

Abstract
Correct operation of neuronal networks depends on the interplay between synaptic excitation and inhibition processes leading to a dynamic state termed balanced network. In the spinal cord, balanced network activity is fundamental for the expression of locomotor patterns necessary for rhythmic activation of limb extensor and flexor muscles. After spinal cord lesion, paralysis ensues often followed by spasticity. These conditions imply that, below the damaged site, the state of balanced networks has been disrupted and that restoration might be attempted by modulating the excitability of sublesional spinal neurons. Because of the widespread expression of inhibitory GABAergic neurons in the spinal cord, their role in the early and late phases of spinal cord injury deserves full attention. Thus, an early surge in extracellular GABA might be involved in the onset of spinal shock while a relative deficit of GABAergic mechanisms may be a contributor to spasticity. We discuss the role of GABA A receptors at synaptic and extrasynaptic level to modulate network excitability and to offer a pharmacological target for symptom control. In particular, it is proposed that activation of GABA A receptors with synthetic GABA agonists may downregulate motoneuron hyperexcitability (due to enhanced persistent ionic currents) and, therefore, diminish spasticity. This approach might constitute a complementary strategy to regulate network excitability after injury so that reconstruction of damaged spinal networks with new materials or cell transplants might proceed more successfully.
Keywords: GABA, Spinal circuits, Spinal cord injury, Spinal shock, Neuroprotection, Spasticity
Synaptic Inhibition Is an Important Component of Spinal Locomotor Networks
In mammals, rhythmic motor tasks such as locomotion require balanced network activity based on the coordinated interaction between synaptic excitation and inhibition [1–3]. While inhibition typically dampens neuronal excitability, its overall impact traditionally depends on the reciprocal coupling to excitation in a “push-pull fashion,” whereby inhibition declines as excitation rises and neuron excitability grows, and vice versa [4]. Studies of spinal networks have, however, indicated that, in certain circuits impinging upon motoneurons, synaptic inhibition remains operative even during excitation, suggesting that there are multiple sources of inhibitory inputs beyond the mutual interaction between excitatory and inhibitory local circuits [3]. These observations support the concept of recurrent connectivity [5] that should include a robust component of recurrent inhibition to prevent network instability and ensure multifunction flexibility [6]. In this framework, an important role is played by the neurotransmitter gamma-aminobutyric acid (GABA) that controls not only locomotor cycles but also network assembly during early development [7]. These properties are particularly expressed by a spinal circuit termed central pattern generator (CPG; [8, 9] that can produce rhythmic locomotor activity independent from sensory inputs). Such a process is readily replicated with a model system like the isolated rodent spinal cord which generates alternating rhythmic patterns termed fictive locomotion because of the absence of muscle targets [10]. While excitation is primarily mediated by glutamate and its pharmacological block arrests locomotion [11], blocking inhibition evoked by amino acid transmitters like GABA and glycine suppresses alternation of motor output by the CPG and replaces it with slow rhythmic motor discharges detected synchronously in ventral roots. This phenomenon is exemplified in Fig. 1 in which the fictive locomotor patterns elicited by co-applied N-methyl-d-aspartate (NMDA) and serotonin (5HT, see Fig. 1a) and recorded from ventral roots (VRs) are converted into slow synchronous discharges (Fig. 1b).
Fig. 1.

During locomotor patterns, fast synaptic transmission is essential to allow the sequential activation of antagonistic motor pools innervating flexor and extensor hindlimb muscles. a A stable locomotor-like rhythm is induced in the spinal cord isolated from a neonatal rat by co-application of the glutamate agonist NMDA plus 5HT. The rhythm reflects the basic pattern of activation of lower limb muscles during real locomotion, which is composed of electrical discharges characteristically alternating between right (r) and left (l) ventral roots (VRs, exemplified in this figure at the second lumbar segment; L2) and between flexor (L2)- and extensor (L5)-related ventral roots on the same side of the cord (shown in this figure as the left L2 and L5). b On the same preparation, strychnine plus bicuculline are further applied to block glycinergic and GABAergic fast inhibitory transmission, respectively. Starting from 30 s after drug application, the double alternating pattern is replaced by a stable and slower rhythm that becomes synchronous among all ventral roots (unpublished traces, replicating results originally reported by Beato and Nistri, [12])
It should be noted that the GABA receptor antagonist bicuculline [13] is selectively blocking a distinct class of GABA receptors termed GABA A receptors (GABA ARs) known to mediate fast synaptic inhibition [14, 15] as well as to modulate neuronal excitability through extrasynaptic GABA receptors [16, 17]. The term “fast” inhibition, therefore, refers to the short time course underlining the loss of excitability mainly caused by hyperpolarization of the neuronal membrane (for less than 100 ms; [18]). Data in Fig. 1b also indicate that strychnine, a potent glycine receptor antagonist, contributes to block fast inhibition and suggests that, in addition to GABA, glycine is an important mediator of locomotor activity [19, 20].
Indeed, intrasegmental GABAergic and glycinergic interneurons with short axons have been found in ventral laminae where locomotor circuits are located [21]. On in vitro spinal networks, application of strychnine alone evokes irregular and asynchronous discharges while application of bicuculline per se produces a more structured repetitive activity [22, 23]. It may also be suggested that when one type of synaptic inhibition is blocked, the other one can at least in part expand its role because the circuitry is not arrested in a state of sustained excitation. It is noteworthy that the persistent rhythmic activity evoked by the convulsants strychnine and bicuculline is not associated with extensive neuronal or glial death [24, 25], indicating that spinal networks are far more resistant than brain networks to seizure-evoked neurodegeneration [26].
Principal Properties of GABAergic Mechanisms in the Spinal Cord
GABA is produced by decarboxylation of l-glutamate by glutamic acid decarboxylase (GAD), of which two isoforms exist: the transiently activated GAD65, which synthesizes vesicular GABA to be released by exocytosis, and the constitutively active GAD67, responsible for cytosolic GABA released by paracrine diffusion [27, 28]. In the spinal cord, GAD67 immunostaining has been found in cell bodies and fibers, while GAD65 is mainly located at synaptic terminals [29]. In addition to GABA locally released by spinal neurons and glial cells, GABAergic descending projections from the ventromedial medulla of the brainstem reach ventral and dorsal horns [30–32]. The development of the spinal GABAergic system is guided by several descending projections and the perinatal interruption of these projections impairs the regulation of GABA synthesizing enzymes [33] and receptors in the spinal cord [34]. For instance, interruption of descending serotoninergic input disrupts maturation of spinal GABAergic systems [34].
GABA acts on multiple ionotropic receptors, namely the A subtype, which drives a fast synaptic inhibition and the C subtype, whose role in the spinal cord is however limited, even if functionally expressed in the postnatal mammalian spinal cord [35]. Moreover, GABA acts as mediator of presynaptic inhibition by activating the G protein–coupled B receptor involved in a slower neuromodulating action particularly at presynaptic level via inhibition of calcium conductances [15, 18].
In adult neurons, GABA A receptor–mediated inhibition is due to the permeation of Cl- (and HCO3-) through an intrinsic channel that drives an influx of Cl- into the postsynaptic cell (Fig. 2a) [36, 37]. Conversely, in the first postnatal days of life, the opening of GABA ARs coincides with the Cl- electrochemical gradient (driving force) set at the less negative value and, thus, it drives Cl- efflux across the neuronal membrane. This phenomenon decreases intracellular negative charges with consequent cell depolarization from resting potential. It should also be noted that the opening of Cl- channels reduces membrane resistance and temporarily determines a conductance short-circuit (shunting), which limits further depolarization by incoming excitatory inputs. Thus, GABA-mediated depolarization exerts an inhibitory function in neonatal spinal neurons [37]. An action similar to GABA on neonatal neurons is displayed by afferent terminals throughout their maturation and adult stages, due to the high concentrations of intracellular Cl- in Dorsal root ganglions (DRGs) [38].
Fig. 2.
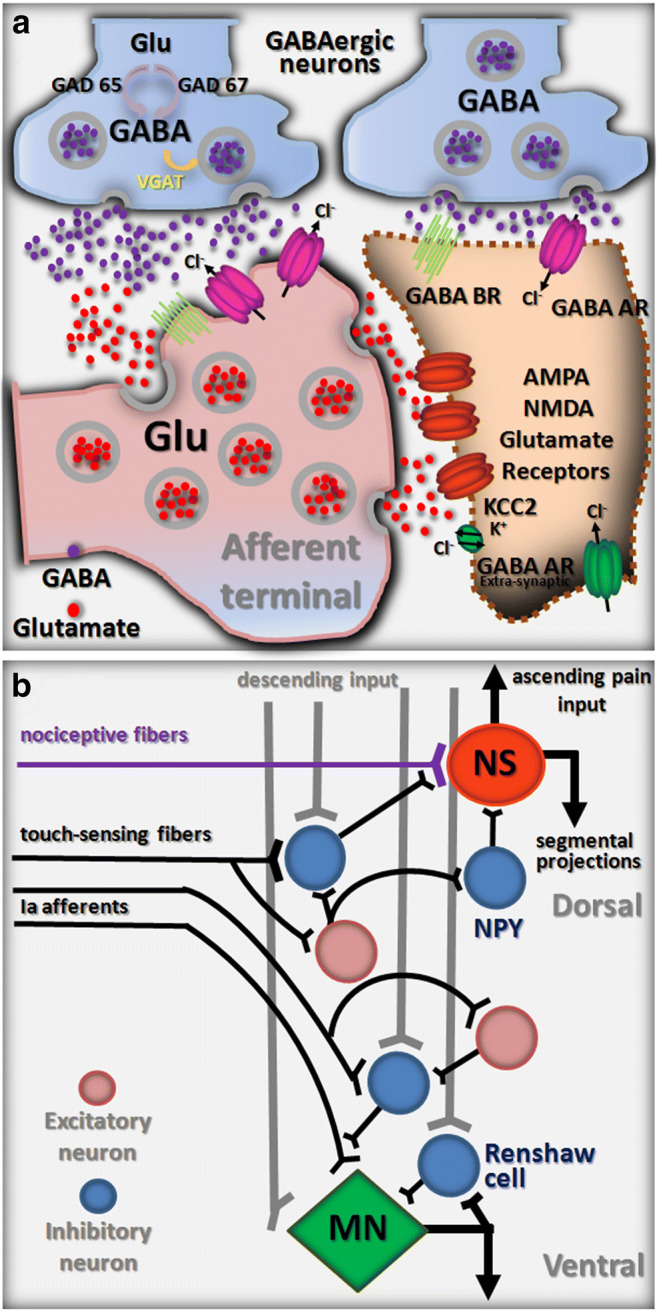
GABA-mediated inhibition at the cellular and network levels. a Schematic representation of two prototypical GABAergic synapses mediating pre (left)- and post (right)-synaptic inhibition, respectively. The main cellular and molecular players relevant to a spinal cord injury are depicted as discussed in this review. b Simplified wiring diagram of the basic GABAergic circuits involved in presynaptic inhibition of afferent input. NS, nociceptive-specific projection neuron; MN, motoneuron
It is important to emphasize that, in the neonatal spinal cord, the functional outcome of GABA-mediated activity may depend on the location of GABAergic synapses on postsynaptic neurons and their Cl- equilibrium potential [39] because the shunting effect is briefer than the membrane depolarization that, if prolonged, may facilitate excitation [39].
Noteworthily, there is also a subpopulation of extrasynaptic GABA ARs with distinct subunit composition and high affinity to GABA [16, 17, 40], generating tonic modulation of sensory transmission [41]. As exemplified in Fig. 3a, b, for the strong distribution of GABAergic GAD67 neurons in the dorsal horn, the corresponding expression of GABA ARs is intense in inner dorsal laminae (II, III), around the central canal (X), and the ventral horns (VII-IX), where GABA ARs are found at axo-axonic contacts and extra-synaptic sites [43, 44].
Fig. 3.

Expression of GABAergic neurons in the spinal cord and real-time glutamate release from spinal cord following experimental spinal cord injury (SCI). a Typical neuronal staining with neuronal nuclear protein (NeuN; red) restricted to the spinal cord tissue region in a spinal cord slice of a GAD67-glial filament protein (GFP) expressing mouse (green). Example of 22 DIV slice with two regions of interest (ROIs), namely a dorsal and a ventral horn, and a dorsal root ganglion (DRG). b Histograms showing the number of GAD67-positive cells (light green columns) or NeuN-positive cells (orange columns) at 22DIV, in control slices. Inset with the circle chart showing the percentage of GAD67 from NeuN-positive cells (redrawn from Mazzone and Nistri, 2019). c Examples of the time-course of endogenous glutamate release detected by glutamate biosensor in cultures that were treated with 0.5 mM kainate (blue traces, mean ± SD, n=5 slices). Glutamate concentrations in microdialysis samples collected after spinal cord injury, filled circles (redrawn from [42])
Neuronal Chloride Homeostasis in the Spinal Cord is Regulated by Two Transporters
The synaptic action of GABA and glycine depends on the intracellular concentration of Cl- that is primarily maintained by cation-chloride co-transporters [45]. Among the most important families of Cl- transporters, the Na+-K+-2Cl− cotransporter 1 (NKCC1) and KCC2 reciprocally control the intracellular Cl- concentration whose efflux causes, for instance, primary afferent-mediated depolarization with depression of excitatory inputs [46, 47]. Cl- transport into the cell is mostly due to NKCC1 activity, whereas KCC2 extrudes Cl- via a fast and concentration-dependent process generated by Na+/K+-ATPase [46, 47]. Previous studies have demonstrated that Cl- transporter expression and Cl- homeostasis are regulated by developmental changes that include gene transcription modification, posttranslational and trafficking alterations [47–49]. NKCC1 expression is widespread in neurons, glial, blood vessels, and other epithelial cells in the developing and mature central nervous system [50]. On the contrary, KCC2 is restricted to the somatodendritic membrane of mature central neurons and is almost absent in neuronal axons, peripheral neurons, and non-neuronal cells [51, 52]. Due to the broad NKCC1 distribution, NKCC1 null mice have been used to examine the transporter expression and its impact to induce abnormal GABA responses by DRG [53] and cortical neurons [54].
The strength of postsynaptic inhibition, related to Cl- homeostasis, is hampered in several pathophysiological conditions [55] such as seizure, epilepsy, stroke, and ischemic injury [33, 56] and proprioception disorders [57]. Indeed, impaired excitation/inhibition balance due to changed NKCC1 or KCC2 expression was also related to chronic stress [58], brain or peripheral injury [47, 59], and locomotor activity after spinal cord injury [60, 61] or developmental changes [62–64].
In rodent models of spinal cord injury, the role of intracellular chloride concentration and the modulation of cation chloride co-transporter expression have been amply investigated [65, 66]. In particular, synaptic inhibition, KCC2 and NKCC1 expression, and functional recovery were reportedly improved by programmed exercise or bumetanide, a pharmacological antagonist of NKCC1, 28 days after spinal cord transection in rats [67]. Similarly, a reduction in tissue damage and edema was observed by using bumetanide in a spinal cord contusion model [68]. A recent study has shown that the application of anodal trans-spinal direct current stimulation plus bumetanide administration downregulated the expression of NKCC1 after spinal cord contusion with significant amelioration of spasticity and locomotor muscle tone [69]. This is strong evidence that modulation of chloride homeostasis by NKCC1 pharmacological regulation during pathological conditions such as spinal cord injury can favor locomotor network improvement.
Presynaptic GABAergic Inhibition and Neuropathic Pain
Depolarizing axo-axonic synapses on primary afferent fibers filter incoming input from the periphery via membrane shunting and Na+ channel inactivation [38]. This basic wiring scheme fulfills multiple functions in sensory-motor networks. Indeed, a first mechanism to gate pain signals is represented by touch-sensing fibers depolarizing nociceptive primary afferents, thus causing pre-synaptic inhibition of nociceptive input. Furthermore, presynaptic primary afferent depolarization also contributes to shaping motor reflexes and efficiently modulates rhythmic motor behaviors, such as stepping and scratching, in response to proprioceptive input about joint position. Descending commands targeted to local interneurons control the efficiency of presynaptic inhibition triggered by peripheral inputs (Fig. 2b). Although this key frame is complicated by additional neuronal elements that release several types of neurotransmitters and neuropeptides onto primary afferents, the role of GABAergic interneurons remains crucial.
Based on the expression of transcription factors, different subtypes of spinal interneuron with distinct settling positions, neurotransmitter expression, and profiles of connectivity have been identified [70], among which a few have an inhibitory phenotype [9]. In particular, an adenovirus vector including a neuropeptide Y promoter has been recently used to discover, in the superficial dorsal horn, a subset of inhibitory GABAergic interneurons (AAV-NpyP) with the ability to prevent the conversion of touch-sensing signals into pain-like behavioral responses [71]. This class of interneuron receives mono- or polysynaptic excitatory inputs from touch-sensing fibers and uses GABA for transmitting inhibitory signals to lamina I neurons that project to the brain, thus avoiding abnormal excitation following innocuous mechanical stimulations (Fig. 2b). Dysfunctions of GABAergic transmission at the level of dorsal microcircuits impair the mechanisms of presynaptic inhibition, resulting in neuropathic pain states [72]. Neuropathic pain is one of the most frequent complications in paraplegics, with an incidence of 53% [73], and is often treated with GABAergic drugs [74, 75].
Indeed, the severity of neuropathic pain states following an experimental SCI [76] and other neurologic disturbances [77] is correlated to a reduced GABAergic tone, as the loss of GABAergic inhibitory interneurons in the superficial dorsal horn is verified by the reduction in GAD65/67 immunostaining. Thus, interventions for restoring the impaired production of GABA and GADs in the dorsal horns also alleviate pain states [77].
NKCC1 is crucial for the accumulation of Cl- in DRG neurons, leading to depolarizing GABA responses on primary afferents. Different studies demonstrated a transient upregulation of NKCC1 at DRG neurons after nerve injury indicating that Cl- efflux contributes to presynaptic inhibition and neuropathic pain induction [78–80]. Consequently, transgenic knockout mice lacking NKCC1 show impairments of presynaptic inhibition and significant alterations in locomotor and pain behaviors [53, 81]. Recently, disruption of NKCC1/KCC2 balance and chloride gradient below the injury site were found after spinal cord cervical contusion demonstrating the contribution of Cl- homeostasis for spasticity and chronic pain [82]. Indeed, in a rat model of neuropathic pain, the use of the extrusion enhancer CLP257, a KCC2-selective analog that lowers Cl- intracellular concentration, can alleviate hypersensitivity [83]. Hyperalgesia and allodynia were improved by using bumetanide for 2 weeks following sciatic nerve lesion, demonstrating the role of cation chloride co-transporter expression to modulate nociceptive pathways [84]. These data demonstrate that neuronal GABA neurotransmission is dependent on precise regulation of the level of intracellular chloride, which is determined by the coordinated activities of cation chloride co-transporters and could open new perspectives to prevent or alleviate neuropathic pain and functional recovery after SCI.
Collectively, these data show notably similar features between SCI and neuropathic pain, as they may both originate from alterations of presynaptic GABAergic mechanisms, which in turn broaden the potential translation of novel approaches to redress the tilted balance between excitation and inhibition in either neurological conditions.
Glycine Is a Fast Inhibitory Transmitter in the Spinal Cord
In adult rats, GABAergic axon terminals represent only 20% of the inhibitory input converging onto lumbar motoneurons, while the remaining 80% are glycinergic [85].
Glycine is a fast inhibitory transmitter on spinal motoneurons [19, 86], and it might be co-released with GABA at certain synapses [87]. However, not all synaptic boutons on motoneurons have both inhibitory neurotransmitters, but rather a strong prevalence of glycine alone [88]. Postsynaptic GABA A and glycine receptors are often, albeit not necessarily, co-localized [89] and aggregated in clusters formed by the submembrane scaffolding protein gephyrin [90, 91].
The glycinergic system is relatively insensitive to spinal transection [92]. Indeed, both the density of glycine receptors on motoneurons and the kinetics of glycine-mediated currents remain unchanged [34]. In accordance with these observations, the concentration of glycine, as determined by HPLC on spinal cord homogenates (2–12 h after spinal cord contusion), is preserved [93]. Only much later (3 weeks from transection), the expression of glycine receptors is temporarily decreased with subsequent recovery and re-emergence of physiological reflexes [94]. After complete spinal transection, the comparatively well-preserved glycinergic system at segmental level below the lesion may represent one significant component for neurorehabilitation protocols [92].
Since the main focus of the present review manuscript is the dysfunction of GABAergic mechanisms in damaged spinal networks, we refer the reader to previous work to examine the role of glycine after SCI [34, 92, 94–97].
Early Peak of GABA Immediately after SCI
Mechanical impact to the spinal cord massively increases the extracellular concentration of several neurotransmitters including GABA. Experimentally, a strong increase of GABA at the lesion site has been observed shortly after an SCI in vivo [42] following the very early rise in glutamate concentration (Fig. 3c). The increased extracellular concentration of GABA rapidly declines following SCI and later recovers to the pre-trauma levels [42, 93, 98]. The peak of GABA after SCI originates from not only the destruction of the membrane of GABAergic and glia cells but also the synaptic release at the site of injury [99] facilitated by spreading depolarization along the injured tissue [100]. The contribution of circulating GABA leaking through the impaired blood-spinal barrier is probably a minor one as GABA concentrations in the plasma [101, 102] are far below the ones found at the lesion site. Nevertheless, there might be enough GABA to activate highly sensitive extra-synaptic GABA receptors such as the ones incorporating the δ subunit [40]. An additional contribution to the peak in extracellular GABA immediately after SCI comes from the reversed function of membrane GABA transporters that depend on Na+ concentrations. In both neurons and glia, physiological reuptake of GABA is coupled to Na+ and Cl- inflow into the cell [103]. The increased concentration of intracellular Na+ (and Cl-) caused by spreading depolarization following an acute injury reverts the transport systems to extrude GABA [104]. At the same time, downregulation of the vesicular GABA transporter caused by SCI [105] increases the amount of cytosolic GABA available for extrusion.
The peak of GABA corresponds to the onset of a transient depression of spinal reflexes below the level of injury named spinal shock [106] typically present after severe spinal contusions in rats [107], although rarely found after surgical transection of the cord [108]. We, therefore, propose a role for GABA in spinal shock alongside a similar role for glycine [96].
Fast Synaptic GABAergic Transmission Is Early Affected by Spinal Cord Injury
The excitation/inhibition balance ensures physiological motor responses executed by healthy spinal cords and may be directly altered by SCI. Future studies are required to clearly identify the components of the locomotor systems primarily altered after SCI and their impact on the excitation/inhibition balance. In broad terms, changes in excitation/inhibition balance might originate from an alteration in cellular mechanisms and/or disruption and rewiring of local networks. Hence, in response to spinal damage, GABAergic cells show particular vulnerability, as their number decreases [109]. One reason for their vulnerability might be their location because important members of the spinal GABAergic population are commissural interneurons, which cross the midline and project ventrally, thus offering a long section liable to injury [110]. Furthermore, the ventral region is vulnerable to SCI because of its dense vascularization prone to produce large hemorrhage and neuronal loss [111]. In addition, in the acute phase of SCI, complex neurodegenerative events develop to generate a secondary injury that amplifies and spreads damage to the neighboring tissue [112]. Our former studies have provided a comparative description of the different neuronal cell types with particular vulnerability to injury [25, 113, 114]. In the early phases of experimental SCI, significant reduction in GABAergic GAD65 expression occurs at the injury site [115].
One important contributor to secondary injury is the over-activation of glutamate receptors, leading to a massive influx of calcium ions into spinal cells and contributing to the release of free radicals from mitochondria, such as reactive oxygen and nitrogen species, in turn triggering intracellular toxic cascades (excitotoxicity; [25, 113, 116–118]).
The oxidative stress occurring during secondary damage is one important cause for the impairment in GABAergic neurotransmission, because reactive oxygen species increase synaptic release of GABA [119, 120] that desensitizes GABA ARs [121]. Reactive oxygen species also alter the function of GABA A receptor-gated Cl- channels due to a reduced driving force for Cl- because of failure of its transport [122]. In addition, free radicals alter the binding characteristics of GABA, possibly by affecting redox-sensitive receptor sites or via peroxidation of membrane lipids surrounding the receptor [122].
GABAergic descending inputs that control motoneuron excitability are also damaged by SCI contributing to functional motor deficits and other disabling consequences. In the majority of people with chronic SCI, paralyzed muscles are often accompanied by involuntary contractions (spasticity), increased resistance to passive stretch (muscle hypertonia), and exaggerated motor responses to light peripheral stimulation (hyperreflexia; [123]). Indeed, despite the reduced excitability of axons at the periphery [124], a brief sensory stimulation (< 20 ms) evokes a prolonged depolarization (~ 1 s) of single motor units apparently without efficient synaptic inhibition. Conversely, the same light afferent stimulus applied to neurologically intact subjects generates a sustained depolarization interposed by an inhibitory phase [125]. The increased amplitude that characterizes motor responses after SCI and the lack of inhibitory contributions have been associated with multiple neuronal mechanisms at both cell and network levels. While the increased excitation should be, at least in part, attributed to the activation of Na+ and Ca2+ persistent inward currents (PICs) in motoneurons [126–130], a pivotal role in reduced inhibition has been ascribed to depression in GABAergic transmission [92, 131]. Indeed, at pre-synaptic level, despite the increased size of GABAergic synapses, the lower number of vesicles in the active zone [132] determines less neurotransmitter available for release. At the same time, an SCI also produces aberrant hyper-connectivity among GABAergic interneurons, with the formation of new axo-axonic synapses [132] that, along with changes in Cl- transporter isoforms, might contribute to the disinhibition reported after SCI [133].
Noteworthily, dysregulation of the balance between excitation and inhibition may also result from changes in other components of the spinal network after injury. For instance, aberrant sprouting of primary afferents or expansion of interneuronal receptive and projective fields after SCI may augment the excitatory drive to spinal networks [134]. On the other hand, inhibition is affected by the interruption of serotoninergic descending tracts, which modulate inhibitory interneurons, like Renshaw cells [135, 136]. Moreover, Renshaw cell recurrent circuitry might become disconnected from motoneurons [137] suppressing their excitatory drive to Renshaw cells, in turn reducing the GABAergic inhibitory feedback. Also, changes in long-term gene expression, such as upregulation and phosphorylation of several signaling proteins in spinal ventral horns, have been linked to early and long-term changes in spinal excitability, leading to spasticity states after spinal trauma [138].
Furthermore, circuit reorganization after spinal cord injury occurs also at the supraspinal level. The strength of brainstem reflexes is enhanced as a result of increased excitability and reduced GABA-mediated inhibition in the brainstem circuits that project to spinal interneurons [139].
Table 1 shows interventions aimed at normalizing the altered excitability after injury from multiple experimental settings. Pharmacological manipulations, transplants of different cell lineages, and activity-dependent protocols have been applied in the acute and chronic phases of SCI to exploit GABA-related mechanisms and rescue homeostasis between excitation and inhibition.
Table 1.
GABAergic mechanisms targeted to rescue altered inhibition
| Intervention | Model | GABAergic mechanisms | Main outcome | Reference | |
|---|---|---|---|---|---|
| Physical exercise | • Treadmill running | • Partial sciatic nerve (PSL) ligation in adult C57BL/6 J mice |
• Restoration GABAergic interneuron numbers • Upregulation of GAD65/67 immunoreactivities |
• Alleviates allodynia and heat hyperalgesia • Positive correlation between GABA levels and the thresholds of von Frey or plantar tests |
Kami et al. (2016) [77] |
| Physical exercise & Pharmacology |
• Cycling exercise • Bumetanide (NKCC1 antagonist) • DIOA([(dihydroindenyl)oxy]alkanoic acid; KCC2 antagonist) |
• SCI complete transection in adult female Sprague Dawley rats |
• Increase in KCC2 levels and decrease in NKCC1 expression levels • Blockage of NKCC1 impacts on reflex recovery • Apparent modulation of KCC2, but not NKCC1, by BDNF |
• Exercise contributes to functional recovery by restoring chloride homeostasis | Côté et al. (2014) [67] |
| Pharmacology |
• CLP290 (KCC2 agonist) • Bumetanide (NKCC1 inhibitor) • 8-OH-DPAT (5HT1A/7 agonist) • Quipazine (5HT2A/C agonist) • CP101606 (NMDA receptor antagonist) • Baclofen • L838414 (GABA A-positive allosteric modulator) |
• SCI bilateral hemisection in adult mice | • Increase in KCC2 function | • Restores inhibition in the injured spinal cord, leading to functional recovery | Chen et al. (2018) [140] |
|
• CLP257 • CLP290 |
• NG-108 cell line and HEK293-cl cells • Horizontal spinal dorsal horn slices obtained from animals with peripheral nerve injury (PNI) • PNI in adult male Sprague-Dawley rats |
• CLP257 and CLP290 enhance Cl− extrusion | • CLP257 has antinociceptive properties in PNI animals | Gagnon et al. (2013) [83] | |
| • Intrathecal administration of brain-derived neurotrophic factor (BDNF) and of BDNF sequestering agent, TrkB-IgG | • SCI transection in adult male Sprague-Dawley rats | • Increase in KCC2 expression post-SCI by BDNF | • BDNF plays an antinociceptive role | Huang et al. (2017) [141] | |
| • Activation of 5-HT2A receptors with TCB-2 |
• SCI hemisection in adult female Wistar rats • Peroneal and tibial nerve injury by ligation and transection • Injection of TCB-2 and intrathecal DIOA injection |
• Increase in membrane KCC2 expression |
• Restores motoneuronal inhibition, and reduces SCI-induced spasticity, mechanical and thermal hyperalgesia • Nerve injury-induced neuropathic pain was not attenuated by TCB-2 |
Sánchez-Brualla et al. (2018) [142] | |
|
• Midazolam (allosteric GABAA modulator) • THIP (GABA agonist) • Bicuculline • Gabazine (antagonist of GABA ARs) • Strychnine • L-Alanine |
• Mouse organotypic spinal slice cultures, excitotoxicity induced by kainate | • Increase in GABA receptor activity through pharmacological GABA agonism | • Decreases excitotoxic death in spinal networks in vitro | Mazzone and Nistri (2019) [143] | |
| • TGN-20 (AQP4 inhibitor) and bumetanide | • SCI contusion rats |
• Upregulation of AQP4 mRNA and reduction of NKCC1 expression |
• Reduces SCI edema and tissue destruction |
Yan et al. (2018) [68] |
|
| • Anodal trans-spinal direct current stimulation and bumetanide | • SCI contusion in CD-1 mice | • Upregulation of NKCC1 | • Reduces spasticity and increases muscle tone | Mekhael et al. (2019) [69] | |
| Transplantation | • Transplantation of MGE-like cells derived from human embryonic stem cells (hESC-MGEs) | • SCI moderate contusion in B6.CB17- Prkdcscid/SzJ transgenic mouse | • Migration and differentiation into GABAergic neurons subtypes |
• Transplanted cells functionally integrate into host’s spinal cord • Attenuate mechanical allodynia of hind paws • Sustained motor recovery |
Fandel et al. (2016) [144] |
| • Transplantation of embryonic precursors of GABAergic neurons from medial ganglionic eminence (MGE) | • Peripheral nerve injury models of neuropathic pain in adult mouse | • Differentiation into GABAergic neurons | • Transplanted cells functionally integrate into host’s dorsal horn circuits | Llewellyn-Smith et al. (2017) [145] | |
| • Transplantation of fetal neural stem cells (fNSC) extracted from the telencephalic vesicles (TV) and the ventral medulla (VM) | • SCI contusion in adult Wistar rats |
• Differentiation into GABAergic neurons • Greater proportion of GABAergic cells from the TV group compared to the VM group |
• Improves from thermal hyperalgesia • Ameliorates mechanical allodynia |
Batista et al. (2019) [146] | |
| • Transplantation of differentiated human induced pluripotent stem cell-derived GABAergic (iGABAergic) neurons | • Peroneal and tibial nerve injury by ligation and transection in adult mice |
• Differentiation into GABAergic neurons. • VGAT and GAD65/67 expression |
• Transplanted cells functionally integrate into host’s dorsal horn active inhibitory circuits • Reduces tactile allodynia |
Manion et al. (2020) [147] | |
| • Transplantation of GABAergic neural progenitor cell and intensive locomotor training (ILT) | • SCI compression in adult male Sprague Dawley rats | • Upregulation of KCC2 |
• Reduces mechanical allodynia and thermal hyperalgesia • Reduces pro-inflammatory markers |
Dugan et al. (2020) [148] | |
| Genetic manipulation |
• NKCC1 gene ablation in DRGs • Bumetanide |
• NKCC1 knockout mice, deletion of exon 9 of the gene |
• Absence of Cl- accumulation in DRGs • Absence of GABA depolarizing responses |
• Alters nociception and motor coordination | Sung et al. (2000) [53] |
Despite the plethora of experimental approaches, restoring physiological spinal inhibition in the clinic remains a timely and demanding challenge that requires further studies. Indeed, potentiating the GABAergic system, when not carefully timed, might even hinder activity-based rehabilitation and electrical neuromodulation protocols for motor recovery, by depressing synaptic transmission [149] and reducing excitability of locomotor spinal circuits [150].
Pharmacological Neuroprotection by GABA Modulation after Experimental Lesion
Several GABAergic mechanisms targeted at restoring functional homeostasis and rescuing neuronal loss after injury have been explored with different experimental models (Table 1). For their part, reduced preparations from neonatal rodents suggest that a large rise in extracellular glutamate is responsible for the excitotoxicity arising early after SCI (Fig. 3c). In this model, excitotoxicity is produced by transient application of the powerful glutamate analog kainate [151]. While glutamate excitotoxicity can be attenuated with agents that decrease its release [152–156], a distinct approach is to boost inhibition to render spinal neurons less excitable. Thus, neuroprotection by general anesthetics like methoxyflurane and propofol indicates that this process effectively counteracts excitotoxicity [157–159] albeit through distinct molecular mechanisms. In fact, while methoxyflurane primarily acts by hyperpolarizing motoneurons via opening a voltage-independent K+ channel [159], propofol enhances GABA ARs activity by binding to a discrete allosteric site [158]. The implication of these results is that neuronal inhibition, regardless of its effector mechanisms, is an important factor to contrast excitotoxicity. Nevertheless, using general anesthetics as a neuroprotective drug is complex and prompts the search for alternative approaches. In line with this strategy, more direct investigation into the effects of GABA receptor agonists and antagonists on experimental spinal damage has shown that modulation of extrasynaptic GABA ARs could prevent excitotoxic death of spinal organotypic cultures [143]. In particular, the allosteric GABA A modulator midazolam and the GABA agonist 4,5,6,7-tetrahydroisoxazolo [5,4-c] pyridin-3-ol (THIP; preferentially acting on extrasynaptic receptors) are powerfully effective [143]. In addition, the GABA AR antagonist bicuculline prevents the neuroprotective effect of propofol via GABA AR function, suggesting the importance of GABA receptor activity in modulating excitotoxicity [157]. Endogenous neurosteroids can also induce neuroprotection by upregulating GAD67 enzyme level [160] or GABA AR function [161]. Thus, even if transient changes in GABAergic synaptic transmission after SCI might not be immediately translated into neuroprotection, other GABAergic targets are available to perform this role. Interestingly, cultured motoneurons show that the excitotoxic action of glutamate is limited by direct application of GABA agonists [162, 163].
The neuroprotective role of GABA as well as the activation of different GABA receptors following insults to the CNS [15] may represent potential targets to limit damage and develop innovative and selective therapeutical approaches.
However, side effects of current pharmacological therapy for other neurological disturbances, as epilepsy, suggest potential risks from potentiating GABAergic mechanisms [164]. Likewise, the use of the anticonvulsant baclofen determines muscle weakness and sedative effects [165], along with a baclofen-withdrawal syndrome, with a psychotic status when the drug is abruptly discontinued [166]. However, since GABA BRs are less prone to receptor desensitization, the abovementioned adverse effects are likely to be more pronounced than interventions targeted to GABA ARs.
Neurons and Astrocytes May Counteract Excitotoxicity via GABAergic Mechanisms
One key element to modulate synaptic transmission and neuronal network activity seems to be the presence of astrocytes and the type of neuron involved [167]. It is now widely accepted that astrocytes can modulate neuronal activity through the tripartite synapse [168]. Thus, cells immunoreactive to S100β (a cytoplasmic calcium-binding protein mainly expressed by glia), may take part in tissue protection and repair, as well as they are useful biomarkers for brain or spinal cord injury [169]. These cells are the most abundant astrocyte cell type in the ventral horn area and less abundant in the dorsal horn [170]. The differential distribution of glial cells within the spinal cord regions might be an important factor in considering the high vulnerability of neurons to excitotoxicity [25, 113, 114]. Accumulating evidence demonstrates the role of astrocytes in GABA synthesis and release, as well as in the activation of GABA receptors on neighboring neurons [60]. During synaptic transmission, GABA release triggers astrocytic release of calcium from the endoplasmic reticulum via the inositol 1, 4, 5-trisphosphate pathway [171]. As pointed out by Christensen and collaborators [172], in the dorsal horn of adult turtle, astrocytes coordinate calcium-mediated excitation and tonic inhibition by GABA ARs to induce phasic release of GABA. Finally, lampreys show spontaneous functional recovery and neuroprotection after complete SCI that depends on astrocytes properties related to GABA accumulation and neurotransmitter uptake [173].
Although promising for the design of novel interventions to rescue cellular loss after spinal damage, these results must be considered with caution and must be supported by compelling new studies to validate any translation to clinical use. Potential limitations can originate when interpreting results coming from different species, genders, age, phases of lesion, and injury protocols (Table 1). In fact, the distribution of GABA ARs and their binding properties might vary among different strains [174], while also circulating sex hormones affect the sensitivity of GABA ARs to the allosteric endogenous modulator allopregnanolone in females [175]. Moreover, mechanical properties of the spinal cord change with size, making it hard to compare the severity of experimental injuries among studies of animals at different developmental stages [176].
Prolonged Dysfunction of Fast GABAergic Transmission after SCI
After spinal cord transection, the number of GABA ARs increases in fast flexor motoneuronal pools and synaptic clustering augments as a consequence of subunit overexpression. This latter feature is reversed to control after step training and aids functional recovery [177]. Furthermore, long-term changes in protein and mRNA levels of GAD67 (but not GAD65) have been found after a chronic transection, possibly leading to increased GABA production in spinal neurons below the site of injury [29]. Interestingly, GAD67 is the predominant form in ventral horn neurons around motoneuronal pools [178] and the recovery of locomotor functions in SCI rats corresponds to a return of GAD67 toward baseline levels [179].
Enhancement in motoneuron excitability stems from their dysregulation of intracellular Cl- caused by the spinal lesion itself [180]. In lumbar motoneurons, thoracic SCI reduces the expression of KCC2 which co-transports potassium and Cl- outside the cell [181]. The switch of GABA A from inhibition to excitation contributes to the spasticity of hind limbs [182]. In fact, upregulation of KCC2 after transection restores some locomotor activity in the mouse [140].
The interaction between excitation and inhibition at chronic stages of SCI remains an incompletely understood process as much as the relative weight of GABA and glycine mediated transmission. In fact, although glycine receptor operation is also sensitive to intracellular Cl- [183, 184], the kinetics of glycinergic currents are not affected after spinal transection [34] and the administration of glycine continues to produce inhibitory effects and limit spasticity after SCI [95]. Pharmacological block of both GABA A and glycine receptors prolongs spasms in chronically transected animals, confirming that a degree of fast inhibition remains efficacious even after lesion [95, 185]. In keeping with these observations, optogenetic activation of spinal inhibitory interneurons silences spasms evoked by electrical afferent stimulation [185]. Conversely, Edgerton and Roy [186] have proposed low doses of pharmacological blockers of Cl--mediated inhibition for recovery of gait in injured animals. Antagonism of inhibitory transmission has been claimed to facilitate locomotion by limiting excessive inhibition following SCI [97, 187, 188].
In sum, after SCI, the excitability of spinal networks at rest is changed at distinct nodes of the pre-motor neuronal circuitry by the appearance of complex contributions with a very fine balance among them. On the one hand, GABA-mediated depolarizing signals result from the reversed Cl- gradient [182, 189]. On the other hand, supplementary GABA-mediated inhibitory input arises from upregulation of GABA synthesis [178], overexpression of GABA AR subunits [34, 177], and a greater activation of inhibitory interneurons [185]. Ultimately, whether synaptically released GABA can either inhibit or facilitate excitatory inputs depends on the time course of the event and its membrane topography on the postsynaptic neuron [39]. Hence, the longer lasting the effect of GABA is, the higher is the likelihood of inducing neuronal excitation.
Factors Regulating the Excitability of Motoneurons after SCI
First, chronic changes in motoneuronal excitability after human SCI depend on how close these cells are to the site of spinal injury. Namely, while perilesional motoneurons are hypo-excitable, those farther from the lesion epicenter show increased excitability [190]. In line with this finding, in subjects with incomplete SCI, corticospinal pathways evoke aberrantly high facilitation of motor output distant from the epicenter of the lesion. Conversely, no change is reported at the level of injury and nearby segments [191]. Animal experiments indicate that sustained depolarization of sacral motoneurons below the lesion [192] is accompanied by hypertonia, hyperreflexia, and clonus [193, 194]. Other studies have demonstrated aberrant membrane properties of lumbar motoneurons underlying hind limb spasticity after thoracic spinal lesions in rodents although direct evidence for the excitability of motoneurons close to the contusion site is still missing [181, 189]. While motoneuron properties (essential to support motoneuron firing) slowly recover to their preinjury state, their corresponding receptive fields remain broad so that sensory input to even a small area of the limb can trigger widespread excitation capable of generating whole-limb spasms [195]. Further studies are eagerly awaited to explore whether different states of excitability of motoneurons proximal and distal to an SCI are related to the early transient changes in extracellular GABA concentrations at the epicenter of injury. Potentially, these findings might bring novel pharmacological interventions to acutely modulate GABAergic transmission below the lesion [196] with the timely goal of preventing the onset of spasticity in addition to the widely-used administration of the GABA BR agonist baclofen [197]. In particular, an important issue is whether activation of spinal GABA ARs may be able to counteract the upregulation of the persistent sodium current of motoneurons typically observed after lesion [198]. This conductance is considered to be the target for neuromodulation, a phenomenon in which GABA is expected to play a role [199]. PICs which comprise sodium as well as calcium conductances [126–130, 200] contribute to the nonlinearity between the level of network excitation and motor output [201]. As spinal neurons possess strong plasticity during recovery after SCI [202], GABA AR currents display more powerful control over PIC activation than glycinergic currents, an effect attributable to their slower kinetics [196]. Additionally, extrasynaptic GABA ARs (with their high sensitivity to even low GABA concentrations) may represent a further mechanism to downplay neuronal excitability even when synaptic transmission has failed after SCI. Nevertheless, the functional outcome of modulation by GABA receptor activity may also depend on the shifting balance between hyperpolarizing and depolarizing action of GABA due to post lesional changes in chloride transmembrane gradient [140, 180–182, 189] and their timing as discussed earlier.
In conclusion, restoration of locomotor network activity after injury depends on the correct interplay between excitation and inhibition and recovery of the fine balance between synaptic and non-synaptic GABA AR activity. These goals are eminently suitable for pharmacological investigations.
We suggest that this is a complementary strategy to concur with the use of new materials and cell transplants to a successful repair or reconfiguration of damaged locomotor networks that need a suitable functional milieu to reestablish their correct operation.
Acknowledgements
The authors are grateful to Dr. Elisa Ius for her excellent assistance in preparing the manuscript.
Availability of Data and Material
Not applicable
Abbreviations
- 5HT
Serotonin
- CPG
Central pattern generator
- DRG
Dorsal root ganglion
- GABA
Gamma-aminobutyric acid
- GAD
Glutamic acid decarboxylase
- GFP
Glial filament protein
- L
Lumbar
- MN
Motoneuron
- NeuN
Neuronal nuclear protein
- NMDA
N-methyl-d-aspartate
- NS
Nociceptive-specific projection neuron
- PICs
Persistent inward currents
- ROIs
Regions of interest
- SCI
Spinal cord injury
- SD
standard deviation
- THIP
4,5,6,7-Tetrahydroisoxazolo [5,4-c] pyridin-3-ol
- VGAT
vesicular GABA transporter
- VRs
ventral roots
Author Contribution
GLM and GT had the idea for the article; GLM, AM, AN, and GT performed the literature search and data analysis; GLM, AM, AN, and GT drafted the work; and GLM, AM, JBA, AN, and GT critically revised the work.
Funding
Open access funding provided by Scuola Internazionale Superiore di Studi Avanzati - SISSA within the CRUI-CARE Agreement. This study was supported by an intramural SISSA grant, CONICET, and Regular Associate Scheme of the Abdus Salam International Centre for Theoretical Physics (ICTP).
Declarations
Consent to Participate
Not applicable
Consent for Publication
Not applicable
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Berg RW, Alaburda A, Hounsgaard J. Balanced inhibition and excitation drive spike activity in spinal half-centers. Science. 2007;315:390–393. doi: 10.1126/science.1134960. [DOI] [PubMed] [Google Scholar]
- 2.Cazalets JR, Borde M, Clarac F. The synaptic drive from the spinal locomotor network to motoneurons in the newborn rat. J Neurosci. 1996;16:298–306. doi: 10.1523/JNEUROSCI.16-01-00298.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petersen PC, Vestergaard M, Jensen KHR, Berg RW. Premotor spinal network with balanced excitation and inhibition during motor patterns has high resilience to structural division. J Neurosci. 2014;34:2774–2784. doi: 10.1523/JNEUROSCI.3349-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson MD, Hyngstrom AS, Manuel M, Heckman CJ. Push-pull control of motor output. J Neurosci. 2012;32:4592–4599. doi: 10.1523/JNEUROSCI.4709-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shu Y, Hasenstaub A, McCormick DA. Turning on and off recurrent balanced cortical activity. Nature. 2003;423:288–293. doi: 10.1038/nature01616. [DOI] [PubMed] [Google Scholar]
- 6.Berkowitz A, Hao Z-Z. Partly shared spinal cord networks for locomotion and scratching. Integr Comp Biol. 2011;51:890–902. doi: 10.1093/icb/icr041. [DOI] [PubMed] [Google Scholar]
- 7.Ziskind-Conhaim L. Neuronal correlates of the dominant role of GABAergic transmission in the developing mouse locomotor circuitry. Ann N Y Acad Sci. 2013;1279:43–53. doi: 10.1111/nyas.12064. [DOI] [PubMed] [Google Scholar]
- 8.Grillner S, Jessell TM. Measured motion: searching for simplicity in spinal locomotor networks. Curr Opin Neurobiol. 2009;19:572–586. doi: 10.1016/j.conb.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiehn O. Decoding the organization of spinal circuits that control locomotion. Nat Rev Neurosci. 2016;17:224–238. doi: 10.1038/nrn.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kudo N, Yamada T. N-methyl-D,L-aspartate-induced locomotor activity in a spinal cord-hindlimb muscles preparation of the newborn rat studied in vitro. Neurosci Lett. 1987;75:43–48. doi: 10.1016/0304-3940(87)90072-3. [DOI] [PubMed] [Google Scholar]
- 11.Cazalets JR, Sqalli-Houssaini Y, Clarac F. Activation of the central pattern generators for locomotion by serotonin and excitatory amino acids in neonatal rat. J Physiol. 1992;455:187–204. doi: 10.1113/jphysiol.1992.sp019296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beato M, Nistri A. Interaction between disinhibited bursting and fictive locomotor patterns in the rat isolated spinal cord. J Neurophysiol. 1999;82:2029–2038. doi: 10.1152/jn.1999.82.5.2029. [DOI] [PubMed] [Google Scholar]
- 13.Curtis DR, Duggan AW, Felix D, Johnston GA. GABA, bicuculline and central inhibition. Nature. 1970;226:1222–1224. doi: 10.1038/2261222a0. [DOI] [PubMed] [Google Scholar]
- 14.Nistri A, Constanti A. Pharmacological characterization of different types of GABA and glutamate receptors in vertebrates and invertebrates. Prog Neurobiol. 1979;13:117–235. doi: 10.1016/0301-0082(79)90016-9. [DOI] [PubMed] [Google Scholar]
- 15.Sivilotti L, Nistri A. GABA receptor mechanisms in the central nervous system. Prog Neurobiol. 1991;36:35–92. doi: 10.1016/0301-0082(91)90036-Z. [DOI] [PubMed] [Google Scholar]
- 16.Baur R, Kaur KH, Sigel E. Structure of alpha6 beta3 delta GABA(A) receptors and their lack of ethanol sensitivity. J Neurochem. 2009;111:1172–1181. doi: 10.1111/j.1471-4159.2009.06387.x. [DOI] [PubMed] [Google Scholar]
- 17.Sigel E, Steinmann ME. Structure, function, and modulation of GABA(A) receptors. J Biol Chem. 2012;287:40224–40231. doi: 10.1074/jbc.R112.386664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis KL, Charney D, Coyle JT, Nemeroff C (2002) Neuropsychopharmacology - 5th Generation of Progress. Lippincott, Williams, & Wilkins, Philadelphia, Pennsylvania, pp 159–168
- 19.Curtis DR, Hösli L, Johnston GA. Inhibition of spinal neurons by glycine. Nature. 1967;215:1502–1503. doi: 10.1038/2151502a0. [DOI] [PubMed] [Google Scholar]
- 20.Davidoff RA, Shank RP, Graham LT, et al. Association of glycine with spinal interneurones. Nature. 1967;214:680–681. doi: 10.1038/214680a0. [DOI] [PubMed] [Google Scholar]
- 21.Liu TT, Bannatyne BA, Maxwell DJ. Organization and neurochemical properties of intersegmental interneurons in the lumbar enlargement of the adult rat. Neuroscience. 2010;171:461–484. doi: 10.1016/j.neuroscience.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 22.Bonnot A, Morin D. Hemisegmental localisation of rhythmic networks in the lumbosacral spinal cord of neonate mouse. Brain Res. 1998;793:136–148. doi: 10.1016/s0006-8993(98)00153-x. [DOI] [PubMed] [Google Scholar]
- 23.Streit J. Regular oscillations of synaptic activity in spinal networks in vitro. J Neurophysiol. 1993;70:871–878. doi: 10.1152/jn.1993.70.3.871. [DOI] [PubMed] [Google Scholar]
- 24.Mladinic M, Nistri A, Taccola G. Acute spinal cord injury in vitro: insight into basic mechanisms. In: Aldskogius H, editor. Animal Models of Spinal Cord Repair. Totowa: Humana Press; 2013. pp. 39–62. [Google Scholar]
- 25.Taccola G, Margaryan G, Mladinic M, Nistri A. Kainate and metabolic perturbation mimicking spinal injury differentially contribute to early damage of locomotor networks in the in vitro neonatal rat spinal cord. Neuroscience. 2008;155:538–555. doi: 10.1016/j.neuroscience.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 26.Ben-Ari Y. Cell death and synaptic reorganizations produced by seizures. Epilepsia. 2001;42(Suppl 3):5–7. doi: 10.1046/j.1528-1157.2001.042suppl.3005.x. [DOI] [PubMed] [Google Scholar]
- 27.Soghomonian JJ, Martin DL. Two isoforms of glutamate decarboxylase: why? Trends Pharmacol Sci. 1998;19:500–505. doi: 10.1016/s0165-6147(98)01270-x. [DOI] [PubMed] [Google Scholar]
- 28.Wei J, Wu J-Y. Post-translational regulation of L-glutamic acid decarboxylase in the brain. Neurochem Res. 2008;33:1459–1465. doi: 10.1007/s11064-008-9600-5. [DOI] [PubMed] [Google Scholar]
- 29.Tillakaratne NJ, Mouria M, Ziv NB, et al. Increased expression of glutamate decarboxylase (GAD(67)) in feline lumbar spinal cord after complete thoracic spinal cord transection. J Neurosci Res. 2000;60:219–230. doi: 10.1002/(SICI)1097-4547(20000415)60:2<219::AID-JNR11>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 30.Antal M, Petkó M, Polgár E, et al. Direct evidence of an extensive GABAergic innervation of the spinal dorsal horn by fibres descending from the rostral ventromedial medulla. Neuroscience. 1996;73:509–518. doi: 10.1016/0306-4522(96)00063-2. [DOI] [PubMed] [Google Scholar]
- 31.Holstege JC. Ultrastructural evidence for GABAergic brain stem projections to spinal motoneurons in the rat. J Neurosci. 1991;11:159–167. doi: 10.1523/JNEUROSCI.11-01-00159.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hossaini M, Goos JAC, Kohli SK, Holstege JC. Distribution of glycine/GABA neurons in the ventromedial medulla with descending spinal projections and evidence for an ascending glycine/GABA projection. PLoS ONE. 2012;7:e35293. doi: 10.1371/journal.pone.0035293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russ JB, Verina T, Comer JD, et al. Corticospinal tract insult alters GABAergic circuitry in the mammalian spinal cord. Front Neural Circuits. 2013;7:150. doi: 10.3389/fncir.2013.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sadlaoud K, Tazerart S, Brocard C, et al. Differential plasticity of the GABAergic and glycinergic synaptic transmission to rat lumbar motoneurons after spinal cord injury. J Neurosci. 2010;30:3358–3369. doi: 10.1523/JNEUROSCI.6310-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rozzo A, Armellin M, Franzot J, et al. Expression and dendritic mRNA localization of GABAC receptor rho1 and rho2 subunits in developing rat brain and spinal cord. Eur J Neurosci. 2002;15:1747–1758. doi: 10.1046/j.1460-9568.2002.02013.x. [DOI] [PubMed] [Google Scholar]
- 36.Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 37.Marchetti C, Pagnotta S, Donato R, Nistri A. Inhibition of spinal or hypoglossal motoneurons of the newborn rat by glycine or GABA. Eur J Neurosci. 2002;15:975–983. doi: 10.1046/j.1460-9568.2002.01927.x. [DOI] [PubMed] [Google Scholar]
- 38.Guo D, Hu J. Spinal presynaptic inhibition in pain control. Neuroscience. 2014;283:95–106. doi: 10.1016/j.neuroscience.2014.09.032. [DOI] [PubMed] [Google Scholar]
- 39.Jean-Xavier C, Mentis GZ, O’Donovan MJ, et al. Dual personality of GABA/glycine-mediated depolarizations in immature spinal cord. Proc Nat Acad Sci U S A. 2007;104:11477–11482. doi: 10.1073/pnas.0704832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benkherouf AY, Taina K-R, Meera P, et al. Extrasynaptic δ-GABAA receptors are high-affinity muscimol receptors. J Neurochem. 2019;149:41–53. doi: 10.1111/jnc.14646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lucas-Osma AM, Li Y, Lin S, et al. Extrasynaptic α5GABAA receptors on proprioceptive afferents produce a tonic depolarization that modulates sodium channel function in the rat spinal cord. J Neurophysiol. 2018;120:2953–2974. doi: 10.1152/jn.00499.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McAdoo DJ, Robak G, Xu GY, Hughes MG. Adenosine release upon spinal cord injury. Brain Res. 2000;854:152–157. doi: 10.1016/s0006-8993(99)02333-1. [DOI] [PubMed] [Google Scholar]
- 43.Sur C, McKernan R, Triller A. Subcellular localization of the GABAA receptor gamma 2 subunit in the rat spinal cord. Eur J Neurosci. 1995;7:1323–1332. doi: 10.1111/j.1460-9568.1995.tb01123.x. [DOI] [PubMed] [Google Scholar]
- 44.Alvarez FJ, Taylor-Blake B, Fyffe RE, et al. Distribution of immunoreactivity for the beta 2 and beta 3 subunits of the GABAA receptor in the mammalian spinal cord. J Comp Neurol. 1996;365:392–412. doi: 10.1002/(SICI)1096-9861(19960212)365:3<392::AID-CNE5>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 45.Viemari J-C, Bos R, Boulenguez P, et al. Chapter 1--importance of chloride homeostasis in the operation of rhythmic motor networks. Prog Brain Res. 2011;188:3–14. doi: 10.1016/B978-0-444-53825-3.00006-1. [DOI] [PubMed] [Google Scholar]
- 46.Nilius B, Droogmans G. Amazing chloride channels: an overview. Acta Physiol Scand. 2003;177:119–147. doi: 10.1046/j.1365-201X.2003.01060.x. [DOI] [PubMed] [Google Scholar]
- 47.Price TJ, Cervero F, Gold MS, et al. Chloride regulation in the pain pathway. Brain Res Rev. 2009;60:149–170. doi: 10.1016/j.brainresrev.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 49.Sun D, Murali SG. Na+-K+-2Cl- cotransporter in immature cortical neurons: a role in intracellular Cl- regulation. J Neurophysiol. 1999;81:1939–1948. doi: 10.1152/jn.1999.81.4.1939. [DOI] [PubMed] [Google Scholar]
- 50.Virtanen MA, Uvarov P, Hübner CA, Kaila K. NKCC1, an Elusive molecular target in brain development: making sense of the existing data. Cells. 2020;9:2607. doi: 10.3390/cells9122607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stein V, Hermans-Borgmeyer I, Jentsch TJ, Hübner CA. Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol. 2004;468:57–64. doi: 10.1002/cne.10983. [DOI] [PubMed] [Google Scholar]
- 52.Tillman L, Zhang J. Crossing the chloride channel: the current and potential therapeutic value of the neuronal K+-Cl− cotransporter KCC2. Biomed Res Int. 2019;2019:8941046. doi: 10.1155/2019/8941046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sung K-W, Kirby M, McDonald MP, et al. Abnormal GABAA receptor-mediated currents in dorsal root ganglion neurons isolated from Na–K–2Cl cotransporter null mice. J Neurosci. 2000;20:7531–7538. doi: 10.1523/JNEUROSCI.20-20-07531.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khirug S, Yamada J, Afzalov R, et al. GABAergic depolarization of the axon initial segment in cortical principal neurons is caused by the Na-K-2Cl cotransporter NKCC1. J Neurosci. 2008;28:4635–4639. doi: 10.1523/JNEUROSCI.0908-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Delpire E, Mount DB. Human and murine phenotypes associated with defects in cation-chloride cotransport. Annu Rev Physiol. 2002;64:803–843. doi: 10.1146/annurev.physiol.64.081501.155847. [DOI] [PubMed] [Google Scholar]
- 56.Hampel P, Römermann K, Gailus B, et al. Effects of the NKCC1 inhibitors bumetanide, azosemide, and torasemide alone or in combination with phenobarbital on seizure threshold in epileptic and nonepileptic mice. Neuropharmacology. 2021;185:108449. doi: 10.1016/j.neuropharm.2021.108449. [DOI] [PubMed] [Google Scholar]
- 57.Willis WD. Dorsal root potentials and dorsal root reflexes: a double-edged sword. Exp Brain Res. 1999;124:395–421. doi: 10.1007/s002210050637. [DOI] [PubMed] [Google Scholar]
- 58.Tsukahara T, Masuhara M, Iwai H, et al. Repeated stress-induced expression pattern alterations of the hippocampal chloride transporters KCC2 and NKCC1 associated with behavioral abnormalities in female mice. Biochem Biophys Res Commun. 2015;465:145–151. doi: 10.1016/j.bbrc.2015.07.153. [DOI] [PubMed] [Google Scholar]
- 59.Jaggi AS, Kaur A, Bali A, Singh N. Expanding spectrum of sodium potassium chloride co-transporters in the pathophysiology of diseases. Curr Neuropharmacol. 2015;13:369–388. doi: 10.2174/1570159x13666150205130359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hsu Y-T, Chang Y-G, Chern Y. Insights into GABAAergic system alteration in Huntington’s disease. Open Biol. 2018;8:180165. doi: 10.1098/rsob.180165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lozovaya N, Ben-Ari Y, Hammond C. Striatal dual cholinergic /GABAergic transmission in Parkinson disease: friends or foes? Cell Stress. 2018;2:147–149. doi: 10.15698/cst2018.06.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Allain A-E, Cazenave W, Delpy A, et al. Nonsynaptic glycine release is involved in the early KCC2 expression. Dev Neurobiol. 2016;76:764–779. doi: 10.1002/dneu.22358. [DOI] [PubMed] [Google Scholar]
- 63.Bos R, Brocard F, Vinay L. Primary afferent terminals acting as excitatory interneurons contribute to spontaneous motor activities in the immature spinal cord. J Neurosci. 2011;31:10184–10188. doi: 10.1523/JNEUROSCI.0068-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Phan H-L, Pflieger J-F. Immunolocalization of cation-chloride cotransporters in the developing and mature spinal cord of opossums, Monodelphis domestica. Front Neuroanat. 2013;7:12. doi: 10.3389/fnana.2013.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cramer SW, Baggott C, Cain J, et al. The role of cation-dependent chloride transporters in neuropathic pain following spinal cord injury. Mol Pain. 2008;4:36. doi: 10.1186/1744-8069-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hasbargen T, Ahmed MM, Miranpuri G, et al. Role of NKCC1 and KCC2 in the development of chronic neuropathic pain following spinal cord injury. Ann N Y Acad Sci. 2010;1198:168–172. doi: 10.1111/j.1749-6632.2010.05462.x. [DOI] [PubMed] [Google Scholar]
- 67.Côté M-P, Gandhi S, Zambrotta M, Houlé JD. Exercise modulates chloride homeostasis after spinal cord injury. J Neurosci. 2014;34:8976–8987. doi: 10.1523/JNEUROSCI.0678-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yan X, Liu J, Wang X, et al. Pretreatment with AQP4 and NKCC1 inhibitors concurrently attenuated spinal cord edema and tissue damage after spinal cord injury in rats. Front Physiol. 2018;9:6. doi: 10.3389/fphys.2018.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mekhael W, Begum S, Samaddar S, et al. Repeated anodal trans-spinal direct current stimulation results in long-term reduction of spasticity in mice with spinal cord injury. J Physiol. 2019;597:2201–2223. doi: 10.1113/JP276952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tanabe Y, Jessell TM. Diversity and pattern in the developing spinal cord. Science. 1996;274:1115–1123. doi: 10.1126/science.274.5290.1115. [DOI] [PubMed] [Google Scholar]
- 71.Tashima R, Koga K, Yoshikawa Y, et al. A subset of spinal dorsal horn interneurons crucial for gating touch-evoked pain-like behavior. Proc Natl Acad Sci U S A. 2021;118:e2021220118. doi: 10.1073/pnas.2021220118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yin Y, Yi M-H, Kim DW. Impaired autophagy of GABAergic interneurons in neuropathic pain. Pain Res Manag. 2018;2018:9185368. doi: 10.1155/2018/9185368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Burke D, Fullen BM, Stokes D, Lennon O. Neuropathic pain prevalence following spinal cord injury: a systematic review and meta-analysis. Eur J Pain. 2017;21:29–44. doi: 10.1002/ejp.905. [DOI] [PubMed] [Google Scholar]
- 74.Kumru H, Benito-Penalva J, Kofler M, Vidal J. Analgesic effect of intrathecal baclofen bolus on neuropathic pain in spinal cord injury patients. Brain Res Bull. 2018;140:205–211. doi: 10.1016/j.brainresbull.2018.05.013. [DOI] [PubMed] [Google Scholar]
- 75.Zarepour L, Gharaylou Z, Hadjighassem M, et al. Preliminary study of analgesic effect of bumetanide on neuropathic pain in patients with spinal cord injury. J Clin Neurosci. 2020;81:477–484. doi: 10.1016/j.jocn.2020.10.010. [DOI] [PubMed] [Google Scholar]
- 76.Meisner JG, Marsh AD, Marsh DR. Loss of GABAergic interneurons in laminae I-III of the spinal cord dorsal horn contributes to reduced GABAergic tone and neuropathic pain after spinal cord injury. J Neurotrauma. 2010;27:729–737. doi: 10.1089/neu.2009.1166. [DOI] [PubMed] [Google Scholar]
- 77.Kami K, Taguchi Ms S, Tajima F, Senba E (2016) Exercise modulates chloride homeostasis after spinal cord injury. Mol Pain 12. 10.1177/1744806916629059
- 78.Chen S-R, Zhu L, Chen H, et al. Increased spinal cord Na+-K+-2Cl− cotransporter-1 (NKCC1) activity contributes to impairment of synaptic inhibition in paclitaxel-induced neuropathic pain. J Biol Chem. 2014;289:31111–31120. doi: 10.1074/jbc.M114.600320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pieraut S, Lucas O, Sangari S, et al. An autocrine neuronal interleukin-6 loop mediates chloride accumulation and NKCC1 phosphorylation in axotomized sensory neurons. J Neurosci. 2011;31:13516–13526. doi: 10.1523/JNEUROSCI.3382-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wei B, Kumada T, Furukawa T, et al. Pre- and post-synaptic switches of GABA actions associated with Cl- homeostatic changes are induced in the spinal nucleus of the trigeminal nerve in a rat model of trigeminal neuropathic pain. Neuroscience. 2013;228:334–348. doi: 10.1016/j.neuroscience.2012.10.043. [DOI] [PubMed] [Google Scholar]
- 81.Laird JMA, García-Nicas E, Delpire EJ, Cervero F. Presynaptic inhibition and spinal pain processing in mice: a possible role of the NKCC1 cation-chloride co-transporter in hyperalgesia. Neurosci Lett. 2004;361:200–203. doi: 10.1016/j.neulet.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 82.Allen LL, Seven YB, Baker TL, Mitchell GS. Cervical spinal contusion alters Na+-K+-2Cl- and K+-Cl- cation-chloride cotransporter expression in phrenic motor neurons. Respir Physiol Neurobiol. 2019;261:15–23. doi: 10.1016/j.resp.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gagnon M, Bergeron MJ, Lavertu G, et al. Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat Med. 2013;19:1524–1528. doi: 10.1038/nm.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mòdol L, Cobianchi S, Navarro X. Prevention of NKCC1 phosphorylation avoids downregulation of KCC2 in central sensory pathways and reduces neuropathic pain after peripheral nerve injury. Pain. 2014;155:1577–1590. doi: 10.1016/j.pain.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 85.Khalki L, Sadlaoud K, Lerond J, et al. Changes in innervation of lumbar motoneurons and organization of premotor network following training of transected adult rats. Exp Neurol. 2018;299:1–14. doi: 10.1016/j.expneurol.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 86.Werman R, Davidoff RA, Aprison MH. Inhibition of motoneurones by iontophoresis of glycine. Nature. 1967;214:681–683. doi: 10.1038/214681a0. [DOI] [PubMed] [Google Scholar]
- 87.Jonas P, Bischofberger J, Sandkühler J. Corelease of two fast neurotransmitters at a central synapse. Science. 1998;281:419–424. doi: 10.1126/science.281.5375.419. [DOI] [PubMed] [Google Scholar]
- 88.Shigenaga Y, Moritani M, Oh SJ, et al. The distribution of inhibitory and excitatory synapses on single, reconstructed jaw-opening motoneurons in the cat. Neuroscience. 2005;133:507–518. doi: 10.1016/j.neuroscience.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 89.Baer K, Waldvogel HJ, During MJ, et al. Association of gephyrin and glycine receptors in the human brainstem and spinal cord: an immunohistochemical analysis. Neuroscience. 2003;122:773–784. doi: 10.1016/s0306-4522(03)00543-8. [DOI] [PubMed] [Google Scholar]
- 90.Pfeiffer F, Simler R, Grenningloh G, Betz H. Monoclonal antibodies and peptide mapping reveal structural similarities between the subunits of the glycine receptor of rat spinal cord. Proc Natl Acad Sci U S A. 1984;81:7224–7227. doi: 10.1073/pnas.81.22.7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Triller A, Cluzeaud F, Pfeiffer F, et al. Distribution of glycine receptors at central synapses: an immunoelectron microscopy study. J Cell Biol. 1985;101:683–688. doi: 10.1083/jcb.101.2.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bras H, Liabeuf S (2020) Differential effects of spinal cord transection on glycinergic and GABAergic synaptic signaling in sub-lesional lumbar motoneurons. J Chem Neuroanat 101847. 10.1016/j.jchemneu.2020.101847 [DOI] [PubMed]
- 93.Diaz-Ruiz A, Salgado-Ceballos H, Montes S, et al. Acute alterations of glutamate, glutamine, GABA, and other amino acids after spinal cord contusion in rats. Neurochem Res. 2007;32:57–63. doi: 10.1007/s11064-006-9225-5. [DOI] [PubMed] [Google Scholar]
- 94.Sadlaoud K, Khalki L, Brocard F, et al. Alteration of glycinergic receptor expression in lumbar spinal motoneurons is involved in the mechanisms underlying spasticity after spinal cord injury. J Chem Neuroanat. 2020;106:101787. doi: 10.1016/j.jchemneu.2020.101787. [DOI] [PubMed] [Google Scholar]
- 95.Simpson RK, Gondo M, Robertson CS, Goodman JC. The influence of glycine and related compounds on spinal cord injury-induced spasticity. Neurochem Res. 1995;20:1203–1210. doi: 10.1007/BF00995384. [DOI] [PubMed] [Google Scholar]
- 96.Simpson RK, Robertson CS, Goodman JC. The role of glycine in spinal shock. J Spinal Cord Med. 1996;19:215–224. doi: 10.1080/10790268.1996.11719437. [DOI] [PubMed] [Google Scholar]
- 97.de Leon RD, Tamaki H, Hodgson JA, et al. Hindlimb locomotor and postural training modulates glycinergic inhibition in the spinal cord of the adult spinal cat. J Neurophysiol. 1999;82:359–369. doi: 10.1152/jn.1999.82.1.359. [DOI] [PubMed] [Google Scholar]
- 98.Panter SS, Yum SW, Faden AI. Alteration in extracellular amino acids after traumatic spinal cord injury. Ann Neurol. 1990;27:96–99. doi: 10.1002/ana.410270115. [DOI] [PubMed] [Google Scholar]
- 99.Demediuk P, Daly MP, Faden AI. Effect of impact trauma on neurotransmitter and nonneurotransmitter amino acids in rat spinal cord. J Neurochem. 1989;52:1529–1536. doi: 10.1111/j.1471-4159.1989.tb09204.x. [DOI] [PubMed] [Google Scholar]
- 100.Gorji A, Zahn PK, Pogatzki EM, Speckmann E-J. Spinal and cortical spreading depression enhance spinal cord activity. Neurobiol Dis. 2004;15:70–79. doi: 10.1016/j.nbd.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 101.Petty F, Sherman AD. Plasma GABA levels in psychiatric illness. J Affect Disord. 1984;6:131–138. doi: 10.1016/0165-0327(84)90018-1. [DOI] [PubMed] [Google Scholar]
- 102.Bhandage AK, Cunningham JL, Jin Z et al (2019) Depression, GABA, and age correlate with plasma levels of inflammatory markers. Int J Mol Sci 20. 10.3390/ijms20246172 [DOI] [PMC free article] [PubMed]
- 103.Willford SL, Anderson CM, Spencer SR, Eskandari S. Evidence for a revised ion/substrate coupling stoichiometry of GABA transporters. J Membr Biol. 2015;248:795–810. doi: 10.1007/s00232-015-9797-6. [DOI] [PubMed] [Google Scholar]
- 104.Raiteri L, Stigliani S, Zedda L, et al. Multiple mechanisms of transmitter release evoked by “pathologically” elevated extracellular [K+]: involvement of transporter reversal and mitochondrial calcium. J Neurochem. 2002;80:706–714. doi: 10.1046/j.0022-3042.2001.00750.x. [DOI] [PubMed] [Google Scholar]
- 105.Song G, Cechvala C, Resnick DK, et al. GeneChip analysis after acute spinal cord injury in rat. J Neurochem. 2001;79:804–815. doi: 10.1046/j.1471-4159.2001.00626.x. [DOI] [PubMed] [Google Scholar]
- 106.Ditunno JF, Little JW, Tessler A, Burns AS. Spinal shock revisited: a four-phase model. Spinal Cord. 2004;42:383–395. doi: 10.1038/sj.sc.3101603. [DOI] [PubMed] [Google Scholar]
- 107.Taccola G, Gad P, Culaclii S, et al. Acute neuromodulation restores spinally-induced motor responses after severe spinal cord injury. Exp Neurol. 2020;327:113246. doi: 10.1016/j.expneurol.2020.113246. [DOI] [PubMed] [Google Scholar]
- 108.Coskun C, Avci B, Ocak N, et al. Effect of repeatedly given CDP-choline on cardiovascular and tissue injury in spinal shock conditions: investigation of the acute phase. J Pharm Pharmacol. 2010;62:497–506. doi: 10.1211/jpp.62.04.0013. [DOI] [PubMed] [Google Scholar]
- 109.Rafati DS, Geissler K, Johnson K, et al. Nuclear factor-kappaB decoy amelioration of spinal cord injury-induced inflammation and behavior outcomes. J Neurosci Res. 2008;86:566–580. doi: 10.1002/jnr.21508. [DOI] [PubMed] [Google Scholar]
- 110.Restrepo CE, Lundfald L, Szabó G, et al. Transmitter-phenotypes of commissural interneurons in the lumbar spinal cord of newborn mice. J Comp Neurol. 2009;517:177–192. doi: 10.1002/cne.22144. [DOI] [PubMed] [Google Scholar]
- 111.Mautes AE, Weinzierl MR, Donovan F, Noble LJ. Vascular events after spinal cord injury: contribution to secondary pathogenesis. PhysTher. 2000;80:673–687. [PubMed] [Google Scholar]
- 112.Tator CH, Fehlings MG. Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J Neurosurg. 1991;75:15–26. doi: 10.3171/jns.1991.75.1.0015. [DOI] [PubMed] [Google Scholar]
- 113.Taccola G, Mladinic M, Nistri A. Dynamics of early locomotor network dysfunction following a focal lesion in an in vitro model of spinal injury. Eur J Neurosci. 2010;31:60–78. doi: 10.1111/j.1460-9568.2009.07040.x. [DOI] [PubMed] [Google Scholar]
- 114.Mazzone GL, Margaryan G, Kuzhandaivel A, et al. Kainate-induced delayed onset of excitotoxicity with functional loss unrelated to the extent of neuronal damage in the in vitro spinal cord. Neuroscience. 2010;168:451–462. doi: 10.1016/j.neuroscience.2010.03.055. [DOI] [PubMed] [Google Scholar]
- 115.Deumens R, Mazzone GL, Taccola G. Early spread of hyperexcitability to caudal dorsal horn networks after a chemically-induced lesion of the rat spinal cord in vitro. Neuroscience. 2013;229:155–163. doi: 10.1016/j.neuroscience.2012.10.036. [DOI] [PubMed] [Google Scholar]
- 116.Faden AI, Simon RP. A potential role for excitotoxins in the pathophysiology of spinal cord injury. Ann Neurol. 1988;23:623–626. doi: 10.1002/ana.410230618. [DOI] [PubMed] [Google Scholar]
- 117.Liu D, Xu GY, Pan E, McAdoo DJ. Neurotoxicity of glutamate at the concentration released upon spinal cord injury. Neuroscience. 1999;93:1383–1389. doi: 10.1016/s0306-4522(99)00278-x. [DOI] [PubMed] [Google Scholar]
- 118.Mailly F, Marin P, Israël M, et al. Increase in external glutamate and NMDA receptor activation contribute to H2O2-induced neuronal apoptosis. J Neurochem. 1999;73:1181–1188. doi: 10.1046/j.1471-4159.1999.0731181.x. [DOI] [PubMed] [Google Scholar]
- 119.Saransaari P, Oja SS. Release of endogenous glutamate, aspartate, GABA, and taurine from hippocampal slices from adult and developing mice under cell-damaging conditions. Neurochem Res. 1998;23:563–570. doi: 10.1023/a:1022494921018. [DOI] [PubMed] [Google Scholar]
- 120.Rego AC, Santos MS, Oliveira CR. Oxidative stress, hypoxia, and ischemia-like conditions increase the release of endogenous amino acids by distinct mechanisms in cultured retinal cells. J Neurochem. 1996;66:2506–2516. doi: 10.1046/j.1471-4159.1996.66062506.x. [DOI] [PubMed] [Google Scholar]
- 121.Masiulis S, Desai R, Uchański T, et al. GABAA receptor signalling mechanisms revealed by structural pharmacology. Nature. 2019;565:454–459. doi: 10.1038/s41586-018-0832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sah R, Galeffi F, Ahrens R, et al. Modulation of the GABA(A)-gated chloride channel by reactive oxygen species. J Neurochem. 2002;80:383–391. doi: 10.1046/j.0022-3042.2001.00706.x. [DOI] [PubMed] [Google Scholar]
- 123.Basmajian JV. New views on muscular tone and relaxation. Can Med Assoc J. 1957;77:203–205. [PMC free article] [PubMed] [Google Scholar]
- 124.Lin CS-Y, Macefield VG, Elam M, et al. Axonal changes in spinal cord injured patients distal to the site of injury. Brain. 2007;130:985–994. doi: 10.1093/brain/awl339. [DOI] [PubMed] [Google Scholar]
- 125.Norton JA, Bennett DJ, Knash ME, et al. Changes in sensory-evoked synaptic activation of motoneurons after spinal cord injury in man. Brain. 2008;131:1478–1491. doi: 10.1093/brain/awn050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schwindt P, Crill WE. A persistent negative resistance in cat lumbar motoneurons. Brain Res. 1977;120:173–178. doi: 10.1016/0006-8993(77)90510-8. [DOI] [PubMed] [Google Scholar]
- 127.Flatman JA, Schwindt PC, Crill WE, Stafstrom CE. Multiple actions of N-methyl-D-aspartate on cat neocortical neurons in vitro. Brain Res. 1983;266:169–173. doi: 10.1016/0006-8993(83)91323-9. [DOI] [PubMed] [Google Scholar]
- 128.Bennett DJ, Hultborn H, Fedirchuk B, Gorassini M. Synaptic activation of plateaus in hindlimb motoneurons of decerebrate cats. J Neurophysiol. 1998;80:2023–2037. doi: 10.1152/jn.1998.80.4.2023. [DOI] [PubMed] [Google Scholar]
- 129.Li Y, Gorassini MA, Bennett DJ. Role of persistent sodium and calcium currents in motoneuron firing and spasticity in chronic spinal rats. J Neurophysiol. 2004;91:767–783. doi: 10.1152/jn.00788.2003. [DOI] [PubMed] [Google Scholar]
- 130.Hamm TM, Turkin VV, Bandekar NK, et al. Persistent currents and discharge patterns in rat hindlimb motoneurons. J Neurophysiol. 2010;104:1566–1577. doi: 10.1152/jn.00380.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kakinohana O, Hefferan MP, Nakamura S, et al. Development of GABA-sensitive spasticity and rigidity in rats after transient spinal cord ischemia: a qualitative and quantitative electrophysiological and histopathological study. Neuroscience. 2006;141:1569–1583. doi: 10.1016/j.neuroscience.2006.04.083. [DOI] [PubMed] [Google Scholar]
- 132.Tai Q, Palazzolo K, Mautes A, et al. Ultrastructural characteristics of glutamatergic and GABAergic terminals in cat lamina IX before and after spinal cord injury. J Spinal Cord Med. 1997;20:311–318. doi: 10.1080/10790268.1997.11719481. [DOI] [PubMed] [Google Scholar]
- 133.Nacimiento W, Sappok T, Brook GA, et al. Structural changes of anterior horn neurons and their synaptic input caudal to a low thoracic spinal cord hemisection in the adult rat: a light and electron microscopic study. Acta Neuropathol. 1995;90:552–564. doi: 10.1007/BF00318567. [DOI] [PubMed] [Google Scholar]
- 134.Ondarza AB, Ye Z, Hulsebosch CE. Direct evidence of primary afferent sprouting in distant segments following spinal cord injury in the rat: colocalization of GAP-43 and CGRP. Exp Neurol. 2003;184:373–380. doi: 10.1016/j.expneurol.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 135.Carr PA, Pearson JC, Fyffe RE. Distribution of 5-hydroxytryptamine-immunoreactive boutons on immunohistochemically-identified Renshaw cells in cat and rat lumbar spinal cord. Brain Res. 1999;823:198–201. doi: 10.1016/s0006-8993(98)01210-4. [DOI] [PubMed] [Google Scholar]
- 136.Jordan LM, McCrea DA. Analysis of the effects of p-methoxy-phenylethylamine on spinal cord neurones. Br J Pharmacol. 1976;57:191–199. doi: 10.1111/j.1476-5381.1976.tb07467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wootz H, Fitzsimons-Kantamneni E, Larhammar M, et al. Alterations in the motor neuron-renshaw cell circuit in the Sod1(G93A) mouse model. J Comp Neurol. 2013;521:1449–1469. doi: 10.1002/cne.23266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kupcova Skalnikova H, Navarro R, Marsala S, et al. Signaling proteins in spinal parenchyma and dorsal root ganglion in rat with spinal injury-induced spasticity. J Proteome. 2013;91:41–57. doi: 10.1016/j.jprot.2013.06.028. [DOI] [PubMed] [Google Scholar]
- 139.Kumru H, Vidal J, Kofler M, et al. Alterations in excitatory and inhibitory brainstem interneuronal circuits after severe spinal cord injury. J Neurotrauma. 2010;27:721–728. doi: 10.1089/neu.2009.1089. [DOI] [PubMed] [Google Scholar]
- 140.Chen B, Li Y, Yu B, et al. Reactivation of dormant relay pathways in injured spinal cord by KCC2 manipulations. Cell. 2018;174:521–535.e13. doi: 10.1016/j.cell.2018.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Huang YJ, Lee KH, Grau JW. Complete spinal cord injury (SCI) transforms how brain derived neurotrophic factor (BDNF) affects nociceptive sensitization. Exp Neurol. 2017;288:38–50. doi: 10.1016/j.expneurol.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 142.Sánchez-Brualla I, Boulenguez P, Brocard C, et al. Activation of 5-HT 2A receptors restores KCC2 function and reduces neuropathic pain after spinal cord injury. Neuroscience. 2018;387:48–57. doi: 10.1016/j.neuroscience.2017.08.033. [DOI] [PubMed] [Google Scholar]
- 143.Mazzone GL, Nistri A. Modulation of extrasynaptic GABAergic receptor activity influences glutamate release and neuronal survival following excitotoxic damage to mouse spinal cord neurons. Neurochem Int. 2019;128:175–185. doi: 10.1016/j.neuint.2019.04.018. [DOI] [PubMed] [Google Scholar]
- 144.Fandel TM, Trivedi A, Nicholas CR, et al. Transplanted human stem cell-derived interneuron precursors mitigate mouse bladder dysfunction and central neuropathic pain after spinal cord injury. Cell Stem Cell. 2016;19:544–557. doi: 10.1016/j.stem.2016.08.020. [DOI] [PubMed] [Google Scholar]
- 145.Llewellyn-Smith IJ, Basbaum AI, Bráz JM. Long-term, dynamic synaptic reorganization after GABAergic precursor cell transplantation into adult mouse spinal cord. J Comp Neurol. 2017;526:480–495. doi: 10.1002/cne.24346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Batista CM, Mariano ED, Dale CS, et al. Pain inhibition through transplantation of fetal neuronal progenitors into the injured spinal cord in rats. Neural Regen Res. 2019;14:2011–2019. doi: 10.4103/1673-5374.259624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Manion J, Khuong T, Harney D, et al. Human induced pluripotent stem cell-derived GABAergic interneuron transplants attenuate neuropathic pain. Pain. 2020;161:379–387. doi: 10.1097/j.pain.0000000000001733. [DOI] [PubMed] [Google Scholar]
- 148.Dugan EA, Jergova S, Sagen J. Mutually beneficial effects of intensive exercise and GABAergic neural progenitor cell transplants in reducing neuropathic pain and spinal pathology in rats with spinal cord injury. Exp Neurol. 2020;327:113208. doi: 10.1016/j.expneurol.2020.113208. [DOI] [PubMed] [Google Scholar]
- 149.Lev-Tov A, Pinco M. In vitro studies of prolonged synaptic depression in the neonatal rat spinal cord. J Physiol. 1992;447:149–169. doi: 10.1113/jphysiol.1992.sp018996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dose F, Taccola G. Coapplication of noisy patterned electrical stimuli and NMDA plus serotonin facilitates fictive locomotion in the rat spinal cord. J Neurophysiol. 2012;108:2977–2990. doi: 10.1152/jn.00554.2012. [DOI] [PubMed] [Google Scholar]
- 151.Mazzone GL, Nistri A. Electrochemical detection of endogenous glutamate release from rat spinal cord organotypic slices as a real-time method to monitor excitotoxicity. J Neurosci Methods. 2011;197:128–132. doi: 10.1016/j.jneumeth.2011.01.033. [DOI] [PubMed] [Google Scholar]
- 152.McLamore ES, Mohanty S, Shi J, et al. A self-referencing glutamate biosensor for measuring real time neuronal glutamate flux. J Neurosci Methods. 2010;189:14–22. doi: 10.1016/j.jneumeth.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 153.Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460:525–542. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- 154.Mazzone GL, Nistri A. Delayed neuroprotection by riluzole against excitotoxic damage evoked by kainate on rat organotypic spinal cord cultures. Neuroscience. 2011;190:318–327. doi: 10.1016/j.neuroscience.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 155.Mazzone GL, Nistri A. Effect of the PARP-1 inhibitor PJ 34 on excitotoxic damage evoked by kainate on rat spinal cord organotypic slices. Cell Mol Neurobiol. 2011;31:469–478. doi: 10.1007/s10571-010-9640-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sámano C, Nistri A. Mechanism of neuroprotection against experimental spinal cord injury by riluzole or methylprednisolone. Neurochem Res. 2019;44:200–213. doi: 10.1007/s11064-017-2459-6. [DOI] [PubMed] [Google Scholar]
- 157.Bajrektarevic D, Nistri A. Delayed application of the anesthetic propofol contrasts the neurotoxic effects of kainate on rat organotypic spinal slice cultures. Neurotoxicology. 2016;54:1–10. doi: 10.1016/j.neuro.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 158.Kaur J, Flores Gutiérrez J, Nistri A. Neuroprotective effect of propofol against excitotoxic injury to locomotor networks of the rat spinal cord in vitro. Eur J Neurosci. 2016;44:2418–2430. doi: 10.1111/ejn.13353. [DOI] [PubMed] [Google Scholar]
- 159.Shabbir A, Bianchetti E, Nistri A. The volatile anesthetic methoxyflurane protects motoneurons against excitotoxicity in an in vitro model of rat spinal cord injury. Neuroscience. 2015;285:269–280. doi: 10.1016/j.neuroscience.2014.11.023. [DOI] [PubMed] [Google Scholar]
- 160.Cheng Q, Sun G-J, Liu S-B, et al. A novel translocator protein 18 kDa ligand, ZBD-2, exerts neuroprotective effects against acute spinal cord injury. Clin Exp Pharmacol Physiol. 2016;43:930–938. doi: 10.1111/1440-1681.12606. [DOI] [PubMed] [Google Scholar]
- 161.Labombarda F, Ghoumari AM, Liere P, et al. Neuroprotection by steroids after neurotrauma in organotypic spinal cord cultures: a key role for progesterone receptors and steroidal modulators of GABA(A) receptors. Neuropharmacology. 2013;71:46–55. doi: 10.1016/j.neuropharm.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 162.Fontana G, Taccola G, Galante J, et al. AMPA-evoked acetylcholine release from cultured spinal cord motoneurons and its inhibition by GABA and glycine. Neuroscience. 2001;106:183–191. doi: 10.1016/s0306-4522(01)00272-x. [DOI] [PubMed] [Google Scholar]
- 163.Cervetto C, Taccola G. GABAA and strychnine-sensitive glycine receptors modulate N-methyl-D-aspartate-evoked acetylcholine release from rat spinal motoneurons: a possible role in neuroprotection. Neuroscience. 2008;154:1517–1524. doi: 10.1016/j.neuroscience.2008.04.066. [DOI] [PubMed] [Google Scholar]
- 164.Rogvi-Hansen B, Gram L. Adverse effects of established and new antiepileptic drugs: an attempted comparison. Pharmacol Ther. 1995;68:425–434. doi: 10.1016/0163-7258(95)02014-4. [DOI] [PubMed] [Google Scholar]
- 165.Montané E, Vallano A, Laporte JR. Oral antispastic drugs in nonprogressive neurologic diseases: a systematic review. Neurology. 2004;63:1357–1363. doi: 10.1212/01.wnl.0000141863.52691.44. [DOI] [PubMed] [Google Scholar]
- 166.Leo RJ, Baer D. Delirium associated with baclofen withdrawal: a review of common presentations and management strategies. Psychosomatics. 2005;46:503–507. doi: 10.1176/appi.psy.46.6.503. [DOI] [PubMed] [Google Scholar]
- 167.Losi G, Gomez-Gonzalo M, Zonta M et al (2019) Cellular and molecular mechanisms of new onset seizure generation. Aging Clin Exp Res. 10.1007/s40520-019-01396-z [DOI] [PubMed]
- 168.Araque A, Parpura V, Sanzgiri RP, et al. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/S0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- 169.Mazzone GL, Nistri A. S100β as an early biomarker of excitotoxic damage in spinal cord organotypic cultures. J Neurochem. 2014;130:598–604. doi: 10.1111/jnc.12748. [DOI] [PubMed] [Google Scholar]
- 170.Cifra A, Mazzone GL, Nani F, et al. Postnatal developmental profile of neurons and glia in motor nuclei of the brainstem and spinal cord, and its comparison with organotypic slice cultures. Dev Neurobiol. 2012;72:1140–1160. doi: 10.1002/dneu.20991. [DOI] [PubMed] [Google Scholar]
- 171.Liu J, McDaid L, Araque A, et al. GABA regulation of burst firing in hippocampal astrocyte neural circuit: a biophysical model. Front Cell Neurosci. 2019;13:335. doi: 10.3389/fncel.2019.00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Christensen RK, Delgado-Lezama R, Russo RE, et al. Spinal dorsal horn astrocytes release GABA in response to synaptic activation. J Physiol Lond. 2018;596:4983–4994. doi: 10.1113/JP276562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Fernández-López B, Valle-Maroto SM, Barreiro-Iglesias A, Rodicio MC. Neuronal release and successful astrocyte uptake of aminoacidergic neurotransmitters after spinal cord injury in lampreys. Glia. 2014;62:1254–1269. doi: 10.1002/glia.22678. [DOI] [PubMed] [Google Scholar]
- 174.Lei Y, Yaroslavsky I, Tejani-Butt SM. Strain differences in the distribution of N-methyl-d-aspartate and gamma (gamma)-aminobutyric acid-A receptors in rat brain. Life Sci. 2009;85:794–799. doi: 10.1016/j.lfs.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Dornellas APS, Macedo GC, McFarland MH, et al. Allopregnanolone decreases evoked dopamine release differently in rats by sex and estrous stage. Front Pharmacol. 2020;11:608887. doi: 10.3389/fphar.2020.608887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Clarke EC, Cheng S, Bilston LE. The mechanical properties of neonatal rat spinal cord in vitro, and comparisons with adult. J Biomech. 2009;42:1397–1402. doi: 10.1016/j.jbiomech.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 177.Khristy W, Ali NJ, Bravo AB, et al. Changes in GABA(A) receptor subunit gamma 2 in extensor and flexor motoneurons and astrocytes after spinal cord transection and motor training. Brain Res. 2009;1273:9–17. doi: 10.1016/j.brainres.2009.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Mackie M, Hughes DI, Maxwell DJ, et al. Distribution and colocalisation of glutamate decarboxylase isoforms in the rat spinal cord. Neuroscience. 2003;119:461–472. doi: 10.1016/s0306-4522(03)00174-x. [DOI] [PubMed] [Google Scholar]
- 179.Tillakaratne NJK, de Leon RD, Hoang TX, et al. Use-dependent modulation of inhibitory capacity in the feline lumbar spinal cord. J Neurosci. 2002;22:3130–3143. doi: 10.1523/JNEUROSCI.22-08-03130.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lu Y, Zheng J, Xiong L, et al. Spinal cord injury-induced attenuation of GABAergic inhibition in spinal dorsal horn circuits is associated with down-regulation of the chloride transporter KCC2 in rat. J Physiol Lond. 2008;586:5701–5715. doi: 10.1113/jphysiol.2008.152348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Boulenguez P, Liabeuf S, Bos R, et al. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat Med. 2010;16:302–307. doi: 10.1038/nm.2107. [DOI] [PubMed] [Google Scholar]
- 182.Boulenguez P, Vinay L. Strategies to restore motor functions after spinal cord injury. Curr Opin Neurobiol. 2009;19:587–600. doi: 10.1016/j.conb.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 183.Beato M. The time course of transmitter at glycinergic synapses onto motoneurons. J Neurosci. 2008;28:7412–7425. doi: 10.1523/JNEUROSCI.0581-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Pitt SJ, Sivilotti LG, Beato M. High intracellular chloride slows the decay of glycinergic currents. J Neurosci. 2008;28:11454–11467. doi: 10.1523/JNEUROSCI.3890-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bellardita C, Caggiano V, Leiras R, et al. Spatiotemporal correlation of spinal network dynamics underlying spasms in chronic spinalized mice. Elife. 2017;6:e23011. doi: 10.7554/eLife.23011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Edgerton VR, Roy RR. Spasticity: a switch from inhibition to excitation. Nat Med. 2010;16:270–271. doi: 10.1038/nm0310-270. [DOI] [PubMed] [Google Scholar]
- 187.Robinson GA, Goldberger ME. The development and recovery of motor function in spinal cats. I. The infant lesion effect. Exp Brain Res. 1986;62:373–386. doi: 10.1007/BF00238857. [DOI] [PubMed] [Google Scholar]
- 188.Edgerton VR, de Leon RD, Tillakaratne N, et al. Use-dependent plasticity in spinal stepping and standing. Adv Neurol. 1997;72:233–247. [PubMed] [Google Scholar]
- 189.Brocard C, Plantier V, Boulenguez P, et al. Cleavage of Na(+) channels by calpain increases persistent Na(+) current and promotes spasticity after spinal cord injury. Nat Med. 2016;22:404–411. doi: 10.1038/nm.4061. [DOI] [PubMed] [Google Scholar]
- 190.Thomas CK, Häger CK, Klein CS. Increases in human motoneuron excitability after cervical spinal cord injury depend on the level of injury. J Neurophysiol. 2017;117:684–691. doi: 10.1152/jn.00676.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bunday KL, Oudega M, Perez MA. Aberrant crossed corticospinal facilitation in muscles distant from a spinal cord injury. PLoS ONE. 2013;8:e76747. doi: 10.1371/journal.pone.0076747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Li Y, Bennett DJ. Persistent sodium and calcium currents cause plateau potentials in motoneurons of chronic spinal rats. J Neurophysiol. 2003;90:857–869. doi: 10.1152/jn.00236.2003. [DOI] [PubMed] [Google Scholar]
- 193.Bennett DJ, Gorassini M, Fouad K, et al. Spasticity in rats with sacral spinal cord injury. J Neurotrauma. 1999;16:69–84. doi: 10.1089/neu.1999.16.69. [DOI] [PubMed] [Google Scholar]
- 194.Bellardita C, Marcantoni M, Löw P, Kiehn O. Sacral spinal cord transection and isolated sacral cord preparation to study chronic spinal cord injury in adult mice. Bio Protoc. 2018;8:e2784. doi: 10.21769/BioProtoc.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Johnson MD, Kajtaz E, Cain CM, Heckman CJ. Motoneuron intrinsic properties, but not their receptive fields, recover in chronic spinal injury. J Neurosci. 2013;33:18806–18813. doi: 10.1523/JNEUROSCI.2609-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Venugopal S, Hamm TM, Crook SM, Jung R. Modulation of inhibitory strength and kinetics facilitates regulation of persistent inward currents and motoneuron excitability following spinal cord injury. J Neurophysiol. 2011;106:2167–2179. doi: 10.1152/jn.00359.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Davidoff RA. Pharmacology of spasticity. Neurology. 1978;28:46–51. doi: 10.1212/wnl.28.9_part_2.46. [DOI] [PubMed] [Google Scholar]
- 198.ElBasiouny SM, Schuster JE, Heckman CJ. Persistent inward currents in spinal motoneurons: important for normal function but potentially harmful after spinal cord injury and in amyotrophic lateral sclerosis. Clin Neurophysiol. 2010;121:1669–1679. doi: 10.1016/j.clinph.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Powers RK, Heckman CJ. Synaptic control of the shape of the motoneuron pool input-output function. J Neurophysiol. 2017;117:1171–1184. doi: 10.1152/jn.00850.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tazerart S, Viemari J-C, Darbon P, et al. Contribution of persistent sodium current to locomotor pattern generation in neonatal rats. J Neurophysiol. 2007;98:613–628. doi: 10.1152/jn.00316.2007. [DOI] [PubMed] [Google Scholar]
- 201.Binder MD, Powers RK, Heckman CJ. Nonlinear input-output functions of motoneurons. Physiology (Bethesda) 2020;35:31–39. doi: 10.1152/physiol.00026.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Johnson MD, Frigon A, Hurteau M-F, et al. Reflex wind-up in early chronic spinal injury: plasticity of motor outputs. J Neurophysiol. 2017;117:2065–2074. doi: 10.1152/jn.00981.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable