

Abstract
Delayed reconstitution of the immune system is a long-recognized complication after allogeneic hematopoietic cell transplantation (HCT). Specifically, loss of T-cell diversity has been thought to contribute to infectious complications, graft versus host disease (GVHD) and disease relapse. We performed serial high-resolution next generation sequencing of TCR-β in 99 related or unrelated donor (51 unrelated, 39 related) HCT (55 reduced intensity conditioning, 44 myeloablative conditioning) recipients during the first 3 months after HCT using the immunoSEQ® Assay. We measured T-cell fraction, clonality (1- Peilou’s eveness) and Daley-Smith richness from recipient samples at multiple time points. In agreement with prior studies, we found that although absolute T-cell numbers recover relatively quickly after transplant, T-cell repertoire diversity remains diminished. Restricted diversity was associated with conditioning intensity, use of ATG, and donor type. Increased number of expanded clones compared to donor T-cell clones at Day +30 was associated with the incidence of acute GVHD (HR=1.11, p=5x10−5). Even after exclusion of the twelve patients who developed acute GVHD before Day +30, the association between acute GVHD and an increased clonal expansion at Day +30 remained (HR=1.098, p=0.041), indicating that increased clonal T-cell expansion preceded the development of acute GVHD. Our results highlight T-cell clonal expansion as a potential novel biomarker for acute GVHD that warrants further study.
Introduction
The therapeutic mechanism underlying allogeneic hematopoietic cell transplantation (HCT) is an immunologically-driven graft-versus-malignancy (GVM) effect. However, the immunogenicity of allogeneic HCT is also responsible for the development of acute and chronic graft-versus-host disease (GVHD), which remain significant post-transplantation complications. The development of effective GVM with or without GVHD is undoubtedly impacted by donor T-cell reconstitution, the specific kinetics of which are poorly understood after HCT.
T-cell receptor (TCR) immunosequencing has the potential to expand our understanding of T-cell repertoire diversification and provides an avenue to gain further insight into immune reconstitution after HCT. The vast majority of TCRs in the human body are heterodimers of an alpha and beta chain, with a minority expressing gamma and delta chains. Between the alpha and beta genes, there are approximately 120 variable (V) exons, 2 diversity (D) exons, and 70 joining (J) segments capable of producing a junctional diversity of ~2x1011. Recognition of antigen and engagement of the TCR leads to antigen-specific stimulation and clonal proliferation.1 Some prior efforts to characterize immune reconstitution focused on flow cytometric evaluation of T-cell subsets, and, while informative, this approach was incapable of revealing T-cell clonal dynamics.2 Early investigations of the T-cell repertoire diversity using CDR3 spectratyping showed markedly skewed repertoires in the early months after HCT that could persist for years.3 More recent evaluations of the T-cell repertoire using next-generation sequencing (NGS) technology have similarly shown restricted TCR clonal diversity early after HCT, and have suggested that correlations may exist between T-cell changes and clinical outcomes, such as development of GVHD and disease relapse.4–8
We used immunosequencing of the TCRβ chain on serial peripheral blood samples to describe longitudinal changes in the reconstitution of T-cell diversity in 99 transplant recipients during the first 3 months after related or unrelated allogeneic HCT and correlated these to patient or transplant-specific characteristics. We also investigated relationships between T-cell repertoire characteristics and clinical outcomes after HCT to assess whether specific T-cell repertoire metrics were associated with significant post-transplant complications.
Methods
Study Design
This study was approved by the institutional review board at the Dana-Farber Harvard Cancer Center. From 2008-2011, allogeneic hematopoietic cell transplant (HCT) recipients from Dana-Farber/Brigham and Women’s Cancer Center and Massachusetts General Hospital Cancer Center were enrolled on separate IRB-approved sample banking protocols. From this cohort, we identified 99 adult patients with acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), or acute lymphoblastic leukemia (ALL), who had cryopreserved peripheral blood mononuclear cells (PBMC). Recipient samples were categorized as being collected at several time points after transplant [Day +15 (+/− 3 days), Day +30 (+/− 3 days), Day +50 (+/− 10 days), and Day 100 (+/− 10 days)]. Additionally, frozen PBMC samples from quality control aliquots from donor stem cell products were obtained for analysis. Clinical data were extracted from the institutional database and individual medical record of each patient.
T-cell receptor variable beta chain sequencing
Immunosequencing of the CDR3 regions of human TCRβ chains was performed using the immunoSEQ® Assay (Adaptive Biotechnologies, Seattle, WA). Extracted genomic DNA was amplified in a bias-controlled multiplex PCR, followed by high-throughput sequencing. Sequences were collapsed and filtered in order to identify and quantitate the absolute abundance of each unique TCRβ CDR3 region for further analysis as previously described.9–11 Downstream analyses utilized only “productive” rearrangements, i.e. only T cells clones with TCRβ sequences expected to generate functional T-cell receptors. For the remainder of this work, we will define a T cell “template” as a single T cell, regardless of the receptor sequence, while a “rearrangement” refers to the unique VDJ recombination. For instance, multiple T cell templates may all contain the same rearrangement, and thus derive from the same progenitor cell.
Statistical analyses of TCR-β sequencing results
Clonality was defined as 1- Peilou’s eveness12 (Supplementary Data). Clonality values range from 0 to 1 and describe the shape of the frequency distribution: clonality values approaching 0 indicate a very even distribution of frequencies, whereas values approaching 1 indicate an increasingly asymmetric distribution in which a few clones are present at high frequencies. Clonality is typically strongly correlated with the top clone frequency in the sample.
Daley-Smith (DS) Richness extrapolates the number of unique rearrangements expected if a sample contained an arbitrarily defined value of 400,000 templates, which represents double the sample size the immunoSEQ Assay is designed to capture, and serves as a common benchmark to compare between samples.13 This non-parametric empirical Bayes estimator of repertoire richness is based on extrapolation of the rarefaction curve to 10 times the actual sample size.
T-cell fraction was calculated by taking the total number of T-cell templates and dividing it by the estimated nucleated cell number based on overall sample DNA input using a conversion factor of 153.846 diploid genomes per nanogram of DNA. Importantly, all statistics were normalized to the number of T cells or nucleated cells sequenced, such that they were not confounded by the amount of DNA into the assay. Statistical analysis was performed in R version 3.2.
Two complementary methods were used to compare individual clones across samples from the same subject. First, the number of expanded clones in a given recipient sample compared to the donor sample was calculated according to a binomial distribution framework as previously described.14 Briefly, a two-sided test of the null hypothesis that the probability of success in a Bernoulli experiment is p is computed for each clone (Supplementary Data). Second, Morisita’s Index, a population overlap metric that compares the presence and frequency of T cells in two repertoires, was also calculated for these comparisons (Supplementary Data). Morisita’s Index varies from 0 to 1, where values approaching 1 indicate highly correlated repertoires with many of the same T cells at similar frequencies and a value of 0 would indicate that the two samples do not share even a single T cell with the same T-cell receptor.
Association of TCR-β sequencing results with clinical variables and outcomes
Analyses were performed to assess for correlations between the T-cell repertoire as assessed by immunosequencing of the TCRβ locus and clinical metrics. For each time point, the immunoSEQ metrics analyzed included: clonality, richness, T-cell fraction, Morisita’s Index, clonal expansion, and fraction of the recipient samples comprised of TCRs from the donor sample. The clinical metrics analyzed included: recipient age, recipient sex, diagnosis, disease risk, donor type, conditioning regimen and intensity and GVHD prophylaxis regimen. Age was analyzed as a continuous variable, while all others were considered discrete.
We first performed pairwise correlations between all immunoSEQ and clinical metrics (Supplementary Figure 1). The choice of statistical test was made according to the classification of variables being analyzed: Fisher’s exact test was used to analyze two categorical variables, Spearman correlation was used to analyze two numerical variables, and Mann-Whitney and Kruskal Wallis tests were used to assess for correlations between categorical and numerical variables. P-values were not corrected for multiple testing due to the high correlation among immune repertoire metrics.
Next, we performed Cox regression to determine if clinical or immunoSEQ metrics were associated with risk of acute GVHD, chronic GVHD, relapse, overall survival or each cause of death (infection, relapse, GVHD, or other) separately. Clonality, T-cell fraction, Morisita’s Index, and donor frequency were scaled to units of 0.1, DS richness was scaled to units of 50,000, and the number of expanded clones was scaled to units of 50. For all immunoSEQ metrics with p<0.01, we performed two tests to determine if confounding clinical variables could account for the observed association. First, we performed multiple Cox regression to determine if a given immunoSEQ metric was still significant after accounting for other correlated clinical variables. Second, we performed likelihood ratio tests to compare nested Cox models, the first of which included only the correlated clinical metrics, and the second of which also included the immunoSeq metric. A clinical metric was considered to be correlated with a given immunoSEQ metric if the two had a p value < 0.05.
Disease risk was characterized according to the Disease Risk Index (DRI).15 Acute GVHD16 and chronic GVHD17 were graded according to established consensus clinical criteria. Acute and chronic GVHD were reported as cumulative incidence from the date of transplant, respectively, with relapse or death as competing risks. Relapse was reported as cumulative incidence from the date of transplant, with death without relapse as a competing risk. The cumulative incidence of non-relapse mortality was calculated from the date of transplant to death, with relapse as a competing risk. Progression-free (PFS) and overall survival (OS) were estimated using the Kaplan-Meier method. PFS was calculated from the date of transplantation to disease progression or death from any cause. Patients who were alive without relapse or progression were censored at the time of last clinical evaluation. OS was calculated from the date of transplantation to death or censored at last clinical evaluation.
Results
Patient and transplant characteristics
Ninety-nine patients were included in the analysis. Patient and transplant characteristics are shown in Table 1. Forty-eight patients were female and 51 were male. The median age at transplant was 55 years (range, 20-74). In regards to DRI, most patients had intermediate (n=61) or high (n=32) risk disease. The most common donor types were matched unrelated (n=51) or matched related (n=39) donors. Fifty-five patients received reduced intensity conditioning while 44 patients received myeloablative conditioning regimens. The most common GVHD prophylaxis regimen consisted of tacrolimus and methotrexate (n=63). Twenty-five patients received anti-thymocyte globulin (ATG, Thymoglobulin) as part of GVHD prophylaxis.
Table 1.
Patient and transplant characteristics
| Characteristic | Value |
|---|---|
| Total patients, n | 99 |
| Sex, n | |
| Female | 48 |
| Male | 51 |
| Median age in years at transplant (range) | 55 (20-74) |
| Diagnosis, n | |
| Acute lymphoblastic leukemia | 13 |
| Acute myeloid leukemia | 61 |
| Myelodysplastic syndrome | 25 |
| Disease Risk Index, n | |
| Low | 3 |
| Intermediate | 61 |
| High | 32 |
| Very High | 3 |
| Donor type, n | |
| Matched related donor | 39 |
| Matched unrelated donor | 51 |
| Mismatched related donor | 3 |
| Mismatched unrelated donor | 6 |
| Graft source, n | |
| Bone marrow | 1 |
| Peripheral blood stem cells | 98 |
| Conditioning intensity, n | |
| Myeloablative | 44 |
| Reduced intensity/non-myeloablative | 55 |
| GVHD prophylaxis backbone, n | |
| Cyclosporine/methotrexate | 8 |
| Cyclosporine/mycophenolate mofetil | 2 |
| Tacrolimus/methotrexate | 63 |
| Tacrolimus/sirolimus | 9 |
| Tacrolimus/sirolimus/methotrexate | 17 |
| ATG for GVHD prophylaxis, n | |
| Yes | 25 |
| No | 74 |
| CMV reactivation | |
| Total, n | 27 |
| Before day +100, n | 18 |
| Before day +30, n | 4 |
Abbreviations: ATG: anti-thymocyte globulin; GVHD: graft-versus-host disease;
Longitudinal changes in T-cell repertoire after allogeneic HCT
Samples from donors (n=83) and transplant recipients (post-transplant Day +15: n=65; Day +30: n=80; Day +50: n=80, Day +100: n=77;) were available for immunosequencing of the TCRβ chains. We inputted a median of 1993.6 nanograms (ng) of DNA into each sequencing reaction, with a range of 66.9 to 2007.2 ng, and obtained a median of 70,825 T-cell templates per sample with a range of 69 to 291,729. These comprised a median of 39,563 unique rearrangements, with a range of 58 to 153,926. Accordingly, we found that the T-cell fraction (percent T cells out of the estimated number of nucleated cells input) ranged from .09% to 97%, with a median of 27.9%. The maximum productive clone frequency ranged from 73% with a clonality of 0.752, to 0.1%, with a clonality of 0.
We found extensive changes in the T-cell repertoire of recipients over the first 100 days after HCT (Figure 1). At Day +15, clonality was similar to the donor sample (p=0.75; Wilcoxon Rank Sum test) (Figure 1A). After this, clonality increased significantly over time (p<2.2x10−16; linear mixed-effects model). Daley-Smith Richness, which represents the number of unique T-cell clones among 400,000 T cells, was already decreased at Day +15 compared to the donor sample (p<2.2x10−16; Wilcoxon Rank Sum test), and continued to decrease across the sampling period (p<2.2x10−16; linear mixed-effects model) (Figure 1B). In effect, this shows that while the distribution of T-cell clonotypes was similar between donor and recipient at Day +15, the total number of unique T-cell clones was decreased. Although T cells comprised a lower proportion of total nucleated cells at Day +15 compared to the donor sample (p=8.85x10−5, Wilcoxon Rank Sum test), this T-cell fraction increased steadily over time (p<2.2x10−16; linear mixed effects model) (Figure 1C).
Figure 1. T-cell Repertoire Dynamics Over Time.
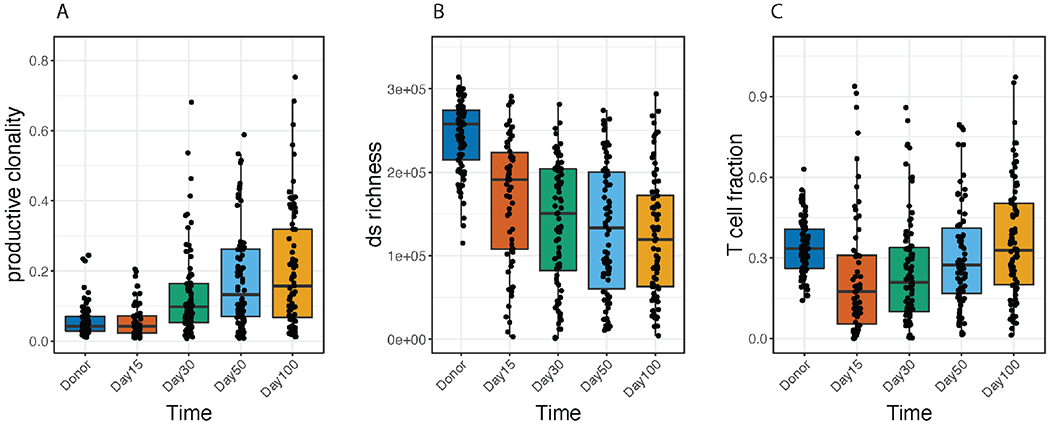
Immunosequencing of the TCRβ chains from transplant using the immunoSEQ Assay. Donor samples are compared to patient samples at Day 15, 30, 50, and 100 after allogeneic transplant. A. T-cell productive clonality, where increasing values indicate a more clonal, or focused repertoire. B. Daley-Smith Richness, where values indicate an estimate of the number of unique rearrangements in a sample of 400,000 T cells. C. T-cell fraction, an estimate of the fraction of T cells out of total nucleated cells. P values in the upper left corners indicate overall differences by Kruskall Wallis test. Horizontal lines indicate significant differences compared to the Day +15 time point (p < 0.05, Wilcoxon Rank Sum test).
These observations indicate that even though T-cell numbers compared to other nucleated cells return to baseline in the early months after transplant, diversity remains relatively low due to the expansion of specific T-cell clones (i.e., oligoclonal expansion). This is exemplified by the median DS Richness estimate for the donor samples which was >250,000, while in transplant recipients at Day +100, the median estimate was 100,000.
Next, we directly compared overlap and frequencies of each clone in post-transplant samples to the donor sample in order to quantify clonal expansion and repertoire turnover (Figure 2A). We found that there was decreasing similarity between the transplant recipient and the donor repertoires over time (indicated by decrease in Morisita’s Index (p<2.2x10−16; linear mixed effects model), driven by clonal expansion in the recipient repertoire (p<2.2x10−16; linear mixed effects model) (Figure 2B). However, longitudinal evaluations of both Morisita’s Index and the number of expanded clones indicate that the rate of repertoire turnover reaches stasis by approximately Day +50 after HCT.
Figure 2. Post-Transplant versus Donor Repertoire.
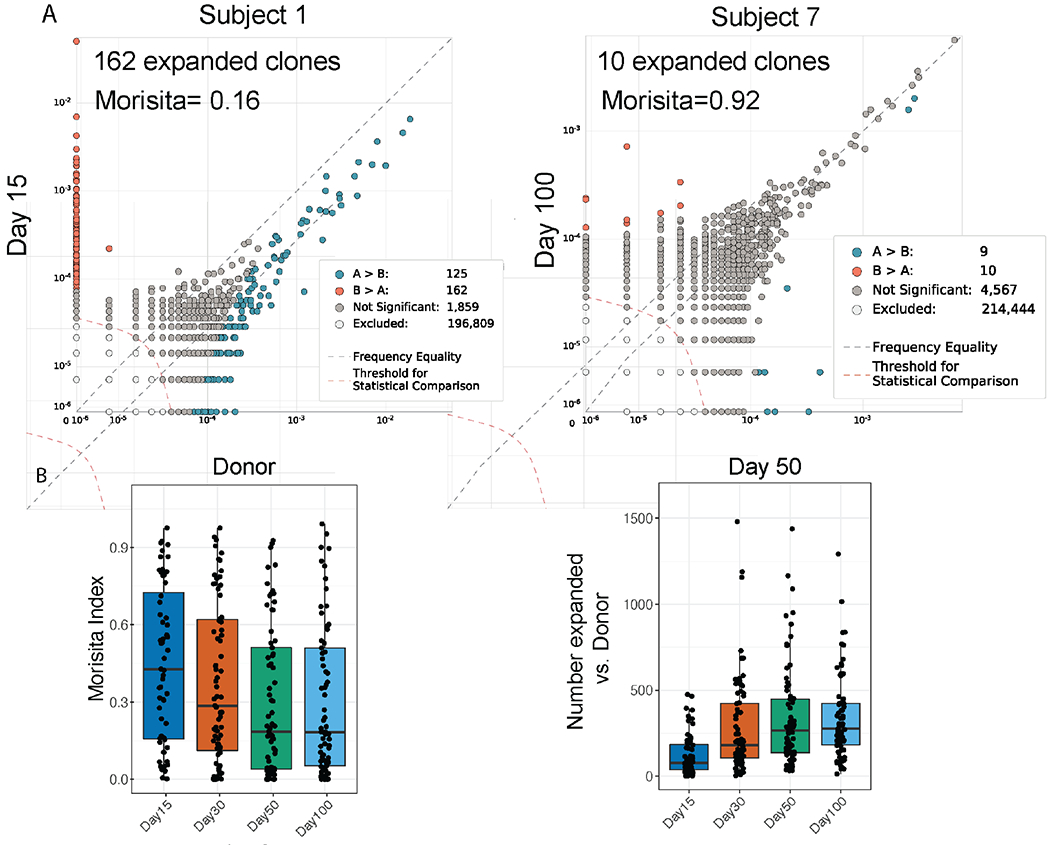
A. Examples of repertoire comparisons between two samples with either extensive turnover (left), or limited turnover (right). The legend quantifies expanded clones (B > A) and contracted clones (A > B). B. Morisita’s Index and number of expanded clones compared to donor samples at the indicated time points. P values in the upper left corners indicate overall differences by Kruskall Wallis test.
Associations between T-cell repertoire characteristics and clinical metrics
We performed univariate analyses to look for associations between the T-cell repertoire of transplant recipients and clinical variables and found multiple significant relationships (Supplementary Figure 1). There is less repertoire turnover at all time points post-transplant (increase in Morisita’s Index) for transplants from HLA-matched related donors, as compared to matched unrelated donors (p<0.05 for all time points). Donor relatedness also plays a role in several other repertoire metrics, including decreased Morisita’s index in matched unrelated donor HCT recipients at Day +30 (p=0.004; Supplementary Figure 2a). Additionally, the use of reduced intensity conditioning was associated with a smaller number of expanded clones compared to donor at Day +15, +30 and +50 compared to that seen in recipients who had received myeloablative conditioning (p<0.05; Supplementary Figure 2b).
We found that use of ATG strongly correlated with changes in the recipient T-cell repertoire. At Day +15, subjects that received ATG had decreased richness (p=2x10−6) and T-cell fraction (p=6x10−5) and increased clonality (p=0.021) compared to other subjects (Wilcoxon rank sum test). By Day +100, richness remained lower in subjects who received ATG (p=4x10−7), and clonality remained higher (p=0.001), while T-cell fraction was only marginally lower indicating notable repertoire skewing (p = 0.064; Wilcoxon rank sum test; Figure 3).
Figure 3. Effects of ATG on the T-cell Repertoire.
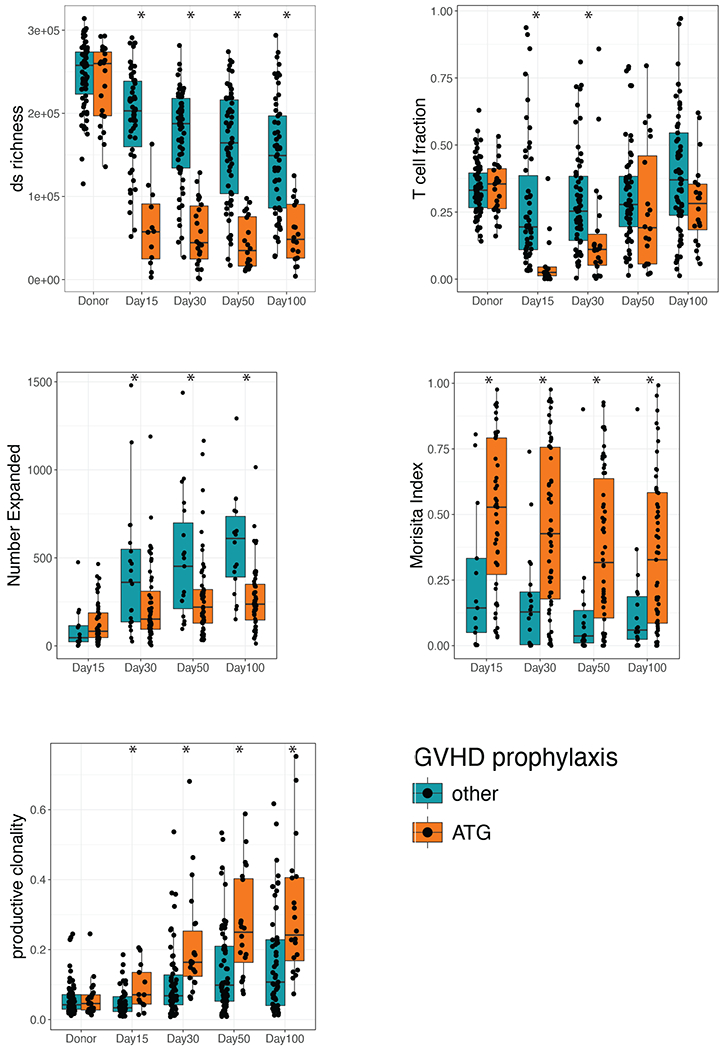
Subjects are grouped into those receiving ATG and those that did not receive ATG with the relevant metrics plotted at the specified time points (ds richness, T-cell fraction, number of clones expanded, Morisita’s Index, and productive clonality) * = p < 0.05 by Mann-Whitney-U test.
Associations between T-cell repertoire characteristics and acute GVHD
We performed univariate Cox regression analysis with all clinical and T-cell repertoire metrics to determine correlations with the incidence of grade II-IV acute GVHD. An increased number of expanded T-cell clones at Day +30 was the only T-cell repertoire metric that was significantly associated with acute GVHD at α=0.01, portending an increased risk of grade II-IV acute GVHD (HR=1.11, 95% CI: 1.06-1.17, p=5x10−5) (Figure 4). Of the 40 subjects that developed acute GVHD, 12 developed acute GVHD prior to Day +30. After removing these subjects from the analysis, we found that the association remained significant, indicating that an increased number of expanded T-cell clones compared to donor can be detected prior to development of clinical manifestations of acute GVHD (HR=1.098, 95%CI 1.003-1.202, p=0.041) (Figure 4).
Figure 4. Associations with Acute GVHD Grade II-IV.
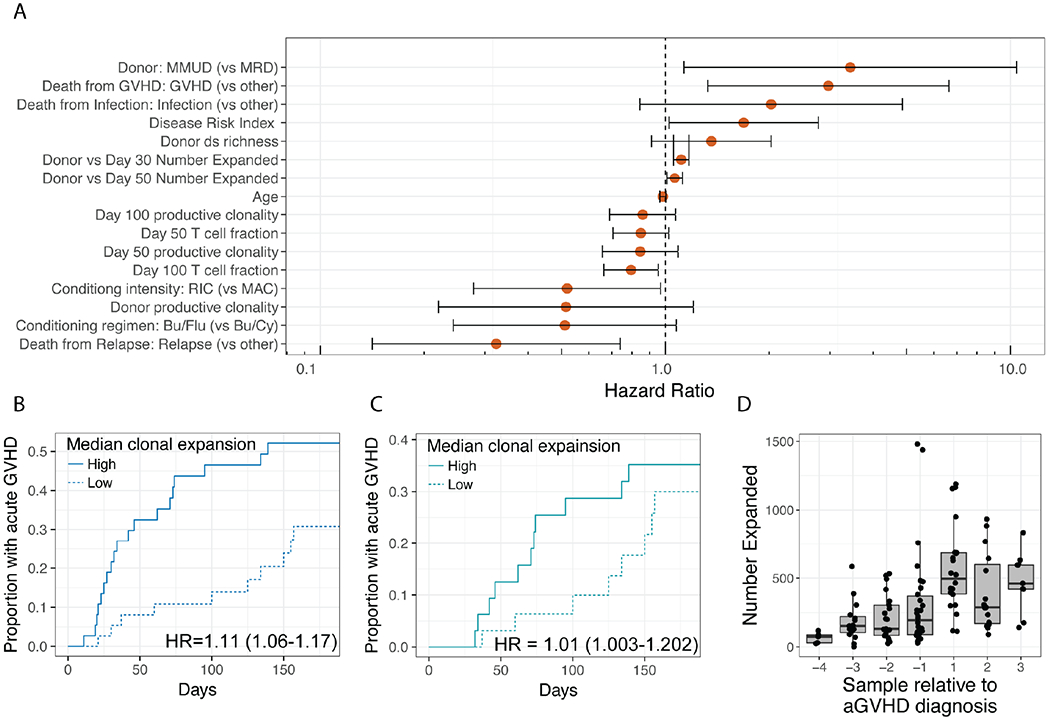
A. TCR repertoire metrics and clinical variables with single variable Cox Regression P < 0.2 are shown with 95% confidence intervals noted. B. Cumulative Incidence of acute GVHD stratified by high (greater than the median) and low (less than the median) clonal expansion at Day +30. C. Same analysis as B, but patients who have already developed aGVHD by Day +30 have been removed. D. The number of expanded clones at each collection time relative to acute GVHD diagnosis (time 0).
Multiple clinical metrics were associated with an increased number of expanded clones at Day +30 (Supplementary Figure 1). Although none of these were independently associated with the incidence of acute GVHD (α=0.01), we performed additional analyses to explore their relationships. In a multiple Cox regression analysis including all clinical variables associated with the number of expanded clones at Day +30, we found it to be significantly associated with incidence of grades II-IV acute GVHD (p=0.003). Additionally, a full model including these clinical variables and the number of expanded clones was significantly better than a reduced model that excluded the number of expanded clones (p= 0.00059; Likelihood ratio test). Although the use of ATG had a significant relationship with the number of expanded clones at Day +30 (p=0.042) and no significant relationship with the incidence of acute GVHD (p=0.93), we excluded the 25 patients given ATG and found that the relationship between the number of expanded clones and acute GVHD remained significant (p=0.07). Cumulative incidence analysis stratifying patients by both the number of expanded clones and ATG revealed that the relationship between acute GVHD and the number of expanded clones was consistent in subjects who had and had not received ATG, respectively (Supplementary Figure 3a). As conditioning intensity was strongly associated with the number of expanded clones at Day +30 (p=8x10−4, Supplementary Figure 1 and 2), and trended towards an association with incidence of acute GVHD (p=0.039, α=0.01, Figure 4), we stratified analysis by conditioning regimen intensity (Supplementary Figure 3b). We found that in subjects who received MAC, the number of expanded clones at Day +30 was still associated with acute GVHD despite the smaller sample size (p=0.003), but in subjects that received RIC, the number of expanded clones at Day +30 was not associated with acute GVHD (p=0.42). Finally, donor type was also strongly associated with the number of expanded clones at Day +30 (p=0.004, Mann-Whitney-U test comparing patients with matched unrelated donors (MUD) to those who with matched related donors (MRD), Supplementary Figure 1 and 2), but was not associated with incidence of acute GVHD (p=0.52, Cox Regression). Restricting analysis of the association between acute GVHD and the number of expanded clones to transplants from matched unrelated donors was still significant (p=0.0015), while analysis was not significant when restricting to matched related donors (p=0.42, Supplementary Figure 3c).
We next sought to determine whether serial monitoring of the number of expanded clones could potentially predict when a patient will develop acute GVHD. We re-aligned this data relative to the time at which each subject developed acute GVHD (Figure 4d). We found that the median number of expanded clones peaked in the first sample taken after the presentation of clinical acute GVHD (median=495 expanded clones relative to the donor sample, range=113-1189). This was greater than the median at any fixed time point in the entire cohort, or any fixed time point when considering only patients that developed acute GVHD (median=277, range=182-1292 and median= 343, range= 149-835, respectively, for Day 100).
Associations between T-cell repertoire characteristics and other clinical transplant outcomes
We performed independent Cox regression analyses with all clinical and T-cell repertoire metrics to determine correlations with subsequent development of moderate to severe chronic GVHD, disease relapse, and overall survival, respectively. We did not find any T-cell repertoire metrics that were significantly associated with underlying disease relapse or the incidence of moderate to severe chronic GVHD. We found that increased T-cell richness at Day +30 was associated with increased risk of death, and this was the only T-cell repertoire metric that was significantly associated with overall survival (Supplementary Figure 4), however after removing subjects given ATG, the association was no longer significant (Supplementary Figure 5).
Associations between T-cell repertoire characteristics and CMV reactivation
CMV infection is known to have a large effect on the T cell repertoire18. Our cohort included 23 patients who had evidence by (tissue or blood testing) of CMV reactivation before day +150, 18 before day +100, and 4 patients prior to day +30. We performed cox regression analysis to explore the relationship between CMV reaction in this cohort and other clinical and immunoSEQ metrics. We found that higher clonality was significantly associated with increased risk of CMV reactivation (p= 8.7x10-4 5.3x10-5, and 7.9x10-4 for Day +30, +50, and +100, respectively; Supplementary figure 6). The number of expanded clones at day 30 showed a trend towards association with CMV reaction (p=0.058). However, CMV reactivation and acute GVHD incidence were not correlated (p=0.32; Chi-squared test).
Discussion
The reconstitution of donor derived T-cells after HCT is a critical step in the establishment of resistance to infections, GVHD and graft versus malignancy effects. While nearly two decades have been spent trying to clarify T-cell clonotype dynamics after transplant, there remains a primitive understanding of this process. An early analysis of the T-cell repertoire used CDR3 spectratyping on a small number of patients after allogeneic T-cell depleted HCT for recipients with chronic myeloid leukemia. Results showed significantly shifted repertoires consisting of absent, monoclonal or oligoclonal profiles for the majority of Vβ subfamilies. Complexity scores (a quantitative estimate of Vβ diversity) remained low in patients with mixed chimerism (6 of 7 who relapsed), but by 24 months after HCT, they had normalized in patients with complete donor hematopoiesis (all four remained in remission). One patient with mixed chimerism continued to have abnormally low complexity scores over five years after transplant.3
More recent evaluations of the T-cell repertoire using NGS-based techniques have confirmed restricted clonal diversity in the post-HCT setting.4–8 Interestingly, one study revealed that patients who received umbilical cord-blood grafts had nearly approximated the T-cell diversity of healthy individuals by six months after HCT, while recipients of T cell-depleted peripheral stem cell grafts had 14-28 fold lower T-cell diversity. By twelve months after HCT, the CD4+ T-cell repertoire continued to increase in diversity, while that of the CD8+ cells remained stagnant. That was also the only study to date to report an increase in T-cell diversity associated with the development of acute GVHD.4
Another more recent analysis of T-cell repertoire after HCT used next-generation sequencing and an RNA PCR system capable of identifying novel TCRα and TCRβ exons in the matched conventional donor and umbilical cord blood setting. By analyzing the combined proportion of the ten most abundant clones of TCRβ occurring in conjunction with the development of acute GVHD in each patient, the authors were able to show a statistically-significant enrichment of the these top ten clones at the time of acute GVHD compared to non-GVHD patients who did not have the same degree of proportional enrichment of the top ten clones. T-cell diversity was also significantly lower among patients with acute GVHD compared to those without acute GVHD. Interestingly, among patients with acute GVHD, those who relapsed had a more diverse TCR spectrum than those who did not. In contrast, in patients who never had acute GVHD, non-relapsed patients had more diversity than relapsed patients.5 The authors concluded that increased T-cell diversity early after HCT may indicate lower risks for both acute GVHD and disease relapse.5 A recent analysis of the T-cell diversity of Swiss allogeneic transplant patients one year after transplant and their respective donors found CMV reactivation and increasing patient age were both associated with reduced T-cell diversity; however, they did not find an association with GVHD, relapse, or mortality.19
In contrast to prior studies interrogating the relationship between T-cell diversity and acute GVHD, we did not find that restricted diversity or increased clonality occurred during the development of acute GVHD by Day 30+. Instead, we found that large expansions of T-cell clones occurred in patients with acute GVHD. Depending on the frequencies of existing clones, these expansions can either increase or decrease repertoire diversity and/or clonality. Additionally, unlike prior studies, we show that even after removing the subjects who had already developed acute GVHD by Day +30, the association of acute GVHD with an increased number of expanded T-cell clones remained significant, potentially providing an early biomarker for impending acute GVHD prior to clinical manifestations. We also demonstrated that the number of expanded T-cell clones progressively increases until the diagnosis of acute GVHD, upon which it decreases, presumably due to the initiation of immunosuppressive therapy. We hypothesize that this increase in the number of expanded T-cell clones reflects donor T-cell recognition of specific host epitopes and subsequent antigen dependent proliferation. Confirmation of these findings in larger cohorts is clearly needed and identification of target peptides would be revealing.
The choice of GVHD prophylaxis regimen can clearly alter the pattern and kinetics of immune reconstitution after HCT. Indeed, in our analysis, we found that the use of ATG affected T-cell fraction and richness. Post-transplant cyclophosphamide (PTCy) based regimens are an increasingly popular choice for GVHD prophylaxis and similar trends in T-cell repertoire diversity have been observed in patients receiving PTCy as assessed by using the immunoSEQ assay.6,7 Furthermore, use of either ATG or PTCy resulted in a delay in the reconstitution of naïve CD8+ repertoire and an increase in TCRα dominant clones.8
Importantly, this study has limitations. Current immunosequencing approaches to understanding immune reconstitution lies in the lack of our interpretation of individual clones. While the field has developed useful, orthogonal methods of characterizing general diversity and repertoire turnover, which we show strongly correlate with clinical correlates and outcomes, future efforts at mapping T cell receptors to the antigens they bind will further elucidate the process of immune reconstitution. For instance, knowing the specificity of expanding clones could allow us to further predict acute GVHD symptoms. Due to the highly private nature of the immune repertoire, and the complexity of TCR-antigen binding solutions, this task remains challenging.
In summary, this study represents the largest cohort of HCT recipients with serial analysis of post-HCT T-cell repertoire changes to date. We found that although absolute T-cell numbers return to baseline in the early months after HCT, T-cell diversity remains relatively low due to the restricted expansion of selected T-cell clones. We observed associations between T-cell repertoire diversity and multiple clinical variables, including donor type, conditioning intensity and the use of ATG. Finally, we identified an association between the number of T-cell clones at Day +30 and the incidence of acute GVHD (HR=1.098, p=0.041), and show that this effect may be particularly strong in patients receiving myeloablative conditioning, and a transplant from a matched, unrelated donor. Restricted T-cell clonal diversity in patients with established acute GVHD has been previously suggested by other groups.7,20–22 We believe that this data confirms that T-cell clonal dynamics can provide valuable insight into immune reconstitution in recipients after HCT and early clonal T-cell expansion may be able to serve as a predictive biomarker for acute GVHD, although this will need to be further investigated prospectively in larger and more homogeneous cohorts of patients.
Supplementary Material
Key Points.
After allogeneic HCT, T-cell repertoire diversity lags behind quantitative T-cell recovery.
Increase in the number of expanded clones at Day +30 is associated with an increased risk of acute GVHD.
Use of ATG results in prolonged T-cell anomalies, including increased clonality (1- Peilou’s eveness) and decreased Daley-Smith richness, despite a nearly equivalent T-cell fraction.
References
- 1.Murphy K, Travers P, Walport M, Janeway C. Janeway’s immunobiology. 7th ed. New York: Garland Science; 2008. [Google Scholar]
- 2.Jacobson CA, Turki AT, McDonough SM, et al. Immune reconstitution after double umbilical cord blood stem cell transplantation: comparison with unrelated peripheral blood stem cell transplantation. Biol Blood Marrow Transplant 2012;18:565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu CJ, Chillemi A, Alyea EP, et al. Reconstitution of T-cell receptor repertoire diversity following T-cell depleted allogeneic bone marrow transplantation is related to hematopoietic chimerism. Blood 2000;95:352–9. [PubMed] [Google Scholar]
- 4.van Heijst JW, Ceberio I, Lipuma LB, et al. Quantitative assessment of T cell repertoire recovery after hematopoietic stem cell transplantation. Nat Med 2013;19:372–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yew PY, Alachkar H, Yamaguchi R, et al. Quantitative characterization of T-cell repertoire in allogeneic hematopoietic stem cell transplant recipients. Bone Marrow Transplant 2015;50:1227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanakry CG, Coffey DG, Towlerton AM, et al. Origin and evolution of the T cell repertoire after post transplantation cyclophosphamide. JCI Insight 2016; 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meier JA, Haque M, Fawaz M, et al. T Cell Repertoire Evolution after Allogeneic Bone Marrow Transplantation: An Organizational Perspective. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Link-Rachner CS, Eugster A, Rucker-Braun E, et al. T-cell receptor-alpha repertoire of CD8+ T cells following allogeneic stem cell transplantation using next-generation sequencing. Haematologica 2019;104:622–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robins HS, Campregher PV, Srivastava SK, et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood 2009;114:4099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carlson CS, Emerson RO, Sherwood AM, et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nat Commun 2013;4:2680. [DOI] [PubMed] [Google Scholar]
- 11.Robins H, Desmarais C, Matthis J, et al. Ultra-sensitive detection of rare T cell clones. J Immunol Methods 2012;375:14–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirsch I, Vignali M, Robins H. T-cell receptor profiling in cancer. Mol Oncol 2015;9:2063–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daley T, Smith AD. Predicting the molecular complexity of sequencing libraries. Nat Methods 2013;10:325–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeWitt WS, Emerson RO, Lindau P, et al. Dynamics of the cytotoxic T cell response to a model of acute viral infection. J Virol 2015;89:4517–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Armand P, Kim HT, Logan BR, et al. Validation and refinement of the Disease Risk Index for allogeneic stem cell transplantation. Blood 2014;123:3664–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris AC, Young R, Devine S, et al. International, Multicenter Standardization of Acute Graft-versus-Host Disease Clinical Data Collection: A Report from the Mount Sinai Acute GVHD International Consortium. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2016;22:4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2015;21:389–401 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindau P, Mukherjee R, Gutschow MV, et al. Cytomegalovirus Exposure in the Elderly Does Not Reduce CD8 T Cell Repertoire Diversity. The Journal of Immunology 2018:ji1800217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buhler S, Bettens F, Dantin C, et al. Genetic T-cell receptor diversity at 1 year following allogeneic hematopoietic stem cell transplantation. Leukemia 2019. [DOI] [PubMed] [Google Scholar]
- 20.Liu C, He M, Rooney B, Kepler TB, Chao NJ. Longitudinal Analysis of T-Cell Receptor Variable β Chain Repertoire in Patients with Acute Graft-versus-Host Disease after Allogeneic Stem Cell Transplantation. Biology of Blood and Marrow Transplantation 2006;12:335–45. [DOI] [PubMed] [Google Scholar]
- 21.Du J-W, Gu J-Y, Liu J, et al. TCR spectratyping revealed T lymphocytes associated with graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Leukemia & Lymphoma 2007;48:1618–27. [DOI] [PubMed] [Google Scholar]
- 22.Alachkar H, Nakamura Y. Deep-sequencing of the T-cell receptor repertoire in patients with haplo-cord and matched-donor transplants. Chimerism 2015;6:47–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.