

SUMMARY
Mismatch repair (MMR) safeguards genome stability through recognition and excision of DNA replication errors.1–4 How eukaryotic MMR targets the newly replicated strand in vivo has not been established. MMR reactions reconstituted in vitro are directed to the strand containing a preexisting nick or gap,5–8 suggesting that strand discontinuities could act as discrimination signals. Another candidate is the proliferating cell nuclear antigen (PCNA) that is loaded at replication forks and is required for the activation of Mlh1-Pms1 endonuclease.7–9 Here, we discovered that overexpression of DNA ligase I (Cdc9) in Saccharomyces cerevisiae causes elevated mutation rates and increased chromatin-bound PCNA levels and accumulation of Pms1 foci that are MMR intermediates, suggesting that premature ligation of replication-associated nicks interferes with MMR. We showed that yeast Pms1 expression is mainly restricted to S phase, in agreement with the temporal coupling between MMR and DNA replication.10 Restricting Pms1 expression to the G2/M phase caused a mutator phenotype that was exacerbated in the absence of the exonuclease Exo1. This mutator phenotype was largely suppressed by increasing the lifetime of replication-associated DNA nicks, either by reducing or delaying Cdc9 ligase activity in vivo. Therefore, Cdc9 dictates a window of time for MMR determined by transient DNA nicks that direct the Mlh1-Pms1 in a strand-specific manner. Because DNA nicks occur on both newly synthesized leading and lagging strands,11 these results establish a general mechanism for targeting MMR to the newly synthesized DNA, thus preventing the accumulation of mutations that underlie the development of human cancer.
Graphical Abstract
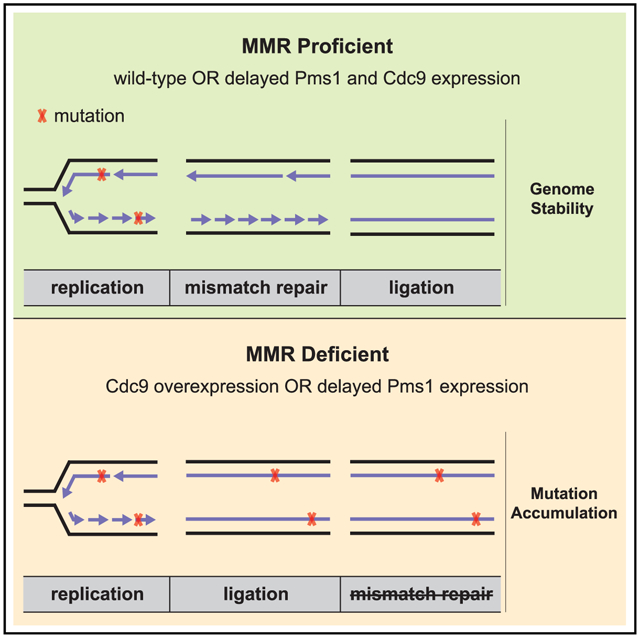
In Brief
The correction of DNA replication errors by the mismatch repair (MMR) machinery requires the discrimination between parental and daughter DNA strands. Reyes et al. provide evidence that DNA replication-associated nicks are used as MMR strand discrimination signals and that DNA ligase I (Cdc9) activity dictates a window of time for MMR.
RESULTS AND DISCUSSION
DNA mismatch repair (MMR) utilizes an excision-resynthesis mechanism to correct mispaired bases that occur in DNA due to errors during DNA synthesis.1–4 To repair these errors, MMR must target only the newly synthesized daughter strand; however, it remains unclear how this machinery discriminates between the parental strand and the daughter strand in eukaryotic living cells. It is known that eukaryotic MMR is temporally coupled to DNA replication, as newly replicated DNA in S. cerevisiae is proficient for MMR for no longer than 10 min during S phase.10 This result suggests that the MMR strand-discrimination signal in eukaryotes involves some aspect of the DNA replication machinery and/or an S-phase-associated property of the daughter strand itself.
Several studies in eukaryotes have investigated transient features of the newly replicated DNA as candidate strand-discrimination signals. Single-strand discontinuities (nicks) present between Okazaki fragments or those generated upon removal of misincorporated ribonucleotides have been proposed to serve as strand-discrimination signals.12,13 However, the inactivation of the ribonucleotide excision repair (RER) pathway causes far lower mutation rates than complete loss of MMR and results in an unusual spectrum of mutations whose formation depends on the activity of topoisomerase I.14 In contrast, mutations affecting MMR strand-discrimination components in Escherichia coli, including the Dam methylase or the MutH endonuclease, result in complete loss of MMR function.15 Another candidate is Proliferating Cell Nuclear Antigen (PCNA), which is loaded asymmetrically at the replication fork,16 interacts with Msh2-Msh6,17 and stimulates the latent endonuclease activity of Mlh1-Pms1 (called MLH1-PMS2 in human).7–9,18,19 PCNA has been proposed to act as a strand-discrimination signal even in the absence of daughter strand discontinuities.20 However, this mechanism is not consistent with the very modest increase in mutation rate caused by loss of the Msh6-PCNA interaction that would be required to target Msh2-Msh6 to newly replicated DNA or retain PCNA at the mispair site.17 Furthermore, the ability of new PCNA trimers to be loaded at nicks in DNA raises the question of whether replication-associated PCNA or replication-associated nicks are ultimately the strand discrimination signal.
If DNA nicks are MMR strand-discrimination signals in vivo, we reasoned that overexpression of the replicative DNA ligase Cdc9 in S. cerevisiae should reduce nick lifetime in newly replicated DNA and potentially cause a mutator phenotype. To increase the expression of Cdc9, we constructed high-copy-number vectors expressing wild-type (WT) CDC9, two CDC9 ligase-defective mutants (cdc9-K419A and cdc9-K598A),21,22 and a Cdc9 variant that does not interact with PCNA in vitro (cdc9-FFAA).23 Next, we examined whether these plasmids could support the growth of a yeast strain in which the endogenous Cdc9 protein was depleted using the auxin-inducible degron (AID) system.24 For this, we generated a yeast strain with a chromosomally encoded version of CDC9 fused to the AID-tag (CDC9-AID), which, in the presence of the plant hormone auxin and the auxin-receptor Afb2, triggers the ubiquitin-mediated proteasomal degradation of the protein fused to the AID-tag. In agreement with previous results,25 WT-CDC9 and the cdc9-FFAA mutant promoted survival of the CDC9-AID strain on auxin-containing medium, whereas the ligase-defective mutants did not (Figure S1A), despite similar levels of expression (Figure S1B). WT Cdc9 overexpression driven by this high-copy-number plasmid caused a modest increase in mutation rates in a WT strain and a strong mutator phenotype in an exo1Δ strain (Figure 1A; Table S1), which lacks the Exo1-dependent MMR pathway.26,27 This increase in the mutation rates was also observed, to a lesser extent, with the cdc9-FFAA allele but was not seen with ligase-defective mutants, indicating that this mutagenic effect requires the overexpression of ligase-proficient Cdc9 and is promoted by an intact PCNA interaction motif. These requirements differ from the trinucleotide repeat (TNR) instability phenotype caused by Cdc9 overexpression, which depends on PCNA recruitment but not on ligase activity and was attributed to a competition between Cdc9 and the flap endonuclease Rad27 for PCNA binding.22
Figure 1. Increased Cdc9 activity results in elevated mutation rates and accumulation of Pms1 foci.
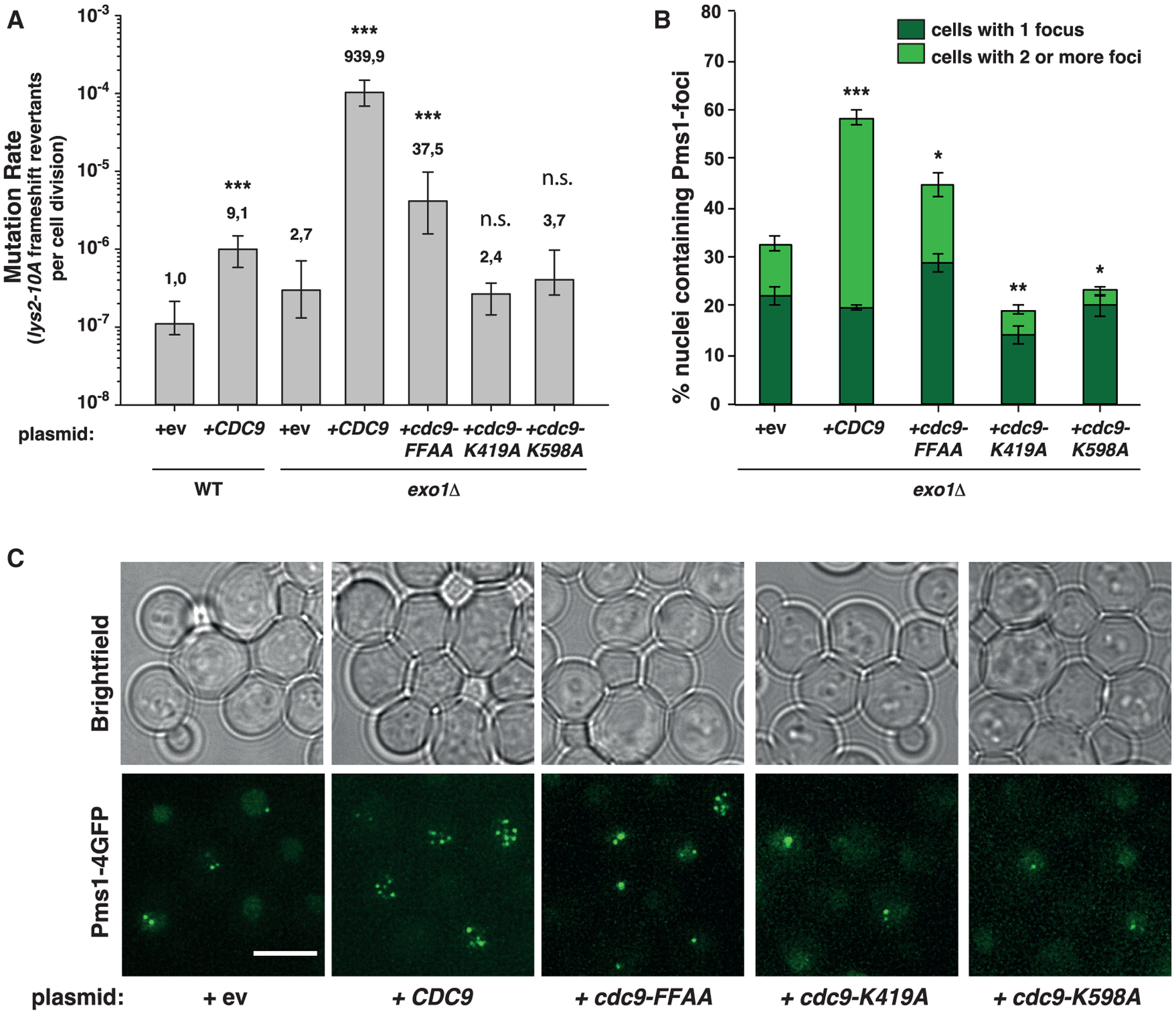
(A) Mutation rate analysis using the lys2-10A frameshift reversion assay in WT or exo1Δ strains carrying plasmids (2μ) bearing WT-CDC9, the PCNA interaction-deficient cdc9-FFAA mutant, the ligase-defective mutants cdc9-K491A and cdc9-K598A, or an empty vector (ev). Bars correspond to the median rate, with error bars corresponding to the 95% confidence interval. Numbers on top of the bars indicate fold increase in mutation rate relative to the WT strain. See also Figure S1 and Table S1.
(B) Percentage of cells containing Pms1-4GFP foci in exo1Δ strains transformed with plasmids shown in (A). Bars represent the average of the percentage of nuclei containing foci; error bars represent standard error of the mean (SEM).
(C) Representative fluorescent microscopy live-cell images of cells containing Pms1 foci used for quantification shown in (B). Brightfield images are shown on top. Scale bar represents 5 μm. p values indicated in (A) and (B) were calculated with the Mann-Whitney rank-sum test using SigmaPlot. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; n.s., not significant.
Mispair recognition by Msh2-Msh6 or Msh2-Msh3 promotes recruitment of the Mlh1-Pms1 endonuclease. Recruitment can be observed as discrete foci in yeast strains expressing a Pms1-4GFP fusion; these Pms1 foci are MMR intermediates that accumulate when downstream MMR functions are compromised (due to mutations affecting Exo1 or preventing the activation of the Mlh1-Pms1 endonuclease) or due to increased levels of mispaired bases.19,28,29 Overexpression of WT-CDC9 or cdc9-FFAA in an exo1Δ strain caused an increase in the percentage of Pms1 foci-containing cells with ⩾2 foci per cell (Figures 1B and 1C). In contrast, overexpression of ligase-defective cdc9 variants modestly reduced Pms1 foci levels. These results suggest that overexpression of functional Cdc9 either slows downstream steps in MMR or inhibits MMR at a step prior to Mlh1-Pms1 turnover.
In agreement with the Cdc9-overexpression plasmid-based results, we found that strains in which we replaced the endogenous CDC9 promoter with the strong constitutive promoter pGPD (CDC9-OE) (causing a ~50-fold increase in Cdc9 levels; Figure S1C) showed a modest mutator phenotype that was greatly exacerbated by mutations that prevent Exo1-dependent MMR (an EXO1 deletion or an exo1-FFAA-Δ571–702 mutant allele, which cannot be recruited by Msh2 or Mlh1 to MMR sites30) (Figure 2A; Table S2). To determine whether MMR defects induced by CDC9-OE were only seen in the presence of defects in Exo1-dependent MMR, we combined the CDC9-OE allele with mutations that disrupt the Exo1-independent MMR pathway: pol30-K217E, which prevents PCNA-dependent activation of the Mlh1-Pms1 endonuclease19 and pms1-A99V (previously called pms1-A130V), which affects a residue in the Pms1 ATPase domain and reduces endonuclease activation.26 We found that CDC9-OE caused a synergistic increase in mutation rates in combination with both the pol30-K217E and pms1-A99V mutations, indicating that Cdc9 overexpression affects both Exo1-dependent and Exo1-independent MMR pathways (Figure 2A). Consistent with this, the CDC9-OE exo1Δ pol30-K217E triple mutant had mutation rates that were significantly higher than those of the exo1Δ pol30-K217E, CDC9-OE exo1Δ and CDC9-OE pol30-K217E double mutants. Even though the CDC9-OE allele in combination with an msh2Δ mutation did not cause a synergistic increase in frameshift mutation rates (Figure 2A), the CDC9-OE exo1Δ pol30-K217E triple mutant showed mutation rates that were modestly higher than that of the MMR-deficient msh2Δ strain (Figure 2A; Table S2). This result raises the possibility that some interaction between Cdc9 overexpression, the absence of Exo1, and disruption of the positively charged surface at the PCNA-DNA interaction surface caused by the pol30-K217E mutation leads to an increased accumulation of mispairs that does not occur when Cdc9 is overexpressed in the absence of MMR.
Figure 2. Cdc9 overexpression interferes with both Exo1-dependent and Exo1-independent MMR pathways.
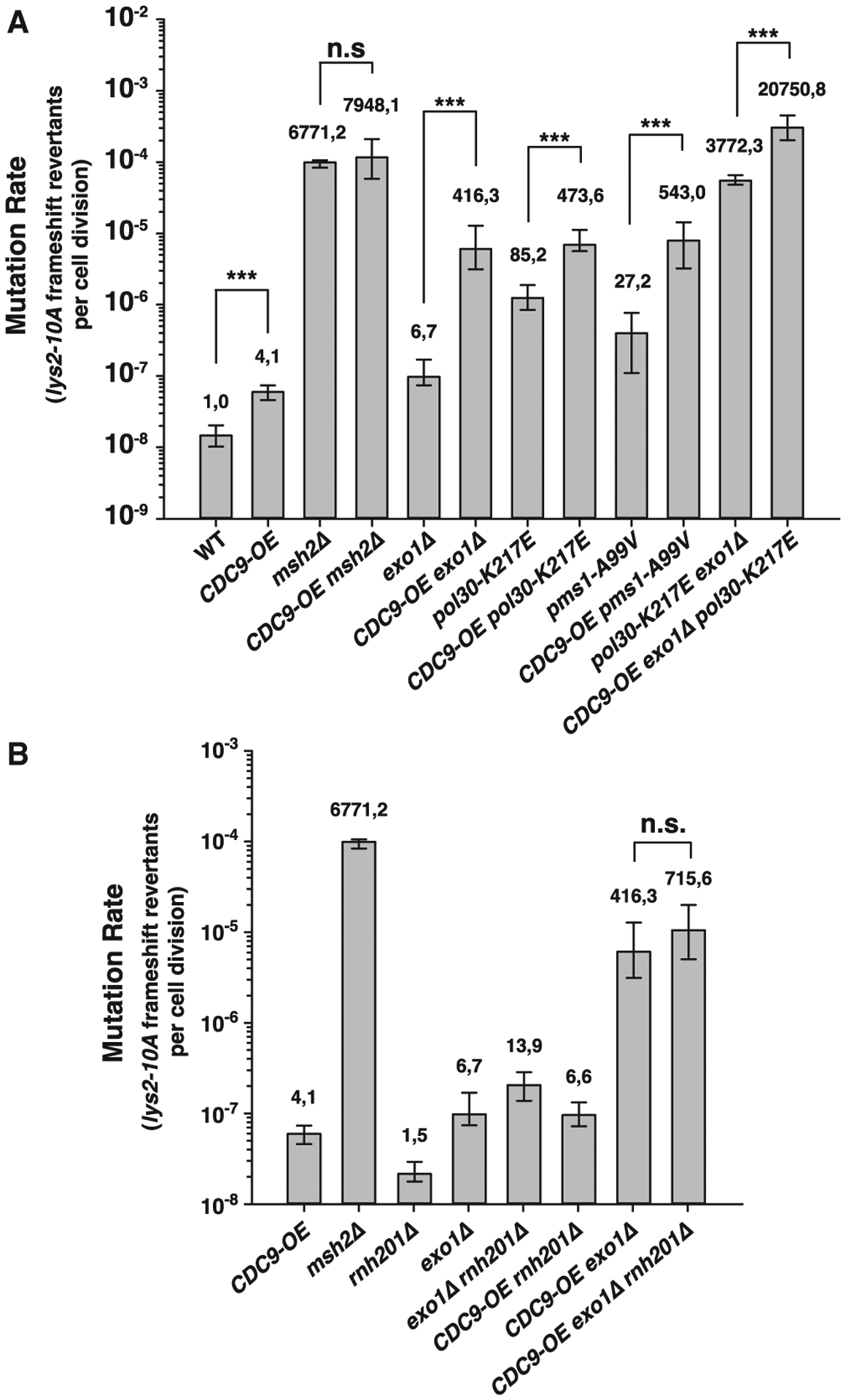
(A and B) Mutation rate analysis using the lys2-10A frameshift reversion reporter in the indicated yeast genetic backgrounds. Bars correspond to the median rate, with error bars representing the 95% confidence interval. Numbers on top of the bars indicate fold increase in mutation rate relative to WT. p values were calculated with the Mann-Whitney rank-sum test using SigmaPlot. ***p ≤ 0.001; n.s., not significant. See also Table S2 for additional mutation rate analysis using two alternative mutational reporters.
We hypothesized that the impaired MMR function caused by CDC9-OE could be the result of the premature ligation of DNA replication-associated nicks. Leading-strand nicks have been suggested to be due to the processing of misincorporated ribonucleotides, as Polε, the leading-strand DNA polymerase, incorporates ribonucleotides four times more frequently than Polδ, the lagging-strand DNA polymerase.31 If ribonucleotide excision plays an important role in MMR strand discrimination, then combining the CDC9-OE allele with a deletion of the RNH201 gene, which encodes the catalytic subunit of RNase H2, should cause increased mutation rates. In contrast, we found that mutation rates in the double-mutant strain CDC9-OE rnh201Δ were similar to that of the CDC9-OE strain; and the CDC9-OE exo1Δ rnh201Δ triple mutant was no more MMR defective than the CDC9-OE exo1Δ strain (Figure 2B; Table S2). These results suggest that RER-associated strand discontinuities are not a major source of MMR strand-discrimination signals.
To further characterize the mutator phenotype associated with Cdc9 overexpression, we analyzed CAN1 mutational spectra. The CAN1 mutation spectrum of the CDC9-OE single mutant showed a similar frequency of base substitutions, a reduced frequency of frameshift mutations, and twice as many complex mutations as the WT strain (Table S3). These complex mutations consisted of duplications or deletions (ranging from 15 to 72 bp) flanked by direct repeats (Table S4) that strongly resemble the mutations that arise in a rad27Δ mutant.32 This observation, together with the TNR-instability phenotype reported for the CDC9-OE strain,22 supports the idea that CDC9-OE interferes with Rad27-dependent Okazaki fragment maturation, resulting in large deletions and duplications that are not restricted to DNA sequences containing TNRs. Consistent with this hypothesis, combining the CDC9-OE allele with an msh2Δ mutation did not cause a synergistic increase in mutation rates using MMR-specific mutation rate assays (lys2-10A and hom3-10 frameshift reversion assays), but it did cause elevated rates in the CAN1 inactivation assay, which detects a broader spectrum of mutations than just those caused by MMR defects (Figure 2A; Table S2). In contrast, the CAN1 mutational spectrum of the exo1-FFAA-Δ571–702 single mutant was indistinguishable from that of the WT strain (Table S3). Remarkably, the mutation spectrum of the CDC9-OE exo1-FFAA-Δ571–702 double mutant was dominated by single-base frameshift mutations, primarily in the three longest mononucleotide runs within the CAN1 gene (six consecutive A or T bases). This spectrum is very similar to that of the msh2Δ strain33 and is characteristic of MMR-deficient strains. The difference between the CAN1 mutational spectra of the CDC9-OE single-mutant and the CDC9-OE exo1-FFAA-Δ571–702 double-mutant strains suggests that the elevated CAN1 mutation rate in the double mutant is caused by an increased MMR defect and not an increased defect in Rad27-dependent Okazaki fragment maturation that would result in the accumulation of large insertion and deletion mutations.
We considered the possibility that the MMR defects in Cdc9-overexpressing strains could be caused by reduced levels of PCNA impairing the activation of the Mlh1-Pms1 endonuclease, as PCNA unloading by the Replication factor C-like complex (RLC)-Elg1 depends on Okazaki fragment ligation.25 We therefore analyzed the effect of a CDC9-OE allele on chromatin-bound PCNA levels (Figures 3A and S1D). As a control, we included strains carrying the pol30-C81R allele or an elg1Δ mutation, which cause reduced or increased levels of chromatin-bound PCNA, respectively.34,35 We found that CDC9-OE caused an increased level of chromatin-bound PCNA (2-fold higher than WT), suggesting that the mutator phenotype and the accumulation of Pms1 foci in CDC9-OE strains are not a consequence of reduced chromatin-bound PCNA. This increased chromatin-bound PCNA is consistent with the results of previous experiments in vitro showing that excesses of PCNA-interacting proteins (e.g., Polδ, FEN1, and DNA ligase I) prevent the RLC-Elg1-dependent unloading of PCNA from DNA, most likely by occluding access to the RLC-Elg1-unloader complex.36 Deletion of ELG1 resulted in the accumulation of PCNA and SUMOylated PCNA (indicated by high-molecular-weight bands recognized by the PCNA-specific antibody) on chromatin (Figure S1D), in accordance with a previous report.35 Similar accumulation of chromatin-bound PCNA was observed in the CDC9-OE elg1Δ double-mutant strain (Figure 3A). In contrast, chromatin-bound PCNA levels were reduced by overexpression of ELG1 (ELG1-OE) in both WT and CDC9-OE strains. Furthermore, ELG1-OE caused a synergistic increase in mutation rate in the CDC9-OE exo1Δ double mutant, resulting in mutation rates comparable to that of an msh2Δ single mutant, but only caused a small effect when combined with an exo1Δ mutation (Figure 3B; Table S2). In addition, ELG1-OE resulted in increased levels of Pms1 foci by itself and in combination with either exo1Δ or CDC9-OE alleles (Figure 3C). Together, these results suggest that ELG1-OE-driven loss of chromatin-bound PCNA interferes with MMR by a different mechanism than CDC9-OE. Interestingly, mutations resulting in a reduction in chromatin-bound PCNA do not strictly correlate with increased mutation rates (e.g., the ELG1-OE allele, which causes reduced chromatin-bound PCNA levels, causes a mild mutator phenotype, even in combination with an exo1Δ mutation) (Figures 3A and 3B; Table S2). Furthermore, it suggests that the subpopulation of PCNA localized at mispair sites—which activates the Mlh1-Pms1 endonuclease—is present at levels that do not directly correlate with the total amount of chromatin-bound PCNA. Deletion of ELG1 (elg1Δ) causes a mild mutator phenotype, even in combination with an exo1Δ mutation or with the CDC9-OE allele, and suppresses the mutator phenotype of a CDC9-OE exo1Δ strain in the lys2-10A assay (Figure 3D; Table S2). This observation suggests that increased chromatin-bound PCNA levels can partially compensate for the effects caused by CDC9-OE and/or EXO1 deletion.
Figure 3. Increased mutagenesis caused by Cdc9 overexpression is not due to the premature unloading of PCNA from DNA.
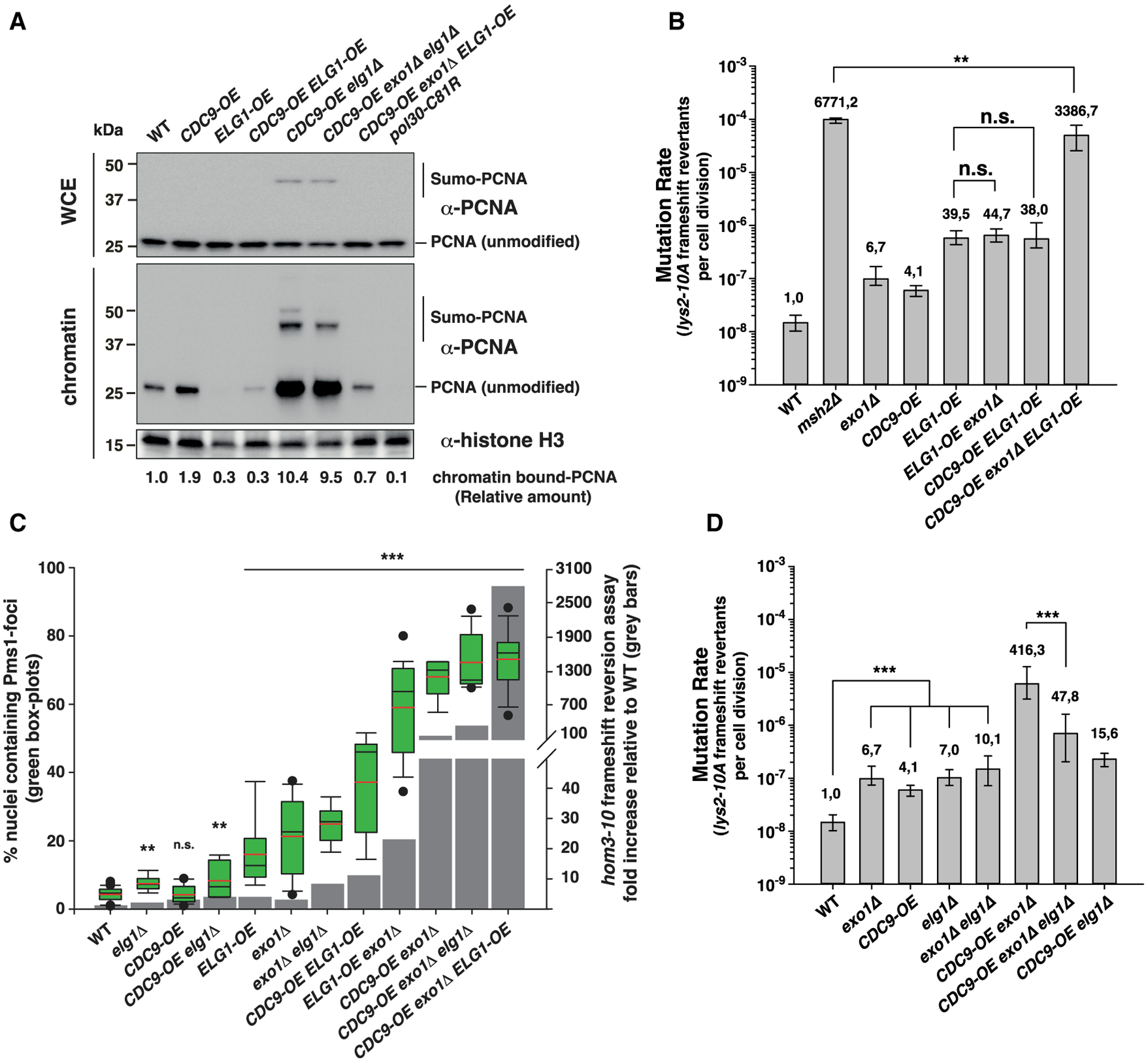
(A) PCNA levels in whole cell extracts (WCE) and chromatin fractions. Histone H3 was the loading control. See also Figure S1D.
(B–D) In (B) and (D): mutation rate analysis using the lys2-10A frameshift reversion reporter in the indicated yeast genetic backgrounds. Bars correspond to the median rate, with error bars indicating the 95% confidence interval. Numbers on top of the bars indicate fold increase in mutation rate relative to WT. See also Table S2 for additional mutation rate analysis and Tables S3 and S4 for CAN1 mutation spectra analysis. (C) Correlation between Pms1-foci abundance and frameshift mutator phenotype (hom3-10 assay). Quantification of Pms1-4GFP foci in boxplot with whiskers; dots represent outliers; black and red lines inside the boxplot represent the median and the average, respectively. Statistical analysis indicated in (C) was performed relative to WT. p values shown in (B)–(D) were calculated with the Mann-Whitney rank-sum test using SigmaPlot. **p ≤ 0.01; ***p ≤ 0.001; n.s., not significant.
We have previously restricted the availability of the Msh2-Msh6 mispair recognition complex to the G2/M phase in living cells by fusing the MSH6 gene to the “G2/M-tag” (derived from the cyclin CLB2 promoter and the Clb2 destruction box; residues 1–181).10 Yeast strains expressing the G2/M-Msh6 fusion protein (in an msh3Δ background to eliminate any repair mediated by the partially redundant Msh2-Msh3 heterodimer) were MMR deficient at loci replicated in mid-S phase but were proficient for MMR at loci replicated in late S phase or at mid-S-phase loci moved to a late-S-phase replication site.10 These previous findings together with the observation that CDC9-OE interferes with MMR suggest that the lifetime of replication-associated nicks control the temporal window for MMR. To test this, we fused the G2/M tag to CDC9 to generate a strain in which CDC9 expression was restricted to G2/M so that replication-associated nicks persist throughout S phase (Figure 4A). Consistent with a recent report,37 strains carrying the G2/M-CDC9 allele were viable and had increased levels of unligated Okazaki fragments relative to WT cells in S phase (Figure S2) but not in G2/M phase when G2/M-Cdc9 is expressed (Figure 4A) or in G1 phase. Moreover, strains expressing G2/M-Cdc9 exhibited DNA-damage checkpoint activation and an accumulation of cells in S phase (Figures S3A–S3C). In contrast, a strain carrying the cdc9-FFAA mutant allele, which is expected to have reduced ligase efficiency but normal expression timing, did not show DNA-damage checkpoint activation or an accumulation of S-phase cells (Figures S3A–S3C) and had less pronounced and slightly larger Okazaki fragments than G2/M-CDC9 when cells were arrested in S or G2/M phase (Figure S2).
Figure 4. Cdc9 activity dictates a window of time for MMR strand discrimination.
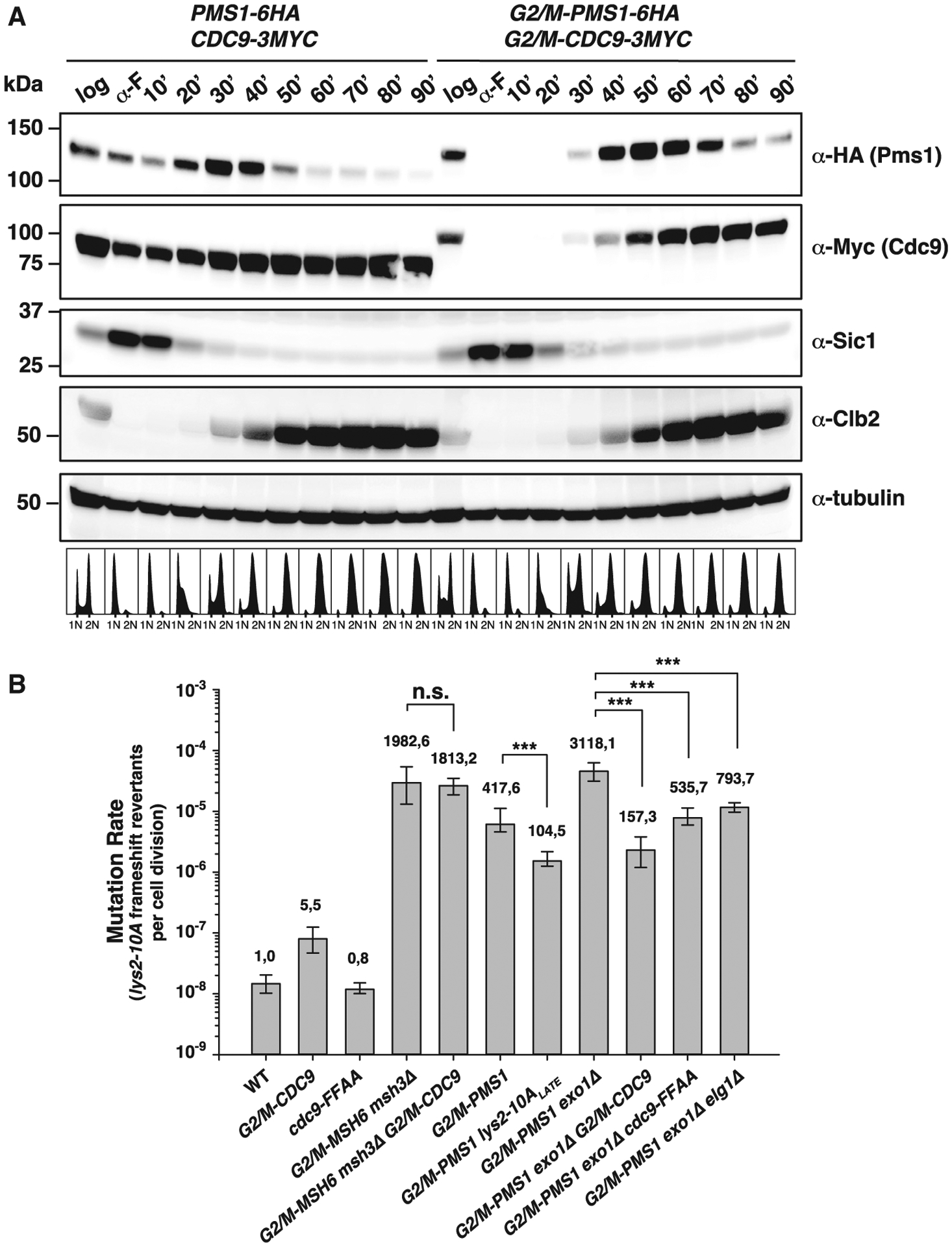
(A) Top: Pms1 and Cdc9 protein expression levels throughout the cell cycle under endogenous regulation (PMS1–6HA and CDC9–9MYC) or under control of the G2/M-tag (G2/M-PMS1–6HA and G2/M-CDC9–9MYC). Bottom: logarithmically growing cells (log), α-factor-arrested cells (α-F), or cells arrested and released from α-F arrest for the indicated time were analyzed by western blotting and DNA content analysis. Sic1, Clb2, and tubulin serve as G1-, G2/M-phase, and loading controls, respectively. See also Figures S2 and S3.
(B) Mutation rate analysis using the lys2-10A frameshift reversion assay in the indicated strains. Bars correspond to the median rate, with error bars representing the 95% confidence interval. Numbers on top of the bars indicate fold increase in mutation rate relative to the WT. p values were calculated with the Mann-Whitney rank-sum test using SigmaPlot. ***p ≤ 0.001; n.s., not significant. See also Table S5 for additional mutation rate analysis using two alternative mutational reporters.
The G2/M-CDC9 allele did not suppress the MMR defect of the G2/M-MSH6 msh3Δ mutant strain measured at three mid-S phase-replicated loci (Table S5), which is consistent with the idea that mispair recognition must occur during DNA replication.10 We therefore constructed the G2/M-PMS1 allele to restrict MMR steps after mispair recognition to the G2/M phase of the cell cycle (Figure 4A). The G2/M-PMS1 allele caused a mutator phenotype that was exacerbated by an exo1Δ mutation at three mid-S phase-replicated loci (replication time, 32–39 min after release from α-factor arrest)38 (Figure 4B; Table S5). In contrast, the G2/M-PMS1 allele was 4 times more proficient at suppressing mutations when the lys2-10A reporter was moved to a late-replicated region (lys2-10ALATE),10 which is replicated 49 min after release from α-factor arrest38 (Figure 4B) and coincides with the time when G2/M-Pms1 reaches the peak level in expression (Figure 4A). If nicks were the daughter-strand discrimination signal, the G2/M-PMS1 mutator phenotype could be due to the temporal uncoupling of this signal and the repair machinery. Thus, we reasoned that mutations that cause the signal to persist should suppress the G2/M-PMS1 mutator phenotype. Remarkably, when the G2/M-PMS1 or the G2/M-PMS1 exo1Δ alleles were combined with the G2/M-CDC9 or the cdc9-FFAA allele, the mutator phenotype was largely suppressed (Figure 4B; Table S5), with G2/M-CDC9 resulting in stronger suppression. This observation is in agreement with the more pronounced and slightly shorter Okazaki fragments (indicative of a stronger Cdc9 ligase defect) in the G2/M-CDC9 strain arrested in S phase, compared to the cdc9-FFAA strain arrested in either S or G2/M phase (Figure S2B). It should be noted that the peak of G2/M-Cdc9 protein expression occurs 20 min after the peak of G2/M-Pms1 protein expression, which could explain why G2/M-Cdc9 protein expression does not inhibit MMR in the G2/M-PMS1 strain. Furthermore, deletion of ELG1, which increases the amount of chromatin-associated PCNA (Figure S1D),35 resulted in a modest rescue of the mutator phenotype of G2/M-PMS1 exo1Δ (Figure 4B; Table S5). The suppression of the G2/M-PMS1 exo1Δ mutator phenotype by the previous mutations is unlikely to be related to changes in cell-cycle progression or activation of the DNA damage response, as log-phase cultures of the G2/M-PMS1 exo1Δ elg1Δ and the G2/M-PMS1 G2/M-CDC9 exo1Δ triple mutants had relatively normal DNA content profiles (Figure S3C). These results, together with the observation that the cdc9-FFAA allele (which affects neither cell-cycle progression nor DNA damage response) also suppressed the G2/M-PMS1 exo1Δ strain mutator phenotype, indicate that the suppressive effects of the G2/M-CDC9 and cdc9-FFAA alleles are a consequence of the inefficient ligation of DNA replication-associated nicks. Thus, by delaying the ligation of DNA replication-associated nicks, it is possible to retain MMR proficiency up to G2/M phase for loci replicated in mid-S phase.
In conclusion, our findings indicate that MMR is sensitive to the timing of DNA ligation and the lifetime of replication-associated nicks in DNA. Increased ligase activity causes a mutator phenotype, whereas reduced or delayed ligase activity prolongs the temporal window in which loci can undergo MMR. Importantly, these effects on MMR did not correlate with the bulk levels of chromatin-bound PCNA and were unaffected by loss of ribonucleotide excision. Thus, these results suggest that replication-associated nicks are the in vivo strand-discrimination signals in eukaryotes, consistent with the requirement of pre-existing nicks in reconstituted MMR reactions in vitro.5–8
If nicks act to direct excision during MMR, then they should be present on both the leading and lagging strands. The presence of these nicks has been recently demonstrated by the high-throughput mapping of DNA strand discontinuities on the S. cerevisiae genome.11 Lagging-strand nicks are transient and accumulate only when Okazaki fragment ligation is inhibited, whereas leading-strand nicks are present at higher steady-state levels than lagging-strand nicks in WT cells. The data currently available are not sufficient to determine the absolute numbers of leading- and lagging-strand nicks, as normalization controls required when comparing different DNA libraries were not included11 (H. Ulrich, personal communication). The minimum density of nicks that is required to promote MMR in vivo is also not currently known. However, the results of the studies presented here are consistent with the hypothesis that the nicks present on the leading and lagging strands are sufficient to direct strand specificity during MMR. In addition, the length of mitotic gene conversion tracks in yeast, which are mediated by the formation of heteroduplex DNA and subsequent MMR and range from 7.3 to 32.3 kb in length,39,40 supports the idea that MMR can catalyze mispair excision tracts that approach the average distance between replication origins (20–40 kb) in S. cerevisiae41 and, hence, even the low density of leading-strand nicks significantly contribute to MMR. In addition, interactions between MMR and the leading- and lagging-strand replication machinery may also contribute to direct the strand specificity of MMR.10,28
The absolute requirement of a pre-existing nick in MMR reactions reconstituted in vitro may explain the fact that Mlh1-Pms1 endonuclease activity is dispensable for many MMR reactions reconstituted in vitro, although it is required for MMR in vivo.30,42 In the context of DNA replication-associated nicks acting as MMR strand-discrimination signals, it can be anticipated that DNA ligase I activity would suppress MMR. This idea is supported by our own results and by the observation that reconstituted MMR reactions using E. coli proteins were inhibited by the presence of DNA ligase I, unless E. coli exonuclease I was added to the reaction,43 which, in E. coli, appears to excise only a small number of nucleotides (<50 nt) from the nick introduced by the MutH endonuclease.44 Given the requirement for pre-existing nicks to support MMR, it is unclear what role the eukaryotic Mlh1-Pms1/PMS2 endonuclease plays in MMR in vivo. Based on our findings, a possible role for the eukaryotic Mlh1-Pms1/PMS2 endonuclease (with some marginal contribution of Exo1) is to preserve DNA strand discontinuities on the newly replicated strands to direct MMR. The Mlh1-Pms1/PMS2 endonuclease might accomplish this function by introducing additional daughter-strand-specific nicks in the proximity of the mispair site to convert ligatable nicks into non-ligatable gaps to prevent the premature ligation of the strand discontinuity before MMR is accomplished.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hans Hombauer (h.hombauer@zmbh.uni-heidelberg.de).
Materials availability
Plasmids and yeast strains generated in this study can be obtained through the Lead Contact.
Data and code availability
The published article includes all datasets generated or analyzed during this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strains
All S. cerevisiae strains used in this study are listed in the Key resources table and were derivatives of the S288C strains RDKY368626 (MATα ura3-52 leu2Δ1 trp1Δ63 his3Δ200 hom3-10 lys2-10A) or RDKY596428 (MATa version of RDKY3686). Strains were grown at 30°C in yeast extract-peptone-dextrose media (YPD) or dextrose synthetic dropout media. Arginine dropout media containing 60 mg/L of canavanine was used for selecting canavanine resistant cells (CanR). Gene deletions, gene tagging, and promoter replacements were done using standard PCR-based recombination methods,50 followed by confirmation by PCR and sequencing. Strains containing two or more genetic modifications were usually obtained by mating and sporulation. All experiments were performed with at least two independent biological isolates. With exception of the pol30-C81R and pol30-K217E alleles described below, point mutations were introduced at their chromosomal loci with URA3-integrative vectors using pop-in/pop-out strategies, and were confirmed by sequencing. Strains containing the lys2-10ALATE frameshift reporter (replicated during late S phase) were previously described.10
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-MYC (clone 4A6) | Millipore | Cat# 05-724; RRID: AB_11211891 |
| Rat monoclonal anti-HA (clone 3F10) | Roche | Cat# 3F10; RRID: AB_2314622 |
| Rabbit polyclonal anti-Clb2 | Santa Cruz | Cat# sc-9071; RRID: AB_667962 |
| Mouse monoclonal anti-PCNA | Abcam | Cat# ab70472; RRID: AB_2160644 |
| Rabbit polyclonal anti-Rnr3 | Agrisera | Cat# AS09 574; RRID: AB_1966947 |
| Rat monoclonal anti-Tubulin & Rnr4 (clone YL1/2) | Millipore | Cat# MAB1864; RRID: AB_2210391 |
| Mouse monoclonal anti-Histone H3 | Abcam | Cat# ab46765; RRID: AB_880439 |
| Guinea pig polyclonal anti-Sic1 | 45 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| α-factor | GenScript | RP01002 |
| Nocodazole | TargetMol | T2802 |
| α-32P-dCTP | PerkinElmer | NEG013H |
| Zymolyase-100T | US Biological | Z1005 |
| Restriction endonucleases (Bgl II, BamH I, Xho I, Not I, Sal I, EcoR V, Kpn I) | New England Biolabs | N/A |
| Sytox Green | Thermo Fisher | S7020 |
| Hybond N+ membrane | Amersham GE Healthcare | RPN303B |
| 3-Indoleacetic acid (Auxin) | Sigma Aldrich | I2886 |
| Critical commercial assays | ||
| Megaprime DNA labeling system | Amersham GE Healthcare | RPN1606 |
| PureGene Yeast/Bact. kit | QIAGEN | 158567 |
| Experimental models: organisms/strains | ||
| MATα ura3-52 leu2Δ1 trp1Δ63 his3Δ200 hom3-10 lys2-10A | 26 | RDKY3686 |
| MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 hom3-10 lys2-10A | 28 | RDKY5964 |
| RDKY5964 msh2::HIS3 | 46 | HHY6505 |
| RDKY5964 elg1::HIS3 | this study | HHY8074 |
| RDKY5964 elg1::HIS3 exo1::hphNT1 | this study | HHY6828 |
| RDKY5964 exo1::hphNT1 | 46 | HHY1794 |
| RDKY5964 exo1::hphNT1 LEU2.pol30-K217E | this study | HHY6035 |
| RDKY5964 exo1-F447A-F448A-Δ571-702.kanMX4 | this study | HHY7017 |
| RDKY5964 exo1-F447A-F448A-Δ571-702.kanMX4 natNT2.pGPD-CDC9 | this study | HHY7010 |
| RDKY5964 natNT2.pGPD-CDC9 | this study | HHY6770 |
| RDKY5964 natNT2.pGPD-CDC9-9MYC.HIS3 | this study | HHY8138 |
| RDKY5964 natNT2.pGPD-CDC9 exo1::hphNT1 | this study | HHY6772 |
| RDKY5964 natNT2.pGPD-CDC9 msh2::HIS3 | this study | HHY6899 |
| RDKY5964 kanMX4.pGPD-CDC9 rad27::hphNT1 | this study | HHY7094 |
| RDKY5964 natNT2.pGPD-ELG1 | this study | HHY8075 |
| RDKY5964 natNT2.pGPD-ELG1 exo1::hphNT1 | this study | HHY6837 |
| RDKY5964 pol2-M644G.natNT2 | 46 | HHY1993 |
| RDKY5964 kanMX4.pGPD-CDC9 natNT2.pGPD-ELG1 | this study | HHY6834 |
| RDKY5964 kanMX4.pGPD-CDC9 exo1::hphNT1 natNT2.pGPD-ELG1 | this study | HHY6831 |
| RDKY5964 kanMX4.pGPD-CDC9 elg1::HIS3 | this study | HHY6913 |
| RDKY5964 kanMX4.pGPD-CDC9 exo1::hphNT1 elg1::HIS3 | this study | HHY6825 |
| RDKY5964 LEU2.pol30-C81R | this study | HHY3234 |
| RDKY5964 LEU2.pol30-K217E | this study | HHY7555 |
| RDKY5964 lys2::kl-TRP1 | this study | HHY7586 |
| RDKY5964 natNT2.pGPD-CDC9 LEU2.pol30-K217E | this study | HHY7362 |
| RDKY5964 pms1-A99V | this study | HHY5554 |
| RDKY5964 rad27::hphNT1 | this study | HHY5082 |
| RDKY5964 natNT2.pGPD-CDC9 pms1-A99V | this study | HHY7375 |
| RDKY5964 natNT2.pClb2-Clb2(1–181, L26A)-MSH6-9MYC.hphNT1 msh3::kanMX4 pGAL-his3::intron::cβ2/cβ2::URA3 | 10 | RDKY7676 |
| RDKY5964 natNT2.pClb2-Clb2(1–181, L26A)-3MYC-CDC9 | this study | HHY6701 |
| RDKY5964 natNT2.pClb2-Clb2(1–181, L26A)-MSH6 msh3::HIS3 natNT2.pClb2-Clb2(1–181, L26A)-3MYC-CDC9 | this study | HHY4983 |
| RDKY5964 hphNT1.pClb2-Clb2(1–181, L26A)-3MYC-PMS1 | this study | HHY7192 |
| RDKY5964 kanMX4.pClb2-Clb2(1–181, L26A)-3MYC-PMS1 exo1::hphNT1 | this study | HHY7202 |
| RDKY5964 hphNT1.pClb2-Clb2(1–181, L26A)-3MYC-PMS1 lys2::kl-TRP1 lys2-10ALATE.kanMX4 | this study | HHY7894 |
| RDKY5964 CDC9-3MYC.HIS3 PMS1-6HA.hphNT1 | this study | HHY7972 |
| RDKY5964 natNT2.pClb2-Clb2(1–181, L26A)-3MYC-CDC9 kanMX4.pClb2-Clb2(1–181, L26A)-3mYc-PMS1 | this study | HHY7708 |
| RDKY5964 natNT2.pClb2-Clb2(1–181, L26A)-3MYC-CDC9 mlh1::kl-TRP1 | this study | HHY7768 |
| RDKY5964 natNT2.pClb2-Clb2(1–181, L26A)-3MYC-CDC9 kanMX4.pClb2-Clb2(1–181, L26A)-3mYc-PMS1-6HA.HIS3 | this study | HHY7979 |
| RDKY5964 kanMX4.pClb2-Clb2(1–181, L26A)-3MYC-PMS1 exo1::hphNT1 natNT2.pClb2-Clb2(1–181, L26A)-3MYC-CDC9 | this study | HHY7660 |
| RDKY5964 cdc9-F44A-F45A | this study | HHY7774 |
| RDKY5964 kanMX4.pClb2-Clb2(1–181, L26A)-3MYC-PMS1 cdc9-F44A-F45A | this study | HHY7839 |
| RDKY5964 kanMX4.pClb2-Clb2(1–181, L26A)-3MYC-PMS1 natNT2.pGPD-CDC9 | this study | HHY7715 |
| RDKY5964 kanMX4.pClb2-Clb2(1–181, L26A)-3MYC-PMS1 exo1::hphNT1 cdc9-F44A-F45A | this study | HHY7835 |
| RDKY5964 kanMX4.pClb2-Clb2(1–181, L26A)-3MYC-PMS1 exo1::hphNT1 elg1::HIS3 | this study | HHY7672 |
| RDKY5964 leu2::ADH-AFB2.hphNT1 CDC9-9MYC-AID*.natNT2 | this study | HHY8076 |
| RDKY5964 PMS1-4GFP.kanMX6 | 28 | RDKY7588 |
| RDKY5964 exo1::hphNT1 PMS1-4GFP.kanMX6 | 28 | RDKY7544 |
| RDKY5964 elg1::HIS3 PMS1-4GFP.kanMX6 | this study | HHY8077 |
| RDKY5964 elg1::HIS3 exo1::hphNT1 PMS1-4GFP.kanMX6 | this study | HHY6982 |
| RDKY5964 natNT2.pGPD-CDC9 PMS1-4GFP.kanMX6 | this study | HHY8078 |
| RDKY5964 exo1::hphNT1 natNT2.pGPD-CDC9 PMS1-4GFP.kanMX6 | this study | HHY6895 |
| RDKY5964 elg1::HIS3 natNT2.pGPD-CDC9 PMS1-4GFP.kanMX6 | this study | HHY6987 |
| RDKY5964 elg1::HIS3 exo1::hphNT1 natNT2.pGPD-CDC9 PMS1-4GFP.kanMX6 | this study | HHY6978 |
| RDKY5964 natNT2.pGPD-ELG1 PMS1-4GFP.kanMX6 | this study | HHY7499 |
| RDKY5964 exo1::hphNT1 natNT2.pGPD-ELG1 PMS1-4GFP.kanMX6 | this study | HHY7505 |
| RDKY5964 kanMX4.pGPD-CDC9 natNT2.pGPD-ELG1 PMS1-4GFP.kanMX6 | this study | HHY7142 |
| RDKY5964 exo1::hphNT1 kanMX4.pGPD-CDC9 natNT2.pGPD-ELG1 PMS1-4GFP.kanMX6 | this study | HHY7132 |
| Recombinant DNA | ||
| URA3 integrative plasmid (pop-in/pop-out) AmpR | 47 | pRS306 |
| ori 2μ URA3 AmpR | 48 | pRS426 |
| pCH1572-pol30-C81R-LEU2 (one-step integrative plasmid) AmpR | 49 | pRDK926 |
| kanMX4 AmpR | 50 | pFA6a-kanMX4 |
| hphNT1 AmpR | 50 | pFA6a-hphNT1 |
| natNT2 AmpR | 50 | pFA6a-natNT2 |
| pGPD kanMX4 AmpR | 50 | pYM-N14 |
| pGPD natNT2 AmpR | 50 | pYM-N15 |
| C-terminal 3xMYC HIS3MX6 AmpR | 50 | pYM5 |
| C-terminal 6xHA hphNT1 AmpR | 50 | pYM16 |
| pCH1572-pol30-K217E-LEU2 (one-step integrative plasmid) AmpR | This study | pHHB252 |
| pRS426-CDC9 2μ URA3 AmpR | This study | pHHB1152 |
| pRS426-cdc9-F44A-F45A 2μ URA3 AmpR | This study | pHHB1163 |
| pRS426-cdc9-K419A 2μ URA3 AmpR | This study | pHHB1164 |
| pRS426-cdc9-K598A 2μ URA3AmpR | This study | pHHB1165 |
| pRS306-cdc9-F44A-F45A URA3 integrative AmpR | This study | pHHB1274 |
| pRS306-exo1-F447A-F448A URA3 integrative AmpR | This study | pHHB1187 |
| pSM409-Nat-AID*-9MYC AmpR | 24 | pNat-AID*-9MYC |
| pFA6a-hphNT1-pADH-AFB2 AmpR | This study | pHHB699 |
| pCLB2-3xMYC-(GA)5-CLB2(1−181+L26A).kanMX4 AmpR | This study | pHHB1220 |
| pCLB2-3xMYC-(GA)5-CLB2(1−181+L26A).hphNT1 AmpR | This study | pHHB1221 |
| pCLB2-3xMYC-(GA)5-CLB2(1−181+L26A).natNT2 AmpR | This study | pHHB1222 |
| pRS426-CDC9-HA 2μ URA3 AmpR | This study | pHHB1194 |
| pRS426-cdc9-F44A-F45A-HA 2μ URA3 AmpR | This study | pHHB1195 |
| pRS426-cdc9-K419A-HA 2μ URA3 AmpR | This study | pHHB1196 |
| pRS426-cdc9-K598A-HA 2μ URA3 AmpR | This study | pHHB1197 |
| Software and algorithms | ||
| SoftWoRx 6.1.1 Release 5 | Applied Precision | N/A |
| SigmaPlot version 10 | Systat Software | N/A |
| ImageJ, FIJI | ImageJ | https://fiji.sc |
| Lasergene 15.1.0 | DNASTAR | N/A |
The pol30-C81R and pol30-K217E alleles were introduced at the POL30 locus by a one-step replacement after transformation with Sac I-digested LEU2 plasmids pRDK92649 or pHHB252, respectively.
The cdc9-FFAA mutant allele that contains two consecutive phenylalanine-alanine substitutions (F44A and F45A) was introduced at the CDC9 chromosomal locus using pop-in/pop-out strategy with the integrative plasmid pHHB1274 linearized with Bgl II.
Strains containing the pms1-A99V mutation were obtained after mating with RDKY417726 (MATa ura3-52 leu2Δ1 trp1Δ63 hom3-10 lys2-10A exo1::URA3 pms1-A99V) and sporulation.
The strain expressing the exo1-F447A-F448A-Δ571–702 allele (referred in the main text as exo1-FFAA-Δ571–702) was generated in two steps: (1) the exo1-F447A-F448A point mutations were introduced by pop-in/pop-out strategy with the pHHB1187 integrative vector linearized with Bgl II; and (2) this strain was transformed with a PCR cassette carrying a kanMX4 module that introduces a STOP codon in Exo1, right after codon 570.
The strain HHY8076, which expresses an auxin-inducible degron fused to the chromosomal copy of CDC9 (CDC9–9MYC-AID*-natNT2) used for cdc9 plasmid complementation experiments (Figure S1A), was constructed by PCR-mediated recombination with the plasmid pNat-AID*−9MYC.24 The strain HHY8076 also contains a pADH-AFB2.hphNT1 cassette (integrated at the LEU2 locus) that expresses the AFB2 F-box gene required for auxin-induced protein degradation. Integration of this cassette was performed by PCR-mediated recombination with a PCR product generated using the plasmid pHHB699 as template and S1 and S2 primers.50
METHOD DETAILS
Plasmids
Plasmids used in this study are listed in the Key resources table. The pHHB1220–1222 plasmids were used as template DNAs in PCR reactions to amplify the G2/M-tag (pClb2-Clb2(1–181+L26A)) linked to a selectable marker cassette (kanMX4, hphNT1 and natNT2, respectively). The G2/M-tag present in these three plasmids is identical to that contained in pRDK159810 plasmid, with the exception that these constructs contain a triple MYC-tag and a (GA)5 linker after the Clb2 N terminus (1–181 aa + L26A). These new plasmids are now compatible with S1 and S4 primer design.50
High copy number plasmids (URA3, 2μ, AmpR) harboring WT-CDC9, cdc9-FFAA, cdc9-K419A and cdc9-K598A were constructed using the pRS42648 backbone, and correspond to pHHB1152, pHHB1163, pHHB1164 and pHHB1165, respectively. These CDC9/cdc9 overexpression plasmids contain the full-length CDC9 gene (including 1 kb of the promoter sequence and 300 bp of the terminator region) cloned into the BamH I and Xho I sites present in the pRS426 polylinker. Cdc9 mutant alleles were generated by site-directed mutagenesis. In addition, we constructed another set of plasmids that were almost identical to pHHB1152, pHHB1163, pHHB1164 and pHHB1165 except that they harbor the WT-CDC9, cdc9-FFAA, cdc9-K419A and cdc9-K598A alleles fused to an HA-tag at the C terminus, and are named pHHB1194, pHHB1195, pHHB1196 and pHHB1197, respectively.
The pHHB252 plasmid used to integrate the pol30-K217E allele was constructed as follows. First, the plasmid pRDK92549 containing the pol30-C22Y mutant allele was reverted back to WT-POL30 by site-directed mutagenesis resulting in pHHB247. Next, the pol30-K217E mutation was introduced by site-directed mutagenesis resulting in pHHB252.
The pHHB1274 plasmid used to integrate the cdc9-FFAA mutation was constructed as follows. The 3.6 kb BamH I-Xho I fragment from pHHB1163, which contains the cdc9-F44A-F45A allele, was subcloned into the BamH I-Xho I sites in pRS306,47 resulting in pHHB1274.
The pHHB1187 plasmid used to integrate the exo1-F447A-F448A mutant allele was constructed as follows. First, the WT-EXO1 gene (including 500 bp of the promoter and 253 bp of the terminator) was amplified by PCR from a WT yeast strain (RDKY5964)28 and cloned into the Hind III and Xho I sites in pRS426, resulting in pHHB245. Next, the exo1-F447A-F448A mutations were introduced in pHHB245 by site-directed mutagenesis resulting in pHHB1186. Last, the 2.9 kb Not I-Xho I fragment from pHHB1186 was subcloned into the Not I-Sal I sites in pRS306, resulting in pHHB1187.
The pHHB699 plasmid harboring the pADH-AFB2.hphNT1 cassette (used in auxin-induced protein degradation experiments) was constructed by subcloning the 2.5 kb Sal I fragment from pRS303-ADH-AFB2 into the Sal I site in pFA6a-hphNT1,50 resulting in pHHB699. Further analysis of this plasmid revealed that the AFB2 gene is in the same orientation as the hphNT1 resistance gene.
Whole cell lysates and western blotting
S. cerevisiae whole cell lysates and western blotting analysis were performed as described.28 Antibodies including catalog number and manufacturer used in this study were: MYC (4A6, Millipore), HA (3F10, Roche), Clb2 (sc-9071, Santa Cruz), PCNA (ab70472, Abcam), Rnr3 (AS09574, Agrisera), tubulin-Rnr4 (YL1/2, Sigma), Histone H3 (ab46765, Abcam) and Sic1 (previously described45). In order to detect PCNA more efficiently with the anti-PCNA antibody (ab70472, Abcam), the membrane was incubated in a mild stripping buffer (0.2 M glycine pH 2.2, 0.1% SDS, 1% Tween 20) prior to blocking.
Cell synchronization and release
S. cerevisiae cells were synchronized by G1-arrest using a 3-hour incubation in the presence of 10 μg/ml α-factor (GenScript). For release from G1-arrest, cells were washed with water and resuspended in YPD medium containing 15 μg/ml nocodazole (Sigma-Aldrich) to prevent cells from entering a second G1 and S phase.
DNA content analysis
Analysis of DNA content in yeast cells was done as previously described.10
Mutation rate analysis
The lys2-10A and hom3-10 frameshift reversion assays, and the CAN1 inactivation assay were used to quantify mutation rates using fluctuation analysis as previously described.26 Statistical significance was evaluated by calculating 95% confidence intervals.
Live cell imaging Pms1 foci
Exponentially growing cells were washed, resuspended and placed on agar pads, covered with a coverslip and sealed with valap (1:1:1 mixture of Vaseline, lanolin and paraffin by weight). Cells were imaged at 30°C using a DeltaVision RT (Applied Precision) based on an inverted microscope (IX70, Olympus) with a camera (CoolSNAP HQ2, Photometrics) and a Plan-Apo 100x (1.4 NA) oil immersion objective lens (Olympus). 20 Z stacks spaced 0.3 μm were deconvolved using SoftWoRx software and projected using the maximum intensity projection.
Detection of Okazaki fragments at the LYS2 locus
Genomic DNA from logarithmic cells, synchronized in G1 phase or G1-synchronized and released for the indicated times, was isolated using the PureGene kit (QIAGEN). Approximately, 6 × 108 cells were used for each time point. DNA was resuspended in 30 μL TE buffer (10 mM Tris pH 7.5, 0.1 mM EDTA) and kept at 4°C or used immediately for Southern blot analysis. Quality, purity and concentration of the DNA were evaluated by gel electrophoresis. Then, equal amounts of purified genomic DNA were treated with EcoR V overnight at 37°C, mixed with loading dye containing 1 M urea, heated at 86°C for 5 min, then loaded in 1.2% agarose gels and resolved under urea/heat denaturing conditions; both the gel and running buffer contained 1 M urea and electrophoresis was for 5 hours at 80 V. The gels were blotted to Hybond N+ membrane (Amersham GE Healthcare Life Science) by capillary blotting. Hybridization with 20 ng of 32P-radiolabelled probe was done in 2xSSC, 7% SDS and shared salmon sperm DNA (0.2 mg/ml) at 58°C. To prepare the radiolabeled probe, the pHHB762 plasmid that harbors the LYS2 genomic sequence (including 600 bp of the promoter sequence and 160 bp of the 3′ UTR) was digested with Kpn I and Xho I and the 1.2 kb fragment was purified and labeled with α−32P-dCTP using the Megaprime DNA labeling system (Amersham GE Healthcare Life Science). The probe was purified from unincorporated nucleotides using Illustra microspin G-50 columns (Amersham GE Healthcare Life Science). Before hybridization the probe was denatured 5 min at 95°C and placed on ice before adding to the hybridization buffer. Membranes were exposed to a phospho-screen, and the radioactive signal was detected with a Phosphor Imaging module (Sapphire Biomolecular Imager).
Whole-cell extracts and chromatin fractions
Whole-cell extracts (WCEs) and chromatin enriched-fractions were prepared essentially as described previously.51 Approximately 3 × 108 logarithmically growing cells were harvested and resuspended in 1 mL of prespheroplasting buffer (100 mM PIPES/KOH, pH 9.4, 10 mM DTT, 0.1% sodium azide) then incubated for 10 min on ice. Cells were then incubated in 1 mL of spheroplasting buffer (50 mM KH2PO4/K2HPO4, pH 7.4, 0.8 M sorbitol, 10 mM DTT, 0.1% sodium azide) containing 200 μg/ml Zymolyase-100T at 30°C for 10 min with occasional mixing. Spheroplasts were washed twice with 1 mL of ice-cold wash buffer (20 mM KH2PO4/K2HPO4, pH 6.5, 0.8 M sorbitol, 1 mM MgCl2, 1 mM DTT, 20 mM β-glycerophosphate, 1 mM PMSF, protease inhibitor tablets (EDTA free, Roche)). Spheroplasts were resuspended in 3 vol. of EB buffer (50 mM HEPES/KOH, pH 7.5, 100 mM KCl, 2.5 mM MgCl2, 0.1 mM ZnSO4, 2 mM NaF, 0.5 mM spermidine, 1 mM DTT, 20 mM β-glycerophosphate, 1 mM PMSF, protease inhibitor tablets (EDTA free)), lysed by addition of Triton X-100 to 0.25%, and incubated on ice for 10 min. WCEs were prepared by mixing 20 μL of the lysate with 20 μL of 2 × Tris-Glycine SDS Sample Buffer and incubated for 3 min at 85°C. The remaining lysate was laid over on 0.4 mL of EBX-S buffer (EB buffer, 30% sucrose, 0.25% Triton X-100), and spun at 12,000 rpm for 10 min at 4°C. The chromatin pellet was washed in 0.5 mL of EBX buffer (EB buffer, 0.25% Triton X-100) and spun at 10,000 rpm for 2 min at 4°C. The chromatin pellet was resuspended in 50 μL of 2 × Tris-Glycine SDS Sample Buffer and incubated for 3 min at 85°C. To detect PCNA, 10 μL of chromatin fractions were analyzed in SDS-PAGE, followed by western blot with anti-PCNA antibody (ab70472, Abcam).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis for mutation rates as well as analysis of Pms1 foci were performed in SigmaPlot. P-values were calculated using the Mann-Whitney rank sum test and were indicated on the graphs and represent statistical significance of the difference between the two data groups. Mutation rates analysis in S. cerevisiae were determined using two independent biological isolates and a total of at least 14 independent cultures. The mutation rate data shown in the graphs correspond to median rates for the indicated mutational reporter, the error bars correspond to the 95% confidence intervals. Quantification of Pms1 foci abundance was performed with at least 1000 cells per genotype, using three independent biological isolates. Data presented in Figure 1B shows the average of the percentage of cells containing foci, error bars indicate SEM. In Figure 3C data is presented as box-plots with whiskers and dots represent outliers. Black and red lines inside the box represent the median and average of cells with foci, respectively.
Supplementary Material
Highlights.
Cdc9 overexpression causes increased mutation rates and accumulation of Pms1 foci
DNA replication-associated nicks are required for MMR strand discrimination
Cdc9 activity dictates a temporal window for MMR
ACKNOWLEDGMENTS
We would like to thank to Dr. Michael Knop for sharing plasmids and infrastructure, for critical comments to the manuscript, and generous support. Thanks to Dr. Hannah Zhao and Dr. Tobias Schmidt for constructing some plasmids/strains used in this study, Dr. Helle Ulrich for valuable discussions and for sharing plasmids (auxin-degron system), and Sandra Ruf and Dr. Nina Papa-vasiliou for technical support/infrastructure with radioisotopes. R.D.K. and C.D.P. were supported by the NIH grant R01 GM50006 and the Ludwig Institute for Cancer Research. G.X.R., A.K., and H.H. were supported by the Deutsches Krebsforschungszentrum and the Deutsche Forschungsgemein-schaft grant HO-5501-1.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cub.2020.12.018.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Kolodner RD, and Marsischky GT (1999). Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev 9, 89–96. [DOI] [PubMed] [Google Scholar]
- 2.Jiricny J (2013). Postreplicative mismatch repair. Cold Spring Harb. Perspect. Biol 5, a012633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fishel R (2015). Mismatch repair. J. Biol. Chem 290, 26395–26403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kunkel TA, and Erie DA (2015). Eukaryotic mismatch repair in relation to DNA replication. Annu. Rev. Genet 49, 291–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dzantiev L, Constantin N, Genschel J, Iyer RR, Burgers PM, and Modrich P (2004). A defined human system that supports bidirectional mismatch-provoked excision. Mol. Cell 15, 31–41. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Yuan F, Presnell SR, Tian K, Gao Y, Tomkinson AE, Gu L, and Li GM (2005). Reconstitution of 5¢-directed human mismatch repair in a purified system. Cell 122, 693–705. [DOI] [PubMed] [Google Scholar]
- 7.Kadyrov FA, Dzantiev L, Constantin N, and Modrich P (2006). Endonucleolytic function of MutLalpha in human mismatch repair. Cell 126, 297–308. [DOI] [PubMed] [Google Scholar]
- 8.Smith CE, Bowen N, Graham WJ 5th, Goellner EM, Srivatsan A, and Kolodner RD (2015). Activation of Saccharomyces cerevisiae Mlh1-Pms1 endonuclease in a reconstituted mismatch repair system. J. Biol. Chem 290, 21580–21590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pluciennik A, Dzantiev L, Iyer RR, Constantin N, Kadyrov FA, and Modrich P (2010). PCNA function in the activation and strand direction of MutLα endonuclease in mismatch repair. Proc. Natl. Acad. Sci. USA 107, 16066–16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hombauer H, Srivatsan A, Putnam CD, and Kolodner RD (2011). Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science 334, 1713–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sriramachandran AM, Petrosino G, Méndez-Lago M, Schäfer AJ, Batista-Nascimento LS, Zilio N, and Ulrich HD (2020). Genome-wide nucleotide-resolution mapping of DNA replication patterns, single-strand breaks, and lesions by GLOE-seq. Mol. Cell 78, 975–985.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghodgaonkar MM, Lazzaro F, Olivera-Pimentel M, Artola-Borán M, Cejka P, Reijns MA, Jackson AP, Plevani P, Muzi-Falconi M, and Jiricny J (2013). Ribonucleotides misincorporated into DNA act as strand-discrimination signals in eukaryotic mismatch repair. Mol. Cell 50, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lujan SA, Williams JS, Clausen AR, Clark AB, and Kunkel TA (2013). Ribonucleotides are signals for mismatch repair of leading-strand replication errors. Mol. Cell 50, 437–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim N, Huang SN, Williams JS, Li YC, Clark AB, Cho JE, Kunkel TA, Pommier Y, and Jinks-Robertson S (2011). Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science 332, 1561–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Putnam CD (2016). Evolution of the methyl directed mismatch repair system in Escherichia coli. DNA Repair (Amst.) 38, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bowman GD, O’Donnell M, and Kuriyan J (2004). Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature 429, 724–730. [DOI] [PubMed] [Google Scholar]
- 17.Flores-Rozas H, Clark D, and Kolodner RD (2000). Proliferating cell nuclear antigen and Msh2p-Msh6p interact to form an active mispair recognition complex. Nat. Genet 26, 375–378. [DOI] [PubMed] [Google Scholar]
- 18.Kadyrov FA, Holmes SF, Arana ME, Lukianova OA, O’Donnell M, Kunkel TA, and Modrich P (2007). Saccharomyces cerevisiae MutLalpha is a mismatch repair endonuclease. J. Biol. Chem 282, 37181–37190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goellner EM, Smith CE, Campbell CS, Hombauer H, Desai A, Putnam CD, and Kolodner RD (2014). PCNA and Msh2-Msh6 activate an Mlh1-Pms1 endonuclease pathway required for Exo1-independent mismatch repair. Mol. Cell 55, 291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawasoe Y, Tsurimoto T, Nakagawa T, Masukata H, and Takahashi TS (2016). MutSα maintains the mismatch repair capability by inhibiting PCNA unloading. eLife 5, e15155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomkinson AE, Totty NF, Ginsburg M, and Lindahl T (1991). Location of the active site for enzyme-adenylate formation in DNA ligases. Proc. Natl. Acad. Sci. USA 88, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Subramanian J, Vijayakumar S, Tomkinson AE, and Arnheim N (2005). Genetic instability induced by overexpression of DNA ligase I in budding yeast. Genetics 171, 427–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vijayakumar S, Chapados BR, Schmidt KH, Kolodner RD, Tainer JA, and Tomkinson AE (2007). The C-terminal domain of yeast PCNA is required for physical and functional interactions with Cdc9 DNA ligase. Nucleic Acids Res 35, 1624–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morawska M, and Ulrich HD (2013). An expanded tool kit for the auxin-inducible degron system in budding yeast. Yeast 30, 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kubota T, Katou Y, Nakato R, Shirahige K, and Donaldson AD (2015). Replication-coupled PCNA unloading by the Elg1 complex occurs genome-wide and requires Okazaki fragment ligation. Cell Rep 12, 774–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amin NS, Nguyen MN, Oh S, and Kolodner RD (2001). exo1-dependent mutator mutations: model system for studying functional interactions in mismatch repair. Mol. Cell. Biol 21, 5142–5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goellner EM, Putnam CD, and Kolodner RD (2015). Exonuclease 1-dependent and independent mismatch repair. DNA Repair (Amst.) 32, 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hombauer H, Campbell CS, Smith CE, Desai A, and Kolodner RD (2011). Visualization of eukaryotic DNA mismatch repair reveals distinct recognition and repair intermediates. Cell 147, 1040–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmidt TT, and Hombauer H (2016). Visualization of mismatch repair complexes using fluorescence microscopy. DNA Repair (Amst.) 38, 58–67. [DOI] [PubMed] [Google Scholar]
- 30.Goellner EM, Putnam CD, Graham WJ 5th, Rahal CM, Li BZ, and Kolodner RD (2018). Identification of Exo1-Msh2 interaction motifs in DNA mismatch repair and new Msh2-binding partners. Nat. Struct. Mol. Biol 25, 650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nick McElhinny SA, Watts BE, Kumar D, Watt DL, Lundström EB, Burgers PMJ, Johansson E, Chabes A, and Kunkel TA (2010). Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc. Natl. Acad. Sci. USA 107, 4949–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tishkoff DX, Filosi N, Gaida GM, and Kolodner RD (1997). A novel mutation avoidance mechanism dependent on S. cerevisiae RAD27 is distinct from DNA mismatch repair. Cell 88, 253–263. [DOI] [PubMed] [Google Scholar]
- 33.Buckland RJ, Watt DL, Chittoor B, Nilsson AK, Kunkel TA, and Chabes A (2014). Increased and imbalanced dNTP pools symmetrically promote both leading and lagging strand replication infidelity. PLoS Genet 10, e1004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson C, Gali VK, Takahashi TS, and Kubota T (2016). PCNA retention on DNA into G2/M phase causes genome instability in cells lacking Elg1. Cell Rep 16, 684–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kubota T, Nishimura K, Kanemaki MT, and Donaldson AD (2013). The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol. Cell 50, 273–280. [DOI] [PubMed] [Google Scholar]
- 36.Kang MS, Ryu E, Lee SW, Park J, Ha NY, Ra JS, Kim YJ, Kim J, Abdel-Rahman M, Park SH, et al. (2019). Regulation of PCNA cycling on replicating DNA by RFC and RFC-like complexes. Nat. Commun 10, 2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kahli M, Osmundson JS, Yeung R, and Smith DJ (2019). Processing of eukaryotic Okazaki fragments by redundant nucleases can be uncoupled from ongoing DNA replication in vivo. Nucleic Acids Res 47, 1814–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raghuraman MK, Winzeler EA, Collingwood D, Hunt S, Wodicka L, Conway A, Lockhart DJ, Davis RW, Brewer BJ, and Fangman WL (2001). Replication dynamics of the yeast genome. Science 294, 115–121. [DOI] [PubMed] [Google Scholar]
- 39.Hum YF, and Jinks-Robertson S (2017). Mitotic gene conversion tracts associated with repair of a defined double-strand break in Saccharomyces cerevisiae. Genetics 207, 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.St Charles J, and Petes TD (2013). High-resolution mapping of spontaneous mitotic recombination hotspots on the 1.1 Mb arm of yeast chromosome IV. PLoS Genet 9, e1003434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barberis M, Spiesser TW, and Klipp E (2010). Replication origins and timing of temporal replication in budding yeast: how to solve the conundrum? Curr. Genomics 11, 199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tishkoff DX, Boerger AL, Bertrand P, Filosi N, Gaida GM, Kane MF, and Kolodner RD (1997). Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proc. Natl. Acad. Sci. USA 94, 7487–7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lahue RS, Au KG, and Modrich P (1989). DNA mismatch correction in a defined system. Science 245, 160–164. [DOI] [PubMed] [Google Scholar]
- 44.Liu J, Lee R, Britton BM, London JA, Yang K, Hanne J, Lee J-B, and Fishel R (2019). MutL sliding clamps coordinate exonuclease-independent Escherichia coli mismatch repair. Nat. Commun 10, 5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmidt TT, Sharma S, Reyes GX, Kolodziejczak A, Wagner T, Luke B, Hofer A, Chabes A, and Hombauer H (2020). Inactivation of folylpolyglutamate synthetase Met7 results in genome instability driven by an increased dUTP/dTTP ratio. Nucleic Acids Res 48, 264–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmidt TT, Reyes G, Gries K, Ceylan CU, Sharma S, Meurer M, Knop M, Chabes A, and Hombauer H (2017). Alterations in cellular metabolism triggered by URA7 or GLN3 inactivation cause imbalanced dNTP pools and increased mutagenesis. Proc. Natl. Acad. Sci. USA 114, E4442–E4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sikorski RS, and Hieter P (1989). A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Christianson TW, Sikorski RS, Dante M, Shero JH, and Hieter P (1992). Multifunctional yeast high-copy-number shuttle vectors. Gene 110, 119–122. [DOI] [PubMed] [Google Scholar]
- 49.Lau PJ, Flores-Rozas H, and Kolodner RD (2002). Isolation and characterization of new proliferating cell nuclear antigen (POL30) mutator mutants that are defective in DNA mismatch repair. Mol. Cell. Biol 22, 6669–6680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, and Knop M (2004). A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21, 947–962. [DOI] [PubMed] [Google Scholar]
- 51.Sheu YJ, and Stillman B (2006). Cdc7-Dbf4 phosphorylates MCM proteins via a docking site-mediated mechanism to promote S phase progression. Mol. Cell 24, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study.