

Abstract
Phase separation of intrinsically disordered proteins (IDPs) is a remarkable feature of living cells to dynamically control intracellular partitioning. Despite the numerous new IDPs that have been identified, progress towards rational engineering in cells has been limited. To address this limitation, we systematically scanned the sequence space of native IDPs and designed artificial IDPs (A-IDPs) with different molecular weights and aromatic content, which exhibit variable condensate saturation concentrations and temperature cloud points in vitro and in cells. We created A-IDP puncta using these simple principles, which are capable of sequestering an enzyme and whose catalytic efficiency can be manipulated by the molecular weight of the A-IDP. These results provide a robust engineered platform for creating puncta with new, phase-separation-mediated control of biological function in living cells.
Intrinsically disordered proteins (IDPs) are receiving recognition for their role in various biological (dys)functions1,2. A subset of IDPs, termed biological condensates, physically separate themselves from the cytoplasm to control the accessibility of a variety of macromolecules3,4. Although our ability to detect protein disorder has advanced rapidly thanks to sophisticated statistical methods, the ability to predict phase separation has lagged behind5. The factors that are broadly believed to control phase separation are: (1) amino acid composition and patterning of the primary protein sequence6–8; (2) heterotypic interactions with RNA or other macromolecules9,10; and (3) solvent quality11–13. Many previous reports note the challenge of predicting IDP phase behaviour, but few studies have directly tackled this problem. Given the recognition of its importance to cellular function, this is now an area of active research and many efforts are ongoing using computational14–17 and experimental approaches18. However, most experimental methods to develop a sequence level understanding of phase behaviour have relied on mutational strategies of native IDPs with sweeping residue substitutions or domain deletions19.
We have taken a different and complementary approach to understand how phase behaviour is encoded in polypeptides. Analogous to (and inspired by) synthetic polymers that exhibit lower and upper critical solution temperature (LCST/UCST) phase behaviour, we began by systematically scanning the sequence space of native IDPs to identify minimal peptide motifs that will confer such phase behaviours when polymerized into a polypeptide20,21. With the greatly reduced sequence complexity of these repetitive polypeptides, we then made rational changes in the amino acid repeat motif that systematically propagate along the sequence. These artificial polypeptides also exhibit phase separation inside of cells22–25.
Informed by a heuristic knowledge of factors that drive phase separation in polypeptides, we set out to create artificial IDPs (A-IDPs) that exhibit phase separation in living cells. Our design began with (G1-R2-G3-D4-S5-P6-Y7-S8)XX (where XX is the number of repeats, between 20 and 80), a sequence that is inspired by Drosophilia Melanogastor Rec-1 resilin, a protein known to exhibit UCST phase behaviour, which is thought to be more common in native IDPs than LCST phase behaviour9,11,12,26 (Fig. 1a). We next created a set of A-IDPs that comprised repeats of this parent (G1-R2-G3-D4-S5-P6-Y7-S8)XX motif and variants with rational amino acid mutations. We characterized the UCST phase behaviour for this set of IDPs to quantify the effect of various amino acid mutations and modifications to the chain architecture on homotypic liquid–liquid phase separation.
Fig. 1 |. Artificial intrinsically disordered polypeptides inspired from native IDPs exhibit reversible UCST phase behaviour.
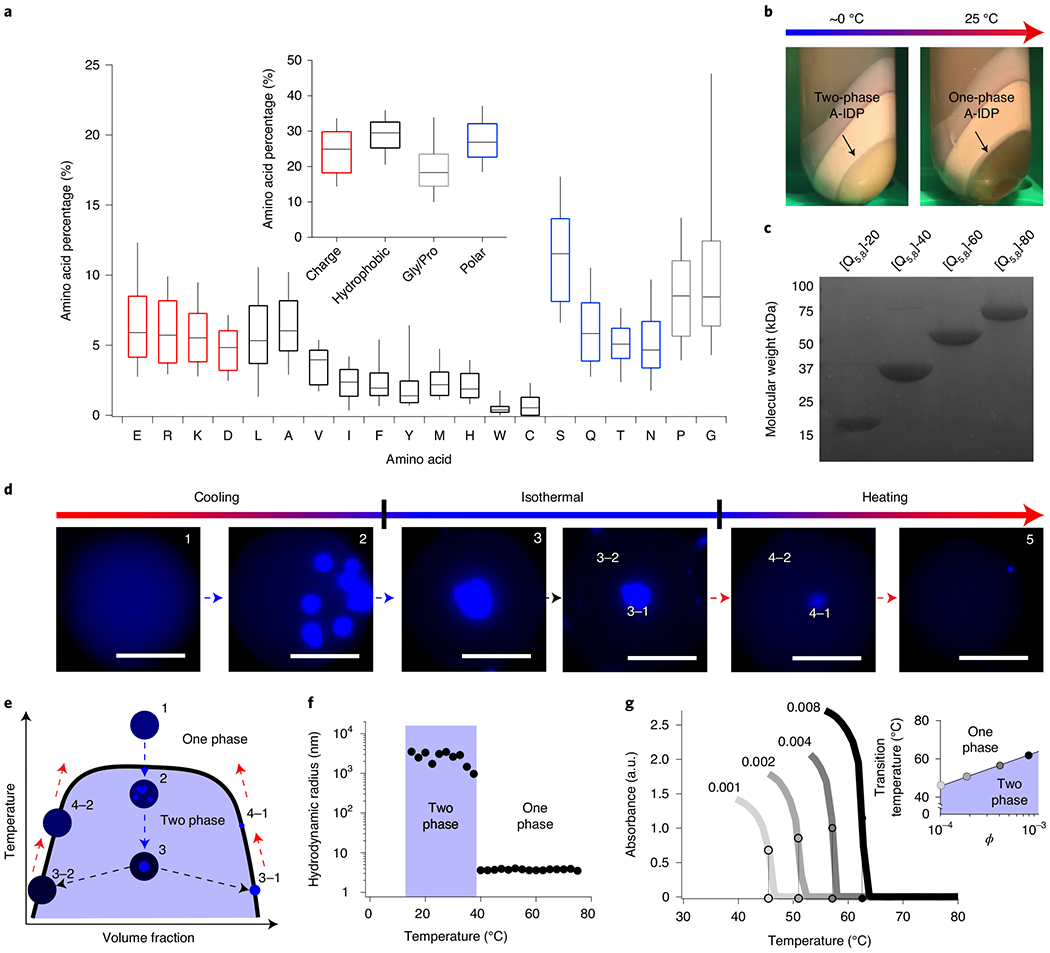
a, Proteomic analysis of native IDPs that form biomolecular condensates reveal that they have an abundance of G/P, charged and uncharged polar residues, yet exhibit a balance of overall charge. b, An example of a dense, exclusionary phase formed by an UCST-exhibiting A-IDP even in the complex medium of bacterial cell lysate. The coacervate shows almost complete separation from all other cellular proteins and debris (non-labelled bands) that are present in the cell lysate after centrifugation, facilitating purification of the A-IDP from the insoluble cell lysate fraction without affinity tags. Even in this concentrated phase, one can still identify the A-IDP with optical turbidity as it transitions between a two- and one-phase solution on heating. c, An example SDS-PAGE gel of a set of A-IDPs ([Q5,8]-20 to [Q5,8]-80) with conserved sequence but increasing molecular weight, showing the high purity of the A-IDPs that is obtained by exploiting their UCST phase behaviour for chromatography-free purification. d, Visualization of UCST phase separation of [Q5,8]-20 in water-in-oil droplets by fluorescence microscopy. On cooling, phase separation in a droplet is initiated at multiple sites; the puncta that grow from each site slowly coalesce with one another into a single dense phase. The equilibrium with the surrounding dilute phase is re-established on reheating, leading to a higher-concentration dilute phase and smaller volume occupied by the dense phase. ϕ = 0.0018 (100 μM); scale bar, 50 μm. e, A schematic UCST phase diagram for a cooling-heating cycle of a UCST polypeptide in a water-in-oil droplet. f, Dynamic light scattering data of [Q5,8]-20, demonstrating the change in the hydrodynamic radius following cooling. On reaching the cloud point, [Q5,8]-20 transitions from soluble unimeric polypeptides with a radius of hydration of 5–6 nm to micron-sized aggregates. Data are collected at ϕ = 0.0043 (245 μM) in 150 mM PBS at pH 7.4. g, Upper critical solution temperature cloud points are affected by ϕ of the polypeptide in solution. The inset shows that this behaviour follows a natural-logarithm dependence in the dilute regime (R2 = 0.98).
We then used a subset of this library to engineer intracellular condensates in living cells. The behaviour of intracellular condensates for these A-IDPs proved to be surprisingly predictable, and enabled dynamic control over their cytoplasmic solubility and their interaction with the surrounding environment. Capitalizing on these observations, we created intracellular droplets capable of sequestering an enzyme whose catalytic efficiency can be genetically encoded by modulating the molecular weight of the A-IDP.
Results
Identification of a minimal IDP repeat from proteomic analysis and sequence heuristics.
We conducted a proteomic analysis of 63 IDPs that form membraneless organelles to investigate their sequence composition3. We were particularly interested in categories of amino acids that were suspected to drive phase behaviour via intrachain interactions, such as charge–charge, cation–π and hydrogen bonding via non-charged polar residues8,19,20,27–30 (Fig. 1a). The composition of these 63 proteins is remarkably similar to previously identified repetitive protein polypeptides that exhibit UCST phase behaviour and their side-chains groups are chemically similar to synthetic UCST polymers20,29,30. Using a combination of our previously developed sequence heuristics and insights from this proteomic analysis, we designed an octapeptide motif that we expected would exhibit UCST phase behaviour when polymerized into a macromolecule under physiological solution conditions.
To manage the vast sequence space of all possible mutations of the octapeptide repeat, we classify each amino acid into categories of intrachain interactions that could contribute to UCST phase behaviour: N, Q, S, T are classified as polar, uncharged amino acids; R–K and D–E are pairs of positively and negatively charged amino acids, respectively; and G and P are placed into a separate category given their unusual structure and importance in promoting a disordered polypeptide backbone (Supplementary Fig. 1)31,32. The remaining amino acids are classified as hydrophobic33,34. To ensure that we modulate the UCST phase behaviour of the wild-type (WT) repeat, but do not abolish it completely, we make mutations where the substituting amino acid only modulates the strength of the interaction within each category. For example, R and K are both positively charged under normal physiological pH; thus, by substituting K for R we maintain the charge neutral state of the polymeric backbone, a parameter known to dramatically affect the observed phase behaviour17,35–37. Similarly, N, Q, S and T are all capable of creating hydrogen bonds with water, and one another, more readily than a hydrophobic amino acid. Each mutant thus has an equal number of residues per chain that are capable of forming each particular type of bond.
The WT repeat unit is (G1-R2-G3-D4-S5-P6-Y7-S8)40, where 40 refers to the number of repeats. The molecular weight of the A-IDPs was varied between ~15 and ~70 kDa (by varying the number of repeat motifs from 20 to 80) to account for observed molecular weights in the intrinsically disordered regions (IDRs) of native IDPs (Supplementary Fig. 1). The parent sequence is referred to as WT in this paper and we use a shorthand notation to refer to sequences throughout the text, where the bracketed letter refers to a specific point mutation. For example, a mutant with a complete substitution of Y7 in the WT repeat unit by V would result in a notation of [V7]-XX, where XX is the number of octapeptide repeats in the A-IDP. When a residue is only partially substituted in the A-IDP (for example, the partial substitution of Y by V), we use the notation [BYo:ZVo], where the B to Z ratio represents the ratio of Y to V in the variant and the subscript o is the position of that residue along the repeat unit. For example, [Y7:V7]-40 would hence represent 50% of all Y replaced with V, whereas [3Y7:V7]-40 would represent a 25% substitution of V for Y. A double mutant, such as 100% substitution of residues at the fifth and eighth position in the octapeptide repeat with Q, would be denoted as [Q5,8]-XX, whereas fractional substitution at these positions with S and Q would be denoted as [BS5,8:ZQ5,8]-XX, where B and Z represent the ratio of S to Q. The sequence descriptions of common A-IDPs studied in the paper are in Table 1, and a full description of all A-IDPs synthesized in this study are in Supplementary Tables 1 and 2.
Table 1 |.
Amino acid sequences of A-IDPs used in this study
| Protein name | Full amino acid sequence | Amino acids (N) | Molecular weight (Da) |
|---|---|---|---|
| [WT]-20 | SKGP-[GRGDSPYS]20-GY | 166 | 17,004 |
| [WT]-40 | SKGP-[GRGDSPYS]40-GY | 326 | 33,400 |
| [WT]-60 | SKGP-[GRGDSPYS]60-GY | 486 | 49,797 |
| [WT]-80 | SKGP-[GRGDSPYS]80-GY | 646 | 66,193 |
| [Q5,8]-20 | SKGP-[GRGDQPYQ]20-GY | 166 | 18,646 |
| [Q5,8]-40 | SKGP-[GRGDQPYQ]40-GY | 326 | 36,685 |
| [3S5,8:Q5,8]-40 | SKGP-[GRGDSPYSGRGDSPYSGRGDSPYSGRGDQPYQ]10-GY | 326 | 34,221 |
| [S5,8:Q5,8]-40 | SKGP-[GRGDSPYSGRGDQPYQ]20-GY | 326 | 35,042 |
| [S5,8:3Q5,8]-40 | SKGP-[GRGDQPYQGRGDQPYQGRGDQPYQGRGDSPYS]10-GY | 326 | 35,863 |
| [3Y7:V7]-40 | SKGP-[GRGDSPYSGRGDSPYSGRGDSPYSGRGDSPVS]10-GY | 326 | 32,760 |
| [Y7:V7]-40 | SKGP-[GRGDSPYSGRGDSPVS]20-GY | 326 | 32,119 |
| [V7]-40 | SKGP-[GRGDSPVS]40-GY | 326 | 30,839 |
| [3R2:K2]-40 | SKGP-[GRGDSPYSGRGDSPYSGRGDSPYSGKGDSPYS]10-GY | 326 | 33,120 |
| [R2:K2]-40 | SKGP-[GRGDSPYSGKGDSPYS]20-GY | 326 | 32,840 |
A-IDPs exhibit robust and reversible UCST phase behaviour in an aqueous environment.
There are two main advantages to using protein polymers to engineer phase-separation behaviour. First, protein polymers have a limited number of unique protein epitopes that could cause heterotypic phase separation with other endogenous cellular components, such as the short linear motifs present in some IDP genes38. Second, this preference for homotypic phase separation allows the A-IDPs to be purified without chromatography by cycling between the one- and two-phase regime of their phase diagram. An example of this purification process is shown in Fig. 1b, where the high titre A-IDP, [Q5,8]-20, completely phase separates from the soluble fraction of the cell lysate and can be isolated by centrifugation. Subsequent removal of the protein-poor supernatant, dissolution of the protein-rich pellet with urea and dialysis of the soluble fraction in water results in 95–99% pure protein as observed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) (Fig. 1c and Supplementary Fig. 2).
A-IDPs [WT]-20 and [Q5,8]-20 exhibit UCST phase behaviour in vitro. For clarity, the UCST of a protein is the unique critical temperature above which the components of a mixture are miscible in all proportions. However, in most cases in this manuscript, we measure the cloud-point temperature; that is, the transition temperature for a specific solution composition (salinity, concentration, pH). We employed three different techniques to characterize these cloud points or transition temperatures (Tt). First, we utilized droplet microfluidics, where monodisperse water droplets are formed in oil that contains the A-IDP solution (Fig. 1d). Phase separation can be directly visualized within the droplets to observe the types of structures formed as one cools the surrounding medium. These A-IDPs exhibit classic liquid–liquid phase separation where, on crossing the phase boundary, multiple nucleation sites of condensates are observed (Fig. 1d, panel 2). These nucleation sites wet one another and quickly coalesce into a single, spherical A-IDP-dense phase that is in equilibrium with the surrounding A-IDP-poor phase (Fig. 1d panel 3 and 3–1/3–2). On reheating, the A-IDP-rich phase shrinks in size, re-establishing equilibrium rapidly with the dilute phase (Fig. 1d, panel 4–1/4–2). A wider field of view is shown in Supplementary Fig. 3. These data clearly show that A-IDPs exhibit reversible UCST phase separation via coalescence and growth kinetics (Fig. 1e).
Second, we employed temperature-dependent dynamic light scattering (DLS). A solution of [Q5,8]-20 is heated to 80 °C and DLS data were collected at each step as the solution was step-wise cooled in 2 °C decrements to 10 °C. We observe a transition from soluble A-IDP molecules, with a hydrodynamic radius (Rh) of 4 nm, to aggregates larger than 1 μm (Fig. 1f and Supplementary Fig. 4). This transition is remarkably sharp, as it occurs within a 2 °C window.
Third, we employed temperature-dependent turbidity measurements at a fixed wavelength of 350 nm to characterize the UCST phase separation (Fig. 1g) and rapidly create partial phase diagrams. At dilute volume fractions (ϕ) of [Q5,8]-40, we observed different UCST cloud points that increase as a function of the natural logarithm of A-IDP concentration. We also confirmed the complete reversibility of the UCST phase behaviour of these A-IDPs, with a <1 °C difference in UCST Tt after ten successive heating and cooling ramps (Supplementary Fig. 5).
Arginine composition, aromatic to aliphatic ratio, charge balance and molecular weight define the UCST cloud point.
To understand the effects of a particular residue substitution in the octapeptide repeat on the phase separation for the A-IDP, we created a set of mutant A-IDPs ranging from 100% of a to 100% of b, where a is the WT repeat unit and b is the mutant repeat unit. The doping scheme—wherein b is periodically inserted into the WT sequence—is visually illustrated by the colour-coded schematic in Fig. 2a. The mutant repeat is well mixed and is distributed along the WT sequence to reduce blockiness of the copolypeptide, which has been known to affect droplet formation in vitro39,40. Due to experimental limitations, loss of function (phase separation that cannot be detected) is operationally defined as a Tt < 4 °C or >90 °C at ϕ less than or equal to 0.1 in a 150 mM salinity aqueous buffer. A total length of 40 repeats was chosen for these A-IDPs to approximate the median length (~320 amino acids) of native IDRs.
Fig. 2 |. Control of the UCST cloud point using main-chain amino acid composition.
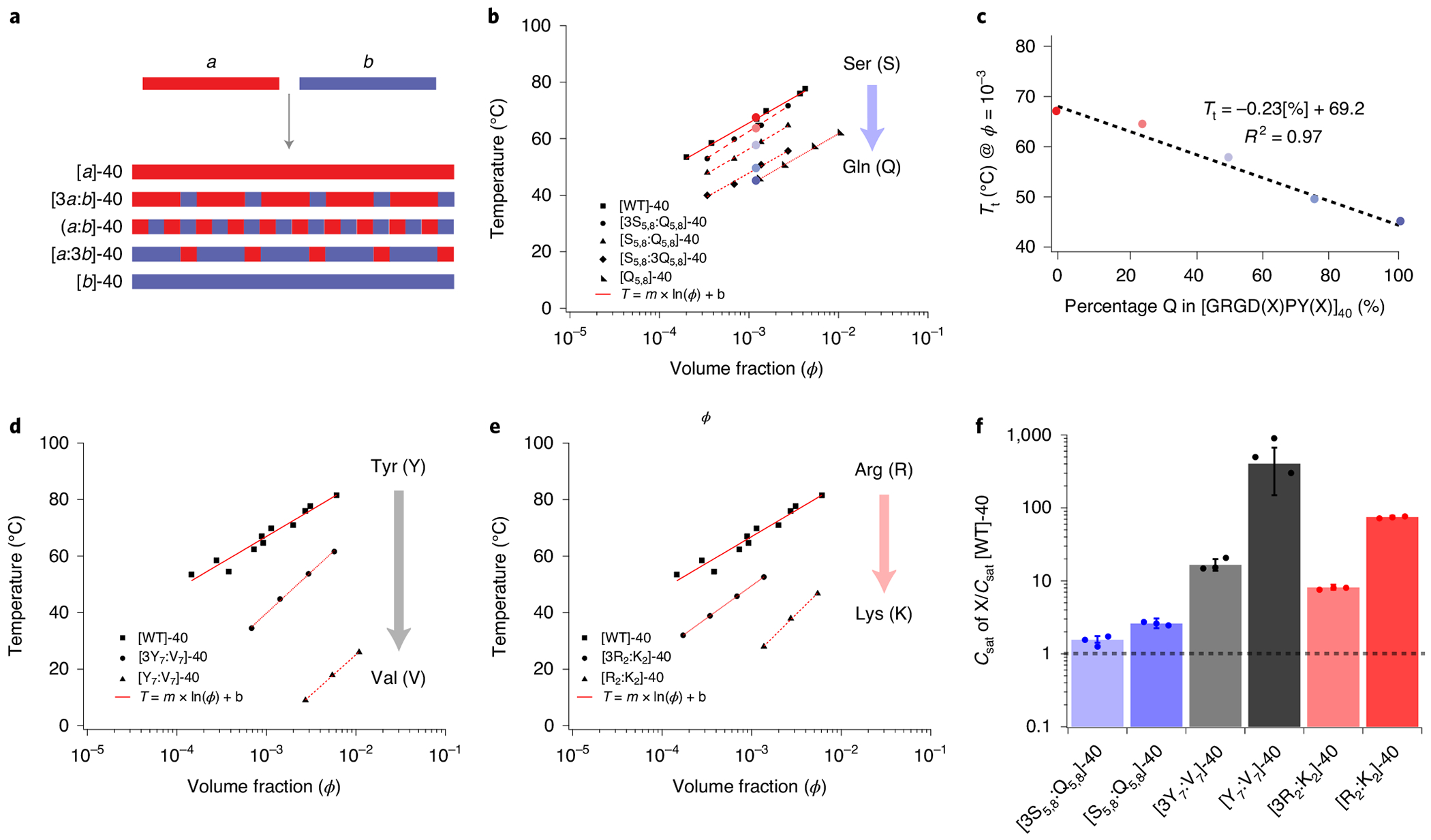
a, A schematic describing the methodology for doping repeat unit b into a homopolymer of a. The WT A-IDP, with a high UCST cloud point comprising 40 repeats of a (GRGDSPYS), is doped with increasing fractions of repeat b (GRGDQPYQ) to probe the loss of function of UCST phase behaviour from polymers of a. The doping of b into a is designed to ensure mixing of the two repeats along the polypeptide chain and minimize blocky behaviour. b, Doping of b into a results in mutant IDPs; the UCST Tt of each mutant IDP is a linear function of the ϕ of the A-IDP. c, The effect of composition (degree of doping) is a linear function of the degree of substitution of b into a at a constant ϕ of 10−3 (25–30 μM) (R2 = 0.97). d, Substitution of aromatic Y residues by aliphatic V dramatically reduces Tt. e, Substitution of R by K dramatically reduces Tt; 50% substitution lowers the Tt by more than 40 °C. f, The chemical composition can affect the saturation concentration by two orders of magnitude at constant molecular weight (Csat @ 37°C = 1–800 μM). This can be visualized by normalizing to the saturation concentration of [WT]-40, which is conveniently ~1 μM and is shown by the dashed horizontal line.
The Tt of each A-IDP mutant is a logarithmic function of its ϕ (Fig. 2b,c). At a specified ϕ, Tt is a function of composition (R2 = 0.97), which demonstrates that the behaviour of the mutant A-IDPs (block copolypeptides of a and b) can be linearly interpolated between that of pure polypeptides of a and b. This linear behaviour allows extrapolation of the UCST phase behaviour for homopolymers that exhibit a UCST cloud point beyond the experimentally observable range of detection, thus putting each point mutation on a single relative scale (Supplementary Fig. 6).
We next tested the effect of fifteen different site-specific substitution mutations of the reference (WT) repeat motif on the saturation concentration (Csat, defined as the concentration at which the Tt is 37 °C) of the A-IDPs. We found that single residue changes in the octapeptide repeat are capable of changing the Csat of the repeat polypeptide by over by two orders of magnitude at constant molecular weight (ranging from 1–800 μM) (Fig. 2d–f). We do not believe that the changes in chain conformation resulting from these mutations are responsible for these effects on the Tt and Csat, as circular dichroism spectroscopy shows that the mutant A-IDPs are structurally disordered, consistent with their G- and P-rich composition41,42 (Supplementary Fig. 7).
These substitutions present quantitative evidence for the importance of interactions between R and aromatic residues in the repeat motif of the A-IDP. When Y7 is substituted, we observe dramatic shifts in the UCST cloud point at ϕ = 10−3 (~25–30 μM), from 66 °C to 123 °C, 59 °C and ~2 °C for W, F and H, respectively (Supplementary Fig. 6). These data indicate that interactions between the cationic side-chain of R and side-chain of W, F, Y and H are important driving forces for phase separation, although the strength of these interactions is side-group dependent with W >> Y > F >> H. Likewise, replacing R with K lowers the UCST cloud-point temperature, and hence the phase boundary (Fig. 2e), and increases the Csat (Fig. 2f).
We next looked at the effect of A-IDP molecular weight on phase behaviour; we chose A-IDPs with molecular weights between ~17 kDa and ~70 kDa, as this molecular weight range covers 75% of native IDRs (Supplementary Fig. 1). Our results indicate that molecular weight exhibits at least as large of an effect on the UCST cloud point as amino acid substitution (Fig. 3a). We observed that the effect of molecular weight on Tt (in the ~17–70 kDa range that we studied) can be approximated with a linear fit to the natural logarithm of the molecular weight (Fig. 3b). By simply doubling the molecular weight of [WT]-40, we were able to create A-IDPs with predicted Csat values in the nanomolar regime (Fig. 3c), similar to the Csat exhibited by some native IDPs11,12. Notably, by varying both the molecular weight and composition we can vary the Csat of A-IDPs by over seven orders of magnitude, ranging from 10−4 to 102 μM.
Fig. 3 |. Control of UCST cloud point by molecular weight of A-IDP.
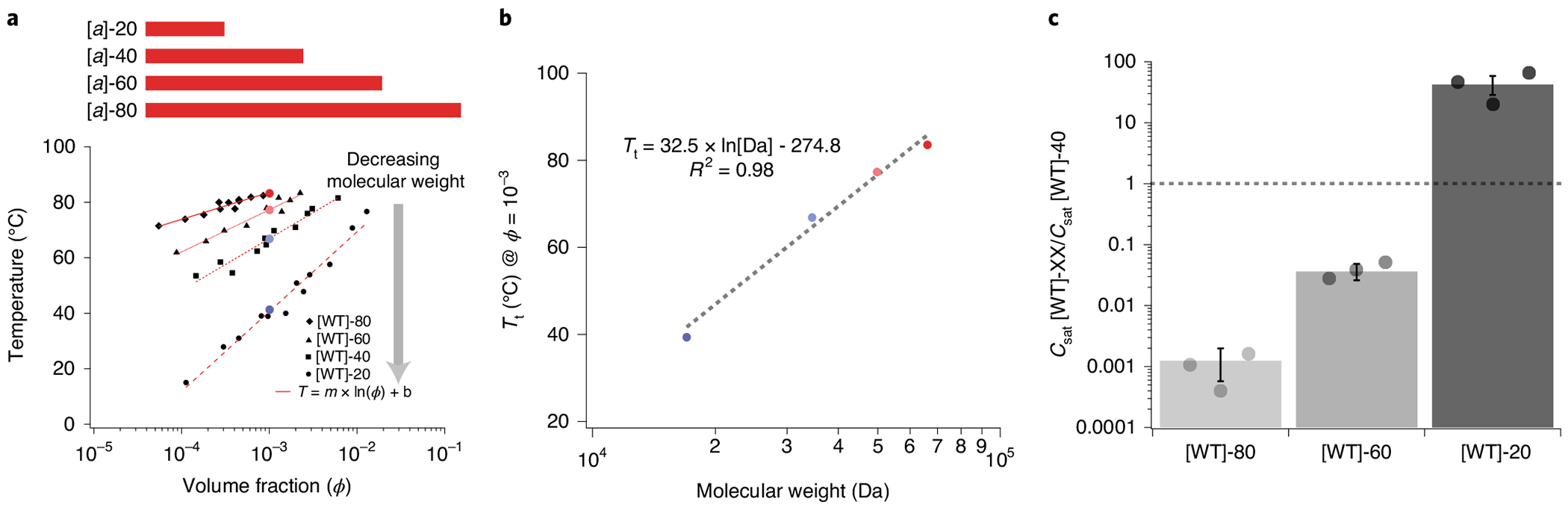
a, The effect of the polypeptide molecular weight on Tt. b, Tt directly scales with the natural logarithm of molecular weight. c. At constant chemical composition, it is possible to modulate Csat by over five orders of magnitude by simply changing the molecular weight of the A-IDP (Csat @ 37°C = 1 nM - 400 μM). [WT]-40 has a Csat of ~1 μM. For reference, ϕ = 0.001 corresponds to concentrations of 58 μM, 30 μM, 20 μM and 15 μM for repeat lengths of 20, 40, 60 and 80 octapeptides, respectively.
In addition to the effect of composition, volume fraction (ϕ) and molecular weight on Tt, there are several other parameters that have a measurable effect on UCST phase behaviour but that do not eliminate UCST phase behaviour under physiologically relevant conditions. Uncharged polar substitutions, the ratio of G/P, the syntax of the repeating polypeptide, solution salt content, pH (in the absence of H) and identity of the negatively charged amino acid (E versus D) all result in smaller changes to the UCST binodal phase boundary than molecular weight, ϕ, aromatic:aliphatic amino acid ratio and R content (Supplementary Fig. 6). The residue N-terminal to P6 seems to have a unique impact on the UCST binodal boundary, where compositionally identical A-IDPs shifted the UCST binodal lines depending on which polar non-charged reside is located at position five of the octapeptide repeat. (Supplementary Fig. 8). We also produced and tested non-repetitive, but compositionally identical versions of [WT]-20 and observed minimal effects of scrambling the amino acid sequence on the UCST binodal (Supplementary Fig. 8). These results collectively indicate that three parameters (aromatic:aliphatic ratio, ϕ and molecular weight) are the most critical for controlling UCST phase boundaries or Csat in vitro.
A-IDPs create dense phase-separated condensates at saturation concentrations mediated by amino acid composition.
Polypeptides [WT]-20 and [Q5,8]-20 express well for recombinant proteins, with yields of ~500 mg l−1 in shaker flask culture, making it easy to purify over one gram of material to directly measure the UCST cloud point behaviour at high volume fractions (ϕ > 0.1). These experiments were performed in a linear-temperature-gradient microfluidic device mounted on an upright light microscope, wherein Tt could be quantified by a visible increase in light scattering intensity. These experiments produce binodal phase boundaries similar to optical turbidity measurements that are typically carried out in a ultraviolet–visible spectrophotometer (Supplementary Fig. 9) and demonstrate that a ~25 °C gap between the two binodal lines of [WT]-20 and [Q5,8]-20 is maintained over the entire phase diagram. This corresponds to an increase in A-IDP volume fraction in the dense phase (ϕ″) 3′ UTR from ϕ″ = 0.4 for [Q5,8]-20 to ϕ″ = 0.55 for [WT]-20 at an isotherm of 37 °C. In addition to these phase diagram descriptions, phase separations in the presence of low- (10 kDa) and high- (40 kDa) molecular weight, fluorescently labelled dextran indicate that both [WT]-20 and [Q5,8]-20 droplets are highly exclusionary, as we observed no fluorescence partitioning of dextran into the dense phase (Supplementary Fig. 10). These data indicate that A-IDPs form highly exclusionary droplets in vitro at physiological solution, temperature and pH conditions (ϕ″ > 0.4).
A-IDPs have controlled Csat in eukaryotic and prokaryotic cell lines.
With a set of A-IDPs that exhibit a range Csat values, we sought to (1) understand the dynamics of droplet assembly in living cells and (2) elucidate whether mutations affect in vivo phase separation similarly to in vitro. We chose a set of IDPs with Csat values ranging from 1 to 815 μM (with molecular weights of either ~17 kDa or ~32 kDa) to explore these two questions. To visualize localization of the A-IDPs within bacterial cells, each A-IDP was genetically fused with a superfolder version of the green fluorescent protein (sfGFP) (Fig. 4a).
Fig. 4 |. A-IDPs exhibit tunable intracellular droplet formation on the basis of the molecular weight and ratio of aromatic to aliphatic content.
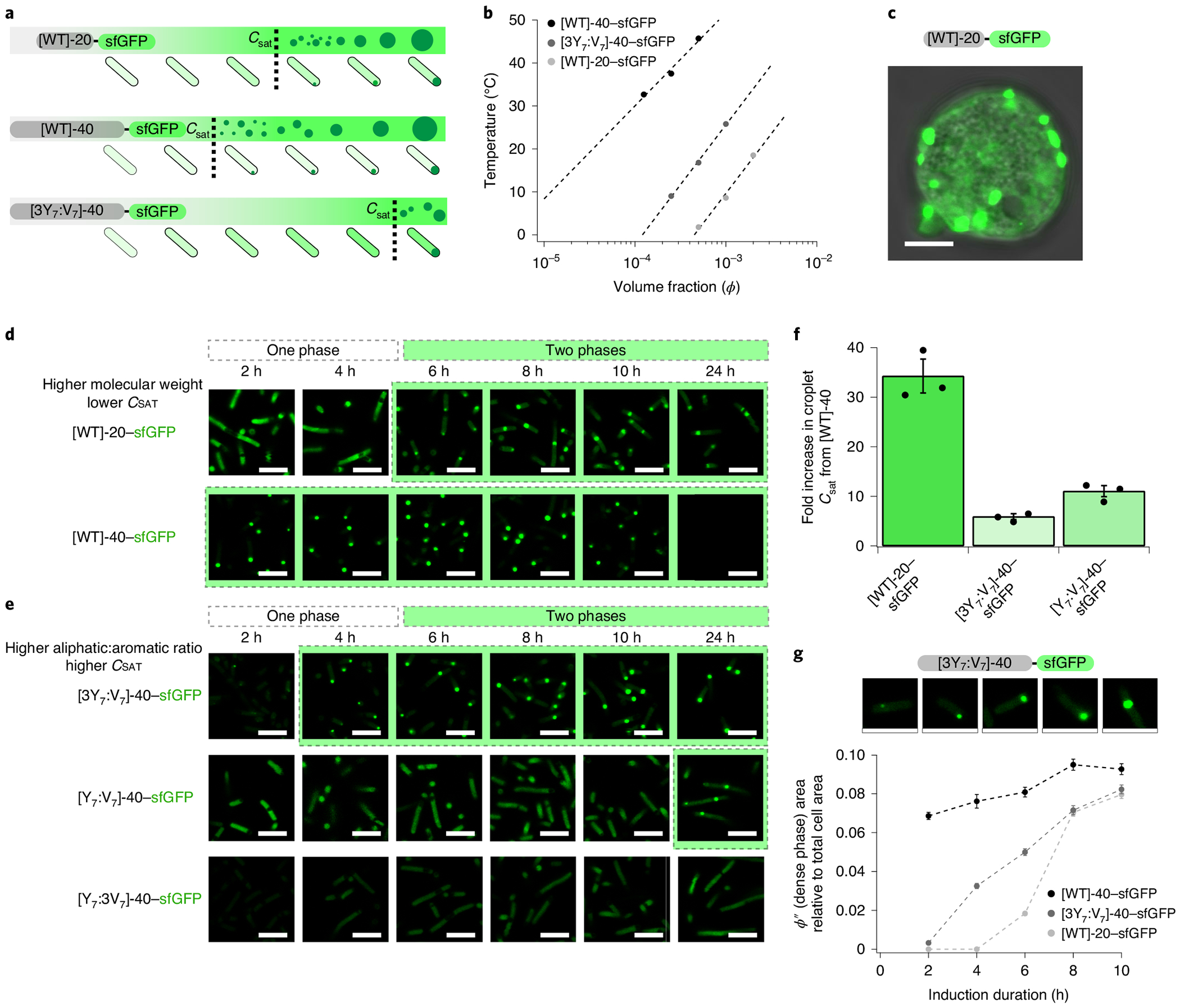
a, A schematic describing the use of two key parameters (the ratio of aromatic:aliphatic content and the molecular weight) to control intracellular droplet formation by modulating Csat. b, A partial in vitro binodal of A-IDP–sfGFP fusions in the dilute regime in 150 mM PBS, pH 7.4. Similar to A-IDPs, A-IDP–GFP fusion proteins exhibit molecular weight and aromatic content dependent phase behaviour. c, The [WT]-20–sfGFP fusion undergoes phase separation in eukaryotic cells (HEK293 cells, day five). Instead of forming a single droplet as seen in vitro in protocells (see Fig. 1c), many distinct droplets are formed that indicate either diffusion- or arrest-limited coalescence. d, Confocal fluorescence images of A-IDP–sfGFP as a function of induction time and molecular weight in E. coli. A higher intracellular concentration is required for [WT]-20 versus [WT]-40 to form intracellular droplets. It is noticeable that [WT]-40 has a lower ϕ′ (A-IDP poor) soluble phase outside of the dense droplet phase compared with [WT]-20. e, Reducing the aromatic content increases Csat in a dose-dependent manner. f, A-IDP–sfGFP fusions exhibit a one order of magnitude shift in their Csat as determined by their molecular weight and ratio of aromatic:aliphatic content. g, The size of the intracellular droplets (ϕ″ or dense phase) grows with induction time. As the concentration of the A-IDP–sfGFP increases inside the cell, the soluble concentration outside of the droplet does not change (Supplementary Fig. 14) but the size of the intracellular droplets grows relative to the total cell area. Images are individual cells from [3Y7:V7]-40–sfGFP cultures at various time points. Error bars represent the standard error of the mean (n = 100 individual cells). All scale bars, 5 μm.
Fusion of sfGFP to A-IDPs and, in turn, to [WT]-20, [WT]-40, [3Y7:V7]-40 and [Y7:V7]-40 did not eliminate the phase behaviour but shifted the phase diagram (Fig. 4b and Supplementary Fig. 11). Despite this shift, we were able to use confocal fluorescence microscopy to observe the formation of intracellular droplets of [WT]-20–sfGFP in both transfected human embryonic kidney (HEK) cells and E. coli (Fig. 4c,d, respectively). By contrast to water-in-oil droplets, in HEK cells these dispersed puncta (2–4 μm) never coalesced into a single coacervate droplet during the experimental timeframe (Supplementary Fig. 12).
The initiation of the UCST phase transition in E. coli is similar to HEK cells (and in vitro) where small densely fluorescent puncta form after the A-IDP concentration in the cell exceeds Csat, and then grow in size over time (Fig. 4g). The growth in the size of these puncta is consistent with increasing sfGFP fluorescence from the bulk E. coli population normalized to the absorbance at 600 nm (OD600) (Supplementary Fig. 13). Unlike HEK cells, but similar to in vitro experiments, these puncta in E. coli coalesce to form a single coacervate droplet per cell (Supplementary Table 4). This result suggests that there are cytoplasmic environment differences between prokaryotic and eukaryotic cells and that the barriers to diffusion and coalescence of coacervate droplets in E. coli are far lower than in HEK cells; the residual dilute regime simultaneously remains at a relatively constant concentration (Supplementary Fig. 14). These results together suggest that as the global concentration of the A-IDP within the cell increases with time, the cytoplastic concentration of the protein is buffered (that is, remains constant), but the volume of the coacervate increases depending on total A-IDP concentration.
Similar to phase separation in vitro, the molecular weight and aromatic:aliphatic content affect droplet formation in E. coli. Doubling the molecular weight of [WT]-20–sfGFP to [WT]-40–sfGFP decreases Csat enough to cause droplet formation even before A-IDP induction, presumably because of leaky transcriptional regulation (Fig. 4d). Similarly, increasing the aliphatic content with V at the expense of Y increases the Csat to a level that is not measurable in the time course of these experiments (Fig. 4e). Although differences in Csat are not as dramatic in vivo as predicted by in vitro experiments with A-IDP–sfGFP fusions (Fig. 4f), perhaps because of the effect of intramolecular crowding within the cell, we can modulate the intracellular Csat by at least an order of magnitude using either the molecular weight or the aromatic:aliphatic ratio of the A-IDP.
A-IDPs exhibit reversible UCST droplet formation in E. coli.
Just as one can cross a binodal line into the two-phase regime under isothermal conditions by increasing polypeptide ϕ, this line may be crossed under constant ϕ by decreasing solvent quality or the chi parameter (χ)6,43. Experimentally, this is most easily accomplished by reducing the temperature of the bulk solution43. Similar to the UCST phase behaviour of A-IDPs in vitro, A-IDPs exhibit reversible UCST phase separation inside cells that is reversible by repeated cooling and heating cycles (Fig. 5a). The phase separation exhibits minimal hysteresis as the difference in the transition temperature of cooling (Tt,C) and the transition temperature of heating (Tt,H) varies by less than 2 °C (Fig. 5b).
Fig. 5 |. A-IDPs exhibit reversible coacervation in E. coli determined by their molecular weight and aromatic:aliphatic ratio.
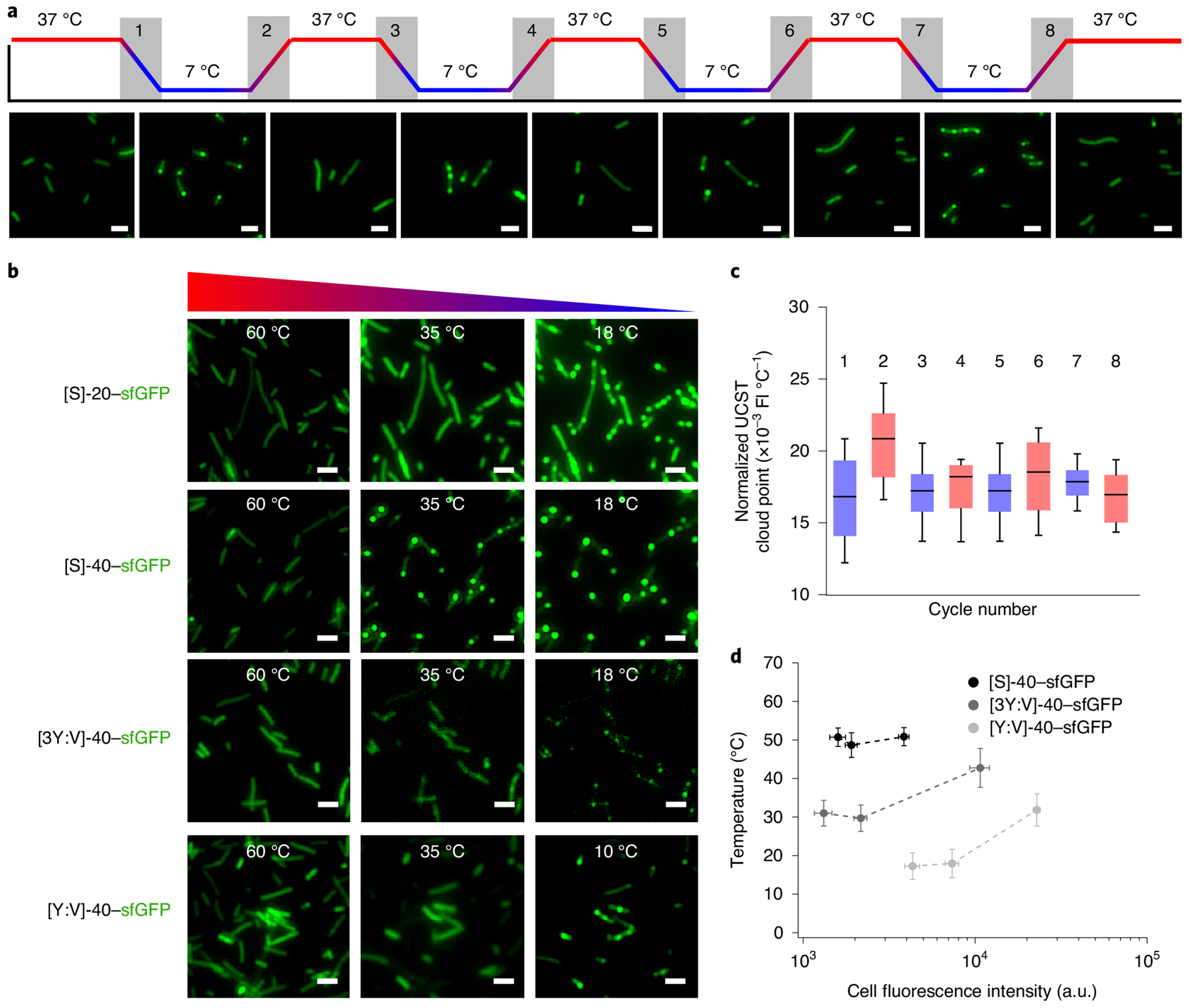
a, Intracellular droplets comprised of [WT]-20–sfGFP can be formed and dissolved reversibly via alternating cooling and heating cycles. This process is completely reversible over four rounds of cooling and heating. Cooling rate = 5°C min−1 with an induction time of 4 h. b, Tt normalized to the intracellular fluorescence of sfGFP in each individual cell (n = 30) does not change substantially over four rounds of heating (red bars) and cooling (blue bars) cycles. c, Intracellular Tt (similar to in vitro) is a function of A-IDP molecular weight and aromatic content. Cooling ramp = 60°C → 10°C; cooling rate = 5°C min−1; A-IDP gene induction time of 8 h. Whiskers indicate 10th-90th percentiles, whereas boxes indicate the 25th-75th percentiles. FI, fluorescence intensity. d, Intracellular binodal lines of various A-IDP–sfGFP fusions. Tt increases as a function of cellular fluorescence (a surrogate of A-IDP concentration) and aromatic content of the A-IDP. Data were analysed at 2, 4 and 8h for [WT]-40–sfGFP and [3Y7:V7]-10, and 4, 8 and 24 h for [Y7:V7]-40 (n = 30). Error bars represent the standard error of the mean.
There are several interesting features about A-IDP puncta formation in E. coli. First, we were able to demonstrate the selective permeability of these droplets in vivo (similar to the in vitro experiment demonstrating dextran exclusion) with sequences of varying aromatic content and hence varying binodals (Supplementary Fig. 15). We found that co-expression of two A-IDPs ([WT]-40–sfGFP and [3Y7:V7]-40–mRuby3) with similar in vitro phase behaviour results in co-localized droplets that contain both IDPs co-localized inside the bacterial cells. By contrast, co-expression of [WT]-40–sfGFP and [Y7:3V7]-40–mRuby3, which exhibit different in vitro phase behaviour, results in the formation of isolated intracellular droplets of [WT]-40–sfGFP. This is because [Y7:3V7]-40–mRuby3 never reaches Csat under experimental conditions and is immiscible with [WT]-40–sfGFP. Second, on multiple heating and cooling cycles, we observed that E. coli exhibit spatial phase separation memory, with puncta forming in the same location as the first cycle (Supplementary Fig. 16). Third, we observed cooling-triggered phase separation over a few minutes results in a higher number of puncta per cell compared to isothermal conditions where the intracellular concentration increases gradually over 24 h (Supplementary Fig. 17). The greater number of puncta observed with higher molecular weight species indicates that the number of puncta formed per cell is a function of their diffusion coefficient, consistent with previous studies44.
Increasing the molecular weight of the A-IDP increases the observed Tt in E. coli (Fig. 5c). By manipulating the aromatic:aliphatic ratio while keeping molecular weight constant and observing the formation of puncta within individual bacterium at various times post induction, we were able to create partial intracellular phase diagrams as a function of Tt and intracellular fluorescence (Fig. 5d). This result is important because it ties the observed behaviour on cooling to a specific concentration for a given construct, essentially normalizing the observed cloud point for differing overall levels of protein expression throughout the cell population and different levels of protein expression which could occur between various A-IDP sequences. Again, with increasing concentration we see an increase in UCST cloud point. What is clear from these studies is modifications to the A-IDP that lower Csat, such as increasing molecular weight or aromatic:aliphatic ratio, also result in a shift upward of the entire phase diagram and vice versa. One can thus use this information to rationally design A-IDPs or rationally mutate existing IDPs to tune the Csats in vivo where the observed change in Csat is strictly driven by a changing homotypic propensity of the A-IDP to phase separate.
De novo design of functional A-IDP droplets in cells.
To understand the potential of using spatially confined intracellular coacervate droplets to carry out new functions, we asked the following questions: (1) could coacervate droplets in cells recruit other molecules, and if so, what, if any, are the size limitations of such molecules? And (2) could these molecules interact with the A-IDP to impart a new function to the droplet?
To answer these questions, we first examined whether a small molecule can diffuse into and react with the A-IDP in a coacervate droplet located within an E. coli cell. We designed and expressed an A-IDP ([3Y7:V7]-40-UAA) that carries three copies of a unique biorthogonal reactive group; that is, an azide (its primary amino acid sequence is listed in Supplementary Table 2). After reaching intracellular concentrations greater than the Csat of [3Y7:V7]-40-UAA, we incubated live E. coli with 1 mg ml−1 dibenzocyclooctyne-dye (DBCO-Alexa488) for 10 min (Fig. 6a). We observed fluorescent condensates in the cells after removing excess dye.
Fig. 6 |. Engineered intracellular droplets with programmable function.
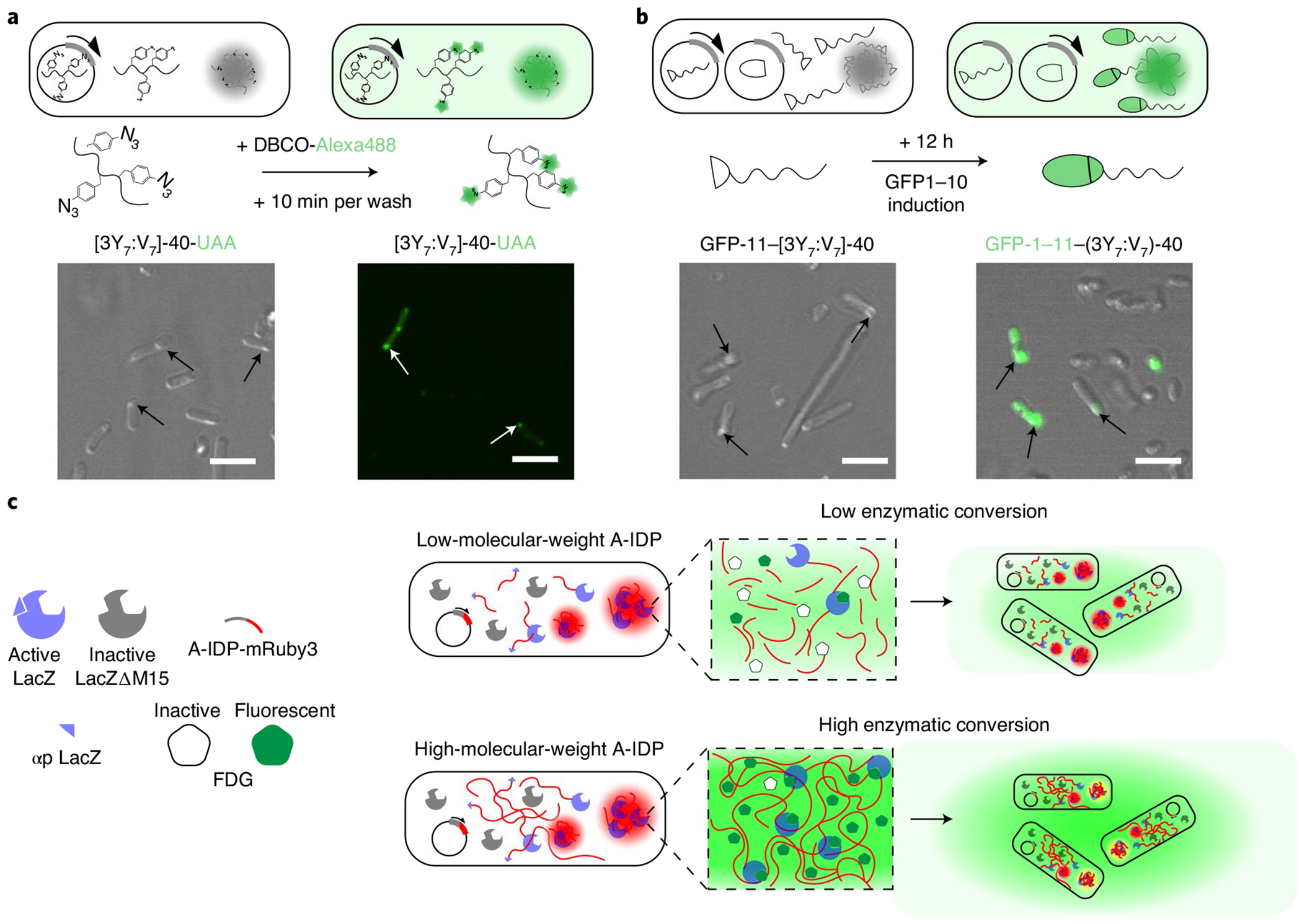
a, Site-specific labelling of droplets with a small molecule fluorescent dye. E. coli cells that contain condensates that are formed by a [3Y7:V7]-40 variant with azidophenylalanine (AzF) residues present a biorthogonal azide that can be labelled in situ with DBCO-Alexa488. DBCO-Alexa488 mixture can diffuse into cells and into the A-IDP condensates within the cell, labelling the azide groups within 10 min of incubation with live E. coli. b, Reconstitution of function GFP in condensates by recruitment of a partner from the cytoplasm using a split GFP system. A GFP-11–[3Y7:V7]-40 fusion protein is able to recruit GFP-1–10 from the surrounding cytoplasm into intracellular droplets. On formation of intracellular condensates after 24 h of IPTG induction of GFP-11–[3Y7:V7]-40 (left panel), induction of GFP-1–10 by arabinose induction enables recruitment of GFP-1–10 into the condensates and reconstitution of functional GFP-1–11 within existing intracellular condensates within 12 h of GFP-1–10 induction (right panel). c, A schematic of the enzyme-condensate experiment. The αp of LacZ is fused with a fluorescent reporter protein (mRuby3) and expressed from an IPTG-inducible gene from a plasmid in the E. coli strain KRX, which has a deletion mutant of the LacZ gene that produces a truncated, catalytically inactive enzyme that lacks the αp. Complementation of LacΔM15 by a αp–A-IDP–mRuby3 fusion creates an active enzyme that converts FDG into fluorescein that is then rapidly exported from the intracellular space into the surrounding medium.
We next questioned whether larger molecules such as proteins are also capable of interacting with an A-IDP puncta. To answer this question, we designed a droplet capture experiment based on split green fluorescent protein (GFP). We first verified if the two components of a split GFP can interact with each other to create a functional GFP molecule if one of the components is fused with an A-IDP GFP-11-[3Y7:V7]-40–mRuby3 was co-expressed in the presence of GFP-1–10; because the IDP is fused with mRuby3, the A-IDP condensates fluoresce red and can be visualized by fluorescence microscopy within the cell. We see fluorescently active GFP only in the interior of the condensates as seen by the co-localization of green fluorescence with the red fluorescence from the A-IDP condensates, indicating that the fragments GFP bind to each other to create an intact and functional GFP molecules (Supplementary Fig. 18). By contrast, in the absence of GFP-1–10 induction, there is minimal green fluorescent inside the red fluorescent condensates (Supplementary Fig. 18).
These results show that two protein fragments of GFP can find and bind to each other in the cell despite the steric hindrance imposed by an A-IDP and a fluorescent reporter. It does not however, prove that a protein can be recruited after an A-IDP condensate has formed, as the protein partners in the previous experiment are co-expressed and could bind in the cytoplasm before phase separation that occurs once the intracellular concentration of GFP-11-[3Y7:V7]-40–mRuby3 exceeds its Csat. To directly answer this question, we co-transformed E. coli with two plasmids: a Lac-operon-regulated plasmid that encodes one fragment of GFP (GFP-11) that is fused with [3Y7:V7]-40 and a second plasmid regulated by araBAD operon that encodes the other fragment of GFP (GFP-1–10). Once expression of GFP-11–[3Y7:V7]-40 at 37 °C proceeds long enough that its intracellular concentrations is greater than its Csat, we removed the isopropyl-β-d-thiogalactoside (IPTG) induction media, and replaced it with arabinose containing media that induces the expression of the larger GFP fragment (GFP-1–10). We observed that subsequent to arabinose induction, both the ϕ′ and ϕ″ fractions of the E. coli contained fluorescently active GFP (Fig. 6b). This result suggests that the large GFP fragment is capable of penetrating the preformed condensate in the cell, finding its binding partner and forming a fully functional molecule, despite the fusion to the A-IDP. Once a fully functional GFP molecule is recruited into the intracellular droplets, it is possible to reversibly modify the solubility of the reconstituted GFP–A-IDP by changing the temperature of the bulk (Supplementary Fig. 19).
These experiments clearly show that small molecules and proteins can be recruited into intracellular coacervate droplets in E. coli.. These results suggested a path for the de novo design of intracellular coacervate droplets with new enzymatic function. We chose biocatalysis as the function of interest, as one of the proposed reasons for the evolutionary development of biomolecular condensates is to modulate the kinetics of various biological functions, including enzymatic reactions45–48; however, there is little experimental evidence demonstrating how the function of enzymes is modulated by phase separation in vivo.
We created an A-IDP fusion that can recruit an enzyme into intracellular droplets to modulate its catalytic activity. We chose β-galactosidase for two reasons: first, it has a range of small molecule substrates, one of which, fluorescein di-β-d-galactopyranoside (FDG), is colourless, but will fluoresce green when cleaved by ß-galactosidase. By using a combination of a red fluorescent protein tagged to our enzyme–A-IDP fusion and the green florescence of fluorescein, we can thus track the co-localization of enzymatic reactions with A-IDPs in real time. Second, we had concerns that a large enzyme fused with a large A-IDP would not express at high enough concentrations in E. coli and thus not phase separate in vivo. To alleviate this concern, we took advantage of the widely used β-galactosidase (LacZ) blue–white screening system, where the so-called alpha peptide (αp) complements the mutated enzyme LacZΔM15 to create a functional β-galactosidase enzyme. In our system, the αp is fused with a A-IDP–mRuby3 construct, such that enzyme activity is physically linked to the A-IDP which in turn is physically linked to red fluorescence.
Our studies with the DBCO-Alexa488 and split GFP provided the basis for this more complicated experiment. The DBCO-Alexa488 experiment suggested that a small molecule such as an enzyme substrate can penetrate bacterial cell walls and puncta (Fig. 6a). The split GFP experiment suggested that relatively large proteins can be recruited to A-IDP condensates to form a functional proteins, suggesting that the same should be possible with the split β-galactosidase system (Fig. 6b).
We thus genetically fused the αp from LacZ β-galactosidase to a A-IDP–mRuby3 construct. Our hypothesis is that the αp–A-IDP–mRuby3 protein can bind and recruit the other inactive fragment of the enzyme (LacZΔM15) that is expressed endogenously in genetically modified E. coli (KRX, Promega) into intracellular droplets. After protein induction and resulting condensate formation, we deliver the substrate FDG to the cell medium where it is trafficked intracellularly, hydrolysed into fluorescein at the sites of active β-galactosidase, and eventually exported outside of the cell (Fig. 6c)49,50. By tracking the onset of the green fluorescence of fluorescein with confocal microscopy we can specifically observe where and when enzymatic activity is occurring and quantitatively track enzyme activity.
In our control experiment (αp–mRuby3) we observe limited persistence of fluorescence within the cells. It is important to note that the αp itself is known to form inclusion bodies51,52 and therefore, even in this control experiment, we observe some puncta inside of the bacterial cells. However, on fusion with [WT]-20, we observe that the fluorescence co-localizes long enough with the A-IDP condensates to be observed with confocal microscopy (Fig. 7a). Despite this increased co-localization, the total fluorescent signal over time is not remarkably different from the αp–mRuby3 control (Fig. 7b).
Fig. 7 |. Engineered intracellular droplets with programmable function.
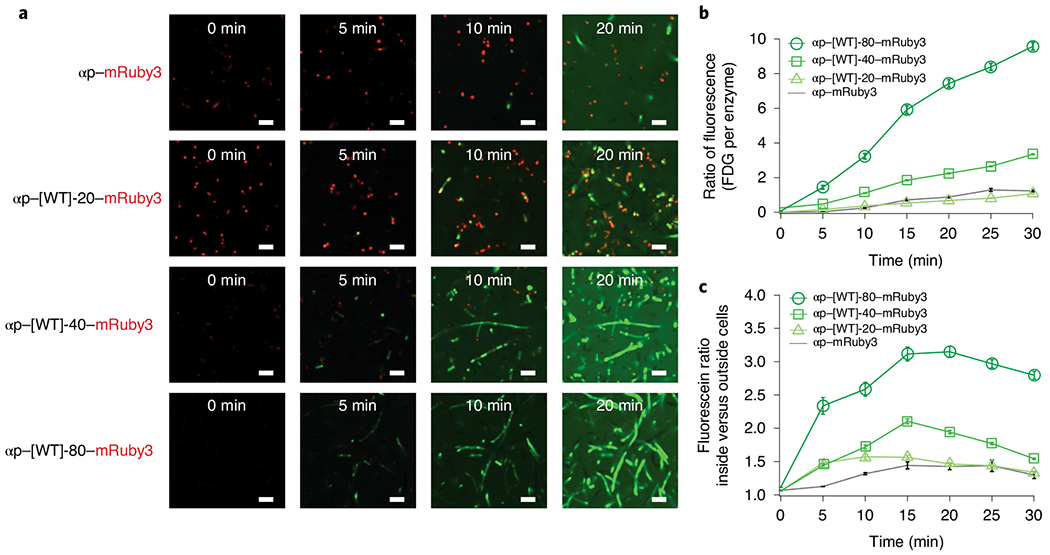
a, Confocal microscopy images showing the fluorescent conversion of fluorescein FDG. Note that the puncta-like structures of αp–mRuby3 in the top panel are due to a fraction of the fusion forming inclusion bodies in cells. When the αp is fused with [WT]-20–mRuby3, the fluorescence is first observed at the sites of intracellular phase transition in coacervate droplets, and the fluorescein then diffuses into the cytosol and then out of the cell into the extracellular space. Increasing the molecular weight of the A-IDP leads to increased FDG conversion at earlier timepoints and higher overall conversion after 20 min. Rebalanced images of αp–[WT]-40–mRuby3 and αp–[WT]-80–mRuby3, with improved visualization of the co-localization of intracellular droplets and converted FDG, can be found in Supplementary Fig. 20. b, Intracellular concentration of fluorescein produced by catalytic conversion of FDG, normalized to the mRuby3 fluorescence of each individual cell (n = 300). The catalytic efficiency increases with A-IDP molecular weight, as seen by the greater ratio of green fluorescence resulting from FDG conversion to fluorescein normalized to the red fluorescence of mRuby3 on a molar basis. Both αp–[WT]-40–mRuby3 and αp–[WT]-80–mRuby3 exhibit statistically significant differences from the control (two-factor ANOVA, F < 2 × 10−13). Error bars indicate standard error of the mean. c, All αp–A-IDP–mRuby3 fusions exhibit a higher ratio of green fluorescence inside the cell, indicating a greater persistence of fluorescent FDG inside the intracellular space compared to the αp–mRuby3 control. Error bars represent the standard error of the mean. n = 300. All scale bars, 5 μm.
When we increase the molecular weight of the A-IDP, and thus decrease Csat, we observe a dose-response effect in the total FDG fluorescence intensity as well as co-localization with the αp–A-IDP–mRuby3 fusion (Fig. 7a). αp–[WT]-40–mRuby3 and αpα[WT]-80–mRuby3 have 2.5- and 7.5-times greater FDG converted at 20 min compared with the αp–mRuby3 control (Fig. 7a and Supplementary Fig. 20). Quantification of the co-localization of green and red fluorescence with Mander’s overlap coefficient53 indicates increased co-localization when the α-peptide is fused with A-IDPs compared to the fluorescent reporter alone (Supplementary Fig. 21). To quantify the observed co-localization, we analysed individual cells within the image frame with green fluorescence that were above the background threshold at each timepoint. Higher-molecular-weight A-IDPs exhibit higher fluorescence inside the cell normalized to the background at each point in time (Fig. 7c).
Quantification of fluorescence production at various substrate concentrations in vitro suggests that the mechanism of this effect is a statistically significant increase in the catalytic constant or turnover efficiency (Kcat) of the enzyme with increasing molecular weight. We observed a 1.4-, 1.6- and 4.2-times increase in the Kcat for αp–[WT]-20–mRuby3 (t-test, P = 0.025), αp–[WT]-40–mRuby3, (t-test, P = 2.9 × 10−4), αp–[WT]-80–mRuby3 (t-test, P = 7.4 × 10−5) compared to the αp–mRuby3 control (Supplementary Fig. 22 and Supplementary Table 5). Considering our previous observation of increased co-localization of product (fluorescein) and the labelled A-IDP as a function of molecular weight, we propose that the observed increase in fluorescence is caused by increased co-localization of the enzyme and substrate in the condensates, leading to a higher measured Kcat. In contrast, the Michaelis–Menten constant (Km) which describes the affinity of the enzyme for the substrate remained constant, suggesting that fusion of the A-IDP does not change the binding kinetics of the enzyme–substrate complex. Using Kcat and Km, we can define a catalytic efficiency which also supports our hypothesis of an increase in the enzymes’ efficiency within condensates with increasing molecular weight of the A-IDP. This enhancement in efficiency is on the order of magnitude of change observed by protein engineering techniques used to improve Kcat (refs. 54–56).
We also fused the LacZ αp to A-IDPs with differing levels of aromatic content at a constant molecular weight (Supplementary Fig. 23). We hypothesized that differing levels of aromatic content would affect FDG uptake into the droplets and therefore affect overall enzymatic activity. Surprisingly, we observed similar overall levels of fluorescence between αp–[WT]-40–mRuby3, αp–[3Y7:V7]-40–mRuby3 and αp–[Y7:V7]-40–mRuby3. However, the dynamics of enzymatic activity are different, with A-IDPs with greater aliphatic content allowing for faster uptake into the condensates (Supplementary Fig. 23). The difference between the ratio of FDG fluorescence inside the cell and outside the cell between αp–A-IDP–mRuby3 fusions with different aliphatic content were not notable, indicating that molecular weight is the primary driving force for fluorescein and/or FDG persistence inside intracellular droplets (Supplementary Fig. 23). Completely deleting the aromatic residues from the repeat unit of the A-IDP results in apparently soluble enzymes that do not form intracellular condensates, and whose activity is lower than the enzyme formed by complementation of LacZΔM15 with the αp–mRuby3 fusion that possess no A-IDP tag (Supplementary Fig. 24).
Discussion
We show A-IDPs comprising repeats of an octapeptide motif inspired by native IDPs exhibit reversible UCST phase separation in aqueous solution. Despite the simplicity of their sequence, they recapitulate many of the features seen in more complex, native IDPs. The formation and dynamics of their phase separation into coacervate droplets are controlled by two simple design parameters (the molecular weight of the A-IDP and the ratio of aromatic:aliphatic residues in the octapeptide repeat) that are genetically encodable at the sequence level. Using these two parameters, we were able to produce A-IDPs with Csats ranging from nanomolar to millimolar concentrations. This work supports the growing evidence of R-aromatic interactions that drive phase behaviour8,11,18,26,57,58 and also provides additional evidence of the molecular hierarchy that exists between the aromatic groups W, Y, F and H in modulating UCST phase behaviour. In particular, W and H require further investigation as they are equally prevalent in IDPs as F and Y, as W exhibits an extremely potent pair-wise interaction with arginine, whereas H exhibits a physiologically relevant shift in phase behaviour between pH 6.0–7.4. Aromatic groups, in particular, are interesting mechanistically for their propensity to promote lower Csats for both UCST and LCST systems whereas polar non-charged residues, charged residues and aliphatic residues affect Csat in UCST or LCST phase behaviour differently. It is possible that the key to intracellular phase separation is the incorporation of these residues and structural disorder into a sequence at a minimum threshold of the total sequence such that they phase separate on expression59. Finally, our results suggest that molecular weight may be more critical than composition in defining the UCST binodal line. These results provide a rational approach to mutate native IDPs and design A-IDPs de novo with bespoke properties.
These design parameters also faithfully translate from in vitro to intracellular environments. The A-IDPs phase separate inside cells by the same principles that drive their UCST phase separation in vitro, indicating that the same thermodynamic driving forces embedded in the sequence and molecular weight also modulate droplet formation dynamics. A-IDPs behave in vivo as their phase diagrams in vitro suggest—as their intracellular concentration increases to a sequence-encoded Csat, small phase-separating droplets form at individual points in space that continue to grow in size with increasing overall A-IDP concentration.
Finally, these proteins can be used for the de novo design of functional intracellular droplets. We rationally designed intracellular puncta capable of binding and recruiting a β-galactosidase deletion mutant, which could modify the catalytic efficiency of the enzyme–substrate complex, a complex that did not evolve to form intracellular condensates. The catalytic efficiency of the reconstituted enzyme in phase-separated coacervate droplets is molecular-weight dependent and increases with the molecular weight of the A-IDP. Higher-molecular-weight A-IDPs more efficiently sequester the substrate in the enzymatically active puncta, which results in a higher catalytic efficiency. These proof-of-concept experiments demonstrate that intracellular droplets can be engineered to have non-canonical functions in live cells and provide a new platform for intracellular material manipulation. In summary, with over 60 A-IDPs synthesized for this study that span a range of Csats, and proof-of-concept experiments recruiting proteins into coacervate droplets, these studies lay the groundwork for the de novo design of functional intracellular condensates. We expect that these A-IDPs will be useful as building blocks from which new biological condensates with emergent behaviours can be constructed within living cells to better study the functional significance of phase separation in living cells and encode for new cell behaviours. We also anticipate that these IDPs will prove useful in other biomedical applications beyond the design of intracellular droplets that can profit from the their tunable UCST phase behaviour. This marriage of soft material science with biophysical characterization of subcellular materials will continue to be an exciting space for engineering cells with new or improved function and new biomaterials.
Methods
Proteomic analysis.
A search of the literature provided an excellent list of intrinsically disordered proteins or protein regions that are present in genes known to form membraneless organelles3. Each gene was divided into disordered regions and ordered regions according to the Predictor of Natural Disordered Regions VSL2 algorithm, which is a metapredictor of protein disorder of various lengths60. Amino acid quantity was normalized to total protein length.
Gene synthesis.
Each octapeptide amino acid motif inspired by our proteomic analysis was propagated twenty times in silico. This repetitive amino acid sequence was fed into an algorithm that creates an optimally non-repetitive DNA template from a repetitive protein gene. This 20-mer repeat gene was then ordered from Integrated DNA technologies with Gibson assembly overhangs for easy insertion into our in-house-modified pet24+ vector61. To increase the number of total repeats of the gene, we performed iterative cloning steps of recursive directional ligation by plasmid reconstruction, adding an extra twenty repeats during each step. This method has been described previously in ref. 62. Transformations were performed in the desired E. coli cell line (BL21 (DE3)) for recombinant expression and single-plasmid confocal experiments, and a modified BL21 (DE3) cell line termed KRX by Promega that contains a mutated LacZ gene for enzymatic experimentation.
In experiments with dual expression, genes were inserted into the pBAD33.1 vector by cutting custom pet24+ vector and pBAD33.1 cut with HindIII and XbaI. Gel purification was used to isolate the gene of interest from the housing pet24+ vector, which was then ligated into the similarly cut pBAD33.1 vector. Co-transformation was performed with ~1 ng final concentration of each plasmid on kanamycin/chloramphenicol dual-selection plates.
Protein expression, purification and characterization.
Individual liquid cultures of BL21 E. coli strains each harbouring our gene of interest from Supplementary Table 1 or 2 were inoculated into 5 ml of Terrific Broth (TB) medium from frozen glycerol stocks and grown to confluence overnight (16–18 h). Cultures were then inoculated at a 1:200 dilution in 1 l TB media supplemented with 45 μg ml−1 kanamycin. Cells were grown at 37 °C in a shaking incubator (~200 r.p.m.) for 9 h, at which time protein expression was induced by the addition of 500 μM IPTG (final concentration). Cells were then incubated at 37 °C (shaking at ~200 r.p.m.) for an additional 18 h. Protein was then purified from the insoluble cell suspension fraction. Cell pellets were isolated by centrifuging cultures at 3,500 RCF and resuspending in 20 ml of milli-Q water. Cells were then lysed by sonicating the cell solutions for 2 min, with 10 s of pulsing followed by 40 s of rest on ice (Misonix)
Centrifuging each lysate suspension at 20,000 RCF for 20 min resulted in a soluble and insoluble fraction. The supernatant was discarded with the insoluble fraction resuspended in an approximately equal volume of 8 M urea + 150 mM PBS buffer (~6–8 ml). For proteins with a fluorescent fusion tag, the insoluble fraction was resuspended in three-times insoluble volume at a final concentration of urea of ~2 M to prevent protein misfolding. This suspension was heated for 10 min in a 37 °C water bath and then centrifuged at 20,000 RCF for 20 min. The supernatant was collected from this suspension and dialysed in a 10 kDa membrane (SnakeSkin, Thermo Fischer Scientific) against a 1:200 milli-Q water solution at 4 °C. The dialysis water was changed twice over a 48 h period. From inside the dialysis bag, both insoluble and soluble components were collected and centrifuged at 3,500 RCF for 10 min and 4 °C. The supernatant was removed and the remaining insoluble pellet containing the protein of interest was lyophilized for a minimum of three days to remove all water from the pellet.
Protein purity was characterized by 4–20% gradient tris-HCl (Biorad) SDS–PAGE and staining with either 0.5 M copper chloride or SimplyBlue SafeStain (Thermo Fischer Scientific). Protein yield was determined by weight after lyophilization.
Creation of water-in-oil droplets with chip microfluidics.
To create water-in-oil emulsion droplets, two liquid phases—a dispersed, aqueous phase containing the protein of interest in 150 mM PBS, and an organic, continuous phase comprised of 75/5/20% vol/vol TEGOSOFT DEC, ABIL EM 90 and mineral oil—were injected into the microfluidic droplet generators at constant flow rates using precision syringe pumps. The flow rates of the dispersed and continuous fluids were tuned to ensure droplet formation in the dripping regime; in these experiments, the dripping regime was achieved using a constant flow rate of 500 μl h−1 for the organic continuous phase and 50–75 μl h−1 for the aqueous, dispersed phase. The production of droplets within the microfluidic device was monitored using a ×5 objective on an inverted microscope (Leica) equipped with a digital microscopy camera (Lumenera Infinity 3-1 charge-coupled device).
Circular dichroism spectroscopy.
Circular dichroism spectroscopy was performed using an Aviv Model 202 instrument and a 1 mm quartz sample cell (Hellma). A-IDPs were prepared by dissolving the purified lyophilized product in 5 mM PBS, pH 7.4 at a final concentration of 10 μM. The circular dichroism spectra were obtained at 50 °C from 260 nm to 180 nm in 1 nm steps at a 0.5 s average time. Data points with a dynode voltage above 500 V were ignored in the analysis. The circular dichroism spectra were corrected for the 5 mM PBS buffer signal at 50 °C. This data collection was repeated in triplicate and the average of the three measurements was represented as molar ellipticity.
Light scattering.
Dynamic light scattering measurements were performed over a temperature range of 10–80 °C using a Wyatt DynaPro temperature-controlled microsampler (Wyatt Technology). Samples for the DLS system were prepared in 150 mM PBS and filtered through 0.02 μm Whatman Anotop sterile syringe filters (GE Healthcare Life Sciences) into a 12 μl quartz crystal cuvette (Wyatt Technology). Five acquisitions were taken at each temperature for a 5 s duration, and the results presented represent the mean Rh of the sample at each temperature.
Temperature-controlled ultraviolet–visible spectrophotometry.
Tt values were determined via temperature-controlled spectrophotometry using a Cary 300 (Agilent Technologies). Samples containing various concentrations of protein in 150 mM PBS were cooled at 1 °C min−1 while the absorbance at λ = 350 nm was recorded at every 1 °C decrement. Absorbance was normalized to the absorbance at the highest temperature point collected, corresponding to the more soluble point during a given experiment. The cloud point was determined as the maximum in the first derivative of the absorbance as a function of temperature.
The transition temperature was calculated by the point of minimum slope. Saturation concentration was defined by the natural logarithm fit line created from a minimum of three ϕ. Error bars represent the standard error of the mean from three repeats of a minimum of three transition temperatures.
Dextran uptake experiments.
The uptake of dextran molecules into the phase-separated space of [WT]–20 and [Q5,8]-20 was performed to quantify the isolation of A-IDPs from their surroundings. Fluorescein-isothiocyanate-labelled dextrans (10 kDa, 40 kDa, Sigma-Aldrich) were added to 4 mg ml−1 solutions of unlabelled [WT]-20 and [Q5,8]-20 at final concentrations of 4 mg ml−1 and 1 mg ml−1, respectively, at 60 °C. Soluble samples were then transferred to room-temperature glass slides and mounted with a 1.5 cover slip. Samples were imaged on an upright Zeiss Axio Imager D2 microscope with a ×20 objective and the appropriate filter set (excitation laser 470/40 nm, emission filter 525/50 nm) after 1 h incubation below the transition temperature. The fluorescent intensity was calculated from background corrected fluorescent intensities inside/outside droplets in ImageJ portioned using bright field images of the phase-separated space.
Sample preparation for temperature-gradient experiments.
High-concentration A-IDP stock solutions (60 wt%) were prepared by resuspending a mass of lyophilized A-IDP pellets with an appropriate volume of PBS at a solution pH of 7.0. The concentration was converted to milligrams per millilitre by assuming that the density of the A-IDP was 1 g ml−1. The A-IDP stock solution was heated in a water bath at 85 °C for 60 min and mixed periodically along with sonication to ensure homogeneity. Lower concentration samples were made by mixing the initial stock solution volumetrically with PBS at a pH of 7. To prepare for temperature-gradient microfluidics measurements, the solutions were loaded into 12 mm ×1 mm × 0.1 mm rectangular borosilicate glass capillary tubes (VitroCom), by capillary action, and sealed with wax to avoid sample evaporation and convection. The capillary tubes were held in contact with a hot plate at 85 °C housed within an incubator at 65 °C during the loading process. The high-temperature environment ensured that the A-IDP solutions were held above the critical phase transition temperature (~85 °C for [WT]-20). Capillary arrays were prepared by taping several capillaries together. The arrays were stored at 85 °C in an oven for 10 min before subjecting them to the temperature-gradient experimentation.
Measuring phase transition temperatures on a temperature-gradient device.
The temperature-gradient device imposed a linear temperature gradient across the A-IDP solutions. This was accomplished by placing the glass capillary array into thermal contact with a heat source on one side and a cold sink on the other. The sample was then bathed in white light. This light was scattered by phase-separated A-IDP droplets at a cold temperature and was imaged via dark-field microscopy. The temperature gradient was calibrated for each experiment using two reference solutions placed alongside the A-IDP samples of interest. The cold temperature calibration reference contained 10 mg ml−1 poly(N-isopropyl acrylamide) with molecular weight = 1.868 × 105 g mol−1 in H2O (Polymer Source). The hot temperature calibration reference contained 10 mg ml−1 poly(ethylene oxide) with a molecular weight of 9 × 105 g mol−1 in a 1 M NaCl aqueous solution (Sigma-Aldrich). The LCST of each reference solution was obtained with a melting point apparatus that measured the light scattering intensity as the temperature was increased at a rate of 0.5 K min−1. When placed onto the temperature-gradient device, the reference solutions became cloudy at temperatures above the LCST. The pixel position of the LCST was obtained by the onset of light scattering intensity relative to the low intensity baseline on the cold side of the capillary. The temperature gradient was calculated using the pixel positions and the LCSTs of the two samples, assuming a linear relationship between position and temperature.
Fitting of phase diagram binodal.
Fits for the roughly dilute, overlap and semidilute regions of our obtained phase diagrams were calculated using fitting methods for LCST polypeptides adopted for UCST polypeptides as described previously63.
Briefly, for low volume fractions (ϕ < 0.1), A-IDPs exhibit roughly a logarithm-normal dependence on UCST cloud point with respect to ϕ as seen with other repeat polypeptides20,21. For the high-density regime (ϕ > ~0.4), using surface tension scaling methods previously described for elastin-like polypeptides64, we determined the coefficients of proportionality (A) and estimated theta temperature (θ) to be A = −0.00092 and θ = 389 K for [WT]-20, and A = −0.00092 and θ = 392 K for [Q5,8]-20. In a poor solvent, the surface tension of a dilute phase globule γ can be written in the form γ ≈ C KTb2 ɸ″2 where b is the polypeptide Kuhn length (b = 2.2 nm as measured by Fluegel and co-workers for other repetitive polypeptides65) and C an adjustable coefficient. On replacing the surface tension γ by kTϕ″ × A × (T − θ), we obtain an equation for the temperature dependence of the coacervate ϕ
Using a least-squares fit in Igor (WaveMetrics), we adjust the coefficient C for this temperature dependence to match the measured [WT]-20 and the [Q5,8]-20 binodal points, determining that C = 0.62 and 1.05, respectively.
Closer to the critical point in the so-called Ginzburg zone43, one needs to use the critical Ising model to describe the phase behaviour of polymer solutions.
The phase boundary in the critical zone varies more gradually than predicted by mean-field theory:
Where Tc = 351.5 K for [WT]-20 and Tc = 332.3 K for [Q5,8]-20; 0.3 is the critical Ising exponent (Flory–Huggins mean-field value is 0.5) for both [WT]-20 and [Q5,8]-20, and Cc is the fitting coefficient. We calculated fitting coefficients in Igor (WaveMetrics) equal to 1.29 and 1.27 for [WT]-20 and [Q5,8]-20, respectively. Note that we calculated ϕ′ explicitly using data collected with ultraviolet–visible spectrophotometry and according to natural logarithm fits described in Supplementary Table 1.
Whole cell fluorescent intensity measurements.
Cells were grown overnight in 5 ml of TB media from glycerol stocks. In conjunction to fluorescent or confocal imaging, cells were analysed for total sfGFP fluorescence and OD600. Briefly, 50 μl of cell culture at various time points was resuspended in 1 ml of 150 mM PBS. Using a combination of a ultraviolet–visible spectrophotometry signal from a NanoDrop 1000 (Thermo Fisher Scientific) and fluorescent spectra from a NanoDrop 3300 (Thermo Fisher Scientific), we calculated the relative ratio of sfGFP fluorescence normalized to cell density. Using this information in conjunction with imaging analysis, we were able to determine the intracellular saturation concentration normalized to cell density.
Temperature-controlled fluorescent microscopy of protocell droplets and E. coli. bacteria.
Water-in-oil droplets were collected on a glass microscope slide and cooled using a precision Peltier heating and cooling stage (Linkam LTS120) equipped with a temperature control unit (Linkam PE95). The spatial distributions of Alexa-Fluor-350-labelled (25% molar fraction N-terminal labelled) [Q5,8]-20 and Alexa-Fluor-594-labelled +4 Net were characterized via fluorescence microscopy using an upright Zeiss Axio Imager D2 microscope with a ×20 objective and the appropriate filter set. Similarly, intracellular pattering of A-IDP-superfolder GFP over time was characterized via fluorescence microscopy using an upright Zeiss Axio Imager D2 microscope with a ×20 objective and the appropriate filter set (excitation laser 470/40 nm, emission filter 525/50 nm). Cell fluorescence was calculated using ImageJ software. Temperature ramps began at various temperatures but always were set to a constant speed of 5 °C min−1.
Transient transfection of [WT]-20–sfGFP in HEK293 cells.
[WT]-20–sfGFP was extracted from the pet24(+) vector using the polymerase chain reaction. Briefly, the forward and reverse primers were resuspended with 1 ng of pet24(+) plasmid containing [WT]-20–sfGFP gene fusion. Using a polymerase-chain-reaction cycle of (98 °C, 1 min; 65 °C, 30 s; 72 °C, 2 min) × 30, followed by gel purification, the gene was finally constructed with Gibson assembly. pcDNA5 vector containing [WT]-20–sfGFP was transfected into HEK293 cells according to manufacturer instructions (Expi293 Expression System, Thermo Fischer Scientific). Cells were spun down at 500 RCF for 10 min at room temperature on day five of transient transfection and resuspended in 150 mM PBS for imaging.
Confocal imaging of A-IDP–sfGFP fusions for puncta formation and co-localization.
Cells were prepared as follows. A tube containing 5 ml of TB media was inoculated overnight with protein of choice from bacterial glycerol stock. After 16 h of growth, cells were induced with 1 mM IPTG and 2% l-rhamnose (Sigma-Aldrich) in each flask of interest. Samples were collected at the indicated time points and prepared for imaging as follows. 50 μl of cell suspension was pelleted under 20,000 RCF for 1 min at room temperature. Cells were resuspended to OD600 = 0.15 at 1 cm path length. 50 μl of resuspended bacterial cells were transferred to a 384-well plated with 1.5 glass bottom (Cellvis). There was a 10 min equilibration period to the incubation chamber before each time point data collection.
Images were collected at different time points with a ×63 oil-immersion objective on a Zeiss 710 inverted confocal with temperature-controlled incubation (Car Zeiss AG). sfGFP fluorescent was detected with a 488 nm excitation laser and 488/594 emission filter. Data was primarily taken at 25 °C unless otherwise noted. All fluorescent quantification and cell portioning analysis was performed in ImageJ.
In co-localization experiments, cells were grown overnight from glycerol stock in dual antibiotic media containing 45 μg ml−1 kanamycin and 25 μg ml−1 chloramphenicol (final concentration). After 16–18 h, pet24(+) expression was induced with 1 mM concentration of IPTG (final concentration). After 24 h of IPTG induction, the medium was replaced with 5 ml of TB supplemented with 1 mM IPTG and 2% arabinose (final concentration) (Sigma-Aldrich). After 9 h of induction with both, cells were prepared for confocal imaging by spinning down 50 μl of culture at room temperature and resuspended in 150 mM PBS to OD600 = 0.15 at 1 cm path length. All imaging details remain the same except that mNeonGreen/sfGFP detection was performed with a 488 nm excitation laser and 488/594 nm emission filter, whereas mRuby3 detection was performed with a 561 nm excitation laser and 488/561 nm emission filter.
Spinning disc confocal imaging of LacZ αp–A-IDP gene fusions for localization and quantification of enzymatic activity.
Cells were prepared as follows. A tube containing 5 ml of TB media was inoculated overnight with the protein of choice from the bacterial glycerol stock. After 16 h of growth, cells were induced with 1 mM IPTG and 2% l-rhamnose (Sigma-Aldrich) in each flask of interest; ~24 h later, 50 μl of cell suspension was pelleted under 20,000 RCF for 1 min at room temperature. Cells were resuspended in 150 mM PBS to OD600 = 0.15 at 1 cm path length. Fifty microliters of sample were added to Culture-Insert 4 Well (1.5 coverslip, Ibidi) petri dishes and allowed to incubate at room temperature for 10 min. After incubation, 2 μl of 1 mg ml−1 FDG resuspended in 98% water, 1% DMSO and 1% EtOH was added. Imaging began immediately (within 20 s) and images were captured every minute for 30 min total.
Imaging was performed on an Andor Dragonfly Spinning Disk 500 series confocal on a LeicaDMi8 microscope stand (Oxford Instruments) with a ×63 water immersion objective and equipped with a Zyla 4.2 series camera. Converted FDG was detected with a 488 nm excitation laser and 525/50 nm emission filter and mRuby3 fluorescence with a 561 nm excitation laser and 600/50 nm emission filter.
Fluorescence spectroscopy for determining Km, Vmax and kcat.
Liquid cultures of KRX E. coli containing plasmid of interest were grown from glycerol stocks overnight (16–18 h). Cells were then induced with 1 mM IPTG and 2% l-rhamnose (Sigma-Aldrich) for 24 h. Cells were pelleted and resuspended at OD600 = ~0.15 in 150 mM PBS. Various concentrations of FDG were added while monitoring fluorescent intensity at 520 nm using NanoDrop 3300 (Thermo Fisher Scientific). The same instrument was used to also calculate the fluorescent intensity of mRuby3 as a relativistic measure of expression level of the various αp fusions. Plotting the observed fluorescent intensity at different times provides a surrogate measure of the rate of hydrolysis at various concentrations of the substrate (Vo). These rates were then converted into typical Lineweaver–Burk conventions to determine Vmax and Km. For consistency in units, [FDG] was converted into fluorescent intensity using a fluorescein standard curve of y = 185919 × [FDG in milligrams] + 1,045. This conversion assumes that converted FDG into fluorescein has similar fluorescent intensity profile to free fluorescein dye.
Supplementary Material
Acknowledgements
A.C. acknowledges support from the National Institutes of Health (NIH) through an MIRA grant R35GM127042 and from the National Science Foundation (NSF) through a DMREF grant DMR-17-29671. P.S.C acknowledges support from the National Science Foundation through CHE-1709735.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41557-020-0511-7.
Reporting summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41557-020-0511-7.
Data availability
The data that support the findings of this study are available within this Article and its Supplementary Information. Material requests should be made to chilkoti@duke.edu.
References
- 1.Babu MM The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem. Soc. Trans 44, 1185–1200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wright PE & Dyson HJ Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell. Biol 16, 18–29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uversky VN, Kuznetsova IM, Turoverov KK & Zaslavsky B Intrinsically disordered proteins as crucial constituents of cellular aqueous two phase systems and coacervates. FEBS Lett. 589, 15–22 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Boeynaems S et al. Protein phase separation: a new phase in cell biology. Trends Cell. Biol 28, 420–435 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sickmeier M et al. DisProt: the database of disordered proteins. Nucleic Acids Res. 35, D786–D793 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brangwynne CP, Tompa P & Pappu RV Polymer physics of intracellular phase transitions. Nat. Phys 11, 899–904 (2015). [Google Scholar]
- 7.Lin Y, Currie SL & Rosen MK Intrinsically disordered sequences enable modulation of protein phase separation through distributed tyrosine motifs. J. Biol. Chem 292, 19110–19120 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pak CW et al. Sequence determinants of intracellular phase separation by complex coacervation of a disordered protein. Mol. Cell 63, 72–85 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elbaum-Garfinkle S et al. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc. Natl Acad. Sci. USA 112, 7189–7194 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin Y, Protter DS, Rosen MK & Parker R Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol. Cell 60, 208–219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nott TJ et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell 57, 936–947 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molliex A et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franzmann TM et al. Phase separation of a yeast prion protein promotes cellular fitness. Science 359, aao5654 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Best RB Computational and theoretical advances in studies of intrinsically disordered proteins. Curr. Opin. Struct. Biol 42, 147–154 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Dignon GL, Zheng W, Best RB, Kim YC & Mittal J Relation between single-molecule properties and phase behavior of intrinsically disordered proteins. Proc. Natl Acad. Sci. USA 115, 9929–9934 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dignon GL, Zheng W, Kim YC, Best RB & Mittal J Sequence determinants of protein phase behavior from a coarse-grained model. PLoS Comput. Biol 14, e1005941 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mao AH, Crick SL, Vitalis A, Chicoine CL & Pappu RV Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl Acad. Sci. USA 107, 8183–8188 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J et al. A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell 174, 688–699 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dzuricky M, Roberts S & Chilkoti A Convergence of artificial protein polymers and intrinsically disordered proteins. Biochemistry 57, 2405–2414 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quiroz FG & Chilkoti A Sequence heuristics to encode phase behaviour in intrinsically disordered protein polymers. Nat Mater. 14, 1164–1171 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyer DE & Chilkoti A Quantification of the effects of chain length and concentration on the thermal behavior of elastin-like polypeptides. Biomacromolecules 5, 846–851 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Li Z, Tyrpak DR, Lien C-L & MacKay JA Tunable assembly of protein-microdomains in living vertebrate embryos. Adv. Biosyst 2, 1800112 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi P, Lin YA, Pastuszka M, Cui H & Mackay JA Triggered sorting and co-assembly of genetically engineered protein microdomains in the cytoplasm. Adv. Mater 26, 449–454 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huber MC et al. Designer amphiphilic proteins as building blocks for the intracellular formation of organelle-like compartments. Nat. Mater 14, 125–132 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Pastuszka MK et al. A tunable and reversible platform for the intracellular formation of genetically engineered protein microdomains. Biomacromolecules 13, 3439–3444 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qamar S et al. FUS phase separation is modulated by a molecular chaperone and methylation of arginine cation-π interactions. Cell 173, 720–734 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shin Y & Brangwynne CP Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Lin YH, Forman-Kay JD & Chan HS Sequence-specific polyampholyte phase separation in membraneless organelles. Phys. Rev. Lett 117, 178101 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Li L, Luo T & Kiick KL Temperature-triggered phase separation of a hydrophilic resilin-like polypeptide. Macromol. Rapid Commun 36, 90–95 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balu R et al. An16-resilin: an advanced multi-stimuli-responsive resilin-mimetic protein polymer. Acta Biomater. 10, 4768–4777 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Uversky VN & Dunker AK Understanding protein non-folding. Biochim. Biophys. Acta 1804, 1231–1264 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Theillet FX et al. The alphabet of intrinsic disorder: I. Act like a Pro: on the abundance and roles of proline residues in intrinsically disordered proteins. Intrinsically Disord. Proteins 1, e24360 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kyte J & Doolittle RF A simple method for displaying the hydropathic character of a protein. J. Mol. Biol 157, 105–132 (1982). [DOI] [PubMed] [Google Scholar]
- 34.Wimley WC & White SH Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Mol. Biol 3, 842–848 (1996). [DOI] [PubMed] [Google Scholar]
- 35.Das RK & Pappu RV Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl Acad. Sci. USA 110, 13392–13397 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin YH & Chan HS Phase separation and single-chain compactness of charged disordered proteins are strongly correlated. Biophys. J 112, 2043–2046 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Urry DW et al. Hydrophobicity scale for proteins based on inverse temperature transitions. Biopolymers 32, 1243–1250 (1992). [DOI] [PubMed] [Google Scholar]
- 38.Van Roey K et al. Short linear motifs: ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem. Rev 114, 6733–6778 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Martin EW et al. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 367, 694–699 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacEwan SR et al. Phase behavior and self-assembly of perfectly sequence-defined and monodisperse multiblock copolypeptides. Biomacromolecules 18, 599–609 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muiznieks LD & Keeley FW Proline periodicity modulates the self-assembly properties of elastin-like polypeptides. J. Biol. Chem 285, 39779–39789 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bochicchio B, Pepe A & Tamburro AM Investigating by CD the molecular mechanism of elasticity of elastomeric proteins. Chirality 20, 985–994 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Rubinstein M & Colby RH Polymer Physics (Oxford Univ. Press, 2003). [Google Scholar]
- 44.Bracha D et al. Mapping local and global liquid phase behavior in living cells using photo-oligomerizable seeds. Cell 175, 1467–1480 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Banani SF, Lee HO, Hyman AA & Rosen MK Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol 18, 285–298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strulson CA, Molden RC, Keating CD & Bevilacqua PC RNA catalysis through compartmentalization. Nat. Chem 4, 941–946 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Banjade S & Rosen MK Phase transitions of multivalent proteins can promote clustering of membrane receptors. eLife 10.7554/eLife.04123 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li P et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hofmann J & Sernetz M A kinetic study on the enzymatic hydrolysis of fluoresceindiacetate and fluorescein-di-β-D-galactopyranoside. Analytical biochemistry 131, 180–186 (1983). [DOI] [PubMed] [Google Scholar]
- 50.Lewis P, Nwoguh C, Barer M, Harwood C & Errington J Use of digitized video microscopy with a fluorogenic enzyme substrate to demonstrate cell-and compartment-specific gene expression in Salmonella enteritidis and Bacillus subtilis. Molecular microbiology 13, 655–662 (1994). [DOI] [PubMed] [Google Scholar]
- 51.Broome AM, Bhavsar N, Ramamurthy G, Newton G & Basilion JP Expanding the utility of β-galactosidase complementation: piece by piece. Mol. Pharm 7, 60–74 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moosmann P & Rusconi S Alpha complementation of LacZ in mammalian cells. Nucleic Acids Res. 24, 1171–1172 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Manders E, Stap J, Brakenhoff G, Van Driel R & Aten J Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J. Cell Sci 103, 857–862 (1992). [DOI] [PubMed] [Google Scholar]
- 54.Duan X, Chen J & Wu J Improving the thermostability and catalytic efficiency of Bacillus deramificans pullulanase by site-directed mutagenesis. Appl. Environ. Microbiol 79, 4072–4077 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goldsmith M & Tawfik DS Enzyme engineering: reaching the maximal catalytic efficiency peak. Curr. Opin. Struct. Biol 47, 140–150 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Nayeem A et al. Engineering enzymes for improved catalytic efficiency: a computational study of site mutagenesis in epothilone-B hydroxylase. Protein Eng. Des. Sel 22, 257–266 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Brady JP et al. Structural and hydrodynamic properties of an intrinsically disordered region of a germ cell-specific protein on phase separation. Proc. Natl Acad. Sci. USA 114, E8194–E8203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoshizawa T et al. Nuclear import receptor inhibits phase separation of FUS through binding to multiple sites. Cell 173, e622 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Holehouse AS & Pappu RV Functional implications of intracellular phase transitions. Biochemistry 57, 2415–2423 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peng K, Radivojac P, Vucetic S, Dunker AK & Obradovic Z Length-dependent prediction of protein intrinsic disorder. BMC Bioinformatics 7, 208 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang NC & Chilkoti A Combinatorial codon scrambling enables scalable gene synthesis and amplification of repetitive proteins. Nat. Mater 15, 419–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McDaniel JR, Mackay JA, Quiroz FG & Chilkoti A Recursive directional ligation by plasmid reconstruction allows rapid and seamless cloning of oligomeric genes. Biomacromolecules 11, 944–952 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Simon JR, Carroll NJ, Rubinstein M, Chilkoti A & Lopez GP Programming molecular self-assembly of intrinsically disordered proteins containing sequences of low complexity. Nat. Chem 9, 509–515 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hassouneh W, Zhulina EB, Chilkoti A & Rubinstein M Elastin-like polypeptide diblock copolymers self-assemble into weak micelles. Macromolecules 48, 4183–4195 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fluegel S, Fischer K, McDaniel JR, Chilkoti A & Schmidt M Chain stiffness of elastin-like polypeptides. Biomacromolecules 11, 3216–3218 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available within this Article and its Supplementary Information. Material requests should be made to chilkoti@duke.edu.