatoid arthritis, juvenile idiopathic arthritis, ankylosing spondylitis, osteopenia, osteoporosis, and osteomyelitis. Indeed, CD4+CD28- T lymphocytes produce higher levels of osteoclastogenic molecular mediators directly related to pathologic bone loss, such as tumor necrosis factor (TNF)-α, interleukin (IL)-17A, and receptor-activator of nuclear factor κB ligand (RANKL), as compared with regular CD4+CD28+ T lymphocytes. In addition, premature senescent CD8+CD28- T lymphocytes have been negatively associated with bone healing and regeneration by inhibiting osteoblast differentiation and mesenchymal stromal cell survival. Therefore, accumulated evidence supports the role of senescent T lymphocytes in osteoimmunology. Moreover, premature senescence of T-cells seems to be associated with the functional imbalance between the osteolytic T-helper type-17 (Th17) and bone protective T regulatory (Treg) lymphocytes, as well as the phenotypic instability of Treg lymphocytes responsible for its trans-differentiation into RANKL-producing exFoxp3Th17 cells, a key cellular phenomenon directly related to bone loss. Herein, we present a framework for the understanding of the pathogenic characteristics of T lymphocytes with a premature senescent phenotype; and particularly, we revise and discuss their role in the osteoimmunology of osteolytic diseases.
Keywords: senescence, T-lymphocytes, CD28, RANKL, osteoimmunology, bone loss
Cellular senescence is defined as a biological process characterized by the arrest of the cell cycle, frequently accompanied by changes in cell phenotype, in response to accumulated DNA damage [1]. Indeed, when cumulative damage to the DNA overwhelms cellular repair mechanisms, cells activate either the program of cell death (apoptosis) or senescence [2]. From an immunological point of view, these processes greatly differ since apoptosis is usually followed by inflammation resolution, while cellular senescence mainly favors inflammation and contributes mostly to immunopathogenic reactions, by prioritizing cell survival instead of cell function [2].
Senescent cells display molecular and morphological changes which translate into heterogeneous functional variations, and these are dependent on the type of affected cell and the type, intensity, and duration of the stimulus that provoked the DNA damage [2, 3]. Even though multiple pathways can lead to cellular senescence and result in equally diverse senescent phenotypes, these pathways seem to converge into a common cell phenotype transversal to the different types of senescent cells (Fig. 1) [2, 3]. After the damage to the genetic material occurs, cells activate the DNA damage response (DDR), which can be detected by the identification of specific markers, such as phosphorylated histone 2AX (γ-H2Ax) [3-5]. In turn, γ-H2Ax positively regulates genes linked to cell cycle arrest, such as p21 and p53, in order to prevent the replication of the defective cell [3-5]. Apart from that, senescent cells accumulate lysosomal content, evidenced by the increase in the activity of senescence-associated-β-galactosidase (SA-βgal), which has been related to lysosomal alterations, autophagy dysfunction, and accumulation of damaged mitochondria [6, 7].
Figure 1.

Cellular senescence as an alternative pathway to apoptosis after accumulated DNA damage. When accumulated DNA damage occurs, the affected cells can repair the DNA damage to continue their programmed functions or, when their repair mechanisms cannot restore the integrity of damaged DNA, cells can initiate either the apoptosis program or senescence program. In fact, cell senescence is an alternative fate to apoptosis that prioritizes cell survival instead of cell function. The programming of the common phenotype among senescent cells is initiated by the DNA damage response (DDR) that leads to the arrest of the cell cycle, in order to avoid the replication of the defective cell. Then, cells present an increase in the activity of senescence-associated-β-galactosidase (SA-βgal), which is linked to lysosomal failure and potential autophagy alterations; however, this link remains unclear. In turn, the defective autophagy observed in senescent cells can lead to the accumulation of dysfunctional mitochondria and consequently to the increase of the production of reactive oxygen species (ROS). Besides, these cells can acquire a senescence-associated secretory phenotype (SASP), producing increased levels of cytokines, chemokines, growth factors, and proteases (Created with BioRender.com).
Functionally, senescent cells acquire pro-inflammatory properties, characterized by increased production of reactive oxygen species (ROS), derived from dysfunctional mitochondria, and the establishment of a senescence-associated secretory phenotype (SASP), responsible for the increased production of cytokines, chemokines, growth factors, and proteases [4, 8]. The SASP-related molecular pattern produced by senescent cells depends on the type of affected cell and the senescence-inducing factor [9]. In addition, it contributes to the autocrine maintenance of the senescent phenotype and the induction of senescence in neighboring cells, a process called paracrine senescence or bystander effect [10, 11]. Therefore, SASP and ROS maintain a vicious circle of inflamm-aging, where chronic inflammation leads to senescence, and senescence contributes even more to unceasing inflammation [12].
Notably, there are no actual unique markers for the identification of cellular senescence [13]; however, the absence of CD28 expression has been proposed as a characteristic that could allow the identification of senescent T lymphocytes [14].
T-cell senescence: CD28- T lymphocytes
During the antigenic presentation, T lymphocytes are activated when their T-cell receptor (TCR) recognizes the antigen/MHC II complex and their CD28 receptor binds to the costimulatory molecules CD80 or CD86, expressed by antigen-presenting cells [15]. The loss of CD28 expression is considered the most consistent biological indicator of cellular senescence in T lymphocytes and the critical aging marker in the human immune system [14]. CD28- T lymphocytes, also called CD28null T lymphocytes, present common characteristics of the senescent cell phenotype, such as the upregulation of γ-H2Ax and p53, and a robust increment in the SA-βgal activity [16-18]. CD28- T lymphocytes are considered dysfunctional, oligoclonal, terminally differentiated, and with limited proliferative capacity, although with an increased capacity to produce pro-inflammatory cytokines [14, 19] and resistance to the suppressive function of T regulatory (Treg) lymphocytes and the effect of steroids [20, 21]. In addition, CD28- T lymphocytes are highly resistant to apoptosis-triggering signals and accumulate in high quantities in the elderly [22, 23].
It is noteworthy that senescent T lymphocytes can also be generated prematurely during chronic diseases regardless of chronological aging [19], since chronic inflammation can lead to immunosenescence, particularly in T lymphocytes [24]. In this sense, T lymphocytes seem to be more susceptible to cellular senescence than other immune cells for several reasons: (1) they are dependent on thymic integrity for cell renewal, (2) they undergo great proliferative stress due to massive clonal expansion during their activation, (3) they have long periods of life as carriers of immune memory, and (4) they are subjected to high oxidative and metabolic stress, which leads to accumulation of unresolved DNA damage [25].
Various pathways can lead to premature senescence of T cells. During repetitive T lymphocyte stimulation involving clonal expansion, the CD28 expression may be irreversibly lost by transcriptional silencing [26]. In addition, the loss of CD28 expression in T lymphocytes is accompanied by a decreased telomerase activity, which can lead to telomere shortening, the reason why T-cell senescence is frequently attributed to a process of replicative senescence [27, 28]. Indeed, somatic cells have a finite number of cell divisions known as the Hayflick limit, which restricts their proliferative capacity to their telomere length, shortened in every replication [5, 29, 30]. However, cells subjected to continuous stress show similar phenotypes to cells with replicative senescence in a matter of hours to days, independent of telomere length, a phenomenon called stress-induced premature senescence or accelerated senescence [3, 5]. Oxidative stress and pro-inflammatory mediators, such as the SASP-related cytokines, can act as cell stressors leading to premature paracrine cellular senescence [31-33]. In particular, ROS, interferon (IFN)-α, tumor necrosis factor (TNF)-α, and prostaglandin (PG)E2 can trigger the loss of CD28 expression and induce the formation of CD28- T lymphocytes with senescent characteristics [34-38].
While CD28 is expressed in most murine T lymphocytes, CD28 is expressed in 90% of CD4+ T lymphocytes and only in 50% of CD8+ T lymphocytes in humans; thus, the loss of CD28 expression seems to be a good senescence marker for CD4+ T lymphocytes but less specific for CD8+ T lymphocytes [15]. In this context, the expression of CD27, another costimulatory receptor constitutively expressed in T lymphocytes, has also been used to differentially identify senescent CD4+ and CD8+ T lymphocytes. Indeed, senescent CD8+ T lymphocytes first lose the CD28 expression and, in a later stage, they lose the CD27 expression, while CD4+ T lymphocytes lose first the CD27 expression and then the CD28 expression [26]. Both CD4+CD27-CD28- and CD8+CD27-CD28- T lymphocytes downregulate the expression of molecules specifically involved in the TCR signaling pathway, such as Zap70 and Lck [18, 39]. Interestingly, downregulation of these signalosome components does not imply a state of anergy or cellular exhaustion. In fact, senescent T lymphocytes can retrieve the expression of CD45RA and express other signaling molecules that contribute to the maintenance of cell functionality, such as CD11b, CD57, and killer cell lectin-like receptor subfamily G member 1 (KLRG1) [40-42]. Therefore, all these markers can be used to identify the different subsets of senescent T lymphocytes (Fig. 2). Recent reports reveal that, in CD8+CD27-CD28- T lymphocytes, sestrins induce natural killer functions [39], while in CD4+CD27-CD28- T lymphocytes, sestrins induce phosphorylation of distinct mitogen-activated protein kinases (MAPKs) [18]. Thus, MAPKs activation mediates different aspects of CD4+ T-cell senescence; for instance, the activity of p38-MAPK has been associated with low telomerase activity, ERK with the activation of DDR and upregulation of γ-H2Ax, and JNK with the signalosome inhibition [18].
Figure 2.

Hallmarks of premature senescent T lymphocytes. Chronic inflammation and persistent antigenic stimulation can induce premature senescence of T cells. Premature senescent T lymphocytes express traditional senescent cell characteristics, such as increased DNA damage response (DDR), increased cell cycle arrest protein levels, such as p53, and senescence-associated-β-galactosidase (SA-βgal) activity. In addition, senescent T lymphocytes express specific cell markers, such as the loss of co-stimulatory receptors CD28 and CD27, as well as the loss of the signalosome molecules Zap70 and Lck, involved in the T-cell receptor (TCR) signaling. Furthermore, premature senescent T lymphocytes have a diminished proliferative capacity, low telomerase activity, and altered apoptosis. However, they maintain potent effector functions, including the expression of innate immunity receptors, such as CD11b and CD57, and the co-inhibitory killer cell lectin-like receptor G1 (KLRG1). Recently, it has been proposed that sestrin proteins could be involved in the development of the T-cell senescence phenotype (Created with BioRender.com).
Osteoimmunology: Role of T lymphocytes
Osteoimmunology studies the relation and cross-communication between the immune and bone systems. Although osteoimmunology initially focused on the influence of the immune response on osteoclast activity, the key cell responsible for bone resorption, nowadays it also involves its effects over osteoblast activity and bone formation [43, 44]. Hence, the immune response is closely related to the maintenance of bone homeostasis, which ultimately depends on the molecularly regulated cellular coupling between osteoclasts and osteoblasts (Fig. 3) [43, 45].
Figure 3.
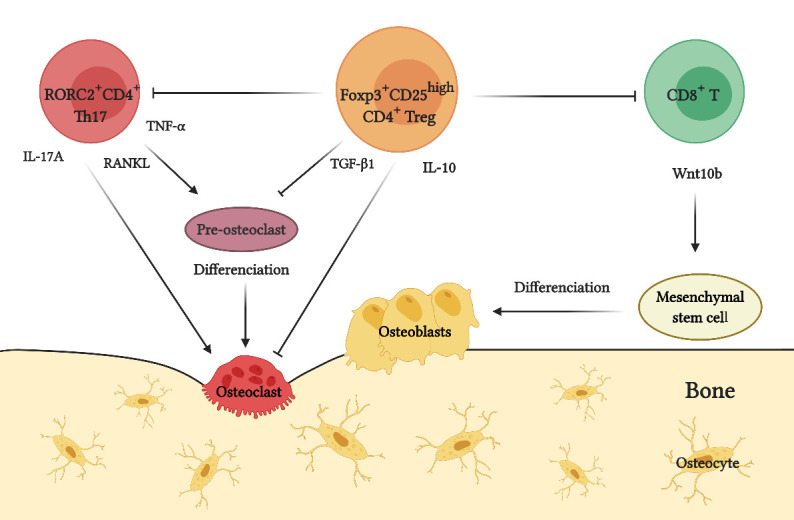
Role of T lymphocytes during osteoimmunology. Under physiological conditions, the balance between the osteolytic activity of T-helper type-17 (Th17) lymphocytes and bone protective activity of T regulatory (Treg) lymphocytes controls the maintenance of bone tissue homeostasis. Th17 lymphocytes express the transcription factor retinoid acid receptor-related orphan nuclear receptor C2 (RORC2) and induce osteoclastogenesis and activation of mature osteoclasts by producing tumor necrosis factor (TNF)-α, interleukin (IL)-17A, and receptor-activator of nuclear factor κB ligand (RANKL). Treg lymphocytes express the transcription factor forkhead box P3 (Foxp3), overexpress the cell surface marker CD25, and inhibit osteoclastogenesis and osteoclast activity mainly by producing transforming growth factor (TGF)-β1 and IL-10. It is noteworthy that Foxp3+CD25high Treg lymphocytes can directly inhibit the differentiation, proliferation, and function of RORC2+ Th17 lymphocytes. Moreover, Foxp3+CD25high Treg lymphocytes can promote the production of Wnt10b by CD8+ T lymphocytes, which induces the differentiation of osteoblasts precursors through the Wnt-β-catenin-signaling pathway and consequently promotes bone formation (Created with BioRender.com).
Osteoimmunological control of osteoclast function comprises the activity of a triad of proteins belonging to the superfamily of TNF ligands and receptors, which are the receptor-activator of nuclear factor κB ligand (RANKL), its functional receptor (RANK), and its soluble decoy receptor osteoprotegerin (OPG) [46]. In general terms, RANKL induces osteoclast differentiation and activation by interacting with its specific receptor RANK, expressed in osteoclast precursors and mature osteoclasts; otherwise, OPG inhibits the RANKL/RANK interaction and arrests osteoclastogenesis and bone loss [46]. The RANKL/RANK signaling induces the activation of the transcription factors termed nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) and nuclear factor of activated T cells-cytoplasmic 1 (NFATc1), considered as the master regulators during osteoclastogenesis [43]. Activation of NFκB and NFATc1 subsequently induces the expression of the osteoclast-characteristic genes implied in the process of bone resorption, including dendritic cell-specific transmembrane protein (DC-STAMP), cathepsin-K, and tartrate-resistant acid phosphatase (TRAP) [43]. Although infrequently, RANKL-independent osteoclast differentiation mechanisms have also been described, such as those dependent on TNF-α or interleukin (IL)-17A signaling. In experimental conditions, the combination of TNF-α and macrophage colony-stimulating factor (M-CSF) induces functional TRAP+ osteoclasts, and this induction can be completely blocked by using neutralizing antibodies against the TNF-α receptor instead of OPG or RANK neutralizing antibodies [47, 48]. Similarly, IL-17A combined with M-CSF triggers the formation of TRAP+ cathepsin-K+ DC-STAMP+ osteoclasts in the absence of exogenous RANKL [49, 50].
At a cellular level, the osteoimmunological regulation of osteoclasts development and activity is mediated by T lymphocytes, particularly T-helper type-17 (Th17) and T regulatory (Treg) lymphocytes, which have opposite roles during immune surveillance, autoimmunity, and inflammation [46, 51]. Th17 lymphocytes express the transcription factor retinoid acid receptor-related orphan nuclear receptor C2 (RORC2), the master-switch gene that determines their differentiation and pro-inflammatory functions [52, 53]. On the other hand, Treg lymphocytes express the transcription factor forkhead box P3 (Foxp3), the master-switch gene that determines their differentiation and regulatory functions, by producing anti-inflammatory cytokines, such as transforming growth factor (TGF)-β1 and IL-10 [51, 54]. Th17 cell pro-inflammatory role has been implicated in many chronic inflammatory and osteolytic diseases, while Treg cell anti-inflammatory role has been associated with the maintenance of immune tolerance and regulation of effector T lymphocyte activity, including Th17 lymphocytes [46, 51]. Under physiological conditions, the balance between the activity of Th17 and Treg lymphocytes allows the dynamic regulation of bone homeostasis. However, chronic inflammation induces a Th17/Treg imbalance, characterized by the increased activity of IL-6, IL-17A, and RANKL-producing Th17 lymphocytes and phenotypically unstable Treg lymphocytes, which lose their regulatory capacity [51, 55]. In addition, this inflammatory milieu further favors the production of pro-inflammatory cytokines, including IFN-γ, TNF-α, and IL-1β, which can induce the expression of RANKL in osteoblasts and fibroblasts with osteoclastogenic capacity, thus generating a complex network of RANKL-producing cells that contribute even more to pathologic bone loss [46].
Otherwise, osteoimmunological control of osteoblast function and bone formation depends on the Wnt signaling pathway. In osteoblasts precursors such as mesenchymal stromal cells, the binding of Wnt10b to its coreceptor, presumably low-density lipoprotein receptor-related protein 5 (LRP5), leads to β-catenin nuclear translocation, which in turn induces the expression of the transcription factors responsible for osteoblastogenesis and necessary for bone formation, such as runt-related transcription factor 2 (Runx2), distal-less homeobox 5 (Dlx5), and osterix (Osx) [56]. At a cellular level, the osteoimmunology of osteoblast function relies on the interaction between Treg and CD8+ T lymphocytes. Indeed, the increase in the number and activity of TGF-β1-producing Treg lymphocytes drives the assembly of the NFAT1/SMAD3 transcription complex in CD8+ T lymphocytes and Wnt10b production, and consequently, osteoblast differentiation and bone formation [44, 57, 58].
Premature senescence of T lymphocytes and pathological bone loss
Despite having different etiologies, chronic diseases characterized by manifesting pathological bone loss usually share osteoimmunological similarities that affect the target tissues. In some of these diseases, premature senescence of T lymphocytes has been related to increased Th17 lymphocyte activity and RANKL production, which induces osteoclast differentiation and provokes adverse effects on bone structure and function (Fig. 4A). In fact, finding an increased number of prematurely senescent CD4+CD28- T lymphocytes has become relatively common in rheumatoid arthritis, juvenile idiopathic arthritis, ankylosing spondylitis, osteopenia, osteoporosis, and osteomyelitis [59-64]. Apart from that, CD4+CD28- T cells have also been reported in HIV-infected individuals and lupus erythematosus-affected patients, in which their activity could explain the increased risk of developing osteoporosis observed in these diseases [65-68].
Figure 4.

Role of premature senescent T lymphocytes during pathological bone loss. (A) Premature senescent CD4+CD28- T lymphocytes promote osteoclastogenesis and osteolysis by producing increased levels of tumor necrosis factor (TNF)-α, interleukin (IL)-17A, and receptor-activator of nuclear factor κB ligand (RANKL). (B) Premature senescent Foxp3+CD28- Treg lymphocytes show phenotypic instability, downregulated CD25 expression, and decreased immunosuppressive capacity. In consequence, these Treg lymphocytes could lose Foxp3 expression and trans-differentiate into exFoxp3Th17 cells, which express killer cell lectin-like receptor G1 (KLRG1) and acquire osteoclastogenic and bone resorptive capacities by producing TNF-α, IL-17A, and RANKL. (C) Premature senescent CD8+CD28-CD45RA+CD57+ T lymphocytes inhibit osteoblastogenesis and bone formation by producing TNF-α and interferon (INF)-γ. In addition, these senescent CD8+ T lymphocytes downregulate their Wnt10b production and consequently affect the Wnt-β-catenin-dependent osteoblastogenesis and bone formation; however, this mechanism has not been entirely demonstrated yet (Created with BioRender.com).
In the case of rheumatoid arthritis, CD4+CD28- T lymphocytes produced higher levels of RANKL as compared with CD4+CD28+ T lymphocytes, and these senescent CD4+CD28- T lymphocytes showed a greater capacity to induce the differentiation of TRAP+ osteoclasts and bone resorption when compared with non-senescent CD4+CD28+ T lymphocytes [62]. Also, CD4+CD28- T lymphocytes isolated from the synovial fluid of rheumatoid arthritis-affected patients were capable of producing higher levels of IL-17A as compared with isolated CD4+CD28+ T lymphocytes [69, 70]. During osteoporosis, CD4+CD28- T lymphocytes expressed higher levels of TNF-α and intensely induced the activation of TRAP in osteoclasts as compared with CD4+CD28+ T lymphocytes [71]. Besides, in an animal model of osteoporosis induced by ovariectomy, when the immunoprotective effect of the neutralizing antibodies anti-RANKL, anti-TNF-α, or anti-IL-17A was compared, IL-17A blockade led to a more potent reduction in the number of CD4+CD28- T lymphocytes as compared with RANKL or TNF-α blockade, which translated into an improvement in skeletal parameters and demonstrated the key role for IL-17A in the CD4+ T-cell senescence [72]. During osteomyelitis, an increased number of CD4+CD28- T lymphocytes was observed in bone tissue, which was associated with long-term activated T cells, CD11b overexpression, and enhanced cytotoxic ability, all characteristics of T-cell senescence [64, 73]. Although the presence and activity of CD4+CD28- T lymphocytes during periodontitis, the most prevalent osteolytic disease in humans, has not been reported, the continuous inflammatory signals and the persistent antigenic stimulus, provided by microbial dysbiosis and the destruction of periodontal tissues, could trigger the loss of CD28 expression and senescence of CD4+ T lymphocytes [74, 75].
Similar to effector CD4+ and CD8+ T lymphocytes, Treg lymphocytes can also lose the CD28 expression and acquire a senescent phenotype and related functions (Fig. 4B). In rheumatoid arthritis-affected patients, an increase in the number of Foxp3-expressing CD4+CD28- T lymphocytes has been reported, and these senescent Treg cells expressed lower levels of CD25, showed higher SA-βgal activity, and produced higher levels of TNF-α, IFN-γ, and IL-17A, as compared with Foxp3+CD4+CD28+ lymphocytes [16]. Interestingly, no differences in telomere length were observed in these Foxp3+CD4+CD28- lymphocytes, suggesting a state of premature senescence rather than replicative senescence [16].
In rheumatoid arthritis and periodontitis, the presence of unstable Treg lymphocytes that have lost the expression of Foxp3 and their regulatory capacities have been described [76, 77]. These lymphocytes, termed exFoxp3Th17 lymphocytes, acquire the capacity to produce a Th17-pattern of cytokines, such as IL-17A and RANKL, and even show a greater osteoclastogenic capacity than regular Th17 lymphocytes [76, 77]. Interestingly, exFoxp3Th17 lymphocytes, apart from presenting the loss of Foxp3 and downregulated CD25 expression, upregulate the expression of the senescence marker KLRG1 [76, 77]. In fact, KLRG1+ Treg lymphocytes show low proliferative capacity and impaired suppressive functions in vitro and in vivo, frequently losing the Foxp3 expression and reprogramming their activity towards a Th17 effector profile [78]. In this context, exFoxp3Th17 lymphocytes could correspond, at least in part, to senescent Treg cells that have lost their CD28 expression, which also has a direct role in the Foxp3 expression and the maintenance of high levels of CD25 expression [79, 80]. Thus, exFoxp3Th17 lymphocytes could lose the ability to regulate Foxp3 and CD25 through the absence of CD28 co-stimulation, favoring the Th17/Treg imbalance, increasing the production of SASP-related cytokines, and inducing osteoclastogenesis and pathological bone resorption.
Premature senescence of T lymphocytes and bone formation
The senescence of CD8+ T lymphocytes has been negatively associated with bone formation and regeneration (Fig. 4C). Senescent CD8+CD28-CD45RA+ CD57+ T lymphocytes inhibit osteoblast differentiation and human mesenchymal stromal cell survival, leading to a fewer number of osteoblast precursors [81]. In addition, CD8+CD28-CD45RA+CD57+ T lymphocytes have been identified as the main producers of pro-inflammatory cytokines, such as TNF-α and IFN-γ, during bone fracture healing failure [81]. Indeed, TNF-α and IFN-γ seem to be part of the SASP in these senescent T lymphocytes [82]. Furthermore, in aged mice, the number and activity of CD8+CD28+ T lymphocytes decreases and associates with the downregulation of Wnt production [83]; however, it is unknown whether the production of Wnt10b by premature senescent CD8+CD28-CD45RA+CD57+ T lymphocytes is altered, which could also affect the process of bone formation. Additionally, CD8+ T lymphocytes must be in an anergic state to produce Wnt10b [84]; thus, senescent Treg lymphocytes with impaired suppressive functions could be unable to induce CD8+ T lymphocytes to an anergic state, affecting osteoblastogenesis and bone formation. Indeed, animal models with deficient Treg function show reduced proliferation, differentiation, and lifespan of osteoblasts, and consequently, less bone formation mediated by CD8+ T lymphocytes [58].
SASP in senescent T lymphocytes
Phosphorylation of p38-MAPK is increased in both senescent CD8+ and CD4+ T lymphocytes [82, 85]. In senescent CD8+ T lymphocytes, p38-MAPK signaling governs SASP and regulates the production of SASP-related cytokines [82, 86]. In addition, p38-MAPK inhibits autophagy independently of mTOR, by keeping the p38-MAPK interacting protein (p38IP) sequestered, which is necessary to translocate the autophagy-related protein (ATG)-9 to the autophagosome [82]. These changes lead to the accumulation of dysfunctional mitochondria and increased amounts of ROS, which favor the production of SASP-related pro-inflammatory cytokines, such as TNF-α and IFN-γ, contributing to the inhibition of bone formation [81, 82].
In senescent CD4+ T lymphocytes, the constitutive phosphorylation of p38-MAPK is determined by a sestrin-mediated intrasensory signaling pathway dependent on the DNA damage [18, 85, 87]. In these cells, it is possible that p38-MAPK signaling can also govern SASP; however, this phenomenon has not been described yet. In elderly people, defective autophagy, accumulation of dysfunctional mitochondria, and increased ROS production in CD4+ T lymphocytes leads to intracellular STAT3 phosphorylation and Th17-type cytokine production [88]. All these mechanisms are dependent on p38-MAPK phosphorylation, and additionally, SASP-related cytokines produced by CD4+CD28- T lymphocytes, including TNF-α, IL-17A, and RANKL, could also contribute to osteoclastogenesis and bone loss.
Taken together, the p38-MAPK phosphorylation could be a conserved mechanism in senescent CD8+ and CD4+ T lymphocytes that contributes to the explaining of the upregulation of SASP-related pro-inflammatory and pro-osteoclastogenic mediators, including RANKL, triggered by the inhibition of autophagy, accumulation of dysfunctional mitochondria, and increased ROS production (Fig. 5).
Figure 5.
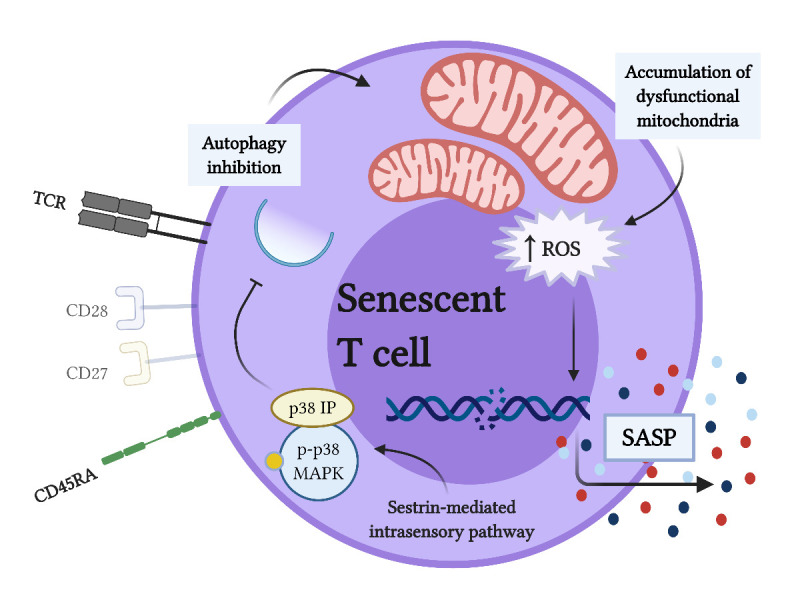
Phosphorylation of p38-MAPK drives SASP-related cytokine production in premature senescent T lymphocytes. Senescent T lymphocytes exhibit phosphorylation of p38-mitogen-activated protein kinase (p38-MAPK) by a sestrin-mediated intrasensory signaling pathway that is maintained over time. Phosphorylation of p38-MAPK (p-p38-MAPK) inhibits autophagy by sequestering the p38-MAPK interacting protein (p38IP), necessary to translocate autophagy-related protein (ATG)-9 to the autophagosome. Inhibition of autophagy results in defective clearance of dysfunctional mitochondria, which accumulate and cause the rise of intracellular reactive oxygen species (ROS) levels that act as inducers of the senescence-associated secretory phenotype (SASP)-related cytokine production (Created with BioRender.com).
Therapeutic strategies aimed at controlling premature senescence of T lymphocytes
Nowadays, different therapeutic strategies have been proposed to specifically target cellular senescence. These emerging senotherapeutic approaches comprise senolytic strategies, which seek to eliminate senescent cells, and senomorphic strategies, which can partially reverse the senescent phenotype, including the establishment of SASP [89]. Although senolytic strategies have been reported to be very beneficial and do not seem to have negative long-term consequences, this approach has not been evaluated in senescent T lymphocytes [89]. On the contrary, different senomorphic strategies have been evaluated in senescent T cells using diverse in vitro and in vivo models, including metformin, sestrin blockade, p38-MAPK inhibitors, and TNF-α1-receptor blockade [18, 37, 39, 82, 88, 90].
In CD4+ T lymphocytes obtained from elderly people, metformin rescued autophagy and mitochondrial function, inhibited the production of ROS, and downregulated the production of Th17-associated cytokines, IL-6, IL-17A, and IL-21 [88]. In addition, blocking sestrins in senescent CD4+ T lymphocytes reduced the damaged DNA accumulation and enhanced the cell proliferative potential and telomerase activity, while its blockade in senescent CD8+ T lymphocytes negatively regulated natural killer functions [18, 39]. Similarly, inhibition of p38-MAPK signaling in senescent CD4+ T lymphocytes significantly improved telomerase activity and cell survival after TCR activation, while its inhibition in senescent CD8+ T lymphocytes increased autophagic activity, restored mitochondrial function, and downregulated the TNF-α expression [82, 90]. Finally, blocking TNF-α1-receptor in senescent CD8+ T lymphocytes increased their proliferative potential, delayed the loss of CD28 expression, and improved telomerase activity [37]. However, to our knowledge, none of these senomorphic strategies have been evaluated in premature senescent T lymphocytes; thus, making the analysis of senotherapeutic approaches for osteolytic diseases, in the context of immunosenescence, novel and attractive.
Conclusion
With age, the inherent cellular mediated immunity flaws related to inflamm-aging increase the vulnerability of the immune system, which can lead to defective bone homeostasis. In addition, senescent T lymphocytes can also be generated prematurely during chronic inflammatory diseases regardless of chronological aging. In the context of osteoimmunology, the activity of T lymphocytes with senescent phenotype contributes to a novel understanding of the cross-communication between the immune response and bone metabolism. During osteolytic diseases, premature senescence of T cells can be crucially involved in the increased activity of RANKL-producing Th17 and exFoxp3Th17 lymphocytes, which cause the Th17/Treg imbalance and bone loss. These new findings uncover the need for the exploration of innovative therapeutic strategies focused on the control of immunosenescence related pathological bone loss. In fact, considering that the loss of CD28 expression and the SASP-related RANKL production are distinctive features of premature senescent T lymphocytes during osteolytic diseases, these could be a focus of therapeutic attention since these changes could potentially be reversible.
Acknowledgments
This study was financially supported by grant FONDECYT 1181780 (RV) from the Agencia Nacional de Investigación y Desarrollo (ANID), Chile. SM-R, EAC, and AS-C were the recipients of Ph.D. scholarships from the Faculty of Dentistry, Universidad de Chile, Chile. LG-O and CR were the recipients of Ph.D. scholarships Fondecyt 21190087 and 21180841, respectively, from ANID.
Footnotes
Conflict of interest
The authors declare that they have no competing interests.
References
- [1].López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013). The hallmarks of aging. Cell, 153:1194-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Soto-Gamez A, Quax WJ, Demaria M (2019). Regulation of survival networks in senescent cells: from mechanisms to interventions. J Mol Biol, 431:2629-2643. [DOI] [PubMed] [Google Scholar]
- [3].Debacq-Chainiaux F, Ben Ameur R, Bauwens E, Dumortier E, Toutfaire M, Toussaint O (2016). Stress-induced (premature) senescence. In: Rattan SI Hayflick L, editors. Cellular ageing and replicative senescence. Switzerland: Springer, 243-262. [Google Scholar]
- [4].Chapman J, Fielder E, Passos JF (2019). Mitochondrial dysfunction and cell senescence: deciphering a complex relationship. FEBS Lett, 593:1566-1579. [DOI] [PubMed] [Google Scholar]
- [5].Fridlyanskaya I, Alekseenko L, Nikolsky N (2015). Senescence as a general cellular response to stress: A mini-review. Exp Gerontol, 72:124-128. [DOI] [PubMed] [Google Scholar]
- [6].Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, et al. (2006). Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell, 5:187-195. [DOI] [PubMed] [Google Scholar]
- [7].Von Zglinicki T, Wan T, Miwa S (2020). Senescence in post-mitotic cells: a driver of aging? Antioxid Redox Signal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Freund A, Patil CK, Campisi J (2011). p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J, 30:1536-1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C, et al. (2020). A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol, 18:e3000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, et al. (2012). A senescent cell bystander effect: Senescence-induced senescence. Aging Cell, 11:345-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. (2013). A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol, 15:978-990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Xia S, Zhang X, Zheng S, Khanabdali R, Kalionis B, Wu J, et al. (2016). An update on inflamm-aging: mechanisms, prevention, and treatment. J Immunol Res, 2016:8426874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dodig S, Čepelak I, Pavić I (2019). Hallmarks of senescence and aging. Biochem Medica, 29:483-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vallejo NA (2005). CD28 extinction in human T cells: Altered functions and the program of T-cell senescence. Immunol Rev, 205:158-169. [DOI] [PubMed] [Google Scholar]
- [15].Acuto O, Michel F (2003). CD28-mediated co-stimulation: A quantitative support for TCR signalling. Nat Rev Immunol, 3:939-951. [DOI] [PubMed] [Google Scholar]
- [16].Fessler J, Raicht A, Husic R, Ficjan A, Schwarz C, Duftner C, et al. (2017). Novel senescent regulatory T-cell subset with impaired suppressive function in rheumatoid arthritis. Front Immunol, 8:300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mondal AM, Horikawa I, Pine SR, Fujita K, Morgan KM, Vera E, et al. (2013). P53 isoforms regulate aging- and tumor-associated replicative senescence in T lymphocytes. J Clin Invest, 123:5247-5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lanna A, Gomes DCO, Muller-Durovic B, McDonnell T, Escors D, Gilroy DW, et al. (2017). A sestrin-dependent Erk-Jnk-p38 MAPK activation complex inhibits immunity during aging. Nat Immunol, 18:354-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vallejo AN, Weyand CM, Goronzy JJ (2004). T-cell senescence: A culprit of immune abnormalities in chronic inflammation and persistent infection. Trends Mol Med, 10:119-124. [DOI] [PubMed] [Google Scholar]
- [20].Thewissen M, Somers V, Hellings N, Stinissen P, Venken K (2007). CD4+CD28null T cells in autoimmune disease: pathogenic features and decreased susceptibility to immunoregulation. Clin Immunol, 179:6514-6523. [DOI] [PubMed] [Google Scholar]
- [21].Hodge G, Hodge S (2016). Steroid resistant CD8+CD28null NKT-like pro-inflammatory cytotoxic cells in chronic obstructive pulmonary disease. Front Immunol, 7:617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Vallejo AN, Schirmer M, Weyand CM, Goronzy JJ (2000). Clonality and longevity of CD4+CD28null T cells are associated with defects in apoptotic pathways. J Immunol, 165:6301-6307. [DOI] [PubMed] [Google Scholar]
- [23].Spaulding C, Guo W, Effros RB (1999). Resistance to apoptosis in human CD8+ T cells that reach replicative senescence after multiple rounds of antigen-specific proliferation. Exp Gerontol, 34:633-644. [DOI] [PubMed] [Google Scholar]
- [24].Johnson ND, Conneely KN (2019). The role of DNA methylation and hydroxymethylation in immunosenescence. Ageing Res Rev, 51:11-23. [DOI] [PubMed] [Google Scholar]
- [25].Weyand CM, Yang Z, Goronzy JJ (2014). T-cell aging in rheumatoid arthritis. Curr Opin Rheumatol, 26:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Moro-García MA, Alonso-Arias R, López-Larrea C (2013). When aging reaches CD4+ T-cells: Phenotypic and functional changes. Front Immunol, 4:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Valenzuela HF, Effros RB (2002). Divergent telomerase and CD28 expression patterns in human CD4 and CD8 T cells following repeated encounters with the same antigenic stimulus. Clin Immunol, 105:117-125. [DOI] [PubMed] [Google Scholar]
- [28].Chou JP Effros RB (2013). T cell replicative senescence in human aging. Curr Pharm Des, 19:1680-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hayflick L, Moorhead OS (1961). The serial cultivation of human diploid cell strains. Exp Cell Res, 37:614-636. [DOI] [PubMed] [Google Scholar]
- [30].Harley CB, Futcher AB, Greider CW (1990). Telomeres shorten during ageing of human fibroblasts. Nature, 256:271-282. [DOI] [PubMed] [Google Scholar]
- [31].Prattichizzo F, Giuliani A, Recchioni R, Bonafè M, Marcheselli F, De Carolis S, et al. (2016). Anti-TNF-α treatment modulates SASP and SASP-related microRNAs in endothelial cells and in circulating angiogenic cells. Oncotarget, 7:11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li P, Gan Y, Xu Y, Song L, Wang L, Ouyang B, et al. (2017). The inflammatory cytokine TNF-α promotes the premature senescence of rat nucleus pulposus cells via the PI3K/Akt signaling pathway. Sci Rep, 7:42938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Polettini J, Richardson LS, Menon R (2018). Oxidative stress induces senescence and sterile inflammation in murine amniotic cavity. Placenta, 63:26-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bryl E, Vallejo AN, Weyand CM, Goronzy JJ (2001). Down-Regulation of CD28 expression by TNF-α. J Immunol, 167:3231-3238. [DOI] [PubMed] [Google Scholar]
- [35].Chou JP, Ramirez CM, Ryba DM, Koduri MP, Effros RB (2014). Prostaglandin E2 promotes features of replicative senescence in chronically activated human CD8+ T cells. PLoS One, 9:e99432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ma S, Ochi H, Cui L, Zhang J, He W (2003). Hydrogen peroxide induced down-regulation of CD28 expression of Jurkat cells is associated with a change of site α-specific nuclear factor binding activity and the activation of caspase-3. Exp Gerontol, 38:1109-1118. [DOI] [PubMed] [Google Scholar]
- [37].Parish ST, Wu JE, Effros RB (2009). Modulation of T lymphocyte replicative senescence via TNF-α inhibition: Role of caspase-3. J Immunol, 182:4237-4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lanna A, Coutavas E, Levati L, Seidel J, Rustin MHA, Henson SM, et al. (2013). IFN-α inhibits telomerase in human CD8+ T cells by both hTERT downregulation and induction of p38 MAPK signaling. J Immunol, 191:3744-3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pereira BI, De Maeyer RPH, Covre LP, Nehar-Belaid D, Lanna A, Ward S, et al. (2020). Sestrins induce natural killer function in senescent-like CD8+ T cells. Nat Immunol, 21:684-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Namekawa T, Snyder MR, Yen J-H, Goehring BE, Leibson PJ, Weyand CM, et al. (2000). Killer cell activating receptors function as costimulatory molecules on CD4+CD28null T cells clonally expanded in rheumatoid arthritis. J Immunol, 165:1138-1145. [DOI] [PubMed] [Google Scholar]
- [41].Scheuring UJ, Sabzevari H, Theofilopoulos AN (2002). Proliferative arrest and cell cycle regulation in CD8+CD28- versus CD8+CD28+ T cells. Hum Immunol, 63:1000-1009. [DOI] [PubMed] [Google Scholar]
- [42].Voehringer D, Koschella M, Pircher H (2002). Lack of proliferative capacity of human effector and memory T cells expressing killer cell lectin-like receptor G1 (KLRG1). Blood, 100:3698-3702. [DOI] [PubMed] [Google Scholar]
- [43].Ponzetti M, Rucci N (2019). Updates on osteoimmunology: What’s new on the cross-talk between bone and immune system. Front Endocrinol (Lausanne), 10:236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tyagi AM, Yu M, Darby TM, Vaccaro C, Li JY, Owens JA, et al. (2018). The microbial metabolite butyrate stimulates bone formation via T regulatory cell-mediated regulation of Wnt10b expression. Immunity, 49:1116-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Weitzmann MN (2017). Bone and the immune system. In: Smith SY Varela A, Samadfam R editors. Bone toxicology. Switzerland: Springer, 363-398. [Google Scholar]
- [46].Alvarez C, Monasterio G, Cavalla F, Córdova LA, Hernández M, Heymann D, et al. (2019). Osteoimmunology of oral and maxillofacial diseases: Translational applications based on biological mechanisms. Front Immunol, 10:1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, et al. (2000). Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med, 191:275-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A (2000). Tumor necrosis factor-α induces differentiation of and bone resorption by osteoclasts. J Biol Chem, 275:4858-4864. [DOI] [PubMed] [Google Scholar]
- [49].Kim H-R, Kim K-W, Kim B-M, Won J-Y, Min H-K, Lee K-A, et al. (2019). Regulation of Th17 cytokine-induced osteoclastogenesis via SKI306X in rheumatoid arthritis. J Clin Med, 8:1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kim KW, Kim HR, Kim BM, Cho M La, Lee SH (2015). Th17 cytokines regulate osteoclastogenesis in rheumatoid arthritis. Am J Pathol, 185:3011-3024 [DOI] [PubMed] [Google Scholar]
- [51].Alvarez C, Rojas C, Rojas L, Cafferata EA, Monasterio G, Vernal R (2018). Regulatory T lymphocytes in periodontitis: a translational view. Mediators Inflamm, 7806912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Díaz-Zúñiga J, Melgar-Rodríguez S, Alvarez C, Monasterio G, Benítez A, Ciuchi P, et al. (2015). T-lymphocyte phenotype and function triggered by Aggregatibacter actinomycetemcomitans is serotype-dependent. J Periodontal Res, 50:824-835. [DOI] [PubMed] [Google Scholar]
- [53].Vernal R, Diaz-Guerra E, Silva A, Sanz M, Garcia-Sanz JA (2014). Distinct human T-lymphocyte responses triggered by Porphyromonas gingivalis capsular serotypes. J Clin Periodontol, 41:19-30. [DOI] [PubMed] [Google Scholar]
- [54].Vernal R, Díaz-Zúñiga J, Melgar-Rodríguez S, Pujol M, Diaz-Guerra E, Silva A, et al. (2014). Activation of RANKL-induced osteoclasts and memory T lymphocytes by Porphyromonas gingivalis is serotype dependant. J Clin Periodontol, 41:451-459. [DOI] [PubMed] [Google Scholar]
- [55].Alvarez C, Suliman S, Almarhoumi R, Vega ME, Rojas C, Monasterio G, et al. (2020). Regulatory T cell phenotype and anti-osteoclastogenic function in experimental periodontitis. Sci Rep, 10:1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bennett CN, Longo KA, Wright WS, Suva LJ, Lane TF, Hankenson KD, et al. (2005). Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci USA, 102:3324-3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Terauchi M, Li JY, Bedi B, Baek KH, Tawfeek H, Galley S, et al. (2009). T lymphocytes amplify the anabolic activity of parathyroid hormone through Wnt10b signaling. Cell Metab, 10:229-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yu M, D’Amelio P, Tyagi AM, Vaccaro C, Li J, Hsu E, et al. (2018). Regulatory T cells are expanded by Teriparatide treatment in humans and mediate intermittent PTH-induced bone anabolism in mice. EMBO Rep, 19:156-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Thewissen M, Somers V, Venken K, Linsen L, Van Paassen P, Geusens P, et al. (2007). Analyses of immunosenescent markers in patients with autoimmune disease. Clin Immunol, 123:209-218. [DOI] [PubMed] [Google Scholar]
- [60].Zhang L, Liang H, Guan H, Liu H (2015). Study of the association between CD28/CTLA-4 expression and disease activity in juvenile idiopathic arthritis. Exp Ther Med, 9:1733-1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Duftner C, Goldberger C, Falkenbach A, Würzner R, Falkensammer B, Pfeiffer KP, et al. (2003). Prevalence, clinical relevance and characterization of circulating cytotoxic CD4+CD28- T cells in ankylosing spondylitis. Arthritis Res Ther, 5:R292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Fessler J, Husic R, Schwetz V, Lerchbaum E, Aberer F, Fasching P, et al. (2018). Senescent T-cells promote bone loss in rheumatoid arthritis. Front Immunol, 9:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Tyagi AM, Srivastava K, Kureel J, Kumar A, Raghuvanshi A, Yadav D, et al. (2012). Premature T cell senescence in Ovx mice is inhibited by repletion of estrogen and medicarpin: A possible mechanism for alleviating bone loss. Osteoporos Int, 23:1151-1161. [DOI] [PubMed] [Google Scholar]
- [64].Kumar G, Roger PM, Ticchioni M, Trojani C, Bernard de Dompsur R, Bronsard N, et al. (2014). T cells from chronic bone infection show reduced proliferation and a high proportion of CD28- CD4+ T cells. Clin Exp Immunol, 176:49-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhao J, Nguyen LNT, Nguyen LN, Dang X, Cao D, Khanal S, et al. (2019). ATM deficiency accelerates DNA damage, telomere erosion, and premature t cell aging in HIV-infected individuals on antiretroviral therapy. Front Immunol, 10:2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Lopez P, Rodriguez-Carrio J, Martinez-Zapico A, Caminal-Montero L, Suarez A (2016). Senescent profile of angiogenic T cells from systemic lupus erythematosus patients. J Leukoc Biol, 99:405-412. [DOI] [PubMed] [Google Scholar]
- [67].Annapoorna N, Rao GV, Reddy NS, Rambabu P, Rao KRSS (2012). An increased risk of osteoporosis during acquired immunodeficiency syndrome. Int J Med Sci, 1:152-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Bultink IEM, Lems WF, Kostense PJ, Dijkmans BAC, Voskuyl AE (2005). Prevalence of and risk factors for low bone mineral density and vertebral fractures in patients with systemic lupus erythematosus. Arthritis Rheum, 52:2044-2050. [DOI] [PubMed] [Google Scholar]
- [69].Pieper J, Johansson S, Snir O, Linton L, Rieck M, Buckner JH, et al. (2014). Peripheral and site-specific CD4+CD28null T cells from rheumatoid arthritis patients show distinct characteristics. Scand J Immunol, 79:149-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Phoksawat W, Jumnainsong A, Sornkayasit K, Srisak K, Komanasin N, Leelayuwat C (2020). IL-17 and IFN-γ productions by CD4+ T cells and T cell subsets expressing NKG2D associated with the number of risk factors for cardiovascular diseases. Mol Immunol, 122:193-199. [DOI] [PubMed] [Google Scholar]
- [71].Tyagi AM, Srivastava K, Sharan K, Yadav D, Maurya R, Singh D (2011). Daidzein prevents the increase in CD4+CD28null T cells and B lymphopoesis in ovariectomized mice: A key mechanism for anti-osteoclastogenic effect. PLoS One, 6:e21216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Tyagi AM, Mansoori MN, Srivastava K, Khan MP, Kureel J, Dixit M, et al. (2014). Enhanced immunoprotective effects by anti-IL-17 antibody translates to improved skeletal parameters under estrogen deficiency compared with anti-RANKL and anti-TNF-α antibodies. J Bone Miner Res, 29:1981-1992. [DOI] [PubMed] [Google Scholar]
- [73].Dapunt U, Giese T, Prior B, Gaida MM, Hänsch GM (2014). Infectious versus non-infectious loosening of implants: Activation of T lymphocytes differentiates between the two entities. Int Orthop, 38:1291-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Corrêa JD, Fernandes GR, Calderaro DC, Mendonça SMS, Silva JM, Albiero ML, et al. (2019). Oral microbial dysbiosis linked to worsened periodontal condition in rheumatoid arthritis patients. Sci Rep, 9:1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Sczepanik FSC, Grossi ML, Casati M, Goldberg M, Glogauer M, Fine N, et al. (2020). Periodontitis is an inflammatory disease of oxidative stress: We should treat it that way. Periodontol 2000, 84:45-68. [DOI] [PubMed] [Google Scholar]
- [76].Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-Hora M, Kodama T, et al. (2014). Pathogenic conversion of Foxp3+ T cells into Th17 cells in autoimmune arthritis. Nat Med, 20:62-68. [DOI] [PubMed] [Google Scholar]
- [77].Tsukasaki M, Komatsu N, Nagashima K, Nitta T, Pluemsakunthai W, Shukunami C, et al. (2018). Host defense against oral microbiota by bone-damaging T cells. Nat Commun, 9:1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Kornete M, Mason E, Istomine R, Piccirillo CA (2017). KLRG1 expression identifies short-lived Foxp3+ Treg effector cells with functional plasticity in islets of NOD mice. Autoimmunity, 50:354-362. [DOI] [PubMed] [Google Scholar]
- [79].Soligo M, Camperio C, Caristi S, Scotta C, Del Porto P, Costanzo A, et al. (2011). CD28 costimulation regulates Foxp3 in a RelA/NF-κB-dependent mechanism. Eur J Immunol, 41:503-513. [DOI] [PubMed] [Google Scholar]
- [80].Tang Q, Henriksen KJ, Boden EK, Tooley AJ, Ye J, Subudhi SK, et al. (2003). Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J Immunol, 171:3348-3352. [DOI] [PubMed] [Google Scholar]
- [81].Reinke S, Geissler S, Taylor WR, Schmidt-Bleek K, Juelke K, Schwachmeyer V, et al. (2013). Terminally differentiated CD8+ T cells negatively affect bone regeneration in humans. Sci Transl Med, 5:177ra36. [DOI] [PubMed] [Google Scholar]
- [82].Henson SM, Lanna A, Riddel NE, Franzese O, Macaulay R, Griffiths SJ, et al. (2014). P38 signaling inhibits mTORC1-independent autophagy in senescent human CD8+ T cells. J Clin Invest, 124:4004-4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Zhou B, Zhao Z, Zhang X, Deng W, Li Y (2020). Effect of allogenic bone marrow mesenchymal stem cell transplantation on T cells of old mice. Cell Reprogram, 22:30-35. [DOI] [PubMed] [Google Scholar]
- [84].Roser-Page S, Vikulina T, Yu K, McGee-Lawrence ME, Weitzmann MN (2018). Neutralization of CD40 ligand costimulation promotes bone formation and accretion of vertebral bone mass in mice. Rheumatol, 57:1105-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Lanna A, Henson SM, Escors D, Akbar NA (2014). The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat Immunol, 15:965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Callender LA, Carroll EC, Beal RWJ, Chambers ES, Nourshargh S, Akbar AN, et al. (2018). Human CD8+ EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK. Aging Cell, 17:e12675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Akbar AN, Henson SM, Lanna A (2016). Senescence of T lymphocytes: Implications for enhancing human immunity. Trends Immunol, 37:866-876. [DOI] [PubMed] [Google Scholar]
- [88].Bharath LP, Agrawal M, McCambridge G, Nicholas DA, Hasturk H, Liu J, et al. (2020). Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab, 32:44-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Di Micco R, Krizhanovsky V, Baker D, Di Fagagna FDA (2020). Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol, 1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Di Mitri D, Azevedo RI, Henson SM, Libri V, Riddell NE, Macaulay R, et al. (2011). Reversible senescence in human CD4+CD45RA+CD27- memory T cells. J Immunol, 187:2093-2100. [DOI] [PubMed] [Google Scholar]