

Abstract
The identification of a gain-of-function mutation in CACNA1C as the cause of Timothy syndrome, a rare disorder characterized by cardiac arrhythmias and syndactyly, highlighted roles for the L-type voltage-gated Ca2+ channel CaV1.2 in nonexcitable cells. Previous studies in cells and animal models had suggested that several voltage-gated Ca2+ channels (VGCCs) regulated critical signaling events in various cell types that are not expected to support action potentials, but definitive data were lacking. VGCCs occupy a special position among ion channels, uniquely able to translate membrane excitability into the cytoplasmic Ca2+ changes that underlie the cellular responses to electrical activity. Yet how these channels function in cells not firing action potentials and what the consequences of their actions are in nonexcitable cells remain critical questions. The development of new animal and cellular models and the emergence of large data sets and unbiased genome screens have added to our understanding of the unanticipated roles for VGCCs in nonexcitable cells. Here, we review current knowledge of VGCC regulation and function in nonexcitable tissues and cells, with the goal of providing a platform for continued investigation.
Keywords: voltage-gated Ca2+ channel, nonexcitable cells, Timothy syndrome
1. INTRODUCTION
Voltage-gated Ca2+ channels (VGCCs) occupy a unique role among ion channels. Not only do their resultant Ca2+ fluxes shape action potentials, but the selective permeability of VGCCs to Ca2+ drives an increase in the cytoplasmic Ca2+ concentration that, exclusively among ions, triggers downstream signaling pathways. If not for VGCCs, membrane excitability serves a futile cycle: Ion fluxes through open channels change membrane potential and thereby control the opening and closing of other voltage-gated ion channels that subsequently affect membrane potential. Breaking this futile cycle is the entry of Ca2+ through VGCCs. The consequent activation of downstream Ca2+-dependent signaling pathways translates electrical activity at the membrane into intracellular actions, such as contraction of a myocyte, release of neurotransmitter, or secretion of a hormone from an endocrine cell. Their lofty position has earned VGCCs the moniker of queen among ion channels (1), and the expression of a VGCC has been considered the sine qua non of an excitable cell (2).
Until the discovery that a gain-of-function mutation in the CaV1.2 L-type VGCC gene CACNA1C caused Timothy syndrome (TS), a multisystem disorder characterized by life-threatening cardiac arrhythmias and fusion of digits, or syndactyly (3), studies of VGCCs focused mainly on excitable tissues such as muscle, brain, and endocrine organs, all of which feature action potentials, characterized by active depolarization and repolarization of membrane potential either spontaneously or in response to a stimulus. Roles for VGCCs in nonexcitable tissue, in which spontaneous or stimulated action potentials are absent, received limited attention, at least in part because there were no obvious mechanisms by which nonexcitable cells controlled membrane voltage to regulate VGCCs. While the TS-associated arrhythmias resulting from abnormal CaV1.2 function fit with the well-characterized functions of CaV1.2 in cardiac myocytes, perturbation of limb development was unexpected. Similarly, TS phenotypes such as craniofacial abnormalities and immunodeficiency pointed to previously unknown and unanticipated VGCC contributions in cells not thought to fire action potentials.
The recent development of new models and tools combined with motivation to explain the contributions of VGCCs in nonexcitable tissues are driving studies that reveal additional unexpected roles for VGCCs. Moreover, these studies are beginning to define the underlying molecular mechanisms by which VGCCs function in nonexcitable cells. Here, we provide the background for these unexpected functions and describe new progress with a goal of fostering continued acceleration of this exciting field.
1.1. Voltage-Gated Ca2+ Channels: Structure and Voltage-Dependent Function
VGCCs are transmembrane proteins with multiple subunits, each with distinct functions (4, 5). Ten homologous genes encode the different pore-forming CaV α1 subunits, which provide the critical distinguishing features among different VGCCs (6). Three of them (CACNA1A, CACNA1B, and CACNA1E), which encode the CaV2 family, are mainly restricted to neurons (excitable cells) and so are not a major focus of this review. CACNA1S and CACNA1F encoding the CaV1.1 and CaV1.4 α1 subunit, respectively, are expressed almost exclusively in a single cell type. CaV1.1 is the VGCC mediating excitation-contraction coupling in muscle, and CaV1.4 is expressed in the retina. Some reports place CaV1.1 or CaV1.4 in subsets of T lymphocytes, in which they are thought to control immune responses, as discussed below. In contrast, CACNA1C and CACNA1D, which encode the CaV1.2 and CaV1.3 α1 subunits, respectively, are broadly expressed and have been found reliably in nonexcitable cells. CaV1.1–CaV1.4 are members of the L-type subfamily. Finally, the CACNA1G, CACNA1H, and CACNA1I genes encode the T-type subfamily members CaV3.1, CaV3.2, and CaV3.3, respectively, which are also expressed in excitable and nonexcitable tissues.
The pore-forming CaV α1 subunits contain 24 transmembrane-spanning segments grouped in four homologous sets of six (DI–DIV) and arranged with a pseudo-fourfold symmetry (Figure 1a). Each set of six transmembrane-spanning segments resembles a potassium channel monomer, for which four subunits assemble as a homomeric or heteromeric tetramer (Figure 1b). Like other cation-selective voltage-gated channels, the first four transmembrane-spanning segments (S1–S4) of each set form a voltage sensor, while the S5 and S6 segments from each set form the ion pore. A cryogenic electron microscopy (cryo-EM) structure (Protein Data Bank: 5GJV) of CaV1.1 from rabbit skeletal muscle (7) shows a typical domain-swapped architecture, in which the voltage sensor domain from one pseudorepeat is next to the pore domain of its neighbor instead of its own pore domain (Figure 1c–e). In VGCCs, four aspartate residues, one from each of the S5–S6 domains (Figure 1f), provide the Ca2+ selectivity (8). VGCC CaV α1 subunits undergo extensive alternative splicing in a tissue-specific manner (9). Some reports suggest that splicing events in nonexcitable immune cells generate unconventional, truncated VGCCs that may be voltage independent (10, 11) and differ from the canonical architecture depicted in Figure 1. Functional assessment of those channels, however, has not reliably indicated that they support Ca2+ influx, and whether they exist in vivo is unclear.
Figure 1.
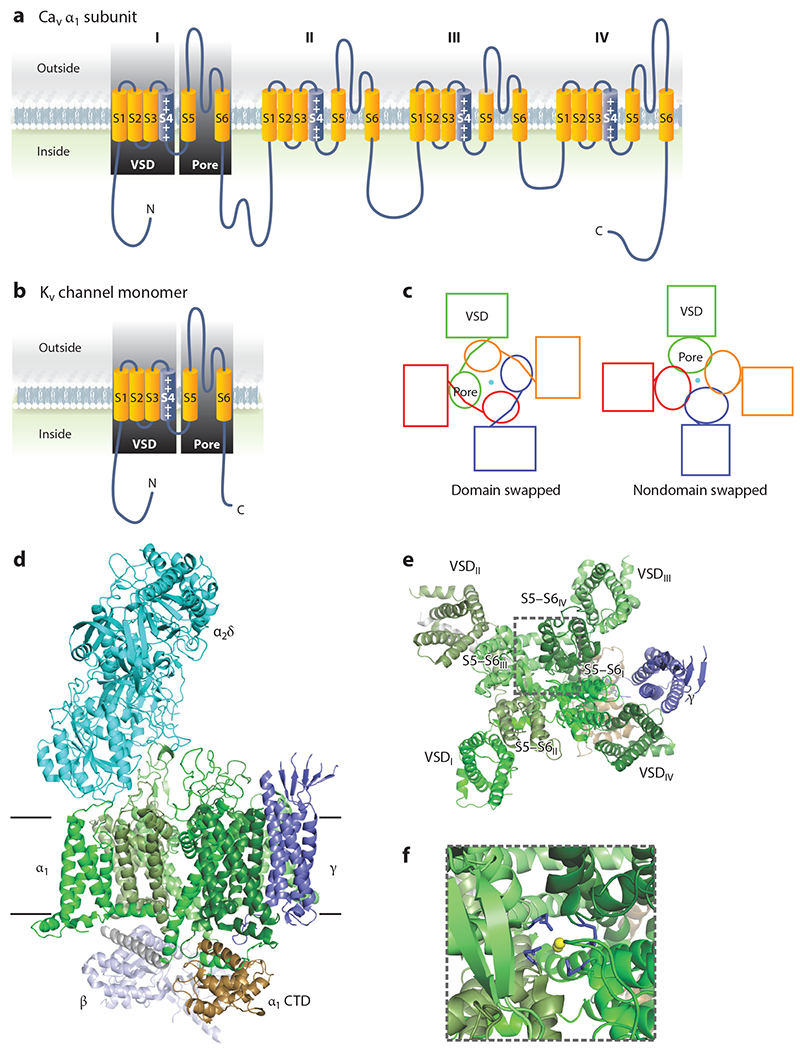
Voltage-gated Ca2+ channel structure. (a) Ribbon diagram of the pore-forming α1 subunit of a voltage-gated Ca2+ channel demonstrating the pseudo-fourfold symmetry and homology of each domain to a Kv channel monomer, as depicted in panel b. (c) Schematic of a domain-swapped architecture, shared by voltage-gated Ca2+ channels, and a nondomain-swapped architecture found in other cation channels for comparison. (d) Structure of rabbit CaV1.1 (Protein Data Bank: 5GVY) shown from the membrane. The pore-forming α1 subunit spans the membrane. The individual domains (DI–DIV) are colored in various shades of green, and the intracellular C-terminal domain (CTD) is brown. The extracellular α2δ subunit is turquoise, the γ subunit is dark purple, and the cytoplasmic β subunit is light purple. (e) View of the α1 and γ subunits from outside of the cell shows the domain-swapped architecture. For clarity, the α2δ subunit is not shown. (f) Zoomed image of the highlighted area in panel e, with the pore glutamate residues colored in purple and the Ca2+ ions in yellow. Abbreviation: VSD, voltage sensor domain.
Associating with the pore-forming CaV α1 subunits of L-type channels, but not with T-type channels, are several auxiliary subunits that regulate VGCC function (Figure 1d). An extracellular CaV α2δ subunit, attached to the plasma membrane by a glycosylphosphatidylinositol (GPI) anchor, enhances channel trafficking and tunes the voltage dependence of channel activation and inactivation (12). A cytoplasmic CaV β subunit binds to the linker between DI and DII. Like the CaV α2δ subunits, CaV β subunits tune channel activation and inactivation (13). In heterologous expression systems, CaV β subunits traffic α1 subunits to the plasma membrane (13) but are not absolutely required in vivo (14). In the heart, CaV β subunits cooperate with the cytosolic VGCC inhibitor Rad to increase Ca2+ influx through CaV1.2 in the “flight or fight” response mediated by β-adrenergic stimulation (14, 15). The Ca2+-binding protein calmodulin interacts with VGCC intracellular C termini to provide Ca2+-dependent feedback regulation (16), but it is not observed in Figure 1 because the cryo-EM structure (7) does not include the part of the C-terminal domain to which calmodulin binds. Finally, VGCCs in a few tissues contain a transmembrane spanning γ subunit that interacts with DIV (Figure 1d). There are multiple γ subunit homologous genes, but—although historically designated by name as Ca2+ channel γ subunits—most of the gene products neither interact with nor regulate VGCCs (17). Rather, they form a family of proteins that regulate the trafficking of AMPA receptors in neurons. Best characterized in terms of regulating VGCCs is γ1, which interacts with CaV1.1 in skeletal muscle and exerts an antagonist function by shifting the voltage dependence of channel inactivation (18), perhaps through its interaction with the DIV voltage sensor (7). Because of this ability to modulate the voltage-dependent behavior of VGCCs, the role of γ subunits in nonexcitable tissues is of potential interest, but there has been limited investigation.
2. VOLTAGE-GATED Ca2+ CHANNEL FUNCTIONS IN NONEXCITABLE CELLS AND TISSUES
2.1. Voltage-Gated Ca2+ Channels Function in Nonexcitable Cells
Attention to VGCCs in nonexcitable cells lagged the first studies of VGCCs in excitable cells that followed the classic demonstration by Fatt & Katz (19) that Ca2+ supported action potentials in muscle even in the absence of Na+. Initial studies defined roles for VGCCs in excitation-contraction coupling in muscle and synaptic transmission in neurons. Subsequently, the massive increase in intracellular Ca2+ ([Ca2+]i) that followed T lymphocyte activation prompted a search for Ca2+ entry pathways. A correlation between Ca2+ influx and changes in membrane potential initially suggested a role for VGCCs (20). Similarly, the inhibition of parathyroid hormone-stimulated [Ca2+]i increase in the rat osteosarcoma (ROS) 17/2.8 osteoblast-like osteosarcoma cell line by Ca2+ channel-blocking drugs (CCBs) implicated a VGCC influx pathway (21), and whole-cell voltage-clamp recordings from osteoblasts isolated from newborn rat calvaria demonstrated inward Ca2+ currents (22). Moreover, Bay K 8644, an L-type Ca2+ channel agonist, augmented 1,25(OH)2D3-stimulated osteocalcin secretion from ROS 17/2.8 cells and parathyroid hormone-stimulated resorption from fetal bones maintained in organ culture (23, 24) while 1,25(OH)2D3 shifted L-type VGCC Ca2+ current activation in ROS 17/2.8 cells to more negative potentials and increased single channel open time (25). These studies, relying on nonspecific pharmacological tools, predated molecular identification of the hypothesized VGCCs, so subsequent cloning of the L-type CaV1.2 gene provided the means to test for and reveal expression of the CaV1.2 in ROS 17/2.8 cells (26), offering necessary evidence that VGCCs may function in osteoblasts. Together, these studies formed a foundation for additional investigations of VGCCs in bone and the immune system, both of which are discussed separately below, but definitive evidence that VGCCs had physiological roles in nonexcitable cells in vivo was lacking.
2.2. Timothy Syndrome: In Vivo Evidence for Voltage-Gated Ca2+ Channels in Nonexcitable Tissues
Identification of a mutation in CACNA1C as the cause for TS provided the strongest and most definitive evidence for physiological or pathological roles for VGCCs outside of excitable tissues. All 17 individuals with life-threatening cardiac arrhythmias and syndactyly had a spontaneous (except for two siblings who inherited the causative variant from their mother who was mosaic) heterozygous G406R mutation in an alternatively spliced, mutually exclusive exon, exon 8a (3). The homologous exons 8 and 8a encode for 35 amino acids. The mutated glycine is the last amino acid encoded by the exon and resides at the cytoplasmic end of the IS6 transmembrane segment (Figure 2a,b). The cryo-EM structure of CaV1.1 (7) reveals that IS6 anchors a long α-helix that extends several helical turns below the membrane. The polypeptide makes a sharp turn and then forms another long α-helix that includes the α interaction domain (AID), which is the binding site in α1 for the CaV β auxiliary subunit (Figure 2b). The AID is a critical regulator of channel inactivation, and the sequence between IS6 and the AID is a key component for proper channel inactivation (27). Indeed, expression of G406R mutant CaV1.2 channels in heterologous expression systems revealed defective voltage-dependent inactivation (3, 28). In vivo, this delays channel closure after openings in response to an action potential. The resulting excess inward-depolarizing Ca2+ current in cardiomyocytes (Figure 2c) readily explains the prolonged electrocardiographic QT interval and the life-threatening arrhythmias in children with TS, but not the invariant syndactyly phenotype. CaV1.2 expression within skin of the developing limb had not been previously identified. Not only did in situ hybridization confirm CaV1.2 expression in an embryonic day (E)12.5 mouse limb (3)—suggesting that the channel directly affects limb development—but phenotypic characterization of this initial TS cohort suggested that several other nonexcitable tissues expressed CaV1.2 and were affected by the mutant channel. For example, baldness at birth in all TS subjects, together with syndactyly, implicates roles for CaV1.2 in the integumentary system; facial dysmorphia (increased protrusion of the mandible compared to maxilla) in over half of the children suggests a role for CaV1.2 in craniofacial development, and recurrent infections or pneumonia/bronchitis in about half of the children indicate immunodeficiency due to the mutant channel. Data from ARCHS4, an online resource of published RNA-seq data (29), confirm that CaV1.2 expression is widespread (Figure 3) and provides post hoc support that the mutant CaV1.2 is causative for many of the phenotypes observed in TS subjects. For example, high expression of CaV1.2 in several cell types within the immune system hints at reasons for the observed infections. The expression in chondrocytes and osteoblasts, at levels similar to values in heart, provides a basis for the observed facial dysmorphia: The affected mandible is one of the few bones in the skull that develops through endochondral ossification (in which a cartilaginous template is replaced by bone during development). Moreover, morpholino knockdown of cacna1c in a zebrafish embryo reduces mandibular size, which is restored by expression of a rabbit Cacna1c cDNA. This process requires Ca2+ influx through the channel. Expression of mutant Cacna1c in which the four glutamates that form the ion selectivity pore (see Figure 1) are changed to lysines fails to restore the mandibular size (30). The reduction of mandibular size in the zebrafish when embryos were treated with the calcineurin inhibitors cyclosporin and tacrolimus indicates that the Ca2+-dependent phosphatase calcineurin may be a critical downstream signaling target for Ca2+ influx through CaV1.2. Moreover, expression of a constitutively active calcineurin mutant restored mandibular size in zebrafish embryos treated with cacna1c morpholinos (30). Still, how CaV1.2, which requires membrane depolarization to open, contributes to these phenotypes in nonexcitable cells is not clear.
Figure 2.
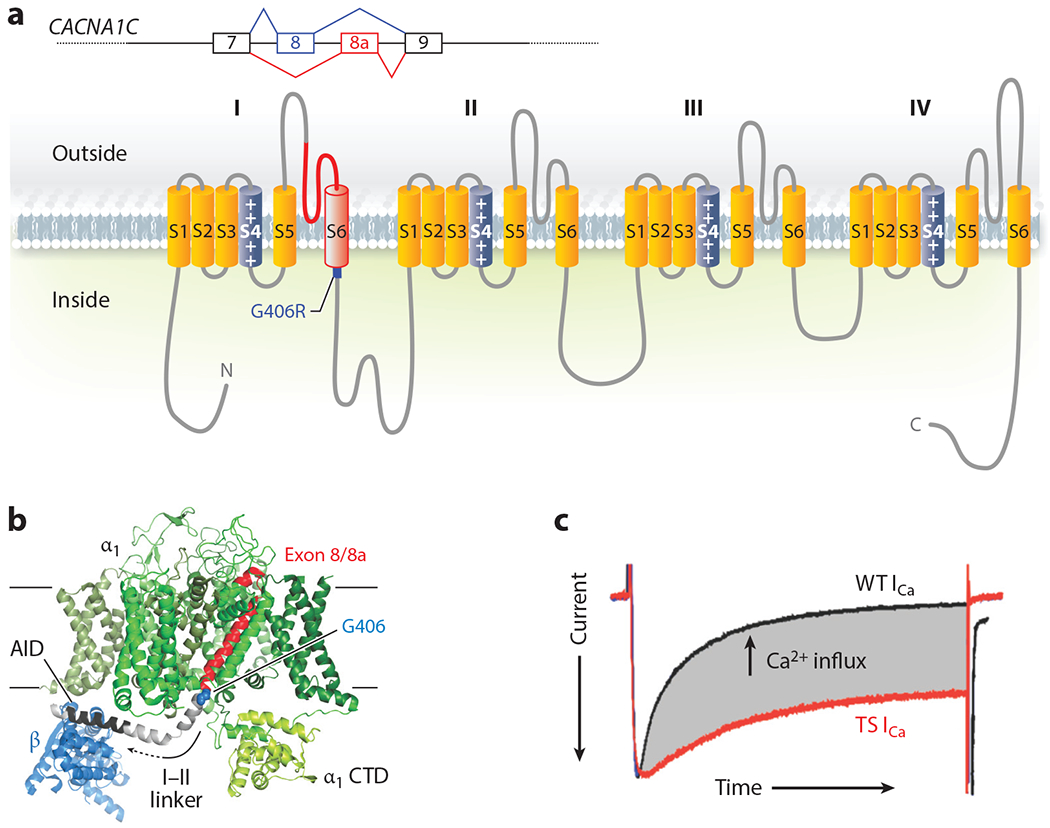
Structure and function of Timothy syndrome (TS) mutation. (a) Schematic of the alternative splicing event for exon 8 or 8a in CACNA1C and the exon in the context of the CaV1.2 α1C subunit. (b) Structure of the homologous rabbit CaV1.1 (Protein Data Bank: 5GVY) shown from the membrane. Only the α1 subunit and the β subunit (blue) are shown. The individual α1 domains (DI–DIV) are colored in various shades of green except for the polypeptide encoded by exon 8 (or exon 8a) in red, with the G406 residue that is mutated to G406R highlighted in blue; the linker between domain I and domain II (I–II linker) is depicted in gray, in which the α interaction domain (AID) is highlighted in black. (c) Voltage-clamp Ca2+ current recording from a cardiomyocyte isolated from a mouse with the TS mutation (red) overlaid with a recording from a cardiomyocyte isolated from a wild-type (WT) littermate. The currents are scaled for comparison, and the enhanced Ca2+ influx through the TS mutant channel is highlighted in gray (E.Q. Wei and G.S. Pitt, unpublished data).
Figure 3.
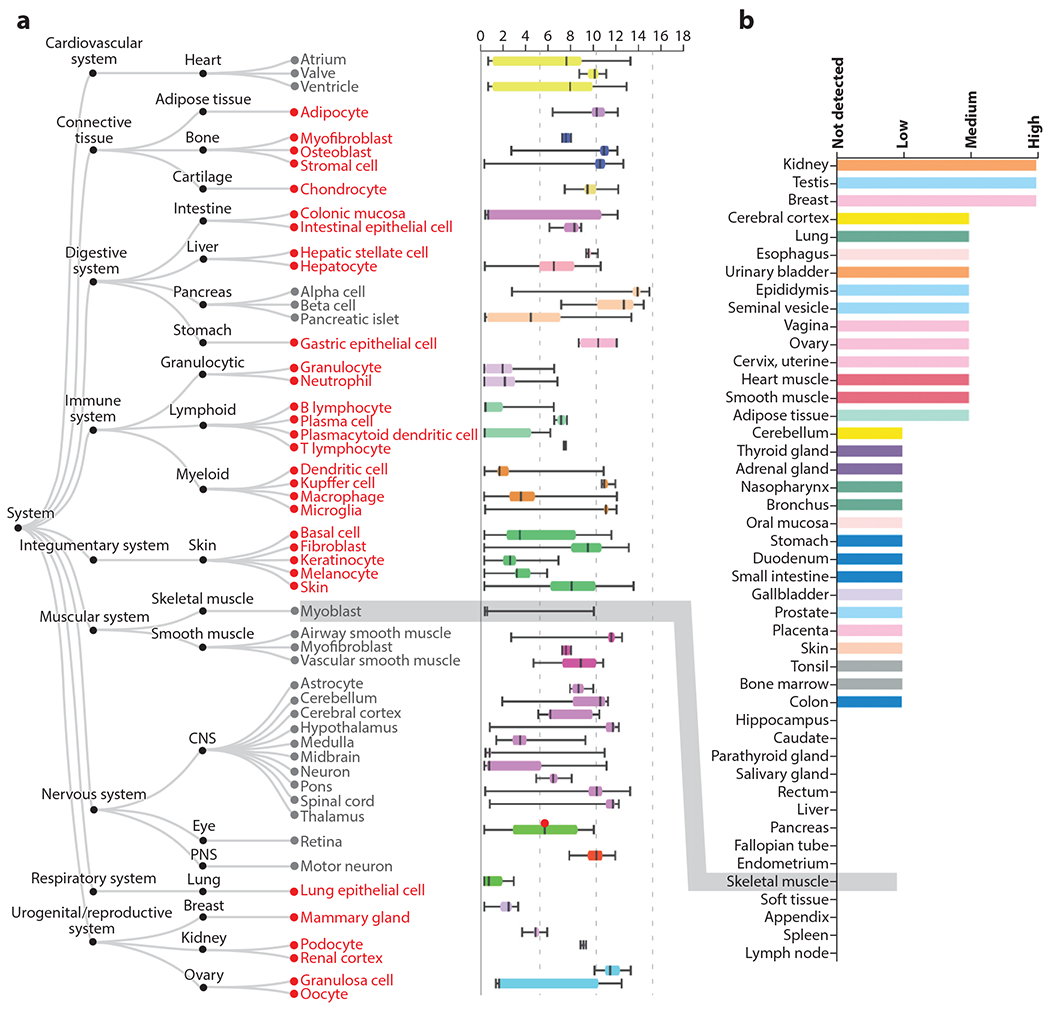
Tissue expression pattern of CaV1.2. (a) Figure and data are adapted from the ARCHS4 web resource (https://amp.pharm.mssm.edu/archs4/gene/CACNA1C) showing CACNA1C expression in various tissues. In red are nonexcitable cell types with expression of CaV1.2. Note that expression levels for many of these cell types are like the expression level in the heart, the canonical CaV1.2-expressing tissue. In contrast, skeletal muscle myoblasts, which express an alternative L-type voltage-gated Ca2+ channel (CaV1.1), show no expression (highlighted by gray box). (b) Figure and data for protein detection (adapted from https://www.proteinatlas.org/ENSG00000151067-CACNA1C/tissue) (55) also demonstrate broad tissue expression, with no CaV1.2 protein detected in skeletal muscle (highlighted by gray box).
Moreover, a homologous G406R mutation in the alternatively spliced exon 8 causes a variant form of TS (TS2, to distinguish it from TS that results from the G406R in exon 8a, which is designated TS1). TS2 is characterized by a markedly prolonged QT electrocardiographic interval and some but not all of the noncardiac phenotypes observed in the originally described cohort (28). Strikingly, the invariant syndactyly that is a hallmark of the TS1 phenotype is absent in TS2 individuals. This highlights that the alternative splicing of CACNA1C is dependent upon the cell type. A recent study suggests that alternative splicing of CaV1.2 during brain development may be temporally regulated as well (31). What controls temporospatial regulation of CACNA1C mRNA splicing outside of the brain is not known, but actions of polypyrimidine tract-binding protein (PTB) and its neuronal homolog nPTB coordinate to control splicing in neurons (32). Temporospatial regulation of CaV1.2 may be even more complex since syndactyly, the hallmark feature of TS1 in humans, is not conserved in mice. We generated mouse models for TS (both TS1 and TS2) by using CRISPR technology to knockin a G406R mutation in either exon 8 or exon 8a (M. Matsui and G.S. Pitt, unpublished data). These models recapitulate many, but not all, of the phenotypes observed in TS human subjects, but most strikingly, neither model displays syndactyly (Figure 4). Even in a homozygous TS1 mouse (these mice are viable and fertile), we do not observe obvious digit abnormalities (Figure 4). Thus, the tissue- or cell-specific regulation of splicing of Cacna1c exon 8 versus exon 8a in mice may differ from the regulation in humans, and the observation that this cardinal feature of TS is not recapitulated in a mouse model suggests an additional layer of complexity to VGCCs in nonexcitable tissues.
Figure 4.
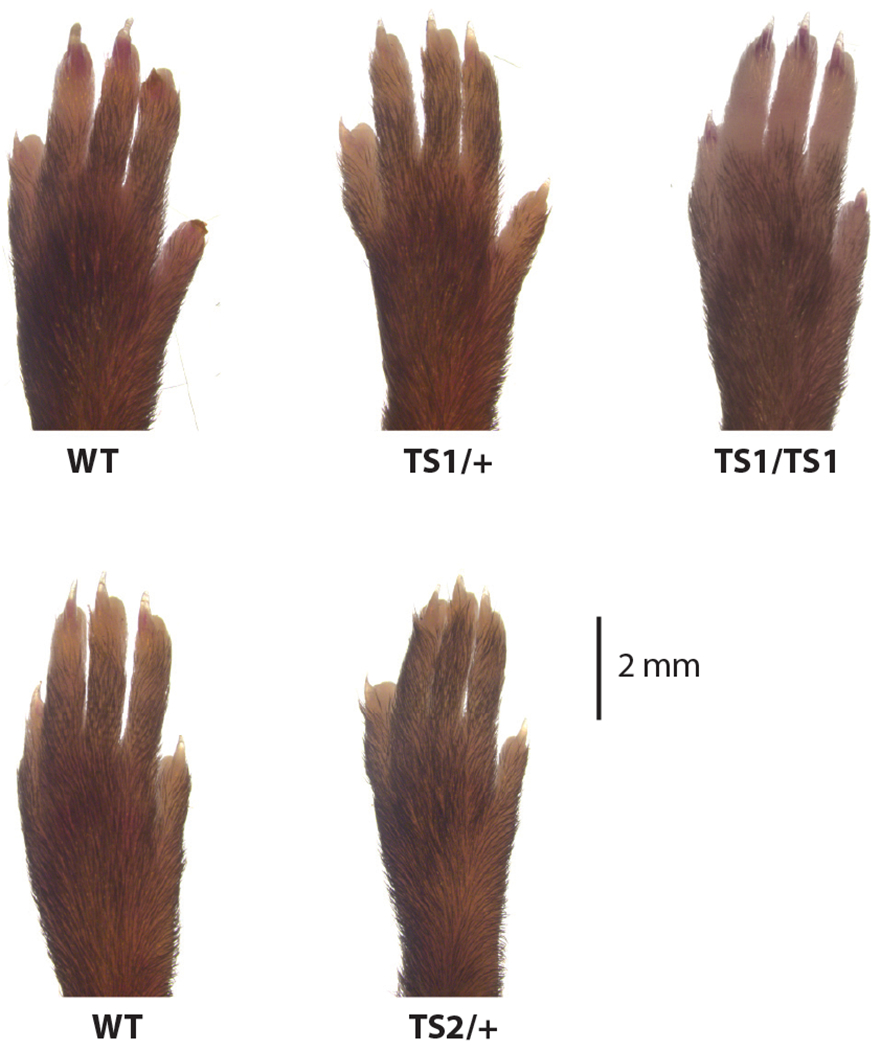
Timothy syndrome (TS) mouse models do not display syndactyly. Images of forelimbs from TS1 and TS2 mice and their respective littermate wild-type (WT) controls.
2.3. Beyond Timothy Syndrome: Human Data Supporting a Role for CaV1.2 in Diseases of Nonexcitable Tissues
Clinical trials with CCBs and, more recently, unbiased transcriptomic studies have revealed potential roles for CaV1.2 in various disease processes involving nonexcitable tissues and thereby provide evidence independent of TS that CaV1.2 exerts effects in nonexcitable cells. Atherosclerosis is a chronic inflammatory disease initiated by oxidation of low-density lipoprotein (LDL) particles, which trigger responses from arterial wall endothelial cells and smooth muscle cells that attract circulating monocytes (33). Subsequent infiltration of macrophages instigates a local immune response, causing vessel damage and development of the atherosclerotic lesion, for which one of the early markers is calcification (34). This prompted investigators to attempt to inhibit calcification with CCBs, reasoning that calcification depends on Ca2+ entry through VGCCs. Suppression by the CCB nifedipine of atherogenesis in cholesterol-fed rabbits (35) led to clinical investigations that demonstrated prevention and regression of coronary artery disease with nifedipine and related dihydropyridine CCBs (36–39). Although hypertension is a major risk factor for coronary artery disease, the beneficial outcome in a trial with the CCB amlodipine even in patients with normal blood pressure suggests that the CCB’s effect was independent of its known antihypertensive actions (37) and thus independent of the excitable vascular smooth muscle cells through which CCBs lower blood pressure. Rather, cell culture and animal studies point to mechanism(s) such as reduction of LDL oxidation by free radicals by CCBs (40) or that blockade of CaV1.2 in macrophages may be anti-inflammatory (41).
A genome-wide association study (GWAS) of patients with calcific aortic stenosis, the most common cardiovascular valve disease, implicates CACNA1C (42) and highlights another unexpected role for VGCCs in nonexcitable tissue. Expression quantitative trait loci mapping suggested that the associated CACNA1C variants correlate with increased CaV1.2 expression, and the calcium signaling pathway (KEGG hsa04020) was the top gene set when considering all loci differentially expressed between normal and calcified valves (42). The bulk of the aortic valve cusps, the site of calcification that leads to the pathological stenosis with clinical consequences, contain fibroblast-like valve interstitial cells and their extracellular matrix surrounded by a single endothelial cell layer. In diseased but not normal valves, macrophages are also abundant (43). Valve interstitial cells, endothelial cells, and macrophages are nonexcitable cells. Which of these cells express CaV1.2 and how CaV1.2 affects valve calcification are not clear. Nevertheless, aortic stenosis shares underlying mechanisms and risk factors with atherosclerosis (44), suggesting parallels between the diseases regarding the roles of CaV1.2.
Use of CCBs in the treatment of hypertrophic disorders of wound healing (e.g., keloid scars) (45) emphasizes roles for VGCCs in skin and echoes the syndactyly seen in TS. Moreover, this may provide further insight into the association between VGCCs and aortic stenosis. Keloids develop due to excessive fibroblast proliferation and increased collagen turnover (46, 47). Dermal fibroblasts have VGCC activity, which is blocked by CCBs (48), and they secrete collagen, also blocked by CCBs (49). Since aortic valve interstitial cells are a fibroblast subset, and calcific aortic stenosis is also characterized by aberrant extracellular matrix secretion (50), keloids and calcific aortic stenosis suggest common roles for VGCCs in nonexcitable fibroblast-like cells. In addition to these observations that VGCCs influence clinical and pathophysiological events, analysis of animal models with informative mutations provides models to characterize these roles in more detail and to define the underlying mechanisms by which VGCC signaling in nonexcitable cells contributes to the observed physiology or pathological phenotypes.
3. ANIMAL MODELS TO DISSECT THE ROLES OF VOLTAGE-GATED Ca2+ CHANNELS IN NONEXCITABLE TISSUES
Like human disease reports, animal models provide powerful means to validate unexpected findings, such as roles for VGCCs in nonexcitable cells. Highlighted here are studies identified by a strategy that included (a) a PubMed search for knockout/knockin animal models for each specific VGCC pore-forming or auxiliary subunit and an associated phenotype that could be attributable to traditionally nonexcitable cells or tissues; and (b) a search in the Online Mendelian Inheritance in Man database for phenotypes associated with each VGCC subunit and for corresponding knockout/knockin animal models for each specific VGCC pore-forming or auxiliary subunit and an associated phenotype that could be attributable to traditionally nonexcitable cells or tissues. In our discussion we prioritize animal models with genetic manipulations over pharmacological or shRNA/siRNA approaches because the genetic manipulations provide greater specificity. Nevertheless, we are cognizant that animal models do not always accurately mimic human disease or pathology, as we already indicated with our example of TS mouse models and the absence of the cardinal human syndactyly phenotype (Figure 4).
With that caveat, we start our review of animal models with an emphasis on the skeleton, as many early studies of VGCCs in nonexcitable cells focus on bone physiology. Ablation of Cacna1d (CaV1.3) in a mouse model reduced cross-sectional area midshaft on the femur and lowered bone mineral density and bone mineral content compared to wild-type animals (51), suggesting that CaV1.3 contributes to bone formation. Because CaV1.3, like CaV1.2, is expressed in several different endocrine tissues (52, 53), it is not clear whether CaV1.3 exerts its role directly in osteoblasts, where it is expressed (51), or by affecting hormone secretion from any one of the endocrine organs with extensive regulation of bone metabolism, where it is also expressed (54). Moreover, CaV1.3 is expressed in jejunal glandular cells (55), in which CaV1.3 mediates transcellular Ca2+ absorption necessary for bone mineralization during the early postnatal period; both knockout of CaV1.3 and reduced Ca2+ absorption from the gut are associated with decreased bone mineralization (56), thus providing an alternative mechanism to an osteoblast-specific effect. In contrast, a recent study suggests a direct effect of CaV1.2 within developing limb structures on bone development (57). First, live-imaging analyses with a genetically encoded Ca2+ indicator in micromass-cultured chicken limb bud cells documented CCB-sensitive Ca2+ transients. Second, conditional ablation of CaV1.2 in osteoblast precursors by activating homozygous Prx1-CreER during embryogenesis caused appendicular skeletal abnormalities, including shortened limbs and missing digits. Third, pharmacological blockade of VGCCs with the CCB nifedipine, albeit at high dose, in micromass-cultured chicken limb bud cells inhibited chondrocyte differentiation. Since Prx1-Cre is an early marker of all cells in the developing limb bud mesenchyme (58), together these data suggest an essential role for CaV1.2 in chondrocytes during limb development mediated by endochondral ossification. Using a CaV1.2 LacZ reporter mouse (The Jackson Laboratory, stock #005783), we also observed CaV1.2 expression during skeletal development, but with expression restricted to the perichondrium/periosteum and very limited expression in chondrocytes (Figure 5a). In addition, when we conditionally ablated CaV1.2 in chondrocytes with a constitutive Col2a-Cre, we observed no effect on limb development (Figure 5b) (C. Cao & G.S. Pitt, unpublished data). The reasons for the discrepant results are unclear, but the recently published study that observed the defects in skeletal development did not utilize a littermate control for the homozygous Prx1-CreER, so effects that are independent of CaV1.2 ablation cannot yet be ruled out. Moreover, toxicology studies of CCBs do not support the observation that CaV1.2 can cause limb defects. Although one study (59) reported that vasodilating CCBs administered to pregnant rabbits produced digital defects in the fetuses, that study reported that an unrelated vasodilator without CCB properties also caused digital defects, suggesting that the underlying mechanism was perturbed placental blood flow rather than a specific effect of the CCB. Moreover, CCBs at clinically appropriate doses are safe for human fetuses, and no limb defects have been reported when pregnant mothers took CCBs (60, 61). Nevertheless, along with previous data showing definitive expression of CaV1.2 in resting and proliferating chondrocytes in the early postnatal period, and the demonstration that CCBs markedly decreased osteogenesis of primary bone marrow stromal cells in culture (62), these data support a role for CaV1.2 in endochondral ossification that was also highlighted by the studies of mandible development in zebrafish and mouse (30) and perhaps hint at compensation by other VGCCs, such as CaV1.3, when CaV1.2 is ablated. In an interesting parallel, ablation of the T-type Ca2+ channel CaV3.2 affects development of the cartilaginous rings of the trachea, and Sox9-dependent chondrogenesis depends on Ca2+ influx through these CaV3.2 channels and resulting calcineurin activation (63).
Figure 5.
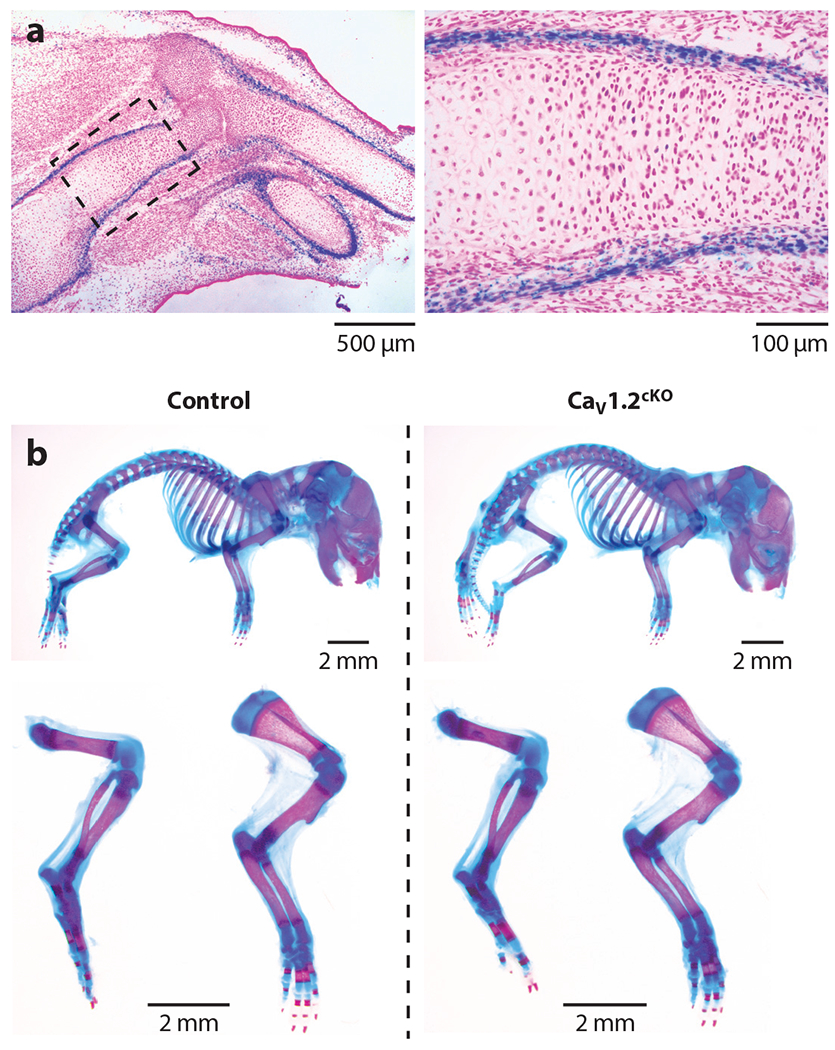
CaV1.2 is expressed during mouse limb development. (a) A CaV1.2lacZ/+ reporter mouse detects CaV1.2 expression during mouse limb development. A frozen tissue section (10 μM) from CaV1.2lacZ/+ forelimb at E14.5 was stained with X-gal and counterstained with nuclear fast red. (b) Normal skeletal development of Col2a1-Cre;Cacna1cflox/flox conditional knockout (cKO) mouse embryos. Double staining with alizarin red and Alcian blue of the whole skeleton (top), hindlimbs and forelimbs (bottom) of Cacna1cflox/flox (control), and cKO littermate mouse embryos at P0.
Those loss-of-function experiments, showing decreased osteogenesis and blocked development of skeletal elements, are complemented by gain-of-function approaches that also suggest how Ca2+ signaling through CaV1.2 affects endochondral ossification and bone mass. Using various Cre lines to drive a CaV1.2 with the TS mutation in mice or microinjection of a cRNA encoding a TS-mutant CaV1.2 into zebrafish embryos led to increased bone mass specifically in the skeletal elements in which the Cre drivers are expressed (30, 62, 64). Indeed, targeted expression of the TS mutant CaV1.2 in the mandible led to a protuberant mandible, phenocopying the feature seen in TS patients (30). Additional analysis showed activation of transcripts associated with osteogenesis downstream of increased intracellular Ca2+ signaling from a TS mutant CaV1.2 (62). Further, the osteoblasts expressing the gain-of-function mutant CaV1.2 secreted higher levels of osteoprotegerin, a cytokine that inhibits osteoclast activity, thus tipping the balance toward bone formation rather than osteoclast-mediated bone resorption (62, 64). Thus, osteoblasts appear to have exploited a VGCC function in excitable cells (i.e., neurons and endocrine cells) to regulate secretion of a critical intercellular signaling molecule.
Gain-of-function CaV1.2 models also suggest that VGCCs affect several processes in the integumentary system. Severe dermatitis presented more than twice as frequently in a TS syndrome knockin mouse model than in wild-type littermates (65). Consistent with TS patients, these TS knockin mice displayed several behaviors associated with autism, including repetitive stereotypical behaviors that echo the autism-associated behaviors observed in TS children. The investigators were thus unable to conclude whether the dermatitis resulted from excessive grooming and/or scratching rather than from a defect within the skin and/or an immune defect, which would echo the recurrent infections experienced by individuals with TS (3).
In another study, based on the observation that individuals with TS show delayed hair growth for the first two years of life (3), Yucel et al. (66) investigated CaV1.2 channels in hair follicle bulge cells and suggested a different mechanism by which the channels affect these nonexcitable cells. Bulge cells form the stem cell niche in the developing hair follicle and express CaV1.2. Transgenic overexpression of the TS gain-of-function mutant CaV1.2 in bulge cells delayed entry into anagen, the growth phase of the hair cycle. However, the authors were unable to detect voltage-gated Ca2+ currents. Moreover, the authors overexpressed a loss-of-function CaV1.2 mutant (one of the four pore loops deleted) and observed a similar delay in entry into anagen, suggesting that increased channel protein—independent of its ability to support Ca2+ permeation—drove the phenotype. Further, treatment with verapamil, a use-dependent CCB that favors the channel’s inactivated state, accelerated entry into anagen. Thus, these authors propose a noncanonical Ca2+-independent function for CaV1.2 in hair follicles. Whether this or a similar mechanism functions in other nonexcitable cells or tissues is not known, but at least one other report suggested that knockdown of the Ca2+ channel auxiliary CaV β4 subunit (see Figure 1) by injection of a morpholino blocks initiation of epiboly, the spread of the epithelial layer during gastrulation, in teleosts. Injection of a cRNA encoding a wild-type CaV β4 subunit as well as injection of a cRNA encoding a mutant CaV β4 that was incapable of binding to, or regulating, a VGCC restored epiboly (67), suggesting that functions of this VGCC subunit in regulation of epiboly are independent of canonical channel activities.
Several knockout models in lymphocytes also hint at potential roles for VGCCs that are independent of Ca2+ influx. First, lethargic mice, named for ataxia, lethargic, and spontaneous seizure phenotypes, are shown to have a mutation in the CaV β4 subunit (68). They also have defective T lymphocyte function (69), suggesting a role for the CaV β4 subunit in T cells. Mouse T cells also express the CaV β3 subunit. T cells isolated from mice lacking either CaV β3 or CaV β4 show deficient increases in intracellular Ca2+ when stimulated with a cross-linking assay and reduced production of interferon-γ and interleukin (IL)-4 cytokines (70). Interestingly, the intracellular Ca2+ increase and downstream generation of cytokines appear to be independent of membrane depolarization (70). Moreover, ablation of CaV β3 decreased CD8+ T cell survival, and CD8+ T cells from CaV β3 knockout mice showed decreased production of key effector cytokines (71). Further, knockout of the CaV β2 subunit in developing thymocytes suggested a role for CaV β2 during the earlier stages of thymocyte development (72). Voltage-gated Ca2+ currents were not recorded in these studies, so the molecular role of the CaV β subunits remained undefined, perhaps suggesting a CaV β function that is independent of VGCCs, mirroring the hypothesized role of CaV β4 in epiboly. Intriguingly, however, knockout of CaV β3 reduced the amount of CaV1.4 protein detected in lymphocytes (71).
CaV1.1 and CaV1.4, almost exclusively expressed in skeletal muscle and retina, respectively (55), are two CaV α1 subunits that CaV β subunits are hypothesized to regulate in lymphocytes (70, 73). Knockout of Cacna1f, which encodes the L-type CaV1.4 channel, affects the generation of antigen-specific cytotoxic T cells (73). While the investigators reported differences in current density between wild-type and mutant T cells, the recorded currents from the wild-type cells were small and displayed unexpected biophysical characteristics, as also noted in an accompanying editorial (74). This raises the possibility of a mechanism independent of Ca2+ influx through CaV1.4. Interestingly, in previous work these authors reported novel spliced CaV1.4 transcripts that lack canonical elements of the DIV voltage sensor and/or pore domain (11), further raising the possibility that any CaV1.4 protein that arises from CaV1.4 transcripts in T cells functions in a manner that does not require Ca2+ influx through the channel’s pore or normal voltage-dependent function. Alternatively, the reported novel splice variants may be cloning or PCR artefacts, as neither are documented in the UCSC genome browser (75). Moreover, although cytotoxic T cells isolated from Cacna1f−/− mice and the two different β subunit ablation models showed deficient cellular T cell function, all experiments were performed with isolated T cells. It is not known if any of CaV β or the Cacna1f−/− knockout mice experienced decreased survival or any other immunological consequences from the observed T cell functional defects. Still, roles for VGCCs in immune system function are suggested by the “pneumonia/bronchitis” and “immunodeficiency/recurrent infections,” both reported in about half of the subjects from the original TS series (3). CaV1.2 channels may also regulate the immune response through actions in macrophages (41), as also noted. Interestingly, differentiation of monocytes to macrophages induced CaV1.2 expression as well as CaV β1 and CaV β2 and upregulated CaV1.2 protein at the cell surface. Further, chronic administration of CCBs reduced the macrophage inflammatory response to a peritonitis model in mice (41). In combination with the data in TS subjects, these results suggest that altered Ca2+ influx through CaV1.2 in macrophages perturbs the appropriate inflammatory response. What controls CaV1.2 function in these nonexcitable cells is not known.
The CaV1.2 channel may regulate roles in nonexcitable cells in the brain. For example, CaV1.2 appears to control remyelination by oligodendrocytes (76). Glia, including oligodendrocytes, are not generally considered excitable cells and do not fire action potentials in their mature state. However, oligodendrocyte precursor cells do (77). Conditional CaV1.2 knockout in oligodendrocyte precursor cells affected maturation and caused a decrease in axon myelination. Thus, CaV1.2 actions in oligodendrocyte precursors appear to fall within the traditional roles ascribed to VGCCs in excitable cells.
4. ROLES OF VOLTAGE-GATED Ca2+ CHANNELS IN NONEXCITABLE CELLS SUGGESTED BY GENETIC STUDIES AND UNBIASED SCREENS
While animal models and manipulation of VGCCs by knockdown, knockout, or transgenic expression provide compelling evidence for VGCCs in nonexcitable tissues, several studies in cells and cell culture systems, as well as emerging large data set analyses such as bulk and single-cell RNA-Seq or GWASs, implicate additional roles for VGCCs in other nonexcitable cells and tissues. Here, we build upon the animal models discussed above and focus especially on data that provide insight into phenotypes documented in subjects with TS because of the particular validation offered by genetic mouse models or the phenotypes in individuals with the documented syndrome caused by a VGCC mutation. The correlations discussed in these studies offer opportunities and motivation for translation into animal models for the discovery of new VGCC signaling paradigms and for new understanding about physiology and disease associated with VGCCs.
The set of studies identifying VGCCs in the skeletal system, particularly osteoblasts and chondrocytes as described above, prompted us to look for the presence of VGCCs in other connective tissues. Our analyses of recent bulk and single-cell RNA-seq data sets (78, 79) suggest a role for VGCCs in tendon function. CaV1.1, CaV1.2, and CaV3.1 are among the highly expressed genes in the bulk RNA-seq data set. Moreover, gene ontology analysis of differentially expressed genes across different tendons identified the calcium signaling pathway. Thus, we hypothesize that VGCCs may play distinct roles in different tendons, perhaps regulated by different loads on the specific tendons. Using the resolution provided by the single-cell RNA-seq data we found that, among the three VGCCs highly expressed in tendons, CaV1.2 and CaV3.1 are prominent only in the pericyte cluster among all the clusters identified after reduction of dimensionality by T-distributed stochastic neighbor embedding (t-SNE). This is significant, as the authors of the single-cell RNA-Seq manuscript showed that the pericytes serve as progenitor cells for the fibroblasts in the adult tendon (78). Since VGCCs appear to exert specific roles in bone development, and those tendon RNA-seq data sets derive from animals ~6 weeks old, we analyzed a separate bulk RNA-seq data set obtained from embryonic cells (80), which showed expression of CaV1.2, CaV3.1, and CaV3.2, with CaV3.1 expression as the most prominent. Thus, there may be additional or alternative roles for VGCCs during embryogenesis and early tendon development.
Another intriguing phenotype in subjects with TS is that they have small teeth at birth (3), and several studies suggest potential roles for VGCCs in tooth development. Odontoblasts, the cells in the dental pulp that secrete dentin (the extracellular matrix beneath the exterior enamel) express CaV1.2 channels (81). A previous study identified dihydropyridine-sensitive voltage-gated Ca2+ currents in isolated dental pulp cultures (82), so the odontoblast progenitors, like osteoblast precursors, may require VGCC currents for differentiation. Interestingly, nerve activity acutely regulates CaV1.2 expression in odontoblasts (81). Thus, the dental phenotype in TS individuals may actually reflect a direct and indirect CaV1.2 role in nonexcitable odontoblasts. On the other hand, the CaV1.1 channel appears to regulate tooth development by an unknown mechanism. A single missense mutation in the CaV1.1-encoding CACNA1S has been associated with a dominantly inherited disorganized supernumerary cusp phenotype (83), and a GWAS for tooth agenesis pinpointed CACNA1S (84). Whether there are functional CaV1.1 channels in any of the cells in the developing tooth bud, however, is not known.
5. POTENTIAL MECHANISMS OF VOLTAGE-GATED Ca2+ CHANNEL REGULATION IN NONEXCITABLE CELLS
While cellular experiments and animal models, supported by clinical correlations, emphasize roles for VGCCs in nonexcitable cells and tissues, the mechanisms by which the channels function in these cells remain unclear. Specifically, how do VGCCs receive the depolarizing stimulus necessary to open and support Ca2+ influx in cells not thought to fire action potentials? Here, we describe some potential mechanisms, but clearly much remains unknown.
One possibility is that the depolarizing drive depends on mechanisms that do not require action potentials, particularly for mechanosensitive tissues such as bone. Among channels that permeate Ca2+, osteocytes express high levels of the mechanosensitive channel Piezo1 in static conditions and the amount of Piezo1 transcripts increase with shear stress (85, 86). Stretch and mechanical forces cause Piezo1 and its homolog Piezo2 to open, providing an inward depolarizing current, including Ca2+ (87). Activation of Piezo1 or Piezo2 by mechanical stimuli may in turn activate VGCCs to amplify Ca2+ influx. The functional coupling of mechanosensitive channels and VGCCs has not yet been tested, but ablation of Piezo1 in osteocytes and osteoblasts reduced the normal increase in bone mass to mechanical load (85, 86). Chondrocytes are also sensitive to mechanical load through actions of Piezo1 as well as the related Piezo2 (88), suggesting that mechanical load during endochondral ossification may likewise trigger membrane depolarization and subsequent activation of VGCCs. This mechanism may also contribute to the hypothesized role of CaV1.2 in calcific aortic stenosis (42), as one of the drivers of the disease process is shear stress (89).
VGCCs in nonexcitable cells may also be activated by membrane depolarization mediated by the TRPC family of transient receptor potential cation channels. TRPC channel activation is downstream of phospholipase C (PLC), so G protein–coupled receptor signaling events that activate PLC are poised to open VGCCs. Of note, the parathyroid hormone that stimulates Ca2+ influx through VGCCs in ROS 17/2.8 activates PLC (90), and ROS 17/2.8 cells have TPRC3 transcripts (91).
The hyperpolarizing effects of 1,25(OH)2D3 on L-type VGCC Ca2+ current activation in ROS 17/2.8 cells suggest another activation mechanism for VGCCs in nonexcitable cells (25) and may explain some actions of other steroid hormones such as estradiol on L-type VGCC Ca2+ currents in osteoblasts (92). Steroid hormones, including 1,25(OH)2D3 and estradiol, alter gene transcription through actions on intracellular steroid hormone receptors, but they can also act rapidly at the plasma membrane—too fast to affect transcription—to affect L-type VGCC Ca2+ currents (93). These nongenomic effects of steroid hormones appear to be regulated by a small fraction of steroid hormone receptors that localize to the plasma membrane through interactions with caveolin-1 (94). For estradiol, activation of a G protein–coupled estrogen receptor, GPER, may also exert some of nongenomic actions at the plasma membrane (95). While the specific mechanisms by which 1,25(OH)2D3 leads to hyperpolarization of VGCC activation are not clear, the resting membrane potential for osteoblast is approximately −43 mV (96), which is more depolarized than most excitable cells and is not far from the V1/2 of activation for L-type VGCCs. Thus, actions of 1,25(OH)2D3 on VGCCs may move the threshold for channel activation closer to that of the resting membrane potential and thereby increase the probability of channel opening.
It is also possible that the VGCCs function in a noncanonical manner. As described for the study investigating the contribution of CaV1.2 in hair follicles, gain-of-function and loss-of-function models produced the same delay to enter anagen, as did a mutant CaV1.2 unable to permeate Ca2+ (66). Together, these results suggest a Ca2+-independent signaling mechanism. Consistent with this possibility, the same investigators found that expression of a TS mutant CaV1.2 in neurons led to dendritic retraction even if the expressed channel also had mutations blocking Ca2+ permeation (97). They therefore hypothesized that a voltage-dependent conformational change in CaV1.2 signals through the small GTPase Gem to activate the RhoA signaling leads to dendritic retraction. Gem, like Rad—the VGCC inhibitor that regulates adrenergic response of CaV1.2 in the heart (15)—is a member of the RGK class of small GTPases that interact with CaV β subunits (98) and also regulates the Rho kinase pathway (99, 100). Because Gem is also expressed in T cells (101) and RhoA signaling is fundamental to T cell development and activation (102), the hypothesized effects of VGCCs modulating T cell function may also be Ca2+ independent and instead be mediated by Gem in a manner analogous to the pathway hypothesized for dendritic retraction in neurons.
6. CONCLUSIONS
Cemented by the striking findings in subjects with TS and aided by emerging unbiased screening approaches, newly discovered roles for VGCCs in nonexcitable cells continue to grow in number. Defining what controls VGCC functions in these cells, however, remains an interesting challenge. Nevertheless, as these broad VGCC roles become more clearly defined and documented, they not only expand our understanding of physiology but also provide motivation for reappraisal of both the consideration and causality of specific genetic variants in VGCCs that are reported to have limited pathological effects. For example, the multiple phenotypes associated with the gain-of-function mutation in CaV1.2 that is causal for TS raise a question of whether reported gain-of-function variants in CaV1.2 associated with “cardiac only Timothy syndrome” (103, 104)—increasingly discovered with whole-exome or whole-genome sequencing diagnostic approaches—are indeed causal for the observed cardiac-specific phenotypes, especially since there are no associated noncardiac phenotypes. While next-generation sequencing technology has led to variant identification in subjects with specific phenotypes, the increased availability of sequenced genomes simultaneously provides more detailed information on population genetic variation and has motivated the reassessment of previously ascribed causal variants in certain loci (105, 106). Because the splicing of VGCCs is complex, and may be tissue specific as previously noted, it is possible that certain CaV1.2 variants could exert effects in exons that are expressed only in the heart. However, the myriad roles for VGCCs generally—and the well-documented roles for CaV1.2 because of TS specifically—make that possibility less likely.
ACKNOWLEDGMENTS
Research presented here was supported by the National Heart, Lung, and Blood Institute (NHLBI) grant R01 HD090132 and National Institute of Child Health and Human Development (NICHD) grant R01 HL146149 (G.S.P.); the Lisa and Sanford B. Ehrenkranz Young Scientist Fund for Women’s Cardiovascular Health from Weill Cornell Medicine (M.M.); and National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) grant R21 AR075214 (C.C.).
DISCLOSURE STATEMENT
G.S.P. has served as a consultant to MyoKardia and Abbott Laboratories and serves on event adjudication committees for the Cardiovascular Research Foundation. G.S.P. and C.C. have received research grant funding from the US National Institutes of Health (NIH). M.M. is not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Yue DT. 2004. The dawn of high-resolution structure for the queen of ion channels. Neuron 42:357–59 [DOI] [PubMed] [Google Scholar]
- 2.Hille B 2001. Ion Channels of Excitable Membranes. Oxford, UK: Oxford Univ. Press [Google Scholar]
- 3.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, et al. 2004. CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119:19–31 [DOI] [PubMed] [Google Scholar]
- 4.Catterall WA. 2011. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol 3:a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Catterall WA, Lenaeus MJ, Gamal El-Din TM. 2020. Structure and pharmacology of voltage-gated sodium and calcium channels. Annu. Rev. Pharmacol. Toxicol 60:133–54 [DOI] [PubMed] [Google Scholar]
- 6.Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, et al. 2000. Nomenclature of voltage-gated calcium channels. Neuron 25:533–35 [DOI] [PubMed] [Google Scholar]
- 7.Wu J, Yan Z, Li Z, Qian X, Lu S, et al. 2016. Structure of the voltage-gated calcium channel Cav1.1 at 3.6 Å resolution. Nature 537:191–96 [DOI] [PubMed] [Google Scholar]
- 8.Yang J, Ellinor PT, Sather WA, Zhang JF, Tsien RW. 1993. Molecular determinants of Ca2+ selectivity and ion permeation in L-type Ca2+ channels. Nature 366:158–61 [DOI] [PubMed] [Google Scholar]
- 9.Lipscombe D, Andrade A. 2015. Calcium channel CaVα1 splice isoforms—tissue specificity and drug action. Curr. Mol. Pharmacol 8:22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma Y, Kobrinsky E, Marks AR. 1995. Cloning and expression of a novel truncated calcium channel from non-excitable cells. J. Biol. Chem 270:483–93 [DOI] [PubMed] [Google Scholar]
- 11.Kotturi MF, Jefferies WA. 2005. Molecular characterization of L-type calcium channel splice variants expressed in human T lymphocytes. Mol. Immunol 42:1461–74 [DOI] [PubMed] [Google Scholar]
- 12.Dolphin AC. 2018. Voltage-gated calcium channel α2 δ subunits: an assessment of proposed novel roles. F1000Research 7. 10.12688/f1000research.16104.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buraei Z, Yang J. 2010. The β subunit of voltage-gated Ca2+ channels. Physiol. Rev 90:1461–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang L, Katchman A, Kushner J, Kushnir A, Zakharov SI, et al. 2019. Cardiac Cav1.2 channels require β subunits for β-adrenergic-mediated modulation but not trafficking. J. Clin. Investig 129:647–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu G, Papa A, Katchman AN, Zakharov SI, Roybal D, et al. 2020. Mechanism of adrenergic Cav1.2 stimulation revealed by proximity proteomics. Nature 577:695–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ben-Johny M, Yue DT. 2014. Calmodulin regulation (calmodulation) of voltage-gated calcium channels. J. Gen. Physiol 143:679–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dolphin AC. 2016. Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J. Physiol 594:5369–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andronache Z, Ursu D, Lehnert S, Freichel M, Flockerzi V, Melzer W. 2007. The auxiliary subunit γ1 of the skeletal muscle L-type Ca2+ channel is an endogenous Ca2+ antagonist. PNAS 104:17885–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fatt P, Katz B. 1953. The electrical properties of crustacean muscle fibres. J. Physiol 120:171–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alcover A, Weiss MJ, Daley JF, Reinherz EL. 1986. The T11 glycoprotein is functionally linked to a calcium channel in precursor and mature T-lineage cells. PNAS 83:2614–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamaguchi DT, Hahn TJ, Iida-Klein A, Kleeman CR, Muallem S.1987. Parathyroid hormone-activated calcium channels in an osteoblast-like clonal osteosarcoma cell line. cAMP-dependent and cAMP-independent calcium channels. J. Biol. Chem 262:7711–18 [PubMed] [Google Scholar]
- 22.Chesnoy-Marchais D, Fritsch J. 1988. Voltage-gated sodium and calcium currents in rat osteoblasts. J. Physiol 398:291–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guggino SE, Lajeunesse D, Wagner JA, Snyder SH. 1989. Bone remodeling signaled by a dihydropyridine- and phenylalkylamine-sensitive calcium channel. PNAS 86:2957–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guggino SE, Wagner JA, Snowman AM, Hester LD, Sacktor B, Snyder SH. 1988. Phenylalkylamine-sensitive calcium channels in osteoblast-like osteosarcoma cells. Characterization by ligand binding and single channel recordings. J. Biol. Chem 263:10155–61 [PubMed] [Google Scholar]
- 25.Caffrey JM, Farach-Carson MC. 1989. Vitamin D3 metabolites modulate dihydropyridine-sensitive calcium currents in clonal rat osteosarcoma cells. J. Biol. Chem 264:20265–74 [PubMed] [Google Scholar]
- 26.Meszaros JG, Karin NJ, Akanbi K, Farach-Carson MC. 1996. Down-regulation of L-type Ca2+ channel transcript levels by 1,25-dihyroxyvitamin D3 osteoblastic cells express L-type α1C Ca2+ channel isoforms. J. Biol. Chem 271:32981–85 [DOI] [PubMed] [Google Scholar]
- 27.Findeisen F, Minor DL Jr. 2009. Disruption of the IS6-AID linker affects voltage-gated calcium channel inactivation and facilitation. J. Gen. Physiol 133:327–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, et al. 2005. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. PNAS 102:8089–9615863612 [Google Scholar]
- 29.Lachmann A, Torre D, Keenan AB, Jagodnik KM, Lee HJ, et al. 2018. Massive mining of publicly available RNA-seq data from human and mouse. Nat. Commun 9:1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramachandran KV, Hennessey JA, Barnett AS, Yin X, Stadt HA, et al. 2013. Calcium influx through L-type Cav1.2 Ca2+ channels regulates mandibular development. J. Clin. Investig 123:1638–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panagiotakos G, Haveles C, Arjun A, Petrova R, Rana A, et al. 2019. Aberrant calcium channel splicing drives defects in cortical differentiation in Timothy syndrome. eLife 8:e51037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang ZZ, Sharma S, Zheng S, Chawla G, Nikolic J, Black DL. 2011. Regulation of the mutually exclusive exons 8a and 8 in the Cav1.2 calcium channel transcript by polypyrimidine tract-binding protein. J. Biol. Chem 286:10007–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Libby P 2002. Inflammation in atherosclerosis. Nature 420:868–74 [DOI] [PubMed] [Google Scholar]
- 34.Kolodgie FD, Burke AP, Nakazawa G, Virmani R. 2007. Is pathologic intimal thickening the key to understanding early plaque progression in human atherosclerotic disease? Arterioscler. Thromb. Vase. Biol 27:986–89 [DOI] [PubMed] [Google Scholar]
- 35.Henry PD, Bentley KI. 1981. Suppression of atherogenesis in cholesterol-fed rabbit treated with nifedipine. J. Clin. Investig 68:1366–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pitt B, Byington RP, Furberg CD, Hunninghake DB, Mancini GB, et al. 2000. Effect of amlodipine on the progression of atherosclerosis and the occurrence of clinical events. Circulation 102:1503–10 [DOI] [PubMed] [Google Scholar]
- 37.Nissen SE, Tuzcu EM, Libby P, Thompson PD, Ghali M, et al. 2004. Effect of antihypertensive agents on cardiovascular events in patients with coronary disease and normal blood pressure. The CAMELOT study: a randomized controlled trial. JAMA 292:2217–25 [DOI] [PubMed] [Google Scholar]
- 38.Weber MA, Jamerson K, Bakris GL, Weir MR, Zappe D, et al. 2013. Effects of body size and hypertension treatments on cardiovascular event rates: subanalysis of the ACCOMPLISH randomised controlled trial. Lancet 381:537–45 [DOI] [PubMed] [Google Scholar]
- 39.Lichtlen PR, Hugenholtz PG, Rafflenbeul W, Hecker H, Jost S, Deckers JW. 1990. Retardation of angiographic progression of coronary artery disease by nifedipine. Results of the International Nifedipine Trial on Antiatherosclerotic Therapy (INTACT). Lancet 335:1109–13 [DOI] [PubMed] [Google Scholar]
- 40.Napoli C, Chiariello M, Palumbo G, Ambrosio G. 1996. Calcium-channel blockers inhibit human low-density lipoprotein oxidation by oxygen radicals. Cardiovasc. Drugs Ther 10:417–24 [DOI] [PubMed] [Google Scholar]
- 41.Das R, Burke T, Van Wagoner DR, Plow EF. 2009. L-type calcium channel blockers exert an antiinflammatory effect by suppressing expression of plasminogen receptors on macrophages. Circ. Res 105:167–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guauque-Olarte S, Messika-Zeitoun D, Droit A, Lamontagne M, Tremblay-Marchand J, et al. 2015. Calcium signaling pathway genes RUNX2 and CACNA1C are associated with calcific aortic valve disease. Circ. Cardiovasc. Genet 8:812–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaden JJ, Dempfle CE, Grobholz R, Fischer CS, Vocke DC, et al. 2005. Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovasc. Pathol 14:80–87 [DOI] [PubMed] [Google Scholar]
- 44.Dorn GW 2nd. 2013. Shared genetic risk for sclerosis of valves and vessels. N. Engl. J. Med 368:569–70 [DOI] [PubMed] [Google Scholar]
- 45.Verhiel S, Piatkowski de Grzymala A, van der Hulst R.2015. Mechanism of action, efficacy, and adverse events of calcium antagonists in hypertrophic scars and keloids: a systematic review. Dermatol. Surg 41:1343–50 [DOI] [PubMed] [Google Scholar]
- 46.Nakaoka H, Miyauchi S, Miki Y. 1995. Proliferating activity of dermal fibroblasts in keloids and hypertrophic scars. Acta Derm. Venereol 75:102–4 [DOI] [PubMed] [Google Scholar]
- 47.Fujiwara M, Muragaki Y, Ooshima A. 2005. Keloid-derived fibroblasts show increased secretion of factors involved in collagen turnover and depend on matrix metalloproteinase for migration. Br. J. Dermatol 153:295–300 [DOI] [PubMed] [Google Scholar]
- 48.Okada Y, Tsuchiya W, Yada T. 1982. Calcium channel and calcium pump involved in oscillatory hyperpolarizing responses of L-strain mouse fibroblasts. J. Physiol 327:449–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee RC, Ping JA. 1990. Calcium antagonists retard extracellular matrix production in connective tissue equivalent. J. Surg. Res 49:463–66 [DOI] [PubMed] [Google Scholar]
- 50.Akat K, Borggrefe M, Kaden JJ. 2009. Aortic valve calcification: basic science to clinical practice. Heart 95:616–23 [DOI] [PubMed] [Google Scholar]
- 51.Li J, Zhao L, Ferries IK, Jiang L, Desta MZ, et al. 2010. Skeletal phenotype of mice with a null mutation in Cav 1.3 L-type calcium channel. J. Musculoskelet. Neuronal Interact 10:180–87 [PubMed] [Google Scholar]
- 52.Yang SN, Berggren PO. 2006. The role of voltage-gated calcium channels in pancreatic β-cell physiology and pathophysiology. Endocr. Rev 27:621–76 [DOI] [PubMed] [Google Scholar]
- 53.Marcantoni A, Vandael DH, Mahapatra S, Carabelli V, Sinnegger-Brauns MJ, et al. 2010. Loss of Cav1.3 channels reveals the critical role of L-type and BK channel coupling in pacemaking mouse adrenal chromaffin cells. J. Neurosci 30:491–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zaidi M, Yuen T, Sun L, Rosen CJ. 2018. Regulation of skeletal homeostasis. Endocr. Rev 39:701–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, et al. 2015. Tissue-based map of the human proteome. Science 347:1260419. [DOI] [PubMed] [Google Scholar]
- 56.Beggs MR, Lee JJ, Busch K, Raza A, Dimke H, et al. 2019. TRPV6 and Cav1.3 mediate distal small intestine calcium absorption before weaning. Cell. Mol. Gastroenterol. Hepatol 8:625–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Atsuta Y, Tomizawa RR, Levin M, Tabin CJ. 2019. L-type voltage-gated Ca2+ channel Cav1.2 regulates chondrogenesis during limb development. PNAS 116:21592–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. 2002. Expression of Cre recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33:77–80 [DOI] [PubMed] [Google Scholar]
- 59.Danielsson BR, Reiland S, Rundqvist E, Danielson M. 1989. Digital defects induced by vasodilating agents: relationship to reduction in uteroplacental blood flow. Teratology 40:351–58 [DOI] [PubMed] [Google Scholar]
- 60.Alabdulrazzaq F, Koren G. 2012. Fetal safety of calcium channel blockers. Can. Fam. Phys 58:746–47 [PMC free article] [PubMed] [Google Scholar]
- 61.Magee LA, Schick B, Donnenfeld AE, Sage SR, Conover B, et al. 1996. The safety of calcium channel blockers in human pregnancy: a prospective, multicenter cohort study. Am. J. Obstet. Gynecol 174:823–28 [DOI] [PubMed] [Google Scholar]
- 62.Cao C, Ren Y, Barnett AS, Mirando AJ, Rouse D, et al. 2017. Increased Ca2+ signaling through Cav1.2 promotes bone formation and prevents estrogen deficiency-induced bone loss. JCI Insight 2:e95512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin SS, Tzeng BH, Lee KR, Smith RJ, Campbell KP, Chen CC. 2014. Cav3.2 T-type calcium channel is required for the NFAT-dependent Sox9 expression in tracheal cartilage. PNAS 111:E1990–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cao C, Oswald AB, Fabella BA, Ren Y, Rodriguiz R, et al. 2019. The CaV1.2 L-type calcium channel regulates bone homeostasis in the middle and inner ear. Bone 125:160–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bett GC, Lis A, Wersinger SR, Baizer JS, Duffey ME, Rasmusson RL. 2012. A mouse model of Timothy syndrome: a complex autistic disorder resulting from a point mutation in Cav1.2. N. Am. J. Med. Sci 5:135–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yucel G, Altindag B, Gomez-Ospina N, Rana A, Panagiotakos G, et al. 2013. State-dependent signaling by Cav1.2 regulates hair follicle stem cell function. Genes Dev. 27:1217–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ebert AM, McAnelly CA, Srinivasan A, Linker JL, Horne WA, Garrity DM. 2008. Ca2+ channel-independent requirement for MAGUK family CACNB4 genes in initiation of zebrafish epiboly. PNAS 105:198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Burgess DL, Jones JM, Meisler MH, Noebels JL. 1997. Mutation of the Ca2+ channel β subunit gene Cchb4 is associated with ataxia and seizures in the lethargic (lh) mouse. Cell 88:385–92 [DOI] [PubMed] [Google Scholar]
- 69.Dung HC. 1977. Deficiency in the thymus-dependent immunity in “lethargic” mutant mice. Transplantation 23:39–43 [DOI] [PubMed] [Google Scholar]
- 70.Badou A, Jha MK, Matza D, Mehal WZ, Freichel M, et al. 2006. Critical role for the β regulatory subunits of Cav channels in T lymphocyte function. PNAS 103:15529–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jha MK, Badou A, Meissner M, McRory JE, Freichel M, et al. 2009. Defective survival of naive CD8+ T lymphocytes in the absence of the β3 regulatory subunit of voltage-gated calcium channels. Nat. Immunol 10:1275–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jha A, Singh AK, Weissgerber P, Freichel M, Flockerzi V, et al. 2015. Essential roles for Cavβ2 and Cav1 channels in thymocyte development and T cell homeostasis. Sci. Signal 8:ra103. [DOI] [PubMed] [Google Scholar]
- 73.Omilusik K, Priatel JJ, Chen X, Wang YT, Xu H, et al. 2011. The Cav1.4 calcium channel is a critical regulator of T cell receptor signaling and naive T cell homeostasis. Immunity 35:349–60 [DOI] [PubMed] [Google Scholar]
- 74.Niemeyer BA, Hoth M. 2011. Excitable T cells: Cav1.4 channel contributions and controversies. Immunity 35:315–17 [DOI] [PubMed] [Google Scholar]
- 75.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, et al. 2002. The human genome browser at UCSC. Genome Res. 12:996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gonzalez DAS, Cheli VT, Zamora NN, Lama TN, Spreuer V, et al. 2017. Conditional deletion of the L-type calcium channel Cav1.2 in NG2-positive cells impairs remyelination in mice. J. Neurosci 37:10038–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fields RD. 2008. Oligodendrocytes changing the rules: action potentials in glia and oligodendrocytes controlling action potentials. Neuroscientist 14:540–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Swanson JB, de Micheli AJ, Disser NP, Martinez LM, Walker NR, et al. 2019. A single-cell transcriptional atlas identifies extensive heterogeneity in the cellular composition of tendons. bioRxiv 801266. 10.1101/801266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Disser NP, Ghahramani GC, Swanson JB, Wada S, Chao ML, et al. 2020. Widespread diversity in the transcriptomes of functionally divergent limb tendons. J. Physiol 598:1537–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu H,Xu J, Liu CF, Lan Y, Wylie C, Jiang R. 2015. Whole transcriptome expression profiling of mouse limb tendon development by using RNA-seq. J. Orthop. Res 33:840–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Westenbroek RE, Anderson NL, Byers MR. 2004. Altered localization of Cav1.2 (L-type) calcium channels in nerve fibers, Schwann cells, odontoblasts, and fibroblasts of tooth pulp after tooth injury. J. Neurosci. Res 75:371–83 [DOI] [PubMed] [Google Scholar]
- 82.Davidson RM, Guo L. 2000. Calcium channel current in rat dental pulp cells. J. Membr. Biol 178:21–30 [DOI] [PubMed] [Google Scholar]
- 83.Laugel-Haushalter V, Morkmued S, Stoetzel C, Geoffroy V, Muller J, et al. 2018. Genetic evidence supporting the role of the calcium channel, CACNA1S, in tooth cusp and root patterning. Front. Physiol 9:1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jonsson L, Magnusson TE, Thordarson A, Jonsson T, Geller F, et al. 2018. Rare and common variants conferring risk of tooth agenesis. J. Dent. Res 97:515–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sun W, Chi S, Li Y, Ling S, Tan Y, et al. 2019. The mechanosensitive Piezo1 channel is required for bone formation. eLife 8:e47454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li X, Han L, Nookaew I, Mannen E, Silva MJ, et al. 2019. Stimulation of Piezo1 by mechanical signals promotes bone anabolism. eLife 8:e49631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu J, Lewis AH, Grandl J. 2017. Touch, tension, and transduction—the function and regulation of Piezo ion channels. Trends Biochem. Sci 42:57–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee W, Leddy HA, Chen Y, Lee SH, Zelenski NA, et al. 2014. Synergy between Piezo1 and Piezo2 channels confers high-strain mechanosensitivity to articular cartilage. PNAS 111:E5114–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Back M, Gasser TC, Michel JB, Caligiuri G. 2013. Biomechanical factors in the biology of aortic wall and aortic valve diseases. Cardiovasc. Res 99:232–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cosman F, Morrow B, Kopal M, Bilezikian JP. 1989. Stimulation of inositol phosphate formation in ROS 17/2.8 cell membranes by guanine nucleotide, calcium, and parathyroid hormone. J. Bone Min. Res 4:413–20 [DOI] [PubMed] [Google Scholar]
- 91.Santillan G, Baldi C, Katz S, Vazquez G, Boland R. 2004. Evidence that TRPC3 is a molecular component of the 1α,25(OH)2D3-activated capacitative calcium entry (CCE) in muscle and osteoblast cells. J. Steroid Biochem. Mol. Biol 89–90:291–95 [DOI] [PubMed] [Google Scholar]
- 92.Lieberherr M, Grosse B, Kachkache M, Balsan S. 1993. Cell signaling and estrogens in female rat osteoblasts: a possible involvement of unconventional nonnuclear receptors. J. Bone Min. Res 8:1365–76 [DOI] [PubMed] [Google Scholar]
- 93.Mermelstein PG, Becker JB, Surmeier DJ. 1996. Estradiol reduces calcium currents in rat neostriatal neurons via a membrane receptor. J. Neurosci 16:595–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Arnal JF, Lenfant F, Metivier R, Flouriot G, Henrion D, et al. 2017. Membrane and nuclear estrogen receptor alpha actions: from tissue specificity to medical implications. Physiol. Rev 97:1045–87 [DOI] [PubMed] [Google Scholar]
- 95.Filardo E, Quinn J, Pang Y, Graeber C, Shaw S, et al. 2007. Activation of the novel estrogen receptor G protein-coupled receptor30 (GPR30) at the plasma membrane. Endocrinology 148:3236–45 [DOI] [PubMed] [Google Scholar]
- 96.Edelman A, Fritsch J, Balsan S. 1986. Short-term effects of PTH on cultured rat osteoblasts: changes in membrane potential. Am. J. Physiol 251:C483–90 [DOI] [PubMed] [Google Scholar]
- 97.Krey JF, Pasca SP, Shcheglovitov A, Yazawa M, Schwemberger R, et al. 2013. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat. Neurosci 16:201–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang T, Colecraft HM. 2013. Regulation of voltage-dependent calcium channels by RGK proteins. Biochim. Biophys. Acta 1828:1644–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hatzoglou A, Ader I, Splingard A, Flanders J, Saade E, et al. 2007. Gem associates with Ezrin and acts via the Rho-GAP protein Gmip to down-regulate the Rho pathway. Mol. Biol. Cell 18:1242–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ward Y, Yap SF, Ravichandran V, Matsumura F, Ito M, et al. 2002. The GTP binding proteins Gem and Rad are negative regulators of the Rho-Rho kinase pathway. J. Cell Biol 157:291–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Maguire J, Santoro T, Jensen P, Siebenlist U, Yewdell J, Kelly K. 1994. Gem: an induced, immediate early protein belonging to the Ras family. Science 265:241–44 [DOI] [PubMed] [Google Scholar]
- 102.Ricker E, Chowdhury L, Yi W, Pernis AB. 2016. The Rho A-ROCK pathway in the regulation of T and B cell responses. F1000Research 5:2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wemhoner K, Friedrich C, Stallmeyer B, Coffey AJ, Grace A, et al. 2015. Gain-of-function mutations in the calcium channel CACNA1C (Cav1.2) cause non-syndromic long-QT but not Timothy syndrome. J. Mol. Cell. Cardiol 80:186–95 [DOI] [PubMed] [Google Scholar]
- 104.Boczek NJ, Ye D, Jin F, Tester DJ, Huseby A, et al. 2015. Identification and functional characterization of a novel CACNA1C-mediated cardiac disorder characterized by prolonged QT intervals with hypertrophic cardiomyopathy, congenital heart defects, and sudden cardiac death. Circ. Arrhythm. Electrophysiol 8:1122–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Adler A, Novelli V, Amin AS, Abiusi E, Care M, et al. 2020. An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation 141:418–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Giudicessi JR, Rohatgi RK, Tester DJ, Ackerman MJ. 2020. Variant frequency and clinical phenotype call into question the nature of minor, nonsyndromic long-QT syndrome-susceptibility gene-disease associations. Circulation 141:495–97 [DOI] [PubMed] [Google Scholar]