

Abstract
DNA interstrand crosslinks (ICLs) induced by endogenous aldehydes or chemotherapeutic agents interfere with essential processes such as replication and transcription. ICL recognition and repair by the Fanconi Anemia pathway require the formation of an X‐shaped DNA structure that may arise from convergence of two replication forks at the crosslink or traversing of the lesion by a single replication fork. Here, we report that ICL traverse strictly requires DNA repriming events downstream of the lesion, which are carried out by PrimPol, the second primase‐polymerase identified in mammalian cells after Polα/Primase. The recruitment of PrimPol to the vicinity of ICLs depends on its interaction with RPA, but not on FANCM translocase or the BLM/TOP3A/RMI1‐2 (BTR) complex that also participate in ICL traverse. Genetic ablation of PRIMPOL makes cells more dependent on the fork convergence mechanism to initiate ICL repair, and PRIMPOL KO cells and mice display hypersensitivity to ICL‐inducing drugs. These results open the possibility of targeting PrimPol activity to enhance the efficacy of chemotherapy based on DNA crosslinking agents.
Keywords: ICL repair, ICL traverse, interstrand crosslink, PrimPol, RPA
Subject Categories: DNA Replication, Repair & Recombination
A specialized human primase involved in DNA damage tolerance facilitates continuous DNA synthesis past ICL lesions, and their post‐replicative repair via the Fanconi Anemia pathway.
Introduction
DNA interstrand crosslinks (ICLs) are cytotoxic lesions induced by endogenous aldehydes or exogenous chemicals such as mitomycin C (MMC), cisplatin, or trimethyl‐psoralen (TMP), some of which are extensively used as chemotherapeutic agents. ICLs are repaired in a DNA replication‐dependent manner by a protein network that, if lost or altered by mutation, results in Fanconi Anemia (FA), a rare but very severe disease (reviewed by Ceccaldi et al, 2016). ICLs are recognized by the FANCM complex (FANCM/MHF1‐2/FAAP24), which assists in the recruitment of the FA core complex responsible for the ubiquitylation and activation of the FANCD2/I heterodimer (Deans & West, 2009). Activated FANCD2/I recruits specific nucleases to one of the DNA strands initiating a complex process that involves homologous recombination (HR), translesion synthesis (TLS), and nucleotide excision repair (NER) mechanisms (reviewed by Deans & West, 2011; Zhang & Walter, 2014; Lopez‐Martinez et al, 2016).
Many mechanistic aspects of the initial steps in ICL repair have been derived from biochemical studies in X. laevis egg extracts (Räschle et al, 2008; Knipscheer et al, 2009). In this system, ICL repair is triggered by the convergence of two forks that reach the ICL from opposite sides, creating an X‐shaped structure that resembles a replication termination event (Zhang et al, 2015a). In the case of TMP‐induced ICLs, fork convergence may be followed by NEIL3 glycosylase‐dependent cleavage of the ICL, avoiding the generation of DSBs (Semlow et al, 2016; Li et al, 2020). Nonetheless, FA‐deficient cells are sensitive to TMP‐induced ICLs, suggesting that the FA pathway is still required for complete repair (Li et al, 2020). The choice between NEIL3‐dependent cleavage or the complete FA repair pathways relies on TRAIP‐ubiquitin ligase (Wu et al, 2019; Li et al, 2020). Mono‐ubiquitylation of the Cdc45‐MCM‐GINS helicase (CMG) by TRAIP triggers the NEIL3 pathway, while poly‐ubiquitylation of the same complex promotes CMG unloading by p97 segregase and favors FA‐dependent repair (Wu et al, 2019).
In addition to fork convergence‐based mechanisms, in avian and mammalian cells ICLs can be “traversed” by the first fork that reaches the lesion, creating a suitable template for ICL repair (Huang et al, 2013). Traverse reactions account for up to 60% of the replication events monitored around ICLs in human cells and require FANCM (Huang et al, 2013), its interaction with PCNA (Rohleder et al, 2016), and the BTR complex (BLM/TOP3A/RMI1‐2; Ling et al, 2016). The loss of BLM helicase activity within the BTR reduced the frequency of ICL traverse in chicken DT40 cells, suggesting that BLM might unwind the DNA at the unreplicated side of the ICL (Ling et al, 2016). Temporary fork reversal and CMG (Cdc45‐MCM‐GINS) unloading appear to be necessary to initiate ICL repair after fork convergence (Amunugama et al, 2018) and also to facilitate ICL traverse in mammalian cells (Mutreja et al, 2018).
The molecular events that take place during the traverse of ICLs are starting to be elucidated. ATR signaling and FANCD2 protein mediate the interaction between FANCM and the CMG helicase and trigger the eviction of its GINS component. CMG remodeling may facilitate the opening of the MCM hexameric ring, a necessary step for the translocation of the replisome across the ICL (Huang et al, 2019). After the replisome is relocated downstream of the ICL, restart of DNA synthesis by DNA polymerases would require repriming events that had not been characterized to date. Besides the canonical Polα/Primase, many archaeal and eukaryotic organisms encode PrimPol, a primase‐polymerase specialized in DNA damage tolerance (Bianchi et al, 2013; García‐Gómez et al, 2013; Mourón et al, 2013; Wan et al, 2013). Human PrimPol is distributed among nuclei, cytosol, and mitochondria but is rapidly redirected to chromatin upon UV‐C irradiation to reprime DNA synthesis at UV‐induced photoproducts. Repriming reactions allow fork progression and leave bypassed lesions to be repaired post‐replicatively (Mourón et al, 2013). PrimPol has a role in mitochondrial DNA maintenance (García‐Gómez et al, 2013) that also relies on repriming (Torregrosa‐Muñumer et al, 2017). The interaction of PrimPol with ssDNA‐binding proteins RPA and mitochondrial SSBP1 facilitates its recruitment to ssDNA generated next to DNA lesions (Wan et al, 2013; Guilliam et al, 2015, 2017). Furthermore, long ssDNA stretches enhance the primase activity of PrimPol in vitro (Martínez‐Jiménez et al, 2017). Of note, PrimPol also operates at natural replication blocks formed by special structures such as G‐quadruplexes (Schiavone et al, 2016) and R‐loops (Šviković et al, 2019).
Here we show that human cells rely on PrimPol primase to mediate the ICL traverse reaction, sustaining DNA synthesis during the cellular recovery from ICL‐inducing agents. In the absence of PRIMPOL, cells become more dependent on the fork convergence mechanism, and the global efficiency of ICL repair is compromised.
Results
PrimPol interacts with ICL recognition and repair factors
A search for PrimPol‐interacting factors was conducted by immunoprecipitation (IP) coupled to mass spectrometry in U2OS cells synchronized in S phase. Isogenic PRIMPOL KO cells were generated by CRISPR/Cas9 technology (Figs 1A and EV1) and used as control for non‐specific binding proteins. A quantitative proteomic analysis (enrichment ratio and statistical significance of proteins identified in WT vs KO cells) confirmed the efficient IP of PrimPol and the co‐precipitation of known interacting factors RPA and SSBP1. Among the new potential PrimPol‐interacting proteins were HERC2, involved in the resolution of G4 quadruplex structures (Wu et al, 2018) and several proteins that participate in the recognition and repair of ICL lesions: MHF1 (part of the FANCM complex; Ciccia et al, 2007; Yan et al, 2010; Wang et al, 2013); RUNX1 (Tay et al, 2018); and BLM, RMI1, and RMI2 (three components of the BTR complex; Manthei & Keck, 2013; Fig 1B and Dataset EV1). Both MHF1 and BTR are required for the ICL traverse reaction (Huang et al, 2013; Ling et al, 2016).
Figure 1. PrimPol interacts with factors involved in ICL traverse.
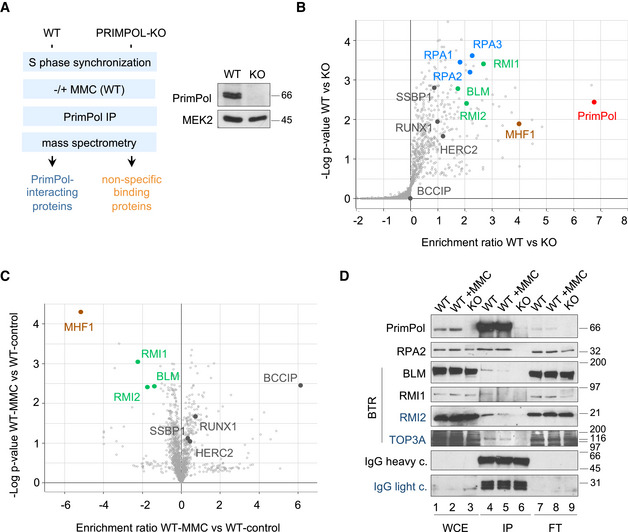
- Experimental design for the proteomic identification of PrimPol‐interacting factors in S phase‐synchronized cells. Immunoblot showing PrimPol levels in WT and KO cells. MEK2 is shown as loading control.
- Volcano plot showing statistical significance (−log P‐value) vs. enrichment ratio (log2) of identified proteins after PrimPol immunoprecipitation in WT and KO cells. The positions of PrimPol, RPA1‐3, BLM, RMI1, RMI2, MHF1, SSBP1, RUNX1, HERC2, and BCCIP proteins are indicated.
- Volcano plot showing statistical significance (−log P‐value) vs. enrichment ratio (log2) of identified proteins after PrimPol immunoprecipitation in WT cells with or without MMC (2 µg/ml; 5 h). BLM, RMI1, RMI2, MHF1, SSBP1, RUNX1, HERC2, and BCCIP proteins are indicated.
- Immunoblot detection of the indicated proteins in whole cell extracts (WCE), PrimPol immunoprecipitates (IP), and non‐precipitated flow‐through fractions (FT). IgG shows the presence of anti‐PrimPol antibody in the IP fractions (lanes 4–6). Protein names in black and blue indicate the two different gels used.
Source data are available online for this figure.
Figure EV1. (related to Fig 1). Allele sequences in CRISPR/Cas9‐derived PRIMPOL KO U2OS cells, cell synchronization, and markers of DNA damage induced by MMC.
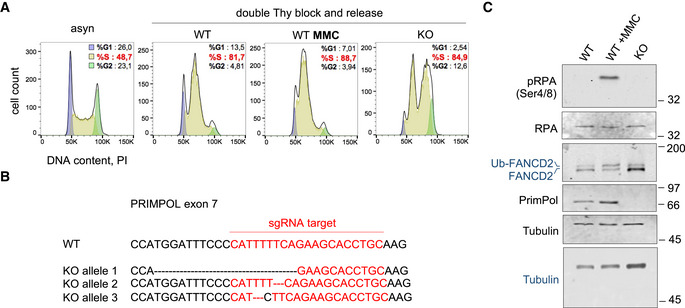
- Flow cytometry profiles of DNA content (PI) in the synchronized cell cultures used for PrimPol IP and mass spectrometry. The percentage of cells in each phase of the cell cycle is indicated.
- Genomic sequences derived from WT and KO alleles in U2OS‐PRIMPOL KO cells obtained with CRISPR/Cas9. The Cas9 guide RNA sequence is marked in red. As the 4q35.1 chromosome locus is amplified in U2OS cells, three PRIMPOL alleles were targeted to achieve a full KO.
- Immunoblots show the levels of the indicated proteins in whole cell extracts from the same samples used in Fig 1C. The position of the ubiquitylated form of FANCD2 (Ub‐FANCD2) is indicated. Tubulin is shown as a loading control. Protein names in black and blue indicate the two different gels used.
Source data are available online for this figure.
The IP‐mass spectrometry assay was also performed in cells treated with mitomycin C (MMC), a DNA crosslinking agent used in chemotherapy. As expected, MMC induced RPA phosphorylation and FANCD2 ubiquitylation (Fig EV1C). The comparison of MMC‐treated vs control cells hinted at dynamic changes in the association between PrimPol and some of its potential partners. Interactions with SSBP1, HERC2, RUNX, and HR protein BCCIP (Lu et al, 2005) were enhanced, whereas the binding to BLM, RMI1, RMI2, and MHF1 was apparently reduced (Fig 1C and Dataset EV1). The interaction between PrimPol and BLM, RMI1, and RMI2 was confirmed using IP‐immunoblot assays, which also revealed the co‐precipitation of TOP3A, the fourth component of the BTR complex (Fig 1D). The dynamic PrimPol‐BTR interaction was also validated (Fig 1D, lanes 4 and 5) and suggests that the physical interaction between PrimPol and the BTR complex is disrupted during the cellular response to MMC.
PrimPol is recruited to chromatin in response to ICL‐inducing agents
Following UV‐C irradiation or treatment with cisplatin, PrimPol accumulates on chromatin to facilitate the replicative bypass of DNA photoadducts or cisplatin‐induced lesions (Mourón et al, 2013; Quinet et al, 2020). To test whether PrimPol is also recruited to chromatin in response to ICLs, its subcellular localization was tested in cells treated with MMC, which creates DNA monoadducts, intra‐strand crosslinks and ICLs; or with UVA‐activated trimethyl‐psoralen (TMP‐UVA), which induces ICLs with high specificity (~90% of total lesions; Lopez‐Martinez et al, 2016). Both MMC and TMP‐UVA caused the ubiquitylation and chromatin association of FANCD2, a marker of ICL repair, and led to the accumulation of PrimPol protein on chromatin (Fig 2A, lanes 9–12). To address whether PrimPol was recruited specifically to the proximity of ICL lesions, TMP‐treated cells were irradiated with an UVA laser beam to generate ICLs in defined areas of the nucleus (Li et al, 2020). DNA damage marker γH2AX was readily detected at the laser path by immunofluorescence (IF; Fig 2B). 1h post‐irradiation, endogenous PrimPol was recruited to the laser path in a large fraction of the cells (44%). Neither γH2AX nor PrimPol was directed to the UVA laser path if TMP was omitted. In addition, PrimPol KO cells confirmed the specificity of the PrimPol antibody used for IF (Fig 2B).
Figure 2. PrimPol is rapidly recruited to ICLs in an RPA‐dependent manner.
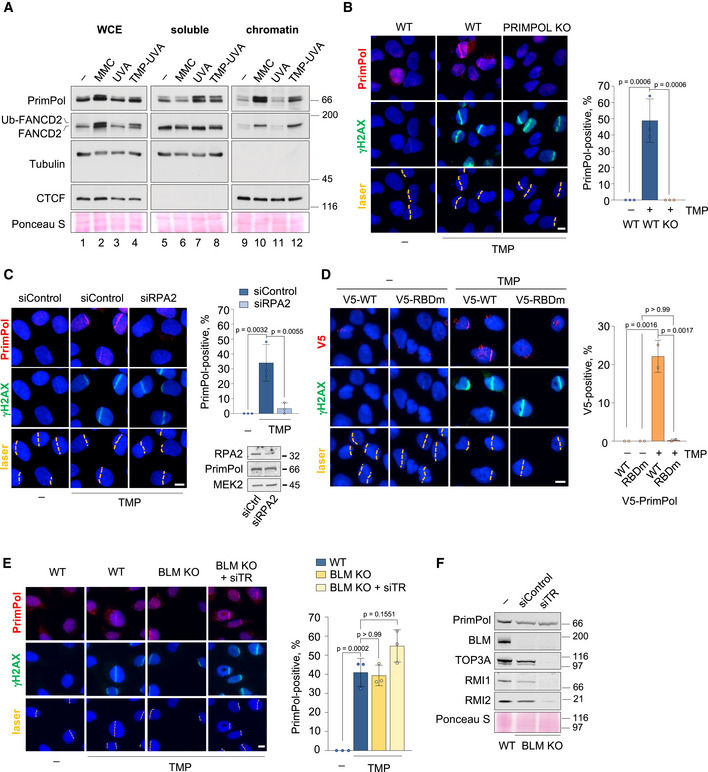
- Immunoblots show the levels of the indicated proteins in whole cell extracts (WCE), soluble and chromatin‐enriched fractions. Tubulin and CTCF are shown as controls for soluble and chromatin‐bound proteins, respectively. The position of the ubiquitylated form of FANCD2 (Ub‐FANCD2) is indicated. Ponceau S staining is shown as a loading control.
- Confocal microscopy images of PrimPol and γH2AX (damage control) immunofluorescence staining in control (UVA) or TMP‐UVA‐laser irradiated (10 µM TMP, 2 h followed by UVA laser irradiation) WT and PRIMPOL KO cells. Nuclear DNA is counterstained with DAPI. UVA laser path is indicated. Scale bar, 10 µm. Histograms (right panel) show the average percentage of PrimPol‐positive cells and SD of three assays (n ≥ 100 cells per TMP‐treated conditions in each replicate). Circle dots in each column represent the values of individual replicates. Statistical analysis was conducted with one‐way ANOVA followed by Bonferroni post‐test. P‐values of individual comparisons are indicated.
- Confocal microscopy images of PrimPol and γH2AX (damage control) immunofluorescence staining in control (UVA) or TMP‐UVA‐treated cells following control or RPA2 downregulation. Nuclear DNA is counterstained with DAPI. UVA laser path is indicated. Scale bar, 10 µm. Histograms (right panel) show the average PrimPol‐positive cells and SD of three assays (n ≥ 100 cells per TMP‐treated conditions in each replicate). Circle dots in each column represent the values of individual replicates. Statistical analysis was conducted with one‐way ANOVA followed by Bonferroni post‐test. P‐values of individual comparisons are indicated.
- Confocal microscopy images of PrimPol and γH2AX (damage control) immunofluorescence staining in control (UVA) or TMP‐UVA‐treated PRIMPOL KO cells stably expressing V5‐tagged WT or RPA binding domain mutant (RBDm) PrimPol versions. Nuclear DNA is counterstained with DAPI. UVA laser path is indicated. Scale bar, 10 µm. Histograms (right panel) show the average percentage of PrimPol‐positive cells and SD of two assays (n ≥ 100 cells per condition in each replicate). Circle dots in each column represent the values of individual replicates. Statistical analysis was conducted with one‐way ANOVA followed by Bonferroni post‐test. P‐values of individual comparisons are indicated.
- Confocal microscopy images of PrimPol and γH2AX (damage control) immunofluorescence staining in control (UVA) or TMP‐UVA‐treated WT and BLM KO cells upon downregulation of TOP3A, RMI1, and RMI2 (siTR) when indicated. Nuclear DNA is counterstained with DAPI. UVA laser path is indicated. Scale bar, 10 µm. Histograms (right panel) show the average percentage of PrimPol‐positive cells and SD of three assays (n ≥ 250 cells per condition). Circle dots in each column represent the values of individual replicates. Statistical analysis was conducted with one‐way ANOVA followed by Bonferroni post‐test.
- Immunoblots show the levels of BLM, TOP3A, RMI1, RMI2, and PrimPol after TOP3A, RMI1, and RMI2 downregulation in WT and BLM KO cells. Ponceau S is shown as loading control.
Source data are available online for this figure.
PrimPol binding to ICLs requires RPA but not BTR or FANCM
RPA mediates the binding of PrimPol to other fork blocks (Guilliam et al, 2017) and enhances its activity in vitro (Martínez‐Jiménez et al, 2017). The recruitment of PrimPol to ICL‐containing DNA is also mediated by RPA, as shown by RPA2 downregulation (Fig 2C) or expression of a mutant PrimPol deficient for RPA binding (RBDm) in PRIMPOL KO cells (Fig 2D). In contrast, the recruitment of PrimPol was not affected by the loss of BTR or FANCM and only modestly by FANCD2 downregulation (Fig EV2A and B). The fact that PrimPol can bind to the ICLs created by the laser path in the absence of BLM was confirmed in U2OS BLM KO cells generated by CRISPR‐Cas9 editing (Loe et al, 2020), even when the rest of the BTR components were silenced with siRNA (Fig 2E and F). In turn, normal loading of BTR, FANCM, and FANCD2 proteins to ICL‐containing DNA was observed in PrimPol KO cells (Fig EV2C–F). These results indicate that PrimPol and the BTR complex can bind to ICL‐containing DNA independently of each other, but PrimPol recruitment requires RPA protein.
Figure EV2. (related to Fig 2). Independent binding of PRIMPOL, BTR, FANCM, and FANCD2 to ICL‐containing DNA.
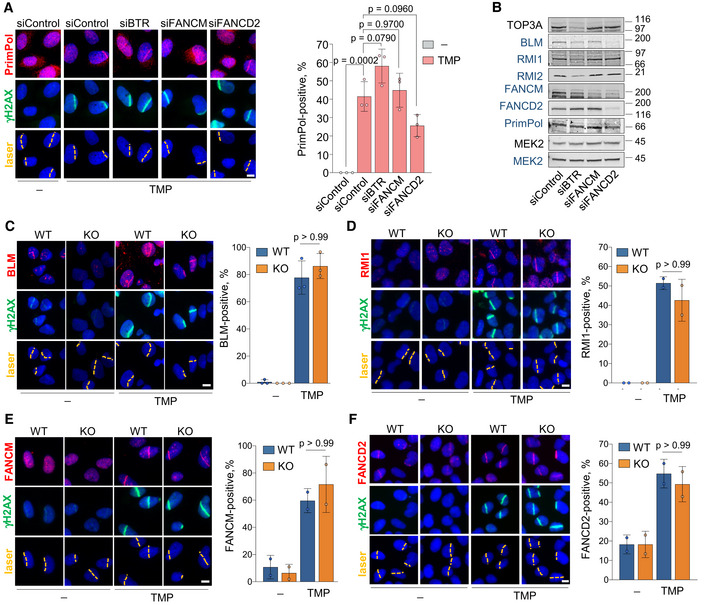
-
AConfocal microscopy images of PrimPol and γH2AX immunofluorescence (IF) in control (UVA) or TMP‐UVA‐treated cells following downregulation of the indicated factors. Nuclear DNA is counterstained with DAPI. The UVA laser path is indicated. Scale bar, 10 µm. Histograms (right panel) show the average and SD of the percentage of cells that are PrimPol‐positive in the laser path in three assays (n ≥ 100 cells per TMP‐treated conditions in each replicate). Circle dots in each column represent the values of individual replicates. Statistical analysis was conducted with one‐way ANOVA followed by Bonferroni post‐test. The p‐values of the indicated comparisons are shown.
-
BImmunoblots showing the levels of the indicated proteins after siRNA‐mediated downregulation. siBTR is a combination of siBLM, siTOP3A, siRMI1, and siRMI2. MEK2 is shown as loading control. Protein names in black and blue indicate the two different gels used.
-
C–FConfocal microscopy images of BLM (C), RMI1 (D), FANCM (E), or FANCD2 (F) together with γH2AX IF staining in control (UVA) or TMP‐UVA‐laser irradiated WT or PRIMPOL KO cells, as in (A). Scale bar, 10 µm. Histograms (right panels) show the average percentage of cells positive for the recruitment of the indicated factor to the laser path. Average and SD of at least two assays are shown (n ≥ 100 cells per TMP‐treated conditions in each replicate). Circle dots in each column represent the values of individual replicates. Statistical analysis was conducted with one‐way ANOVA followed by Bonferroni post‐test. P‐values of the indicated comparisons are included.
Source data are available online for this figure.
PrimPol engages in repriming reactions at ICLs
Given the biochemical function of PrimPol at UV‐induced DNA lesions (Mourón et al, 2013), we hypothesized that it may reprime DNA synthesis at ICLs. Primases require a ssDNA stretch that may be formed by the uncoupling of polymerase and helicase activities in the replisome or by an specialized helicase. We observed that TMP‐UVA induced the formation of ssDNA foci using BrdU IF in native conditions (Fig EV3A–C). ssDNA was not generated after downregulation of BTR or FANCM, which contributes to recruit BTR to ICLs (Fig EV3A–C; Deans & West, 2009; Ling et al, 2016). These observations support the notion that BLM helicase unwinds DNA at the unreplicated side of ICLs, as suggested (Ling et al, 2016). RPA downregulation also prevented ssDNA foci formation, probably because of rapid reannealing of ssDNA strands (Xue et al, 2019). In PRIMPOL KO cells, ssDNA foci were formed as in WT cells, indicating that PrimPol protein itself is not involved in the unwinding of dsDNA upon TMP‐UVA.
Figure EV3. (related to Fig 3). Generation of ssDNA by TMP‐UVA, and effects of RBDm PRIMPOL mutation and FANCM downregulation in ICL traverse.
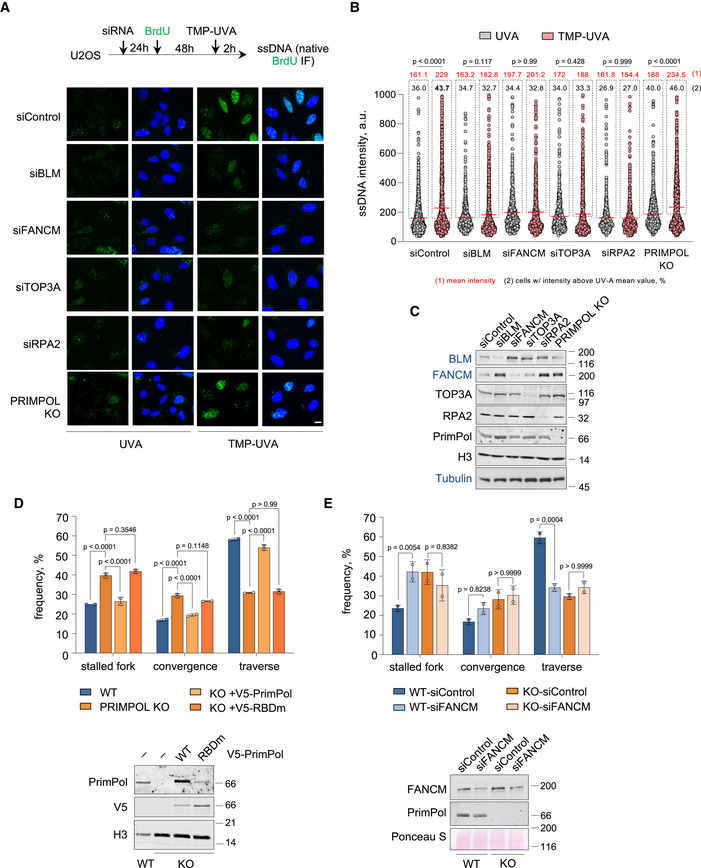
- Top, experimental design. Bottom, confocal microscopy images of ssDNA (anti‐BrdU in native conditions; green color) IF in control (UVA) or TMP‐UVA‐treated cells following downregulation of the indicated factors. Nuclear DNA is counterstained with DAPI (blue color). Scale bar, 10 µm.
- Dot plot shows the distribution and mean value (horizontal red line) of ssDNA intensity in individual cells in each condition. Mean intensity values are indicated in red, outside dashed boxes that include individual events above the mean intensity in the UVA control. The percentage of these positive cells is also indicated inside the dashed box. Data pooled from three replicates are represented (n ≥ 200 cells per condition). Statistical analysis was conducted with one‐way ANOVA followed by Tukey’s multiple comparison post‐test. P‐values of the indicated comparisons are shown.
- Immunoblots showing the levels of the indicated proteins corresponding to the experiments shown in panels A–B. H3 and Tubulin are shown as loading controls. Protein names in black and blue indicate the two different gels used.
- ICL‐localized DNA fiber experiments in the indicated conditions. Top, histograms show the percentage of the different replication patterns in each condition (average and SD of two assays). Circle dots in each column represent the values of individual replicates. Statistical analysis: two‐way ANOVA followed by Bonferroni post‐test. P‐values are indicated. Bottom, immunoblots showing the levels of endogenous PrimPol or exogenous V5‐PrimPol (WT and RBDm versions). H3 is shown as loading control.
- ICL‐localized DNA fiber experiments in the indicated conditions. Top, histograms show the percentage of the different replication patterns observed in each experimental condition (average and SD of two assays). Circle dots in each column represent the values of individual replicates. Statistical analysis: two‐way ANOVA followed by Bonferroni post‐test. P‐values are indicated. Bottom, immunoblots showing the levels of FANCM and PrimPol proteins. Ponceau S is shown as loading control.
Source data are available online for this figure.
In order to test directly whether PrimPol re‐primes DNA synthesis at ICLs, we used a modification of the stretched DNA fiber assay in which TMP molecules linked to digoxigenin (Dig‐TMP) serve to localize ICLs in the DNA fibers (Huang et al, 2013; Mutreja et al, 2018). Combined with CldU/IdU labeling, this approach allows the identification of three main types of replication tracks around ICLs: (i) single forks stalled at the ICL; (ii) two forks converging at the ICL; (iii) single forks that have traversed the lesion (Fig 3A). In U2OS cells, the majority of labeled tracks corresponded to ICL traverse reactions (60%), followed by single stalled forks (25%) and converged forks (15%), in agreement with previous reports (Fig 3C; Huang et al, 2013, 2019). In PRIMPOL KO cells, however, the frequency of traverse reactions was drastically reduced (26%, less than half than control cells), while fork convergence events increased to 34%. This effect could be entirely attributed to PrimPol protein, as reintroduction of exogenous PrimPol in KO cells restored the proportions observed in WT cells. Neither the catalytic mutant (AxA) nor the primase‐deficient ∆Zn version of PrimPol rescued the frequency of ICL traverse reactions (Fig 3B and C), underscoring the importance of the primase activity. ∆Zn was expressed at lower levels than WT PrimPol (Fig 3C), but the result was confirmed using PrimPol‐CH, a full‐length protein carrying two mutations in the Zn‐finger domain (C419G/H426Y) that inactivate the primase (Mourón et al, 2013). Consistent with the role of RPA in recruiting PrimPol to ICLs, reintroduction of PrimPol RBDm in KO cells also failed to rescue the rate of ICL traverse (Fig EV3D).
Figure 3. PrimPol mediates the ICL traverse reaction through its primase activity.
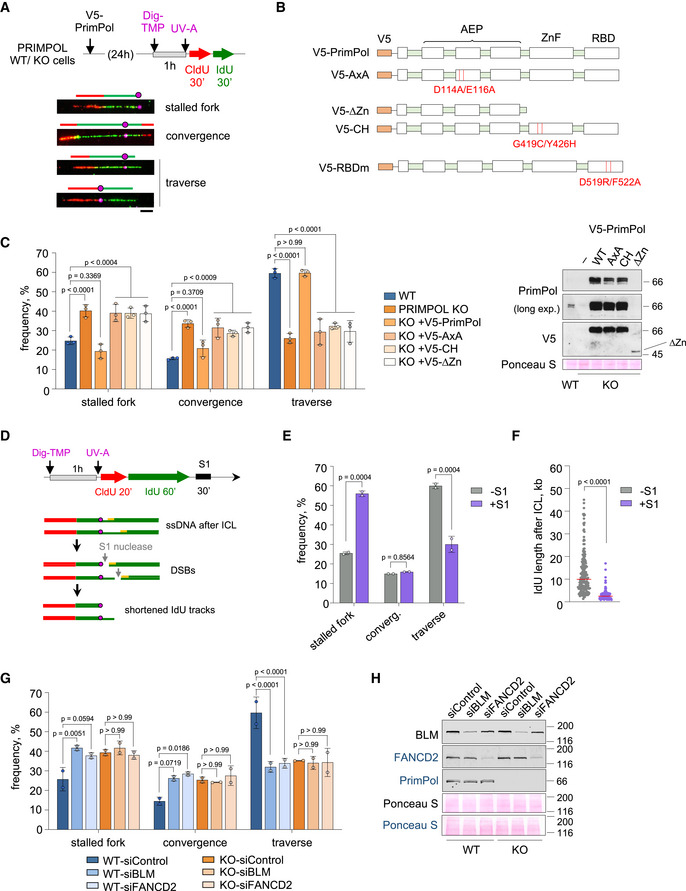
- Experimental design and examples of stretched DNA fibers with different patterns of DNA synthesis around the ICL lesion. Fibers were stained with anti‐CldU (red), anti‐IdU (green), and anti‐Dig (magenta). Schematics over the fiber images highlight the position of the ICL (circle) in each replicative structure. Scale bar, 10 µm.
- PrimPol mutants used in the experiments. AxA is a PrimPol catalytic mutant. ∆Zn and CH are primase‐null, polymerase‐proficient PrimPol mutants. RBDm is a PrimPol mutant defective for RPA‐binding.
- Histograms show the percentage of the different replication patterns observed in each experimental condition (average and SD of three assays). Circle dots in each column represent the values of individual replicates. Statistical analysis: two‐way ANOVA followed by Bonferroni post‐test comparing each condition to the WT. P‐values of individual comparisons are indicated. Immunoblots showing the levels of V5‐PrimPol proteins (WT and mutant derivatives). Ponceau S staining is shown as loading control.
- Experimental design of ICL‐localized DNA fibers followed by S1 nuclease incubation. Hypothetical fiber containing ssDNA downstream of the ICL (circle) turns into a DSB after S1 nuclease action. This would lead to an increase in apparent stalled forks or shortening of IdU length downstream of the lesion.
- Histograms show the percentage of the different replication patterns observed in each experimental condition (average and SD of two assays). Circle dots in each column represent the values of individual replicates. Statistical analysis: two‐way ANOVA followed by Bonferroni post‐test. P‐values of individual comparisons are indicated.
- Dot plot shows the distribution and median (horizontal red line) of IdU track length after an event of ICL traverse in each condition. Data pooled from two replicates are represented (n ≥ 100 cells per condition). Statistical analysis was conducted with Kruskal–Wallis test and Dunn’s post‐test. P‐values of individual comparisons are indicated.
- Histograms show the percentage of the different replication patterns observed in each experimental condition (average and SD of two assays). Circle dots in each column represent the values of individual replicates. Statistical analysis: two‐way ANOVA followed by Bonferroni post‐test. P‐values of individual comparisons are indicated.
- Immunoblots showing the levels of BLM, FANCD2, and PrimPol after BLM or FANCD2 downregulation in WT and PRIMPOL KO cells for experiments in (G). Ponceau S is shown as loading control. Protein names in black and blue indicate the two different gels used.
Source data are available online for this figure.
The ICL‐localized DNA fiber assay can be combined with S1 nuclease, which cleaves at ssDNA gaps and has been used to evaluate repriming events in other contexts (e.g. Bai et al, 2020; Piberger et al, 2020; Quinet et al, 2020). As expected if ssDNA was generated after the lesion, S1 eliminated many IdU tracks downstream of ICLs, resulting in an apparent decrease of traverse reactions and a concomitant increase in the frequency of apparently stalled forks (Fig 3D and E). In the remaining traverse structures, the length of IdU tracks located after ICL lesions was strongly decreased upon S1 incubation (Fig 3F). Combined, these assays provide molecular evidence that the ICL traverse reaction requires ssDNA, likely generated by BLM helicase, which is covered by RPA and followed by PrimPol primase activity.
Epistatic relation between PrimPol and ICL traverse factors
We next tested the potential epistasis between PrimPol and other factors involved in ICL traverse. Downregulation of BLM or FANCD2 in WT cells led to a clear decrease in the frequency of ICL traverse events, as expected (Fig 3G and H; Huang et al, 2013, 2019). Of note, the drop in traverse reactions caused by siBLM or siFANCD2 was similar to that caused by the loss of PrimPol alone, and downregulation of these factors in PRIMPOL KO cells did not further enhance the traverse phenotype (Fig 3G and H). A similar set of results was obtained after downregulation of FANCM translocase (Fig EV3E). Combined, these results suggest an epistatic relationship between PrimPol, BTR, FANCD2, and FANCM in the ICL traverse reaction.
PrimPol promotes DNA synthesis in response to ICL‐inducing agents
Given the participation of PrimPol in the ICL traverse reaction, we assessed its relevance in the global response to ICLs. To this aim, WT and PRIMPOL KO cells were treated with MMC or TMP‐UVA for 2h and kept in regular media for 6 days. Cell survival at the experimental endpoint was significantly lower in PRIMPOL KO cells, reflecting their higher sensitivity to either treatment. The drop in viability was not observed with UVA in the absence of TMP (Fig 4A). Hypersensitivity to MMC and TMP‐UVA was confirmed after PRIMPOL downregulation with shRNA, excluding the possibility of off‐target effects caused by CRISPR/Cas9 editing (Fig EV4A and B).
Figure 4. PrimPol ablation impairs cell proliferation and DNA synthesis in the presence of ICLs.
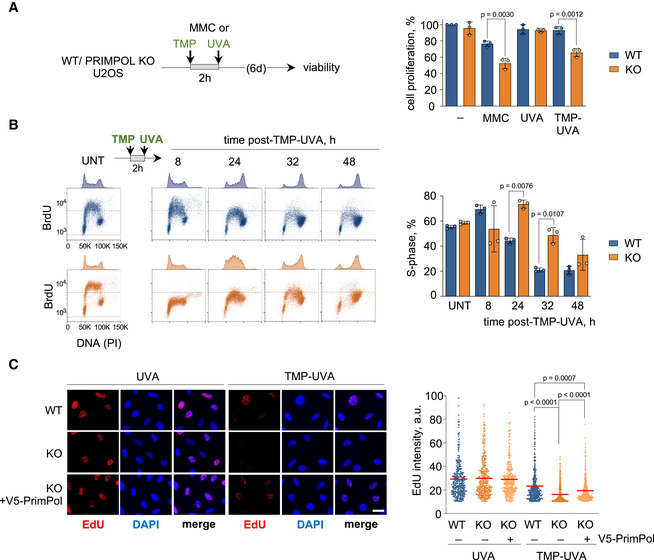
- CellTiter‐Glo viability assays in WT and PRIMPOL KO cells 6 days after treatment with 1 µg/ml MMC (2 h) or TMP‐UVA (2 µM TMP, 2 h followed by 5 s irradiation). Histograms represent the total number of viable cells (average and SD of three assays) at the end of the experiment, expressed as percentage of the number of untreated cells. Circle dots in each column represent the values of individual replicates. Statistical analysis was conducted with one‐way ANOVA and Bonferroni post‐test. P‐values of individual comparisons are indicated.
- Flow cytometry profiles of BrdU incorporation (y‐axis) vs DNA content (PI; x‐axis) in WT (blue) and PRIMPOL KO (orange) cells, either collected without treatment (UNT) or at the indicated times after exposure to TMP‐UVA (2 µM TMP for 2 h followed by 5 s irradiation). DNA content profiles are shown on top of the boxes (cell count). Dashed horizontal lines inside panels are included for comparative purposes between WT and KO BrdU incorporation levels. Histograms (right panel) represent the percentage of cells in S‐phase in each condition (average and SD of three assays). Circle dots in each column represent the values of individual replicates. Statistical analysis in all cases was conducted with one‐way ANOVA and Bonferroni post‐test. P‐values of individual comparisons are indicated.
- Representative confocal microscopy images of EdU staining in WT and PRIMPOL KO cells, 8 h after UVA (control) or TMP‐UVA treatment (2 µM TMP, 2 h followed by 5 s irradiation). Nuclear DNA is counterstained with DAPI. When indicated, V5‐PrimPol was reintroduced into KO cells. Scale bar, 25 µm. Dot plots indicate the distribution of nuclear EdU intensity, and the mean values are indicated by horizontal red lines. Data from the combination of three replicates (n ≥ 200 cells per condition). Statistical analysis was conducted with Kruskal–Wallis test and Dunn’s post‐test. P‐values of individual comparisons are indicated.
Source data are available online for this figure.
Figure EV4. (related to Fig 4). PRIMPOL downregulation or genetic ablation impairs cell proliferation and DNA synthesis in the presence of ICLs.
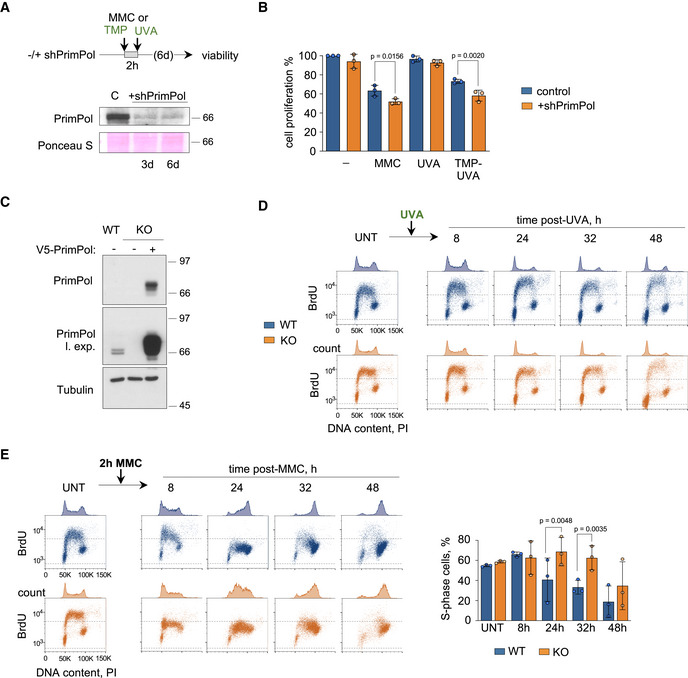
- Top, experimental design of the CellTiter‐Glo viability assays in U2OS‐shPRIMPOL cells after treatment with 1 µg/ml MMC (2 h) or TMP‐UVA (2 µM TMP, 2 h followed by 5 s irradiation). When indicated, PRIMPOL expression was downregulated by a specific shRNA. Bottom, immunoblot shows the levels of PrimPol in control cells or after the expression of shPRIMPOL for 3 or 6 days. Ponceau S staining is shown as loading control.
- Histograms represent the total number of viable cells (average and SD of three assays) at the end of the experiment, expressed as percentage of the number of untreated cells. Circle dots in each column represent the values of individual replicates. Statistical analysis was conducted with one‐way ANOVA and Bonferroni post‐test. P‐values are indicated.
- Immunoblots show levels of endogenous PrimPol and exogenous V5‐PrimPol expressed in KO cells. Tubulin is shown as loading control.
- Flow cytometry profiles of BrdU incorporation (y‐axis) vs. DNA content (PI; x‐axis) in WT (blue) and PRIMPOL KO (orange) cells, either collected without treatment (UNT) or at the indicated times after exposure to UVA (5 s irradiation). The DNA content profiles are shown on top of the boxes (cell count). Dashed horizontal lines inside panels are included for comparative purposes between WT and KO BrdU incorporation levels.
- Same as in D, in cells collected without treatment (UNT) or at the indicated times after exposure to 1 µg/ml MMC (2 h). Histograms (right panel) represent the percentage of cells in S‐phase in each condition (average and SD of three assays). Circle dots in each column represent the values of individual replicates. Statistical analysis in all cases was conducted with one‐way ANOVA and Bonferroni post‐test. P‐values are indicated.
Source data are available online for this figure.
The sensitivity of PRIMPOL KO cells is likely related to the dynamics of DNA replication following exposure to TMP‐UVA. While untreated WT and PRIMPOL KO cells displayed virtually identical DNA content and BrdU incorporation profiles (Fig 4B, left panels), administration of TMP‐UVA for 2 h slowed down cell cycle progression, particularly in PRIMPOL KO cells that required up to 48 h for full completion of S phase, as judged by DNA content and BrdU incorporation (Fig 4B, right panels). This effect was confirmed by confocal microscopy analysis of EdU incorporation, which was reduced in KO cells and partially recovered by reintroduction of exogenous V5‐PrimPol (Figs 4C and EV4C). As expected, UVA without TMP did not affect cell cycle progression (Fig EV4D). Similar results were obtained when WT and PRIMPOL KO cells were treated with MMC for 2h (Fig EV4E). These experiments indicate that PrimPol promotes continuous DNA synthesis during the initial response to DNA damage induced by ICL‐generating agents.
Delayed ICL repair and chromosomal instability in PRIMPOL KO cells
We hypothesized that the lower frequency of ICL traverse and delayed progression through S‐phase in PRIMPOL KO cells could affect the dynamics of ICL repair. Using the formation of nuclear foci of FANCD2 protein as a readout (Garcia‐Higuera et al, 2001; Liang et al, 2015), we observed FANCD2 foci formation as early as 8 h post‐TMP‐UVA (average 19 foci/cell) in WT cells, but not in KO cells. By 24 h, both WT and KO cells accumulated 35–40 FANCD2 foci/cell. By 48 h, the number of foci was significantly reduced in WT cells (av. 15 foci/cell) but remained higher in KO cells (av. 25 foci/cell; Fig 5A and B). The patterns of FANCD2 ubiquitylation and deubiquitylation followed a similar trend (Fig EV5A) and were also observed after MMC treatment (Fig EV5B–D). Furthermore, karyotypic analyses in metaphase spreads revealed the accumulation of chromosomal breaks, gaps, and fusions preferentially in PRIMPOL KO cells treated with TMP‐UVA (Fig 5C and D), which could result from cells having entered mitosis with unrepaired ICLs (Akkari et al, 2000; Mutreja et al, 2018). Combined, these results indicate a delayed initiation and apparently incomplete ICL repair process in the absence of PrimPol protein.
Figure 5. PrimPol deficiency leads to ICL repair defects.
- Confocal microscopy images of FANCD2 immunofluorescence staining in control (UVA) or TMP‐UVA‐treated (2 µM TMP, 2 h followed by 5 s irradiation) WT and PRIMPOL KO cells. Nuclear DNA is counterstained with DAPI. Scale bar, 25 µm.
- Dot plot (left panel) shows the distribution and mean (horizontal red line) of FANCD2 foci number per cell in each condition. Data pooled from three replicates are represented (n ≥ 100 cells per condition). Statistical analysis was conducted with Kruskal–Wallis test and Dunn's post‐test. P‐values of individual comparisons are indicated. Histograms (right panel) show the average foci number and SD of three assays in each case. Circle dots in each column represent the values of individual replicates. Statistical analysis was conducted with one‐way ANOVA followed by Bonferroni post‐test. P‐values of individual comparisons are indicated.
- Metaphase spreads stained with Leishman, obtained from control (‐) or TMP‐UVA‐treated cells (2 µM TMP, 2 h followed by 5 s irradiation). Scale bar, 10 µm. Representative images of chromosomal breaks and fusions are shown at higher magnification.
- Histograms show the number of chromosomal aberrations per metaphase (average and SEM) in each condition. Data taken from three replicates are combined (50 metaphases per replicate). Statistical analysis was conducted with one‐way ANOVA followed by Bonferroni post‐test. P‐values of individual comparisons are indicated.
Source data are available online for this figure.
Figure EV5. (related to Fig 5). Delayed clearance of FANCD2 mono‐ubiquitylation in PRIMPOL KO cells.
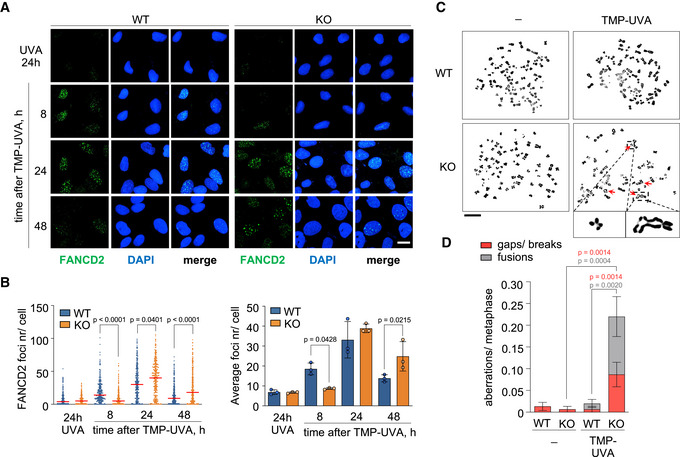
- Immunoblots showing the ubiquitylation of FANCD2 and the presence of PrimPol in WT and PRIMPOL KO cells at the indicated time points after treatment with TMP‐UVA (2 µM TMP for 2 h followed by 10 s irradiation). MEK2 and Ponceau S are shown as loading controls. To evaluate FANCD2 monoubiquitylation, the intensity of the upper and lower bands was calculated with ImageJ. The intensity of the top band was divided by the sum of upper and lower bands. Histograms (right panel) represent the percentage of FANCD2 mono‐ubiquitylation in each condition (average and SD of two replicates). Circle dots in each column represent the values of individual replicates.
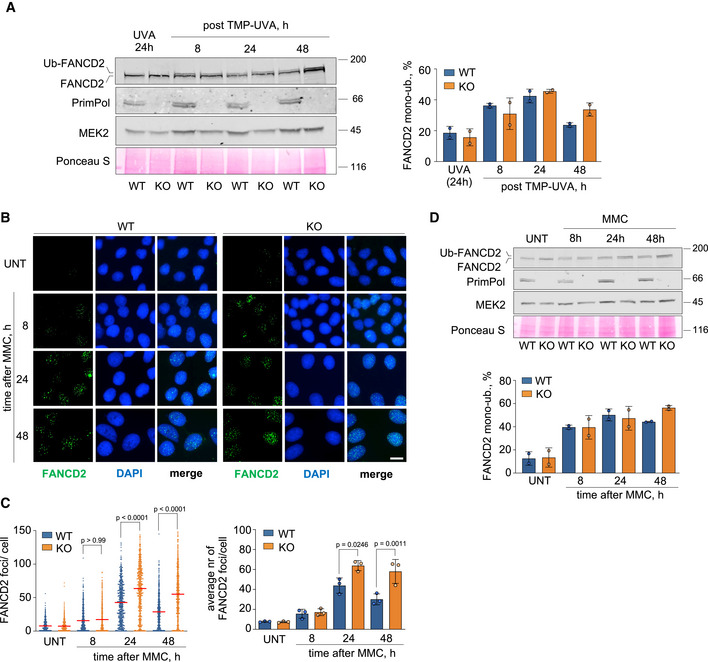
- Confocal IF microscopy images of FANCD2 staining at the indicated times in control or MMC‐treated (0.5 µg/ml; 2 h) WT and PRIMPOL KO cells. Nuclear DNA is counterstained with DAPI. Scale bar, 25 µm.
- Dot plot (left panel) shows the distribution and mean (horizontal red line) of FANCD2 foci number per cell in each condition. The pool of three replicates is represented (n ≥ 100 cells per condition). Statistical analysis was conducted with Kruskal–Wallis test and Dunn’s post‐test. P‐values of the indicated comparisons are shown. Histograms (right panel) show the average foci number and SD of three assays in each case. Circle dots in each column represent the values of individual replicates. Statistical analysis was conducted with one‐way ANOVA followed by Bonferroni post‐test. P‐values are indicated.
- Immunoblots showing the levels and ubiquitylation of FANCD2 and the presence of PrimPol in WT and PRIMPOL KO cells at the indicated time points after treatment with 0,5 µg/ml MMC (2 h). MEK2 and Ponceau S are shown as loading controls. Histograms (bottom panel) represent the percentage of FANCD2 mono‐ubiquitylation in each condition (average and SD of two replicates), calculated as in A. Circle dots in each column represent the values of individual replicates.
Source data are available online for this figure.
Hypersensitivity of PRIMPOL KO mice to MMC
To test the relevance of PrimPol function in response to ICLs in vivo, WT and PRIMPOL KO mice were treated with MMC, which rapidly depletes hematopoietic progenitor cells in the bone marrow (BM). MMC‐induced BM failure is exacerbated in strains deficient for FAN1, MUS81, SNM1, and FAAP20, all of which are required for ICL repair (Dronkert et al, 2000; McPherson et al, 2004; Dendouga et al, 2005; Zhang et al, 2015b; Thongthip et al, 2016). A dose of 7.5 mg MMC/kg of body weight was innocuous for WT mice and lethal only for a small percentage (20%) of KO mice. At 10 mg/kg MMC, median survival was 13d for WT and 10d for KO mice, and at 15 mg/kg, median survival of KO mice was 4 days shorter than WT (7 vs 11d), reaching statistical significance (Fig 6A).
Figure 6. PRIMPOL KO mice are hypersensitive to MMC.
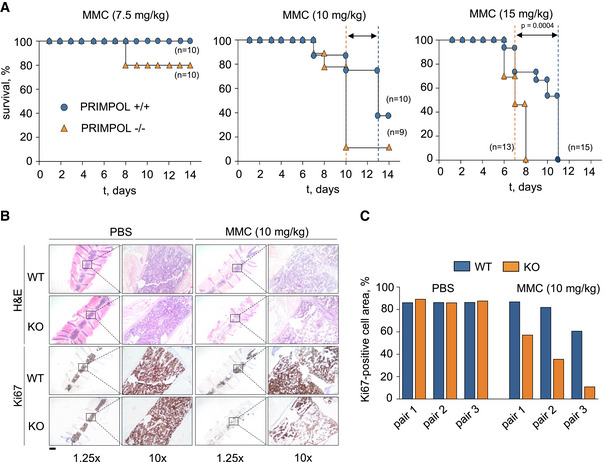
- Kaplan–Meier survival curves of WT and PRIMPOL KO mice after the administration of a single dose of MMC (7.5, 10, or 15 mg/kg). Dashed vertical lines represent median survival values for WT (blue) and KO (orange) mice. Statistical analysis was conducted with Log‐rank (Mantel–Cox) test; P‐value for the highest dose of MMC (15 mg/kg) is indicated.
- Hematoxylin–eosin (H&E) and Ki‐67 immunohistochemistry (IHC) stainings of bone marrow sections derived from WT and PRIMPOL KO mice following the administration of 10 mg/kg MMC. Control mice (PBS) were sacrificed when MMC‐injected mice reached the humane endpoint. Scale bar, 2.5 mm. In each image, a relevant area is shown at higher magnification (10×).
- Histograms show Ki‐67‐positive cellular area in the conditions shown in (B). Results are shown with three pairs of age‐ and gender‐matched WT and KO littermates. When combined, the differences observed between MMC‐treated WT and KO mice were statistically significant (two‐way ANOVA and Bonferroni post‐test, P‐value 0.0044).
Source data are available online for this figure.
To compare the proliferation status of BM cells, three pairs of age‐ and gender‐matched WT and KO mice were treated with 10 mg/kg MMC. WT individuals were euthanized when their paired KO mice had reached the humane endpoint, and their BMs were analyzed in parallel (Fig 6B). While the BM of untreated WT and KO mice displayed similar cellularity and proliferation rate, after exposure to MMC the percentage of Ki67‐positive BM cells was reduced in KO mice compared to WT (Fig 6C). This result indicates a protective function of PrimPol in response to DNA crosslinking agents in vivo.
NEIL3 downregulation further sensitizes PRIMPOL‐deficient cells to ICLs
In addition to the FA pathway, some ICLs may be directly processed by a separate pathway involving Neil3 glycosylase (Semlow et al, 2016; Li et al, 2020). As shown above, PRIMPOL KO cells are sensitive to an acute (2 h) treatment with TMP‐UVA (Fig 4A and B). Hypersensitivity was confirmed with different concentrations of TMP and it was further compromised by NEIL3 downregulation (Fig EV6A–C), suggesting that PrimPol and Neil3 operate in complementary pathways. On the other hand, both WT and KO cells displayed the same sensitivity to FANCA downregulation (Fig EV6D), suggesting that PrimPol operates in the same ICL repair pathway as FANCA.
Figure EV6. PRIMPOL and NEIL3 are not epistatic for ICL repair.
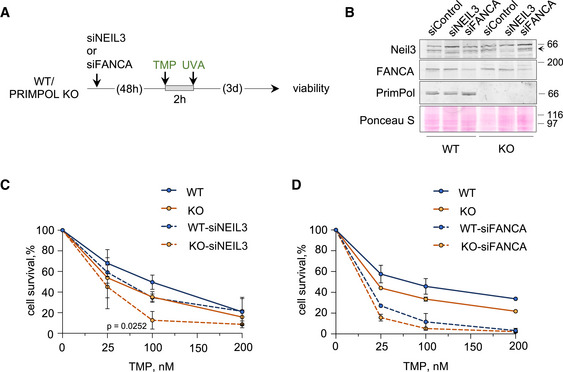
-
AExperimental design of CellTiter‐Glo viability assays in WT and PRIMPOL KO cells 3 days after treatment with different doses of TMP for 2 h followed by irradiation with UVA (90 s).
-
BImmunoblots showing the levels of Neil3, FANCA, and PrimPol proteins in WT and KO cells after siRNA‐mediated downregulation of NEIL3 or FANCA. The arrow indicates the band corresponding to Neil3 protein. Ponceau S is shown as loading control.
-
C, DCell survival assays with WT and PRIMPOL KO cells following NEIL3 (C) or FANCA (D) downregulation with siRNA after a 2 h exposure to increasing amounts of TMP (followed by UVA irradiation, 90 s). The control curves with WT and PRIMPOL KO cells without Neil3 or FANCA downregulation are the same as in Fig 7G. Each point represents the percentage of surviving cells (average and SD of three [NEIL3] or two [FANCA] assays). Statistical analysis was conducted with two‐way ANOVA followed by Bonferroni post‐test. The P‐value of PRIMPOL KO vs KO‐siNEIL3 comparison is indicated.
Source data are available online for this figure.
Activation of extra origins alleviates the loss of PrimPol
Because the ICL traverse reaction is impaired, PRIMPOL KO cells may also become more dependent on the fork convergence mechanism to trigger ICL repair. The probability of a second fork reaching the ICL is increased by the activation of dormant origins, whose availability depends on the cellular concentration of MCM proteins (Ge et al, 2007; Ibarra et al, 2008). To identify possible synthetic effects between the loss of PrimPol and the defective activation of dormant origins, MCM3 expression was downregulated with RNAi (Fig 7A). MCM3 downregulation limited the activation of backup origins, as determined by the abundance of origin tracks relative to other replicative structures such as forks and termination events (Fig 7B and C). Even in an unchallenged S phase, PRIMPOL KO cells display a slightly higher frequency of origin activation, as previously reported (Fig 7C, columns 1 and 5; Rodriguez‐Acebes et al, 2018). MMC increased origin activity in both WT and KO cells, reflecting the activation of backup, dormant origins (Fig 7C, columns 1–2 and 5–6). As expected, MCM3 downregulation did not affect origin activation in unchallenged conditions (Fig 7C, compare columns 1 with 3 and 5 with 7), but completely prevented the activation of additional origins in both WT and KO cells upon MMC treatment (Fig 7C, compare columns 1–2 with 3–4, and 5–6 with 7–8). The loss of dormant origins reduced the frequency of convergence events and increased the frequency of stalled forks in KO cells, as determined by ICL‐localized DNA fiber experiments (Fig 7D). MCM3 downregulation enhanced the phenotype of persistent FANCD2 foci 48 h after TMP‐UVA in PRIMPOL KO cells (Fig 7E and F) and increased their sensitivity to MMC and TMP‐UVA to a higher extent than WT cells (Fig 7G), reflecting their dependency on backup origins to establish new replication forks and maximize fork convergence at ICLs.
Figure 7. Loss of backup origins exacerbates the effect of PrimPol loss in ICL repair.
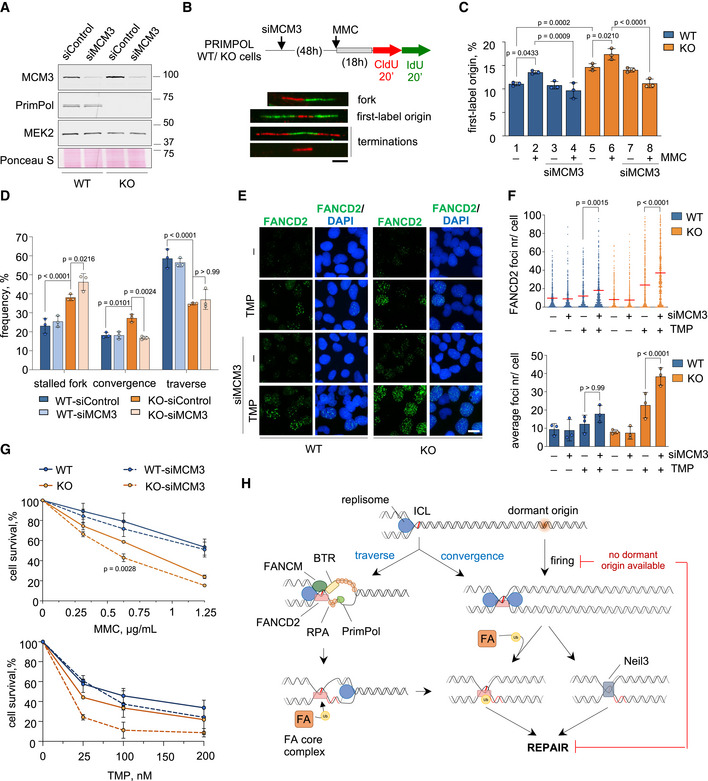
- Immunoblots showing the levels of MCM3 and PrimPol proteins in PRIMPOL WT and KO cells upon MCM3 downregulation. MEK2 and Ponceau S are shown as loading controls.
- Schematic of experimental design and examples of replicative structures (fork/ first‐label origin/ terminations) in stretched DNA fibers. Scale bar, 10 µm.
- Percentage of first‐label origins in each condition (average and SD of three assays). Circle dots in each column represent the values of individual replicates. Statistical analysis: one‐way ANOVA followed by Bonferroni post‐test. P‐values are indicated.
- Histograms show the percentage of the different replication patterns observed in each experimental condition (average and SD of three assays). Circle dots in each column represent the values of individual replicates. Statistical analysis: two‐way ANOVA followed by Bonferroni post‐test. P‐values of the indicated comparisons are shown.
- IF detection of FANCD2 foci in WT and PRIMPOL KO cells 48 h after TMP‐UVA (2 µM TMP, 2 h followed by 5 s irradiation) with or without MCM3 downregulation. Nuclear DNA is counterstained with DAPI. Scale bar, 25 µm.
- Top, dot plot shows the distribution and mean value (horizontal red line) of the number of FANCD2 foci per cell in each condition. The pool of three replicates is represented (n ≥ 100 cells per condition). Statistical analysis was conducted with Kruskal–Wallis test and Dunn’s post‐test. P‐values are indicated. Bottom, histograms show the average foci number and SD of three assays. Circle dots in each column represent the values of individual replicates. Statistical analysis was conducted with one‐way ANOVA followed by Bonferroni post‐test. P‐values of the indicated comparisons are shown.
- Cell survival assays of WT and PRIMPOL KO cells with or without MCM3 downregulation in the presence of increasing amounts of MMC or TMP (followed by 90 s UVA irradiation) for 72 h. The same control curves without MCM3 downregulation are also shown as part of Figure EV6D. Each point in the curve represents the percentage of surviving cells (average and SD of three assays for MMC and two assays for TMP). Statistical analysis was conducted with two‐way ANOVA followed by Bonferroni post‐test. P‐values of comparisons between KO and KO‐siMCM3 are indicated.
- Model for the participation of PrimPol in the ICL traverse reaction (left branch), and the alternative fork convergence mechanism (right branch), both of which generate a suitable template for FA‐mediated ICL repair. In the absence of PrimPol, ICL traverse is much less efficient and cells rely on dormant origin activation to promote fork convergence at the ICL. When dormant origins are not available, a fraction of the ICLs remain unrepaired, generating chromosomal instability. See text for details.
Source data are available online for this figure.
Discussion
Repriming of DNA synthesis downstream of lesions in the template is now recognized as a prominent mechanism of DNA damage tolerance (DDT), next to those involving translesion synthesis by specialized DNA polymerases or the temporary invasion of the complementary strand (template switch). DDT pathways allow the passage of replication forks through damaged DNA, avoiding interruptions in DNA synthesis that could increase genome instability while leaving the lesions to be repaired post‐replicatively (reviewed by Branzei & Szakal, 2016; Muñoz & Méndez, 2017; Vaisman & Woodgate, 2017; Pilzecker et al, 2019).
Interstrand crosslinks are among the most cytotoxic DNA lesions and, at least in theory, represent an absolute roadblock for both replication and transcription. This fits with the model in which ICL repair is initiated by the convergence of two forks arriving from opposite sides at the lesion (Zhang et al, 2015a). However, at least in avian and mammalian cells, the first fork arriving at the lesion may “traverse” the ICL (Huang et al, 2013, 2019; Ling et al, 2016; Mutreja et al, 2018). Both scenarios (fork convergence and traverse) result in similar X‐shaped replicated DNA structures that can be processed for repair by the FA pathway. The choice between traverse or convergence likely depends on two factors: (i) the distance between adjacent origins, which is proportional to the time needed for a second fork to reach the ICL after the first one has been stalled; (ii) the availability of a primase to reinitiate DNA synthesis downstream of the lesion (see model in Fig 7H).
At the mechanistic level, ICLs are initially recognized by the FANCM translocase and the BTR complex (Deans & West, 2009; Hoadley et al, 2012; Huang et al, 2013; Ling et al, 2016). Within the BTR, BLM displays robust helicase activity on dsDNA templates (Xue et al, 2019) and is likely capable of generating a stretch of ssDNA downstream of the ICL, creating an entry point for RPA and PrimPol. FANCM also interacts with replisome components PCNA (Rohleder et al, 2016) and MCM (Huang et al, 2019). The FANCM‐MCM interaction requires ATR function and is mediated by FANCD2. ATR and FANCD2 trigger the release of the GINS component of CMG, presumably to open the MCM hexameric ring and facilitate its passage through the ICL (Huang et al, 2019). This notion is also supported by the ability of CMG helicase to switch from ssDNA to dsDNA and back when a replication fork is stalled in vitro (Wasserman et al, 2019). Of note, the ability of helicases to operate across an ICL may have an interesting antecedent in bacterial DnaB helicase/translocase (Bastia et al, 2008).
Besides the relocation of the replisome, a new primer needs to be synthesized downstream of the ICL to provide a starting point for DNA polymerase ε in the leading strand. We have here identified PrimPol as the main primase involved in this step. PrimPol is rapidly localized to ICL‐containing chromatin upon exposure to DNA crosslinking agents MMC and TMP‐UVA. The recruitment of PrimPol to the vicinity of ICLs is mediated by RPA, which performs a similar role at other polymerase‐stalling lesions (Wan et al, 2013; Guilliam et al, 2015, 2017). PrimPol recruitment to ICLs was not affected by downregulation of BTR, FANCM, or FANDC2, and vice versa. It is therefore possible that the interaction between PrimPol and BTR identified by mass spectrometry is mediated by RPA, known to interact with BTR (Wu et al, 2018). The fact that the PrimPol‐BTR interaction is reduced during the response to MMC, as also reported for the RPA‐BLM interaction (Wu et al, 2018), suggests that PrimPol is released from its interacting factors to synthesize a new primer downstream of the ICL.
The genetic ablation of PRIMPOL caused a striking reduction in the frequency of traverse reactions but did not prevent them completely. Recently, an alternative pathway for ICL traverse involving the microcephaly associated DONSON protein instead of FANCM has been described (Zhang et al, 2020). We have shown here that PrimPol is epistatic within FANCM, so the DONSON pathway could be responsible in part for the remaining ICL traverse reactions in PRIMPOL KO cells. Regarding the actual repriming reaction, because none of the known TLS polymerases can synthesize DNA through ICLs (reviewed by Roy & Schärer, 2016), we favor the simple explanation that in the absence of PrimPol, the replicative Polα/Primase partially covers the DDT function. It is worth noting that biochemical reconstitution of a yeast replication fork stalled at an UV‐photoproduct shows that repriming mediated by Polα/Primase is possible but inefficient in the leading strand (Taylor & Yeeles, 2018). Fungi do not have PrimPol, but most other eukaryotes have maintained this second primase through evolution, probably because it provides an efficient alternative for repriming DDT reactions in the leading strand.
In PRIMPOL KO cells, the impairment of traverse reactions was partially compensated by a higher frequency of fork convergence events. PrimPol also mediates fork progression across endogenous DNA obstacles (Schiavone et al, 2016; Šviković et al, 2019), and its downregulation triggers the activation of backup origins (Mourón et al, 2013; Rodriguez‐Acebes et al, 2018). In the context of ICL repair, the establishment of new forks from these origins increases the chance of two forks converging at the crosslink. Consistent with this notion, the downregulation of MCM proteins to prevent dormant origin activation, reduced the frequency of fork convergence at ICLs and further sensitized PRIMPOL KO cells to ICL‐inducing agents (see model in Fig 7H).
The possibility of targeting DDT proteins as an adjuvant to standard chemotherapy is currently under investigation. For instance, the combination of cisplatin with a chemical inhibitor of Polζ suppressed tumor growth of human melanoma xenografts in mice (Wojtaszek et al, 2019). Our results hint at the possibility of targeting PrimPol to enhance the efficacy of crosslinking‐based chemotherapy. As shown in a recent study, PrimPol expression is upregulated in response to multiple doses of cisplatin as part of an adaptive response in BRCA1‐deficient cells that have already lost the ability to stabilize stalled forks (Quinet et al, 2020). Another recent report shows that the loss of HLTF, a mediator of fork reversal, allows cells treated with HU to undergo unrestrained replication that is partially mediated by PrimPol (Bai et al, 2020). Hence, targeting PrimPol to restrict the tolerance of chemotherapy‐induced ICLs could be clinically relevant in specific backgrounds such as BRCA1/2‐null tumors treated with cisplatin or MMC. Future studies will determine the effectiveness of these approaches, which could be complemented by preventing the activation of backup origins using Cdc7 kinase inhibitors (Rainey et al, 2017; Iwai et al, 2019).
Materials and Methods
Cell lines and manipulations
U2OS‐shPRIMPOL cell line has been described (Mourón et al, 2013). Cells were grown in DMEM supplemented with 10% FBS plus penicillin‐streptomycin and tested monthly for mycoplasma contamination. In U2OS‐shPRIMPOL cells, expression of the shRNA targeting PRIMPOL was induced with 1 μg/ml doxycycline (dox). U2OS PRIMPOL KO cells were generated as described (Ann et al, 2013) with minor modifications. U2OS cells were transfected with pSpCas9(BB)‐2A‐GFP plasmid (Addgene #48138) containing a short guide RNA targeting exon 7 in PRIMPOL gene, built with sgPRIMPOL_Fw (5’‐CACCGGAGGTGCTTCTGAAAAATG) and sgPRIMPOL_Rv (5’‐AAACCATTTTTCAGAAGCACCTCC) oligonucleotides. After 24h, transfected cells were seeded at low confluency to allow single‐clone colony formation. Isolated colonies were tested for PrimPol expression. To test putative PRIMPOL KO clones, the CRISPR/Cas9 target site was amplified with specific primers PRIMPOL_seq_Fw (5’‐GACCTTAAGATGCGGTGTGT) and PRIMPOL_seq_Rv (5’‐TGAACTCACTGGTTGCATCG), cloned into pJET1.2/blunt vector (ThermoFisher, K1232) and sequenced. To reintroduce PrimPol protein in WT or KO cells, pcDNA3.1/nV5 plasmids encoding WT, AxA, ∆Zn, CH (Mourón et al, 2013), or RBDm (generated for this study). PRIMPOL derivatives were transiently transfected using Lipofectamine 2000 (Invitrogen). PRIMPOL KO cells stably expressing WT or RBDm PrimPol were generated by transfection with pcDNA3.1/nV5 plasmids and selection with 400 µg/ml G418 (Sigma #A1720) for 14 days. Cell clones were isolated and tested for WT or RBDm PrimPol expression by IF with V5 antibody. To synchronize cells in S phase, cultures were kept in medium supplemented with 2.5 mM thymidine (Sigma #T9250) for 16 h and then transferred to regular medium for 8 h. The thymidine “block” was performed a second time, and cells were released in the absence or presence of 2 µg/ml MMC for 5 h. For siRNA‐mediated downregulation of gene expression, cells were transfected twice (with a 24 h gap) with the indicated 100 nM siRNA using Oligofectamine (Invitrogen). A list of the siRNA molecules used in the study is included in Table 1. Mitomycin C was obtained from Panreac (#A2190); trimethyl‐psoralen (TMP) was from Sigma (#T6137). UVA irradiation was performed in a BS‐02 irradiation chamber (Opsytec Dr. Gröbel, Ettlingen, Germany). Cell viability was measured with CellTiter‐Glo luminescent assay (Promega) in 96‐well plates.
Table 1.
siRNA molecules used in this study.
| siRNA | Source | Sequence/Reference |
|---|---|---|
| siRPA2 | Dharmacon | AACCUAGUUUCACAAUCUGUU |
| siFANCM | Dharmacon | ON‐TARGET plus, L‐021955‐00‐0010 |
| siBLM | Dharmacon | UGCAAAUCACAUCGCUGCU |
| siTOP3A | Dharmacon | GGACAAAUUUGUGGUUCUA |
| siRMI1 | Dharmacon | AGCCUUCACGAAUGUUGAU |
| siRMI2 | Dharmacon | UGUUGGAACUGUCGUUAAAUU |
| siFANCD2 | Dharmacon | UCAAGUGUAUCCAUGGCUGUU |
| siNEIL3 | Dharmacon | GGGUGGAUCAUGUUAUGGAUU |
| siFANCA | Dharmacon | GGACAUCACUGCCCACUUCUU |
| siMCM3 | Dharmacon | GCAUUGUCACUAAAUGUUCUCUAGU |
Flow cytometry analyses of DNA content and BrdU incorporation
10 μM 5‐bromo‐2’‐deoxyuridine (BrdU; Sigma #B9285) was added to the cell medium for 30 min before harvesting. Cells were fixed with ice‐cold 70% ethanol for 2 h and incubated with 2 M HCl for 20 min. BrdU was immunolabeled with FITC BrdU Flow Kit (BD Bioscience #559619). DNA was stained with 50 µg/ml propidium iodide (PI; Sigma #P4864). Cells were processed in a FACSCanto cytometer (BD Biosciences), and data were analyzed with FlowJo software (Tree Star Inc).
Confocal fluorescence microscopy
For 5‐ethynyl 2’‐deoxyuridine (EdU) detection, cells were grown on μCLEAR polylysine‐treated plates (Greiner Bio‐One) and pulse‐labeled with 10 µM EdU for 30 min. Labeled cells were fixed with 4% paraformaldehyde (PFA) for 10 min and permeabilized with 0.2% Triton X‐100 in PBS for 5 min on ice. EdU incorporation was visualized with Click‐it EdU Alexa Fluor 647 flow cytometry assay (Thermo Fisher). Nuclear DNA was stained with 100 µg/ml 4',6‐Diamidino‐2‐phenylindole (DAPI; Sigma #D9542). Images were acquired in an Opera High‐Content Screening System with an APO 20x, 0.7 numerical aperture (NA) objective and analyzed with Acapella software (PerkinElmer). For FANCD2 immunodetection, cells growing on μCLEAR polylysine‐treated plates were treated before fixation with 0.5% Triton X‐100 in CSK buffer (10 mM Pipes‐KOH pH 7.0, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2) to pre‐extract soluble proteins. Cells were fixed in 4% PFA for 10 min and incubated with blocking solution (3% bovine serum albumin (BSA), 0.05% Tween‐20 in PBS) for 30 min. Incubations with anti‐FANCD2 primary antibody and the corresponding secondary antibody (Table 2) were performed sequentially for 1h at RT. Cell nuclei were counterstained with DAPI as before. Images were acquired in a Leica‐TCS SP5‐MP confocal microscope with a HCX PL APO 40× 1.4 NA oil‐immersion objective and LAS AF (v. 2.5.1) software. Images were analyzed with Definiens software (PerkinElmer). To monitor protein recruitment to TMP‐UVA‐induced ICLs, U2OS cells were seeded in µ‐slide 8‐well plates (ibidi, Gräfelfing, Germany), transfected with siRNAs when indicated, incubated with 10 µM TMP for 2 h, and irradiated with a 360 nm UV laser (36% energy dose) in a Zeiss PALM microdissection microscope. 1h after irradiation, cells were incubated with 0.5% Triton X‐100 in CSK buffer, fixed with 4% PFA, and blocked with 3% BSA in PBS before IF immunodetection with the indicated antibodies. Protein recruitment to the DNA lesions was calculated as the percentage of irradiated cells with positive IF signal in the laser path.
Table 2.
Antibodies used in this study.
| Antibodies | Source | Reference | Application (Dilution) |
|---|---|---|---|
| anti‐PrimPol, polyclonal | Generated in house | Mourón et al, (2013) |
WB (1:1,000) IP (neat serum) |
| anti‐PrimPol, monoclonal | Generated in house | this work |
WB (neat tissue culture supernatant, TCSN) IF (neat TCSN) |
| anti‐CTFC | Millipore | 07‐729 | WB (1:1,000) |
| anti‐TUBULIN | Sigma | T6199 | WB (1:1,000) |
| anti‐H3 | Abcam | 1791 | WB (1:10,000) |
| anti‐MEK2 | BD Biosciences | 610236 | WB (1:1,000) |
| anti‐FANCD2 | Fanconi Anemia Research Fund (FARF) | B9699 | IF (1:100) |
| anti‐FANCD2 | Novus Biologicals | NB100‐182 |
WB (1:10,000) IF (1:300) |
| anti‐BLM | Bethyl Laboratories | A300‐110A | WB (1:1,000) |
| anti‐BLM | GeneTex | GTX25446 | WB (1:100) |
| anti‐RMI1 | Abcam | Ab70525 | WB (1:1,000) |
| anti‐RMI1 | Novus Biologicals | NB100‐1720 | WB (1:1,000) |
| anti‐RMI2 | Novus Biologicals | NBP1‐89962 | WB (1:1,000) |
| anti‐TOP3A | Proteintech | 14525‐1‐AP | WB (1:1,000) |
| anti‐RPA2 | Cell Signaling | 2208S | WB (1:1,000) |
| anti‐phospho‐RPA2 ser4/8 | Cell Signaling | 54762S | WB (1:1,000) |
| anti‐MCM3 | Méndez Laboratory | Ibarra et al, (2008) | WB (1:1,000) |
| anti‐V5 | Invitrogen | 46‐0705 |
WB (1:1,000) IF (1:500) |
| anti‐SMC1 | Losada’s laboratory | Kojic et al, (2018) | WB (1 μg/ml) |
| anti‐FANCA | Bethyl Laboratories | A301‐980A | WB (1:2,000) |
| anti‐FANCM | Bethyl Laboratories | A302‐637A | WB (1:4,000) |
| anti‐FANCM | Sigma | SAB1407805 | IF (1:100) |
| anti‐Neil3 | Proteintech | 11621‐1‐AP | WB (1:1,000) |
| anti‐γH2AX | Merk Millipore | 05‐636 | IF (1:300) |
| anti‐Ki67 | Cell Signaling | 12202 | IHC |
| anti‐CldU (rat monoclonal anti‐ BrdU) | Abcam | ab6326 | IF (1:100) |
| anti‐IdU (mouse monoclonal anti‐BrdU) | BD biosciences | 347580 | IF (1:100) |
| anti‐ssDNA | Millipore | MAB3034 | IF (1:300) |
| anti‐Dig | Abcam | ab76907 | IF (1:1,000) |
| anti‐rabbit IgG AF‐488 (chicken) | Invitrogen Molecular Probes | A‐21441 | IF (1:300) |
| anti‐rat IgG AF‐594 (goat) | Invitrogen Molecular Probes | A‐11007 | IF (1:300) |
| anti‐mouse IgG2a AF‐647 (goat) | Invitrogen Molecular Probes | A‐21241 | IF (1:300) |
| anti‐goat IgG AF‐647 (donkey) | Invitrogen Molecular Probes | A‐21447 | IF (1:1,500) |
To monitor ssDNA foci, cells were grown on μCLEAR polylysine‐treated plates (Greiner Bio‐One) and labeled with 10 µM BrdU for 48 h. 4 h before collection, 5 µM TMP was added to the media for 2 h and UVA irradiation was administered for 1 min. Cells were fixed (4% PFA/ 10 min/ RT), incubated in cold methanol (20 min/−20°C), blocked with 1% BSA in PBS, and stained with anti‐BrdU antibody without DNA denaturation with HCl, as described (Huang et al, 2010). Images were acquired in an Opera High‐Content Screening System with an APO 20×, 0.7 numerical aperture (NA) objective and analyzed with Acapella software (PerkinElmer).
Biochemical fractionation, immunoprecipitation, and immunoblots
Whole cell extracts were prepared by suspension of cells in Laemmli buffer (50 mM Tris–HCl pH 6.8, 10% glycerol, 3% SDS, 0.006 w/v bromophenol blue and 5% 2‐mercaptoethanol) followed by 30 s sonication in a Branson Digital Sonifier at 15% amplitude. Biochemical fractionation was performed as described (Mendez & Stillman, 2000). SDS–PAGE, protein transfer to nitrocellulose, and immunoblots were performed using standard protocols. Primary antibodies are listed in Table 2. LI‐COR secondary antibodies: donkey anti‐rabbit IgG (H + L) conjugated to IRDye 800CW; donkey anti‐mouse/rat IgG (H + L) conjugated to IRDye 680CW (LI‐COR, Bad Homburg, Germany); HRP‐labeled secondary antibodies for chemiluminescence detection were obtained from GE Healthcare (#NA931/4/5). For immunoprecipitation assays, either 5 × 106 (for IP‐immunoblot) or 20 × 106 cells (for mass spectrometry) were lysed in NP40 buffer (150 mM NaCl, 50 mM Tris pH 8, 1% NP40, 0.2 mM PMSF, 5 mM NaF, 5 mM NaVO4, 1 mM DTT) supplemented with protease inhibitors (Roche #11836153001). Anti‐PrimPol antibody (Table 2) was coupled to magnetic M‐270 Epoxy Dynabeads® (7.5 μg antibody/ mg beads; Life Technologies), and IPs were performed following manufacturer instructions. Cell lysates were incubated with 1.5 mg antibody‐coupled beads for 45 min at 4°C. Protein‐antibody‐bead complexes were washed 3x with lysis buffer and collected for immunoblot or mass spectrometry assays.
Mass spectrometry
PrimPol immunoprecipitates were eluted in 8 M urea, 100 mM Tris/HCl pH 8.0. Eluates were incubated with 15 mM Tris(2‐carboxyethyl)phosphine (TCEP), 45 mM chloroacetamide for 1 h at 25°C in the dark. Protein samples digested with 100 ng of endopeptidase LysC for 4 h/ 25°C and subsequently with 100 ng of trypsin (16 h/ 37°C). Samples were analyzed in LTQ Orbitrap Velos (Thermo Fisher Scientific) coupled to a nanoLC Ultra 1D+ system (Eksigent), 5 µl of each sample were loaded onto a reversed‐phase C18, 5 µm, 0.1 × 20 mm trapping column (NanoSeparations) and washed for 10 min at 2.5 µl/min with 0.1% FA. The peptides were eluted at a flow rate of 250 nl/min onto an analytical column packed with ReproSil‐Pur C18‐AQ beads, 2.4 μm, 75 μm × 50 cm, heated to 45°C. Solvent A was 4% ACN in 0.1% FA and Solvent B acetonitrile in 0.1% FA. Peptides were separated using a lineal gradient from 4% to 33% B in 100 min. Total run time, including washing and re‐equilibrating, was 120 min.
Data were analyzed with MaxQuant (Cox & Mann, 2008) v1.5.3.30 with Andromeda (Cox et al, 2011) as the search engine against a Homo sapiens database supplemented with the sequences of the contaminants most frequently detected in the proteomics laboratory (UniProtKB/Swiss‐Prot, 20,599 sequences). Minimal peptide length was set to 6 amino acids, and a maximum of two missed cleavages was allowed. Peptides and proteins were filtered at 1% false discovery rate (FDR). Student’s t‐test statistics were used for determining the significance of the intensity of each protein.
Single‐molecule analysis of DNA replication in stretched fibers
To measure frequency of origin activation, cells were pulse‐labeled sequentially with 50 μM 5‐Chloro‐2'‐deoxyuridine (CldU; 20 min) followed by 250 μM 5‐iodo‐2'‐deoxyuridine (IdU; 20 min). Labeled cells were harvested and resuspended in 0.2 M Tris pH 7.4, 50 mM EDTA, 0.5% SDS. For immunodetection of labeled tracks, fibers were incubated with anti‐CldU and anti‐IdU primary antibodies (1 h/ RT) and the corresponding secondary antibodies (30 min/RT) in a humidity chamber. Fiber integrity was assessed by staining with anti‐ssDNA. Microscopy images were obtained in a DM6000 B Leica unit equipped with an HCX PL APO 40×, 0.75 NA objective. A standard conversion rate of 1 μm = 2.59 kb was used (Jackson & Pombo, 1998). For origin firing assessment, at least 500 structures were evaluated in each condition and replicate.
For the visualization of ICLs in DNA fibers, we used a variation (Mutreja et al, 2018) of the original protocol (Huang et al, 2013). In brief, cells were incubated with 5 μM Dig‐TMP in phenol‐free, FBS‐free, Pen‐Strep‐free DMEM media for 1 h in the dark and irradiated with UV‐A (3 J/cm2). Fibers were incubated anti‐Dig antibody for 2 h and the corresponding secondary antibody for 1 h. For S1 nuclease experiments, cells were treated with Dig‐TMP‐UVA, labeled with CldU for 20 min, labeled with IdU for 60 min, and incubated with S1 nuclease as described (Quinet et al, 2020). In all cases, at least 100 tracks were measured in each condition.
Chromosome stability
Cells in culture were treated with 0.1 μg/ml colcemide for 4 h, harvested by trypsinization, incubated in 75 mM KCl (30 min/ 37°C), fixed in 3:1 methanol:acetic acid, and dropped onto slides to obtain chromosome spreads. Chromosomes were stained with Leishman stain (Sigma #L6254) and mounted with ProLong‐Gold (Invitrogen).
Sensitivity of mouse strains to MMC and histopathology analyses
Mice were housed at the CNIO Animal Facility in accordance with “Federation for Laboratory Animal Science Associations” (FELASA) guidelines. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) from Instituto de Salud Carlos III (Spain). PRIMPOL KO mice have been described (García‐Gómez et al, 2013; Mourón et al, 2013). For survival assays, cohorts of 9–15 mice of both sexes (8–14 weeks old) were injected with MMC at either 7.5, 10, or 15 mg/kg of body weight. Survival was monitored for 2 weeks. Mice were sacrificed if they reached the humane endpoint as defined in the ethical permit. For histopathology analyses, age‐ and gender‐matched pairs of mice from WT or KO genotype were injected with 10 mg/kg MMC. Mice tissues were fixed in 10% buffered formalin (Sigma) and embedded in paraffin using standard procedures. 3‐µm sections were stained with hematoxylin and eosin (H&E). Ki‐67 staining was performed in an automatic Ventana Discovery XT platform (Roche). Tissue slides were digitalized in a Mirax scan or Axio Scan.Z1 (Carl Zeiss). Staining was analyzed using AxioVision digital image software (Carl Zeiss). Areas of positive staining were normalized to the total cellular area in the tissue.
Author contributions
DG‐A, EB‐R, PU‐C, KM, SM, and SL performed the experiments. FG and JMu carried out mass spectrometry analyses. JMé designed and supervised the study, with contributions from LB and ML. DG‐A and JMé wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We are grateful to all members of the CNIO DNA Replication and Chromosome Dynamics Groups for helpful discussions. Thanks to Kurt Jacobs and Jana Krietsch (Lopes lab) for their comments on the manuscript, Michael M. Seidman (NIH, Baltimore, USA) for discussions on the ICL traverse mechanism, Annabel Quinet (Washington U., St Louis, USA) for technical advice with S1 nuclease, and Eros Lazzerini‐Denchi (NIH, Bethesda, USA) for kindly providing BLM KO cells. We are grateful to Diego Megías and Sandra Rodríguez, Heads of the CNIO Confocal Microscopy and Cytogenetics Units, respectively, and laboratory technician Sara San José for excellent assistance. Anti‐FANCD2 antibody was kindly provided by the Fanconi Anemia Research Fund (FARF, http://www.fanconi.org). Research was supported by the Spanish Ministry of Science, Innovation and Universities (grants BFU2016‐80402R and BFU2019‐106707‐RB to JM and PGC2018.093576‐B‐C21 to LB, co‐sponsored by EU ERDF funds), the Swiss National Science Foundation (grant 31003A_169959 to ML), and an ERC Consolidator Grant (617102 to ML).
The EMBO Journal (2021) 40: e106355.
Data availability
Proteomics data have been uploaded at the ProteomeXchange Consortium. https://www.ebi.ac.uk/pride/archive/projects/PXD017951 (identifier PXD017951).
References
- Akkari YMN, Bateman RL, Reifsteck CA, Olson SB, Grompe M (2000) DNA replication is required to elicit cellular responses to psoralen‐induced DNA interstrand cross‐links. Mol Cell Biol 20: 8283–8289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amunugama R, Willcox S, Wu RA, Abdullah UB, El‐Sagheer AH, Brown T, McHugh PJ, Griffith JD, Walter JC (2018) Replication fork reversal during DNA interstrand crosslink repair requires CMG unloading. Cell Rep 23: 3419–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ann FR, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai G, Kermi C, Stoy H, Schiltz CJ, Bacal J, Zaino AM, Hadden MK, Eichman BF, Lopes M, Cimprich KA (2020) HLTF Promotes fork reversal, limiting replication stress resistance and preventing multiple mechanisms of unrestrained DNA synthesis. Mol Cell 78: 1237–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastia D, Zzaman S, Krings G, Saxena M, Peng X, Greenberg MM (2008) Replication termination mechanism as revealed by Tus‐mediated polar arrest of a sliding helicase. Proc Natl Acad Sci USA 105: 12831–12836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi J, Rudd SG, Jozwiakowski SK, Bailey LJ, Soura V, Taylor E, Stevanovic I, Green AJ, Stracker TH, Lindsay HD et al (2013) Primpol bypasses UV photoprodcts during eukaryotic chomosomal DNA replication. Mol Cell 52: 566–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Szakal B (2016) DNA damage tolerance by recombination: molecular pathways and DNA structures. DNA Repair 44: 68–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Sarangi P, D’Andrea AD (2016) The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol 17: 337–349 [DOI] [PubMed] [Google Scholar]
- Ciccia A, Ling C, Coulthard R, Yan Z, Xue Y, Meetei AR, Laghmani EH, Joenje H, McDonald N, de Winter JP et al (2007) Identification of FAAP24, a fanconi anemia core complex protein that interacts with FANCM. Mol Cell 25: 331–343 [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805 [DOI] [PubMed] [Google Scholar]
- Deans AJ, West SC (2009) FANCM connects the genome instability disorders bloom’s syndrome and Fanconi anemia. Mol Cell 36: 943–953 [DOI] [PubMed] [Google Scholar]
- Deans AJ, West SC (2011) DNA interstrand crosslink repair and cancer. Nat Rev Cancer 11: 467–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dendouga N, Gao H, Moechars D, Janicot M, Vialard J, McGowan CH (2005) Disruption of murine Mus81 increases genomic instability and DNA damage sensitivity but does not promote tumorigenesis. Mol Cell Biol 25: 7569–7579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dronkert MLG, de Wit J, Boeve M, Vasconcelos ML, van Steeg H, Tan TLR, Hoeijmakers JHJ, Kanaar R (2000) Disruption of mouse SNM1 causes increased sensitivity to the DNA interstrand cross‐linking agent Mitomycin C. Mol Cell Biol 20: 4553–4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Gómez S, Reyes A, Martínez‐Jiménez MI, Chocrón ES, Mourón S, Terrados G, Powell C, Salido E, Méndez J, Holt IJ et al (2013) PrimPol, an archaic primase/polymerase operating in human cells. Mol Cell 52: 541–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D’Andrea AD (2001) Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 7: 249–262 [DOI] [PubMed] [Google Scholar]
- Ge XQ, Jackson DA, Blow JJ (2007) Dormant origins licensed by excess Mcm2‐7 are required for human cells to survive replicative stress. Genes Dev 21: 3331–3341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliam TA, Brissett NC, Ehlinger A, Keen BA, Kolesar P, Taylor EM, Bailey LJ, Lindsay HD, Chazin WJ, Doherty AJ (2017) Molecular basis for PrimPol recruitment to replication forks by RPA. Nat Commun 8: 15222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliam TA, Jozwiakowski SK, Ehlinger A, Barnes RP, Rudd SG, Bailey LJ, Skehel JM, Eckert KA, Chazin WJ, Doherty AJ (2015) Human PrimPol is a highly error‐prone polymerase regulated by single‐stranded DNA binding proteins. Nucleic Acids Res 43: 1056–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley KA, Xue Y, Ling C, Takata M, Wang W, Keck JL (2012) Defining the molecular interface that connects the Fanconi anemia protein FANCM to the Bloom syndrome dissolvasome. Proc Natl Acad Sci USA 109: 4437–4442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Liu S, Bellani MA, Thazhathveetil A, Ling C, deWinter JP, Wang Y, Wang W, Seidman MM (2013) The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol Cell 52: 434–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Zhang J, Bellani MA, Pokharel D, Gichimu J, James RC, Gali H, Ling C, Yan Z, Xu D et al (2019) Remodeling of interstrand crosslink proximal article remodeling of interstrand crosslink proximal replisomes is dependent on ATR, FANCM, and FANCD2. Cell Rep 27: 1794–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Kim JM, Shiotani B, Yang K, Zou L, D'Andrea AD (2010) The FANCM/FAAP24 complex is required for the DNA interstrand crosslink‐induced checkpoint response. Mol Cell 39: 259–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra A, Schwob E, Méndez J (2008) Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc Natl Acad Sci USA 105: 8956–8961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai K, Nambu T, Dairiki R, Ohori M, Yu J, Burke K, Gotou M, Yamamoto Y, Ebara S, Shibata S et al (2019) Molecular mechanism and potential target indication of TAK‐931, a novel CDC7‐selective inhibitor. Sci Adv 5: eaav3660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DA, Pombo A (1998) Replicon clusters are stable units of chromosome structure: Evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol 140: 1285–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P, Räschle M, Smogorzewska A, Enoiu M, Ho TV, Schärer OD, Elledge SJ, Walter JC (2009) The fanconi anemia pathway promotes replication‐dependent DNA interstrand cross‐link repair. Science 326: 1698–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojic A, Cuadrado A, De Koninck M, Giménez‐Llorente D, Rodríguez‐ Corsino M, Gómez‐López G, Le Dily F, Marti‐Renom MA, Losada A (2018) Distinct roles of cohesin‐SA1 and cohesin‐SA2 in 3D chromosome organization. Nat Struct Mol Biol 25: 496–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Wang J, Wallace SS, Chen J, Zhou J, D'Andrea AD (2020) Cooperation of the NEIL3 and Fanconi anemia/BRCA pathways in interstrand crosslink repair. Nucleic Acids Res 48: 3014–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang CC, Zhan B, Yoshikawa Y, Haas W, Gygi SP, Cohn MA (2015) UHRF1 Is a sensor for DNA interstrand crosslinks and recruits FANCD2 to initiate the Fanconi Anemia pathway. Cell Rep 10: 1947–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling C, Huang J, Yan Z, Li Y, Ohzeki M, Ishiai M, Xu D, Takata M, Seidman M, Wang W (2016) Bloom syndrome complex promotes FANCM recruitment to stalled replication forks and facilitates both repair and traverse of DNA interstrand crosslinks. Cell Discov. 2: 16047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loe TK, Li JSZ, Zhang Y, Azeroglu B, Boddy MN, Lazzerini‐Denchi E (2020) Telomere length heterogeneity in ALT cells is maintained by PML‐dependent localization of the BTR complex to telomeres. Genes Dev 34: 650–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Martinez D, Liang C‐C, Cohn MA (2016) Cellular response to DNA interstrand crosslinks: the Fanconi anemia pathway. Cell Mol Life Sci 73: 3097–3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Guo X, Meng X, Liu J, Allen C, Wray J, Nickoloff JA, Shen Z (2005) The BRCA2‐interacting protein BCCIP functions in RAD51 and BRCA2 focus formation and homologous recombinational repair. Mol Cell Biol 25: 1949–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manthei KA, Keck JL (2013) The BLM dissolvasome in DNA replication and repair. Cell Mol Life Sci 70: 4067–4084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐Jiménez MI, Lahera A, Blanco L (2017) Human PrimPol activity is enhanced by RPA. Sci Rep 7: 783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson JP, Lemmers B, Chahwan R, Pamidi A, Migon E, Matysiak‐Zablocki E, Moynahan ME, Essers J, Hanada K, Poonepalli A et al (2004) Involvement of mammalian Mus81 in genome integrity and tumor suppression. Science 304: 1822–1826 [DOI] [PubMed] [Google Scholar]
- Mendez J, Stillman B (2000) Chromatin association of human origin recognition complex, Cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol 20: 8602–8612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourón S, Rodriguez‐Acebes S, Martínez‐Jiménez MI, García‐Gómez S, Chocrón S, Blanco L, Méndez J (2013) Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol 20: 1383–1389 [DOI] [PubMed] [Google Scholar]
- Muñoz S, Méndez J (2017) DNA replication stress: from molecular mechanisms to human disease. Chromosoma 126: 1–15 [DOI] [PubMed] [Google Scholar]
- Mutreja K, Krietsch J, Hess J, Ursich S, Berti M, Roessler FK, Zellweger R, Patra M, Gasser G, Lopes M (2018) ATR‐mediated global fork slowing and reversal assist fork traverse and prevent chromosomal breakage at DNA interstrand cross‐links. Cell Rep 24: 2629–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piberger AL, Bowry A, Kelly R, Walker AK, González‐Acosta D, Bailey LJ, Doherty AJ, Méndez J, Morris JR, Bryant HE et al (2020) PrimPol‐dependent single‐stranded gap formation mediates homologous recombination at bulky DNA adducts. Nat Commun 11: 5863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilzecker B, Buoninfante OA, Jacobs H (2019) DNA damage tolerance in stem cells, ageing, mutagenesis, disease and cancer therapy. Nucleic Acids Res 47: 7163–7181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Tirman S, Jackson J, Šviković S, Lemaçon D, Carvajal‐Maldonado D, González‐Acosta D, Vessoni AT, Cybulla E, Wood M et al (2020) PRIMPOL‐mediated adaptive response suppresses replication fork reversal in BRCA‐deficient cells. Mol Cell 77: 461–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey MD, Quachthithu H, Gaboriau D, Santocanale C (2017) DNA replication dynamics and cellular responses to ATP competitive CDC7 kinase inhibitors. ACS Chem Biol 12: 1893–1902 [DOI] [PubMed] [Google Scholar]
- Räschle M, Knipsheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Schärer OD, Walter JC (2008) Mechanism of replication‐coupled DNA interstrand crosslink repair. Cell 134: 969–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Acebes S, Mourón S, Méndez J (2018) Uncoupling fork speed and origin activity to identify the primary cause of replicative stress phenotypes. J Biol Chem 293: 12855–12861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohleder F, Huang J, Xue Y, Kuper J, Round A, Seidman M, Wang W, Kisker C (2016) FANCM interacts with PCNA to promote replication traverse of DNA interstrand crosslinks. Nucleic Acids Res 44: 3219–3232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy U, Schärer OD (2016) Involvement of translesion synthesis DNA polymerases in DNA interstrand crosslink repair. DNA Repair (Amst) 44: 33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavone D, Jozwiakowski SK, Romanello M, Guilbaud G, Guilliam TA, Bailey LJ, Sale JE, Doherty AJ (2016) PrimPol is required for replicative tolerance of G Quadruplexes in vertebrate cells. Mol Cell 61: 161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semlow DR, Zhang J, Budzowska M, Drohat AC, Walter JC (2016) Replication‐dependent unhooking of DNA interstrand cross‐links by the NEIL3 glycosylase. Cell 167: 498–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šviković S, Crisp A, Tan‐Wong SM, Guilliam TA, Doherty AJ, Proudfoot NJ, Guilbaud G, Sale JE (2019) R‐loop formation during S phase is restricted by PrimPol‐mediated repriming. EMBO J 38: e99793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay LS, Krishnan V, Sankar H, Chong YL, Chuang LSH, Tan TZ, Kolinjivadi AM, Kappei D, Ito Y (2018) RUNX Poly(ADP‐Ribosyl)ation and BLM interaction facilitate the fanconi anemia pathway of DNA repair. Cell Rep 24: 1747–1755 [DOI] [PubMed] [Google Scholar]
- Taylor MRG, Yeeles JTP (2018) The Initial response of a eukaryotic replisome to DNA damage. Mol Cell 70: 1067–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thongthip S, Bellani M, Gregg SQ, Sridhar S, Conti BA, Chen Y, Seidman MM, Smogorzewska A (2016) Fan1 deficiency results in DNA interstrand cross‐link repair defects, enhanced tissue karyomegaly, and organ dysfunction. Genes Dev 30: 645–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torregrosa‐Muñumer R, Forslund JME, Goffart S, Pfeiffer A, Stojkovič G, Carvalho G, Al‐Furoukh N, Blanco L, Wanrooij S, Pohjoismäki JLO (2017) PrimPol is required for replication reinitiation after mtDNA damage. Proc Natl Acad Sci USA 114: 11398–11403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisman A, Woodgate R (2017) Translesion DNA polymerases in eukaryotes: what makes them tick? Crit Rev Biochem Mol Biol 52: 274–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Lou J, Xia Y, Su B, Liu T, Cui J, Sun Y, Lou H, Huang J (2013) HPrimpol1/CCDC111 is a human DNA primase‐polymerase required for the maintenance of genome integrity. EMBO Rep 14: 1104–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Leung JW, Jiang Y, Lowery MG, Do H, Vasquez KM, Chen J, Wang W, Li L (2013) FANCM and FAAP24 maintain genome stability via cooperative as well as unique functions. Mol Cell 49: 997–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman MR, Schauer GD, O'Donnell ME, Liu S (2019) Replication fork activation is enabled by a single‐stranded DNA gate in CMG helicase. Cell 178: 600–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszek JL, Chatterjee N, Najeeb J, Ramos A, Lee M, Bian K, Xue JY, Fenton BA, Park H, Li D et al (2019) A small molecule targeting mutagenic translesion synthesis improves chemotherapy. Cell 178: 152–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RA, Semlow DR, Kamimae‐Lanning AN, Kochenova OV, Chistol G, Hodskinson MR, Amunugama R, Sparks JL, Wang M, Deng L et al (2019) TRAIP is a master regulator of DNA interstrand crosslink repair. Nature 567: 267–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Rokutanda N, Takeuchi J, Lai Y, Maruyama R, Togashi Y, Nishikawa H, Arai N, Miyoshi Y, Suzuki N et al (2018) HERC2 facilitates BLM and WRN helicase complex interaction with RPA to suppress G‐quadruplex DNA. Cancer Res 78: 6371–6385 [DOI] [PubMed] [Google Scholar]
- Xue C, Daley JM, Xue X, Steinfeld J, Kwon Y, Sung P, Greene EC (2019) Single‐molecule visualization of human BLM helicase as it acts upon double‐ and single‐stranded DNA substrates. Nucleic Acids Res 47: 11225–11237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Delannoy M, Ling C, Daee D, Osman F, Muniandy PA, Shen X, Oostra AB, Du H, Steltenpool J et al (2010) A histone‐fold complex and FANCM form a conserved DNA‐remodeling complex to maintain genome stability. Mol Cell 37: 865–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Bellani MA, James RC, Pokharel D, Zhang Y, Reynolds JJ, McNee GS, Jackson AP, Stewart GS, Seidman MM (2020) DONSON and FANCM associate with different replisomes distinguished by replication timing and chromatin domain. Nat Commun 11: 3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Dewar JM, Budzowska M, Motnenko A, Cohn MA, Walter JC (2015a) DNA interstrand cross‐link repair requires replication‐fork convergence. Nat Struct Mol Biol 22: 242–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Walter JC (2014) Mechanism and regulation of incisions during DNA interstrand cross‐link repair. DNA Repair (Amst) 19: 135–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Wilson AF, Mahmood Ali A, Namekawa SH, Andreassen PR, Ruhikanta Meetei A, Pang Q (2015b) Loss of Faap20 causes hematopoietic stem and progenitor cell depletion in mice under genotoxic stress. Stem Cells 33: 2320–2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
Proteomics data have been uploaded at the ProteomeXchange Consortium. https://www.ebi.ac.uk/pride/archive/projects/PXD017951 (identifier PXD017951).