

ABSTRACT
Serotype M28 isolates of the group A Streptococcus (GAS; Streptococcus pyogenes) are nonrandomly associated with cases of puerperal sepsis, a potentially life-threatening infection that can occur in women following childbirth. Previously, we discovered that the 36.3-kb RD2 pathogenicity island, which is present in serotype M28 isolates but lacking from most other isolates, promotes the ability of M28 GAS to colonize the female reproductive tract. Here, we performed a gain-of-function study in which we introduced RD2 into representative serotype M1, M49, and M59 isolates and assessed the phenotypic consequences of RD2 acquisition. All RD2-containing derivatives colonized a higher percentage of mice, and at higher CFU levels, than did the parental isolates in a mouse vaginal colonization model. However, for two additional phenotypes, survival in heparinized whole human blood and adherence to two human vaginal epithelial cell lines, there were serotype-specific differences from RD2 acquisition. Using transcriptomic comparisons, we identified that such differences may be a consequence of RD2 altering the abundance of transcripts from select core genome genes along serotype-specific lines. Our study is the first that interrogates RD2 function in GAS serotypes other than M28 isolates, shedding light on variability in the phenotypic consequences of RD2 acquisition and informing on why this mobile genetic element is not ubiquitous in the GAS population.
KEYWORDS: Streptococcus pyogenes, mobile genetic elements, virulence
INTRODUCTION
Isolates of a given pathogenic bacterial species often show phenotypic heterogeneity from one another that manifests as differences in disease potential, which, in turn, can negatively impact the diagnosis, treatment, and prevention of infections by these pathogens (1–4). The molecular bases behind this phenotypic heterogeneity include differences in gene expression, for example, due to mutations in regulator-encoding genes such as those described for the Agr system in Staphylococcus aureus (5–9). Differences in gene content due to gene gain or loss can also impact phenotypic heterogeneity, as described for the different virulence factor-encoding mobile genetic elements and deletions that distinguish Escherichia coli pathotypes (10, 11).
The group A Streptococcus (GAS; Streptococcus pyogenes) isolates are human-specific pathogens that cause a wide array of human diseases, from mild and self-limiting (e.g., pharyngitis; also called strep throat) to severe and life-threatening (e.g., necrotizing fasciitis; also called a flesh-eating infection) (12). GAS isolates are divided into serotypes based upon the sequence of the hypervariable region of the classical GAS virulence factor the M protein, encoded by the emm gene (13). Studies have identified nonrandom associations between certain GAS serotypes and particular diseases, such as the association of serotype M3 isolates with particularly severe and lethal invasive infections (4, 14, 15). In addition to serotyping, several other typing schemes also exist for GAS, for example, the chromosomal arrangement of the emm subfamily genes reveals five major emm patterns, denoted patterns A through E (16). GAS strains with emm patterns A to C are usually recovered from cases of pharyngitis, strains with emm pattern D are most often isolated from impetigo lesions, and strains with emm pattern E are readily recovered from both the pharynx and the skin (17). Thus, the emm pattern serves as a genotypic marker for tissue site preference (16, 18).
Previously, we identified that the 36.3-kb RD2 pathogenicity island present in serotype M28 GAS isolates, of apparent group B Streptococcus (GBS) origin (Fig. 1) (19), promotes the ability of these isolates to colonize the murine female reproductive tract (20). Our working hypothesis is that the enhanced colonization phenotype, along with other uncharacterized phenotypes, imparted onto M28 isolates by RD2 are behind the epidemiologically identified association of M28 GAS isolates with cases of puerperal sepsis (19, 21, 22). Infections that originate in the reproductive tract of women ante- or postpartum, so-called puerperal infections, cause morbidity in ∼10% of new mothers globally and can spread to the blood, resulting in a life-threatening sepsis, puerperal sepsis (23). While several pathogens can cause puerperal infections, including GBS, GAS isolates are the most common causes of severe maternal puerperal infections and death worldwide (24, 25).
FIG 1.
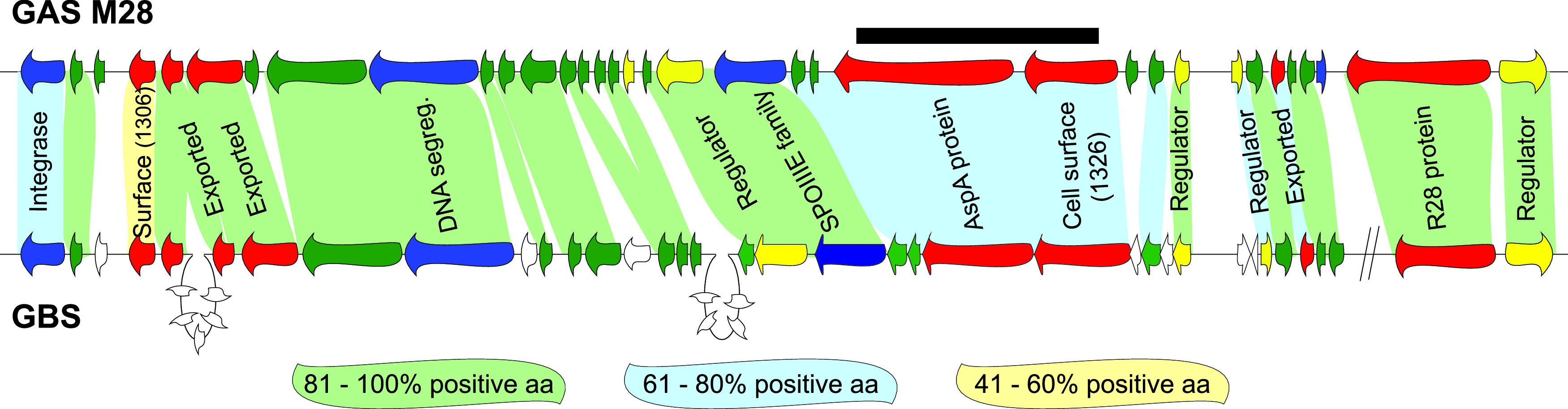
Comparison of genes within RD2 of M28 GAS with genes present in GBS. Genes are represented by arrows and are color-coded based on putative open reading frame (ORF) function (blue, mobility; yellow, gene regulators; red, extracellular proteins; green, hypotheticals). Genes unique to GBS are colored white. Shading between GAS and GBS genes is based upon homology at the protein level. Percent positive amino acids were calculated by adding the percentage of identical and strongly similar amino acids together for each of the compared protein pairs. Note that the two diagonal lines on the GBS part of the figure signify that the two genes are not contiguous with the other genes in GBS but they are in the GAS. The black bar highlights the region of RD2 that was replaced with a spectinomycin resistance cassette in the RD2 derivative used in this study.
RD2 is almost universally present within the genomes of serotype M28 GAS isolates and is present in a significant percentage of serotype M2 isolates, but it is absent from the tested isolates of most other serotypes (7, 19, 26, 27). Of note, while M2 isolates are not associated with puerperal sepsis cases, they are associated with cases of vaginitis and urinary tract infections (15, 28). The question of why RD2 is not more widely distributed in the GAS population has not been adequately explored, but reasons may include serotype-specific barriers to RD2 transfer and/or activity. To initiate an investigation of this, we present data generated from gain-of-function studies in which we introduced RD2 into representative isolates of serotypes that typically lack this element, M1, M49, and M59. While all three RD2-containing isolates showed enhanced vaginal colonization abilities relative to their parental (RD2-lacking) isolates, differences were observed with regard to adherence to vaginal epithelial cells and to survival in whole human blood. Furthermore, we uncovered transcriptomic differences between the presence and absence of RD2 among our tested M1 and M49 strains. The data expand our knowledge of RD2-mediated activity and regulation in GAS and indicate that strain- and/or serotype-specific differences in these functions play a role in the restricted distribution of RD2 among GAS serotypes.
RESULTS
Introduction of RD2 into representative serotype M1, M49, and M59 GAS isolates.
To gain insight into whether RD2 influences the virulence characteristics of isolates from a wider range of GAS serotypes than just M28 isolates, we set out to perform a series of gain-of-function studies. The first step in these studies required the introduction of RD2 into representative isolates from serotypes that naturally lack this element, enabling subsequent comparisons to be made between the parental and RD2-containing derivatives. We chose three RD2-lacking GAS isolates to serve as our parental isolates (Table 1), with these isolates being of serotypes M1, M49, and M59. Isolates of these serotypes were chosen because they are representatives of different emm pattern types (M1 isolates have patterns A to C, M49 isolates have pattern E, and M59 isolates have pattern D) (17), and we wished to investigate whether the emm pattern correlated with any RD2-specific phenotypes. Several RD2 derivatives tagged with an antibiotic resistance cassette (to enable selection upon introduction into our parental isolates) were created, but only one, a derivative that harbored a spectinomycin resistance cassette in place of the aspA and spy1326 genes (Fig. 1), could be transformed into each of the three parental isolates (data not shown). To verify that the resultant strains (M1::RD2, M49::RD2, and M59::RD2) harbored the modified RD2, we used a set of 13 primer pairs in a tiling PCR approach (Fig. 2A). PCR-generated bands of the expected size were observed for strains M1::RD2, M49::RD2, and M59::RD2, while no consistently obtained PCR products were seen for their respective parental isolates, indicating that RD2 had been successfully introduced into the derivative strains (Fig. 2B). The presence of RD2 in strains M1::RD2, M49::RD2, and M59::RD2 was also assessed by Western blotting, assaying for the presence of the RD2-encoded protein R28 within GAS cell wall protein fractions (20). Reactivity to the anti-R28 antibody was not observed for any of the protein samples isolated from the parental isolates but was observed for those from each of the three RD2-containing derivatives (Fig. 2C).
TABLE 1.
GAS strains used in this study
| GAS strain | Description | Disease isolate sourcea | Reference or source |
|---|---|---|---|
| M1 (MGAS2221) | Representative serotype M1 isolate that naturally lacks RD2 | Scarlet fever | 9 |
| M1::RD2 (PGAS885) | MGAS2221 derivative in which RD2 has been introduced | NA | This work |
| M49 (591 and PGAS812) | Representative serotype M49 isolate that naturally lacks RD2 | Acute poststreptococcal glomerulonephritis | 9 |
| M49::RD2 (PGAS890) | 591 derivative in which RD2 has been introduced | NA | This work |
| M59 (MGAS15252 and PGAS535) | Representative serotype M59 isolate that naturally lacks RD2 | Soft tissue infection | 9 |
| M59::RD2 (PGAS891) | MGAS15252 derivative in which RD2 has been introduced | NA | This work |
| M28 (MGAS6180) | Representative serotype M28 isolate that naturally has RD2 | Invasive infection | 19 |
| M28::Spec | MGAS6180 derivative in which a spectinomycin resistance cassette replaced the aspA and M28_Spy1326 genes within RD2 | NA | This work |
NA, not applicable.
FIG 2.

RD2 can be maintained in serotype M1, M49, and M59 GAS isolates. The presence of RD2 in strains M1::RD2, M49::RD2, and M59::RD2 was verified by PCR and Western blot approaches. (A) Schematic showing the locations of the 13 PCR products that were used to tile along RD2. (B) Results of the 13 RD2 PCRs for each of the three tested parental isolates (M1, M49, and M59) and their RD2-containing derivatives are shown. Note that the weak signal with PCR 11 is due to this region containing the r28 gene, which is difficult to amplify due to multiple repeat sequences. (C) Western blot analysis of cell wall-associated R28 expression in the six indicated GAS strains. Note the characteristic laddering pattern, which occurs due to the partial hydrolysis of acid‐labile Asp‐Pro sites within the repeat regions of R28 (47). The loading control was generated by Coomassie staining a gel that was identical to that used in the Western analysis.
Genomic verification of RD2-containing M1, M49, and M59 derivative strains.
To ensure that M1::RD2, M49::RD2, and M59::RD2 differ from their respective parental strains only by the presence/absence of RD2 and do not contain spurious mutations, such as single-nucleotide polymorphisms (SNPs) or insertions/deletions (InDels) that may confound data interpretation, we performed an Illumina-based whole-genome sequencing analysis of each of the six GAS isolates. This analysis identified that, as expected, the presence/absence of RD2 was the only genetic difference between the strains in each strain pair (see Fig. S1 in the supplemental material).
RD2 promotes GAS adherence to human epithelial cell lines in a strain- or serotype-specific manner.
Our previous loss-of-function study identified that the deletion of RD2 from the genome of a serotype M28 GAS isolate resulted in a reduction in adherence (20) and, hence, that RD2 positively influences adherence in M28 GAS. To assess whether RD2 enhances GAS adherence in serotype M1, M49, and M59 strain backgrounds, we utilized a tissue culture-based adherence assay. To reduce the possibility of our data being confounded by cell line-specific differences in adherence, we used two distinct human vaginal epithelial cell lines, A431 and VK2/E6E7 (29, 30). The data gained from each cell line were essentially superimposable and showed that the representative M49 and M59 isolates that harbored RD2 adhered to the epithelial cells at higher rates (∼2.2-fold for strain M49::RD2 and ∼3-fold for strain M59::RD2) than their parental, RD2-lacking strains (Fig. 3A and B). While strain M1::RD2 also showed an increase in adherence relative to its parental strain, the difference was more modest and was not statistically significant (Fig. 3A and B). The data are consistent with RD2 promoting GAS adherence to human vaginal epithelial cell lines regardless of serotype but with the level of enhancement differing along strain- or serotype-specific lines.
FIG 3.
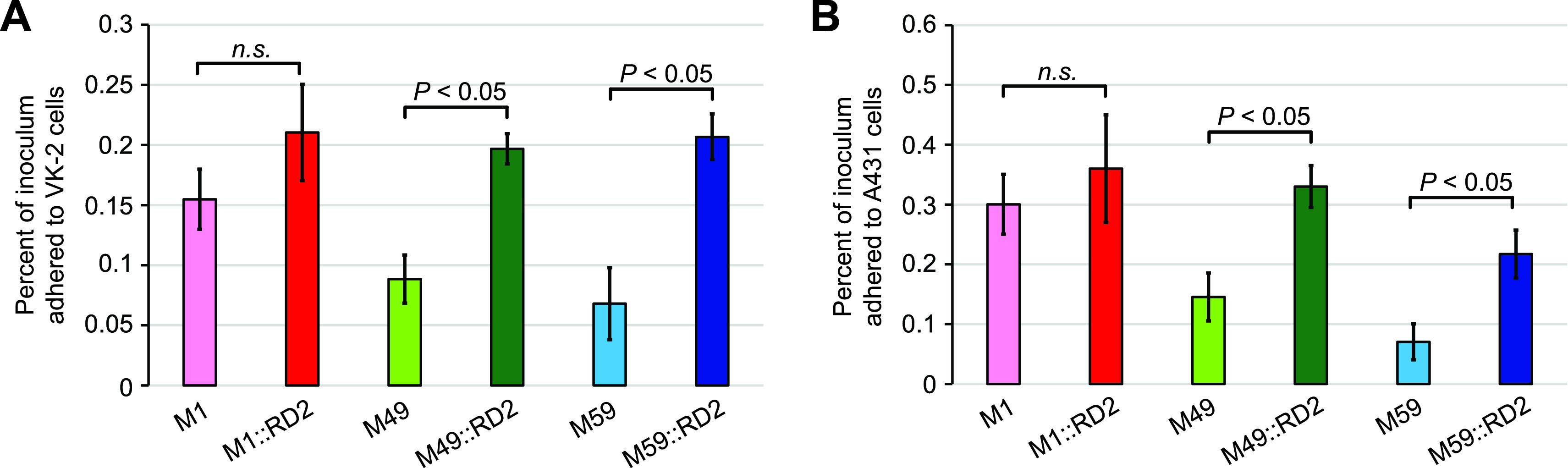
Acquisition of RD2 leads to a serotype-specific enhancement in the ability of GAS to adhere to human epithelial cell lines. The six indicated strains were compared in tissue culture-based assays of adherence using VK2/E6E7 (A) and A431 (B) human cell lines. The experiment was performed on six occasions with each cell line, and mean values (± standard errors of the means) are shown. Statistical significance was determined via t test.
RD2 promotes colonization of M1, M49, and M59 GAS strains in a mouse vaginal colonization model.
Consistent with our hypothesis that RD2 is a key factor behind the association of serotype M28 GAS isolates with cases of puerperal sepsis, our previous loss-of-function study identified that removal of RD2 from an M28 isolate decreased colonization in a mouse vaginal colonization model (20). To investigate whether the enhanced colonization phenotype attributed to RD2 is also observed in non-serotype M28 isolates, we used the paired M1, M49, and M59 strains in the mouse colonization model. Regardless of serotype, higher CFU numbers were recovered from vaginal swabs of the mice infected with the RD2-containing derivative strains relative to the mice infected with the parental strains (Fig. 4A to C). In addition, a higher percentage of mice overall were colonized by the RD2-containing derivatives than by the RD2-lacking parental strains (Fig. 4D). These data show that RD2 promotes vaginal colonization across all tested strains and serotypes.
FIG 4.

RD2 promotes the ability of serotype M1, M49, and M59 GAS isolates to colonize the murine vagina. Groups of 10 estradiol-treated mice (0.5 mg) were vaginally challenged with 1 × 104 CFU of the indicated GAS strains. Shown is the mean number (± standard errors of the means) of CFU recovered from vaginal swabs over time for the two M1 isolates (A), the two M49 isolates (B), and the two M59 isolates (C). Repeated-measure analyses indicated that a statistically significantly higher number of CFU were recovered over time for each of the RD2-containing strains than their parental strains (the P values are shown on each graph). (D) Cumulative comparison (i.e., the 30 mice infected with a parental [M1, M49, or M59] GAS strain against the 30 mice infected with the RD2-containing derivatives) showing the percentage of mice that were colonized over time. Differences between the two data sets were tested for significance at individual time points (days 1, 3, 5, 7, and 10) by Fisher’s exact test (**, P < 0.01; *, P < 0.05; n.s., not significant).
RD2 impacts the ability of GAS to survive in human blood in a strain- or serotype-specific manner.
In addition to promoting vaginal colonization by GAS, we hypothesized, but have not previously tested in any GAS strain, that RD2 also promotes the ability of GAS to survive and proliferate in human blood. To test this, we compared each of our strain pairs in standard bactericidal assays using heparinized whole human blood. We identified that the acquisition of RD2 resulted in an enhanced survival rate for the M49 and M59 derivative strains (∼4-fold for M49:RD2 and ∼2-fold for M59::RD2) relative to their parental strains (Fig. 5). However, the opposite phenotype was observed for the M1 derivative containing RD2 (an ∼2-fold reduction in survival; Fig. 5). Along with our tissue culture-based adherence assays, these data are consistent with there being strain- or serotype-specific factors that influence the phenotypic consequences of RD2 acquisition.
FIG 5.

In a serotype-specific manner, RD2 modifies the ability of GAS to survive and proliferate in human blood. The ability of the indicated strains to survive and replicate in human blood was tested. The data are shown as percentages of the number of CFU following 3 h of growth in blood relative to the inoculum (which is represented as the dashed line). The experiment was performed on six occasions, with mean values ± standard deviations being shown. Statistical significance was determined for pairs of isolates via t test.
RD2-acquisition impacts the transcriptome of GAS isolates in a strain- or serotype-specific manner.
To investigate the possibility that the different phenotypic consequences of RD2 acquisition in strain M1::RD2 relative to strains M49::RD2 and M59::RD2 are a consequence of differential gene expression in these strains, either via core genome genes being differentially regulated by RD2-encoded factors or via RD2 genes being differentially regulated by core genome-encoded factors, we performed a transcriptomic comparison of the M1 and M49 strain pairs. Triplicate cultures of each strain were grown and compared via transcriptome sequencing (RNA-seq). Principal-component analyses of the resultant data identified that the parental and RD2-containing derivative strains could be separated based upon their transcriptomes, with the strains being separated by the first principle component (PC1; Fig. 6A and B). Thus, gain of RD2 alters the transcriptome in the M1 and M49 strain backgrounds.
FIG 6.
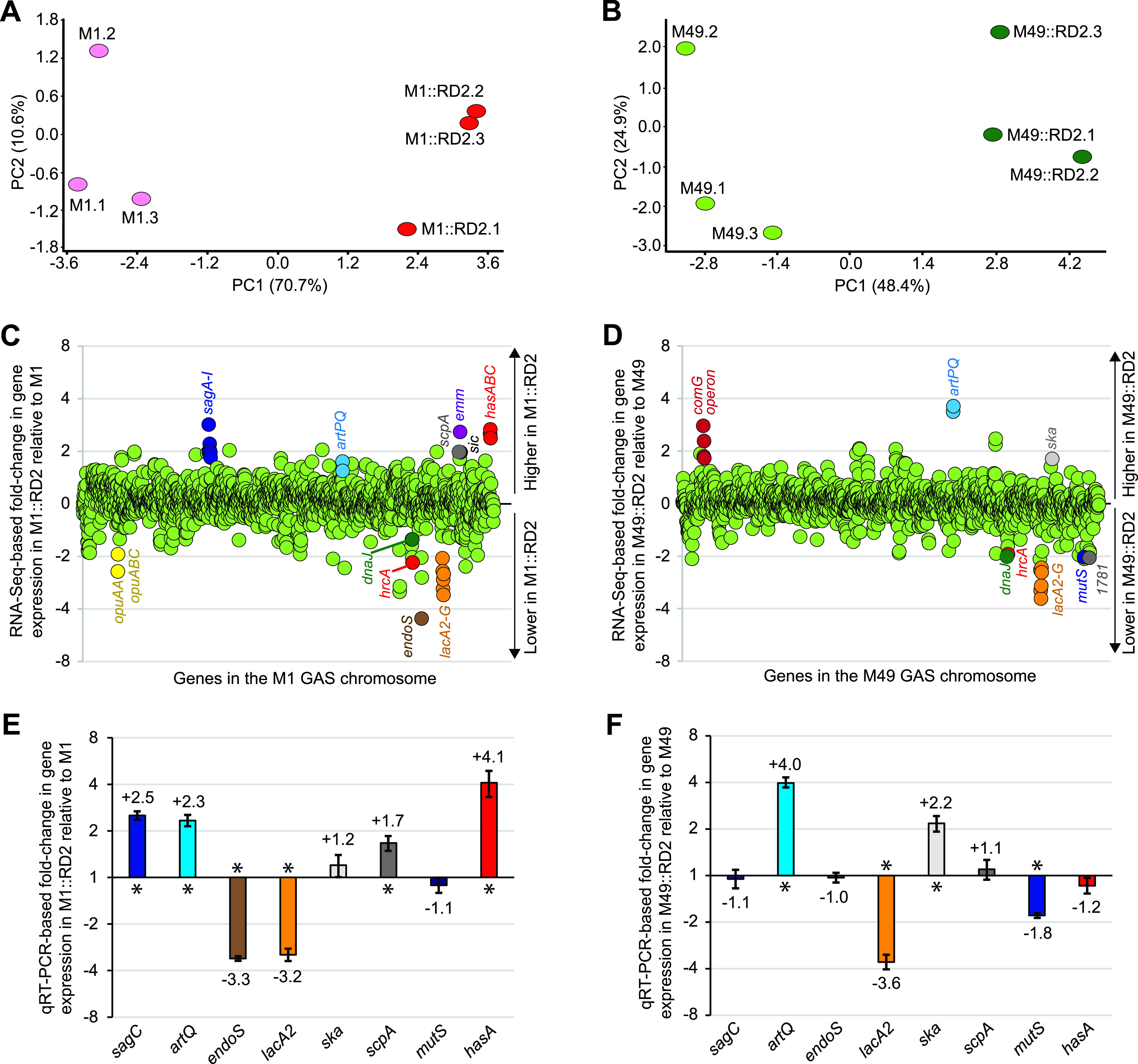
RD2 variably modifies the transcriptomes of representative M1 and M49 GAS isolates. (A and B) Principal-component analysis (PCA) plots showing overall transcriptome variation between parental (pink/light green) and RD2-containing derivatives (red/dark green) of the M1 (A) and M49 (B) strains. PCA assesses the variance in a data set in terms of principal components. The percentages of the total variation that are accounted for by the 1st and 2nd principal components are shown on the x and y axis labels, respectively. Each oval represents one of the three triplicate samples grown and analyzed for each GAS strain. (C and D) Summary of RNA-seq data. Shown are pairwise comparisons of the transcriptomes between the parental and RD2-containing derivative M1 (C) and M49 (D) strains. The relative expression levels of genes present in the parental strains are graphed, with each gene represented by a circle. Select mRNAs of interest are colored and labeled. (E and F) TaqMan-based quantitative RT-PCR (qRT-PCR) data confirming the M1 (E) and M49 (F) RNA-seq data for the same set of select transcripts. Transcript abundances were determined from triplicate exponential-phase GAS cultures run in duplicate, with the mean (± standard deviation) fold change in transcript levels in the RD2-containing strains relative to their parental strains shown. Asterisks highlight data that are statistically significant (P < 0.05), as determined by t test.
Looking at the M1 versus M1::RD2 transcriptome comparison in more detail, 100 genes were identified as being statistically significantly different between the two strains (t test followed by a false discovery rate [FDR] correction; P < 0.05) using a minimum cutoff level of ±1.5-fold (Fig. 6C and Table S2). For the M49 versus M49::RD2 transcriptome comparison, 58 genes were identified using the same parameters (Fig. 6D and Table S3). Several genes, such as those encoding putative arginine transport (artPQ) (31) and carbohydrate utilization (lacA2B2C2D2FG) (32) proteins, were similarly regulated in both data sets, consistent with these genes being part of a core RD2-controlled regulon. Strain- or serotype-specific differences in the regulatory consequences of RD2 acquistion were also apparent. In the M1 strain background, gain of RD2 enhanced expression of the virulence factor genes hasABC (encoding proteins that synthesize the antiphagocytic hyaluronic acid capsule) (33) and sagABCDEFGHI (encoding the streptolysin S biosynthesis proteins) (34), among others, while also reducing expression of the immunomodulatory endoglycosidase gene endoS (Fig. 6C) (35). In the M49 strain background, gain of RD2 resulted in a reduction of mutS transcript levels, encoding a DNA mismatch repair protein, and an enhancement of ska transcript levels, encoding the thrombolytic factor streptokinase (Fig. 6D) (36). Select genes from the RNA-seq data were verified via TaqMan-based quantitative reverse transcription-PCR (RT-PCR), confirming their differential regulation between the presence and absence of RD2 in cross-serotype (artQ and lacA2) or serotype-specific (sagC, endoS, ska, scpA, mutS, and hasA) manners (Fig. 6E and F).
DISCUSSION
Mobile genetic elements such as plasmids, lysogenic bacteriophage, and genomic islands often bestow virulence-altering characteristics on their host bacterium (37–39). In some cases, the phenotypic changes brought about by these elements are positively selected for, often resulting in their presence in a high percentage of the population (40, 41). The high prevalence of a mobile genetic element within a population can be a consequence of the rapid expansion of the initial strain that gained this element, with this strain outcompeting other circulating strains. Alternatively, the genetic element could pass between strains circulating in the population, which requires a combination of high transfer activity and low barriers to transfer (e.g., physiological barriers, geographic barriers, active restriction-modification systems, active CRISPR-Cas systems, etc.) (42–44). Looking at the RD2 pathogenicity island in GAS, this element is found in >99% of tested M28 and ∼70% of tested M2 GAS isolates but is mostly absent or present at low (<5%) frequency in isolates of other tested serotypes (7, 19). The distribution of RD2, combined with our previous and current data (20), is consistent with this element being positively selected for in M2 and M28 isolates. The relative lack of RD2 in most other serotypes may be due to barriers to the transfer of RD2 but, as our data here show, may also be a result of serotype-specific differences in the regulatory and/or phenotypic consequences of RD2 acquisition.
For two of the tested phenotypes, adherence to human epithelial cell lines and colonization of the murine vagina, there was mostly consensus on how gain of RD2 impacted the M1, M49, and M59 strains in our study. Strains M1::RD2, M49::RD2, and M59::RD2 all could colonize the murine vagina at higher numbers of CFU over time than the respective parental isolates (Fig. 4) and also resulted in higher levels of adherence in our tissue culture-based assay (although only the M49 and M59 data were statistically significant; Fig. 3). The molecular basis behind the RD2-mediated increase in adherence and colonization for the M1, M49, and M59 isolates has not been determined, but the simplest explanation is that one or more of the known (R28 and AspA) or putative (Spy1306 and Spy1326) RD2-encoded adhesins are responsible (45, 46). However, the fact that the modified RD2 used in our study lacks the aspA and spy1326 genes (Fig. 1) removes them as potentially being involved while at the same time raising the possibility that even more striking phenotypic consequences of RD2 gain would have been attained had we been able to introduce a full-length RD2 into our tested strains. R28 is an integrin-binding cell surface protein that promotes GAS adherence to some cell lines (e.g., ME180) (47) while having no apparent adherence-enhancing role to others (e.g., VK2 and A431) (20). As we used VK2 and A431 cells in our study, our enhanced adherence phenotype observed upon gain of RD2 would not appear to be a consequence of R28. Thus, Spy1306 is the most likely candidate RD2 gene responsible for the enhanced colonization and adherence phenotype. While Spy1306 has not been studied in detail, it does appear to be produced during human infection and located extracellularly, as evidenced by convalescent (but not acute-phase) patient sera reacting with this protein (19). It should be noted, however, that at least under the conditions used for RNA-seq (exponential-phase growth in Todd-Hewitt broth containing 2% yeast extract [THY broth]), spy1306 transcripts were not present in high abundance (see Table S4 in the supplemental material).
Our bactericidal assays generated the first data showing that RD2 influences GAS survival in human blood. The direction of influence, however, was not the same for all of the tested isolates, with RD2 reducing survival in the M1 GAS background and enhancing survival in the M49 and M59 strain backgrounds (Fig. 5). The RD2-encoded factors responsible for these phenotypes are currently unknown. Our transcriptome data are consistent with the differential regulation of core genome genes by RD2 as playing a role (Fig. 6C and D). For example, the reduction in endoS transcripts by RD2, seen in M1::RD2 but not M49::RD2, may contribute to the reduced survival of M1::RD2 in human blood, given the IgG inactivating function of EndoS (35). Conversely, however, genes encoding several antiphagocytic virulence factors (e.g., the hyaluronic acid capsule and streptolysin S) are upregulated in M1::RD2 relative to the parental isolate, which would be expected to enhance, not inhibit, blood survival (Fig. 6E). Further study is required to parse out the relative contributions of each of these changes to the altered blood survival of strain M1::RD2. Given the importance of the blood survival phenotype and the negative effect of RD2 on this in the M1 GAS background, perhaps it is not surprising that RD2 is not found at high frequency in M1 isolates.
Gain of RD2 in the M49 and M59 isolate backgrounds resulted in enhanced adherence (Fig. 3), colonization (Fig. 4), and blood survival (Fig. 5) phenotypes, consistent with the gain of RD2 having positive effects on the pathogenesis of M49 and M59 GAS isolates. However, since RD2 is not found at an appreciable frequency from isolates of these two serotypes, it is likely that there are other, untested phenotypes that are negatively influenced by RD2. It is also true that higher adherence does not necessarily translate into higher virulence, for example, the greater adherence of strains M49::RD2 and M59::RD2 relative to their parental strains may inhibit their ability to disseminate during infection.
Our transcriptome studies identified that gain of RD2 results in the metabolic adaptation of GAS cells, as exemplified by a reduction in lacA2B2C2D2FG transcripts and an increase in artPQ transcripts (Fig. 6C to F). Of note, these genes were regulated in a similar manner, in terms of the direction of regulation (i.e., plus or minus) if not magnitude, in a previous study investigating the transcriptional consequences of GAS growth in human amniotic fluid relative to THY broth (48). Whether RD2 influenced the regulation of these genes in amniotic fluid was not investigated but is a possibility given our data. Reducing the expression of carbohydrate utilization genes, and increasing arginine uptake, fit with the lower carbohydrate/higher protein abundance encountered during infection of the female reproductive tract relative to growth in THY broth. While regulation of the lac and art gene transcripts occurred in both the M1 and M49 data sets, other transcripts were differentially regulated only in one, such as sagABCDEFGHI and hasABC in the M1 data and ska and M49_1781 in the M49 data. M49_1781 encodes a putative uncharacterized transcriptional regulator, the transcript levels of which were previously identified as being reduced in response to vaginal colonization (in an M49 strain that lacks RD2) (49). Our observation that M49_1781 transcripts are also reduced in the presence of RD2 (Fig. 6D) is consistent with RD2 modulating the core GAS transcriptome into a form that promotes colonization of the female reproductive tract. While modulation of M49_1781 transcript levels may facilitate vaginal colonization, it does not appear to be required for it, as the M1 GAS equivalent to M49_1781 (M5005_Spy_1825) is not modified in its transcript abundance by RD2 (Table S2). It is possible that these serotype-specific differences in regulatory pathways influence additional, unidentified infection attributes, such as the ability to cause ascending infections. Of potential significance, serotype M49 strains, similar to serotype M2 and M28 strains, are generalist strains (i.e., they are of emm pattern E) (50). Thus, it may be that emm pattern E isolates share commonalities in overall transcriptional patterns, relative to emm pattern A-C or D isolates, and are more amenable to rewiring by RD2 into a form that promotes infection of the female reproductive tract.
With regard to how RD2 influences gene expression on a genome-wide scale, there is one known (M28_Spy1337) and four putative transcriptional regulators encoded by this element (Fig. 1), each of which may contribute. It is also possible, however, that the transcriptome changes do not occur due to the action of one or more RD2-encoded regulators but rather occur indirectly due to RD2 gain. RD2 (36.3 kb) appears to be present in approximately eight copies per cell (27), which calculates as 290 kb of additional DNA to be replicated, which equates to ∼15% of the size of the GAS chromosome. Thus, the additional energy demands that replication of RD2 places on a cell may, in itself, explain why RD2 is not universally found in GAS isolates. The altered transcriptomes observed following gain of RD2 may at least in part arise as a consequence of the physiological stress RD2 places on cells, with differences in regulated genes between serotypes being a consequence of serotype-specific variations in stress response pathways (51).
In conclusion, we have shown that the virulence-altering characteristics of the 36.3-kb RD2 pathogenicity island apply not only to serotype M28 GAS isolates but also to isolates of additional serotypes. This mobile genetic element modulates both the adherence and vaginal colonization ability of GAS isolates as well as the ability of these isolates to survive the bactericidal properties of whole human blood. Further study of RD2 is warranted to characterize which RD2-encoded factors are responsible for the observed phenotypes, whether there are barriers to the transfer of RD2 from donor to recipient cells, whether we can establish a stronger link between the presence of RD2 and clinical observations, and whether we can create prophylactic measures that inhibit the activity of key RD2-encoded factors that can be used by women ante- or postpartum.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Representative serotype M1, M28, M49, and M59 clinical GAS isolates were used in this study. Information about these isolates and their derivatives is presented in Table 1 (9, 19, 20, 52, 53). GAS isolates were grown in Todd-Hewitt broth containing 2% yeast extract (i.e., THY broth), with spectinomycin (150 μg/ml) added when needed.
Creation of an RD2 derivative containing a spectinomycin resistance cassette.
The introduction of RD2 into isolates of different GAS serotypes required the creation of an RD2 derivative that contained a selectable marker. Thus, we created a derivative of the M28 strain MGAS6180 in which a nonpolar spectinomycin resistance cassette had been inserted into RD2 (Fig. 1) (54). The strain we created, M28::Spec, was essentially identical to the previously described strain MGAS6180Δ1325-1326spcR (27), with the spectinomycin resistance cassette replacing the aspA and spy1326 genes (Fig. 1). The PCR primers used in the creation of M28::Spec are listed in Table S1 in the supplemental material, and the strain was verified by PCR and targeted sequencing.
Introduction of RD2 into serotype M1, M49, and M59 strains.
Genomic DNA from strain M28::Spec was used to transform competent cells of strains M1, M49, and M59, with transformants being selected for on THY agar plates containing spectinomycin. That the resultant strains (M1::RD2, M49::RD2, and M59::RD2) contained RD2 was verified via a tiling PCR approach in which 13 primer pairs flanking RD2 were used (Table S1). Verification was also performed via whole-genome sequencing and Western blot analyses.
Whole-genome sequencing-based verification of the RD2-containing M1, M49, and M59 derivative strains.
Genomic DNA was isolated from all six of the tested strains, M1, M1::RD2, M49, M49::RD2, M59, and M59::RD2. The isolated genomic DNAs were diluted to a concentration of 0.2 ng/μl and used to prepare paired-end sequencing libraries according to the Illumina Nextera XT kit instructions. Sequencing libraries were used in conjunction with an Illumina NextSeq 550 instrument (Nevada Genomics Center). Geneious Prime (Biomatters Ltd.) software was used to map reads to reference M1 (MGAS5005; CP000017), M49 (NZ131; CP000829), and M59 (MGAS15252; CP003116) genomes and to search for genetic variation (e.g., SNPs and InDels) relative to the reference genomes. Lists of genetic variants were subsequently manually curated to verify the presence of these variants and to assess whether these variants were present in one or both strains of each strain pair (i.e., with or without RD2).
Isolation of GAS cell wall proteins and Western blot analysis.
Cell wall protein fractions were isolated from mid-exponential-phase (optical density at 600 nm [OD600], 0.5) GAS cultures as previously described (55). Proteins were separated on 7.5% Tris-HCl gels before being transferred to membrane and use in Western blot analysis with a custom-made, affinity-purified, anti-R28 rabbit polyclonal antibody (raised against the R28-specific peptide KVVDPRTDADKNDPAGKDQTVK [Pacific Immunology]). After washing, Alexa Fluor 680 donkey anti-rabbit IgG secondary antibody (Molecular Probes) was used (1:10,000 dilution), and the fluorescent signal was detected using a Li-Cor Odyssey near-infrared imaging system. An identical protein gel was stained with Coomassie blue to generate a loading control. This experiment was repeated five times, and representative data are shown.
Tissue culture adherence assays.
Cells of the human epithelial cell line A431 were cultured using Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS), 50 μg/ml streptomycin, and 50 U/ml penicillin. Cells of the human epithelial cell line VK2/E6E7 were cultured using keratinocyte serum-free medium (KSF; Gibco) containing 0.1 ng/ml human recombinant epidermal growth factor 1-53 (EGF 1-53) and 0.05 mg/ml bovine pituitary extract, calcium chloride at 44.1 mg/liter (final concentration, 0.4 mM), 50 μg/ml streptomycin, and 50 U/ml penicillin. Upon nearing confluence, A431 and VK2/E6E7 cells were resuspended in their respective media without antibiotics, seeded into 12 wells of a 12-well tissue culture plate, and then incubated for 24 h at 37°C in a 5% CO2 atmosphere. To prepare GAS isolates for this assay, the strains were grown to an OD600 of 0.4, 1 ml of culture was pelleted, and the cell pellets were resuspended in 1 ml of phosphate-buffered saline (PBS). One hundred microliters of each GAS solution was added to separate wells of the seeded 12-well tissue culture plates, resulting in a multiplicity of infection of ∼50. At the same time, 100 μl of the PBS-diluted GAS cultures (1:1,000,000) was plated onto blood agar plates to enable accurate calculation of the initial inocula. The tissue culture plate was incubated for 5 min (37°C in 5% CO2), the liquid in the wells was removed, and the wells were washed five times with 1 ml of PBS. The tissue culture cells were then lysed by incubating them for 15 min in 1% saponin in PBS at room temperature. The lysed tissue culture cells and adhered GAS cells were scraped from the bottom of each well. The lysates were serially diluted (1:100 and 1:1,000), and 100 μl of each dilution was plated onto blood agar plates in duplicate. The average number of bacteria was determined, and the percentage of adhering bacteria relative to the size of the inoculum was calculated. Statistical significance was determined for pairs of isolates via two-tailed t tests.
Mouse model of vaginal colonization.
The mouse model of vaginal colonization experiment was performed as previously described (20). Briefly, 2 days prior to streptococcal infection, female CD-1 mice were estrogenized by intraperitoneal injection of 0.5 mg β-estradiol 17-valerate (Sigma-Aldrich) dissolved in 0.1 ml sterile sesame oil (Sigma-Aldrich). On the day of inoculation (day 0), mice were inoculated in the vaginal lumen with 10 μl of GAS suspension that had been diluted to 1 × 106 CFU/ml. Ten mice were infected per GAS strain tested. Colonization levels were assayed on days 1, 3, 5, 7, and 10 via vaginal swabs that were vortexed in PBS, and the PBS serially diluted and plated onto Selective Strep agar plates (Becton, Dickinson) to enable determination of CFU numbers. This experiment was performed in accordance with the guidelines set forth in a protocol approved by the Institutional Animal Care and Use Committee of the University of Nevada, Reno.
Bactericidal assays.
To test the ability of GAS strains to survive in human blood, we performed Lancefield bactericidal assays (55). Briefly, cultures of each strain were grown to an OD600 of 0.15 to 0.20 and diluted in sterile PBS. Two 50-μl aliquots of each sample were taken, one of which was mixed with 450 μl of whole heparinized human blood and incubated for 3 h at 37°C with end-over-end rotation. The second 50-μl aliquot was serially diluted and plated on blood agar plates to allow determination of the number of CFU inoculated. Following the 3 h of incubation, the GAS-blood cultures were diluted and plated on blood agar plates. All samples were incubated overnight at 37°C in a 5% CO2 atmosphere. The number of CFU was calculated, and the data are presented as the percentage of CFU relative to the inoculum [(number of CFU after 3 h of blood incubation/number of CFU in initial inoculum) × 100]. Statistical significance was determined for pairs of isolates via two-tailed t tests.
RNA-seq-based transcriptome analyses.
Triplicate cultures of each of the four tested GAS strains were grown to the exponential phase (OD600 of 0.5) in THY broth. Recovered GAS samples were incubated at room temperature for 5 min following the addition of 2 volumes of RNAprotect (Qiagen) to maintain RNA integrity. Total RNA was isolated using the RNeasy minikit (Qiagen). The Ribo-Zero bacterial rRNA removal kit (Illumina) was used to remove rRNAs, and the rRNA-depleted RNA was then used to create cDNA libraries using the ScriptSeq v2 kit (Illumina). Generated libraries were barcoded and run on an Illumina NextSeq 550 system (Nevada Genomics Center). CLC Genomics Workbench (Qiagen) was used to map reads to either the reference M1 (CP000017), M49 (CP000829), or M28 (CP000056; to assess RD2 genes) genomes and to perform the gene expression comparisons. Genes with an overall average expression level below a minimal cutoff level of 5 were removed as a method to reduce the variability in the data. Statistical significance between gene expression levels of strain pairs was tested using the t test with FDR correction.
TaqMan-based quantitative RT-PCR analyses.
Triplicate THY broth cultures of each GAS strain were grown to mid-exponential phase (corresponding to an OD600 of 0.5). GAS were pelleted by centrifugation, mechanically disrupted, and harvested for RNA using an RNeasy minikit (Qiagen). cDNA was synthesized using the SuperScript III kit (Invitrogen) per the manufacturer's protocols. The generated cDNA was analyzed via TaqMan-based quantitative RT-PCR analysis using a CFX Connect real-time system (Bio-Rad). TaqMan primers and probes are shown in Table S1. Data were normalized to the respective parental GAS strain and to the internal control gene proS and were tested for statistical significance via a two-tailed t test.
Data availability.
The generated raw sequence data are available for download from the NCBI Sequence Read Archive under the BioProject no. PRJNA704666. The RNA-seq data have been deposited in the Gene Expression Omnibus (GEO) database at the National Center for Biotechnology Information and are accessible through accession number GSE167343.
ACKNOWLEDGMENTS
The research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) under award numbers R03AI128290 (to P.S.) and R21AI148813 (to P.S.) and by a grant from the Nevada Women’s Health Initiative (to P.S.).
We thank James M. Musser and Bernd Kreikemeyer for providing clinical GAS isolates.
Footnotes
Supplemental material is available online only.
Contributor Information
Paul Sumby, Email: psumby@med.unr.edu.
Nancy E. Freitag, University of Illinois at Chicago
REFERENCES
- 1.Nicol MP, Wilkinson RJ. 2008. The clinical consequences of strain diversity in Mycobacterium tuberculosis. Trans R Soc Trop Med Hyg 102:955–965. 10.1016/j.trstmh.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 2.Croucher NJ, Harris SR, Fraser C, Quail MA, Burton J, van der Linden M, McGee L, von Gottberg A, Song JH, Ko KS, Pichon B, Baker S, Parry CM, Lambertsen LM, Shahinas D, Pillai DR, Mitchell TJ, Dougan G, Tomasz A, Klugman KP, Parkhill J, Hanage WP, Bentley SD. 2011. Rapid pneumococcal evolution in response to clinical interventions. Science 331:430–434. 10.1126/science.1198545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heithoff DM, Shimp WR, House JK, Xie Y, Weimer BC, Sinsheimer RL, Mahan MJ. 2012. Intraspecies variation in the emergence of hyperinfectious bacterial strains in nature. PLoS Pathog 8:e1002647. 10.1371/journal.ppat.1002647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller EW, Danger JL, Ramalinga AB, Horstmann N, Shelburne SA, Sumby P. 2015. Regulatory rewiring confers serotype-specific hyper-virulence in the human pathogen group A Streptococcus. Mol Microbiol 98:473–489. 10.1111/mmi.13136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gor V, Takemura AJ, Nishitani M, Higashide M, Medrano Romero V, Ohniwa RL, Morikawa K. 2019. Finding of Agr phase variants in Staphylococcus aureus. mBio 10:e00796-19. 10.1128/mBio.00796-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheung GY, Wang R, Khan BA, Sturdevant DE, Otto M. 2011. Role of the accessory gene regulator agr in community-associated methicillin-resistant Staphylococcus aureus pathogenesis. Infect Immun 79:1927–1935. 10.1128/IAI.00046-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eraso JM, Kachroo P, Olsen RJ, Beres SB, Zhu L, Badu T, Shannon S, Cantu CC, Saavedra MO, Kubiak SL, Porter AR, DeLeo FR, Musser JM. 2020. Genetic heterogeneity of the Spy1336/R28-Spy1337 virulence axis in Streptococcus pyogenes and effect on gene transcript levels and pathogenesis. PLoS One 15:e0229064. 10.1371/journal.pone.0229064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sarkar P, Sumby P. 2017. Regulatory gene mutation: a driving force behind group a Streptococcus strain- and serotype-specific variation. Mol Microbiol 103:576–589. 10.1111/mmi.13584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. 2006. Genome-wide analysis of group a streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog 2:e5. 10.1371/journal.ppat.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denamur E, Clermont O, Bonacorsi S, Gordon D. 2021. The population genetics of pathogenic Escherichia coli. Nat Rev Microbiol 19:37–54. 10.1038/s41579-020-0416-x. [DOI] [PubMed] [Google Scholar]
- 11.Ahmed N, Dobrindt U, Hacker J, Hasnain SE. 2008. Genomic fluidity and pathogenic bacteria: applications in diagnostics, epidemiology and intervention. Nat Rev Microbiol 6:387–394. 10.1038/nrmicro1889. [DOI] [PubMed] [Google Scholar]
- 12.Cunningham MW. 2000. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev 13:470–511. 10.1128/cmr.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bessen DE, Smeesters PR, Beall BW. 2018. Molecular epidemiology, ecology, and evolution of group a streptococci. Microbiol Spectr 6:CPP3-0009-2018. 10.1128/microbiolspec.CPP3-0009-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beres SB, Sylva GL, Sturdevant DE, Granville CN, Liu M, Ricklefs SM, Whitney AR, Parkins LD, Hoe NP, Adams GJ, Low DE, DeLeo FR, McGeer A, Musser JM. 2004. Genome-wide molecular dissection of serotype M3 group A Streptococcus strains causing two epidemics of invasive infections. Proc Natl Acad Sci U S A 101:11833–11838. 10.1073/pnas.0404163101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colman G, Tanna A, Efstratiou A, Gaworzewska ET. 1993. The serotypes of Streptococcus pyogenes present in Britain during 1980-1990 and their association with disease. J Med Microbiol 39:165–178. 10.1099/00222615-39-3-165. [DOI] [PubMed] [Google Scholar]
- 16.McGregor KF, Spratt BG, Kalia A, Bennett A, Bilek N, Beall B, Bessen DE. 2004. Multilocus sequence typing of Streptococcus pyogenes representing most known emm types and distinctions among subpopulation genetic structures. J Bacteriol 186:4285–4294. 10.1128/JB.186.13.4285-4294.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bessen DE. 2016. Tissue tropisms in group A Streptococcus: what virulence factors distinguish pharyngitis from impetigo strains? Curr Opin Infect Dis 29:295–303. 10.1097/QCO.0000000000000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bessen DE, Lizano S. 2010. Tissue tropisms in group A streptococcal infections. Future Microbiol 5:623–638. 10.2217/fmb.10.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Green NM, Zhang S, Porcella SF, Nagiec MJ, Barbian KD, Beres SB, LeFebvre RB, Musser JM. 2005. Genome sequence of a serotype M28 strain of group a streptococcus: potential new insights into puerperal sepsis and bacterial disease specificity. J Infect Dis 192:760–770. 10.1086/430618. [DOI] [PubMed] [Google Scholar]
- 20.Jain I, Sarkar P, Danger JL, Medicielo J, Roshika R, Calfee G, Ramalinga A, Burgess C, Sumby P. 2019. A mobile genetic element promotes the association between serotype M28 group A Streptococcus isolates and cases of puerperal sepsis. J Infect Dis 220:882–891. 10.1093/infdis/jiz195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Byrne JL, Aagaard-Tillery KM, Johnson JL, Wright LJ, Silver RM. 2009. Group A streptococcal puerperal sepsis: initial characterization of virulence factors in association with clinical parameters. J Reprod Immunol 82:74–83. 10.1016/j.jri.2009.06.126. [DOI] [PubMed] [Google Scholar]
- 22.Chuang I, Van Beneden C, Beall B, Schuchat A. 2002. Population-based surveillance for postpartum invasive group a streptococcus infections, 1995-2000. Clin Infect Dis 35:665–670. 10.1086/342062. [DOI] [PubMed] [Google Scholar]
- 23.Mason KL, Aronoff DM. 2012. Postpartum group a Streptococcus sepsis and maternal immunology. Am J Reprod Immunol 67:91–100. 10.1111/j.1600-0897.2011.01083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamilton SM, Stevens DL, Bryant AE. 2013. Pregnancy-related group a streptococcal infections: temporal relationships between bacterial acquisition, infection onset, clinical findings, and outcome. Clin Infect Dis 57:870–876. 10.1093/cid/cit282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deutscher M, Lewis M, Zell ER, Taylor TH, Jr, Van Beneden C, Schrag S, Active Bacterial Core Surveillance Team.. 2011. Incidence and severity of invasive Streptococcus pneumoniae, group A Streptococcus, and group B Streptococcus infections among pregnant and postpartum women. Clin Infect Dis 53:114–123. 10.1093/cid/cir325. [DOI] [PubMed] [Google Scholar]
- 26.Green NM, Beres SB, Graviss EA, Allison JE, McGeer AJ, Vuopio-Varkila J, LeFebvre RB, Musser JM. 2005. Genetic diversity among type emm28 group A Streptococcus strains causing invasive infections and pharyngitis. J Clin Microbiol 43:4083–4091. 10.1128/JCM.43.8.4083-4091.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sitkiewicz I, Green NM, Guo N, Mereghetti L, Musser JM. 2011. Lateral gene transfer of streptococcal ICE element RD2 (region of difference 2) encoding secreted proteins. BMC Microbiol 11:65. 10.1186/1471-2180-11-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vlaminckx BJ, Mascini EM, Schellekens J, Schouls LM, Paauw A, Fluit AC, Novak R, Verhoef J, Schmitz FJ. 2003. Site-specific manifestations of invasive group a streptococcal disease: type distribution and corresponding patterns of virulence determinants. J Clin Microbiol 41:4941–4949. 10.1128/jcm.41.11.4941-4949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wizemann TM, Moskovitz J, Pearce BJ, Cundell D, Arvidson CG, So M, Weissbach H, Brot N, Masure HR. 1996. Peptide methionine sulfoxide reductase contributes to the maintenance of adhesins in three major pathogens. Proc Natl Acad Sci U S A 93:7985–7990. 10.1073/pnas.93.15.7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fichorova RN, Rheinwald JG, Anderson DJ. 1997. Generation of papillomavirus-immortalized cell lines from normal human ectocervical, endocervical, and vaginal epithelium that maintain expression of tissue-specific differentiation proteins. Biol Reprod 57:847–855. 10.1095/biolreprod57.4.847. [DOI] [PubMed] [Google Scholar]
- 31.Chaussee MS, Somerville GA, Reitzer L, Musser JM. 2003. Rgg coordinates virulence factor synthesis and metabolism in Streptococcus pyogenes. J Bacteriol 185:6016–6024. 10.1128/jb.185.20.6016-6024.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loughman JA, Caparon MG. 2007. Comparative functional analysis of the lac operons in Streptococcus pyogenes. Mol Microbiol 64:269–280. 10.1111/j.1365-2958.2007.05663.x. [DOI] [PubMed] [Google Scholar]
- 33.Wessels MR, Moses AE, Goldberg JB, DiCesare TJ. 1991. Hyaluronic acid capsule is a virulence factor for mucoid group A streptococci. Proc Natl Acad Sci U S A 88:8317–8321. 10.1073/pnas.88.19.8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Datta V, Myskowski SM, Kwinn LA, Chiem DN, Varki N, Kansal RG, Kotb M, Nizet V. 2005. Mutational analysis of the group A streptococcal operon encoding streptolysin S and its virulence role in invasive infection. Mol Microbiol 56:681–695. 10.1111/j.1365-2958.2005.04583.x. [DOI] [PubMed] [Google Scholar]
- 35.Sjögren J, Okumura CY, Collin M, Nizet V, Hollands A. 2011. Study of the IgG endoglycosidase EndoS in group A streptococcal phagocyte resistance and virulence. BMC Microbiol 11:120. 10.1186/1471-2180-11-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramirez-Pena E, Trevino J, Liu Z, Perez N, Sumby P. 2010. The group A Streptococcus small regulatory RNA FasX enhances streptokinase activity by increasing the stability of the ska mRNA transcript. Mol Microbiol 78:1332–1347. 10.1111/j.1365-2958.2010.07427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu M, Gong T, Zhang A, Tang B, Chen J, Zhang Z, Li Y, Zhou X. 2019. Mobile genetic elements in streptococci. Curr Issues Mol Biol 32:123–166. 10.21775/cimb.032.123. [DOI] [PubMed] [Google Scholar]
- 38.Novick RP, Ram G. 2017. Staphylococcal pathogenicity islands-movers and shakers in the genomic firmament. Curr Opin Microbiol 38:197–204. 10.1016/j.mib.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Faruque SM, Mekalanos JJ. 2012. Phage-bacterial interactions in the evolution of toxigenic Vibrio cholerae. Virulence 3:556–565. 10.4161/viru.22351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu J, Chen D, Peters BM, Li L, Li B, Xu Z, Shirliff ME. 2016. Staphylococcal chromosomal cassettes mec (SCCmec): a mobile genetic element in methicillin-resistant Staphylococcus aureus. Microb Pathog 101:56–67. 10.1016/j.micpath.2016.10.028. [DOI] [PubMed] [Google Scholar]
- 41.Brüssow H, Canchaya C, Hardt WD. 2004. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev 68:560–602. 10.1128/MMBR.68.3.560-602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fineran PC. 2019. Resistance is not futile: bacterial “innate” and CRISPR-Cas “adaptive” immune systems. Microbiology 165:834–841. 10.1099/mic.0.000802. [DOI] [PubMed] [Google Scholar]
- 43.Gardner SP, Olson JW. 2012. Barriers to horizontal gene transfer in Campylobacter jejuni. Adv Appl Microbiol 79:19–42. 10.1016/B978-0-12-394318-7.00002-4. [DOI] [PubMed] [Google Scholar]
- 44.Goldberg GW, Marraffini LA. 2015. Resistance and tolerance to foreign elements by prokaryotic immune systems–curating the genome. Nat Rev Immunol 15:717–724. 10.1038/nri3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weckel A, Ahamada D, Bellais S, Méhats C, Plainvert C, Longo M, Poyart C, Fouet A. 2018. The N-terminal domain of the R28 protein promotes emm28 group A Streptococcus adhesion to host cells via direct binding to three integrins. J Biol Chem 293:16006–16018. 10.1074/jbc.RA118.004134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang S, Green NM, Sitkiewicz I, Lefebvre RB, Musser JM. 2006. Identification and characterization of an antigen I/II family protein produced by group A Streptococcus. Infect Immun 74:4200–4213. 10.1128/IAI.00493-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stålhammar-Carlemalm M, Areschoug T, Larsson C, Lindahl G. 1999. The R28 protein of Streptococcus pyogenes is related to several group B streptococcal surface proteins, confers protective immunity and promotes binding to human epithelial cells. Mol Microbiol 33:208–219. 10.1046/j.1365-2958.1999.01470.x. [DOI] [PubMed] [Google Scholar]
- 48.Sitkiewicz I, Green NM, Guo N, Bongiovanni AM, Witkin SS, Musser JM. 2010. Adaptation of group A Streptococcus to human amniotic fluid. PLoS One 5:e9785. 10.1371/journal.pone.0009785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cook LCC, Chatterjee N, Li Y, Andrade J, Federle MJ, Eichenbaum Z. 2019. Transcriptomic analysis of Streptococcus pyogenes colonizing the vaginal mucosa identifies hupY, an MtsR-regulated adhesin involved in heme utilization. mBio 10:e00848-19. 10.1128/mBio.00848-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bessen DE, McShan WM, Nguyen SV, Shetty A, Agrawal S, Tettelin H. 2015. Molecular epidemiology and genomics of group A Streptococcus. Infect Genet Evol 33:393–418. 10.1016/j.meegid.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sarkar P, Danger JL, Jain I, Meadows LA, Beam C, Medicielo J, Burgess C, Musser JM, Sumby P. 2018. Phenotypic variation in the group A Streptococcus due to natural mutation of the accessory protein-encoding gene rocA. mSphere 3:e00519-18. 10.1128/mSphere.00519-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fittipaldi N, Beres SB, Olsen RJ, Kapur V, Shea PR, Watkins ME, Cantu CC, Laucirica DR, Jenkins L, Flores AR, Lovgren M, Ardanuy C, Linares J, Low DE, Tyrrell GJ, Musser JM. 2012. Full-genome dissection of an epidemic of severe invasive disease caused by a hypervirulent, recently emerged clone of group A Streptococcus. Am J Pathol 180:1522–1534. 10.1016/j.ajpath.2011.12.037. [DOI] [PubMed] [Google Scholar]
- 53.Kreikemeyer B, Nakata M, Koller T, Hildisch H, Kourakos V, Standar K, Kawabata S, Glocker MO, Podbielski A. 2007. The Streptococcus pyogenes serotype M49 Nra-Ralp3 transcriptional regulatory network and its control of virulence factor expression from the novel eno ralp3 epf sagA pathogenicity region. Infect Immun 75:5698–5710. 10.1128/IAI.00175-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lukomski S, Hoe NP, Abdi I, Rurangirwa J, Kordari P, Liu M, Dou SJ, Adams GG, Musser JM. 2000. Nonpolar inactivation of the hypervariable streptococcal inhibitor of complement gene (sic) in serotype M1 Streptococcus pyogenes significantly decreases mouse mucosal colonization. Infect Immun 68:535–542. 10.1128/IAI.68.2.535-542.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Calfee G, Danger JL, Jain I, Miller EW, Sarkar P, Tjaden B, Kreikemeyer B, Sumby P. 2017. Identification and characterization of serotype-specific variation in group A Streptococcus pilus expression. Infect Immun 86:e00792-17. 10.1128/IAI.00792-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1; Tables S1 to S4. Download IAI00722-20_Supp_1_seq9.pdf, PDF file, 2.0 MB (2MB, pdf)
Data Availability Statement
The generated raw sequence data are available for download from the NCBI Sequence Read Archive under the BioProject no. PRJNA704666. The RNA-seq data have been deposited in the Gene Expression Omnibus (GEO) database at the National Center for Biotechnology Information and are accessible through accession number GSE167343.