

Abstract
Mitochondria contain ribosomes (mitoribosomes) specialized in the synthesis of a handful of proteins essential for oxidative phosphorylation. Therefore, mitoribosome integrity and function are essential for the life of eukaryotic cells and lesions that affect them result in devastating human disorders. To broadly analyze the integrity and assembly state of mitoribosomes it is useful to start by determining the sedimentation profile of these structures by sucrose gradient centrifugation of mitochondrial extracts. During centrifugation, mitoribosome subunits, monosomes and polysomes, and potentially accumulated assembly intermediates will sediment through the gradient at different rates. Sedimentation will depend on the centrifugal force applied and the density and viscosity of the gradient. Importantly, it will also depend on the size, shape, and density of the mitoribosome particles present in the samples under study. Variations of this technique, often coupled with additional downstream approaches, have been used to analyze the process of mitoribosome biogenesis, the composition of assembly intermediates, or to monitor the interaction of extraribosomal proteins with individual mitoribosome subunits or monosomes.
Keywords: Sucrose gradient, Mitochondrial ribosome, Mitoribosome profile, Gradient maker, Gradient master, Gradient fractionation, Immunoblotting
1. Introduction
1.1. Mitochondrial ribosomes
Mitochondria are vital eukaryotic organelles thought to be derived from a prokaryotic ancestor based on the presence of numerous remnants of their once independent life (1). These vestiges include a compacted mitochondrial genome (mtDNA) maintained in the mitochondrial matrix and a mitochondrial ribosome (mitoribosome), which is more closely related to the bacterial ribosome than to its cytosolic counterpart (2,3). The mitoribosome is solely dedicated to the translation of a handful of mtDNA-encoded genes; 8 in yeast Saccharomyces cerevisiae and 13 in mammals. With the exception of a single yeast mtDNA product that is a ribosomal protein, all other mtDNA-encoded proteins in yeast and mammalian cells are subunits of the enzymes that form the oxidative phosphorylation (OXPHOS) system. These proteins are co-translationally inserted into the membrane where they assemble with nuclear DNA-encoded proteins to form four of the OXPHOS enzymes.
Though involved in numerous critical cellular functions, mitochondria are best known for their role in the production of ATP through OXPHOS. This pathway is the primary source of cellular ATP, and it is therefore not surprising that OXPHOS dysfunction is causative or strongly associated with a plethora of human diseases. Mutations in either the nuclear or mitochondrial genome that produce primary defects in OXPHOS are responsible for the relatively rare, highly heterogeneous, and often devastating collection of syndromes known as mitochondrial diseases (4). Further, mitochondrial dysfunction and the accompanying decline in OXPHOS efficiency is implicated in the progression and phenotype of aging-related diseases including societally costly and increasingly prevalent neurodegenerative diseases like Alzheimer’s and Parkinson’s (5). Relevant to the topic of this chapter, mutations in mitochondrial translation factors and ribosome components have also been revealed to be causal of mitochondrial disorders, mainly producing cardio- and encephalomyopathies (6). Furthermore, mitoribosomes and mitochondrial translation have emerged as a target for therapeutic interventions to combat several types of cancer (7).
The interest in mitochondria and their role in disease have grown steadily in recent decades. However, much remains to be discovered about the underlying processes that directly influence productive assembly and regulation of the OXPHOS complexes, such as the assembly and structure-function relationship of the mitoribosome and its protein components. The complete mitoribosome is a massive complex of two distinct large (LSU) and small (SSU) subunits composed of a total of eighty (mammals) proteins and three rRNAs, which have been resolved to a high resolution by cryo-EM (2,3). These structures provide a trove of invaluable information about the fully assembled mitoribosome but reveal little about how it progresses from individual proteins to a structured multimeric complex. Recently, studies tracking the effects on assembly of systematic deletion of each LSU protein in yeast (8) and incorporation kinetics of labeled mammalian mitoribosomal proteins (MRPs) into the mitoribosome (9) have revealed that mitoribosome assembly initiates near the inner mitochondrial membrane (8,10) and proceeds by incorporation of preassembled modules with groups of proteins preferentially incorporating at early, middle, or late stages of the overall assembly process (8,9). These studies provide a broad foundation from which a more specific understanding of how mitoribosome assembly occurs can be built.
1.2. Density gradient centrifugation: General concepts and applications to the study of mitoribosomes
The biological and biomedical arguments provided in the previous paragraphs highlight the importance of establishing standardized methods to analyze mitoribosome profiles and assess their assembly status. For this chapter, we focus on a classical approach to the study of mitoribosome assembly: density gradient sedimentation analysis.
Density gradient ultracentrifugation is a powerful technique for the fractionation of particle mixtures, purification of subcellular organelles, or isolation of macromolecules. The method involves the layering of a sample containing a mixture of macromolecules of different size and mass on the surface of a vertical column of liquid (e.g., an ultracentrifuge tube containing a solution of sucrose, glycerol, or the chemical of choice) whose density increases from top to bottom forming a gradient. The two main types of density gradient centrifugation are rate-zonal separation and isopycnic separation. In rate-zonal separation, particles are separated based on their size and mass, such as in most sucrose gradient sedimentation analyses. The density of the particles is greater than the density of the gradient, and if centrifugation is continued long enough, all particles will end up as one pellet at the bottom of the tube. In isopycnic separation, the particles migrate through the solvent gradient until they reach the point where their buoyant density is equal to that of the gradient, such as in the separation of nucleic acids in cesium chloride gradients, or of complex macromolecular protein mixtures using gradients of iodixanol, a non-ionic polymer. In this case, the density of the gradient medium must be higher than the density of the particles to be separated. According to the means of preparation, density gradients may be either step gradients or continuous gradients. Step gradients are prepared by successively layering solutions of different density in the centrifuge tube and then layering the sample to be fractionated on top of the last “step”. Sucrose or Nicodenz step gradients are useful for example to purify organelles of similar density. In continuous density gradients, the density changes smoothly and continuously, either linearly or exponentially, from one extreme to another. Continuous gradients can be produced from step gradients by allowing sufficient time for diffusion to smooth out the steps, but they are usually prepared directly by using dedicated gradient makers.
Rate-zonal separation in linear sucrose gradients (11–13) and isopycnic separation in iodixanol gradients (9) are both commonly used for the study of mitoribosome assembly profile. Here, we will focus exclusively on explaining the experimental details of sucrose density gradient sedimentation analysis. This method is prevalent in current literature, and we have used it extensively to analyze mammalian (11,13,14) and, with some modifications, S. cerevisiae mitoribosomes (8,15,16). It is useful in identifying factors associating with the mitoribosome in wild-type cells and to determine the consequences on mitoribosome assembly of gene ablation and/or complementation of cells depleted of individual MRPs or mitoribosome assembly factors with modified versions. The body of the method is written for use with mammalian cell lines and is based in large part on previously published methods (17,18) with minor modifications. Here, we have combined those methods to a single protocol, which covers mitochondrial isolation, sucrose gradient formation, sedimentation, and subsequent analysis by immunoblotting to determine mitoribosome composition through immunoblot of candidate proteins. Though not covered in this method, the total composition of co-sedimenting mitoribosomal proteins and/or associated factors can then be identified in an unbiased manner by mass spectrometry analysis. Due to the availability of a large number of approaches and commercial options for gene editing in mammalian cell lines that frequently require extensive optimization, the generation of gene-edited cell lines is beyond the scope of this method, and it assumes the user has already procured cell lines with genetic modifications of interest. S. cerevisiae yeast cells are far more amenable to genetic modification for which we suggest the approaches described in a recent publication by our group (8). Overall, the method yields consistent results and is simple and manageable with numerous potential pause points. In addition to the elaboration of individual steps, the notes section details alternative approaches useful in fitting the protocol to the question, timing, and economic constraints of the user.
The usefulness of the methodology is exemplified with the recent observation by our group that the mitochondrial DEAD-box helicase DDX28 co-sediments with the mitoribosome large subunit and the monosome in a salt-dependent manner (Fig 1). This information was an essential step in guiding subsequent experiments that led us to conclude that DDX28 resides in mitochondrial RNA granules and functions in mitoribosome large subunit biogenesis (11).
Figure 1. Sucrose gradient sedimentation analyses of the mtLSU assembly factor DDX28 and mitoribosomal proteins from wild-type (WT) HEK293T mitochondrial extracts.
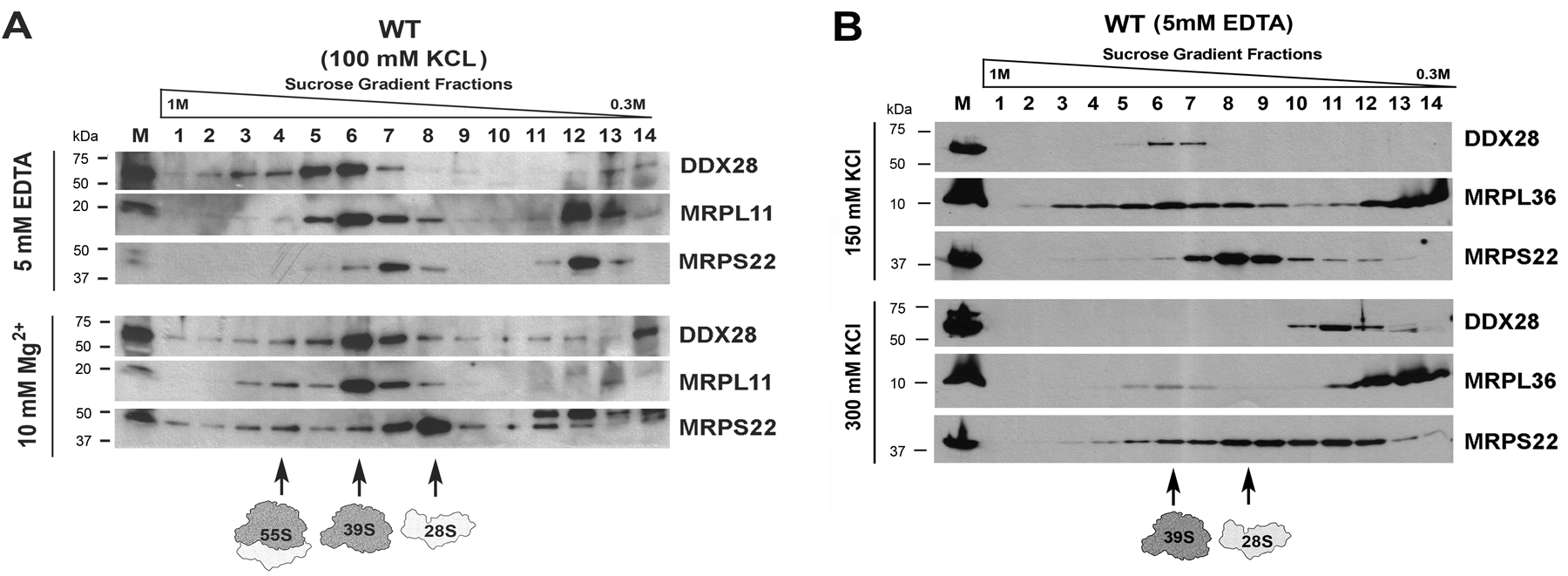
(A) Sedimentation profile of mitoribosomes extracted in 5 mM EDTA or 10 mM MgCl2, (B) Sedimentation profile of mitoribosomes extracted in 5 mM EDTA and 150 or 300 mM KCl. Reproduced and modified from Tu and Barrientos, 2015 (11) with permission from Elsevier (license # 4470451296140).
2. Materials
All solutions are prepared in double distilled water (ddH2O) and should be made fresh unless otherwise noted. Chill solutions to 4 °C before use to minimize potential protein degradation. MgCl2 or EDTA, as well as KCL or other salts, are used at different concentrations in extraction buffers and sucrose solutions depending on the purpose of the experiment (see Notes 1 and 2).
2.1. Isolation of Mitochondria
T-K-Mg Buffer: 10 mM Tris-HCl pH 7.4, 10 mM KCl, 0.5 mM MgCl2 (Stock solution can be filtered and stored at room temperature (RT)).
Trypsin-EDTA solution: 0.5 g/l porcine trypsin and 0.2 g/L EDTA•4Na in Hank’s Balanced Salt Solution with phenol red (commercially available and can be stored long-term at −20° C or a few weeks at 4° C).
1M Sucrose Stock solution: 1 M sucrose, 10 mM Tris-HCl pH 7,4.
2.2. Preparation of a 10–30% Linear Sucrose Gradient with Biocomp Gradient Master
10 % sucrose solution: (10 mM MgCl2 or 5 mM EDTA), 20 mM HEPES pH 7.4, 100 mM KCl, 0.1 M PMSF, 0.1% Digitonin, 1x EDTA-free protease inhibitor, 10 % w/v Sucrose.
30 % sucrose solution: (10 mM MgCl2 or 5 mM EDTA), 20 mM HEPES pH 7.4, 100 mM KCl, 0.1 M PMSF, 0.1 % Digitonin, 1x EDTA-free protease inhibitor, 30 % w/v Sucrose.
Polypropylene ultra-centrifuge tube (13 × 51 mm).
Marker block (Provided with Gradient Master).
Four mm tube caps (Provided with Gradient Master).
Sucrose layering cannula – syringe attachment (Provided with Gradient Master).
Tube holder (Provided with Gradient Master).
Air bubble leveling tool.
2.3. Mitoribosome Extraction and Sedimentation Analysis
2.4. Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Immunoblotting
12 % SDS-PAGE polyacrylamide gel (home-made gels or gels commercially available from multiple sources – e.g., Biorad, GeneScript- can be used).
Wash Buffer: 10 mM Trish pH 8, 1 mM EDTA, 150 mM NaCl, 0.1 % (v/v) Triton X-100.
Blocking Buffer: 5 % (w/v) non-fat dry milk in Wash Buffer.
Ponceau protein staining solution: 0.2 g Ponceau in 3 % (w/v) TCA (Trichloroacetic acid).
Laemmli sample buffer 4x (LB 4x): 200mM Tris-HCl pH 6.8, 4 % SDS, 40 % glycerol, 4 % β-mercaptoethanol, 0.05 % (w/v) bromophenol blue.
Antibodies against mitoribosome large and small subunit proteins: A large number of antibodies raised against each mitoribosomal protein are commercially available. A few high quality antibodies we have used are listed here: anti-mL45 (Abcam-ab113786), anti-uL16m (Sigma-HPA054133), anti-mS27 (Proteintech-17280-1-AP), anti-mS29 (ab155499).
3. Methods
3.1. Isolation of Mitochondria
Minimum starting material ranges between 2 – 4 × 108 adherent mammalian cells grown on 15 × 150 mm cell culture plates (see Note 4).
Aspirate culture media from plates, wash with 10 ml PBS and add 1.5 mL trypsin to each plate. Tilt plates back and forth to ensure trypsin has passed over entire plate surface area. Incubate for 2 min. (see Note 5).
Completely dislodge adherent cells from plate surface by firmly tapping each plate against the hand.
Inactivate trypsin by re-suspending cells in 5 mL complete DMEM and combine cell suspension in 50 mL conical tubes.
Pellet cells by centrifugation at 600 × g for 3 min. and aspirate media. All subsequent steps are performed on ice, at 4° C, and in pre-chilled buffers.
Resuspend cell pellet in 10 mL PBS (If cell suspension is spread across multiple tubes, combine to a single 50 mL conical tube).
Pellet cells by centrifugation at 600 × g for 3 min. and aspirate PBS.
Repeat Steps # 6–7.
Determine the weight of cell pellet and gently re-suspend cells in 1 mL of the hypotonic T-K-Mg buffer per 0.15 g cells.
Incubate cells for 5 min. to allow swelling of the cells. Avoid incubation over 10 min. (see Note 6).
Homogenize cells with a glass/Teflon tissue grinder Dounce homogenizer equipped with a serrated PTFE (polytetrafluoroethylene, a.k.a. Teflon) pestle (10 mL capacity). Homogenize the cells using 10–25 strokes of the pestle of a tight-fitting Dounce (see Note 7).
Immediately bring cell homogenate to 0.25 M sucrose with 1 M Sucrose Stock (1:3 homogenate: 1 M Sucrose Stock) to make it isotonic.
Return homogenate to 50 mL conical tube and centrifuge for 3 min. at 1,200 × g to pellet unbroken cells, nuclei, and other cellular debris.
Collect supernatant in a new 50 mL conical tube and centrifuge again for 3 min. at 1,200 × g. If any pellet is observed, repeat this step (see Notes 8 and 9).
Collect supernatant (containing mitochondria) and aliquot into 1.5mL microcentrifuge tubes with 1 mL per tube (see Note 10).
Centrifuge tubes at 15,000 × g for 5 min.
Wash pellets by re-suspending in STE buffer and combine suspension into a single tube.
Pellet suspension at 15,000 × g for 5 min.
Optional: Repeat steps # 17–18 for increased mitochondrial purity.
Re-suspend pellet in minimal STE buffer (Use a volume of STE buffer roughly equivalent to the volume of the mitochondrial pellet). This sample contains isolated mitochondria.
Determine mitochondrial protein concentration by Lowry, Bradford, or preferred method (see Note 11).
Keep mitochondria on ice until used or immediately flash freeze aliquots by submerging in liquid N2 and storing at −80 °C for long-term storage (see Note 12).
3.2. Preparation of a 10–30% Linear Sucrose Gradient with Biocomp Gradient Master
Prepare the master sucrose solutions in the same buffer as the mitochondrial extraction is performed, although devoid of detergent. The two sucrose solutions must be of the maximum and minimum concentrations wanted (e.g. 10–30% is useful to fractionate the mitoribosome components and the monosome) (see Note 13).
To prepare the gradient using the Gradient Master from Biocomp Instruments, start by reading the Operator’s Manual at <http://www.biocompinstruments.com>.
Place the ultra-centrifuge tube inside the marker block and use a fine tip marker to mark at the upper, 4 mm cap level, which will indicate the “stop point” when filling the tube with sucrose solution (Fig. 2A). See Notes 14 and 15 for alternative methods to gradient formation requiring little to no equipment.
Using the cannula attached to a syringe or simply by pipetting, fill the ultra-centrifuge tube with 0.3 M sucrose solution until the volume of solution in the tube is just past the stop point.
Using a cannula-attached syringe filled with 1 M sucrose solution, insert the cannula tip to the bottom of the tube and slowly inject the 1 M sucrose solution. An interface between the 0.3 M and 1 M sucrose solutions should be visible. Continue adding 1 M sucrose solution until the interface is just past the stop point.
Remove the cannula quickly but smoothly.
Cap the ultra-centrifuge tube with the 4 mm tube cap. Use a pipette to remove any excess sucrose from the top of the cap. Ensure no bubble is present between the sucrose solution and the cap. If a bubble is present, remove the cap, add sufficient 0.3 M sucrose solution on top of the gradient and recap the tube.
Insert the tube into the gradient holder.
Ensure the Gradient Master base plate is level using the digital leveling controls and an air bubble level.
Gently place the gradient tube holder containing the sucrose gradient directly onto the center of the Gradient Master base plate.
“Balance” the tube holder as you would a centrifuge, such that each tube is placed directly across from a tube filled with similar density material.
Select on digital gradient master controls: “Grad” --> “List” --> “SW50”, scroll down until “10–30 %” is displayed then select “Use” --> “Run”.
The Gradient Master will now mix the sucrose solutions to produce the 0.3–1 M linear sucrose gradient in approximately 1 min. (see Note 16).
Remove the cap. The gradient is now ready and should be kept on ice until used (see Note 17).
Figure 2. Preparation of a continuous sucrose gradient using gradient makers.
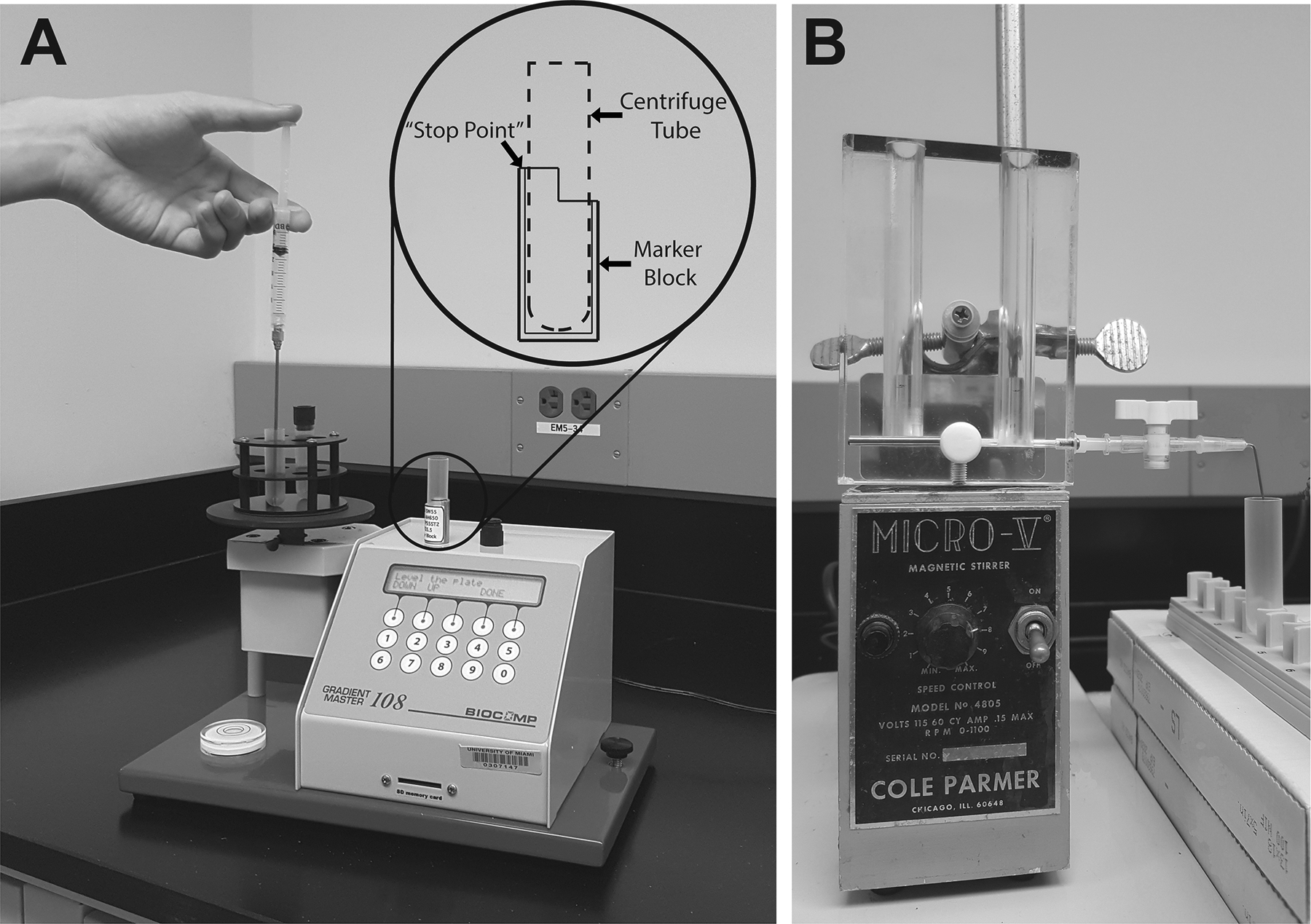
(A) The automatized BioComp Instruments Gradient Master. This instrument is programmable, capable of producing 6 identical, linear gradients in less than 1 min. by utilizing tilted tube rotation technology. Inset depicts marker block described in section 3.2, step 3. (B) A classical manual gradient maker (C.B.S. Scientific). The device is linear with side outlets and has two chambers where the two sucrose solutions (of maximum and minimum concentrations) are placed. Gradient flow is controlled by a valve centered between the two chambers. The outflow channel is fitted with a male luer adapter, which can be used with any needle or luer valve to facilitate continuous and smooth flow into the ultracentrifugation tube. Only one gradient can be prepared at a time in some 10 min. The system includes a tubing adapter kit with a valve, tubing adapters, butterfly set, and silicone tubing. This kit can be used to adapt your gradient maker for either a gravity gradient or a pumping gradient.
3.3. Mitoribosome Extraction and Sedimentation Analysis
All steps are performed on ice or at 4 °C in pre-chilled buffers.
Pellet 4 mg of isolated mitochondria at 10,000 × g for 5 min. (see Note 18).
Remove supernatant and re-suspend mitochondrial pellet in 400 μL of extraction buffer and incubate for 10 min. (see Notes 1-3 and 19).
Centrifuge extract at 24,000 × g for 15 min. to pellet mitochondrial membrane components.
Collect the supernatant containing the mitoribosome and soluble mitochondrial proteins and aliquot 40 μL (10 %) for later use in Western blot analysis as “total extract”. The pellet can either be discarded or re-suspended in sample buffer as an alternative “total extract”.
Gently add the supernatant onto the top of the 10–30 % sucrose gradient. If the volume of sample is large, 100–200 μL of sucrose solutions can be removed from the top of the sucrose gradient to make space (see Note 20).
Place the sucrose gradient tubes into an SW55Ti swinging rotor bucket and cap the bucket tightly.
Hook the bucket into the SW55Ti rotor.
Ensure the rotor is balanced by ultra-centrifuge tubes filled with sucrose stock solution loaded into rotor buckets.
Centrifuge at 40,000 rpm (Average RCF = 151,693) for 3 hr. and 10 min. (see Note 21).
Cut the tip of a standard 1000 μL micropipette tip so that the tip mouth is approximately 0.3–0.4 cm wide.
Using the modified tip and a 1000 μL pipette, place the top just under the gradient surface at the middle of the tube and collect 260 μL into a microcentrifuge tube.
Repeat step #11 until the entire gradient has been collected for a total of ~14 fractions.
Consecutively label the collected fractions.
The fraction samples can then be immediately used for western blot, protein precipitation, or stored at −80 °C for later use (see Note 22).
3.4. Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Immunoblotting
Prepare samples for western blot analysis by adding 13 μL of 4x LB to 40 μL “Total extract” and 37.5 μL 4x LB to 112.5 μL from each fraction # 1–14 in microcentrifuge tubes and mix thoroughly.
Load a 12 % SDS-PAGE gel starting with a pre-stained protein ladder, total extract, and then 100 μL of each of the 14 fractions in sequential order.
Separate sample proteins by electrophoresis at 80 V for 30 min. to allow samples to enter the resolving portion of the gel, and then increase the voltage to 150 V until Laemmli sample buffer dye front reaches the bottom of the gel.
Transfer proteins from gel to nitrocellulose membrane using, for example, an Owl™ HEP Series Semidry Electroblotting System (ThermoFisher Scientific). Any alternative semidry or wet transfer systems can be used.
To assess a normal distribution of proteins throughout the gradient and the absence of protein degradation, incubate membrane in Ponceau protein staining solution for 15 min. or until protein bands are visible.
Using the protein ladder and protein staining as a guide, cut out horizontal strips of membrane corresponding to the molecular weight of proteins to be blotted against.
Wash membrane strips by incubation in washing buffer for 5 min. with agitation on a rocker to remove the Ponceau protein stain.
Incubate membrane strips in blocking buffer for 30 at room temperature with agitation.
Incubate membranes with antibodies against mtLSU or mtSSU markers, or mitoribosome interacting factors of interest at a dilution of 1:250–1000 (depending on manufacturer recommendations or optimization) for 3 hr. at RT or 4 °C overnight with agitation (see Note 23).
Wash membrane for 5 min. with agitation in washing buffer three times.
Incubate membrane with horseradish peroxidase (HRP)-conjugated secondary antibodies with agitation for 1–3 hr. at RT or overnight at 4 °C.
Wash membrane for 15 min. in washing buffer three times.
Develop membrane by enhanced chemiluminescence (ECL) reagents followed by exposition X-ray films or luminescence detection of choice. See Figure 1 for representative mitoribosome sedimentation profiles.
4. Notes
MgCl2 (usually 10–20 mM) maintains mitoribosome LSU and SSU interactions to preserve the fully formed monosome while EDTA chelates Mg2+ causing disassociation of the monosome into separated LSU and SSU. In some mutant cell lines, EDTA could disrupt unstable intermediates and labile interactions; therefore, simply 0–1 mM should be used instead. Salt concentration in extraction buffer and sucrose gradient solutions can be varied between 10–300 mM KCl to preserve or interrupt a substantial number of mitoribosome-protein interactions. Integral mitoribosome components are not disrupted even at 300 mM KCl (19). Figure 1 portrays an example of how the associations of mitoribosome subunits and interacting proteins are affected by inclusion of EDTA, Mg2+, or increasing salt concentrations in the extraction buffer and sucrose solutions.
Efficient purification of intact mitoribosomes requires optimized conditions in which the ratio of monovalent to divalent cations is crucial (20, 21). For that purpose, the ideal buffer would contain 50 mM NH4Cl and 10 mM MgCl2.
The choice of detergent in the mitochondrial extraction buffer is essential. Avoid strong detergents that can disrupt mitoribosome integrity. Non-ionic detergents such as n-Dodecyl-β-D-Maltoside (lauryl maltoside) or digitonin are especially useful for solubilizing membrane proteins or large complexes such as mitoribosomes to preserve their integrity and activity. It is strongly advised to perform preliminary mitoribosome extraction tests in-house by titrating the detergent of choice to the optimal concentration. 0.5–0.8 % (w/v) digitonin or 0.5 % (w/v) lauryl maltoside are useful reference concentrations. Further, some optimization is necessary for every detergent batch, as purity can vary between lots.
For larger scale preparations of isolated mitochondria, cells capable of non-adherent growth (like 293T) can be grown in suspension by liquid culture methods, which can be less laborious and make more efficient use of culture media. Our usual yield is 4 g of cells per liter. If using liquid cultures, start at step 5 of the Mitochondrial Isolation section.
Trypsinization efficiency can be increased by placing cells in 37 °C incubator or rinsing cells with PBS an extra time before adding trypsin. As an alternative to trypsinization, after washing adherent cells in PBS, they can also be collected by adding 5 ml PBS, scraping cells from culture plate surface with a cell scraper, collecting PBS with the scraped cell suspension, and continuing at step 5 of the Mitochondrial Isolation section.
Do not allow excessive incubation of cells in the T-K-Mg buffer to avoid rupture of the outer mitochondrial membrane.
Appropriate homogenization technique involves full insertion of a pestle to bottom of a homogenizer vessel and slow withdrawal of pestle such that a vacuum is generated just below the pestle which forces the sample volume past pestle. Do not overload the homogenizer, as the entire sample will not be exposed to vacuum homogenization with each stroke. Sample volume should not exceed 75 % nominal capacity of the homogenizer. If the sample volume is large, sample aliquots can be homogenized in multiple rounds or in a larger volume homogenizer.
To increase the total mitochondrial yield, the pellet from step 13 can be re-suspended, re-homogenized, cleared according to steps 13–14, and the supernatant combined with the original low-speed supernatant.
If working with large volumes of homogenate (>10 mL) the given spin times are insufficient. It is advised to increase spin time to 10–15 min. or aliquot 10 mL homogenate per 50 mL conical tube before spinning.
Larger 50 mL tubes can be used (for substantial reduction of labor) if your lab possesses centrifuges/rotors capable of loading large tubes, refrigeration, and speeds of 15,000 × g.
Isolated mitochondria can be further purified using Sucrose (22) or Nicodenz (23) step gradients. For example, use the following steps based on the protocol described by Meisinger et al. (22). Prepare a 60 % / 32 % / 23 % / 15 % sucrose step gradient in TE buffer (10 mM Tris-HCl, 1 mM EDTA pH 7.4) by gently layering 1 mL 60 % sucrose, 2 mL 32 % sucrose, 1 mL 23 % sucrose, and 1mL 15% sucrose on top of one another (densest to lightest: bottom to top) in a 13 × 51 mm ultracentrifuge tube. Load the isolated mitochondria onto the top of the gradient and centrifuge for 1 h at 4 °C at 134,000 × g. Collect mitochondria from a minimal volume at the 60 % / 32 % sucrose interface which should contain 70–80% of the starting material (at least 2 mg starting material is recommended). Dilute sample with 2–3 volumes of STE buffer. Pellet mitochondria at 10,000 × g for 10 min, discard sup, and re-suspend pellet in a minimal volume of STE buffer (~2–3 packed pellet volume). Measure protein concentration of the sample and use it immediately or flash-freeze in liquid nitrogen for long-term storage.
Mitochondria prepared and stored in this way have intact outer and inner membranes, are electrochemically competent, and capable of trans-membrane protein transport. They can be used for measurements of oxygen consumption and oxidative phosphorylation, in organello DNA synthesis, in organello transcription, in organello tRNA aminoacylation, in organello protein synthesis, in organello protein import, and in organello mtDNA footprinting as described by Fernandez-Vizarra et al. (17). If you intend to perform any of these assays use of freshly isolated mitochondria is highly recommended as mitochondrial competence can decrease dramatically with freezing. Further, 0.1 % fatty acid-free BSA should be added to homogenization media to prevent fatty acid-induced mitochondrial membrane uncoupling as electrochemically competent mitochondria are required for normal mitochondrial function.
After preparing the sucrose solutions, allow to sit for 5–10 min. to allow any air bubbles to dissipate from the sucrose solution. Air bubbles are disruptive to the gradient making process. The solutions could also be degassed by vacuum. For that purpose, place the solution in a side-arm flask and use a rubber stopper to seal off the top. Place the flask on a stir plate and turn the plate on so the stir bar is spinning at a medium speed. Apply vacuum until all the gas is released.
A gradient maker (like Thomas-Scientific CAT# 1186V98; Fig. 2B) can also be used to generate a 10–30 % gradient. In this case, the addition of 0.1 % Digitonin to both 10 % and 30 % sucrose solutions is recommended to lubricate the passage of solutions through this apparatus and into the ultracentrifuge tube with a constant and smooth flow.
Gradients can also be formed by simple diffusion with no additional equipment (24, 25). In detail: 2.5 mL 30 % sucrose solution is added to the 13 × 51 mm ultra centrifuge tube. 2.5 mL of 10 % solution is then layered on top. An interface between the two sucrose solutions should be visible. The tube is then sealed with a cap or parafilm and slowly and gently inverted horizontally. The solutions then mix by diffusion and the gradient forms throughout 1 hr. at room temperature or 3 hr. at 4 °C. The tube is then slowly and gently returned to a vertical position, the seal removed, and stored at 4 °C until used, preferably on the same day. Gradients formed in this way are not perfectly linear but are consistent between batches as well as cost and time efficient.
If an air bubble is present and moves through the gradient during the mixing process, the gradient should be discarded, as moving bubbles will disrupt the gradient.
Handle gradients with care to prevent disturbing the contents.
Significantly less isolated mitochondria (1–2 mg) can be used if a protein precipitation step is added after gradient fraction collection. For a preliminary and cost-effective alternative to using isolated mitochondria, whole cells collected from a single 80–90 % confluent 150 mm culture plate can be extracted and used for sedimentation analysis exactly as described for isolated mitochondria. A minimum of 1 mg whole cell extract should be loaded onto the sucrose gradient for sedimentation. Following gradient fractionation, fraction proteins should be precipitated before immunoblot analysis. Proteins loosely associated with the mitoribosome or with low stoichiometry may not be detectable with this approach. Further, large protein complexes from other cellular compartments will be present in extract and may confound results.
RNase inhibitors can be added to the extraction buffer if excessive mitoribosome degradation is observed. A cost-effective option is to use ribonucleoside vanadyl complexes at 10 mM.
The sample volume to be loaded onto the gradient is approximately 10% of the total gradient volume, since deviations of this proportion can compromise the fractionation resolution.
Slow acceleration and deceleration centrifuge settings are recommended to avoid disturbing gradient.
Protein precipitation can be performed on each fraction for greater resolution in immunoblotting or subsequent analysis by mass spectrometry. For immunoblotting, immunoprecipitation can be performed with final 25 % trichloroacetic acid (TCA) followed by washes of the precipitate first with 0.5 M Tris-Base and subsequently with water, before solubilization of the protein pellet in 25 μL of loading Laemmli buffer for loading onto an SDS-polyacrylamide gel. For mass spectrometry analysis of fraction protein composition, the samples are best precipitated by a method based on a defined methanol-chloroform-water mixture for the quantitative precipitation of soluble as well as hydrophobic proteins from dilute solutions (26). Practically, 4 volumes (V) methanol and 1 V chloroform are added to 1 V of the sample. A phase separation is achieved by the addition of 3 V water whereby the protein is precipitated at the chloroform-methanol-water interphase. The addition of an excess (4 V) of methanol and subsequent centrifugation results in a protein pellet which is free of interfering substances (26).
Primary antibodies can be kept in the immunoblotting blocking solution at −20 °C and reused extensively until no longer effective, or solution spoils.
Acknowledgements
We thank Dr. Priyanka Maiti for critical reading of the manuscript. This research was supported by NIH R35 Grant GM118141 (to A.B.) and MDA Grant MDA-381828 (to A.B.).
References
- 1.Margulis L (1975) Symbiotic theory of the origin of eukaryotic organelles; criteria for proof. Symp. Soc. Exp. Biol, 29, 21–38. [PubMed] [Google Scholar]
- 2.Amunts A, Brown A, Toots J, Scheres SHW and Ramakrishnan V (2015) The structure of the human mitochondrial ribosome. Science, 348, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greber BJ, Bieri P, Leibundgut M, Leitner A, Aebersold R, Boehringer D and Ban N (2015) Ribosome. The complete structure of the 55S mammalian mitochondrial ribosome. Science, 348, 303–308. [DOI] [PubMed] [Google Scholar]
- 4.Lightowlers RN, Taylor RW and Turnbull DM (2015) Mutations causing mitochondrial disease: What is new and what challenges remain? Science, 349, 1494–1499. [DOI] [PubMed] [Google Scholar]
- 5.Murphy MP and Hartley RC (2018) Mitochondria as a therapeutic target for common pathologies. Nature reviews. Drug discovery, 5, 174. [DOI] [PubMed] [Google Scholar]
- 6.De Silva D, Tu YT, Amunts A, Fontanesi F and Barrientos A (2015) Mitochondrial ribosome assembly in health and disease. Cell Cycle, 14, 2226–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim HJ, Maiti P and Barrientos A (2017) Mitochondrial ribosomes in cancer. Semin. Cancer Biol, DOI: 10.1016/j.semcancer.2017.1004.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeng R, Smith E and Barrientos A (2018) Yeast mitoribosome large subunit assembly proceeds by hierarchical incorporation of protein clusters and modules on the inner membrane. Cell Metab, 27, 645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bogenhagen DF, Ostermeyer-Fay AG, Haley JD and Garcia-Diaz M (2018) Kinetics and mechanism of mammalian mitochondrial ribosome assembly. Cell Rep, 22, 1935–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bogenhagen DF, Martin DW and Koller A (2014) Initial steps in RNA processing and ribosome assembly occur at mitochondrial DNA nucleoids. Cell Metab, 19, 618–629. doi: 6 10.1016/j.cmet.2014.1003.1013. [DOI] [PubMed] [Google Scholar]
- 11.Tu YT and Barrientos A (2015) The human mitochondrial DEAD-box protein DDX28 resides in RNA granules and functions in mitoribosome assembly. Cell Rep, 10, 854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lavdovskaia E, Kolander E, Steube E, Mai MM, Urlaub H and Richter-Dennerlein R (2018) The human Obg protein GTPBP10 is involved in mitoribosomal biogenesis. Nucleic Acids Res, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maiti P, Kim HJ, Tu YT and Barrientos A (2018) Human GTPBP10 is required for mitoribosome maturation. Nucleic Acids Res, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim H-J and Barrientos A (2018) MTG1 couples mitoribosome large subunit assembly and intersubunit bridge formation. Nucleic Acid Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Silva D, Fontanesi F and Barrientos A (2013) The DEAD-Box protein Mrh4 functions in the assembly of the mitochondrial large ribosomal subunit. Cell Metab, 18, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Silva D, Poliquin S, Zeng R, Zamudio-Ochoa A, Marrero N, Perez-Martinez X, Fontanesi F and Barrientos A (2017) The DEAD-box helicase Mss116 plays distinct roles in mitochondrial ribogenesis and mRNA-specific translation. Nucleic Acids Res, 45, 6628–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez-Vizarra E, Ferrin G, Perez-Martos A, Fernandez-Silva P, Zeviani M and Enriquez JA (2010) Isolation of mitochondria for biogenetical studies: An update. Mitochondrion, 10, 253–262. [DOI] [PubMed] [Google Scholar]
- 18.Horn D, Fontanesi F and Barrientos A (2008) Exploring protein-protein interactions involving newly synthesized mitochondrial DNA-encoded proteins. Methods Mol. Biol, 457, 125–139. [DOI] [PubMed] [Google Scholar]
- 19.Kehrein K, Schilling R, Moller-Hergt BV, Wurm CA, Jakobs S, Lamkemeyer T, Langer T and Ott M (2015) Organization of mitochondrial gene expression in two distinct ribosome-containing assemblies. Cell Rep, 12, S2211–1247. [DOI] [PubMed] [Google Scholar]
- 20.Couvillion MT, Soto IC, Shipkovenska G and Churchman LS (2016) Synchronized mitochondrial and cytosolic translation programs. Nature, 533, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vignais PV, Stevens BJ, Huet J and Andre J (1972) Mitoribosomes from Candida utilis. Morphological, physical, and chemical characterization of the monomer form and of its subunits. J. Cell Biol, 54, 468–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meisinger C, Pfanner N and Truscott KN (2006) Isolation of yeast mitochondria. Methods Mol. Biol, 313, 33–39. [DOI] [PubMed] [Google Scholar]
- 23.Glick BS and Pon LA (1995) Isolation of highly purified mitochondria from Saccharomyces cerevisiae. Methods Enzymol, 260, 213–223. [DOI] [PubMed] [Google Scholar]
- 24.Davies E and Abe S (1995) Methods for isolation and analysis of polyribosomes. Methods Cell Biol, 50, 209–222. [DOI] [PubMed] [Google Scholar]
- 25.Stone AB (1974) A simplified method for preparing sucrose gradients. Biochem J, 137, 117–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wessel D and Flugge UI (1984) A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem, 138, 141–143. [DOI] [PubMed] [Google Scholar]