

Abstract
There have been recent significant advances in short-term outcomes in renal transplantation, however, long-term allograft survival remains a challenge. With reported incidences as high of 74.5% of chronic graft loss in patients with biopsies showing transplant glomerulopathy (TG), this syndrome represents an important factor for chronic allograft complications. In this review we show an overview of the novel mechanistic insights into pathogenesis of TG, as well as a brief description of the pathology, diagnosis and newer prognostic indices within TG diagnosis. These data raise intriguing roles for cell-mediated immunity and podocyte stress in TG as well as reinforce previous associations of TG with ABMR. We also delve into management strategies for TG and report the paucity of existing clinical trial data for this prevalent condition in renal transplants.
Keywords: allograft failure, proteinuria, transplant glomerulopathy
1 |. INTRODUCTION
Combined advances in our understanding of transplant immunology, immunosuppression, donor selection and surgical techniques have led to significant improvements in short-term outcomes in renal transplantation.1,2 However, long-term graft survival remains a challenge.3 Chronic transplant glomerulopathy (cg), also known as Transplant Glomerulopathy (TG), initially described in 1970 refers to a unique glomerular lesion that is now defined as duplication of the glomerular basement membranes leading to formation of double contours. It accompanies chronic allograft injury (CAI), and represents an important contributing factor in late allograft loss. TG lesions, when encountered in allograft biopsies have been repeatedly associated with poor graft outcomes.4–6 There have been important advances in the classification, pathogenesis, and potential mechanisms involved in the development of TG and its treatment over the last decade. This review will highlight novel advances in our understanding of the pathogenesis of TG, and discuss recent work on the diagnosis, prognosis and management of TG.
2 |. EPIDEMIOLOGY
From multiple case series, TG is a common lesion identified in 5%–20% of all studied biopsies7–10 (see Table 1). In data from the Mayo clinic, TG, defined pathologically as Cg > 0 on light microscopy, was seen in 20% of all biopsies by 5-years post-transplant and accounted for 36% of biopsies with any glomerular disease with proteinuria post-transplant.8 Another study from the United States (US) examined 525 clinically indicated allograft biopsies reporting a TG incidence of 10%, with 32% of TG patients losing their allografts.7 Among 1606 clinically indicated and surveillance transplant biopsies in a large US dataset, the incidence of TG was found to be 6%.9
TABLE 1.
Summary of case series showing sample size, nature of biopsies, median follow up, prevalence of TG, graft loss
| Sr no. | Series reference | Sample size | Nature of biopsies | Median follow up (months) | Prevalence of TG; N and percentage | Graft loss rate; percentage | Notes |
|---|---|---|---|---|---|---|---|
| 1. | Suri et al4 | 406 | Retrospective review of biopsies with TG | Review of biopsies with TG in a course of 4 years in a single center | 25 (6.1%) | 12% (4 years) | TG patients had increased rate of graft loss when compared to chronic rejection |
| 2. | Banfi et al6 | 666 | Clinically indicated | 120 months | 28 (5.6%) | 52% (120 months) | Incidence and clinical course of TG is not modified by CNI based immunosuppression |
| 3. | Kamal et al7 | 525 | Clinically indicated | 23 (1–46) months after biopsy | 52 (10%) | 32% (23 months after diagnosis) | Graft loss is associated with increased expression of ENDAT |
| 4. | Gloor et al8 | 582 | Surveillance and clinically indicated | 41–61 months after transplant | −55 (9.5%) at 2 years −117 (20%) at 5 years |
27% (3½ years) | Subclinical TG had similar poor prognosis as TG highlighting importance of subclinical TG in graft dysfunction |
| 5. | Patri et al9 | 1606 | Surveillance and clinically indicated | 60 months from diagnosis | 92 (6%) | 70% (60 months after diagnosis) | Development of prognostic index to predict prognosis of TG |
| 6. | Aubert et al10 | 8207 | Surveillance and clinically indicated | 33 months from time of transplant to development of TG | 552 (6.7%) | 74.5% (10 years) | Probabilistic data-driven archetype analysis approach refines the diagnostic and prognostic features associated with cases of TG |
| 7. | Sharif et al13 | 124 | Surveillance and clinically indicated | 42 months post biopsy | 31 (25%) in desensitized patients | 33.3 (42 months after diagnosis) | Incidence of TG was 25% biopsies from patients with HLA-incompatible desensitized renal transplant patients |
| 8. | Shimizu et al16 | 86 | Clinically indicated | Retrospective analysis of TG | only TG biopsies reported in study | 22% (72 months) | Most common pathologic finding was peritubular capillaritis |
| 9. | Sis et al17 | 1036 | Clinically indicated | Retrospective review | 53 (5.1%) | NA | Transplant glomerulopathy has evidence of alloantibody-mediated injury |
| 10 | Lesage et al18 | 61 | Clinically indicated | Retrospective review | Only TG biopsies reported in study | 16% (48 months) | TG is associated with poor prognosis whether there is evidence of tissue or peripheral alloantibody reactivity |
| 11. | Issa et al23 | 598 | Surveillance and clinically indicated | 54 months | 73 (12%) | 25% TG/C4D- and 80% TG/C4d+ (54 months) | Higher anti-HLA-II antibody levels are related to increase risk of developing TG |
| 12. | Joosten et al32 | 16 | Surveillance and clinically indicated | Retrospective Review | only TG biopsies with similar controls reported in study | NA | Humoral response of Glomerular Basement heparan sulfate proteoglycans agrin (GBM-HSPG) may play a role in pathogenesis of TG |
| 13. | Akalin et al39 | 428 | Clinically indicated | Retrospective Review | 36 (7%) TG + CAN | NA | Non-alloantibody-mediated process may be involved in the development of TGP in some patients |
| 14. | Homs et al41 | 35 | Clinically indicated | Retrospective Review | Only TG biopsies with similar controls reported in study | NA | Results indicate a role of an active T-mediated inflammatory and cytotoxic process in the pathogenesis of TGP |
| 15. | Li et al50 | 1587 | Surveillance and clinically indicated | 60 months | 180 (11%) | 65% (60 months after diagnosis) | ci + ct and cg at biopsy were predictors of unfavorable prognosis |
Note: The percentage of graft loss is reported from time of transplant unless otherwise indicated.
Surveillance biopsies are thought to be better estimates of the true prevalence of disease burden, as they identify subclinical lesions. In our own multi-center biopsy cohort including Australian and US centers, the cross-sectional incidence of subclinical TG was found to be 3% by the end of 3 months, and 5% by 2 years.11 Another study of protocol and clinically indicated biopsies showed that cumulative incidence of TG increased over 20% at five years.8 In a detailed biopsy study from France and Canada, involving 8207 post-transplant biopsies performed over 10 years, 552 biopsies (6.7%) had evidence of TG.10 Most interestingly, the median time to development of TG in this study was estimated to be 33 months post- transplantation. In an intriguing study from Australia that examined the development of TG using sequential surveillance biopsies, a small proportion of cases who developed TG lesions later had ultrastructural evidence of endothelial injury even at implantation biopsy, vs non-TG controls.12 An immunologic basis for the development of TG is also suggested by data examining TG prevalence in HLA-incompatible kidney transplants, where, in 124 one-year post-transplant surveillance biopsies in desensitized recipients, TG lesions were present in 25% (vs ~5% in all transplants) and resulted in worse graft survival.13 Together, these reports show that TG is a common lesion in both clinically indicated and surveillance biopsies, with an increasing prevalence over time post-transplantation, increasing alloimmune risk, and the presence of coexisting proteinuria in the patient (vs no proteinuria).
3 |. PATHOGENESIS
The histopathologic changes that are now considered emblematic of TG were first described in association with rejection by Busch et al14 in 1971, and thought to be due to chronic, persistent endothelial damage secondary to immunological mechanisms. That repetitive injury to endothelium may be the precursor to the development of TG is also supported by more recent studies where significantly increased expression of endothelial cell-associated genes (ENDATs) have been identified in transcriptomes of TG biopsies (vs non-TG biopsies).7 The pathogenic basis of such persistent endothelial injury that leads to development of TG is likely multifactorial, including donor-specific anti-HLA antibodies (HLA-DSA), non-HLA antibodies, cell-mediated rejection mechanisms, thrombotic microangiopathy and hepatitis C infection (reviewed in Filippone et al).15
3.1 |. Anti-HLA antibodies
Among all the proposed mechanisms, the most frequently reported has been the association of anti-Human Leukocyte antigen donor-specific antibody (anti-HLA-DSA) and antibody-mediated mediated rejection (ABMR) and endothelial damage leading to the development of TG.15–18 Experimentally, the development of TG was shown elegantly in an allotransplantation model in Cynomolgus monkeys, where the animals developed chronic ABMR with coexistent anti-Major Histocompatibility Complex (anti-MHC) DSA in sera and C4d staining in renal allograft biopsies along with TG.19 In this study the time frame of development of TG ranged from 82–818 days with median first appearance of TG at 161 days. TG lesions have also been recently reported in a novel murine model of antibody-mediated injury characterized by high titers of DSA.20,21 In this model, CCR5-knockout B6 recipient mice develop exceedingly high titers of anti-MHC DSA after allogenic A/J donor transplants and acute ABMR, while F1 donors (ie, A/J xB6 offspring) with similarly high DSA titers developed chronic ABMR and TG.21 As discussed later, the model used in this study demonstrated that diminished NK cell activation in F1 donors allowed sustained chronic TG like injury from antibodies and effector mechanisms, but prevented acute ABMR.
The association with humoral alloimmunity and TG has also been evident in clinical studies. First, TG is considered to be the defining lesion of chronic ABMR. The prevalence of TG is higher in HLA-incompatible transplants requiring desensitization versus others.13 A higher incidence of antecedent or concurrent ABMR and increased prevalence of DSA has been consistently reported in patients with TG.22–24 In addition to preformed DSA, the development of de novo DSA has been associated with TG, suggesting that either new or recurrent endothelial damage from de novo DSA also has pathogenetic relevance to TG.25 Numerous series have also demonstrated the association of anti-HLA-DSA and/or C4d with TG, and have been discussed in detail in prior reviews.15,26 Histopathological data have also consistently reported a greater association of TG with glomerular or peritubular capillaritis, that is, Banff g/ptc scores (microvascular inflammation (MVI) considered to be associated with ABMR), rather than with Banff i/t scores (associated with TCMR).16 It is important to note here that recent work has raised questions regarding the pathogenesis of MVI and the requirement (if any), of DSA or ABMR for the development of MVI27 (discussed below). The association of humoral alloimmunity with TG also extends to subclinical ABMR, where development of TG has been associated with subclinical ABMR diagnosis in surveillance biopsies.24
3.2 |. Role of non-HLA antibodies
The hypothesis that non-HLA antibodies might contribute to ABMR has been most clearly shown in HLA identical renal allografts where recipients developed anti-donor endothelial reactive antibodies.28 Recent evidence suggests a role of non-HLA auto and alloantibodies in chronic graft loss either through independent cytotoxicity or with concurrent anti-HLA antibodies.29 These non-HLA antibodies can again be either preformed or de novo. The milieu of tissue injury in a renal allograft as a result of ischemia-reperfusion injury and/or episodes of rejection may unmask polymorphic donor antigens causing antibody formation against non-HLA proteins.30 Non-HLA antibody-mediated injury can also arise from new exposure of auto-antigens by HLA-DSA-mediated or cell-mediated injury. Although many non-HLA antibodies have been reported in renal transplant patients, we will focus on those that have been specifically associated with TG.
Dinavahi et al31 used protein microarrays to compare antibody panels in pre- and post-transplant sera from patients with and without transplant glomerulopathy. The study showed that reactivity against peroxismal-trans-2-enoyl-coA-reductase was strongly associated with development of TG. Experimentally, Joosten et al32 showed in mouse models the development of antibodies to glomerular basement membrane components, perlecan, collagen type IV and type VI and association with the development of TG, which suggested role of non-HLA antibodies in causing endothelial damage and resultant changes of chronic renal allograft rejection. Subsequently, sera of 16 patients with TG demonstrated the presence of antibodies to glomerular basement membrane heparan sulfate proteoglycans, specifically Agrin. In clinical data, the presence of anti-Agrin antibodies was identified in 44% TG cases and associated with rejection episodes prior to the diagnosis of TG.33 Angaswamy et al34 examined sera from kidney transplant patients who developed TG for association of antibodies to renal tissue restricted self-antigens, particularly fibronectin and collagen type IV, and found that the presence of these antibodies increased the odds of TG. Here, non-HLA antibodies were detected irrespective of the presence of anti-HLA antibodies, while MVI features and C4d deposition were equivalent in patients with non-HLA or HLA antibodies, suggesting an independent role for non-HLA antibodies in the development of TG. Anti-endothelial cell antibodies (AECAs) detected by ELISA were demonstrably increased with ABMR along with MVI lesions and TG. However, a higher presence of anti-HLA-DSA (75%) was also seen in AECA-ELISA positive patients, possibly suggesting a highly sensitized study cohort.35 The most widely studied non-HLA antigen target implicated in ABMR is the angiotensin receptor (AT1R).36–38 However, the association of anti-AT1R antibodies with TG independent of HLA-DSA is yet unclear. Regardless, these studies suggest that HLA or non-HLA antibodies are associated with TG clinically.
3.3 |. Role of cell-mediated mechanisms
Clinical biopsy studies demonstrating TG lesions without evidence of recognizable acute or chronic ABMR have suggested that the pathogenesis of TG in these cases may be caused by non-humoral mechanisms. Akalin et al39 reported that 60% of TG cases in their biopsy cohort developed in the absence of anti-HLA antibodies, both DSA and non-DSA. Furthermore, C4d in peritubular capillaries (either diffuse or focal) was found in only 14% of biopsies with TG, implying that non-humoral mechanisms were involved in the development of TG. Recently, novel data from this group also examined gene expression profiles of DSA negative/C4d negative TG biopsies. They identified increased allograft expression of cytotoxic T cell-associated transcripts in the subset of DSA negative/C4d negative TG biopsies vs controls/DSA-positive TG biopsies. Upregulation of transcripts associated with macrophages and NK cells was also identified in DSA-negative TG biopsies, supporting a role for cell-mediated mechanisms in the development of TG.40 Similarly, increased activity of CD4 and CD8 T cells within TG biopsies were also identified by measuring increased expression of IFN-y, T-bet and Granzyme B supporting cell-medicated mechanisms in the pathogenesis of TG.41
MVI, an accompaniment of ABMR and TG has been conventionally ascribed to humoral alloimmunity. However, the pivotal role of NK cell-mediated injury in MVI and ABMR was demonstrated experimentally using CCR5-knockout recipient B6 mice that develop high DSA titers after allotransplantation. Here, depletion of NK cells prevented ABMR lesions in spite of DSA.42 Furthermore, high levels of IFN-y, perforin and granzyme B were found 3 days after transplantation suggesting T cell/NK cell activation in the allograft occurred before DSA was detectable in recipient mice with MVI.
Analogous exciting clinical data also suggest that NK cell-mediated injury has an important role in the etio-pathogenesis of MVI. Koenig et al27 performed genetic analysis of donor- and recipient samples of 129 transplant biopsies with MVI showing that ~half of MVI biopsies had anti-donor antibody-independent MVI (41.1% of biopsies were MVI+DSA−). In biopsies with MVI+DSA+C3d- as well as in MVI+DSA− there was a significantly higher prevalence of mismatches between donor class-I HLA and recipient KIRs (inhibitory killer cell immunoglobulin-like receptor). KIRs are expressed on NK cells and crucial for modulating “non-self” and “missing self” responses by transmitting activating or inhibitory signals by interacting with specific donor Class-I HLA molecules. Hence, mismatches between donor Class-I and recipient NK cells seen in MVI+DSA− may result in increased NK activation, that is, “non-self” recognition by NK cells. Allograft endothelium would also be unable to inhibit NK cell activation and promoting “missing self” signals. Together these would activate recipient NK cells independent of DSA. Additionally, when comparing antibody-dependent and antibody-independent MVI, there was no difference noticed in graft survival, both having poor outcomes. Allograft gene expression data examining ABMR biopsies to estimate cell fractions also identified activated NK cell signatures in ABMR biopsies.43 Here too, the NK cell activation signature associated with graft loss independent of DSA.
Whether NK cell-mediated injury could aggravate DSA-mediated endothelial injury in MVI, as well as initiate DSA-negative MVI needs to be further examined in clinical data. In sum, these provocative data suggest that a common pathway in TG & MVI is NK cell-mediated injury either facilitated by the humoral mechanisms or initiated by antibody-independent “missing self” recognition.
3.4 |. Role of timing from transplantation: podocytes and TG
Although endothelial damage initiates the cascade of injury culminating in characteristic glomerular changes of TG, the final common pathway could be podocyte depletion leading to graft loss. In seminal work to study the development of TG, Wiggins et al44 tested the roles of the “two kidneys to one kidney transition” occurring in all allografts as well as immunologic injury. They examined the urine podocin/creatinine ratio as a marker of podocyte depletion after transplantation and correlated these data with morphometric measurements (glomerular volume fractions and podometrics), and with time from transplantation. Surprisingly, while allograft recipients can reasonably be expected to have half the number of nephrons (ie, two kidney to one transition), and therefore podocytes, this study demonstrated a significantly increased rate of podocyte depletion in all allograft recipients vs binephric controls. This observation suggested excessive podocyte stress even at baseline in uninephric transplant recipients, potentially explained by the two kidney to one transition.
Furthermore, they identified that urinary podocyte loss was markedly higher in patients with TG vs patients with non-glomerular allograft pathology (tubular injury or IFTA). The steepest decline in podocyte density, however, occurred in the subset of TG patients diagnosed within 2-years of transplant (early TG). The early TG group also demonstrated the greatest association with Banff rejection episodes and HLA-DSA, while later TG (>2 years) was less associated with biopsy confirmed rejection episodes, and had a distinct podometric profile.44 Subsequent data from the same group suggested that such early loss of podocytes in urine from any cause was associated with later graft loss.45 Together, these data may suggest that early TG could result from early persistent endothelial injury (mostly immunologic) that serves to accentuate the podocyte stress necessitated by the two kidney to one transition, thus promoting podocyte depletion and progressive allograft loss.
4 |. DIAGNOSIS
4.1 |. Histology
The term chronic transplant glomerulopathy (cg) is a histopathological entity defined as the duplication and double contouring of glomerular basement membranes. The earliest form (graded as cg1a) can only be diagnosed by electron microscopy. The updated ultrastructural diagnostic criteria46 are the formation of new basement membranes (new lamina densa) accompanied by subendothelial translucent material and endothelial swelling (Figure 1). C4d staining may be present (Figure 2). No immune complex deposits should be present or else the diagnosis defaults to membranoproliferative glomerulonephritis. Diagnosing the earliest stage of the disease by light microscopy (grade cg1b) requires that 1%–25% of the glomerular capillary loops show double contours in the most affected glomerulus. The PAS stain can be quite helpful in recognizing this lesion when the Jones stain is technically suboptimal. Grades cg2 and cg3 are respectively assigned when 26%–50% and >50% of the capillary loops show duplication in the maximally affected glomerulus. Glomeruli with ischemic changes, collapsed capillary loops and >50% segmental sclerosis should not be scored. Concurrent changes of chronic ABMR and/or chronic TCMR may be present, and include glomerulitis, ultrastructural multilayering of peritubular capillary basement membranes, tubulitis, and chronic allograft arteriopathy. Non-specific changes like the degree of interstitial fibrosis and tubular atrophy must also be considered in the overall assessment of the severity of antibody-mediated injury.9 Clinical-pathologic correlations in TG: The earliest form of cg is asymptomatic and only identified by kidney biopsy as evidenced by multiple case series of TG in surveillance biopsies.8 The histologic changes precede clinical manifestations and can occur as early as 3-months. Later stages of cg are accompanied by slowly progressive allograft dysfunction elevated creatinine and worsening proteinuria. The morphologic spectrum of cg is much wider than can be summarized by a three-tier grading system that takes only the maximally involved glomerulus into account. For example, a biopsy can be designated a cg2 even if a single glomerulus has 30% of the capillary loops affected, whereas all other glomeruli may be normal or mildly affected. It is essential for clinicians to review the biopsy directly with a pathologist to better appreciate the extent of disease in any given patient. While interpreting studies showing the overall 53.7%–80.6% agreement rates between cg scores assigned by different pathologists,47 it should be kept in mind that cg0 and cg3 can be quite consistently identified by the trained observer. It is the intermediate stages where exact biopsy classification is imprecise. Taking into account the clinical severity of graft dysfunction can minimize the effect of this imprecision on patient management. Lack of sufficient attention to these variables while building cg-based machine learning classifiers has contributed to the poor correlations between histologic and molecular methods of diagnosing antibody-mediated injury. It would be apparent that imprecision in machine learning input will significantly affect the accuracy of any resulting molecular test. Some molecular tests (such as the Molecular Microscope) have additional issues that they are performed on as little as 3 mm of tissue, which is insufficient to capture the variation seen in 1–2 cm long core biopsies. Cg probability scores are included in these molecular reports but the inherent variation in the generation of these scores has not yet been rigorously determined, and systematic correlations with clinical parameters remain to be performed.
FIGURE 1.
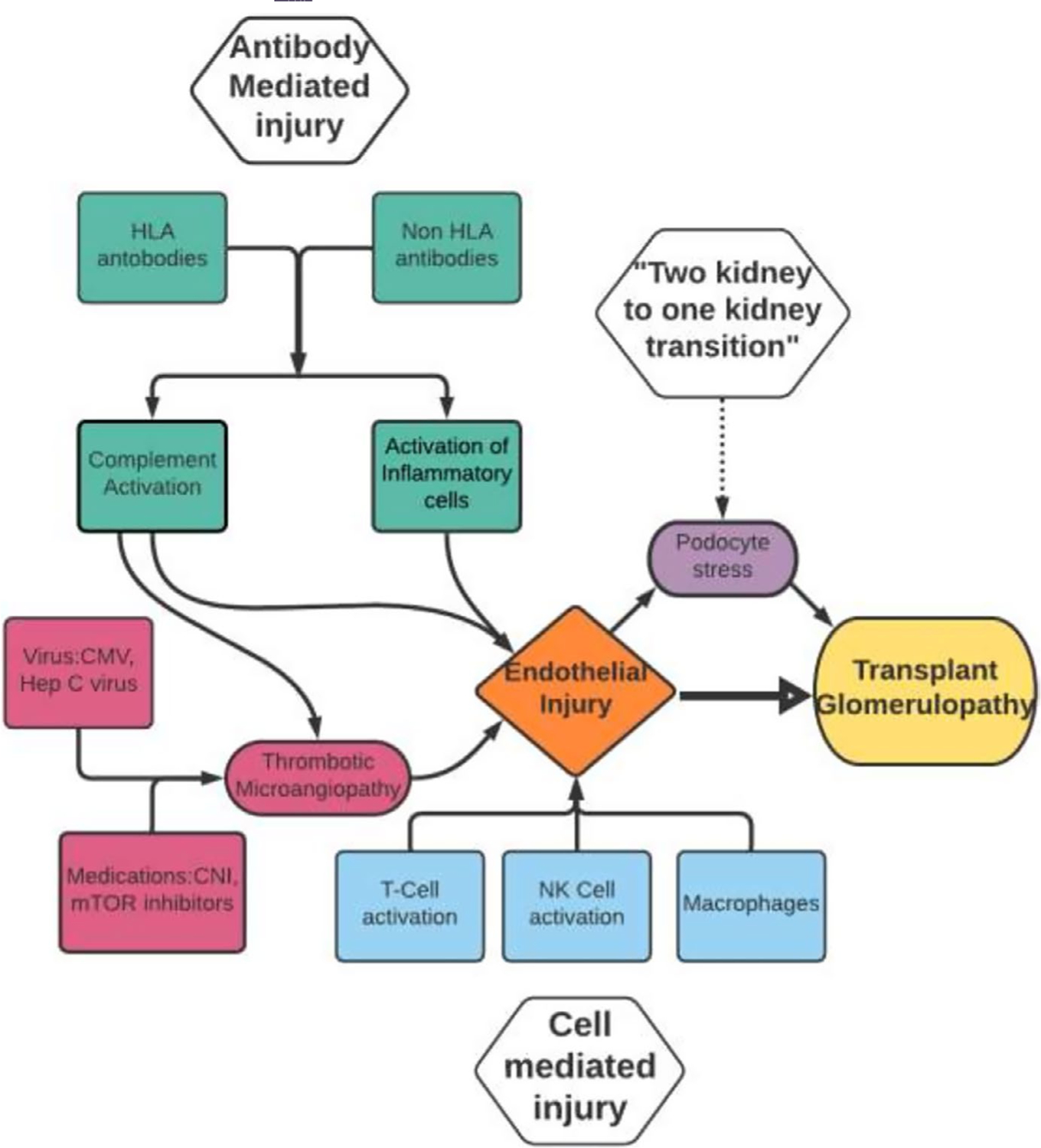
Proposed pathogenesis flow chart of Transplant Glomerulopathy. There are 3 main areas: Antibody-mediated injury, cell-mediated injury and “Two kidney to one Kidney Transition”, all leading to a common pathway which is endothelial injury
FIGURE 2.
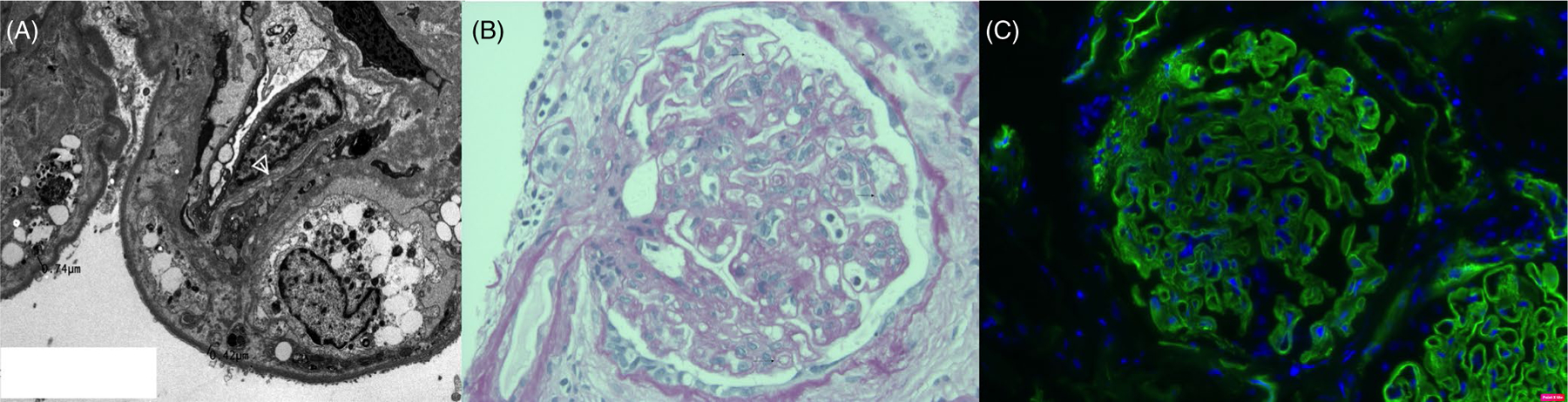
(A) Electron microscopy features of cg. Note multiple layers of new glomerular basement membrane formation (arrow head). The capillary wall has been insinuated by a mesangial cell and it cytoplasmic processes (so-called mesangial interposition). Since, electron microscopic changes precede histologic abnormalities, they have the potential to act as surrogate markers in clinical trials seeking to evaluate treatment regimens to prevent chronic ABMR. (B) Light microscopy: PAS image showing TG graded as cg1b in the Banff Schema. This finding is subtle and the earliest findings can be over-interpreted. Pathologist agreement between cg0 and cg1b is of the order of 45%. Accordingly, molecular classifiers developed by including biopsies graded as cg1b have the potential to generate false positive diagnoses of ABMR. (C) C4d staining in the glomerular basement membranes can be indicative of cg. However, as opposed to peritubular capillary staining glomerular basement membranes staining it is not a specific marker ABMR. It has been described in other forms of injury that lead to basement membrane remodeling such as ischemic glomerulopathy, membranoproliferative glomerulonephritis, thrombotic microangiopathy and eclampsia of pregnancy.64 Some non-specific linear C4d staining in the glomerular basement membranes is seen in virtually all biopsies, and follow up electron microscopy studies are performed only on tissues with distinctly granular staining
5 |. PROGNOSIS
Overall, a diagnosis of TG has been consistently associated with reduced long-term allograft survival.4,6,48 Recently, Loupy et al49 developed IBOX, a comprehensive prognostic index to estimate the risk of allograft loss in kidney transplant recipients with clinical and histologic parameters where the presence of TG independently conferred a hazard ratio of 5–6 for graft loss. However, within the TG diagnosis, several individual parameters have been associated with graft loss in studies. For instance, time of onset of TG (early vs late), TG severity (by Banff) and amount of proteinuria have all been associated with poor long-term allograft survival.6,18,50 Further, differential gene expression of complement cascade, interleukins, granulysin and ENDATs were associated with allograft loss, suggesting that gene expression profiles might help in predicting allograft loss within this syndrome.7
Patri et al,9 in 2016, developed a novel prognostic index in patients with TG by analyzing 92 TG patients. To prognosticate allograft loss, Patri et al51 used the following parameters: chronic inflammation score (combination of Banff ci, ct and ti scores), proteinuria and serum creatinine at the time of biopsy. Patients were then divided into low risk, medium risk and high risk for allograft failure. This score was externally validated in French and US patients. Similarly, Aubert et al10 identified distinct profiles in renal transplant recipients with TG to better explain the heterogeneity in the allograft survival in TG. Archetype 1 with the best allograft survival in the groups was characterized by the highest mean eGFR, low grade proteinuria, lowest cg, lowest IFTA score and lowest microcirculation inflammation and tubulointerstitial inflammation Banff score. Archetype 4 with the lowest allograft survival had the highest level of proteinuria, highest mean cg score, higher IFTA but low microcirculation inflammation and tubulointerstitial inflammation score. Hence, these data suggest new methods to quantify variability of graft survival within TG albeit only partially explaining the underlying biologic processes causing the variability.
6 |. TREATMENT
TG has posed a significant challenge as far as treatment is concerned. Strategies employed before the development of TG to address causative mechanisms may be more effective than treatment of TG after the usually inexorable chronic allograft changes have already occurred. There are however, no completed randomized controlled trials specifically designed to prevent or treat TG reported at this time. We briefly outline generally utilized measures and specific therapies in clinical use to treat TG below.
6.1 |. General management
General management of patients with TG include strategies to reduce intra-glomerular pressure by weight loss, blood pressure and diabetes control, as well as renin-angiotensin-aldosterone (RAAS) blockade. Based on data from proteinuria in native kidney diseases, RAAS blockade could be effective in slowing graft loss in patients with TG, although it has not been specifically studied in this population. These measures have not been studied exclusively in patients with TG.
6.2 |. Management directed toward cause
6.2.1 |. Antibody-mediated rejection
The most common pathogenic mechanism for development of TG is ABMR. A non-randomized controlled trial to study the treatment of TG during chronic ABMR examined the use of intravenous immunoglobulin (IVIG) with Rituximab in 24 patients with TG and compared to 10 patients with TG (standard-of-care). This trial however, did not show any difference in graft survival at 2 years follow up with graft survival of 40%–47% in the two groups.52 A randomized trial of clazakizumab to treat chronic ABMR is enrolling.
6.2.2 |. Complement targeted therapy
Complement targeted therapies like Eculizumab and C1 esterase inhibitors have been studied for prevention of IRI and DGF in order to improve graft survival. These agents have not been studied to prevent TG specifically. In an observational study done by Schinstock et al,53 long-term outcomes of eculizumab-treated, positive crossmatch kidney transplant recipients were compared to age-matched (a) untreated +XM and (b) negative crossmatch (-XM) controls. Five-year protocol biopsy findings showed chronic glomerulopathy (cg) in 57% of Eculizumab-treated patients as compared to 71% untreated patients.
6.2.3 |. Hepatitis C virus (HCV) treatment
The association of HCV with TG is likely to be less important in the current era due to highly effective anti-viral agents (DAAs) and direct data regarding TG and HCV has not been reported from DAA clinical trials.
6.2.4 |. Thrombotic microangiopathy (TMA)
Development of de novo TMA right after transplantation is a rare but serious condition that is associated with later TG. Calcineurin inhibitors may cause TMA, where treatment involves changing immunosuppressant. TMA may also be triggered by ABMR/cell-mediated rejection and/or infections like CMV. Treatment of the cause has been shown to be successful in preventing graft loss in certain cases.54–57 The use of complement antagonism for TMA and TG outcomes will need further data.
6.3 |. Novel agents
6.3.1 |. Tocilizumab
Interleukin 6 (IL-6) regulates maturation/activation of T cells, B Cells and plasma cells.58,59 Blockade of IL-6 receptor results in direct inhibition of plasma cell anti-HLA antibody production, induction of T regulatory cells and T follicular helper cells.58,60 Tocilizumab is a monoclonal antibody against IL-6 receptor which has showed efficacy in a study to desensitize patients, and subsequently in small series to treat DSA-positive chronic ABMR with or without TG who failed standard treatment.61,62
6.3.2 |. Acthar gel
A single center report described the potential benefit of Acthar gel in TG.63 Further controlled trials are required to test its use in TG.
7 |. SUMMARY
In summary, we review here recent data regarding the novel mechanistic insights into pathogenesis of TG. These data suggest intriguing roles for cell-mediated immunity and podocyte stress in TG as well as reinforce previous associations of TG with ABMR. We briefly describe the pathology and diagnosis of TG, and incorporate newer prognostic indices within TG diagnosis that impact graft survival. Finally, we summarize the management strategies for TG and report the paucity of existing clinical trial data for this prevalent condition in renal transplants, reflecting an important and urgent need for the transplant community.
ACKNOWLEDGEMENTS
MCM is supported by National Institute of Health award number: ROIDK122164.
Funding information
National Institute of Diabetes and Digestive and Kidney Diseases, Grant/Award Number: DK122164
Footnotes
CONFLICT OF INTEREST
None.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1.Guild WR, Harrison JH, Merrill JP, Murray J. Successful homotransplantation of the kidney in an identical twin. Trans Am Clin Climatol Assoc. 1955;67:167–173. [PMC free article] [PubMed] [Google Scholar]
- 2.Coemans M, Susal C, Dohler B, et al. Analyses of the short- and long-term graft survival after kidney transplantation in Europe between 1986 and 2015. Kidney Int. 2018;94(5):964–973. [DOI] [PubMed] [Google Scholar]
- 3.Hariharan S Long-term kidney transplant survival. Am J Kidney Dis. 2001;38(6 Suppl 6):S44–S50. [DOI] [PubMed] [Google Scholar]
- 4.Suri DL, Tomlanovich SJ, Olson JL, Meyer TW. Transplant glomerulopathy as a cause of late graft loss. Am J Kidney Dis. 2000;35(4):674–680. [DOI] [PubMed] [Google Scholar]
- 5.Kieran N, Wang X, Perkins J, et al. Combination of peritubular c4d and transplant glomerulopathy predicts late renal allograft failure. J Am Soc Nephrol: JASN. 2009;20(10):2260–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banfi G, Villa M, Cresseri D, Ponticelli C. The clinical impact of chronic transplant glomerulopathy in cyclosporine era. Transplantation. 2005;80(10):1392–1397. [DOI] [PubMed] [Google Scholar]
- 7.Kamal L, Broin PO, Bao Y, et al. Clinical, histological, and molecular markers associated with allograft loss in transplant glomerulopathy patients. Transplantation. 2015;99(9):1912–1918. [DOI] [PubMed] [Google Scholar]
- 8.Gloor JM, Sethi S, Stegall MD, et al. Transplant glomerulopathy: subclinical incidence and association with alloantibody. Am J Transplant. 2007;7(9):2124–2132. [DOI] [PubMed] [Google Scholar]
- 9.Patri P, Seshan SV, Matignon M, et al. Development and validation of a prognostic index for allograft outcome in kidney recipients with transplant glomerulopathy. Kidney Int. 2016;89(2):450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aubert O, Higgins S, Bouatou Y, et al. Archetype analysis identifies distinct profiles in renal transplant recipients with transplant glomerulopathy associated with allograft survival. J Am Soc Nephrol: JASN. 2019;30(4):625–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Menon MC, Chuang PY, Li Z, et al. Intronic locus determines SHROOM3 expression and potentiates renal allograft fibrosis. J Clin Invest. 2015;125(1):208–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wavamunno MD, O’Connell PJ, Vitalone M, et al. Transplant glomerulopathy: ultrastructural abnormalities occur early in longitudinal analysis of protocol biopsies. Am J Transplant. 2007;7(12):2757–2768. [DOI] [PubMed] [Google Scholar]
- 13.Sharif A, Kraus ES, Zachary AA, et al. Histologic phenotype on 1-year posttransplantation biopsy and allograft survival in HLA-incompatible kidney transplants. Transplantation. 2014;97(5):541–547. [DOI] [PubMed] [Google Scholar]
- 14.Busch GJ, Galvanek EG, Reynolds ES Jr. Human renal allografts. Analysis of lesions in long-term survivors. Hum Pathol. 1971;2(2):253–298. [DOI] [PubMed] [Google Scholar]
- 15.Filippone EJ, McCue PA, Farber JL. Transplant glomerulopathy. Mod Pathol. 2018;31(2):235–252. [DOI] [PubMed] [Google Scholar]
- 16.Shimizu T, Ishida H, Toki D, et al. Clinical and pathological analyses of transplant glomerulopathy cases. Nephrology. 2014;19(Suppl 3):21–26. [DOI] [PubMed] [Google Scholar]
- 17.Sis B, Campbell PM, Mueller T, et al. Transplant glomerulopathy, late antibody-mediated rejection and the ABCD tetrad in kidney allograft biopsies for cause. Am J Transplant. 2007;7(7):1743–1752. [DOI] [PubMed] [Google Scholar]
- 18.Lesage J, Noel R, Lapointe I, et al. Donor-specific antibodies, C4d and their relationship with the prognosis of transplant glomerulopathy. Transplantation. 2015;99(1):69–76. [DOI] [PubMed] [Google Scholar]
- 19.Smith RN, Kawai T, Boskovic S, et al. Four stages and lack of stable accommodation in chronic alloantibody-mediated renal allograft rejection in Cynomolgus monkeys. Am J Transplant. 2008;8(8):1662–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abe T, Ishii D, Gorbacheva V, et al. Anti-huCD20 antibody therapy for antibody-mediated rejection of renal allografts in a mouse model. Am J Transplant. 2015;15(5):1192–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yagisawa T, Tanaka T, Miyairi S, et al. In the absence of natural killer cell activation donor-specific antibody mediates chronic, but not acute, kidney allograft rejection. Kidney Int. 2019;95(2):350–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gloor JM, Cosio FG, Rea DJ, et al. Histologic findings one year after positive crossmatch or ABO blood group incompatible living donor kidney transplantation. Am J Transplant. 2006;6(8):1841–1847. [DOI] [PubMed] [Google Scholar]
- 23.Issa N, Cosio FG, Gloor JM, et al. Transplant glomerulopathy: risk and prognosis related to anti-human leukocyte antigen class II antibody levels. Transplantation. 2008;86(5):681–685. [DOI] [PubMed] [Google Scholar]
- 24.Loupy A, Suberbielle-Boissel C, Hill GS, et al. Outcome of subclinical antibody-mediated rejection in kidney transplant recipients with preformed donor-specific antibodies. Am J Transplant. 2009;9(11):2561–2570. [DOI] [PubMed] [Google Scholar]
- 25.Wiebe C, Gibson IW, Blydt-Hansen TD, et al. Evolution and clinical pathologic correlations of de novo donor-specific HLA antibody post kidney transplant. Am J Transplant. 2012;12(5):1157–1167. [DOI] [PubMed] [Google Scholar]
- 26.Husain S, Sis B. Advances in the understanding of transplant glomerulopathy. Am J Kidney Dis. 2013;62(2):352–363. [DOI] [PubMed] [Google Scholar]
- 27.Koenig A, Chen CC, Marcais A, et al. Missing self triggers NK cell-mediated chronic vascular rejection of solid organ transplants. Nat Commun. 2019;10(1):5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahern AT, Artruc SB, DellaPelle P, et al. Hyperacute rejection of HLA-AB-identical renal allografts associated with B lymphocyte and endothelial reactive antibodies. Transplantation. 1982;33(1):103–106. [DOI] [PubMed] [Google Scholar]
- 29.Opelz G, Collaborative Transplant Study. Non-HLA transplantation immunity revealed by lymphocytotoxic antibodies. Lancet. 2005;365(9470):1570–1576. [DOI] [PubMed] [Google Scholar]
- 30.Cardinal H, Dieude M, Hebert MJ. The emerging importance of non-HLA autoantibodies in kidney transplant complications. J Am Soc Nephrol: JASN. 2017;28(2):400–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dinavahi R, George A, Tretin A, et al. Antibodies reactive to non-HLA antigens in transplant glomerulopathy. J Am Soc Nephrol: JASN. 2011;22(6):1168–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joosten SA, van Dixhoorn MG, Borrias MC, et al. Antibody response against perlecan and collagen types IV and VI in chronic renal allograft rejection in the rat. Am J Pathol. 2002;160(4):1301–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Joosten SA, Sijpkens YW, van Ham V, et al. Antibody response against the glomerular basement membrane protein agrin in patients with transplant glomerulopathy. Am J Transplant. 2005;5(2):383–393. [DOI] [PubMed] [Google Scholar]
- 34.Angaswamy N, Klein C, Tiriveedhi V, et al. Immune responses to collagen-IV and fibronectin in renal transplant recipients with transplant glomerulopathy. Am J Transplant. 2014;14(3):685–693. [DOI] [PubMed] [Google Scholar]
- 35.Jackson AM, Sigdel TK, Delville M, et al. Endothelial cell antibodies associated with novel targets and increased rejection. J Am Soc Nephrol: JASN. 2015;26(5):1161–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dragun D, Muller DN, Brasen JH, et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N Engl J Med. 2005;352(6):558–569. [DOI] [PubMed] [Google Scholar]
- 37.Reinsmoen NL, Lai CH, Heidecke H, et al. Anti-angiotensin type 1 receptor antibodies associated with antibody mediated rejection in donor HLA antibody negative patients. Transplantation. 2010;90(12):1473–1477. [DOI] [PubMed] [Google Scholar]
- 38.Lefaucheur C, Viglietti D, Bouatou Y, et al. Non-HLA agonistic anti-angiotensin II type 1 receptor antibodies induce a distinctive phenotype of antibody-mediated rejection in kidney transplant recipients. Kidney Int. 2019;96(1):189–201. [DOI] [PubMed] [Google Scholar]
- 39.Akalin E, Dinavahi R, Dikman S, et al. Transplant glomerulopathy may occur in the absence of donor-specific antibody and C4d staining. Clin J Am Soc Nephrol. 2007;2(6):1261–1267. [DOI] [PubMed] [Google Scholar]
- 40.Hayde N, Bao Y, Pullman J, et al. The clinical and genomic significance of donor-specific antibody-positive/C4d-negative and donor-specific antibody-negative/C4d-negative transplant glomerulopathy. Clin J Am Soc Nephrol. 2013;8(12):2141–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Homs S, Mansour H, Desvaux D, et al. Predominant Th1 and cytotoxic phenotype in biopsies from renal transplant recipients with transplant glomerulopathy. Am J Transplant. 2009;9(5): 1230–1236. [DOI] [PubMed] [Google Scholar]
- 42.Kohei N, Tanaka T, Tanabe K, et al. Natural killer cells play a critical role in mediating inflammation and graft failure during antibody-mediated rejection of kidney allografts. Kidney Int. 2016;89(6):1293–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yazdani S, Callemeyn J, Gazut S, et al. Natural killer cell infiltration is discriminative for antibody-mediated rejection and predicts outcome after kidney transplantation. Kidney Int. 2019;95(1):188–198. [DOI] [PubMed] [Google Scholar]
- 44.Yang Y, Hodgin JB, Afshinnia F, et al. The two kidney to one kidney transition and transplant glomerulopathy: a podocyte perspective. J Am Soc Nephrol: JASN. 2015;26(6):1450–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Naik AS, Afshinnia F, Aqeel J, et al. Accelerated podocyte detachment early after kidney transplantation is related to long-term allograft loss of function. Nephrol Dial Transplant. 2019;34(7):1232–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loupy A, Haas M, Roufosse C, et al. The Banff 2019 Kidney Meeting Report (I): updates on and clarification of criteria for T cell- and antibody-mediated rejection. Am J Transplant. 2020;20(9):2318–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith B, Cornell LD, Smith M, et al. A method to reduce variability in scoring antibody-mediated rejection in renal allografts: implications for clinical trials - a retrospective study. Transpl Int. 2019;32(2):173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lopez Jimenez V, Fuentes L, Jimenez T, et al. Transplant glomerulopathy: clinical course and factors relating to graft survival. Transplant Proc. 2012;44(9):2599–2600. [DOI] [PubMed] [Google Scholar]
- 49.Loupy A, Aubert O, Orandi BJ, et al. Prediction system for risk of allograft loss in patients receiving kidney transplants: international derivation and validation study. BMJ. 2019;366:l4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li X, Chen J, Cheng D, et al. Histopathologic features that predict transplant glomerulopathy progression in a Chinese cohort. Am J Nephrol. 2019;49(6):425–434. [DOI] [PubMed] [Google Scholar]
- 51.Talwar M, Balaraman V, Bhalla A, et al. Validation of prognostic index for allograft outcome in kidney transplant recipients with transplant glomerulopathy. Kidney Int Rep. 2020;5(6):915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bachelet T, Nodimar C, Taupin JL, et al. Intravenous immunoglobulins and rituximab therapy for severe transplant glomerulopathy in chronic antibody-mediated rejection: a pilot study. Clin Transplant. 2015;29(5):439–446. [DOI] [PubMed] [Google Scholar]
- 53.Schinstock CA, Bentall AJ, Smith BH, et al. Long-term outcomes of eculizumab-treated positive crossmatch recipients: allograft survival, histologic findings, and natural history of the donor-specific antibodies. Am J Transplant. 2019;19(6):1671–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patel A, Knorr JP, Campos S, Khanmoradi K, Zaki RF, Bradauskaite G. De Novo thrombotic microangiopathy immediately after kidney transplant in patients without apparent risk factors. Exp Clin Transplant. 2016;14(2):230–234. [DOI] [PubMed] [Google Scholar]
- 55.Satoskar AA, Pelletier R, Adams P, et al. De novo thrombotic microangiopathy in renal allograft biopsies-role of antibody-mediated rejection. Am J Transplant. 2010;10(8):1804–1811. [DOI] [PubMed] [Google Scholar]
- 56.Ozdemir BH, Ok Atilgan A, Yilmaz Akcay E, et al. De novo thrombotic microangiopathy in renal transplant patients. Exp Clin Transplant. 2018;16(Suppl 1):131–135. [DOI] [PubMed] [Google Scholar]
- 57.Akl A, Alobaidi S, Aboalsamh G. Living-donor kidney transplant-associated thrombotic microangiopathy successfully treated with thymoglobulin: a case report. Exp Clin Transplant. 2019;17(Suppl 1): 175–177. [DOI] [PubMed] [Google Scholar]
- 58.Kim I, Wu G, Chai NN, Klein AS, Jordan S. Anti-interleukin 6 receptor antibodies attenuate antibody recall responses in a mouse model of allosensitization. Transplantation. 2014;98(12): 1262–1270. [DOI] [PubMed] [Google Scholar]
- 59.Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014;6(10):a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao X, Boenisch O, Yeung M, et al. Critical role of proinflammatory cytokine IL-6 in allograft rejection and tolerance. Am J Transplant. 2012;12(1):90–101. [DOI] [PubMed] [Google Scholar]
- 61.Choi J, Aubert O, Vo A, et al. Assessment of tocilizumab (anti-interleukin-6 receptor monoclonal) as a potential treatment for chronic antibody-mediated rejection and transplant glomerulopathy in HLA-sensitized renal allograft recipients. Am J Transplant. 2017;17(9):2381–2389. [DOI] [PubMed] [Google Scholar]
- 62.Vo AA, Choi J, Kim I, et al. A phase I/II trial of the interleukin-6 receptor-specific humanized monoclonal (tocilizumab) + intravenous immunoglobulin in difficult to desensitize patients. Transplantation. 2015;99(11):2356–2363. [DOI] [PubMed] [Google Scholar]
- 63.Markell M, Brar A, Bhela S, Patel A, Salifu M. Use of repository corticotropin gel (Acthar) in progressive nephrotic syndrome secondary to transplant glomerulopathy: a report of three cases. Kidney Med. 2019;1(1):31–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gasim AH, Chua JS, Wolterbeek R, Schmitz J, Weimer E, Singh HK, Nickeleit V. Glomerular C4d deposits can mark structural capillary wall remodelling in thrombotic microangiopathy and transplant glomerulopathy: C4d beyond active antibody-mediated injury: a retrospective study. Transpl Int. 2017;30:519–532. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.