

Abstract
Background
Mitochondrial alanyl-tRNA synthetase 2 gene (AARS2) related disease is a rare genetic disorder affecting mitochondrial metabolism, leading to severe cardiac disease in infants or progressive leukodystrophy in young adults. The disease is considered ultra-rare with only 39 cases of AARS2-leukodystrophy previously reported.
Case presentation
We present the case of a young man of consanguineous heritage suffering from cognitive decline and progressive spasticity as well as weakness of the proximal musculature. Utilizing MRI and whole genome sequencing, the patient was diagnosed with a homozygous AARS2 missense variant (NM_020745.3:c.650C > T; p.(Pro217Leu)) and a homozygous CAPN3 variant (NM_000070.2: c.1469G > A; p.(Arg490Gln)), both variants have previously been identified in patients suffering from AARS2 related leukodystrophy and limb-girdle muscular dystrophy, respectively.
Conclusions
This case report presents a case of homozygous AARS2 leukodystrophy and serves to highlight the importance of whole genome sequencing in diagnosing rare neurological diseases as well as to add to the awareness of adult onset leukodystrophies.
Keywords: AARS2, Adult onset leukodystrophies, Limb-girdle muscular dystrophy, Whole genome sequencing, Case report, Inborn errors of metabolism
Abbreviations: AARS2, Mitochondrial alanyl-tRNA synthetase 2 gene; Mt-aaRS, Mitochondrial aminoacyl-tRNA synthetase; AARS2-L, Mitochondrial alanyl-tRNA synthetase 2 gene leukodystrophy; DARS2, Deficiency of aspartyl-tRNA; EARS2, Deficiency of glutamate-tRNA synthetase; MMSE, Mini-Mental State Examination; CSF, Cerebrospinal fluid; ADLs, activities of daily living; mtDNA, Mitochondrial DNA; LGMD R1, Limb-girdle muscular dystrophy R1 calpain3-related; IEM, Inborn errors of metabolism; HDLS, Hereditary Diffuse Leukodystrophy with axonal Spheroids; ALSP, Adult-Onset Leukoencephalopathy With Axonal Spheroids and Pigmented Glia; CSF1R, Colony stimulating factor-1 receptor
1. Background
Mitochondrial alanyl-tRNA synthetase 2 gene (AARS2) related (ovario)leukodystrophy (MIM #615889) is a rare subtype of adult onset leukodystrophies with autosomal recessive inheritance. Adult onset leukodystrophy is in itself a rare condition, showing an incidence of 2/100.000 in a German population [1]. AARS2 is a nuclear gene encoding mitochondrial alanyl-tRNA synthetase which is responsible for transferring the amino acid alanine to the accepting mitochondrial tRNA in the mitochondrial matrix. In addition, the enzyme has an editing domain, which can deacylate tRNAs, charged with an incorrect amino acid [2,3]. AARS2 leukodystrophy (AARS2-L) thus belongs to the group of genetic diseases of mitochondrial metabolism caused by deficiency of mitochondrial aminoacyl-tRNA synthetase (mt-aaRS) proteins. The literature describes three well-defined mt-aaRS related leukodystrophy syndromes with deficiency of aspartyl-tRNA (DARS2) and glutamate-tRNA synthetase (EARS2), respectively, in addition to AARS2 leukodystrophy [4]. Despite a relatively well-defined disease phenotype and cerebral MRI pattern, diagnosis can be challenging due to the rarity of the disease, the potential lack of awareness and several less rare differential diagnoses.
Pathogenic AARS2 variants were first described in infants suffering from progressive fatal cardiomyopathy in 2011. Later cases were also described with lung hypoplasia and skeletal muscle myopathy [2,5]. The slightly more prevalent adult phenotype, first described in 2014 [6], is characterized by adult-onset premature ovarian failure in females and progressive leukodystrophy [4]. Age at onset is typically around 25 years with patients displaying cognitive decline, neuropsychiatric symptoms along with pyramidal and extrapyramidal signs with rapid progression [4,7,8]. Some cases have been described with preexisting developmental delay or motor problems in childhood [8]. However, the spectrum of AARS2-related phenotypes has recently been widened. There are various symptoms and their prevalence differ among patients. Cases with ataxia, demyelinating polyneuropathy and tremor, basal ganglia and cerebellar symptoms as well as a case with progressive optic neuropathy have also been described [[9], [10], [11]]. There is currently no treatment for the condition.
With this case report, we wish to contribute to the understanding of the natural course of AARS2 leukodystrophy, raise awareness for adult onset leukodystrophies, and, not least, illustrate substantial benefit of a diagnostic approach using next generation sequencing in a selected group of patients at the adult neurology department suspected of inborn errors of metabolism.
2. Case presentation
In our clinic we were presented to a 27-year old male of Afghan heritage showing signs of cognitive decline. The patient's gait was reportedly altered after a falling accident in his early teens, but he did not otherwise suffer from any other known medical conditions. The patient showed no recognition of his own shortcomings. However, his next of kin reported that over a two months period, the patient had developed increasing forgetfulness and decreasing ability to carry out basic activities of daily living. The next-of-kin reported that the patient was an only-child and that his parents were first cousins, with no first- or second-degree relatives exhibiting signs of neurological disease.
On initial exam the patient appeared unkempt and with a decreased nutritional level. He was unaware where he was or why, and had great trouble following instructions. MMSE (Mini-Mental State Examination) was 13/30. He was unable to recall three words after five minutes. He showed slightly increased muscle tone in his left side, mainly in his lower limb. He also presented with a bizarre, waddling gait with his body tilted back and dragging feet. The left arm was held in a spastic flexed posture with decreased pendulation of his right arm and en-bloc turns. There was no history of drug abuse, mood changes, hallucinations or psychosis.
The remainder of the exam was unremarkable, with only a minor degree of scapular winging. His blood biochemistry was completely normal, including s-lactate and creatine kinase. A CAT-scan done on suspicion of intracranial mass, showed mild cortical atrophy along with widespread leukoencephalopathy (see Fig. 1).
Fig. 1.
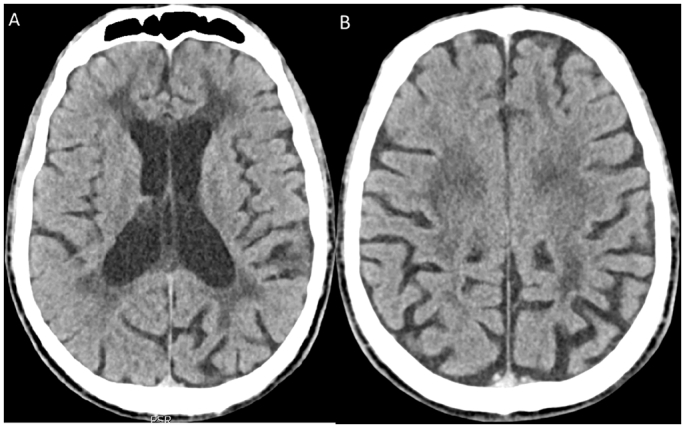
CAT scan of the brain showing bilateral, symmetrical periventricular and deep white matter leukoencephalopathy, central atrophy with moderate atrophy of corpus callosum and the basal ganglia, and slight cortical atrophy, with parietal predominance.
A subsequent MRI revealed a leukoencephalopathy suggestive of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) (see Fig. 2).
Fig. 2.

Brain MRI. Axial T2 and coronal FLAIR sequences show extensive bilateral, grossly symmetrical and predominantly confluent hyperintensities of periventricular, deep and subcortical white matter, with spared U fibers (A-B). Axial T1W image shows corresponding low signal in the affected areas (C). The hyperintensities are slightly inhomogeneous, with some patchy areas of yet spared white matter in the deep white matter (D-E, arrows). There are no signs of white matter rarefaction. The hyperintensities show slight fronto-parietal predominance, but the occipital lobes and the posterior parts of the temporal lobes are also affected (F-G, arrows). DW images show few, punctate and linear areas of restricted diffusion, bilaterally aligned in abnormal white matter in centrum semiovale and corona radiata, with intermediate low signal on the ADC map (H-I-J, arrows). Corpus callosum shows substantial diffuse atrophy, with segmental signal abnormalities in both genu, body and splenium, corresponding to connections between the abnormal white matter in the two hemispheres (K) with some even more atrophic areas (K, arrows). There is symmetrical, mild cerebral atrophy with parietal predominance, corresponding to the more severe white matter volume loss in the parietal lobes (L). There is no cerebellar atrophy (K-L). There are very discrete signal abnormalities along the corticospinal tracts in the posterior limbs of the internal capsules on both sides, descending through the brain stem, including pons, indicating involvement of descending tracts (M-N, arrows). There are no signal abnormalities in the basal ganglia and cerebellum.
Cerebrospinal fluid (CSF) analysis showed a normal cell count and protein levels. Tests for very long chain fatty acids in plasma, phytanic acid as well as lysosomal enzyme activities in leukocytes (beta galactosidase, arylsulfatase A, hexosaminidase A and B, galaktocerebrosidase, alpha fucosidase, alpha mannosidase, tripeptidyl-peptidase, palmitoyl protein thioesterase) were negative, ruling out X-linked adrenoleukodystrophy and several lysosomal leukodystrophies.
Whole genome sequencing (paired-end 2x150bp, ~30× coverage, Illumina platform) revealed a homozygous AARS2 missense variant (NM_020745.3:c.650C > T; p.(Pro217Leu)) and a homozygous CAPN3 missense variant (NM_000070.2: c.1469G > A; p.(Arg490Gln)), both variants previously identified in patients suffering from AARS2 related leukodystrophy [12] and limb-girdle muscle dystrophy [13], respectively.
The patient was tested by occupational therapists who found him able to perform general activities of daily living (ADLs) such as bathroom visits, showering etc. He was, however, completely unable to perform more advanced functions. The patient was discharged to rehabilitation.
On follow-up 8 months later, the patient still displayed severe deficit of self-awareness and reported to feel fine. However, rehabilitation staff reported, that the patient had further deteriorated; now being unable to navigate within the rehabilitation center and he furthermore developed fecal and urinal incontinence. The objective examination of the patient showed further worsening of anterograde memory with a score of 7/30 on MMSE. The patient had developed dysarthria and positive primitive reflexes including a positive snout reflex, grasp reflexes, bilateral positive Rossolimo's sign, further increase in general spasticity with a left-sided predominance as well as positive Hoffmans reflex on both upper limbs. Muscle reflexes were generally brisk (grade 3) and unchanged, as was his strength. The staff from the patients care center described the patient to be increasingly impulsive and disinhibited in his behavior.
On his last follow up 19 months after initial admission the patient's condition had worsened further. According to staff from his care center, he had become increasingly unresponsive with apathy and required continuous prompting when eating. Frontal release signs and spasticity in upper and lower extremities had increased. However, the most striking difference consisted of a severely reduced attention span and loss of ability to walk unaided (see Fig. 3 for timeline).
Fig. 3.

Timeline of events.
ECG and transthoracic echocardiography showed no signs of cardiac disease. Due to advanced cognitive impairment at onset, we felt it was unethical to do a muscle biopsy for further investigation of limb girdle muscular dystrophy, since there would be no therapeutic consequences.
3. Discussion and conclusions
We report the case of a young Afghan man of consanguineous heritage presenting with rapid cognitive decline, a mixture of spastic and myopathic gait as well as upper motor neuron signs with right sided predominance, progressing with further cognitive impairment and development of primitive reflexes over a course of 18 months. Cerebral MRI showed symmetrical leukodystrophy, with similarities to ALSP (Fig. 2) and other genetic leukoencephalopathies. Initial metabolic screening tests could not establish a specific diagnosis. In this situation, we applied a rapid whole genome sequencing panel comprising 2136 nuclear genes, all variants annotated as pathogenic in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) and the entire mitochondrial DNA (mtDNA). The analysis revealed that the patient is homozygous for a pathogenic AARS2 missense variant as well as a pathogenic CAPN3 missense variant. The patient was thus diagnosed with two rare diseases, namely AARS2 leukodystrophy and limb-girdle muscular dystrophy R1 calpain3-related (LGMD R1, formerly known as LGMD2a) [14,15]. Whole genome sequencing is currently increasingly employed in the pediatric setting, e.g. in neonatal and pediatric intensive care, when inborn errors of metabolism (IEM) are possible differential diagnoses, but also in patients with signs of pediatric dementia or other severe neurodegenerative presentations [16]. The background for this is the need to establish IEM diagnoses in a timely manner, as increasing numbers of IEM can be treated with specific interventions [17]. It has been shown that this approach can reduce time to diagnosis and treatment, and by this decrease the following socio-economic burden [18]. Even if specific therapeutic intervention is not possible, a shorter time to diagnosis and prognostic information for guiding of further patient management, e.g. towards palliative care, as in this case, is helpful. On the background of the lower incidence of IEM, first line extended genetic screening is not widely used in neurological departments, but could potentially have a significant role, as the present report underlines.
The initial presentation of our patient's symptoms was atypical, this was later explained by the fact, that the patient displayed signs of two rare genetic diseases [19]. The age of onset for the cognitive decline is close to the reported median age of onset in AARS2 leukodystrophy [4]. Also, the onset of gait affection corresponds to the typical age of onset for LGMD R1 [14]. In fact, it has been reported, that 4,9% of patients with autosomal recessive disease, suffer from more than one autosomal recessive disease [20]. The prevalence is presumably even higher in consanguineous individuals, such as the present patient. This is an important message especially in cases of unusual phenotypes or if one genetic diagnosis does not seem to explain all aspects of the phenotype.
Whilst the patient originally presented to our clinic due to cognitive decline, his phenotypical presentation was highly marked by decreased strength in his pelvic area, contributing strongly to his abnormal gait. There was only a minor degree of scapular winging, a non-specific sign of LGMD in general. However, the disparity between the marked myopathy of his pelvic area and the relatively intact shoulder girdle, is a classic hallmark of LGMD R1, as the pelvic girdle is usually more affected than the shoulders [14]. In the initial stages of LMGD R1, creatinine kinase levels are usually elevated, but decrease along with loss of muscle mass, which is probably the case in our patient [14]. The combination of LGMD 1R and AARS2-L is complicating care for our patient, as the usual course of treatment for LGMD is physiotherapy and occupational therapy in order to prevent contractures and hinder muscle wasting. As our patient was already highly marked by his cognitive impairment by the time of presentation, he was unable to participate in active goal-directed therapy. His muscular dystrophy is also a highly contributing factor to the impairment of mobility seen at follow-up visits.
The MRI findings in our patient are highly consistent with those previously described in patients with AARS2-L [6,7,21]. For these patients extensive, bilateral, variably inhomogeneous, grossly symmetrical T2 and Flair hyperintensities with mild atrophy of the affected cerebral areas; variable thinning of corpus callosum with pathological signal in areas connecting the abnormal hemispheric white matter; small areas of restricted diffusion in affected white matter, and signs of involvement of descending tracts have been described. Within the group of mt-aaRS – related leukodystrophies (DARS2, EARS2, AARS2) this MRI pattern seems to be specific for AARS2-L [4]. Within the larger group of genetic leukodystrophies the described MRI findings are not completely specific for AARS2-L. A very similar MRI pattern is seen in patients suffering from the well-defined Hereditary Diffuse Leukodystrophy with axonal Spheroids (HDLS), also known as Adult-Onset Leukoencephalopathy With Axonal Spheroids and Pigmented Glia (ALSP) related to genetic variants in colony stimulating factor-1 receptor (CSF1R) gene. These patients also share very similar neurological phenotype [22]. Two previous cases of AARS2 leukodystrophy have had a pathological examination of their brain performed. Both cases displayed accumulation of axonal spheroids, gliosis, myelin loss as well as an overrepresentation of CD68-positive immune cells [7,23]. In addition, pathogenic variants in AARS, encoding the cytosolic paralogue of AARS2, have been identified as the likely cause of Swedish type hereditary diffuse leukoencephalopathy with spheroids sharing clinical and histological features with both AARS2-L and ALSP [24]. A recent review of adult onset leukodystrophies highlights the small areas of restricted diffusion in affected deep white matter, which are also present in our patient, as highly specific for AARS2-L and HDSL/ALSP. In another article Lakshmannan et al. point out white matter rarefaction as a distinctive feature of AARS2-L compared to HDSL/ALSP, which is not observed in our patient. On the other hand, recent case reports [9,10] show that AARS2 mutations not always give rise to leukodystrophy at all, and previous reports have shown cases with very few, and asymmetric white matter changes [6,22]. Thus the group of leukodystrophies, even narrowed within a single gene mutation, is heterogeneous, and from a neuroradiological point of view, the process of diagnosing adult leukodystrophies will require a systematic approach [[25], [26], [27]] and supplementation by next generation genetic sequencing diagnostics.
With only 39 patients with the AARS2 leukodystrophic phenotype previously reported in the scientific literature and only three with homozygous genotype [8,28,29], it would be inaccurate to describe the present patient as a”classic” case of the disease. However, our case presents with several similarities compared to previous cases of AARS2-L. Firstly, symptom-onset in our patient was cognitive decline in his 20's conforming with the mean age-of-onset in other cases [8]. Secondly, our patient presented with an abnormal gait, dysarthria, primitive reflexes and universal spasticity with a one-sided lower limb predominance, thus pyramidal signs were present as in many previous cases. Thus, our case conforms well to the predominant presentation of AARS2 leukodystrophy as reported in the literature [8]. Absent pharyngeal reflexes, involuntary hand tremor, ophthalmoplegia and psychiatric symptoms such as depression and psychosis were also described in earlier cases of AARS2 leukodystrophy [8], these were, however, not present in our patient.
With this case report we hope to add to the growing understanding of AARS2 leukodystrophy, and advocate for the use of whole-genome sequencing as an invaluable tool in the diagnosis of neurometabolic conditions.
Consent for publication
Written informed consent was obtained from the patient and his next-of-kin for publication of this case report and any accompanying images. A copy of the written consent is available for review by the editor of this journal.
Availability of data and materials
Relevant genetic data can be made available upon request to the authors.
Funding
Not applicable.
Declaration of Competing Interest
The authors declare that they have no competing interests.
Acknowledgements
The authors wish to thank the patient and his next-of-kin as well as the reviewers of the paper.
References
- 1.Heim P., Claussen M., Hoffmann B., Conzelmann E., Gärtner J., Harzer K. Leukodystrophy incidence in Germany. Am. J. Med. Genet. 1997 Sep;71(4):475–478. [PubMed] [Google Scholar]
- 2.Euro L., Konovalova S., Asin-Cayuela J., Tulinius M., Griffin H., Horvath R. Structural modeling of tissue-specific mitochondrial alanyl-tRNA synthetase (AARS2) defects predicts differential effects on aminoacylation. Front. Genet. 2015;6:21. doi: 10.3389/fgene.2015.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tawfik D.S., Gruic-Sovulj I. How evolution shapes enzyme selectivity - lessons from aminoacyl-tRNA synthetases and other amino acid utilizing enzymes. FEBS J. 2020 Apr;287(7):1284–1305. doi: 10.1111/febs.15199. [DOI] [PubMed] [Google Scholar]
- 4.Fine A.S., Nemeth C.L., Kaufman M.L., Fatemi A. Mitochondrial aminoacyl-tRNA synthetase disorders: an emerging group of developmental disorders of myelination. J. Neurodev. Disord. 2019 Dec;11(1):29. doi: 10.1186/s11689-019-9292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Götz A., Tyynismaa H., Euro L., Ellonen P., Hyötyläinen T., Ojala T. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am. J. Hum. Genet. 2011 May;88(5):635–642. doi: 10.1016/j.ajhg.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dallabona C., Diodato D., Kevelam S.H., Haack T.B., Wong L.-J., Salomons G.S. Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology. 2014 Jun;82(23):2063–2071. doi: 10.1212/WNL.0000000000000497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lynch D.S., Zhang W.J., Lakshmanan R., Kinsella J.A., Uzun G.A., Karbay M. Analysis of mutations in AARS2 in a series of CSF1R-negative patients with adult-onset Leukoencephalopathy with axonal spheroids and pigmented glia. JAMA Neurol. 2016 Dec;73(12):1433–1439. doi: 10.1001/jamaneurol.2016.2229. [DOI] [PubMed] [Google Scholar]
- 8.Wang X., Wang Q., Tang H., Chen B., Dong X., Niu S. Novel Alanyl-tRNA Synthetase 2 pathogenic variants in Leukodystrophies. Front. Neurol. 2019;10:1321. doi: 10.3389/fneur.2019.01321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuo M.E., Antonellis A., Shakkottai V.G. Alanyl-tRNA Synthetase 2 (AARS2)-related Ataxia without Leukoencephalopathy. Cerebellum. 2020 Feb;19(1):154–160. doi: 10.1007/s12311-019-01080-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srivastava S., Butala A., Mahida S., Richter J., Mu W., Poretti A. Expansion of the clinical spectrum associated with AARS2-related disorders. Am. J. Med. Genet. A. 2019 Aug;179(8):1556–1564. doi: 10.1002/ajmg.a.61188. [DOI] [PubMed] [Google Scholar]
- 11.Peragallo J.H., Keller S., van der Knaap M.S., Soares B.P., Shankar S.P. Retinopathy and optic atrophy: expanding the phenotypic spectrum of pathogenic variants in the AARS2 gene. Ophthalmic Genet. 2018;39(1):99–102. doi: 10.1080/13816810.2017.1350723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong Q., Long L., Chang Y.-Y., Lin Y.-J., Liu M., Lu Z.-Q. An adolescence-onset male leukoencephalopathy with remarkable cerebellar atrophy and novel compound heterozygous AARS2 gene mutations: a case report. J. Hum. Genet. 2018 Jul;63(7):841–846. doi: 10.1038/s10038-018-0446-7. [DOI] [PubMed] [Google Scholar]
- 13.Dinçer P., Leturcq F., Richard I., Piccolo F., Yalnizoglu D., de Toma C. A biochemical, genetic, and clinical survey of autosomal recessive limb girdle muscular dystrophies in Turkey. Ann. Neurol. 1997 Aug;42(2):222–229. doi: 10.1002/ana.410420214. [DOI] [PubMed] [Google Scholar]
- 14.Gallardo E., Saenz A., Illa I. Limb-girdle muscular dystrophy 2A. Handb. Clin. Neurol. 2011;101:97–110. doi: 10.1016/B978-0-08-045031-5.00006-2. [DOI] [PubMed] [Google Scholar]
- 15.Straub V., Murphy A., Udd B. Vol. 28. Neuromuscular Disorders: NMD; England: 2018. 229th ENMC International Workshop: Limb Girdle Muscular Dystrophies - Nomenclature and Reformed Classification Naarden, the Netherlands, 17–19 March 2017; pp. 702–710. [DOI] [PubMed] [Google Scholar]
- 16.Yubero D., Brandi N., Ormazabal A., Garcia-Cazorla À., Pérez-Dueñas B., Campistol J. Targeted next generation sequencing in patients with inborn errors of metabolism. PLoS One. 2016;11(5) doi: 10.1371/journal.pone.0156359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boycott K.M., Vanstone M.R., Bulman D.E., MacKenzie A.E. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat. Rev. Genet. 2013 Oct;14(10):681–691. doi: 10.1038/nrg3555. [DOI] [PubMed] [Google Scholar]
- 18.Farnaes L., Hildreth A., Sweeney N.M., Clark M.M., Chowdhury S., Nahas S. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genom. Med. 2018;3:10. doi: 10.1038/s41525-018-0049-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu W., Pajusalu S., Lake N.J., Zhou G., Ioannidis N., Mittal P. Estimating prevalence for limb-girdle muscular dystrophy based on public sequencing databases. bioRxiv [Internet] 2019 Jan;1:502708. doi: 10.1038/s41436-019-0544-8. http://biorxiv.org/content/early/2019/03/28/502708.abstract Available from: [DOI] [PubMed] [Google Scholar]
- 20.Posey J.E., Harel T., Liu P., Rosenfeld J.A., James R.A., Coban Akdemir Z.H. Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J. Med. 2017 Jan;376(1):21–31. doi: 10.1056/NEJMoa1516767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamatani M., Jingami N., Tsurusaki Y., Shimada S., Shimojima K., Asada-Utsugi M. The first Japanese case of leukodystrophy with ovarian failure arising from novel compound heterozygous AARS2 mutations. J. Hum. Genet. 2016 Oct;61(10):899–902. doi: 10.1038/jhg.2016.64. [DOI] [PubMed] [Google Scholar]
- 22.Lakshmanan R., Adams M.E., Lynch D.S., Kinsella J.A., Phadke R., Schott J.M. Redefining the phenotype of ALSP and AARS2 mutation-related leukodystrophy. Neurol. Genet. 2017 Apr;3(2) doi: 10.1212/NXG.0000000000000135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang D., Yu M., Zhang W., Wang Z., Yuan Y. AARS2 compound heterozygous variants in a case of adult-onset Leukoencephalopathy with axonal spheroids and pigmented glia. J. Neuropathol. Exp. Neurol. 2018 Nov;77(11):997–1000. doi: 10.1093/jnen/nly087. [DOI] [PubMed] [Google Scholar]
- 24.Sundal C., Carmona S., Yhr M., Almström O., Ljungberg M., Hardy J. An AARS variant as the likely cause of Swedish type hereditary diffuse leukoencephalopathy with spheroids. Acta Neuropathol. Commun. 2019 Nov;7(1):188. doi: 10.1186/s40478-019-0843-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lynch D.S., Wade C., de Paiva A.R.B., John N., Kinsella J.A., Merwick Á. Practical approach to the diagnosis of adult-onset leukodystrophies: an updated guide in the genomic era. J. Neurol. Neurosurg. Psychiatry. 2019 May;90(5):543–554. doi: 10.1136/jnnp-2018-319481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Knaap M.S., Bugiani M. Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol. 2017 Sep;134(3):351–382. doi: 10.1007/s00401-017-1739-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Köhler W., Curiel J., Vanderver A. Adulthood leukodystrophies. Nat. Rev. Neurol. 2018 Feb;14(2):94–105. doi: 10.1038/nrneurol.2017.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu T.-H., Peng J., Zhang C.-L., Wu L.-W., Yang L.-F., Peng P. Mutations in aminoacyl-tRNA synthetase genes: an analysis of 10 cases. Zhongguo Dang Dai Er Ke Za Zhi. 2020 Jun;22(6):595–601. doi: 10.7499/j.issn.1008-8830.1912040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Felhi R., Charif M., Sfaihi L., Mkaouar-Rebai E., Desquiret-Dumas V., Kallel R. Mutations in aARS genes revealed by targeted next-generation sequencing in patients with mitochondrial diseases. Mol. Biol. Rep. 2020 May;47(5):3779–3787. doi: 10.1007/s11033-020-05425-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Relevant genetic data can be made available upon request to the authors.