

Abstract
Bax proteins form pores in the mitochondrial outer membrane to initiate apoptosis. This might involve their embedding in the cytosolic leaflet of the lipid bilayer, thus generating tension to induce a lipid pore with radially arranged lipids forming the wall. Alternatively, Bax proteins might comprise part of the pore wall. However, there is no unambiguous structural evidence for either hypothesis. Using NMR, we determined a high‐resolution structure of the Bax core region, revealing a dimer with the nonpolar surface covering the lipid bilayer edge and the polar surface exposed to water. The dimer tilts from the bilayer normal, not only maximizing nonpolar interactions with lipid tails but also creating polar interactions between charged residues and lipid heads. Structure‐guided mutations demonstrate the importance of both types of protein–lipid interactions in Bax pore assembly and core dimer configuration. Therefore, the Bax core dimer forms part of the proteolipid pore wall to permeabilize mitochondria.
Keywords: bax core dimer, functional mutagenesis, membrane lipid bilayer, NMR structure, pore formation
Subject Categories: Autophagy & Cell Death, Membrane & Intracellular Transport, Structural Biology
NMR structures of membrane‐bound Bax core dimer reveal dual interactions with non‐polar tails and charged heads of bilayer lipids contributing directly to apoptotic mitochondrial permeabilization.
Introduction
Apoptotic cell death initiates when the mitochondrial outer membrane (MOM) is permeabilized by Bax or Bak, two pore‐forming proteins in the Bcl‐2 family. Bax and Bak share the same structural fold with the colicine family of α‐helical pore‐forming bacterial toxins (Parker et␣al, 1989; Suzuki et␣al, 2000; Moldoveanu et␣al, 2006). Despite more than three decades of intensive investigation, it is still unclear how these proteins form pores in various membranes and what are the pore structures that allow molecules of various sizes to cross diverse membranes (Andrews, 2014; Cosentino & Garcia‐Saez, 2017; Dal Peraro & van der Goot, 2016; Uren et␣al, 2017). In particular, it is unclear how Bax can form giant pores in the MOM that can be tens to hundreds of nanometers in diameter (Grosse et␣al, 2016; Salvador‐Gallego et␣al, 2016). The most popular proteolipid pore model requires Bax to fulfill two functions (Cosentino & Garcia‐Saez, 2017). First, Bax would embed in the cytosolic leaflet of the MOM lipid bilayer, asymmetrically expanding it to bend the bilayer and generate membrane tension that eventually breaks the lipid bilayer. The lesion in the membrane exposes the nonpolar lipid acyl tails to aqueous milieu, and hence, is thermodynamically unstable. An immediate solution is to cover the exposed acyl tails of the bilayer lipids with non‐bilayer lipids, for example, lipids that are radially arranged to a micellar structure, resulting in an aqueous pore lined by the polar or charged heads of bent lipids. However, bending the lipids at the pore rim generates line tension that is proportional to the pore radius, and hence, opposes pore opening. Thus, the second function of Bax is to reduce the line tension by intercalating between the bent lipids and reinforcing the pore rim around the bilayer lipids.
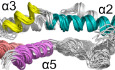
In healthy cells, Bax is mostly a soluble cytoplasmic protein but occasionally visits the mitochondria as a peripherally bound protein (Edlich et␣al, 2011). To become a membrane protein and fulfill the pore‐inducing and stabilizing functions in stressed cells, Bax must change conformation. The conformation change is triggered by BH3 proteins of the Bcl‐2 family (Fig 1A). In particular, transient binding by Bid or Bim unfolds the soluble and globular Bax structure of a bundle of nine α‐helices (α1 to α9) (Fig 1A, soluble Bax) (Suzuki et␣al, 2000), resulting in an exposed α9 at the C‐terminus that can insert into the MOM, and an exposed α2 to α5 region that can interact with its counterpart in another activated and unfolded Bax protein (Suzuki et␣al, 2000; Gavathiotis et␣al, 2008; Lovell et␣al, 2008; Gavathiotis et␣al, 2010; Chi et␣al, 2020). According to a crystal structure, the two α2 to α5 regions can form a symmetric and amphipathic dimer with a polar and charged α2–α3 surface on the top and a concave nonpolar α4–α5 surface at the bottom (Fig 1A, Bax (α2–α5) dimer) (Czabotar et␣al, 2013). This structure would fulfill the two functions if the sidechains of the nonpolar α4–α5 residues could not only insert into the cytosolic leaflet of the MOM lipid bilayer to induce the pore opening, as the “in‐plane” insertion model postulated (Fig 1A, in‐plane model) (Westphal et␣al, 2014), but also intercalate between the bent lipids at the pore rim to stabilize the opening, as the “clamp” to rim model suggested (Fig 1A, clamp model) (Bleicken et␣al, 2014).
Figure 1. Models for the membrane topology of Bax and NMR spectra of Bax (α2–α5).
-
AThe “in‐plane” and the “clamp” models for the membrane topology of pore‐forming Bax protein. After activation by BH3 proteins, the soluble Bax protein unfolds the α‐helical bundle structure and inserts the α9 helix into the MOM. The tail‐anchored Bax proteins dimerize via their α2, α3, α4, and α5 helices resulting in an amphipathic structure (shown as Bax (α2–α5) dimer with the protomers in cyan or gray color) with a polar surface containing positive and negative charged α2 and α3 residues (blue and red sticks) and a nonpolar surface containing aromatic α4 and α5 residues (orange rings). According to the in‐plane model, the α2–α5 dimer engages the planer cytosolic surface of the MOM with the aromatic α4 and α5 residues projected into the cytosolic leaflet of the lipid bilayer to generate membrane tension thereby inducing a lipid pore with radially arranged lipids at the rim. In contrast, the clamp model positions the α2–α5 dimer at the lipid pore rim with the aromatic α4 and α5 residues intercalated between the radiated lipids to reduce line tension thereby stabilizing the pore. As illustrated, the membrane topologies of other helices are also different between the models. The structures of the soluble Bax protein and the α2–α5 dimer were generated from PDB files 1F16 and 4BDU, respectively, using PyMOL program. The structures of other helices are modeled based on the corresponding structures in the soluble Bax protein.
-
B2D 1H–15N TROSY‐HSQC spectra of DMPC/DHPC bicelle‐bound Bax (α2–α5) (blue) and soluble Bax (α2–α5) (red) at 600 MHz. The magnified regions highlight the spectral changes for some residues in the bicelle‐bound and soluble proteins.
Accumulating biochemical and biophysical evidence has supported these structure‐based functions. (i) Site‐specific crosslinking, double electron–electron resonance (DEER) spectroscopy and Förster resonance energy transfer have verified the dimeric interaction between the α2 and α5 regions of two full‐length Bax proteins integrated into the native or model mitochondrial membrane (Dewson et␣al, 2012; Bleicken et␣al, 2014; Gahl et␣al, 2014; Zhang et␣al, 2016). (ii) Accessibility of cysteine residues placed in the nonpolar surface of the α2 to α5 dimer to a charged sulfhydryl reactive labeling agent (IASD), electron paramagnetic resonance (EPR) detected accessibility of spin probes attached to the nonpolar surface to molecular oxygen enriched in the lipid bilayer or paramagnetic quenchers soluble in water, fluorescence emission and quenching of environment sensitive dyes attached to the nonpolar surface, and H‐D exchange of the α4 to α5 region in a membrane‐bound Bax oligomer are consistent with their locations within the membrane (Annis et␣al, 2005; Bleicken et␣al, 2010; Westphal et␣al, 2014; Pogmore et␣al, 2016; Zhang et␣al, 2016; Flores‐Romero et␣al, 2017; Bleicken et␣al, 2018). (iii) In contrast to the stable conformation of the α2 to α5 dimer, the DEER spectral data from the α6 to α9 region indicate a dynamic conformation that would allow the α6 from one protomer to engage the cytosolic surface of the MOM, and the α6 from the other protomer to engage the intermembrane surface, with the two α9 membrane anchors in opposite orientations (Bleicken et␣al, 2014). This topographic model provides a mechanism that clamps the α2 to α5 dimer to the pore rim using the α6 to α9 regions and fits the concave nonpolar surface of the dimer nicely to the convex micellar lipid surface (Fig 1A, clamp model) (Bleicken et␣al, 2014).
Therefore, both of the proposed functions for Bax have been supported by experimental evidence. As illustrated in Fig 1A, the pore‐inducing function would be fulfilled by the nonpolar surface of the α2 to α5 dimer if it engages the cytosolic leaflet of the MOM lipid bilayer. In contrast, if the α2 to α5 dimer intercalates between the bent lipids surround the opening of a lipid pore, it would stabilize the pore. However, the structure of the α2 to α5 dimer was determined by crystallography in the absence of membranes. Thus, we do not know where and how the dimer engages the membrane. On the other hand, although the IASD‐labeling, EPR, fluorescence, and H‐D exchange data were obtained with full‐length Bax proteins bound to membranes, they do not possess enough structural resolution to differentiate the two potential membrane topologies of the α2 to α5 regions proposed by the in‐plane and the clamp models (Andrews, 2014; Bleicken et␣al, 2018). To fill the knowledge gap, we first determined the structure of the α2 to α5 region bound to a lipid bicelle using NMR. The resulting atomic resolution model for how the α2 to α5 dimer interacts with a model lipid bilayer is more consistent with the clamp model than the in‐plane model. We then performed structure‐guided mutagenesis to assess how distinct interactions between specific residues around or within the nonpolar surface of the α2 to α5 dimer and heads or tails of lipids in the bilayer contribute to the pore‐forming function of intact Bax protein in model and native mitochondrial membranes and to the dimer configuration in the native mitochondrial membrane. Our study demonstrates that the α2 to α5 dimer forms part of the wall that separates the nonpolar lipid bilayer from the aqueous pore via ionic or polar interactions with the negatively charged or polar lipid headgroups and hydrophobic interactions with the nonpolar lipid acyl chains. Thereby, we propose a model for the mitochondrial Bax pore, which is at least partially lined by a series of α2 to α5 dimers that are stitched together by other Bax regions.
Results
Structure determination of Bax (α2–α5) in lipid bicelles
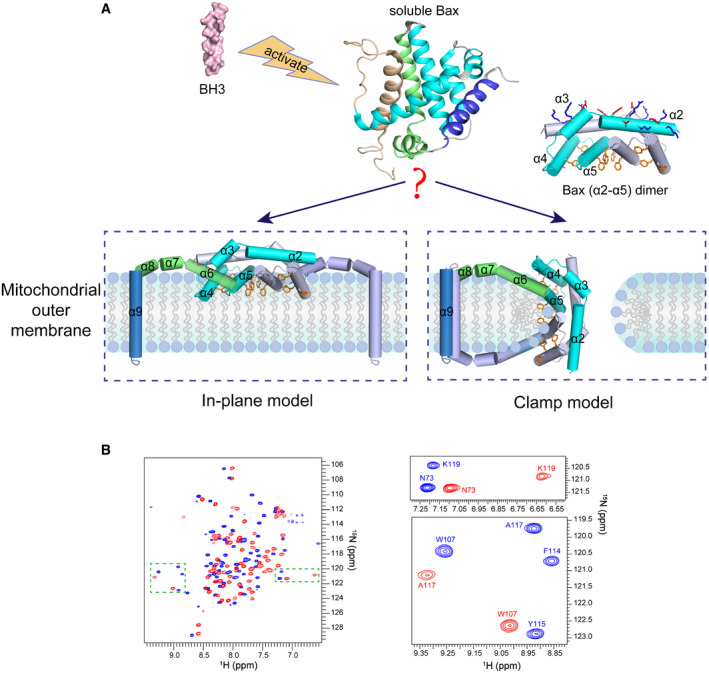
To study the structural properties of the Bax core domain in membranes, we determined the structure of the bicelle‐bound Bax α2 to α5 region (α2–α5) following an NMR strategy used for oligomeric membrane proteins (Fu et␣al, 2019). We made bicelles with 1,2‐dimyristoyl‐sn‐glycero‐3‐phosphocholine (DMPC) and 1,2‐dihexanoyl‐sn‐glycero‐3‐phosphocholine (DHPC) at a molar ratio (q) ≥ 0.5 to yield lipid discs with a radius of ˜ 25 Å for the bilayer region (Appendix␣Fig S1A), large enough to be certain that the Bax protein can be embedded in a lipid bilayer environment. These lipid discs also have radially arranged DHPC lipids at their rim (Chen et␣al, 2018), similar to the lipid pore rim to which the Bax protein may bind. We expressed and purified the isotope‐labeled human Bax (α2–α5) protein (residues D53 to K128 with C62S and C126S mutations). The resulting soluble Bax (α2–α5) is stable enough to generate a two‐dimensional (2D) 1H–15N correlation spectrum with good resolution and dispersion (Fig 1B). Addition of the DMPC/DHPC bicelles dramatically changed the NMR spectrum of the Bax (α2–α5) (Fig 1B), indicating that the protein undergoes large conformational changes due to the interaction with membranes. The same spectral changes were observed when cardiolipin was included in the lipid bicelles (Appendix␣Fig S1B), suggesting that the conformational changes of Bax (α2–α5) are not dependent on this mitochondrial lipid. This is in contrast to Bax protein whose conformational changes are triggered by the mitochondrial membranes and activator proteins such as tBid that require cardiolipin to be fully active (Lovell et␣al, 2008; Shamas‐Din et␣al, 2015). SEC‐MALS analysis showed that the soluble Bax (α2–α5) is mainly a tetramer mixed with lower order complexes (Appendix␣Fig S1C). Consistent with␣this observation, crosslinking with bis(sulfosuccinimidyl)‐suberate (BS3) detected tetramers alongside dimers and trimers (Appendix␣Fig S1D). The crosslinking efficiency was low for the bicelle‐bound Bax (α2–α5) (Appendix␣Fig S1D), possibly because the crosslinker is membrane impermeable. In addition, the efficiency (E) of each bimolecular crosslinking reaction is expected to be less than 1, and hence, the crosslinking efficiency of two or three crosslinking reactions that must occur to a trimer or tetramer to result in a covalently linked trimer or tetramer, respectively, is expected to be much lower since it equals to E2 or E3. Nonetheless, it is clear that a small fraction of the bicelle‐bound protein was crosslinked as a dimer.
To determine the structure, nearly all (97%) of the backbone resonances in the bicelle‐bound Bax (α2–α5) were assigned using standard TROSY‐based triple‐resonance experiments (Fig EV1A). Most resonances are clearly shifted from the assigned backbone resonances of the soluble protein (Figs 1B and EV1, EV2, EV3, EV4, EV5). In addition, the resonances of F114, Y115, and F116 that were missing from the soluble Bax (α2–α5) appeared for the bicelle‐bound Bax (α2–α5). The secondary structures of the bicelle‐bound Bax (α2–α5) were derived from analysis of the backbone chemical shifts using the TALOS+ program (Shen et␣al, 2009). Overall the helical regions of Bax (α2–α5) in bicelles correlate well with those in the crystal structure (PDB code: 4BDU) (Czabotar et␣al, 2013). We then determined the local structures of the bicelle‐bound Bax (α2–α5) monomers and assembled the dimer structures by using 702 local and 60 long‐range distance constraints derived from nuclear Overhauser enhancement (NOE) measurements (Appendix␣Fig S2A–B; Table 1). The 15 lowest energy structures (PDB code: 6L8V) out of 200 calculated dimer structures converged to a root mean squared deviation (RMSD) of 1.058 and 1.504 Å for backbone and all heavy atoms, respectively (Fig 2A; Table 1). Furthermore, a sulfhydryl reactive nitroxide spin label, MTSL (Fu et␣al, 2019), was introduced into two single‐Cys Bax (α2–α5) mutants (A82C and S126C) to collect the paramagnetic resonance enhancement (PRE) data that provided the␣inter‐monomer distance constraints confirming the structures of the symmetric antiparallel Bax (α2–α5) dimers in the bicelles (Appendix␣Fig S2C).
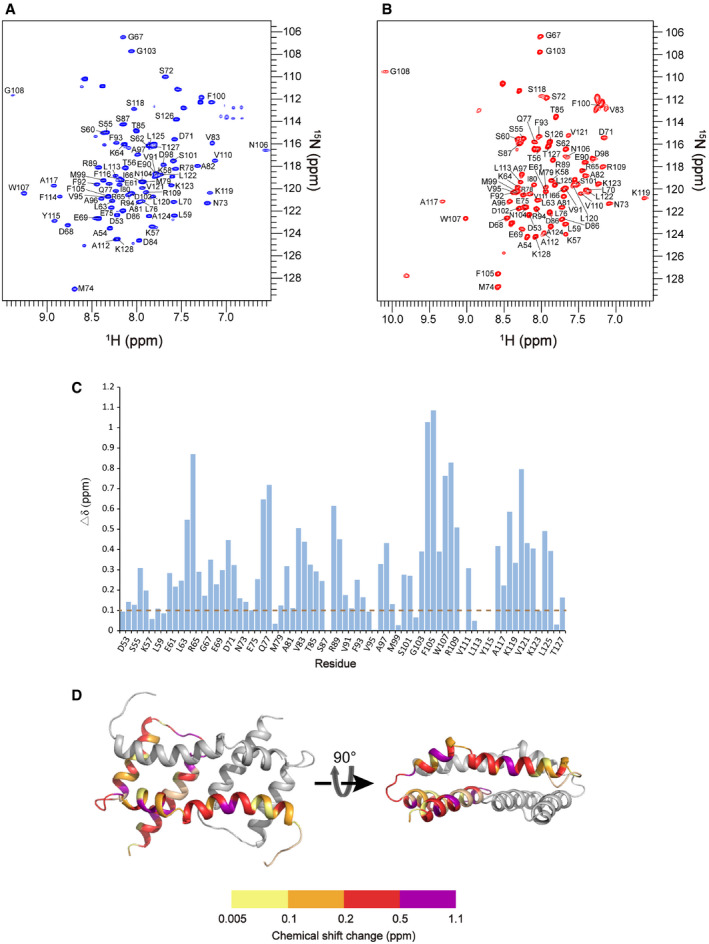
Figure EV1. Related to Figs 1 and 2. NMR spectra of Bax (α2–α5) in bicelles and solution with assignments.
-
A2D 1H–15N TROSY‐HSQC spectrum of the uniformly [15N, 13C, 2H]‐labeled Bax (α2–α5) protein in DMPC/DHPC bicelles (q = 0.5) recorded at 1H frequency of 600 MHz. The backbone amide resonances were assigned to the indicated residues.
-
B2D 1H–15N TROSY‐HSQC spectrum of the uniformly [15N, 13C, 2H]‐labeled soluble Bax (α2–α5) protein recorded at 1H frequency of 600 MHz. The backbone amide resonances were assigned to the indicated residues.
-
CNMR chemical shift changes of Bax (α2–α5) upon the addition of the bicelles. The plot of chemical shift changes between the spectra of bicelle‐bound Bax (α2–α5) and soluble Bax (α2–α5). The combined chemical shift changes (Δδ) were calculated by using the equation: where ∆δH and ∆δN are chemical shift changes (in ppm) in the 1H and 15N dimensions, respectively, and ω H and ω N are normalization factors (ω H = 1.00, ω N = 0.15).
-
DThe chemical shift changes of Bax (α2–α5) were mapped to the structure of the bicelle‐bound Bax (α2–α5), colored in one chain according to the scale of chemical shift changes shown below.
Source data are available online for this figure.
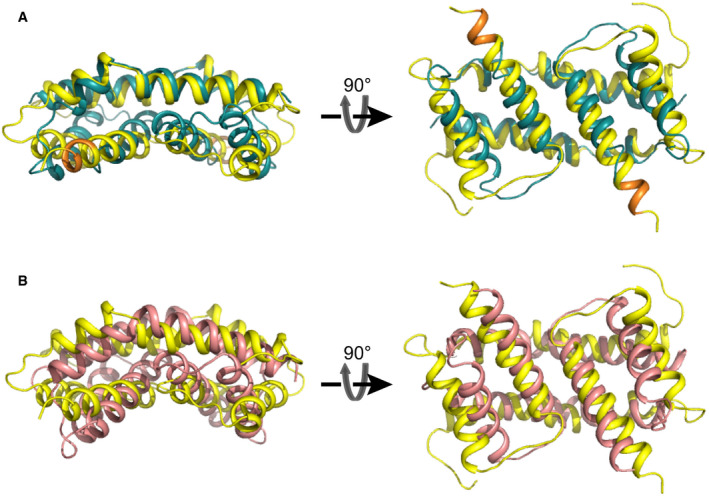
Figure EV2. Related to Fig 2. Comparison of Bax (α2–α5) dimer structures in bicelles (yellow) to crystal structures of Bax (α2–α5) (cyan) and Bak (α2–α5) (salmon) dimers.
-
AThree‐dimensional structure superposition of the structure of Bax (α2–α5) in bicelles determined by NMR (yellow; PDB code: 6L8V) and the structure determined by crystallography in the absence of membranes (cyan; PDB code: 4BDU).The RMSD value between the superimposed protein backbones is 3.878 Å. Note that the residues from K123 to S126 form an extra α helical turn (orange) in the NMR structure.
-
BThree‐dimensional structure superposition of the bicelle‐bound structure of Bax (α2–α5) (yellow; PDB code: 6L8V) and the Bak (α2–α5) dimer structure determined by crystallography in the absence of membranes (salmon; PDB code: 4U2V). The RMSD value between the superimposed protein backbones is 5.177 Å.
Figure EV3. Interactions of Bax (α2–α5) mutants with lipid bicelles.
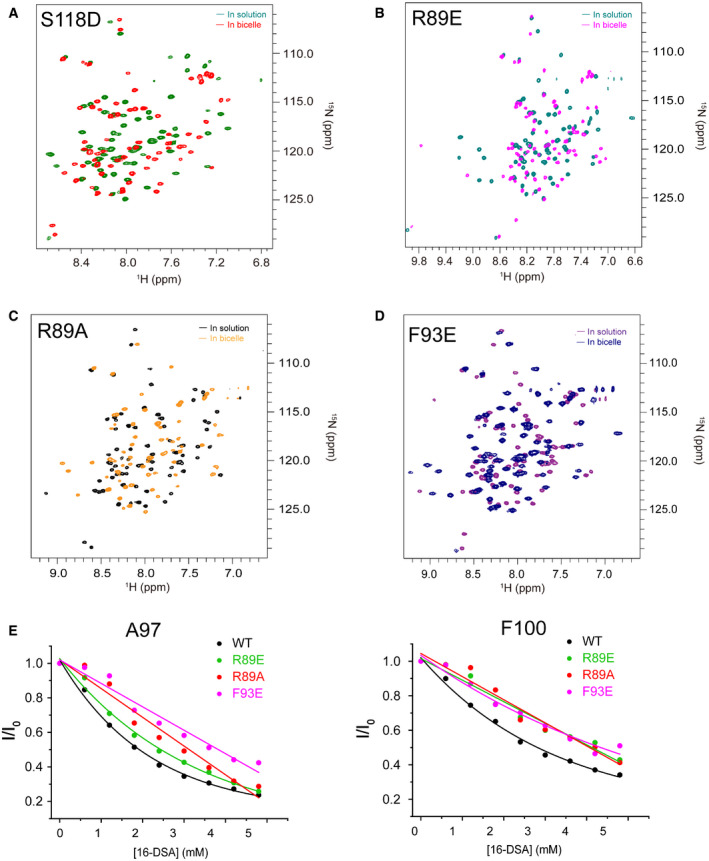
-
A–DSuperimposed 2D 1H–15N TROSY‐HSQC spectra of (A) bicelle‐bound (green) and soluble (red) Bax (α2–α5) S118D, (B) bicelle‐bound (teal) and soluble (magenta) Bax (α2–α5) R89E, (C) bicelle‐bound (yellow) and soluble (black) Bax (α2–α5) R89A, and (D) bicelle‐bound (blue) and soluble (purple) Bax (α2–α5) F93E recorded at 600 MHz.
-
ESpectral peak intensity decay (I/I0) for A97 and F100 caused by PRE agent 16‐DSA, determined from a series of 1H–15NTROSY‐HSQC spectra of bicelle‐bound Bax (α2–α5) (black) or the R89E (green), R89A (red) and F93E (magenta) mutants recorded at different [16‐DSA]. I/I0 = the peak intensity in the presence 16‐DSA/that in the absence of 16‐DSA.
Source data are available online for this figure.
Figure EV4. Related to Fig 4. Pore‐forming activity of Bax mutants at low or high concentration.
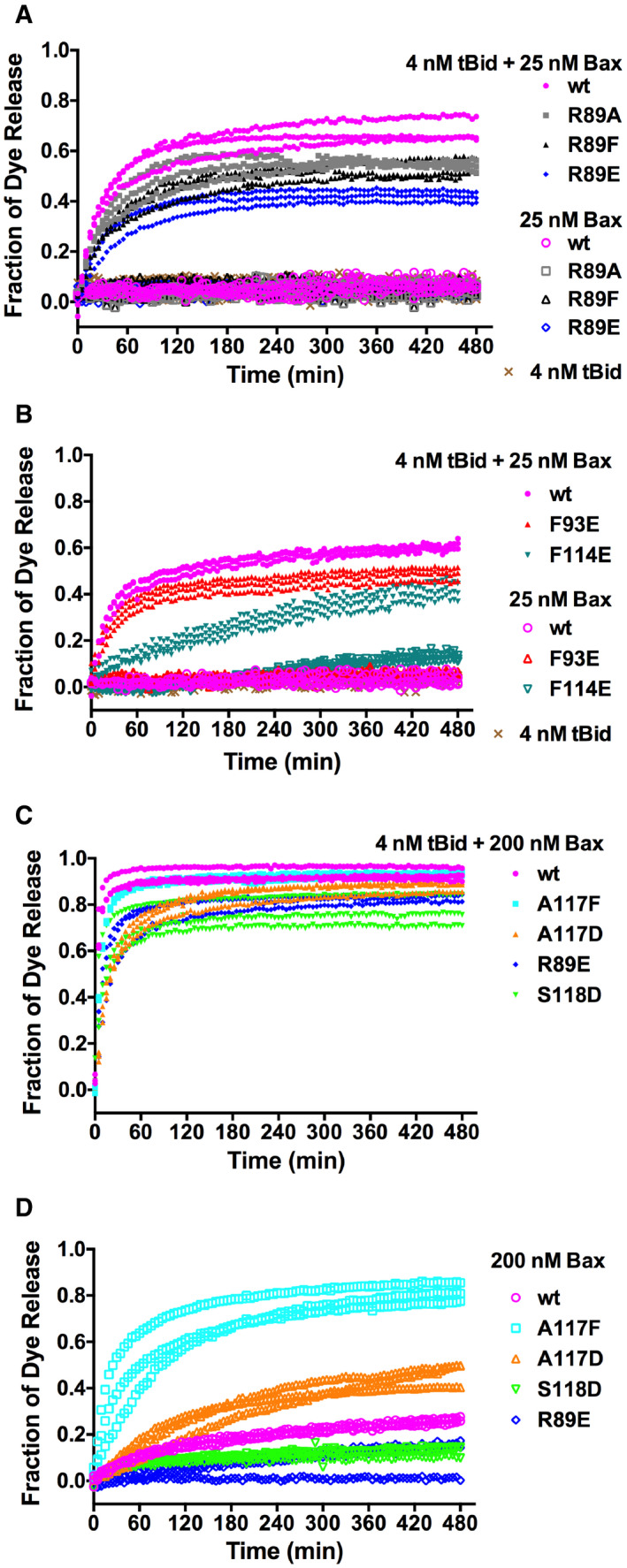
-
A–DThe fluorescent dye release from mitochondrion‐mimic liposomes by 25 or 200 nM wt or mutant Bax in the presence or absence of 4 nM tBid as indicated was measured during a time course by fluorescence quenching. The fraction of dye release was normalized to that by detergent. The data shown were obtained from n = 3 independent replicates using the same preparations of the proteins and liposomes.
Source data are available online for this figure.
Figure EV5. Related to Fig 4. Effect of membrane‐binding defective mutations on the Bax core dimerization.
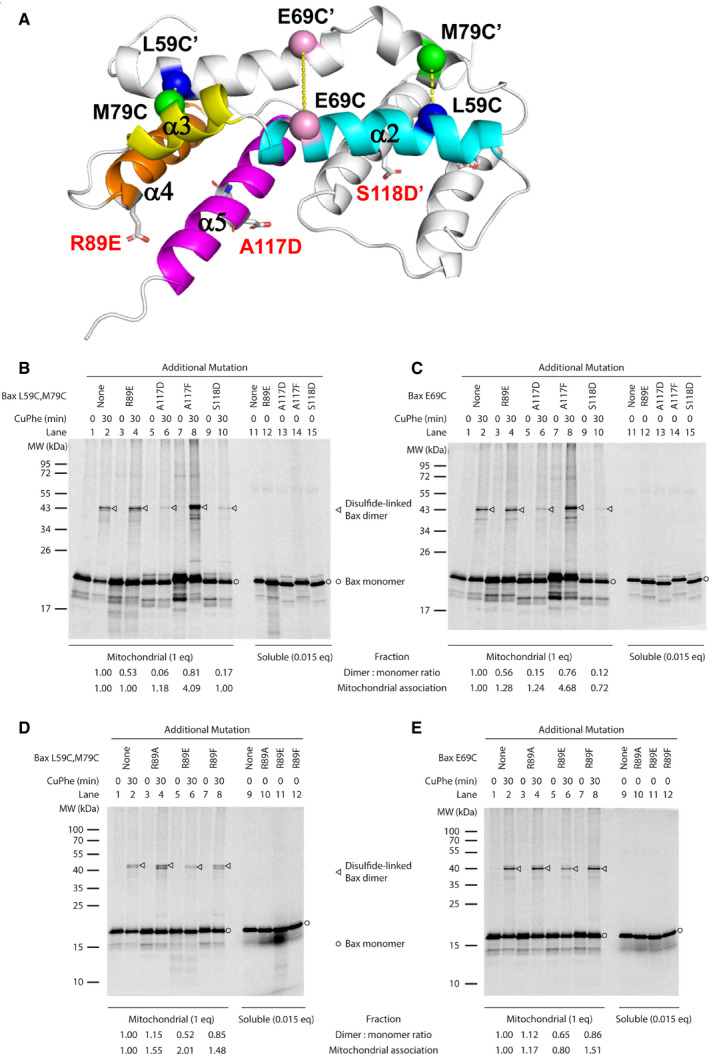
-
AA ribbon diagram of the NMR structure of bicelle‐bound Bax (α2–α5) dimer. One protomer is colored with the helices labeled. The Cα carbons of the residue pairs that were replaced by Cys pairs for disulfide crosslinking are shown as spheres linked by dashed lines. The R89E, A117D, and S118D mutations are represented by sticks.
-
B–E[35S]Met‐labeled Bax proteins with two Cys at position 59 and 79 (B, D) or one Cys at 69 (C, E) and an additional mutation, if indicated, to disrupt the α2–α5 core dimer interaction with membranes were synthesized, activated by Bax BH3 peptide, and targeted to the mitochondria lacking endogenous Bax and Bak proteins. The mitochondria‐bound proteins were oxidized by CuPhe for 30 min to induce disulfide crosslinking of the two protomers via the Cys pair(s) in the core dimer interface as shown in (A). The radioactive crosslinked Bax dimer was then separated from the monomer by non‐reducing SDS–PAGE and detected by phosphor‐imaging. The representative data from two to four independent experiments are shown. The relative dimer:monomer ratio shown at the bottom of the phosphor‐images was determined by the intensities of the dimer and monomer bands of the Cys mutant with the additional mutation, normalized to that of the corresponding Cys mutant without the additional mutation. The mitochondrial association efficiency of each Bax mutant (E) was determined by the summed intensity of the monomer and dimer bands in the mitochondrial fraction that was divided and treated with CuPhe for 0 or 30 min (IM), the intensity of the monomer band in the corresponding soluble fraction that was divided by 0.015 to adjust the inequivalent loading between the mitochondrial (1 equivalent (eq)) and soluble (0.015 eq) fractions (IS), and the following equation, . The relative mitochondrial association efficiency of each Bax Cys mutant with the additional mutation (ER) to that of the Bax Cys mutant without the additional mutation was obtained by dividing the E of the former with the E of the latter and shown at the bottom of the phosphor‐images.
Source data are available online for this figure.
Table 1.
NMR and refinement statistics for bicelle‐bound Bax (α2–α5) structures.
| NMR distance and dihedral constraints | |
| Distance constraints from NOE | 762 |
| Intra‐chain NOEs | 702 |
| Inter‐chain NOEs | 60 |
| Total dihedral angle constraints | 260 |
| Φ (TALOS) | 130 |
| Ψ (TALOS) | 130 |
| Structure statistics a | |
| Violations (mean ± SD) | |
| Distance constraints (Å) | 0.077 ± 0.005 |
| Dihedral angle constraints (°) | 0.653 ± 0.014 |
| Deviations from idealized geometry | |
| Bond lengths (Å) | 0.006 ± 0.000 |
| Bond angles (°) | 0.310 ± 0.057 |
| Impropers (°) | 0.419 ± 0.018 |
| Average pairwise RMSD (Å) b | |
| Heavy atoms | 1.504 |
| Backbone | 1.058 |
| PDB ID | 6L8V |
Statistics are calculated and averaged over an ensemble of the 15 lowest energy structures of the 200 calculated structures.
The precision of the atomic coordinates is defined as the average RMSD between the 15 lowest energy structures and their mean coordinates.
Figure 2. Structures of Bax (α2–α5) in lipid bicelles determined by NMR.
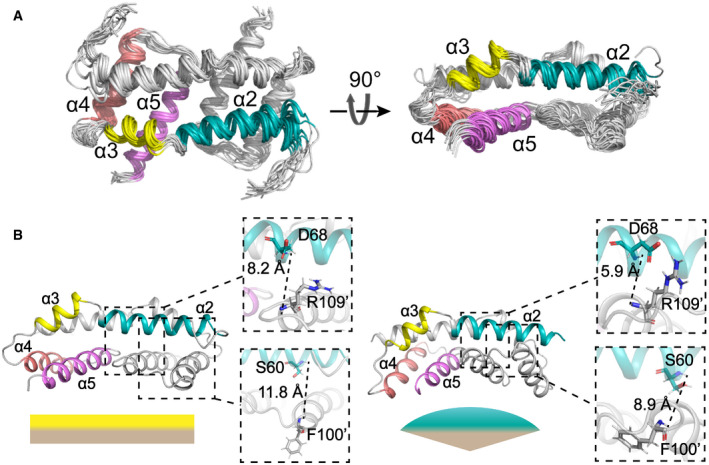
-
AEnsemble of 15 lowest energy structures for Bax (α2–α5) dimer in bicelles calculated from the geometry constraints obtained from the NMR experiments summarized in Table 1. The backbone structures are shown as thin ribbons.
-
BComparison of the NMR structure of Bax (α2–α5) in bicelles (left; PDB code: 6L8V) and the crystal structure (right; PDB code: 4BDU). The NMR or crystal structure has a flat or concave bottom surface formed by the α4–α5 regions, respectively, as indicated by the complimentary flat or convex shapes underneath. The zoom‐in regions display the distances between the amide protons of D68‐R109’ and S60‐F100’, which are longer in the NMR structure than the crystal structure.
The bicelle‐bound Bax (α2–α5) dimer structure is different from the previously reported crystal structure in a GFP‐Bax (α2–α5) tetramer (PDB code: 4BDU) (Fig EV2A) (Czabotar et␣al, 2013). The calculated single‐point energy of the bicelle‐bound Bax (α2–α5) dimer is −4,911.54 kcal/mol, lower than −3,156.73 kcal/mol, the calculated energy of a Bax (α2–α5) dimer in the crystal structure, indicating the binding of Bax (α2–α5) to the bicelles stabilizes the structure. Alignment of the two structures shows a 3.878‐Å backbone RMSD (Fig EV2A). In both structures, the two α2 helices are antiparallel and each interacts with the α3, α4, and α5 helices of the other protomer (Fig 2B). However, upon the bicelle binding, the C‐terminus of the α4 helix and the N‐terminus of the α5 helix tilt away from the α2 helix to yield a larger gap in the dimerization interface with less intermolecular interactions (Fig 2B). As a result, the distances between the amide protons of S60‐F100’ and D68‐R109’ are increased in the bicelle‐bound structure (Fig 2B). Furthermore, the α4 and α5 helices in one protomer form a hydrophobic surface with their counterparts in the other protomer. The angles between the two α4 helices and between the two α5 helices in the dimeric Bax (α2–α5) are reduced from ˜ 82° and ˜ 26o in the crystal structure to ˜ 12° and ˜ 11° in the bicelle‐bound structure, respectively, thereby changing the concave surface to a flat surface (Fig 2B). Finally, the α5 helix is elongated in the bicelle‐bound structure with residues K123 to S126 forming an additional α helical turn, while the structural information of these residues is missing in the crystal structure (Fig EV2A). Our high‐resolution structure for the α5 helix in the bicelle‐bound Bax (α2–α5) dimer is consistent with the α5 helix structure in the clamp model that is in part based on the notion that the DEER‐derived distance distributions could be better fit by the crystal structure after the α5 helix was elongated to S126 (Bleicken et␣al, 2014).
The crystal structure of Bak (α2–α5) dimer (PDB code: 4U2V) (Brouwer et␣al, 2014) shows a different curvature in the α4–α5 surface than the analogous Bax surface (PDB code: 4BDU) (Czabotar et␣al, 2013). The structural comparison between the Bak (α2–α5) dimer and the bicelle‐bound Bax (α2–α5) dimer results in a 5.177‐Å backbone RMSD and the α4–α5 surface is flatter in the latter than the former (Fig EV2B). Recently published crystal structures of lipid‐ or detergent‐bound Bak (α2–α5) dimers also showed flatter α4–α5 surfaces than the free Bak (α2–α5) dimer (Cowan et␣al, 2020). Overall, these structure comparisons suggest that the (α2–α5) dimers adjust their structures to better accommodate the membranes or lipids that they bind.
Interaction of Bax (α2–α5) with lipid bicelles
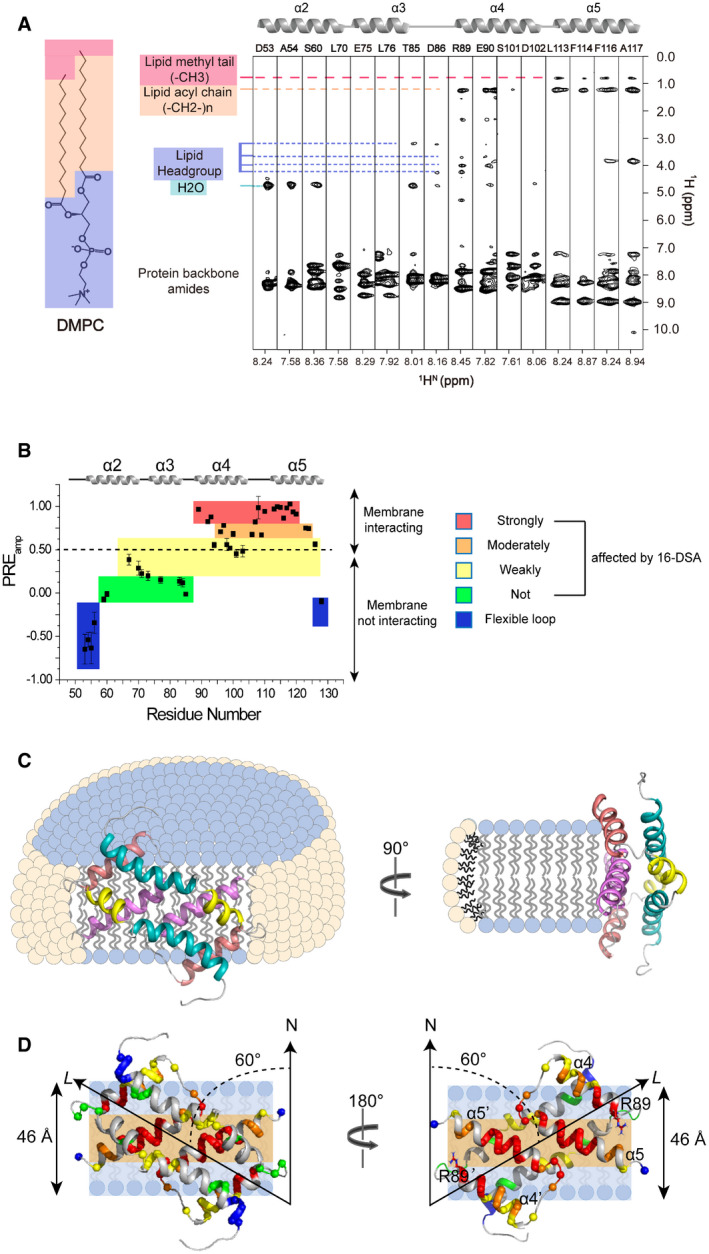
To determine how the Bax (α2–α5) dimer interacts with a membrane, we performed protein‐lipid NOE experiments to examine the interaction between the dimer and the lipid bicelle. We recorded an 15N‐edited NOE spectroscopy (NOESY) spectrum using the [15N, 2H]‐labeled Bax (α2–α5) protein reconstituted in the bicelles composed of regular DMPC and deuterated DHPC. The crosspeaks in the NOESY spectrum appear when two protons are close to each other, typically within 5 Å (Skinner & Laurence, 2008). The NOE strip plot taken from the 3D 15N‐edited NOESY‐TROSY‐HSQC spectrum shows that the residues in α2 and α3 helices have only water crosspeaks from either NOE or proton exchange or both and no NOEs to the DMPC lipids, indicating that these helices are mainly exposed to water and not interacting with the lipid bilayer (Fig 3A). Residues T85 and D86 in the loop between α3 and α4 helices show NOEs only to the headgroups of DMPC lipids, whereas R89 and E90 in α4 helix show NOEs to both headgroups and acyl chains of DMPC lipids, indicating their shallow immersion in the lipid bilayer. In contrast, residues L113, F114, F116, and A117 in α5 helix display strong NOEs to the entire acyl chains of DMPC lipids including the methyl groups at the ends, indicating deep immersion in the lipid bilayer.
Figure 3. Interaction of Bax (α2–α5) with lipid bicelles.
-
ANOE strips taken from 3D 15N‐edited NOESY‐TROSY‐HSQC spectrum (200‐ms NOE mixing time) recorded at 900 MHz using the [15N, 2H]‐labeled Bax (α2–α5) in DMPC/DHPC bicelles with q = 0.55. The indicated crosspeaks in the aliphatic regions are the NOEs between the protein backbone amide protons and the lipid aliphatic protons.
-
BResidue‐specific PREamp of Bax (α2–α5) in DMPC/DHPC bicelles with q = 0.6 determined from the lipophilic PRE analysis. The PREamp values were derived from the 16‐DSA titration. The plot is colored according to the PREamp values that are proportional to the effects of 16‐DSA on the individual residues of Bax (α2–α5). The residues with the least PREamp values in the blue colored areas are in the flexible terminal regions.
-
CSchematics of a Bax (α2–α5) dimer structure in ribbon representation relative to a bicelle with DMPC (blue) in the lipid bilayer and DHPC (pale yellow) in the rim. The protein structure contacts the DMPC lipid bilayer directly with a 60° tilt angle, which is illustrate more clearly by the schematic (D).
-
DPosition of a Bax (α2–α5) dimer structure in ribbon representation relative to the lipid bilayer of a bicelle. The protein structure is placed against the lipid bilayer with a 60° tilt angle between its longest axis (L) and the bilayer normal axis (N) as derived from the best sigmoidal fitting. The hydrophobic α4–α5 surface contacts the lipid bilayer. Amide protons are shown as spheres color‐coded similarly as in panel B. The polar headgroup region and the nonpolar acyl tail region of the lipid bilayer are indicated by light blue and light orange colored strips, respectively. The estimated thickness for the DMPC lipid bilayer is indicated on the left side. The Bax (α2–α5) dimer structure on the left is rotated 180° to generate the structure on the right showing the nonpolar residues in the α4–α5 surface, most of which contact the nonpolar core of the lipid bilayer. Note that the two positively charged R89 residues on the boundary of the nonpolar surface are close to the polar lipid headgroups.
Source data are available online for this figure.
To analyze the depth of immersion of the Bax (α2–α5) dimer along the bicelle normal axis, we used the paramagnetic probe titration method that has been applied to study the transmembrane domains of immune receptors and viral proteins (Piai et␣al, 2017; Chen et␣al, 2018). Following a published protocol (Fu et␣al, 2019), we first titrated the water‐soluble paramagnetic probe gadolinium (III) 1,4,7,10‐tetraazacyclododecane‐1,4,7,10‐tetraacetate (Gd‐DOTA) into the bicelle‐bound Bax (α2–α5) to measure residue‐specific PRE amplitudes (PREamp). The PREamp values derived from the analysis of the peak intensity decay versus the paramagnetic probe concentration report residue‐specific membrane immersion depths. Unexpectedly, a plot of PREamp versus residue number shows close PREamp values ∼ 0.9–1.0 for all the residues (Appendix␣Fig S3A), which is not a typical bowl‐shaped plot expected for transmembrane proteins (Piai et␣al, 2017). However, the residues in the α4 and α5 helices display slower intensity decay rate as the concentration of Gd‐DOTA increases than the residues in the α2 and α3 helices (examples shown in Appendix␣Fig S3B), indicating that the α4 and α5 helices are less exposed to the aqueous solvent.
To clearly define the position of Bax (α2–α5) relative to the bilayer, we acquired a set of lipophilic PRE data. The bicelle‐bound Bax (α2–α5) was titrated with the membrane‐embedded paramagnetic agent 16‐doxyl‐stearic acid (16‐DSA) that places the paramagnetic probe in the center of the lipid bilayer. The residue‐specific PREamp was derived using the same approach as that employed for the hydrophilic PRE. Instead of observing a flat hydrophilic PREamp plot, the lipophilic PREamp plot shows an overall bell‐shaped profile (Fig 3B). The N‐ and C‐termini of Bax (α2–α5) exhibit negative PREamp values, indicating that they are very flexible. Most of α2 and α3 helices exhibit very small PREamp values (< 0.25), confirming that they are not in direct contact with the bicelle. In contrast, the PREamp values for part of α4 helix and entire α5 helix are above 0.5 (Fig 3B), indicating that the residues in these helices are close to the lipid acyl tails, likely interacting with the lipid bilayer directly.
Taken together, the lipophilic PRE and lipid NOE data demonstrate that the α4 and α5 helices in Bax (α2–α5) dimer are directly associated with the DMPC lipid bilayer. However, the flat PREamp (residue number) plot from hydrophilic PRE indicates that no protein region is in a conventional transmembrane topology which could shield the effect of the hydrophilic paramagnetic agent. To fit all sets of data, we propose a model in which the hydrophobic α4–α5 surface of the Bax (α2–α5) dimer binds to the edge of the lipid bilayer of a bicelle with the α2–α3 hydrophilic surface facing the aqueous solvent (Fig 3C). Given the unconventional membrane–protein interaction, the PRE effect of Gd‐DOTA in the aqueous phase is so strong that it affects all the residues to the maximum extent independently of their positions. In contrast, the PRE effect of the lipophilic 16‐DSA in the lipid bilayer is more localized and generates a gradient. Since lipophilic PREamp is proportional only to the residue position along the bicelle normal, the relative position of each residue to the bilayer center could be estimated by fitting a structural model to the experimental PREamp values as we described before (Piai et␣al, 2017; Chen et␣al, 2018). Thus, we calculated the distance rz for each residue in the Bax (α2–α5) dimer structure to an arbitrary point on the bilayer normal axis (examples shown in Appendix␣Fig S3C). Then, we repeated the sigmoidal fitting of PREamp (rz) data while rotating the protein structure until finding the best‐fit model. The R2 adj versus tilt angle plot (Appendix␣Fig S3D) indicates that the best‐fit model has an angle of ˜ 60° between the protein axis (L) and the bilayer normal (N) (Appendix␣Fig S3E), suggesting that the protein is docked to the membrane in an orientation illustrated in Fig 3D. This orientation also agrees with the protein hydrophobicity map, as it places most of the hydrophobic residues on the α4–α5 surface in contact with the hydrophobic core of the lipid bilayer.
Structure validation by functional mutagenesis
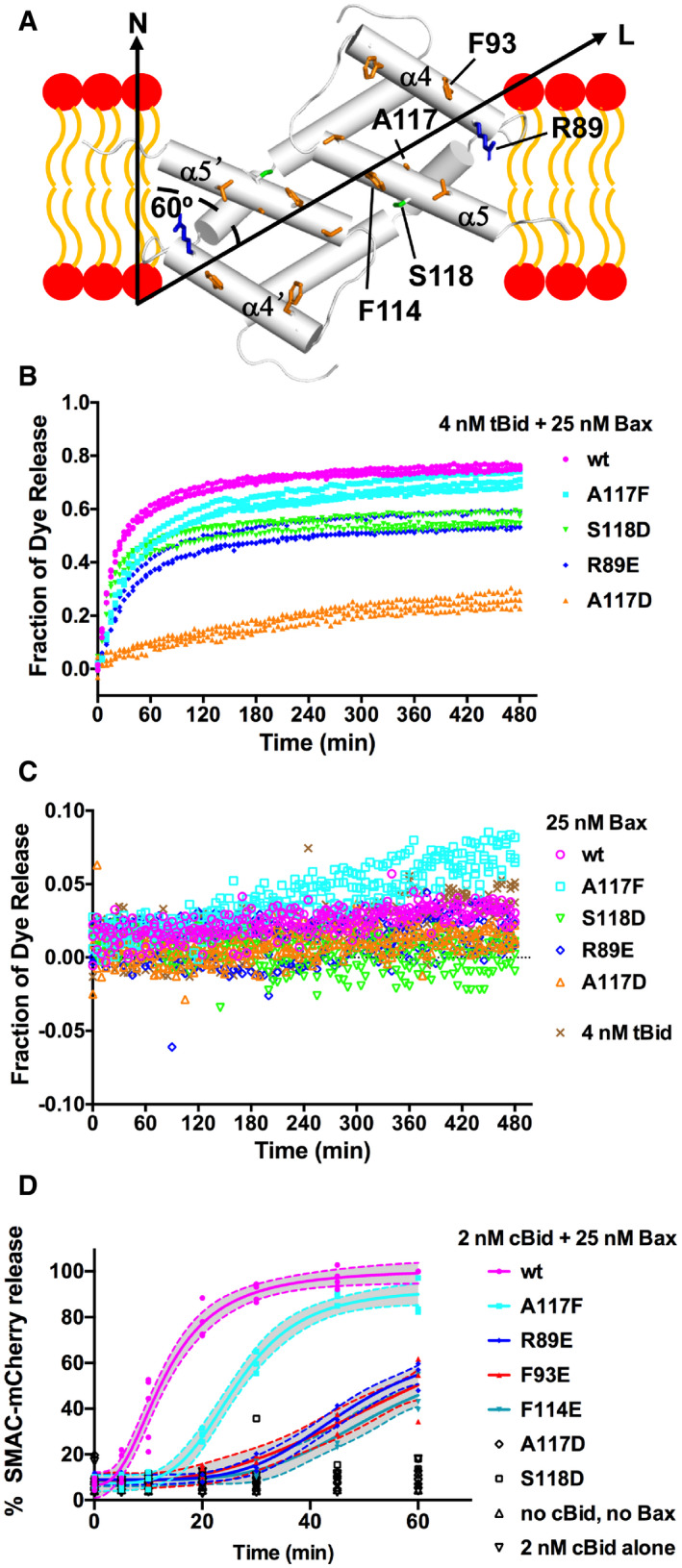
The NMR structure of Bax (α2–α5) dimer bound to the lipid bicelle provides a high‐resolution model for how this Bax core dimer interacts with the lipid bilayer. In particular, many nonpolar residues throughout the α4 and α5 helices and a polar residue, S118, at the middle of α5 helix interact with the nonpolar lipid acyl chains, whereas a positively charged residue, R89, at the N‐terminus of α4 helix interacts with the negatively charged lipid head groups (Fig 4A). These protein–lipid interactions would allow the core dimer to form part of the wall that separates the nonpolar lipid bilayer from the aqueous pore. Mutations of these residues to negatively charged residues are expected to reduce these protein‐lipid interactions, thereby impeding the pore‐forming function of Bax protein. To test this structure‐based functional model for the Bax (α2–α5) dimer, we mutated a representative residue of each category to a negatively charged residue and determined the interaction of the resulting mutants, R89E, F93E, A117D, and S118D, with the lipid bilayer (Fig 4A). In addition, we made R89A mutant to not only eliminate the positive charge but shorten the 3‐carbon aliphatic side chain. While we were unable to purify Bax (α2–α5) with A117D, we successfully purified Bax (α2–α5) with R89E, R89A, F93E, or S118D and reconstituted each into lipid bicelles. Most of the resonances from the α4–α5 residues in both soluble and bicelle‐bound S118D mutant were missing in the NMR spectra (Fig EV3A), preventing us from determining how the mutant interacts with the membrane since these helices are the major contacting region. For the R89E, R89A, and F93E mutants, most of the resonances were preserved including those from the α4–α5 residues (Fig EV3, EV4, EV5). We thus implemented the same lipophilic PRE approach and observed a slower PRE‐induced NMR spectral peak intensity decay for these three mutants in bicelles (Fig EV3E). These data indicate that the replacement of R89 or F93 with a glutamic acid weakened the core dimer interaction with the lipid bilayer. Presumably, the reduced membrane binding is due to the introduction of repulsive negative charges close to negative charges in lipid headgroups (Fig 4A). The membrane binding was also reduced by R89A mutation that eliminates the positive charge and two methylene groups which would interact with the negative charged heads and nonpolar tails of lipids, respectively.
Figure 4. Pore‐forming activity of Bax proteins with mutations that would reduce the α2–α5 dimer interaction with membranes.
-
ANMR structure of Bax (α2–α5) dimer and lipid bilayer‐interacting residues. α helices are shown as cylinders. In the α4 and α5 helices, some of the lipid bilayer‐interacting nonpolar, polar, and positively charged residues are shown as orange, green, and blue sticks, respectively, including the R89, F93, F114, A117, and S118 that were mutated to negatively charged residues to disrupt the interaction with lipid headgroups (red ovals) or acyl chains (orange curves). The longest axis (L) of the dimer is tilted 60° from the bilayer normal (N) to maximize the nonpolar interaction with the lipid acyl chains while allowing R89 to interact with the polar lipid headgroups.
-
B, CFluorescent dye release from the mitochondria‐mimic liposomes by the indicated wild‐type (wt) or mutant Bax in the presence (B) or absence (C) of tBid or by tBid alone (C) was measured by quenching the fluorescence of the released dyes by the dye‐specific antibodies outside the liposomes during a time course. The fraction of dye release was normalized to that by detergent. The data shown were obtained from n = 3 independent replicates using the same preparations of the proteins and liposomes.
-
DDigitonin‐permeabilized BMK Bax−/−/Bak−/− cells expressing SMAC‐mCherry in the mitochondria intermembrane space (50 μl, 500,000 total cells) were incubated with 25 nM of Bax and 2 nM cBid for the indicated time at 37°C in a 96‐well plate. The samples were centrifuged for 10 min and separated into supernatant and pellet fractions. SMAC‐mCherry release was calculated as the fraction of total (supernatant + pellet) mCherry fluorescence coming from the supernatant fraction. The data were normalized to the percent SMAC‐mCherry release of wt Bax and cBid at 60 min. Each symbol represents the normalized SMAC‐mCherry release for one single replicate (n = 3 or more independent replicates). Where applicable the data were fit with the [Agonist] vs. response ‐ Variable slope equation␣in GraphPad Prism 8.0.1. The dotted lines represent the 95% confidence interval of the fit.
Source data are available online for this figure.
We further determined the pore‐forming activity of the resulting full‐length mutant Bax proteins in liposomes with mitochondria‐characteristic lipids in the membrane and fluorescent dye conjugated dextran molecules of 10 kDa in the lumen to mimic the mitochondrial protein cytochrome C. The release of the fluorescent molecules from the liposomes was monitored by quenching of the fluorescence by the anti‐fluorescent dye antibodies located outside of the liposomes. As shown in Fig 4B, in the presence of 4 nM of tBid protein, 25 nM of wild‐type Bax protein released ˜ 75% of the dyes by the end of a 480‐min time course. In comparison, the R89E or S118D mutant released ˜ 55% of the dyes, while the A117D mutant released ˜ 25% of the dyes. In contrast, the A117F mutant, as a control for increasing the size of the side chain but not introducing a negative charge, released a slightly lower amount (˜ 70%) of dye than the wild‐type protein with a slightly slower kinetics. Similarly, the effect of R89A mutation on the dye release is less than R89E mutation (Fig EV4A), which is expected since R89A does not introduce the repulsive negative charge and hence would be less detrimental to the membrane interaction. The effect of R89F on the dye release is similar to R89A (Fig EV4A), further testifying the importance of the positive charge of R89 to the membrane interaction since this mutation eliminates the positive charge but may retain some of the nonpolar interaction of R89 with the lipid acyl tails.
In the absence of tBid, all the proteins released < 10% of the dyes (Figs 4C and EV4A), demonstrating that at this low nM concentration the Bax mutants require activation by tBid to form pores in the membranes like the wild‐type protein. On the other side, increasing tBid concentration did not restore the dye release by the Bax mutants (Appendix␣Fig S4), suggesting that the defect is at a step after tBid activation of Bax. These results clearly show that the mutations which are predicted by our structure‐based model and/or confirmed by our PRE data (Fig EV3E) to reduce the core dimer interaction with the lipid bilayer reduced the pore formation by the Bax protein.
Since the hydrophobic interaction of the core dimer with the lipid bilayer is extensive and mediated by many residues, mutations of individual residues were expected to have small effects and thereby partially reduce the pore‐forming activity. Consistent with this interpretation two additional mutations in the nonpolar α4–α5 surface, F93E and F114E, partially inhibited membrane permeabilization (Fig EV4B). Furthermore, the effects of the R89E, S118D, and A117D mutations, detected at 25 nM Bax were greatly reduced by increasing the concentration of Bax. Indeed, the difference between the wild‐type and mutant proteins in dye release was less when 200 nM Bax were used with 4 nM tBid (Fig EV4C). At this high nM concentration, in the absence of tBid, the wild‐type protein displayed some auto‐activity as it released ˜ 25% of the dyes by the end of the time course (Fig EV4D). Compared to the wild‐type protein, the S118D and R89E mutants displayed lower auto‐activity, whereas A117D and A117F displayed higher auto‐activity (Fig EV4D). Note that the auto‐activity of the A117F mutant is higher than the A117D mutant, consistent with the membrane interaction mediated by the core dimer being important for the pore formation by Bax in the absence of tBid.
The pore‐forming activity of the full‐length Bax mutant proteins in the native mitochondrial membranes was determined by measuring the amount of SMAC‐mCherry protein released from the mitochondria in digitonin‐permeabilized cells that express the SMAC‐mCherry protein localized to the mitochondrial intermembrane space (Fig 4D). In this more physiologically relevant assay, the Bax mutant proteins showed similar activity profiles when compared to the liposome permeabilization data where A117F was the most active mutant followed by R89E, F93E, F114E, S118D, and A117D. Also consistent with the liposome data, none of the mutants at 25 nM showed auto‐activity in the SMAC‐mCherry release assay (Appendix␣Fig S5A). Since 25 nM A117D or S118D released ˜ 25 or 55% of the dyes from the liposomes compared to releasing < 10% of the SMAC‐mCherry proteins from the mitochondria, these Bax mutants were further characterized by titrating them in the SMAC‐mCherry release assay. Bax S118D was less active than WT Bax at lower concentrations but showed similar activity to wild‐type Bax at 100 nM, whereas Bax A117D was mostly inactive at the concentrations tested (Appendix␣Fig S5B). These data corroborate the conclusions from the liposome permeabilization data, further demonstrating the importance of the core dimer interaction with the membrane in mediating the native mitochondrial membrane permeabilization.
Bax activation by BH3 proteins is thought to involve unfolding of the soluble structure. Since the A117 and S118 residues are located in the hydrophobic core of soluble Bax protein structure, changing them to bulky and/or charged residues may unfold the structure thereby activating the protein. To test this possibility, we measured the intrinsic fluorescence from the six Trp residues in the Bax protein that are sensitive to their local environment which in turn indicates the folding state of the protein (Appendix␣Fig S6A). As shown in Appendix␣Fig S6B, the A117D or A117F mutation decreased the Trp fluorescence, suggesting that each mutation increases the access of one or more Trp to water, consistent with the mutation unfolding the protein structure. This unfolding provides an explanation for the high auto‐activity of these mutants in the pore‐forming assay. In contrast, the S118D mutation increased the Trp fluorescence, suggesting that this mutation does not unfold the protein but changes the conformation thereby reducing the Trp exposure to water. As expected, the R89E mutation on the surface of soluble Bax structure only slightly altered the Trp fluorescence, indicating that this mutation mostly does not change the access of water within the protein structure. Consistent with the impact of the S118D or R89E mutation on the protein conformation, these mutations do not increase auto‐activation of the protein.
The NMR structure of Bax (α2–α5) dimer bound to the lipid bicelle is different from the crystal structure, suggesting that the core dimer structure is altered by the interaction with the lipid bilayer. To determine the relationship between the core dimer structure and its interaction with membranes, we monitored the dimerization of full‐length Bax proteins via the core region in the isolated mitochondria by disulfide crosslinking of Bax mutants with cysteine residues positioned in the core dimer interface (Fig EV5A) as we designed before (Zhang et␣al, 2016). As shown in Fig EV5B and C, the R89E, A117D, or S118D mutation reduced the crosslinking of Bax proteins via the L59C in the α2 helix of one protomer and the M79C’ in the α3 helix of the other protomer, and via the two E69C, one in the α2 helix of each protomer. In contrast, the control mutation A117F reduced the Bax crosslinking to a less extent. Of the other two R89 mutations, R89F also reduced the crosslinking but to a less degree than R89E, whereas R89A slightly increased the crosslinking (Fig EV5D–E). These data suggest that most of the mutations that alter the core dimer‐membrane interaction also alter the core dimer configuration such that the L59C and M79C or the two E69C are separated by too large a distance to be linked by a disulfide. Because these mutations are not in the core dimer interface (Fig EV5A), they are not expected to reduce the dimerization directly. Note that the mitochondrial association of Bax was largely unaffected by the mutations that reduce the core dimer interaction with the mitochondria (Fig EV5B–E), which is expected since the interactions of other Bax regions with the mitochondria would be retained, in particular, the insertion of the α9 helix into the MOM. Thus, the reduction of Bax crosslinking cannot be attributed to the reduction of Bax concentration in the mitochondria. Therefore, the alteration of the dimer configuration detected by the crosslinking is most likely due to the alteration of the dimer interaction with the membrane.
Discussion
Since the crystal structure of the Bax core dimer was determined in 2013, different models for its function in Bax pore formation have been proposed (Czabotar et␣al, 2013; Bleicken et␣al, 2014; Cosentino & Garcia‐Saez, 2017; Uren et␣al, 2017; Bleicken et␣al, 2018). In this study, we obtained multiple lines of evidence strongly suggesting that the core dimer forms part of the wall around the pore in the membrane as shown in Fig 5.
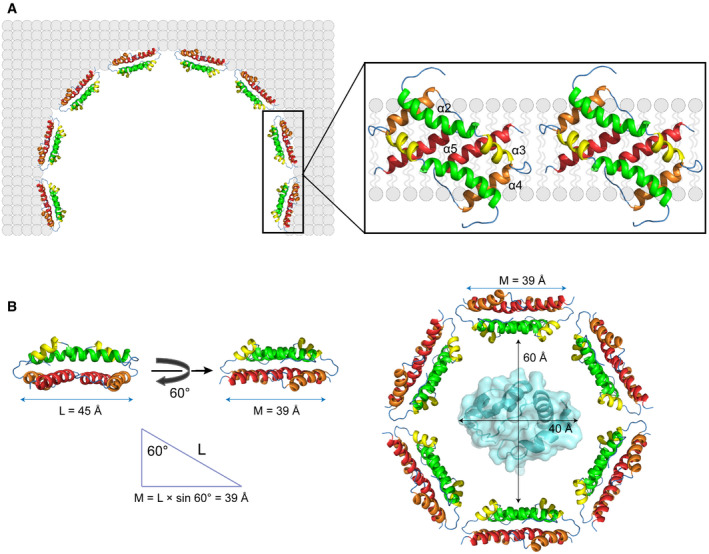
Figure 5. A structural model for an oligomeric Bax pore in the mitochondrial outer membrane.
-
AAs demonstrated by our study and shown in the model, Bax core dimers form parts of the proteolipid wall that separates the nonpolar membrane from the aqueous pore. Other regions of the Bax oligomer and radially arranged lipids may form other parts of the wall. The diameter of the pore will be determined by the stoichiometry of the Bax oligomer and the number of the radially arranged lipids.
-
BIn order to release a hydrated cytochrome c from the mitochondria, the diameter of the pore must be > 60 Å, the diameter of cytochrome c plus a water shell. The length of a core dimer along its longest axis (L) = 45 Å. Since the axis tilts 60° from the bilayer normal, a core dimer can cover 39 Å of the lipid bilayer around the pore (M). Assume the pore diameter = 60 Å, and the pore wall only consists of Bax core dimers, six Bax core dimers or 12 Bax proteins are required to form a hexagonal pore that can release cytochrome c. The structure of cytochrome C in the pore was generated from PDB file 3ZCF using PyMOL program.
First, we determined the first structure of the core dimer interacting with a model lipid bilayer using NMR. This structure demonstrates that the core dimer can cover a side of the lipid bilayer using the nonpolar surface formed by the α4–α5 helices that is flattened compared to the crystal structure. This flattening increases the contacting area with the lipid bilayer (Fig 5A, magnified region). In addition, the longest dimer axis is tilted ˜ 60° from the bilayer normal maximizing the interaction with the lipids, including not only the hydrophobic interactions of multiple nonpolar residues and two polar serines (one from each protomer) with nonpolar lipid acyl chains, but also the ionic or polar interactions of two positively charged arginines (one from each protomer) with the negatively charged or polar lipid headgroups. Since the two arginines are located on the boundary of the nonpolar surface, their interaction with the lipid head groups may dictate the relative orientation of the core dimer to the bilayer normal. While the hydrophobic match between the core dimer structure and the 46‐Å thick lipid bilayer formed by DMPC is maximized at the 60° tilt angle, this dimer could match with other bilayers of more or less thickness by a decrease or increase in the tilt angle, respectively. This may explain how Bax permeabilizes multiple membranes of different lipid compositions in␣vitro (Shamas‐Din et␣al, 2015), and may also permeabilize endoplasmic reticulum, lysosome and other organelles in addition to mitochondria in cells (Kagedal et␣al, 2005; Bove et␣al, 2014; Kanekura et␣al, 2015).
Second, the high‐resolution structure of the lipid bilayer‐bound core dimer structure enabled us to perform site‐specific mutagenesis experiments to examine the contributions of different interactions to the overall pore‐forming activity of intact Bax protein in a model mitochondrial membrane that releases a molecule with a size similar to cytochrome C as well as in native mitochondrial membrane that releases a fluorescent protein with a size larger than cytochrome C. The results clearly show that not only the hydrophobic interactions of two nonpolar alanines and two polar serines near the center of the nonpolar surface with the lipid acyl chains but the ionic and polar interactions of the two positively charged arginines on the boundary with the lipid headgroups are important driving forces for the Bax pore formation. The functional impact of these site‐specific mutations on the intact Bax protein further supports the pore wall‐forming function that we proposed based on the NMR structure of the bilayer‐bound core dimer. The involvement of the core dimer in assembly of the pore wall as proposed here is consistent with that proposed by the clamp model based on the low‐resolution distance distribution data from EPR (Fig 1A) (Bleicken et␣al, 2014). However, the EPR data did not reveal the tilt angle or identify which residues interact with the lipid acyl chains versus the lipid headgroups. Furthermore, our structural‐guided mutagenesis study provides the first functional validation of our refined model that places the amphipathic core dimer into the wall that separates the hydrophobic membrane from the aqueous pore (Fig 5A). Our NMR structure has a flat nonpolar surface that covers the flat edge of the lipid bilayer (Fig 5A, magnified region). This contrasts with the clamp model in which the α2–α5 dimer has a concave nonpolar surface, proposed to cover the complementary convex surface formed by the micellar lipids at the pore rim (Fig 1A). Although the micellar lipids were not visible in our NMR analyses, our model suggests that some of them must be displaced by the core dimer, while the remainders localize on both sides of the core dimer covering the rest of membrane edge.
Third, in the in‐plane model for the function of the core dimer the nonpolar surface of the dimer contacts the surface of lipid bilayer in order for the bulky side chains of nonpolar residues to wedge through the charged and polar region of the lipid bilayer to reach the hydrophobic core (Fig 1A). Intercalation of these bulky side chains between the lipids in one leaflet of the bilayer was proposed to asymmetrically expand the leaflet generating sufficient membrane tension to induce a lesion in the membrane that is lined by the radially arranged micellar lipids. While this model satisfies the pore‐induction role of Bax protein, there is a thermodynamic barrier to overcome. For the nonpolar bulky amino acid side chains to reach the hydrophobic core of a lipid bilayer, they have to first contact and then penetrate through the charged and polar region formed by the lipid head groups. For this to happen, there must be enough free energy to complete this thermodynamically unfavorable process. Moreover, it would result in the charged or polar lipid headgroups being located in the space between the α4 and α5 helices that is mostly filled by interacting nonpolar amino acid side chains. It is not clear how such a structure would be thermodynamically stable. Although our study cannot rule out this in‐plane membrane topology of the core dimer, we did not detect such a topology for the core dimer reconstituted into the lipid bicelle that has more than enough surface area for the core dimer to land “in plane”, indicating that if it does exist, the in‐plane membrane topology is likely a transient state during pore formation, rather than a final stable structure around the pore. Recent crystal structures of Bak core dimers in complex with lipids or detergents suggest another function for the core dimer (Cowan et␣al, 2020). Thus, the dimers contain pockets for lipids allowing the dimers to extract the lipids from the membrane thereby destabilizing the membrane and inducing pore formation. Clearly the more structures we solve with the pore‐forming proteins and the interacting lipids or membranes, the closer we are to the whole truth about how the deadly pores are induced, stabilized, and expanded by the proteins.
Finally, the configuration of the core dimer appears to be induced upon interaction with the membrane. This conformational change was revealed by comparison of the NMR spectra of the core dimer in the absence and the presence of the lipid bicelles (Fig 1B) and by comparing the NMR structure of the bicelle‐bound core dimer with the crystal structures (Figs 2B and EV2). The conformational change is further supported by our core dimer configuration‐specific crosslinking of the Bax proteins, since the mutations that reduce the core dimer interaction with the lipid bilayer also reduce the efficiency of the crosslinking via different points in the dimer interface.
In conclusion, we have determined the first membrane‐bound Bax core dimer structure and validated its function in the pore formation by oligomeric full‐length Bax proteins in the mitochondrial membrane. We propose a model (Fig 5A) that incorporates our structural data and more accurately describes the pore structure as composed of a string of the core dimers as part of the wall encircling the large aqueous conduit that permeabilizes the outer mitochondrial membrane and commits the cell to undergoing apoptosis. We further propose that a minimal of six core dimers formed by 12 Bax proteins are required to form a pore that can release cytochrome C, assuming that the pore is entirely lined by the core dimers (Fig 5B). The challenge ahead is to determine how Bax dimers are organized along the lining of the pore and the possible distribution of lipids between and or around the dimers. Moreover, structure of the entire pore remain unclear particularly how large diameter pores are formed and maintained and to what extent the pore diameter is variable in mitochondrial outer membranes. Such determinations are critical to finding pharmacologically tractable ways to modulate the pore formation thereby enhancing the death of cancer cells while preserving the life of normal cells (Gavathiotis et␣al, 2012; Niu et␣al, 2017; Reyna et␣al, 2017; Garner et␣al, 2019).
Materials and Methods
Protein expression and purification
Escherichia coli‐codon optimized DNA encoding residues 53–128 (α2–α5) of human Bax was synthesized (Genewiz), in which two cysteine codons at positions 62 and 126 are replaced by serine codons to improve the stability of the encoded protein. The resulting Bax (α2–α5) DNA was fused with a DNA encoding a His8‐tag followed by a 3C protease cleavage site (LEVLFQGP) and cloned into a pET28a (+) vector. The resulting plasmid was transformed into E.␣coli BL21 (DE3) cells. The cells were grown in isotopically labeled M9 minimal media at 37°C until optical density at 600 nm reached 0.8–0.9. The fusion proteins were expressed for 18 h at 25°C after induction with 0.5 mM isopropyl‐β‐d‐thiogalactopyranoside. The cells were collected by centrifugation at 8,000 g for 30 min at␣25°C and suspended in Lysis Buffer (150 mM NaCl, 1% 3‐[(3‐cholamidopropyl)dimethylammonio]‐1‐propanesulfonate (CHAPS), 20 mM Tris–HCl pH 8.0). After the cells were disrupted by sonication, the lysate was centrifuged at 40,000 g for 30 min at 4°C. The supernatant with the soluble fusion protein was loaded onto a Ni‐NTA column, which was sequentially washed with 20 mM imidazole, 40 mM imidazole, and 60 mM imidazole in Lysis Buffer. The fusion protein was then eluted from the column with 400 mM imidazole in Lysis Buffer. The elute was dialyzed against Dialysis Buffer (200 mM NaCl, 20 mM MES pH 6.0) at 4°C for 12 h using 3.5 kDa MWCO Slide‐A‐Lyzer MINI Dialysis Devices (Thermo Scientific) to remove imidazole and CHAPS. The fusion protein was cleaved by a His8‐tagged 3C protease in Dialysis Buffer containing 1 mM PMSF, 200 mM Na2SO4 at 4°C overnight. The resulting tag‐free Bax (α2–α5) protein was separated from the His8‐tag and the protease by Ni‐NTA chromatography. The Bax (α2–α5) protein was concentrated and further purified by size exclusion chromatography using a HiLoad® 16/600 Superdex 200 pg column (GE Healthcare) in Dialysis Buffer. Based on SDS–PAGE analysis, the peak fractions containing homogenous Bax (α2–α5) protein were pooled and concentrated for NMR measurements. Typical yield of this protein prep was ˜ 12 mg/l of cell culture. The mutant Bax (α2–α5) proteins were similarly expressed and purified.
Plasmids encoding mutant human Bax proteins were generated from the pTYB1 vector‐based plasmid encoding the wild‐type protein (Suzuki et␣al, 2000) using QuickChange (Thermo Fisher Scientific). These plasmids were used to prepare the recombinant proteins as described (Niu et␣al, 2017) for liposome membrane permeabilization experiments. The recombinant truncated murine Bid (tBid) protein was also prepared as described (Niu et␣al, 2017).
Sample preparation of Bax (α2–α5) in bicelles
A 6.78 mg of 1,2‐Dimyristoyl‐sn‐Glycero‐3‐Phosphocholine (DMPC) (Avanti Polar Lipids) was mixed with 51 μl of 200 mg/ml 1,2‐Dihexanoyl‐sn‐Glycero‐3‐Phosphocholine (DHPC) (Avanti Polar Lipids) in water to reach a ˜ 1:2 DMPC:DHPC molar ratio. The lipid mixture was subjected to cycles of freeze–thaw, pipetting and vortex until the lipids were fully dissolved, and the bicelles were formed homogeneously. The purified Bax (α2–α5) (0.1 mM) was incubated with the bicelles under 20‐rpm rotation at 25°C for 30 min. Five to eight aliquots of 0.1 mM Bax (α2–α5) in bicelles were mixed and concentrated to ˜ 500 μl using Centricon concentrator (EMD Millipore; MWCO, 3.0 kDa). Accordingly, the final sample for NMR measurements contains 0.5‐ 0.8 mM (monomer concentration) Bax (α2–α5), 50–80 mM DMPC, 100–160 mM DHPC, 100 mM NaCl, 10 mM MES pH 6.0, 0.1% NaN3, and 10% D2O. The DMPC: DHPC ratio was monitored by 1D NMR and the molar ratio (q) was kept 0.5–0.6 for all the NMR samples. DHPC and DMPC with deuterated acyl chains were used to prepare the samples for protein–protein NOESY experiments.
SEC‐MALS analysis of Bax (α2–α5)
The molecular weight of Bax (α2–α5) protein in solution was determined by size exclusion chromatography‐multiple angle light scattering (SEC‐MALS) using Agilent 1,260 Infinity Isocratic Liquid Chromatography System connected to Wyatt Dawn Heleos II MALS␣detector, Wyatt Optilab T‐rEX Refractive Index detector, and ViscoStar III differential viscometer (Wyatt Technology). The chromatography was performed at 25°C using Superdex 200 10/300 GL column (GE Healthcare) equilibrated with Dialysis Buffer. 100 μl of 8.0 mg/ml of Bax (α2–α5) was injected into the column and eluted with Dialysis Buffer at a flow rate of 0.5 ml/min. The light absorption at 280 nm, light scattering at 660 nm, and refractive index of the elute were simultaneously monitored with the three inline detectors. The data were analyzed by ASTRA software, and the molecular weight of Bax (α2–α5) peak fraction was calculated using the three‐detector method.
Characterization of the Bax (α2–α5) oligomeric states by␣crosslinking
The oligomeric states of the soluble and bicelle‐bound Bax (α2–α5) protein were examined by chemical crosslinking using bissulfosuccinimidyl suberate (BS3) (Thermo Scientific). The Bax (α2–α5) was first dialyzed overnight against Crosslinking Buffer (150 mM NaCl, 100 mM sodium phosphate pH 7.0) at 4°C. 4 μl of 50 mM BS3 solution in water was added to 50 μl of 80 μM soluble or bicelle‐bound Bax (α2–α5) protein, respectively. The crosslinking reaction was performed at 25°C for 30 min and then quenched by 50 mM Tris–HCl pH 8.0.
NMR data acquisition, processing, and analysis
All NMR experiments were conducted on Bruker spectrometers operating at 1H frequency of 600 or 900 MHz and equipped with cryogenic probes. NMR spectra were processed using NMRpipe (Delaglio et␣al, 1995) and analyzed using XEASY (Bartels et␣al, 1995) and CcpNmr (Vranken et␣al, 2005). Sequence‐specific assignment of backbone amide resonance (1HN, 15N, 13Cα, and 13C’) was accomplished using a series of gradient‐selected, TROSY‐enhanced triple‐resonance experiments, including HNCO, HN(CA)CO, HNCA, HN(CO)CA, and HNCACB (Salzmann et␣al, 1998). The NMR data were collected with a uniformly [15N, 13C, 2H]‐labeled Bax (α2–α5) protein at 600 MHz. In addition, 15N‐edited NOESY‐TROSY‐HSQC experiments (150‐ms NOE mixing time) were performed to validate the assignment. Protein aliphatic and aromatic side chain resonances were assigned using a combination of 15N‐edited NOESY‐TROSY‐HSQC spectrum and 13C‐edited NOESY‐HSQC spectrum recorded at 900 MHz for a [15N, 13C]‐labeled protein. Stereospecific assignments of methyl groups of leucine and valine were performed for a 1H–13C HSQC spectrum obtained from a 15% [13C]‐labeled protein.
To determine the intermolecular distance constraints, a sample containing 1:1 molar ratio of [15N, 2H] and [13C]‐labeled Bax (α2–α5) monomer was used to record a 15N‐edited NOESY‐TROSY‐HSQC spectrum (250‐ms NOE mixing time) to obtain the exclusive NOEs between the 15N‐attached protons of one monomer and aliphatic protons of the neighboring monomer.
All the NMR experiments were conducted at 25 or 32°C for soluble or bicelle‐bound isotope‐labeled Bax (α2–α5) sample, respectively, with both samples buffered to pH 6.0. All the lipids were deuterated for the NOESY experiments.
Structure calculation
Structure was calculated using the program XPLOR‐NIH (Schwieters et␣al, 2018). The crystal structure of Bax (α2–α5) dimer (PDB code: 4BDU) was used to construct an initial model that was then refined against a complete set of NOE constraints (including intramolecular and intermolecular distance constraints) and dihedral constraints using a standard simulated annealing protocol (Bassolino‐Klimas et␣al, 1996). A total of 200 dimer structures were calculated, and 15 lowest energy structures were selected as the structural ensemble.
Structure validation by PRE measurement
To verify the structures calculated above, PRE analysis was performed to obtain intermolecular distance constraints independent of NOEs. An A82C or S126C mutation was made in Bax (α2–α5) for labeling the protein with MTSL ((1‐oxyl‐2,2,5,5‐tetramethyl pyrroline‐3‐methyl) methanethiosulfonate). 40 μl of 100 mM MTSL in DMSO was mixed with 1 ml of 0.4 mM [14N]‐labeled single‐Cys Bax (α2–α5), and the labeling reaction was conducted at 4°C for 12 h. The sample was then dialyzed in Dialysis Buffer to remove free MTSL. A sample containing ˜ 1:1 molar ratio of [15N] and [14N]‐labeled Bax (α2–α5) in DMPC/DHPC bicelles (q = 0.5) was prepared as mentioned above in the sample preparation section. The 1H–15N TROSY‐HSQC spectra were recorded before and after the addition of 10 mM sodium ascorbate. The peak intensity ratios of the paramagnetic (I) to the diamagnetic (I0) state were calculated for all spectrally dispersed residues.
Single‐point energy calculation
The single‐point energies of the bicelle‐bound Bax (α2–α5) dimer and the crystalized Bax (α2–α5) dimer were computed using the Calculate Energy protocol embedded in Discovery Studio 4.0. The simulated systems were put in the CHARMM Force field (Vanommeslaeghe & MacKerell, 2012) in vacuo. The Particle Mesh Ewald method (Norberto de Souza & Ornstein, 1999) was used to treat the long‐range electrostatics with Non‐bond Lower cutoff distance at 10 Å. The protein structures were allowed to move freely.
Detection of protein‐lipid NOE
A 0.8 mM [15N, 2H]‐labeled Bax (α2–α5) in bicelles containing regular DMPC and deuterated DHPC (q = 0.55) was used to measure protein‐lipid NOEs. A 15N‐edited NOESY‐TROSY‐HSQC spectrum (200‐ms NOE mixing time) was recorded at 32°C and 900 MHz. The NOE spectrum was analyzed using XEASY (Bartels et␣al, 1995).
Solvent PRE analysis
The solvent PRE measurements were performed as previously described (Piai et␣al, 2017). Briefly, a sample of 0.8 mM [15N, 85% 2H)‐labeled Bax (α2–α5) mixed with DMPC/DHPC bicelle (q = 0.6) was titrated with the water‐soluble and membrane inaccessible paramagnetic agent Gd‐DOTA of 0, 0.5, 2.5, 4.5, 6.5, 8.5, 13.5, 23.5, 28.5, 33.5, and 38.5 mM. A series of 15N TROSY‐HSQC spectra with 3.5‐s recovery delay were recorded at 600 MHz. The spectra were analyzed by CcpNmr (Vranken et␣al, 2005) to get the ratio of peak intensity in the presence (I) and absence (I0) of the paramagnetic agent. The residue‐specific PREamp values were derived by fitting I/I0 vs. [Gd‐DOTA] data to an exponential decay (Equation 1), where τ is decay constant.
| (1) |
Lipophilic PRE analysis
The Bax (α2–α5) in bicelle sample was prepared as described above for the solvent PRE experiment. The sample was titrated with the membrane‐embedded paramagnetic agent 16‐DSA of 0, 0.6, 1.2, 1.8, 2.4, 3.0, 3.6, 4.2, and 4.8 mM. TROSY‐HSQC spectra were recorded at each [16‐DSA]. The residue‐specific PREamp value was derived by fitting the spectral peak intensity decay as a function of [16‐DSA] using a modified (Equation 1) where [Gd‐DOTA] is replaced by [16‐DSA]. The standard paramagnetic probe titration analysis as described before (Piai et␣al, 2017) was performed until finding the best fit between the PREamp values and the sigmoidal function (Equation 2). Briefly, the projected positions of the amide protons of Bax (α2–α5) protein on the bilayer normal axis were obtained, and the distances of these positions to an arbitrary point (O) on the axis (rz) were determined (Appendix␣Fig S3C). The PREamp vs. rz data were plotted and fit by (Equation 2) to determine the position of the Bax (α2–α5) protein relative to the lipid bilayer, in particular, the␣angle between the longest axis (L) of the Bax (α2–α5) dimer to the bilayer normal axis (N) (Appendix␣Fig S3D). The lipophilic PRE measurement for Bax (α2–α5) R89E was performed similarly.
| (2) |
where and are the minimal and maximal PREamp possible for a particular protein system, is the distance from the bilayer center to the inflection point at which the PREamp is halfway between and ), and SLOPE is a parameter for the steepness of the PREamp curve at the inflection point.
Liposome permeabilization by Bax
Liposomes were prepared with chicken egg phosphatidylcholine and trans phosphatidylethanolamine, porcine brain phosphatidylserine, soy phosphatidylinositol, and bovine heart cardiolipin (Avanti Polar Lipids) of 47:28:9:9:7 mol%, a lipid composition found in Xenopus oocyte mitochondria (Kuwana et␣al, 2002), using an extrusion method as described (Tan et␣al, 2006). Fluorescent dye Cascade blue (CB)‐labeled dextrans of 10 kDa (Thermo Fisher Scientific) were encapsulated in the liposomes. To monitor the liposomal membrane permeabilization by Bax proteins, the liposomes with 50 μM total lipids were mixed with 12 μg/ml anti‐CB antibody (Thermo Fisher Scientific) in 250 μl of buffer A (50 mM Na2HPO4 adjusted to pH 7.4 using ˜ 3 mM citric acid) in a 4 × 4‐mm quartz microcell. The initial CB fluorescence intensity F0 was measured at 37°C using ISS PC1 photon‐counting spectrofluorometer with excitation and emission wavelengths set at 400 and 430 nm, respectively. The slit widths of excitation and emission monochromators were 2.4 and 2.0 mm, corresponding to the spectral resolutions of 19.2 and 16 nm, respectively. 25 or 200 nM Bax protein and/or 4, 8 or 16 nM tBid protein were added to the microcell. After 3 min of mixing and equilibrating the sample to 37°C, the CB fluorescence intensity F(t) was measured for 3 s every 5 min for 480 min or after 480‐min incubation to monitor the quenching of the released CB‐dextrans by the anti‐CB antibodies located outside of the liposomes. At the end, 0.1% Triton X‐100 was added to lyse all the liposomes, releasing all the CB‐dextrans and quenching all the fluorescence. The final intensity FT was then measured at 37°C. The fraction of CB‐dextran release caused by the proteins equals to the extent of fluorescence quenching by the anti‐CB antibody in the presence of the proteins normalized to that in the presence of Triton, i.e., [F0‐F(t)]/[F0‐FT].
For the fraction of dye release data shown in Figs 4B–C and EV4, we did not perform statistical analyses. This is because for each data point shown in the figures, an emission intensity of a fluorescent sample was measured by a photomultiplier tube (PMT) in ISS PC1 photon‐counting spectrofluorimeter for 3 s during which the PMT collected the emitted photons and converted to an electrical current every 0.1 s. The resulting 30 electrical currents were integrated to give an emission intensity with a standard error, both of which were recorded. The values of the 30 electrical currents were not recorded so that we could not perform an independent statistical analysis. The emission intensity was used to calculate a fraction of dye release as described above, resulting in a data point in the figures. For each sample, the same measurement was independently replicated with two more samples that were prepared from the same preparations of proteins and dye‐loaded liposomes, resulting in the other two data points for this condition in the figures. The three data points per condition were insufficient for a statistical analysis. However, we repeated all the measurements with all the samples that were prepared from different preparations of proteins and dye‐loaded liposomes and obtained similar data.
For the fraction of dye release data shown in Appendix␣Fig S4, we did not perform statistical analyses due to the same reasons. Each data point in the figure was obtained using the same method described above, except that an emission intensity was measured for 10 s and the intensity and standard error were recorded.
Mitochondrial permeabilization assay
To assay the function of purified recombinant Bax and cBid proteins, we adapted our previously published SMAC‐mCherry release assay to use digitonin‐permeabilized cells (Iyer et␣al, 2019) instead of isolated mitochondria (Shamas‐Din et␣al, 2014). BMK (baby mouse kidney) Bax−/− Bak−/− cells expressing SMAC(1‐56)‐mCherry fusion protein, which is localized to the mitochondrial intermembrane space, were grown to 60–80% confluency in a 10‐cm tissue culture treated dish (Sarstedt), washed with 5 ml PBS, trypsinized with 1× trypsin‐EDTA, collected with 1 ml DMEM complete (DMEM, 10% FBS, 1× MEM NEAA, 1× Penicillin/Streptomycin), and centrifuged at 800 g for 5 min at 4°C. The supernatant was discarded and the cells were resuspended in 1 ml of ice‐cold PBS and the total number of cells were determined and then centrifuged again at 800 g for 5 min at 4°C. The supernatant was discarded, and the cells were resuspended in cell buffer (20 mM HEPES‐KOH pH 7.4, 250 mM sucrose, 150 mM KCl, 2 mM MgCl2, 1 mM EDTA) supplemented with protease inhibitor (Pierce protease inhibitor mini tablets, EDTA‐free) and 0.015 to 0.025% (w/v) digitonin (the lowest concentration of digitonin that permeabilized > 95% of cells was used for each preparation of digitonin‐permeabilized cells).
After cells were incubated on ice for 10 min, the permeabilization of cells was determined by staining the cells with trypan blue and visualizing the cells with a microscope; if permeabilization was not > 95%, 10% w/v digitonin was added to the resuspended cells in 0.25 μl increments until the permeabilization was > 95%. The permeabilized cells were then centrifuged at 13,000 g for 5 min at 4°C, and the supernatant was discarded. The cell pellet was resuspended in cell buffer + protease inhibitor at a concentration of 107 cells/ml.
To assay Bax function, 50 μl of permeabilized cells (107 cells/ml, 500,000 cells total) was incubated with 25 nM of Bax wild‐type or mutants and 2 nM cBid for various time points at 37°C in a 96‐well plate (Corning). The plate was centrifuged in a Thermo Scientific M‐20 Microplate Swinging Bucket Rotor at 4,000 rpm and room temperature for 10 min, and the supernatant and pellet fractions were separated. The pellet was resuspended with 50 μl of cell buffer + protease inhibitor. SMAC‐mCherry fluorescence in the supernatant and pellet fractions was determined by exciting the samples at 587 nm (6.0 nm bandwidth) and collecting emission at 610 nm (12.0 nm bandwidth) using a Tecan M1000 plate reader. SMAC‐mCherry release was calculated by dividing the mCherry fluorescence of the supernatant fraction by the total mCherry fluorescence of the supernatant and pellet fractions. The data were normalized by dividing the absolute % SMAC‐mCherry release of each samples by the % SMAC‐mCherry release of wild‐type Bax and cBid at 60 min.
Tryptophan fluorescence emission spectrum of Bax proteins
The tryptophan fluorescence spectrum of 1 μM wild‐type or mutant Bax protein in buffer A was measured in a 4 × 4‐mm quartz microcell at 37°C with the excitation and emission wavelengths set at 295 nm and from 315 to 385 nm, respectively, and the same slit widths of excitation and emission monochromators described above. The fluorescence intensity at each nm of emission wavelength was measured for 1 s. Three technical repeats of the spectral measurement were performed for each sample.
For the Trp emission spectral data shown in Appendix␣Fig S6, we did not perform statistical analyses due to the same reasons described above for the liposome dye release data. Each data point in the figure was obtained using the same method described above for the liposome dye release data, except that an emission intensity was measured for 1 s and the intensity and standard error were recorded, and that we did not repeat the measurements with another set of samples that were prepared from different preparations of proteins.
Disulfide crosslinking of Bax in mitochondria
[35S]Met‐labeled Bax L59C/M79C or E69C protein without or with the additional mutation as indicated in Fig EV5 was synthesized in␣vitro using TNT coupled wheat germ extract system (Promega). As previously described (Pogmore et␣al, 2016; Zhang et␣al, 2016), the resulting proteins were targeted to the isolated Bax−/−/Bak−/− mitochondria by Bax BH3 peptide. The mitochondria‐bound proteins were separated from the soluble proteins by centrifugation and oxidized with copper (II) (1,10‐phenanthroline)3 (CuPhe) to induce disulfide crosslinking. The crosslinked Bax dimer was separated from the monomer by non‐reducing SDS–PAGE and detected by phosphor‐imaging using Fuji FLA‐9000 multi‐purpose image scanner. Intensities of both dimer and monomer bands in the phosphor‐image were measured using Fuji Image Analysis Software Multi Gauge. The dimer to monomer ratio was calculated from the intensities. The mitochondrial association efficiency was also calculated from intensities of Bax proteins in the mitochondrial and soluble fractions.
For the crosslinking data shown in Fig EV5B–E, we did not perform statistical analyses due to the inherent difficulty to precisely quantify the phosphor‐imaging data. Each phosphor‐imaging data shown in the figure were obtained from an experiment as described above. Each experiment was replicated two times for Fig EV5B–C, and one time for Fig EV5D–E, using the same method with the proteins produced by different TNT reactions and the mitochondria isolated from the same group of mice. Similar data were obtained, which were not shown in the manuscript.
Author contributions
BO, JL, and DWA conceived and directed the project; BO designed NMR experiments; BO and JL designed mutations and functional assays; FL, YY, LZ, and LD prepared Bax (α2–α5) proteins for NMR; FL, ZL, and MW collected NMR data; FL, MW, and BW analyzed the NMR data and determined the Bax (α2–α5) structure with the help of JJC and BO; FL and AP analyzed PRE data and built the structural model for bicelle‐bound Bax (α2–α5) with JJC; SW performed the single‐point energy calculations; FQ and JDR prepared Bax proteins, performed liposomal dye release experiments, measured Trp emission spectra, and analyzed the data with JL; ZZ generated Bax‐mutant plasmids, performed crosslinking experiments and analyzed the data with JL; JK performed mitochondrial protein release experiments and analyzed and interpreted the data; JP purified mouse liver mitochondria; BO, JL, and DWA interpreted the data and wrote the paper; All authors edited the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View/Appendix
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
We thank the staffs from Large‐scale Protein Preparation, Nuclear Magnetic Resonance and Mass Spectrometry Systems at National Facility for Protein Science in Shanghai, Zhangjiang Laboratory, China for technical support and assistance in data collection and analysis. This work was supported by grants from National Key R&D Program of China (2017YFA0504804), Key Research Program of Frontier Sciences, CAS (QYZDB‐SSW‐SMC043) to B. O., by grants from US National Institutes of Health (R01GM062964), OCAST (HR16‐026) and Presbyterian Health Foundation to J. L., by an Institutional Development Award from the National Institute of General Medical Sciences of US National Institutes of Health (P20GM103640), and by a foundation grant from the Canadian Institutes of Health Research (FDN 143312) to D. W. A. D. W. A. holds the Tier 1 Canada Research Chair in Membrane Biogenesis.
The EMBO Journal (2021) 40: e106438.
Contributor Information
David W Andrews, Email: David.Andrews@sri.utoronto.ca.
Jialing Lin, Email: jialing-lin@ouhsc.edu.
Bo OuYang, Email: ouyang@sibcb.ac.cn.
Data availability
NMR structure coordinates have been deposited in Protein Data Bank under accession number 6L8V (https://www.rcsb.org/structure/6L8V). 1H, 13C, and 15N chemical shifts have been deposited in Biological Magnetic Resonance Bank under accession number 36294 (https://bmrb.io/data_library/summary/index.php?bmrbId=36294).
References
- Andrews DW (2014) Pores of no return. Mol Cell 56: 465–466 [DOI] [PubMed] [Google Scholar]
- Annis MG, Soucie EL, Dlugosz PJ, Cruz‐Aguado JA, Penn LZ, Leber B, Andrews DW (2005) Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J 24: 2096–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels C, Xia TH, Billeter M, Guntert P, Wuthrich K (1995) The program XEASY for computer‐supported NMR spectral analysis of biological macromolecules. J Biomol NMR 6: 1–10 [DOI] [PubMed] [Google Scholar]
- Bassolino‐Klimas D, Tejero R, Krystek SR, Metzler WJ, Montelione GT, Bruccoleri RE (1996) Simulated annealing with restrained molecular dynamics using a flexible restraint potential: theory and evaluation with simulated NMR constraints. Protein Sci 5: 593–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleicken S, Assafa TE, Stegmueller C, Wittig A, Garcia‐Saez AJ, Bordignon E (2018) Topology of active, membrane‐embedded Bax in the context of a toroidal pore. Cell Death Differ 25: 1717–1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleicken S, Classen M, Padmavathi PV, Ishikawa T, Zeth K, Steinhoff HJ, Bordignon E (2010) Molecular details of Bax activation, oligomerization, and membrane insertion. J Biol Chem 285: 6636–6647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleicken S, Jeschke G, Stegmueller C, Salvador‐Gallego R, Garcia‐Saez AJ, Bordignon E (2014) Structural model of active Bax at the membrane. Mol Cell 56: 496–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bove J, Martinez‐Vicente M, Dehay B, Perier C, Recasens A, Bombrun A, Antonsson B, Vila M (2014) BAX channel activity mediates lysosomal disruption linked to Parkinson disease. Autophagy 10: 889–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer JM, Westphal D, Dewson G, Robin AY, Uren RT, Bartolo R, Thompson GV, Colman PM, Kluck RM, Czabotar PE (2014) Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol Cell 55: 938–946 [DOI] [PubMed] [Google Scholar]
- Chen W, Dev J, Mezhyrova J, Pan L, Piai A, Chou JJ (2018) The unusual transmembrane partition of the hexameric channel of the hepatitis C virus. Structure 26: 627–634.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi X, Nguyen D, Pemberton JM, Osterlund EJ, Liu Q, Brahmbhatt H, Zhang Z, Lin J, Leber B, Andrews DW (2020) The carboxyl‐terminal sequence of bim enables bax activation and killing of unprimed cells. elife 9: e44525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosentino K, Garcia‐Saez AJ (2017) Bax and bak pores: are we closing the circle? Trends Cell Biol 27: 266–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan AD, Smith NA, Sandow JJ, Kapp EA, Rustam YH, Murphy JM, Brouwer JM, Bernardini JP, Roy MJ, Wardak AZ et␣al (2020) BAK core dimers bind lipids and can be bridged by them. Nat Struct Mol Biol 27: 1024–1031 [DOI] [PubMed] [Google Scholar]
- Czabotar P, Westphal D, Dewson G, Ma S, Hockings C, Fairlie W, Lee E, Yao S, Robin A, Smith B et␣al (2013) Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152: 519–531 [DOI] [PubMed] [Google Scholar]
- Dal Peraro M, van der Goot FG (2016) Pore‐forming toxins: ancient, but never really out of fashion. Nat Rev Microbiol 14: 77–92 [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J␣Biomol NMR 6: 277–293 [DOI] [PubMed] [Google Scholar]
- Dewson G, Ma S, Frederick P, Hockings C, Tan I, Kratina T, Kluck RM (2012) Bax dimerizes via a symmetric BH3: groove interface during apoptosis. Cell Death Differ 19: 661–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C, Neutzner A, Tjandra N, Youle RJ (2011) Bcl‐x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 145: 104–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores‐Romero H, Garcia‐Porras M, Basanez G (2017) Membrane insertion of the BAX core, but not latch domain, drives apoptotic pore formation. Sci Rep 7: 16259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, Piai A, Chen W, Xia K, Chou JJ (2019) Structure determination protocol for transmembrane domain oligomers. Nat Protoc 14: 2483–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahl RF, He Y, Yu S, Tjandra N (2014) Conformational rearrangements in the pro‐apoptotic protein, Bax, as it inserts into mitochondria: a cellular death switch. J Biol Chem 289: 32871–32882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner TP, Amgalan D, Reyna DE, Li S, Kitsis RN, Gavathiotis E (2019) Small‐molecule allosteric inhibitors of BAX. Nat Chem Biol 15: 322–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavathiotis E, Reyna DE, Davis ML, Bird GH, Walensky LD (2010) BH3‐triggered structural reorganization drives the activation of proapoptotic BAX. Mol Cell 40: 481–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavathiotis E, Reyna DE, Bellairs JA, Leshchiner ES, Walensky LD (2012) Direct and selective small‐molecule activation of proapoptotic BAX. Nat Chem Biol 8: 639–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu H‐C, Kim H, Cheng E‐Y, Tjandra N et␣al (2008) BAX activation is initiated at a novel interaction site. Nature 455: 1076–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse L, Wurm CA, Bruser C, Neumann D, Jans DC, Jakobs S (2016) Bax assembles into large ring‐like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J 35: 402–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer S, Uren RT, Kluck RM (2019) Probing BAK and BAX activation and pore assembly with Cytochrome c release, limited proteolysis, and oxidant‐induced linkage. Methods Mol Biol 1877: 201–216 [DOI] [PubMed] [Google Scholar]
- Kagedal K, Johansson AC, Johansson U, Heimlich G, Roberg K, Wang NS, Jurgensmeier JM, Ollinger K (2005) Lysosomal membrane permeabilization during apoptosis–involvement of Bax? Int J Exp Pathol 86: 309–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekura K, Ma X, Murphy JT, Zhu LJ, Diwan A, Urano F (2015) IRE1 prevents endoplasmic reticulum membrane permeabilization and cell death under pathological conditions. Sci Signal 8: ra62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD (2002) Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111: 331–342 [DOI] [PubMed] [Google Scholar]
- Lovell JF, Billen LP, Bindner S, Shamas‐Din A, Fradin C, Leber B, Andrews DW (2008) Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135: 1074–1084 [DOI] [PubMed] [Google Scholar]
- Moldoveanu T, Liu Q, Tocilj A, Watson M, Shore G, Gehring K (2006) The X‐ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol Cell 24: 677–688 [DOI] [PubMed] [Google Scholar]
- Niu X, Brahmbhatt H, Mergenthaler P, Zhang Z, Sang J, Daude M, Ehlert FGR, Diederich WE, Wong E, Zhu W et␣al (2017) A small‐molecule inhibitor of Bax and Bak oligomerization prevents genotoxic cell death and promotes neuroprotection. Cell Chem Biol 24: 493–506.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norberto de Souza O, Ornstein RL (1999) Molecular dynamics simulations of a protein‐protein dimer: particle‐mesh Ewald electrostatic model yields far superior results to standard cutoff model. J Biomol Struct Dyn 16: 1205–1218 [DOI] [PubMed] [Google Scholar]
- Parker MW, Pattus F, Tucker AD, Tsernoglou D (1989) Structure of the membrane‐pore‐forming fragment of colicin A. Nature 337: 93–96 [DOI] [PubMed] [Google Scholar]
- Piai A, Fu Q, Dev J, Chou JJ (2017) Optimal bicelle size q for solution NMR studies of the protein transmembrane partition. Chemistry 23: 1361–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogmore JP, Pemberton JM, Chi X, Andrews DW (2016) Using forster‐resonance energy transfer to measure protein interactions between Bcl‐2 family proteins on mitochondrial membranes. Methods Mol Biol 1419: 197–212 [DOI] [PubMed] [Google Scholar]
- Reyna DE, Garner TP, Lopez A, Kopp F, Choudhary GS, Sridharan A, Narayanagari S‐R, Mitchell K, Dong B, Bartholdy BA et␣al (2017) Direct activation of BAX by BTSA1 overcomes apoptosis resistance in acute myeloid leukemia. Cancer Cell 32: 490–505.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvador‐Gallego R, Mund M, Cosentino K, Schneider J, Unsay J, Schraermeyer U, Engelhardt J, Ries J, Garcia‐Saez AJ (2016) Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J 35: 389–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzmann M, Pervushin K, Wider G, Senn H, Wuthrich K (1998) TROSY in triple‐resonance experiments: new perspectives for sequential NMR assignment of large proteins. Proc Natl Acad Sci USA 95: 13585–13590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieters CD, Bermejo GA, Clore GM (2018) Xplor‐NIH for molecular structure determination from NMR and other data sources. Protein Sci 27: 26–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamas‐Din A, Bindner S, Chi X, Leber B, Andrews DW, Fradin C (2015) Distinct lipid effects on tBid and Bim activation of membrane permeabilization by pro‐apoptotic Bax. Biochem J 467: 495–505 [DOI] [PubMed] [Google Scholar]
- Shamas‐Din A, Satsoura D, Khan O, Zhu W, Leber B, Fradin C, Andrews DW (2014) Multiple partners can kiss‐and‐run: Bax transfers between multiple membranes and permeabilizes those primed by tBid. Cell Death Dis 5: e1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Delaglio F, Cornilescu G, Bax A (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J␣Biomol NMR 44: 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner AL, Laurence JS (2008) High‐field solution NMR spectroscopy as a tool for assessing protein interactions with small molecule ligands. J␣Pharm Sci 97: 4670–4695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Youle RJ, Tjandra N (2000) Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 103: 645–654 [DOI] [PubMed] [Google Scholar]
- Tan C, Dlugosz PJ, Peng J, Zhang Z, Lapolla SM, Plafker SM, Andrews DW, Lin J (2006) Auto‐activation of the apoptosis protein Bax increases mitochondrial membrane permeability and is inhibited by Bcl‐2. J Biol Chem 281: 14764–14775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren RT, Iyer S, Kluck RM (2017) Pore formation by dimeric Bak and Bax: an unusual pore? Philos Trans R Soc Lond B Biol Sci 372: 20160218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K, MacKerell AD Jr (2012) Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J Chem Inf Model 52: 3144–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59: 687–696 [DOI] [PubMed] [Google Scholar]
- Westphal D, Dewson G, Menard M, Frederick P, Iyer S, Bartolo R, Gibson L, Czabotar PE, Smith BJ, Adams JM et␣al (2014) Apoptotic pore formation is associated with in‐plane insertion of Bak or Bax central helices into the mitochondrial outer membrane. Proc Natl Acad Sci USA 111: E4076–4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Subramaniam S, Kale J, Liao C, Huang B, Brahmbhatt H, Condon SG, Lapolla SM, Hays FA, Ding J et␣al (2016) BH3‐in‐groove dimerization initiates and helix 9 dimerization expands Bax pore assembly in membranes. EMBO J 35: 208–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View/Appendix
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Data Availability Statement
NMR structure coordinates have been deposited in Protein Data Bank under accession number 6L8V (https://www.rcsb.org/structure/6L8V). 1H, 13C, and 15N chemical shifts have been deposited in Biological Magnetic Resonance Bank under accession number 36294 (https://bmrb.io/data_library/summary/index.php?bmrbId=36294).