

Abstract
Human cardiac β myosin undergoes the crossbridge cycle as part of the force-generating mechanism of cardiac muscle. The recovery stroke is considered one of the key steps of the kinetic cycle as it is the conformational rearrangement required to position the active site residues for hydrolysis of ATP and interaction with actin. We explored the free-energy surface of the transition and investigated the effect of the genetic cardiomyopathy causing mutations R453C, I457T, and I467T on this step using metadynamics. This work extends previous studies on Dictyostelium myosin II with engineered mutations. Here, like previously, we generated an unbiased thermodynamic ensemble of reactive trajectories for the chemical step using transition path sampling. Our methodologies were able to predict the changes to the dynamics of the recovery stroke as well as predict the pathway of breakdown of ATP to ADP and HPO42− with the stabilization of the metaphosphate intermediate. We also observed clear differences between the Dictyostelium myosin II and human cardiac β myosin for ATP hydrolysis as well as predict the effect of the mutation I467T on the chemical step.
Graphical Abstract
1. INTRODUCTION
The myosin superfamily is a collection of molecular motors able to perform various biological functions through interaction with actin filaments regulated by hydrolysis of adenosine triphosphate (ATP).1,2 The myosins are diverse in their primary sequence and dynamics and are mainly divided into two broad groups: conventional myosin mainly associated with muscle functions and the unconventional myosin playing a key role in intracellular processes.3
Human cardiac β myosin is a type II conventional myosin forming an integral part of the cardiac thick filament that interacts with the cardiac thin filament to cause cardiac muscle contraction. It is a two-headed hexameric protein comprising a pair of myosin heavy-chain (MHC) and two pairs of myosin light-chain (MLC) subunits. Each myosin heavy chain folds to form a large globular head and an α helical tail, while the two light chains bind to the heavy chain at its α-helical neck region in an antiparallel orientation. The myosin head is the main motor region as it contains the binding site of the nucleotide as well as actin (thin filament)-binding loops to allow force generation.4,5 The functioning of the myosin motor can be understood through its interactions with the thin filament coupled with ATP hydrolysis.
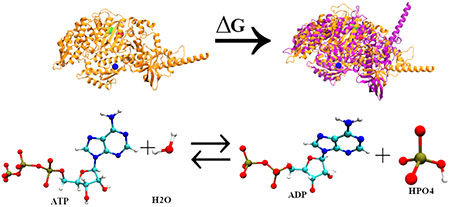
Myosin undergoes a sequence of biochemical steps termed the Lymn–Taylor cycle6 to hydrolyze ATP and reversibly bind actin, eventually leading to muscle contraction.7–9 In the absence of ATP, the myosin is strongly bound to actin and is said to be in its rigor conformation. With ATP binding to myosin, modulation of the nucleotide-binding site occurs leading to the opening of the actin-binding cleft and detachment of actin from myosin. This new conformation is termed as postrigor state. Again, to regain actin-binding affinity, myosin needs to prepare for ATP hydrolysis and hence position its catalytic residues in their active positions. The recovery stroke or swinging of the lever arm from the down to the up state takes place to organize the active residues to hydrolyze ATP leading to pre-powerstroke state. After this conformational rearrangement, ATP hydrolysis takes place leading to myosin rebinding to actin. The recovery stroke10–15 and ATP hydrolysis16–20 are considered as significant steps of the mechanochemical cycle between myosin and actin.
There have been experimental and computational studies on Dictyostelium myosin II, where specifically, engineered mutations have been designed to observe the effect on recovery stroke and influence on the ATP hydrolysis rate.13 There have been several quantum mechanics/molecular mechanics (QM/MM) simulations following minimum potential energy pathways (mPEPS) to determine the mechanism of the decomposition of ATP to ADP and phosphate ion.16–21 Recently, computational models of Dictyostelium myosin II were developed to explore the free-energy profile for the recovery stroke and deviations in these profiles due to the engineered point mutations using metadynamics.22–24 The transition path sampling (TPS) method25 has been employed on the same system to find a thermodynamically valid ensemble of trajectories to provide information on how the active site residues are coupled to the chemical step.26
The recovery stroke is well defined in Dictyostelium myosin II but not much is explored about the same in the human cardiac β myosin. Hence, in this study, we extend the previous computational work on Dictyostelium myosin II.26 We developed computational models of human cardiac β myosin, replicating the recovery stroke by sampling the transition from postrigor state (PR) to pre-powerstroke state (PPS) and calculate the free-energy profile to explore how point mutations associated with familial cardiomyopathy R453C, I457T, and I467T affect the same. We also aim to study the ATP hydrolysis step and predict the isoformic differences as well as the effect of the point mutation I467T on the same.
2. SIMULATION METHODS
2.1. Simulation Design for Metadynamics.
As there is no X-ray diffraction structure for human cardiac β myosin, bovine cardiac β myosin crystal structures with a 98% sequence identity postrigor (PDB ID: 6FSA)27 and the pre-powerstroke (PDB ID: 5N6A)28 were utilized to model the structure of the human isoform. The first 782 amino acid residues of the S1 domain were chosen to be modeled as they contained all of the interactions and binding sites required for the simulations. Homology modeling with the SWISS MODEL29 program was used with both the postrigor and the pre-powerstroke structures of the human isoform as the target structures and with the bovine isoform being the template structure for the primary sequence. The addition and correct placement of missing hydrogen atoms to each structure were accomplished using the CHARMM4230 program. The original substrates were modified to ATP, and a magnesium ion was incorporated at the active site of both the postrigor and the pre-powerstroke structures. Each of the structures was then explicitly solvated using a water box of TIP3P water with the edges at least 15 Å away from the protein surface. After solvation, neutralization was performed by placing counterions of K+ and Cl− throughout the water box at a physiological concentration of 0.15 mol/L.
After the preparation of the neutralized postrigor and pre-powerstroke structures, minimization, heating, and equilibration were performed with the NanoScale Molecular Dynamics (NAMD)31 program version 2.12. All simulations were carried out with the CHARMM36 force field to simulate the protein environment. Minimization of the protein structures was performed with the conjugate gradient method for 5000 steps followed by a slow heating phase to 300 K at a rate of 1 K/ps. Once heating is complete, the system is equilibrated for 2 ns in an isobaric–isothermal (NPT) ensemble at 300 K and 1 atm, respectively. After equilibration, three 10 ns runs were performed with a time step of 2 fs using the NPT ensemble.
For the metadynamics simulation, the equilibrated postrigor structure was the initial structure, while the equilibrated pre-powerstroke structure was the target structure. The most important criterion in a metadynamics simulation is to choose an appropriate collective variable, which can be defined as the slow relevant degrees of freedom able to clearly distinguish the nature of states of interest. We have used the projection of the difference vector between the two coordinate systems as the collective variable for our study. This collective variable was able to sample and clearly differentiate between the defined initial and the final conformations of the recovery stroke. The collective variable was also seen to be robust enough to predict the similar free-energy trend observed in the experimental results for Dictyostelium myosin II (wild type and mutants) in the previous metadynamics study carried out by our group,26 and hence, we have extended the approach to our system as well.
The projection of the difference vector can be defined as the projection of the deviation of the group of atoms from their reference coordinates at any time onto the initial deviation of the same and is expressed as26
where xref and xcog define the coordinates for the reference and the center of geometries of the group of atoms, respectively, U is the optimal rotation matrix, which allows the collective variable to be constant with respect to rotations and translations, and x’ is another set of variables. Our chosen collective variable is analogous to the root-mean-square deviation (RMSD) between two sets of coordinates but does not have a boundary at 0 and hence can have negative values and prevents “hard” boundaries as well. A normalized p reflects the difference between two coordinate states (xref and x’). When the difference between the two states is maximum, p gives a value of 1, while when the two states overlap, p returns a value of 0. The collective variable is supported in NAMD through the collective variable module.32 In our case, postrigor structure is designated by the value 1, while the value 0 represents the pre-powerstroke structure. We also imposed minor constraints on the protein to avoid undesirable translational and rotational degrees of freedom. In conventional metadynamics, the potential of mean force does not converge asymptotically but oscillates about the mean,33 and hence, convergence criteria must be specified. For this study, we observed the progress of the collective variable along the range from 0 to 1 through histogram distributions and convergence was determined when the distribution was its flattest.
All of the metadynamics simulations were performed using the collective variable module associated with NAMD.32
2.2. Simulation Design for ATP Hydrolysis.
For this simulation, we began with the preparation of structure for the pre-powerstroke state of myosin with ATP + MG bound, choosing the first 800 amino acid residues following the same procedure as described previously. Since this part of the study focuses on the chemical step, ATP along with its surrounding protein environment was treated quantum mechanically, while the remaining portion was treated classically according to the CHARMM36 force field. The quantum mechanical/molecular mechanical (QM/MM) regions were partitioned according to the previous investigation on ATP hydrolysis within Dictyostelium myosin II.21,26 The sequentially similar residues for human cardiac β myosin were chosen as the active site residues, which included residues listed as SER180, THR185, ASN238, SER241, SER242, ARG243, and GLU466. Additionally, four water molecules along with the magnesium ion and the triphosphate chain of ATP were included in the QM region (Figure 1). The residues present in the QM region are linked with the classical region through α carbon atoms designated as boundary atoms and are treated with the generalized hybrid orbital (GHO) method.34 The PM3 semiempirical method was implemented in the CHARMM program to treat the QM atoms. After solvation, the system was neutralized with potassium and chloride ions. The system was then minimized initially with 50 steps of steepest descent (SD) and followed by 200 steps with the adopted basis Newton–Raphson (ABNR) algorithm with 10 kcal/mol constraints being exerted on the QM region. After minimization, the system was heated to 300 K with temperature steps of 5 K every 100 steps and the cycle being repeated with decreasing constraints on the QM region and then an unconstrained equilibration was done at 300 K for 20 ps. The active site residues were treated with quantum mechanics throughout all of the three processes.
Figure 1.
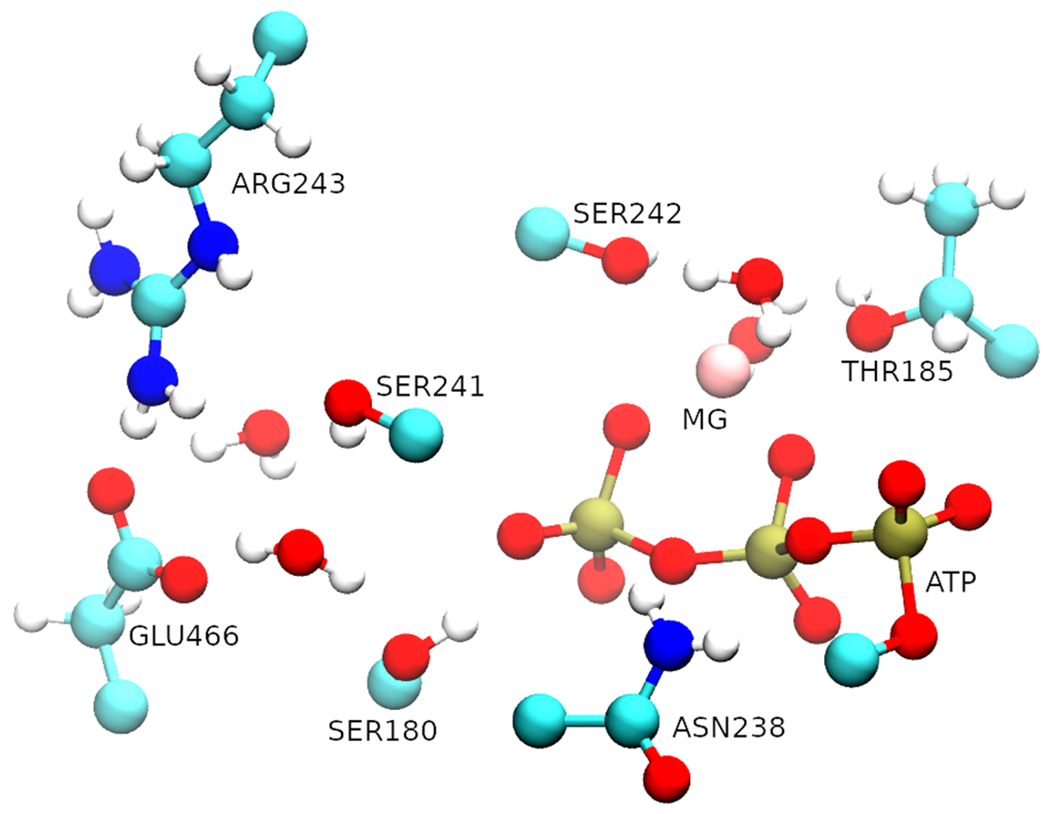
Representation of the QM region. The triphosphate chain (red and gold) of the ATP along with the magnesium ion and four water molecules were treated quantum mechanically as well.
The equilibrated structure was then studied with transition path sampling (TPS)25 to generate statistically unbiased trajectories. Order parameters were assigned to distinguish between the reactant myosin with the ATP moiety and the product state myosin with ADP and phosphate. The three order parameters established were (1) the distance between the γ oxygen and the γ phosphate of ATP, (2) the distance between the γ phosphate with the oxygen of the attacking water, and finally (3) the hydrogen of the attacking water and oxygen of the glutamate. The distances specified for reactant and product states were, for the first two parameters, less than and greater than 2.0 Å, respectively, and the distance specified for the third parameter was greater than 1.1 Å in the reactant state, while the opposite was true for the product state. TPS trajectories were run for 250 fs, which was sufficient to allow barrier crossing. An ensemble of 150 trajectories was created, with the initial 70 trajectories being excluded from the analysis to allow decorrelation from the initial constrained trajectory.
After creating an ensemble of reactive trajectories, committor analysis was performed to generate a transition-state ensemble and to define the stochastic separatrix. Committor values were determined by picking a slice within a trajectory and creating further trajectories from this initial point, each with random momenta. From this, one determines the probability of whether a certain configuration is committed to the reactant or product states. The slices with 50% commitment toward the product or the reactant state were identified as the transition states. After the stochastic separatrix was defined, we identify the reaction coordinate defined as the degrees of freedom responsible for the barrier crossing, which are orthogonal to the stochastic separatrix.35
3. RESULTS AND DISCUSSION
3.1. Effect of the Perturbations on the Recovery Stroke.
We use the method of metadynamics to sample the underlying potential energy surface for the transition of postrigor to pre-powerstroke state of myosin to calculate the overall free-energy change and model how the mutations R453C, I457T, and I467T might perturb the conformational change. Similar to the previous study of free-energy profile of the recovery stroke in Dictyostelium myosin II,26 we defined a unique collective variable for each subdomain of myosin: N-terminal (residues 1–129), U50 (residues 130–460), relay-helix (residues 461–505), L50 (residues 506–710), and converter (711–782). The five, one-dimensional free-energy contributions were determined simultaneously, and the combination of all of them depicts the overall free-energy surface, as shown in Figure 2.
Figure 2.
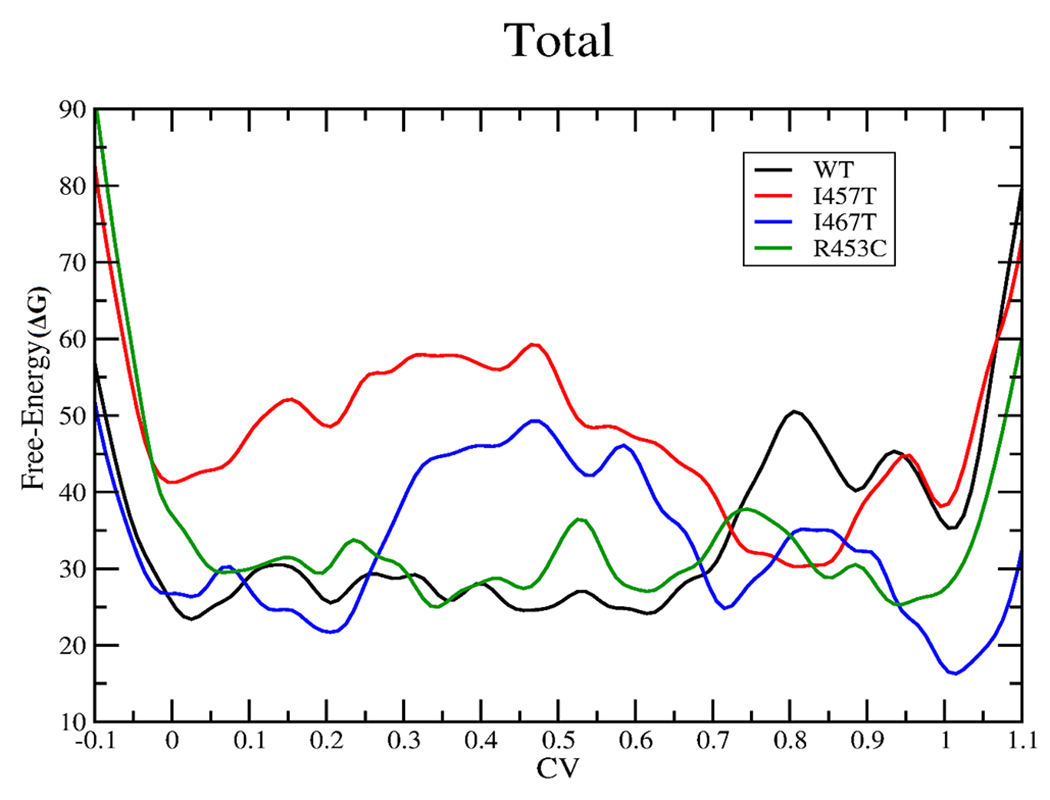
Total free-energy change (kcal/mol) for the recovery stroke in the case of wild type (black), I457T (red), I467T (blue), and R453C (green). The graph should be read from right to left.
ARG453 is found in the helix O linker, and residue ILE457 is in the transducer region of the U50 subdomain, while the residue ILE467 is found in the switch II loop in the relay-helix subdomain. All of the three residues directly interact with the nucleotide-binding pocket, and hence, mutating these three residues is expected to affect the recovery stroke. For the mutations I457T and I467T, a polar residue is placed in a highly hydrophobic region, whereas for R453C, the replacement of a charged residue by a noncharged residue takes place.
From the free-energy profile (Figure 2) for the wild type, we observe that the transition from the postrigor state (CV value 1) to the pre-powerstroke state (CV value 0) for the wild type is fairly smooth after the initial move away from the postrigor structure (0.7–1), with a well at around the pre-powerstroke state predicting a spontaneous transition.
In the case of the R453C mutation, replacement with cysteine leads to the loss of charge and loss of interactions mainly with Asp262, Ala259, Glu448, and Thr449 in the U50 subdomain of the myosin due to smaller side chain of cysteine, as observed from the molecular dynamics simulations. The free-energy plot for R453C (Figure 2) shows that there is no significant free-energy difference for the initial (CV value 1) and the final (CV value 0) states, while we do observe a notable stabilization of the postrigor state (CV value 1) with respect to the postrigor state of the wild type. This overall stabilization of the postrigor state can be attributed to the observed large stabilization of the postrigor state for R453C in the free-energy profiles of the U50, N-terminal, and the converter subdomains, all with respect to that of the wild type (Figure S1). Hence, comparing the free-energy profiles of R453C mutation and wild type, we may predict an increase in the free-energy difference and a subsequent decrease in the equilibrium constant for the transition of the postrigor state to the pre-powerstroke state for the R453C mutation than the wild type (Figure 2).
The free-energy profile (Figure 2) for R453C also suggests smaller energy barriers for the transition between the two conformations with respect to the free-energy barrier observed for that of the wild type. Thus, our results predict rapid interconversion between the postrigor and the pre-powerstroke states for R453C mutation when compared to that of the wild type. The experimental result for R453C suggested an ~3-fold decrease in the rate of the recovery stroke,36 which may be associated with a very little increase in the free-energy barrier (~0.65 kcal/mol) for the recovery stroke with respect to the wild type. Therefore, our results for the R453C mutation do not agree with the current experimental results, but the ~3-fold decrease may be beyond the accuracy of this calculation.
For the I467T mutation, replacement of isoleucine by threonine leads to the disruption of the hydrophobic interactions mainly with Gly584, Asp587, Lys572, and Ile585. In the case of the pre-powerstroke state, only an additional hydrogen bonding was observed between Arg237 and Thr467 in the molecular dynamics simulations. From the free-energy plot for I467T (Figure 2), we observe that the postrigor state (CV value 1) is highly stabilized with respect to the wild type. In the case of I467T, we also observe that all of the subdomains of the postrigor state (Figure S1) are fairly stabilized with respect to that of the wild type and hence contribute to the overall large stabilization of the same. Hence, we can see a significant increase in the free-energy difference for the transition from the postrigor to the pre-powerstroke state for I467T in comparison to that of the wild type, which can further speculate a reduced equilibrium constant for the same (Figure 2).
The free-energy plot for I467T (Figure 2) also predicts a much slower conversion of the postrigor to the pre-powerstroke state, compared to that of the wild type as a much larger free-energy barrier (CV value 0.2–0.7) is observed in the case of former. The minima at a CV value of 0.2 and 0.7 can be assigned to intermediate structures neither resembling the pre-powerstroke nor the postrigor conformation completely. Thus, we may speculate a decrease in the rate of the recovery stroke for the I467T mutation in comparison to that of the wild type.
In the case of the I457T mutation,37 replacement of the hydrophobic residue with a polar residue leads to the disruption of the extensive hydrophobic pocket and hence loss of hydrophobic interactions with Met165, Ala161, Ile174, Val191, Phe195, and Val459, as observed from the molecular dynamics simulations. For the I457T mutation, the free-energy profile (Figure 2) predicts a negligible free-energy difference between the initial (CV value 1) and the final states (CV value 0), which may lead to a nearly equal number of myosin heads with the postrigor and the pre-powerstroke configurations. However, in comparison to the wild type, we can observe that the I457T mutation highly destabilizes the pre-powerstroke state (CV value 0) leading to the speculation that the postrigor state is more favorable for the latter. Thus, comparing the free-energy profiles of the I457T mutation and the wild type, we may predict a lower equilibrium constant for the recovery stroke step for the former than for the latter (Figure 2).
For the I457T mutation, we also observe a large free-energy barrier (CV value 0–0.8) between the postrigor and the pre-powerstroke states compared to that of the wild type (Figure 2). The deep well at around CV value 0.8 cannot be designated as either of the known configurations as observed from the metadynamics trajectories. It is observed to be an intermediate structure formed during the transition, where the N-terminal and the relay-helix domain resemble that of the postrigor conformation but the lever arm in the converter domain is primed up similar to that of the pre-powerstroke configuration. Though the free-energy plot speculates a rapid conversion from the initial state at CV value 1 to the intermediate state at 0.8 but owing to the large free-energy barrier observed in between CV values 0.8 and 0, we can predict an overall decrease in the rate of the recovery stroke compared to that of the wild type.
There are currently no experimental results on how the mutations I457T and I467T perturb the recovery stroke. We can observe additional wells other than the minima observed at the initial and final states on the free-energy surface for the wild type as well as the mutant simulations such as the well observed around the CV value 0.85 for the wild type and the wells observed around CV values 0.2 and 0.7 for I467T and the deep well at around CV value 0.8 for the I457T simulation. The path we have chosen has the CV defined as a generalized reaction coordinate and hence cannot be treated as the ideal path. Thus, though these wells are physiological in nature, we cannot determine whether they can be considered as dominant populations.
3.2. ATP Hydrolysis: Formation of HPO42−.
Previously, the transition path sampling study of the decomposition of ATP to ADP and phosphate for Dictyostelium myosin II26 observed that the proton from the attacking water was abstracted by the glutamate to position the newly formed hydroxide for a nucleophilic attack on the γ phosphate of ATP. We used the same order parameters to create our initial biased trajectory and created an ensemble of unbiased trajectories from it. After examining the ensemble of trajectories, it is evident that the mechanism is similar for human cardiac β myosin as well. The bond-breaking and bond-forming events from a representative trajectory are shown in Figure 3.
Figure 3.
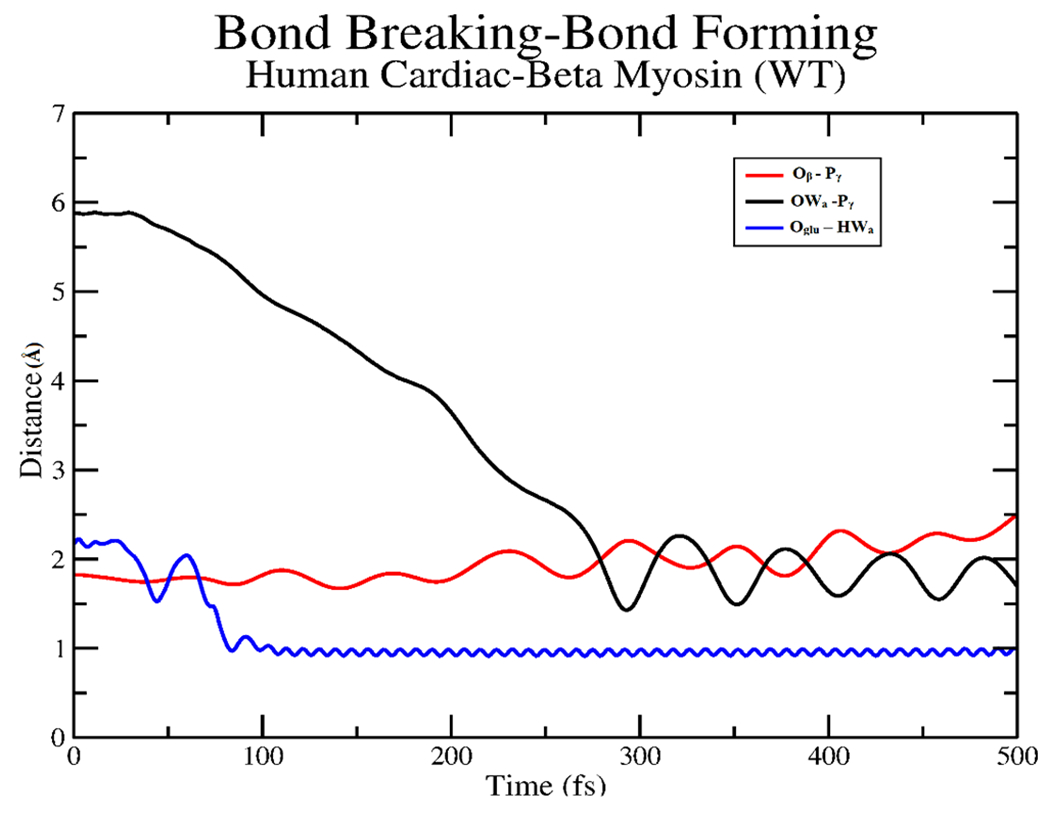
Plot of atomic distances for the γ phosphate (Pγ), β oxygen (Oβ) bond of ATP along with the deprotonation of the attacking water (HWa) by GLU466 (Oglu) and subsequent attack of the newly formed hydroxide (OWa) to the γ phosphate from a representative trajectory.
At first, GLU466 abstracted the proton from the attacking water, while the phosphoanhydride bond of ATP fluctuated until the bond broke around 200 fs. After the formation of ADP + PO3−, the attacking hydroxide comes in and forms the HPO42− intermediate at around 280 fs. Thus, the bond-breaking event takes place before bond formation, which is like that observed in the previous studies with Dictyostelium myosin II20,21,26 The transition states that are determined from these trajectories exhibit the stabilization of the metaphosphate intermediate. The mechanistic summary of the formation of HPO42− observed throughout the ensemble of trajectories is shown in Figure 4.
Figure 4.
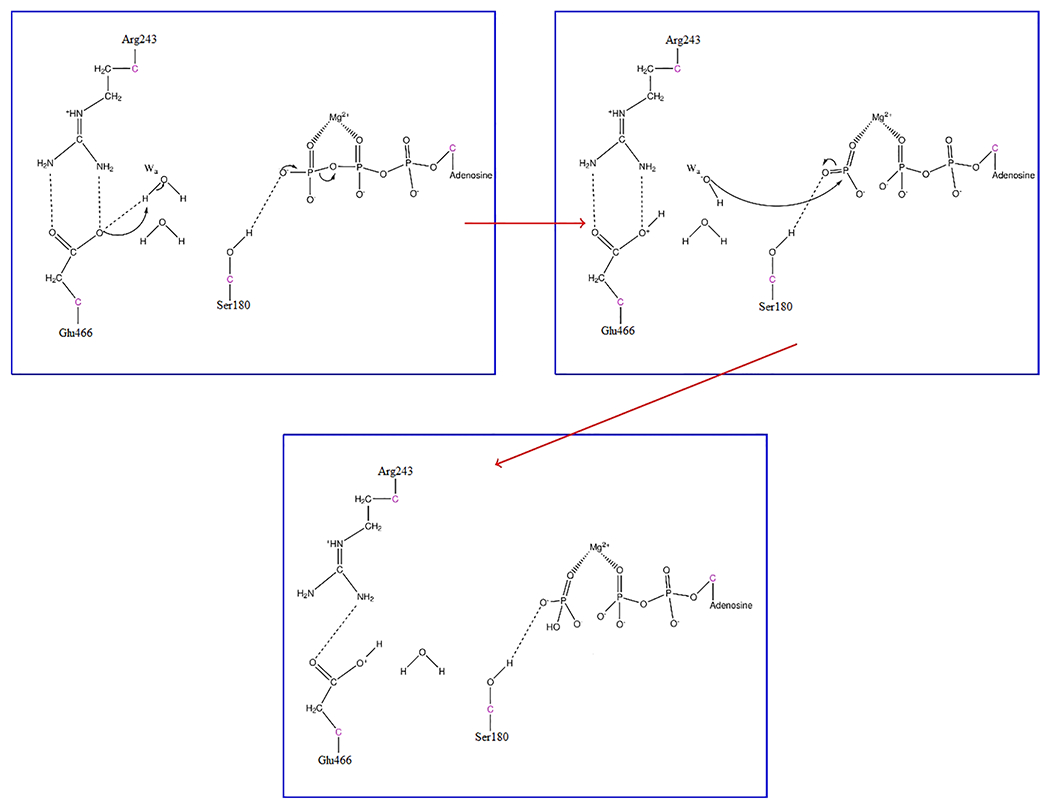
Mechanistic view of the formation of HPO42− in human cardiac β myosin. Hydrogen bonds are shown as dashed lines to demonstrate protein–substrate interactions.
The residues SER180, SER242 (classical portion), LYS184, and GLY464 are involved in stabilizing the PO3− moiety after the bond-breaking and before the hydroxide attack, as shown in Figure 5. We determined a total of four transition states having very little variation for the QM region.
Figure 5.
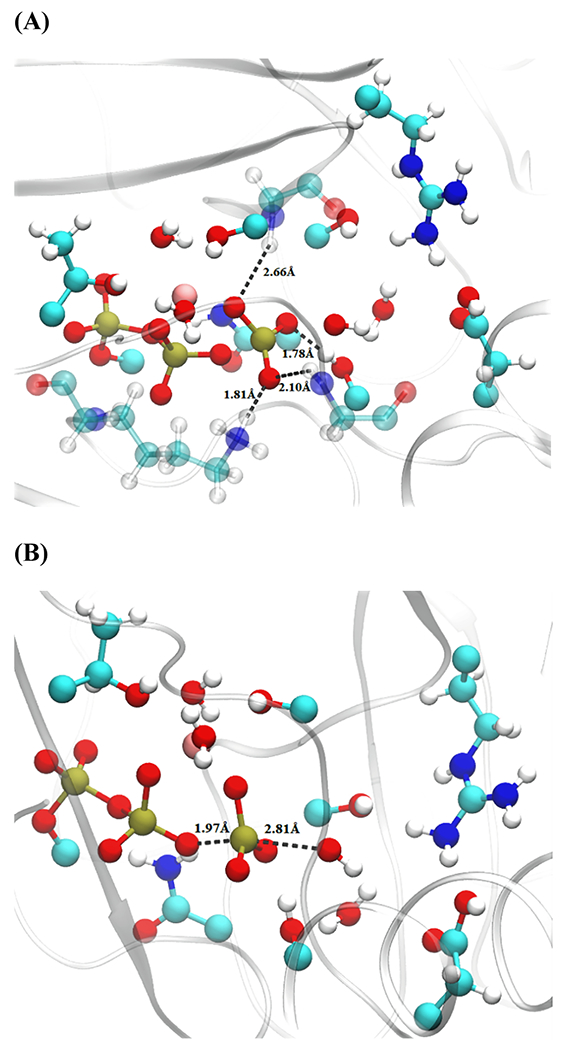
Transition-state distances from a representative structure of wild-type human cardiac β myosin. (A) Residues stabilizing PO3− intermediate and (B) attacking OH−–Pγ and Oβ–Pγ distances. QM-treated residues as well as the additional classical residues (transparent) GLY464, LYS184, and SER242 are shown with a ball and stick representation.
To determine the minimum degrees of freedom associated with the barrier crossing event, we generated constrained ensembles. The first constrained trajectory had only the QM region fixed, while another constrained trajectory was created with the QM region as well as the classical residues GLY464 and LYS184 frozen. The first ensemble showed a lot of committance to the reactant state, while the committor distribution plot of the second ensemble peaked at 0.5, as shown in Figure 6. Thus, the residues included in the QM region were not enough to remain on the stochastic separatrix, and this determined that the degrees of freedom correlated with transition from the reactant to product regions are mainly the QM residues augmented with the classical residues LYS184 and GLY464. Thus, electrostatic stabilization has a large contribution to the barrier crossing events and hence the positions of the active site residues are essential for the ATP hydrolysis mechanism.
Figure 6.
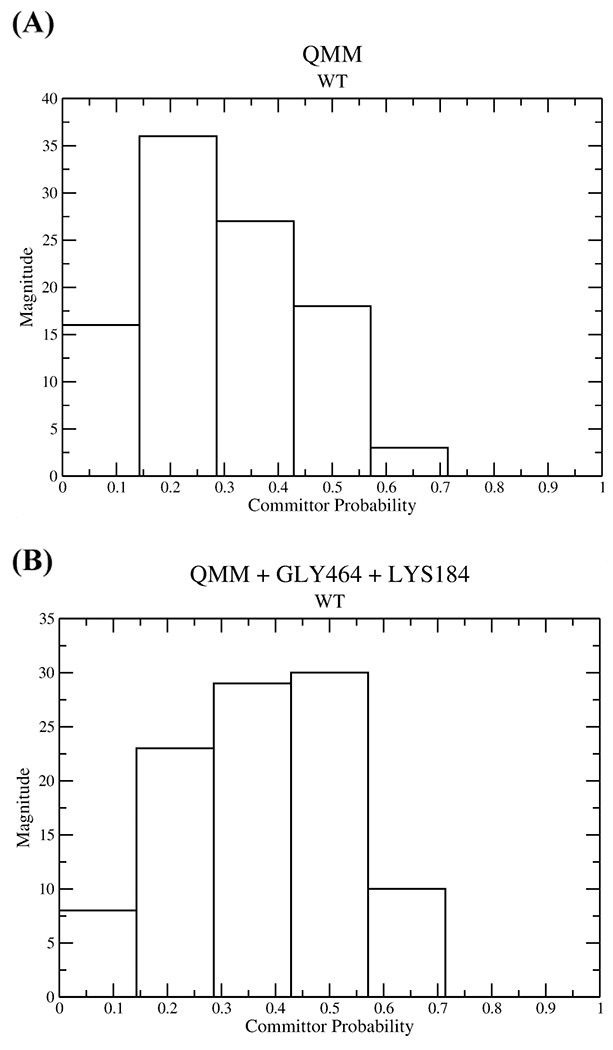
Committor distributions of constrained ensembles for wild-type human cardiac β myosin in which (A) only the QM region was constrained and (B) the classical residues LYS184 and GLY464 along with the entire QM region were constrained.
3.3. Isoformic Differences between Dictyostelium Myosin II and Human Cardiac β Myosin.
Our study on the human cardiac β myosin revealed a similar mechanism for the decomposition of ATP to ADP and phosphate to that observed during the TPS study conducted on the Dictyostelium myosin II. The bond-breaking and bond-formation plot for the human cardiac β myosin (Figure 3) when compared with that observed in the case of the dictyostelium myosin II26 showed that after the bond-breaking event, the Oβ–Pγ distance is much greater in the latter case than in the former. Hence, one may infer that there is a higher chance of the process being reversible in the case of human cardiac β myosin than that of Dictyostelium myosin II, as the phosphate remains more closer to ADP. This fits with the fact that the human cardiac β myosin is considered as a slow myosin. Moreover, residues stabilizing the metaphosphate intermediate for the Dictyostelium myosin II are SER181, SER236, SER237, and GLY457 (classical residues),26 while those involved in the case of the human cardiac β myosin are SER180 (QM residue), Ser242, GLY464, and LYS184 (Figure 5), and hence, comparing, we observe the participation of more classical residues in stabilizing the metaphosphate intermediate in the case of the latter. Also, we observed a strong involvement of the lysine NH3+ with the γ phosphate group in the case of the human isoform, which added to the stability of the PO3− moiety for the same.38 For the Dictyostelium myosin II, it has been observed that the degrees of freedom involved in the barrier crossing event are those associated with the entire QM region,26 while those responsible in the case of human cardiac β myosin are those associated with the entire QM region along with the classical residues LYS184 and GLY464 (Figure 6). Thus, the degrees of freedom involved in the case of the former are different from those of the latter as well as the number of degrees of freedom for the Dictyostelium myosin II are less than those of the human cardiac β myosin.
Due to more reversibility of the reaction as well as due to the more degrees of freedom involved for the barrier crossing event, we can make a theoretical prediction that there is lower probability of ATP hydrolysis from occurring and hence one might observe a lower activity in the case of human cardiac β myosin.
There have been experimental studies on the ATP hydrolysis step of the Dictyostelium myosin II, which shows that the basal ATPase rate for the wild type is ~0.05 s−1,13 while the apparent rate constant of the ATP hydrolysis is ~32 s−1.13 There are also well-defined experimental studies for the wild-type human cardiac β myosin, which suggest that the rate constant for the ATP hydrolysis step is around 14 s−1.36,39 Also, a recent study on the cardiac inhibitor mavacamten promoting the formation of sequestered, super relaxed state of myosin (SRX) has shown that the wild-type human β cardiac myosins in three different constructs, 25-heptad heavy meromyosin (HMM), 2-heptad heavy meromyosin (HMM), and short subfragment-1 (S1), have basal ATPase rates ranging between 0.01 and 0.03 s−1.40
Our study does not aim to predict the rates of the ATP hydrolysis step, but we may infer that the observed variations in the chemical step may at least be partially responsible for the differences in the ATPase activity of the Dictyostelium myosin II and the human cardiac β isoform.
3.4. Effect of the Mutation I467T on the Decomposition of ATP to ADP and Phosphate.
An initial constrained trajectory was created for the mutated myosin using the same order parameters used to create that of the wild-type myosin. The ensemble of trajectories revealed that the mechanism for the breakdown of ATP is like that observed in the wild type. The bond-breaking and bond-forming events from a representative trajectory are shown in Figure S2. Initially, GLU466 abstracted the proton from the attacking water, while the phosphoanhydride bond of ATP broke around 165 fs. Then, the formation of the HPO42− intermediate takes place at around 280 fs. The transition states obtained from these trajectories determined that the residues SER241, SER242, QM water (hydrating water of magnesium), and LYS184 are involved in stabilizing the PO3− in a trigonal planar conformation, as shown in Figure 7.
Figure 7.
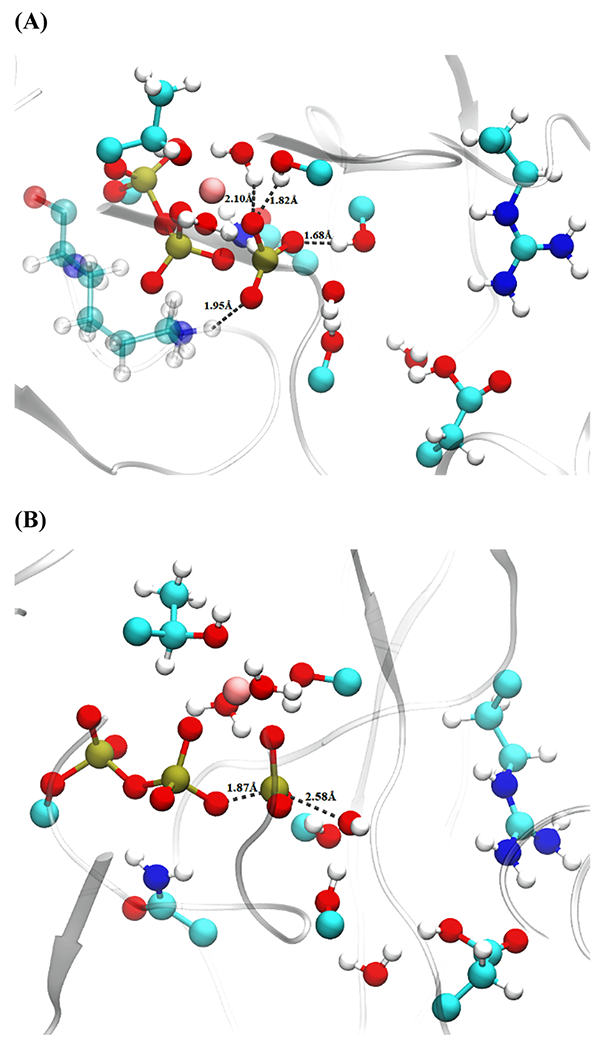
Transition-state distances from a representative structure of the mutated myosin. (A) Residues stabilizing PO3− intermediate and (B) attacking OH−–Pγ and Oβ–Pγ distances. QM-treated residues as well as an additional classical residue LYS184 (transparent) are shown with a ball and stick representation.
Thus, it is seen that the residues observed to stabilize the metaphosphate intermediate in the case of the mutated structure are different from those of the wild type and there are more classical residues involved in the case of the latter.
We tested reaction coordinates with two sets of constrained coordinates: the entire QM region constrained and a second ensemble with the entire QM region along with the classical residue LYS184 fixed. While the first ensemble had a shifted peak toward the product, the second ensemble had the histograms largely weighting around 0.5, as shown in Figure 8. Therefore, the reaction coordinates for I467T are the residues associated with the entire QM region and an additional classical residue LYS184. These residues are also involved in stabilizing the PO3− moiety as seen earlier. Thus, the number of degrees of freedom required in the case of I467T is less than that observed in the case of the wild type. Given that the precise placement of fewer degrees of freedom is necessary for barrier passage in the mutant myosin, we may hypothesize that it is possible that the chemical step in this system is actually faster than that of the wild type.
Figure 8.
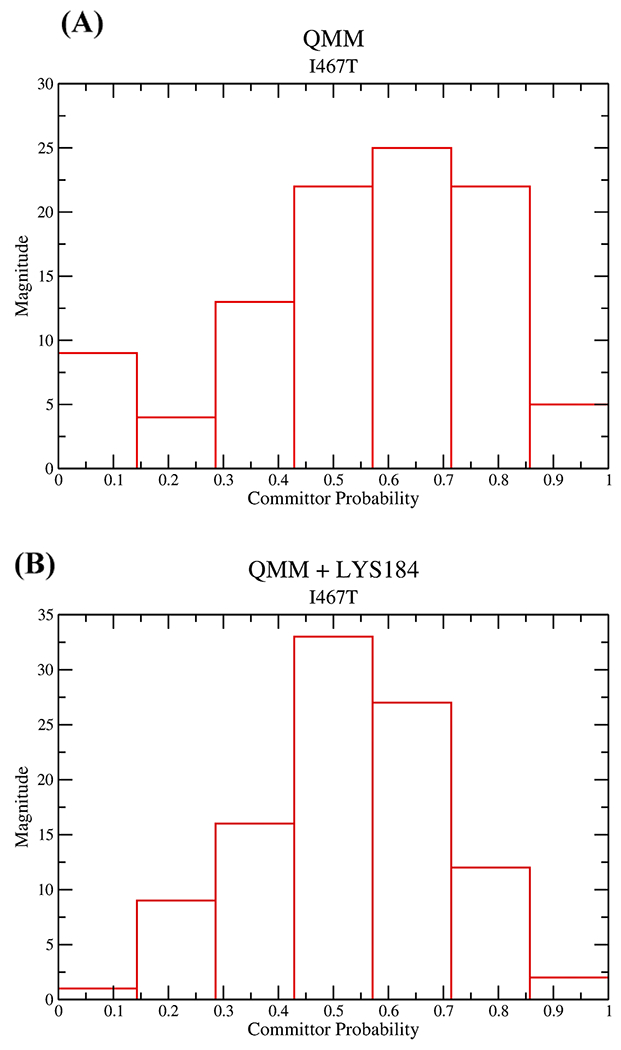
Committor distribution plot for mutated myosin, where (A) the entire QM region was constrained and (B) the entire QM region and LYS184 are constrained.
4. CONCLUSIONS
We have used the enhanced sampling methods, metadynamics, and transition path sampling to sample rare events such as conformational rearrangement and the chemical event, respectively, in both wild type and a mutant human β myosin. All of the three mutations R453C, I457T, and I467T are observed to affect the recovery stroke step. Though R453C and I467T mainly stabilized the initial postrigor state, the I457T mutation highly destabilized the final pre-powerstroke state, predicting an increase in the free-energy difference and a reduction in the equilibrium constant between the initial and the final states compared to that of the wild type for all of the three mutants. Furthermore, the mutation R453C decreased the free-energy barrier for the transition of the postrigor state to the pre-powerstroke state, while the mutations I457T and I467T increased the free-energy barrier for the same, all with respect to the wild type. It will be interesting to further extend this approach in theoretically observing the effect of a small therapeutic drug such as omecamtiv mecarbil27,28,41 on the free-energy profile of the wild type as well as the three mutants, as this drug is known to stabilize the pre-powerstroke state during the recovery stroke. In the case of the chemical event, TPS was used to generate statistically unbiased trajectories of the breakdown of the ATP to ADP and HPO42− through the stabilization of PO3− intermediate. We observed the isoformic differences between the Dictyostelium myosin II26 and the human cardiac β myosin as well as the effect of the mutation I467T on the transition states and the reaction coordinate coupled with the chemical step. These methods can be further augmented to other transitions involved in the mechanochemical cycle such as the powerstroke and present a complete computational picture of the effect of mutations on human cardiac β myosin.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the National Institute of Health Grant R01HL107046 to J.C.T. and S.D.S. All computer simulations were performed on a Lenovo NeXtScale nx360 M5 supercomputer at the University of Arizona High Performance Computing Center.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.1c03144.
Contributions of each of the individual subdomains to the free-energy profile of the recovery stroke and bond-breaking bond-forming versus time plot for the formation of HPO42− for I467T mutation (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jpcb.1c03144
The authors declare no competing financial interest.
Contributor Information
Ananya Chakraborti, Department of Chemistry and Biochemistry, University of Arizona, Tucson, Arizona 85721, United States.
Anthony P. Baldo, Department of Chemistry and Biochemistry, University of Arizona, Tucson, Arizona 85721, United States
Jil C. Tardiff, Department of Biomedical Engineering, University of Arizona, Tucson, Arizona 85724, United States
Steven D. Schwartz, Department of Chemistry and Biochemistry, University of Arizona, Tucson, Arizona 85721, United States.
REFERENCES
- (1).Hartman MA; Spudich JA The Myosin Superfamily at a Glance. J. Cell Sci 2012, 125, 1627–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kolomeisky AB Motor Proteins and Molecular Motors: How to Operate Machines at the Nanoscale. J. Phys.: Condens. Matter 2013, 25, 463101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Sweeney HL; Holzbaur ELF Motor Proteins. Cold Spring Harbor Perspect. Biol. 2018, 10, a021931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).England J; Loughna S Heavy and Light Roles: Myosin in the Morphogenesis of the Heart. Cell. Mol. Life Sci. 2013, 70, 1221–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Rayment I; Rypniewski W; Schmidt-Bäse K; Smith R; Tomchick DR; Benning MM; Winkelmann DA; Wesenberg G; Holden HM Three-Dimensional Structure of Myosin Subfragment-1: A Molecular Motor. Science 1993, 261, 50–58. [DOI] [PubMed] [Google Scholar]
- (6).Lymn RW; Taylor EW Mechanism of Adenosine Triphosphate Hydrolysis by Actomyosin. Biochemistry 1971, 10, 4617–4624. [DOI] [PubMed] [Google Scholar]
- (7).Sweeney HL; Houdusse A Structural and Functional Insights into the Myosin Motor Mechanism. Annu. Rev. Biophys. 2010, 39, 539–557. [DOI] [PubMed] [Google Scholar]
- (8).Houdusse A; Sweeney HL How Myosin Generates Force on Actin Filaments. Trends Biochem. Sci. 2016, 41, 989–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Geeves MA; Holmes KC The Molecular Mechanism of Muscle Contraction. Adv. Protein Chem. 2005, 71, 161–193. [DOI] [PubMed] [Google Scholar]
- (10).Koppole S; Smith JC; Fischer S Simulations of the Myosin II Motor Reveal a Nucleotide-State Sensing Element That Controls the Recovery Stroke. J. Mol. Biol. 2006, 361, 604–616. [DOI] [PubMed] [Google Scholar]
- (11).Koppole S; Smith JC; Fischer S The Structural Coupling between ATPase Activation and Recovery Stroke in the Myosin II Motor. Structure 2007, 15, 825–837. [DOI] [PubMed] [Google Scholar]
- (12).Mesentean S; Koppole S; Smith JC; Fischer S The Principal Motions Involved in the Coupling Mechanism of the Recovery Stroke of the Myosin Motor. J. Mol. Biol. 2007, 367, 591–602. [DOI] [PubMed] [Google Scholar]
- (13).Málnási-Csizmadia A; Tóth J; Pearson DS; Hetényi C; Nyitray L; Geeves MA; Bagshaw CR; Kovács M Selective Perturbation of the Myosin Recovery Stroke by Point Mutations at the Base of the Lever Arm Affects ATP Hydrolysis and Phosphate Release. J. Biol. Chem. 2007, 282, 17658–17664. [DOI] [PubMed] [Google Scholar]
- (14).Nagy NT; Chakraborty S; Harami GM; Sellers JR; Sakamoto T; Kovács M A Subdomain Interaction at the Base of the Lever Allosterically Tunes the Mechanochemical Mechanism of Myosin 5a. PLoS One 2013, 8, e62640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bloemink MJ; Melkani GC; Bernstein SI; Geeves MA The Relay/Converter Interface Influences Hydrolysis of ATP by Skeletal Muscle Myosin II. J. Biol. Chem. 2016, 291, 1763–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Okimoto N; Yamanaka K; Ueno J; Hata M; Hoshino T; Tsuda M Theoretical Studies of the ATP Hydrolysis Mechanism of Myosin. Biophys. J. 2001, 81, 2786–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Li G; Cui Q Mechanochemical Coupling in Myosin: A Theoretical Analysis with Molecular Dynamics and Combined QM/MM Reaction Path Calculations. J. Phys. Chem. B 2004, 108, 3342–3357. [Google Scholar]
- (18).Grigorenko BL; Kaliman IA; Nemukhin AV Minimum Energy Reaction Profiles for ATP Hydrolysis in Myosin. J. Mol. Graphics Model. 2011, 31, 1–4. [DOI] [PubMed] [Google Scholar]
- (19).Kiani FA; Fischer S Catalytic Strategy Used by the Myosin Motor to Hydrolyze ATP. Proc. Natl. Acad. Sci U.S.A. 2014, 111, E2947–E2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lu X; Ovchinnikov V; Demapan D; Roston D; Cui Q Regulation and Plasticity of Catalysis in Enzymes: Insights from Analysis of Mechanochemical Coupling in Myosin. Biochemistry 2017, 56, 1482–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kiani FA; Fischer S Advances in Quantum Simulations of ATPase Catalysis in the Myosin Motor. Curr. Opin. Struct. Biol. 2015, 31, 115–123. [DOI] [PubMed] [Google Scholar]
- (22).Laio A; Parrinello M Escaping Free-Energy Minima. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 12562–12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Laio A; Gervasio FL Metadynamics: A Method to Simulate Rare Events and Reconstruct the Free-Energy in Biophysics, Chemistry and Material Science. Rep. Prog. Phys. 2008, 71, 126601. [Google Scholar]
- (24).Barducci A; Bonomi M; Parrinello M Metadynamics. Wiley Interdiscip Rev.: Comput. Mol. Sci 2011, 1, 826–843. [Google Scholar]
- (25).Bolhuis PG; Dellago C Practical and Conceptual Path Sampling Issues. Eur. Phys. J.: Spec. Top 2015, 224, 2409–2427. [Google Scholar]
- (26).Baldo AP; Tardiff JC; Schwartz SD Mechanochemical Function of Myosin II: Investigation into the Recovery Stroke and ATP Hydrolysis. J. Phys. Chem. B 2020, 124, 10014–10023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Robert-Paganin J; Auguin D; Houdusse A Hypertrophic Cardiomyopathic Disease Results from Disparate Impairments of Cardiac Myosin Function and Auto-Inhibition. Nat. Commun. 2018, 9, 4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Planelles-Herrero VJ; Hartman JJ; Robert-Paganin J; Malik FI; Houdusse A Mechanistic and Structural Basis for Activation of Cardiac Myosin Force Production by Omecamtiv Mecarbil. Nat. Commun. 2017, 8, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Schwede T; Kopp J; Guex N; Peitsch MC SWISS-MODEL: An Automated Protein Homology-Modeling Server. Nucleic Acids Res. 2003, 31, 3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Brooks BR; Brooks CL; Mackerell AD; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kalé L; Schulten K Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Fiorin G; Klein ML; Hénin J Using Collective Variables to Drive Molecular Dynamics Simulations. Mol. Phys. 2013, 111, 3345–3362. [Google Scholar]
- (33).Barducci A; Bussi G; Parrinello M Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603. [DOI] [PubMed] [Google Scholar]
- (34).Gao J; Amara P; Alhambra C; Field MJ A Generalized Hybrid Orbital (GHO) Method for the Treatment of Boundary Atoms in Combined QM/MM Calculations. J. Phys. Chem. A 1998, 102, 4714–4721. [Google Scholar]
- (35).Schwartz SD Protein Dynamics and the Enzymatic Reaction Coordinate. In Dynamics in Enzyme Catalysis. Topics in Current Chemistry; Klinman J; Hammes-Schiffer S, Eds.; Springer: Berlin, 2013; Vol. 337, pp 189–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bloemink M; Deacon J; Langer S; Vera C; Combs A; Leinwand LA; Geeves MA The Hypertrophic Cardiomyopathy Myosin Mutation R453C Alters ATP Binding and Hydrolysis of Human Cardiac β-Myosin. J. Biol. Chem. 2014, 289, 5158–5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Adhikari AS; Trivedi DV; Sarkar SS; Song D; Kooiker KB; Bernstein D; Spudich JA; Ruppel KM β-Cardiac Myosin Hypertrophic Cardiomyopathy Mutations Release Sequestered Heads and Increase Enzymatic Activity. Nat. Commun. 2019, 10, 2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kiani FA; Fischer S Stabilization of the ADP/ Metaphosphate Intermediate During ATP Hydrolysis in Pre-Powerstroke Myosin: Quantitative Anatomy of an Enzyme. J. Biol. Chem. 2013, 288, 35569–35580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Deacon JC; Bloemink MJ; Rezavandi H; Geeves MA; Leinwand LA Identification of Functional Differences between Recombinant Human α and β Cardiac Myosin Motors. Cell. Mol. Life Sci. 2012, 69, 2261–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Anderson RL; Trivedi DV; Sarkar SS; Henze M; Ma W; Gong H; Rogers CS; Gorham JM; Wong FL; Morck MM; et al. Deciphering the Super Relaxed State of Human β-Cardiac Myosin and the Mode of Action of Mavacamten from Myosin Molecules to Muscle Fibers. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, E8143–E8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Malik FI; Hartman JJ; Elias KA; Morgan BP; Rodriguez H; Brejc K; Anderson RL; Sueoka SH; Lee KH; Finer JT; et al. Cardiac Myosin Activation: A Potential Therapeutic Approach for Systolic Heart Failure. Science 2011, 331, 1439–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.