

Abstract
The heterodimers of propargylbenzene (PrBz) with phenylacetylene (PHA) and monosubstituted fluorophenylacetylenes (FPHAs) were investigated using electronic and vibrational spectroscopic methods. The vibrational spectra in the acetylenic C–H stretching region show a marginal shift (0–4 cm–1) upon dimer formation, which suggests minimal perturbation of the acetylenic group. The M06-2X/aug-cc-pVDZ calculations indicate that the π-stacked structures are the most stable, followed by other structures. In general, structures incorporating aromatic C–H···π interactions are much higher in energy. The appearance of the spectra and the energy considerations clearly indicate the preference for the π-stacked structures. Furthermore, the observed trend in the stabilization energies for heterodimers with the three FPHAs is inversely proportional to the dipole moments of FPHAs. On the other hand, the absence of any clear trends in the electrostatic component of the interaction energy is attributed to the presence of the methylene group in PrBz.
1. Introduction
Much interest has been evinced on the properties of weakly bound complexes because of their utility in diverse fields.1−3 The presence of non-covalent interactions in the weakly bound complexes can be generally classified into two qualitatively different interaction motifs, viz., the planar hydrogen bonding and stacking. These interactions are significant, especially in molecular systems incorporating aromatic units, both in chemistry and in biology, and control a diverse range of phenomena such as the packing of aromatic molecules in crystals,4,5 tertiary structures of proteins,6,7 the vertical base-to-base interaction in DNA,8−10 and several others. Hydrogen bonding and its consequences on the vibrational spectra are well understood.11,12 However, the molecular level understanding of factors that influence π-stacking are still a subject of debate.13 Moreover, the usage of the term “π-stacking” is itself a subject of debate.14,15 Experimental investigations on gas-phase π-stacked dimers provide an excellent opportunity to address electronic effects and can be compared directly with the ab initio calculations. However, such reports are comparatively much sparser in comparison to hydrogen bonding for both the homo- and heterodimers.16−30 In this regard, it is important to recognize the difference between “π-stacked” and “parallel-displaced” structures. The π-stacked structures correspond to an interaction between the two aromatic rings directly placed over one another, such as the phenylacetylene (PHA) dimer.28 In a recent work, authors have shown that in the case of homo- and heterodimers of PHA and fluorophenylacetylenes (FPHAs), the dipole moment plays a pivotal role in the formation of the π-stacked structures,20,21 with the anti-parallel π-stacked dimeric structure being the global minimum, especially in the case of homodimers.20,28 On the other hand, the parallel-displaced dimers involve the presence of an additional X–H···π interaction of the substituents on the ring, as observed in the case of dimers of propargyl benzene,19 toluene,23 anisole,25 aniline,31 and others. Because PHA and FPHAs form π-stacked homodimers20,28 while PrBz forms a C–H···π-assisted anti-parallel displaced homodimer,19 the present investigations on the heterodimers of PrBz with PHA and FPHAs are on evaluating the balance between π-stacked and parallel-displaced structures. In this work, electronic and vibrational spectroscopic techniques in combination with dispersion-corrected density functional theory (DFT) calculations are used to probe the heterodimers of PrBz with PHA, 2-fluorophenylacetylene (2FPHA), 3-fluorophenylacetylene (3FPHA), and 4-fluorophenylacetylene (4FPHA), the structures of which are depicted in Figure 1.
Figure 1.

Structures of propargylbenzene (PrBz), phenylacetylene (PHA), 2-fluorophenylacetylene (2FPHA), 3-fluorophenylacetylene (3FPHA), and 4-fluorophenylacetylene (4FPHA) used in the present investigation.
2. Methods
The heterodimers of PrBz with PHA, 2FPHA, 3FPHA, and 4FPHA were synthesized in situ by a supersonic expansion of the desired reagents (at 298 K) doped in helium gas at a 4 atm pressure through a 0.5 mm diameter pulsed nozzle (Series 9, Iota One; General Valve Corporation) operating at 10 Hz. The electronic excitation spectra of the monomers were recorded using the laser-induced fluorescence (LIF) method by monitoring the undispersed total fluorescence with a photomultiplier tube (9780SB + 1252-5F; Electron Tubes Limited) and a filter (BG3 + WG-305) combination. On the other hand, the electronic excitation spectra of the heterodimers were recorded following the one-color resonant two-photon ionization (1C-R2PI) method and monitoring the parent mass ion signal using a two-stage Wiley–McLaren time-of-flight mass spectrometer32 fitted with a channel electron multiplier (CEM-KBL-25RS; Sjuts Optotechnik) and a preamplifier (SR445A; Stanford Research Systems).33 For recording the spectra, the delay time between the valve opening and the laser was optimized separately for the monomer and the dimer to ensure that the signal is maximized. The LIF and the 1C-R2PI signals were digitized by a digital storage oscilloscope (TDS-1012; Tektronix) that was interfaced to a personal computer using a data acquisition program written in LabView. To selectively record the IR spectra of the monomer and the heterodimers in the acetylenic C–H stretching region, the IR–UV double resonance spectroscopic method was used34,35 using either the fluorescence or the ion-detection technique. For this purpose, both the IR and UV lasers focused using a 35 mm plano-convex lens interact with the supersonic free-jet in a counterpropagating manner, with the IR laser arriving about 100 ns prior to the UV laser. In our experiments, the tunable UV laser used was a frequency-doubled output of a dye laser (Narrow Scan GR; Radiant Dyes) operating with the rhodamine-19 dye pumped with the second harmonic of a Nd:YAG laser (Brilliant-B; Quantel). The tunable IR light was generated by LiNbO3 OPO (Custom IR OPO; Euroscan Instruments), as an idler component, pumped with an injection-seeded Nd:YAG laser (Brilliant-B; Quantel). The typical bandwidth of both UV and IR lasers is ∼1 cm–1, and the absolute frequency calibration is within ±2 cm–1.
A detailed conformational search for various dimers was performed by carrying out molecular dynamics (MD) sampling using the MM2 force field36,37 as implemented in the Chem3D Ultra 19.1 software package. The MM2 force field was used for the calculation because it was developed mainly for the conformational analysis of hydrocarbons and other small organic molecules and thus reproduces the equilibrium geometry in very good agreement with the experiment.38 The energy-minimized structure at the same level of theory was used as a starting geometry for the MM2-MD trajectory. The trajectories were calculated for 1 ns with a time step of 2 fs and the intermediate structures were saved at each 10 ps. Subsequently, the selected structures from the MM2-MD trajectory sampling were optimized using the M06-2X/aug-cc-pVDZ level of theory with ultrafine integration grids, followed by frequency calculations. The stabilization energy was determined as the difference between the dimer energy and the sum of monomer energies. The stabilization energies were corrected for the difference in the vibrational zero-point energies (ΔZPE) and the basis–set superposition error (BSSE) using the counterpoise method,39 which was made after geometry optimization. Further, the stabilization energies of the heterodimers were also estimated from the single-point calculation at the MP2/aug-cc-pVDZ level. The justification for the theoretical methods used in the present work is that the M06-2X/aug-cc-pVDZ method is adequate to reliably calculate the geometries and relative energies of various types of intermolecular interactions including π-stacking interaction20 and to further compare the present set of results with our earlier work on heterodimers of PHA with FPHA.21 The analysis of the interaction energies of various structures of heterodimers was carried out using the symmetry-adapted perturbation theory (SAPT).40 The simplest of the SAPT approaches, SAPT0, was performed using the cc-pVTZ basis set along with the cc-pVTZ-JKFIT basis for the Hartree–Fock and cc-pVTZ-RI basis for the SAPT procedure.41 The density-fitting approach was used to reduce the computational expense. The perturbative method SAPT0 treats the monomers at the Hartree–Fock level and then applies the second-order perturbation theory to separate the overall intermolecular interaction energy into the different components. Geometry optimization and frequency calculations were carried out using the Gaussian-16 suite of programs,42 and SAPT0 calculations were carried out using the PSI4 ab initio package.43 The structures and the vibrations were visualized by and GaussView 544 and Chemcraft.45
3. Results and Discussion
The electronic spectra of all the monomers (PrBz, PHA, 2FPHA, 3FPHA, and 4FPHA) have been reported earlier and show sharp bands corresponding to the 000 (band-origin) excitation of the S0→S1 transition (Figure S1, see the Supporting Information).19,21 The electronic spectra of the heterodimers of PrBz with PHA, 2FPHA, 3FPHA, and 4FPHA, recorded using the one color resonant two-photon ionization (1C-R2PI) method by monitoring the parent mass signal, are depicted in Figure 2. The electronic spectra of the PrBz heterodimers with PHA, 2FPHA, 3FPHA, and 4FPHA are very generally red-shifted and show broad bands, similar to the homodimers of PHA and FPHAs, and can be attributed to several reasons which include (i) excitonic coupling, (ii) presence of multiple isomers, (iii) distributed Franck–Condon factors, and combinations thereof.29,30
Figure 2.

Electronic (1C-R2PI) spectra of heterodimers of PrBz with PHA (blue trace), 2FPHA (green trace), 3FPHA (red trace), and 4FPHA (purple trace). For each trace, the color-coded arrow represents the band-origin transition of the corresponding PHA/FPHA monomer. The electronic spectra of all the monomers are shown in Figure S1.
To probe the structural characteristics of the heterodimers of PrBz with PHA, 2FPHA, 3FPHA, and 4FPHA, the IR spectra in the acetylenic C–H stretching region were recorded using the IR–UV double resonance spectroscopic method and are depicted in Figure 3. The IR spectrum of PrBZ in the acetylenic C–H stretching region shows a single band at 3336 cm–1. On the other hand, the corresponding spectrum of PHA shows multiple bands with two primary bands at 3329 and 3344 cm–1, which have been assigned to be originating from Fermi resonance coupling localized on the acetylenic moiety.33 The IR spectrum of the PrBz-PHA heterodimer appears to be convoluted with the transitions of the two constituent monomers and shows two broader bands at 3317 and 3337 cm–1, which appear to be marginally red-shifted relative to both the monomers. To deconvolute the IR spectrum, the PHA monomer in the heterodimer was replaced with its monodeuterated isotopomer (C6H5CCD, PHAD), wherein the acetylenic C–H group was substituted with a C–D group. The resulting IR spectrum of the PrBz-PHAD heterodimer shows a single band at 3334 cm–1 corresponding to the PrBz moiety in the dimer, which is red-shifted by 2 cm–1 from its position in the monomer. This once again suggests that the formation of the dimer marginally red-shifts, by 2 cm–1, the acetylenic C–H oscillator. The IR spectra of the PrBz heterodimers with 2FPHA, 3FPHA, and 4FPHA are also shown in Figure 3. The IR spectra of the heterodimers are convolution of the spectra of both the constituent monomers and show marginal shifts in the band positions and intensities, relative to the monomers, which can be attributed to the changes in the Fermi resonance conditions. In general, the IR spectra of the heterodimers in the acetylenic C–H stretching region show marginal shifts (0–4 cm–1) corresponding to the constituent monomers, which indicates that the acetylenic groups are minimally perturbed due to dimer formation. Similar observations were made in the case of homodimers of PrBz,19 PHA,28 2FPHA, 3FPHA, and 4FPHA20 and heterodimers of PHA with 2FPHA, 3FPHA, and 4FPHA.21
Figure 3.

IR spectra in the acetylenic C–H stretching region. (A) PrBz, PHA, PrBz–PHA, and PrBz–PHAD. (B) PrBz, 2FPHA, and PrBz–2FPHA. (C) PrBz, 3FPHA, and PrBz–3FPHA. (D) PrBz, 4FPHA, and PrBz–4FPHA.
In an effort to assign the structures responsible for the observed spectra, DFT calculations were carried out for all four sets of heterodimers. A structural search followed by geometry optimization using the M06-2X/aug-cc-pVDZ level of theory (see the Methods section) resulted in 13, 19, 23, and 14 structures for the heterodimers of PrBz with PHA (Figure S2, Tables S1), 2FPA (Figure S3, Table S2), 3FPHA (Figure S4, Table S3), and 4FPHA (Figure S5, Table S4), respectively. In general, the structures of the heterodimers of PrBZ with PHA and FPHAs can be categorized into three sets, the π-stacked structures, parallel-displaced structures, and non-stacked structures. In all the cases, the π-stacked structures are the global minima along with several local minima, within an energy band of about 10 kJ mol–1, which differ in the orientation of the two rings in the dimer. On the other hand, the second set consists of parallel-displaced structures in which the two ring planes are almost parallel, additionally incorporating the C–H···π interaction with the methylene group of PrBz, and are marginally (about 1–2 kJ mol–1) less stabilized compared to the π-stacked structures. The third set of structures consist of C–H···π interactions involving either acetylenic C–H or aromatic C–H groups and are at least 6 kJ mol–1 less stabilized compared to the π-stacked global minimum. Unlike the homo- and heterodimers of FPHAs, the heterodimer of PrBz with FPHAs did not yield any structures with C–H···F interactions. The MP2 stabilization energies, calculated at the M06-2X geometries, are in general marginally higher than M06-2X energies consistently for all structures and show the same energy ordering trend, except in a few cases. Figure 4 depicts the six lowest energy structures for the four sets of heterodimers considered in the present work, all of which are π-stacked structures.
Figure 4.

Six lowest energy structures of heterodimers of PrBz with PHA (row 1), 2FPHA (row 2), 3FPHA (row 3), and 4FPHA (row 4) calculated at the M06-2X/aug-cc-pVDZ level of theory. The absolute values of ZPE- and BSSE-corrected stabilization energies (kJ mol–1) are given in parenthesis for each structure. The coloring scheme for the two constituent monomers is different. The structure with carbon atoms in yellow is the PrBz, while the carbon atoms of FPHAs are shown in black and the fluorine atoms in blue.
The IR spectra shown in Figure 3 indicate that the acetylenic C–H oscillator of both the constituent monomers is minimally perturbed due to dimer formation, with shifts of about 0–4 cm–1 that are accompanied by some changes in the intensity redistribution in the Fermi resonance bands. Based on these results, the formation of structures that involve acetylenic C–H···π/F interactions can be ruled out. On the other hand, structures that are π-stacked/parallel-displaced and/or those incorporating aromatic C–H···π/F and interactions would lead to the observed spectra. However, the stabilization energies obtained from the electronic structure calculations indicate π-stacked structures are energetically favorable over other possible structural motifs by at least 6 kJ mol–1. Further, the broadening of the electronic spectra (see Figure 1) in combination with marginal red shifts in the acetylenic C–H stretching vibrations (see Figure 2) have earlier been assigned to (π-)stacked structures of homo- and heterodimers of PHA and FPHAs along with the homodimer of PrBz.19−21,28 Therefore, based on the observed spectra, electronic structure calculations, and comparison with similar systems, the present set of heterodimers of PrBz with PHA and FPHAs are assigned to a set of π-stacked structures within an energy band of 1.5 kJ mol–1 (the mean error for π-stacking interaction energy at the M06-2X level) from the global minima. It must be pointed out that “π-stacking” in the heterodimer structures is loosely defined based on the geometrical criterion.14,21
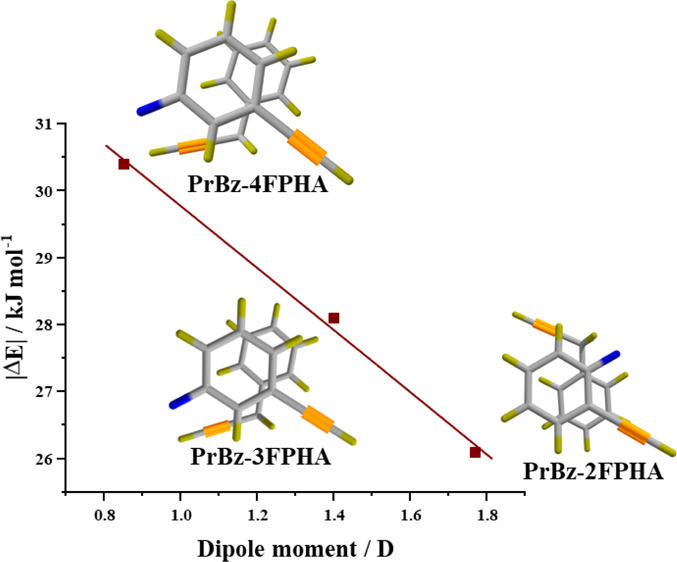
The stacking of heterodimers is further investigated with the energy decomposition analysis using the SAPT0 method, which calculates the contribution of various energy components such as the electrostatic (EElec), induction (EInd), dispersion (EDisp), and exchange (EExch) to the total interaction energy (ESAPT0). The energy components from SAPT0 analysis for all calculated structures of the heterodimers of PrBz are listed in Tables S1–S4 (see the Supporting Information). For all the structures (stacked and non-stacked), the dispersion is the dominant stabilizing component interaction without any exceptions. The ratio of the electrostatic to dispersion components for the six most stable structures for each set of heterodimers, shown in Figure 3, is in the range 0.32–0.46. The stabilization energies of the most stable heterodimers of PrBz in each case are in the order PHA (−24.7 kJ mol–1) < 2FPHA (−26.1 kJ mol–1) < 3FPHA (−28.1 kJ mol–1) < 4FPHA (−30.4 kJ mol–1). The observed trend in the stabilization energies for the three FPHAs is inversely proportional to the dipole moments of FPHAs (see the TOC graphics). This observation is in contrast to the stabilization energies of homodimers of PHA and FPHAs, wherein the stabilization energies are directly proportional to the dipole moments of FPHAs.20 Additionally, the stabilization energies of the present set of PrBz heterodimers are higher than the homo- and heterodimers of PHA and FPHAs and lower than the PrBz homodimer. Surprisingly, the interaction between PrBz and PHA/FPHAs is maximized without incorporating C–H···π interactions due to the methylene group. Further, unlike the case of homo- and heterodimers of PHA and FPHAs,20,21 no clear trends in the stabilization energy or the electrostatic component of the interaction energy with the dipole moments of the PHA and FPHAs were observed for the PrBz heterodimers. This observation is attributed to the presence of the methylene group in PrBz, which leads to structural deformation, relative to homo- and heterodimers of PHA and FPHAs.
4. Conclusions
In summary, the heterodimers of PrBz with PHA and monosubstituted FPHAs were investigated using a combination of electronic and vibrational spectroscopic techniques and electronic structure calculations. The IR spectra of PHA and FPHAs in the acetylenic C–H stretching region show the presence of Fermi resonance bands which get marginally perturbed in terms of both band positions and intensities due to their interaction with PrBz. On the other hand, the acetylenic C–H stretching band of PrBz shows a marginal red shift of 0–2 cm–1. The IR spectra indicate that the formation of the heterodimers does not involve interaction with the acetylenic group, which remains almost unperturbed. The electronic structure calculations indicate that the π-stacked structures are the most stable and could be assigned to the observed spectra based on the energy considerations and circumstantial evidence obtained from the IR spectra. Interestingly, all of the four heterodimers form π-stacked structures in the absence of secondary C–H···π interactions, as was observed in the case of the PrBz dimer. Finally, the interaction energy for the three PrBz heterodimers with FPHAs decreases with an increase in the dipole moment of FPHA, a trend opposite to that of homodimers of FPHAs.
Acknowledgments
This material is based upon work supported in part by the Science and Engineering Research Board, Department of Science and Technology (SERB grant no. EMR/2016/000362) and Board of Research in Nuclear Sciences (grant no. 58/14/18/2020) to G.N.P. The authors gratefully acknowledge the SpaceTime-2 supercomputing facility at IIT Bombay for the computing time.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c02385.
Plot of electronic spectra of the monomers; optimized structures of the PrBz heterodimers with PHA and FPHAs; stabilization energies and SAPT0 energy decomposition analysis; and the coordinates of all the optimized structures (PDF)
Author Present Address
† Present Address: Department of Technical and Applied Chemistry, Veermata Jijabai Technological Institute, Mumbai 400019, India
Author Contributions
The problem was formulated by G.N.P., A.K. performed all of the experiments, and S.S. performed all the calculations. The results were interpreted jointly by all the authors (A.K., S.S., and G.N.P).
The authors declare no competing financial interest.
Notes
This paper is dedicated to the memory of Professor Naohiko Mikami.
Supplementary Material
References
- Jeffrey G. A.An Introduction to Hydrogen Bonding; Oxford University Press: New York, 1997. [Google Scholar]
- Hobza P.; Muller-Dethlefs K.. Non-Covalent Interactions; Theoretical and Computational Chemistry Series; The Royal Society of Chemistry, 2009. [Google Scholar]
- Desiraju G.; Steiner T.. The Weak Hydrogen Bond: In Structural Chemistry and Biology; Oxford University Press: Oxford, 2001. [Google Scholar]
- Dikundwar A. G.; Sathishkumar R.; Guru Row T. N.; Desiraju G. R. Structural Variability in the Monofluoroethynylbenzenes Mediated through Interactions Involving “Organic” Fluorine. Cryst. Growth Des. 2011, 11, 3954–3963. 10.1021/cg2005453. [DOI] [Google Scholar]
- Rubes M.; Bludský O. Intermolecular p–p Interactions in Solids. Phys. Chem. Chem. Phys. 2008, 10, 2611–5. 10.1039/b718701g. [DOI] [PubMed] [Google Scholar]
- Marsili S.; Chelli R.; Schettino V.; Procacci P. Thermodynamics of Stacking Interactions in Proteins. Phys. Chem. Chem. Phys. 2008, 10, 2673–2685. 10.1039/b718519g. [DOI] [PubMed] [Google Scholar]
- Chelli R.; Gervasio F. L.; Procacci P.; Schettino V. Stacking and T-Shape Competition in Aromatic-Aromatic Amino Acid Interactions. J. Am. Chem. Soc. 2002, 124, 6133–6143. 10.1021/ja0121639. [DOI] [PubMed] [Google Scholar]
- Rezác J.; Hobza P. On the Nature of DNA-Duplex Stability. Chem.—Eur J. 2007, 13, 2983–2989. 10.1002/chem.200601120. [DOI] [PubMed] [Google Scholar]
- Hobza P. Stacking Interactions. Phys. Chem. Chem. Phys. 2008, 10, 2581–2583. 10.1039/b805489b. [DOI] [PubMed] [Google Scholar]
- Šponer J.; Riley K. E.; Hobza P. Nature and Magnitude of Aromatic Stacking of Nucleic Acid Bases. Phys. Chem. Chem. Phys. 2008, 10, 2595–2610. [DOI] [PubMed] [Google Scholar]
- Dey A.; Mondal S. I.; Sen S.; Ghosh D.; Patwari G. N. Electrostatics Determine Vibrational Frequency Shifts in Hydrogen Bonded Complexes. Phys. Chem. Chem. Phys. 2014, 16, 25247–25250. 10.1039/c4cp04617j. [DOI] [PubMed] [Google Scholar]
- Sen S.; Patwari G. N. Electrostatics and Dispersion in X–H···Y (X = C, N, O; Y = N, O) Hydrogen Bonds and Their Role in X–H Vibrational Frequency Shifts. ACS Omega 2018, 3, 18518–18527. 10.1021/acsomega.8b01802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-Fenk K.; Herbert J. M. Electrostatics Does Not Dictate the Slip-Stacked Arrangement of Aromatic π–π Interactions. Chem. Sci. 2020, 11, 6758–6765. 10.1039/d0sc02667k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimme S. Do Special Noncovalent π-π Stacking Interactions Really Exist?. Angew. Chem. Int. Ed. 2008, 47, 3430–3434. 10.1002/anie.200705157. [DOI] [PubMed] [Google Scholar]
- Martinez C. R.; Iverson B. L. Rethinking the Term “Pi-Stacking”. Chem. Sci. 2012, 3, 2191–2201. 10.1039/c2sc20045g. [DOI] [Google Scholar]
- Steed J. M.; Dixon T. A.; Klemperer W. Molecular Beam Studies of Benzene Dimer, Hexafluorobenzene Dimer, and Benzene-Hexafluorobenzene. J. Chem. Phys. 1979, 70, 4940–4946. 10.1063/1.437383. [DOI] [Google Scholar]
- Lahmani F.; Lardeux-Dedonder C.; Solgadi D.; Zehnacker A. Spectroscopic Study of the Anisole-Benzene Complex Formed in a Supersonic Free Jet. J. Phys. Chem. 1989, 93, 3984–3989. 10.1021/j100347a024. [DOI] [Google Scholar]
- Capello M. C.; Hernández F. J.; Broquier M.; Dedonder-Lardeux C.; Jouvet C.; Pino G. A. Hydrogen Bonds vs. π-Stacking Interactions in the ρ-Aminophenol. ρ-Cresol Dimer: An Experimental and Theoretical Study. Phys. Chem. Chem. Phys. 2016, 18, 31260–31267. 10.1039/c6cp06352g. [DOI] [PubMed] [Google Scholar]
- Kundu A.; Sen S.; Patwari G. N. The Propargylbenzene Dimer: C-H···π Assisted π-π Stacking. Phys. Chem. Chem. Phys. 2015, 17, 9090–9097. 10.1039/c5cp00162e. [DOI] [PubMed] [Google Scholar]
- Mondal S. I.; Sen S.; Hazra A.; Patwari G. N. π-Stacked Dimers of Fluorophenylacetylenes: Role of Dipole Moment. J. Phys. Chem. A 2017, 121, 3383–3391. 10.1021/acs.jpca.7b00209. [DOI] [PubMed] [Google Scholar]
- Kundu A.; Sen S.; Patwari G. N. Dipole Moment Propels π-Stacking of Hetero-Dimers of Fluorophenylacetylenes. J. Phys. Chem. A 2020, 124, 7470–7477. 10.1021/acs.jpca.0c04005. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Das A. Observation of Exclusively π-Stacked Heterodimer of Indole and Hexafluorobenzene in the Gas Phase. J. Chem. Phys. 2013, 139, 104311. 10.1063/1.4820532. [DOI] [PubMed] [Google Scholar]
- Ishikawa S.; Ebata T.; Ishikawa H.; Inoue T.; Mikami N. Hole-Burning and Stimulated Raman-UV Double Resonance Spectroscopies of Jet-Cooled Toluene Dimer. J. Phys. Chem. 1996, 100, 10531–10535. 10.1021/jp960267d. [DOI] [Google Scholar]
- Goly T.; Spoerel U.; Stahl W. The Microwave Spectrum of the 1,2-Difluorobenzene Dimer. Chem. Phys. 2002, 283, 289–296. 10.1016/s0301-0104(02)00500-1. [DOI] [Google Scholar]
- Pietraperzia G.; Pasquini M.; Schiccheri N.; Piani G.; Becucci M.; Castellucci E.; Biczysko M.; Bloino J.; Barone V.; Barone V.; Ii F. The Gas Phase Anisole Dimer : A Combined High-Resolution Spectroscopy and Computational Study of a Stacked Molecular System. J. Phys. Chem. A 2009, 113, 14343–14351. 10.1021/jp903236z. [DOI] [PubMed] [Google Scholar]
- Guin M.; Patwari G. N.; Karthikeyan S.; Kim K. S. A π-Stacked Phenylacetylene and 1,3,5-Triazine Heterodimer: A Combined Spectroscopic and Ab Initio Investigation. Phys. Chem. Chem. Phys. 2009, 11, 11207–11212. 10.1039/b911640k. [DOI] [PubMed] [Google Scholar]
- Guin M.; Patwari G. N.; Karthikeyan S.; Kim K. S. Do N-Heterocyclic Aromatic Rings Prefer π-Stacking?. Phys. Chem. Chem. Phys. 2011, 13, 5514–5525. 10.1039/c0cp02015j. [DOI] [PubMed] [Google Scholar]
- Maity S.; Patwari G. N.; Sedlak R.; Hobza P. A π-Stacked Phenylacetylene Dimer. Phys. Chem. Chem. Phys. 2011, 13, 16706–16712. 10.1039/c1cp20677j. [DOI] [PubMed] [Google Scholar]
- Muzangwa L.; Nyambo S.; Uhler B.; Reid S. A. On π-Stacking, C-H/π, and Halogen Bonding Interactions in Halobenzene Clusters: Resonant Two-Photon Ionization Studies of Chlorobenzene. J. Chem. Phys. 2012, 137, 184307. 10.1063/1.4765102. [DOI] [PubMed] [Google Scholar]
- Reid S. A.; Nyambo S.; Muzangwa L.; Uhler B. π-Stacking, C-H/π, and Halogen Bonding Interactions in Bromobenzene and Mixed Bromobenzene-Benzene Clusters. J. Phys. Chem. A 2013, 117, 13556–13563. 10.1021/jp407544c. [DOI] [PubMed] [Google Scholar]
- Sugawara K.-i.; Miyawaki J.; Nakanaga T.; Takeo H.; Lembach G.; Djafari S.; Barth H.-D.; Brutschy B. Infrared Depletion Spectroscopy of the Aniline Dimer. J. Phys. Chem. 1996, 100, 17145–17147. 10.1021/jp961901q. [DOI] [Google Scholar]
- Wiley W. C.; McLaren I. H. Time-of-Flight Mass Spectrometer with Improved Resolution. Rev. Sci. Instrum. 1955, 26, 1150–1157. 10.1063/1.1715212. [DOI] [Google Scholar]
- Singh P.; Ganpathi N. P. Infrared-Optical Double Resonance Spectroscopy: A Selective and Sensitive Tool to Investigate Structures of Molecular Clusters in the Gas Phase. Curr. Sci. 2008, 95, 469–474. [Google Scholar]
- Page R. H.; Shen Y. R.; Lee Y. T. Infrared-Ultraviolet Double Resonance Studies of Benzene Molecules in a Supersonic Beam. J. Chem. Phys. 1988, 88, 5362–5376. 10.1063/1.454574. [DOI] [Google Scholar]
- Tanabe S.; Ebata T.; Fujii M.; Mikami N. OH Stretching Vibrations of Phenol-(H2O)n(N=1-3) Complexes Observed by IR-UV Double-Resonance Spectroscopy. Chem. Phys. Lett. 1993, 215, 347–352. 10.1016/0009-2614(93)85726-5. [DOI] [Google Scholar]
- Allinger N. L. Conformational Analysis. 130. MM2. A Hydrocarbon Force Field Utilizing V1 and V2 Torsional Terms. J. Am. Chem. Soc. 1977, 99, 8127–8134. 10.1021/ja00467a001. [DOI] [Google Scholar]
- Burkert U.; Allinger N. L..Molecular Mechanics. ACS Monograph; American Chemical Society: Washington, DC, 1982; Vol. 177. [Google Scholar]
- Jensen F.Introduction to Computational Chemistry; 2nd ed.; John Wiley & Sons Ltd: West Sussex, 2007. [Google Scholar]
- Boys S. F.; Bernardi F. The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. 10.1080/00268977000101561. [DOI] [Google Scholar]
- Szalewicz K. Symmetry-Adapted Perturbation Theory of Intermolecular Forces. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 254–272. 10.1002/wcms.86. [DOI] [Google Scholar]
- Hohenstein E. G.; Sherrill C. D. Density Fitting and Cholesky Decomposition Approximations in Symmetry-Adapted Perturbation Theory: Implementation and Application to Probe the Nature of π-π. J. Chem. Phys. 2010, 132, 184111. 10.1063/1.3426316. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford CT, 2016.
- Turney J. M.; Simmonett A. C.; Parrish R. M.; Hohenstein E. G.; Evangelista F. A.; Fermann J. T.; Mintz B. J.; Burns L. A.; Wilke J. J.; Abrams M. L.; Russ N. J.; Leininger M. L.; Janssen C. L.; Seidl E. T.; Allen W. D.; Schaefer H. F.; King R. A.; Valeev E. F.; Sherrill C. D.; Crawford T. D. Psi4: An Open-Source Ab Initio Electronic Structure Program. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 556–565. 10.1002/wcms.93. [DOI] [Google Scholar]
- Dennington T.; Keith T.; Millam J.. GaussView, version 5, Semichem Inc.: Shawnee Mission KS, 2009.
- ChemCraft. https://www.chemcraftprog.com (accessed August-22-2020).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.