

Summary
Although angiogenesis-osteogenesis coupling is important in ankylosing spondylitis (AS), therapeutic agents targeting the vasculature remain elusive. Here, we identified activating transcription factor 6 (ATF6) as an important regulator of angiogenesis in the pathogenesis of AS. First, we found that ATF6 and fibroblast growth factor 2 (FGF2) levels were higher in SKG mice and in cartilage of pateints with AS1. The proangiogenic activity of human chondrocytes was enhanced by the activation of the ATF6-FGF2 axis following 7 days of stimulation with inflammatory factors, e.g., tumor necrosis factor alpha (TNF-α), interferon-γ (IFN-γ) or interleukin-17 (IL-17). Mechanistically, ATF6 interacted with the FGF2 promotor and promoted its transcription. Treatment with the ATF6 inhibitor Ceapin-A7 inhibited angiogenesis in vitro and angiogenesis-osteogenesis coupling in vivo. ATF6 may aggravate angiogenesis-osteogenesis coupling during AS by mediating FGF2 transcription in chondrocytes, implying that ATF6 represents a promising therapeutic target for AS.
Subject areas: Biological sciences, Physiology, Molecular biology
Graphical abstract
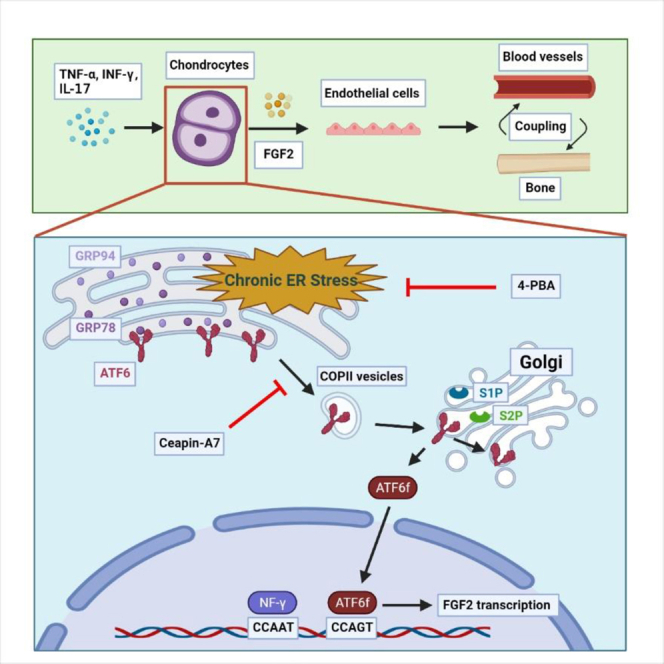
FGF2 is an effective factor that promotes angiogenesis-osteogenesis coupling and plays an important role in AS. Chondrocytes stimulated for 7 days with proinflammatory cytokines undergo chronic ERS with ATF6 pathway activation. By directly binding to the FGF2 promoter, ATF6 increases FGF2 expression. Treatment of chondrocytes with the ATF6 inhibitor Ceapin-A7 effectively inhibits FGF2 expression.
Highlights
-
•
Prolonged inflammatory stimulation triggers ERS in human chondrocytes
-
•
ERS upregulated FGF2 through ATF6 pathway, thereby promoting angiogenesis
-
•
Ceapin-A7 decreases the number of vessels and osteophytes in the AS mouse model
-
•
ATF6 may be a promising therapeutic target for AS
Biological sciences; Physiology; Molecular biology
Introduction
Ankylosing spondylitis (AS) is a rheumatic disease characterized by chronic and repeated inflammation, primarily affecting the axial skeleton and joints (Sieper and Poddubnyy, 2017). Nearly 40% of patients progress to functional deformities, including joint deformities and rigid spines (Landewé et al., 2009). Previous studies have shown that anti-TNF-α treatment is beneficial for patients with AS in relieving signs and symptoms and improving physical functions but has no effect on delaying radiographic progression (Van der Heijde et al., 2008a, 2008b, 2009). Some researchers hypothesized that the process of pathologic bone formation was independent of inflammation as AS progressed (Poddubnyy and Sieper, 2017). However, recent studies have shown that long-term treatment with anti-TNF-α for 8 years delays radiographic progression in patients with AS (Baraliakos et al., 2014; Molnar et al., 2018). A similar result was observed in a clinical study on anti-IL-17 treatment in patients with AS (Wendling et al., 2019). These results indicate that inflammation plays an important role in pathologic bone formation in patients with AS (Lories and Haroon, 2017). Further study on the relationship between the process of pathological bone formation and inflammation is needed.
Cartilage degradation and replacement by granulation tissue containing vessels is considered an important pathological change before joint ankylosis during AS (Lories, 2018; Sieper and Poddubnyy, 2017). The normal cartilage of an adult is avascular due to the presence of many factors that inhibit angiogenesis, including antiangiogenic protease inhibitors and chondromodulin-I, in the extracellular matrix (Krishnan and Grodzinsky, 2018). However, previous studies showed that the amount of collagen is lower in the articular cartilage of patients with AS than in non-AS individuals, indicating that the barrier function of cartilage to vessels might be altered in patients with AS (Bleil et al., 2015). This type of change might allow endothelial cells, osteoblasts, and other cells to invade cartilage, followed by granulation tissue formation and active osteogenesis (Bleil et al., 2014, 2016). Thus, to interfere with pathologic osteogenesis in AS, it is necessary to determine how other cell types, such as endothelial cells, invade cartilage.
Numerous studies have shown that ER stress (ERS) has a significant regulatory effect on angiogenesis (Binet and Sapieha, 2015). ERS is a self-protective response that mainly stabilizes the function of the ER by inducing the unfolded protein response (UPR) (Bettigole and Glimcher, 2015; Hetz and Papa, 2018). Among the 3 UPR pathways, the roles of the inositol-requiring enzyme 1 a (IRE1a) and protein kinase RNA-like ER kinase (PERK) pathways in angiogenesis are clear (Binet and Sapieha, 2015). However, the role of the activating transcription factor 6 (ATF6) pathway in angiogenesis has not been fully elucidated (Ghosh et al., 2010). The authors interfered with ATF6 in the hepatocellular carcinoma cell line HepG2 and observed a significant decrease in vascular endothelial growth factor (VEGF) expression, suggesting that ATF6 bound to the VEGF promoter region. ATF6 is important in cartilage development (Guo et al., 2016; Hillary and FitzGerald, 2018), but whether ATF6 regulates cartilage by regulating angiogenesis is unclear.
In this study, we show that ATF6 aggravated angiogenesis-osteogenesis coupling during AS by mediating FGF2 expression in chondrocytes. Our results indicate that ATF6 represents a promising therapeutic target for AS.
Results
Chondrocytes in AS-associated extended inflammation increase FGF2 expression
Whether chondrocytes in patients with AS undergo functional changes after extended inflammation remains unclear. Previous studies have shown that proinflammatory factors, such as TNF-α, IFN-γ, and IL-17, are significantly increased in the serum and joint fluid of patients with AS (Ranganathan et al., 2017). Thus, we extracted human chondrocytes from hip joints and identified them by immunofluorescence (IF) staining of collagen II (Figure S1). We then stimulated chondrocytes with TNF-α, IFN-γ, and IL-17 to observe the changes in angiogenic factor expression. After TNF-α stimulation of chondrocytes for 1 day (1-day stimulation) or 7 days (7-day stimulation), the expression of various angiogenic factors was upregulated (Figure 1A, 1B, and S2). However, 1-day stimulated chondrocytes tested 7 days after stimulation exhibited significantly decreased expression of angiogenic factors, close to the level before TNF-α stimulation (Figure 1A). In contrast, chondrocytes stimulated for 7 days still expressed FGF2 and VEGF at high levels (Figure 1B). Similarly, chondrocytes stimulated for 7 days with IFN-γ or IL-17 also expressed FGF2 at high levels that persisted for 7 days (Figure 1C). Western blotting (WB) and enzyme-linked immunosorbent assay (ELISA) showed increased FGF2 expression in chondrocytes after 7 days of stimulation with TNF-α, IFN-γ, or IL-17 (Figures 1D and 1E).
Figure 1.

Seven-day stimulation with inflammatory factors increased FGF2 expression in chondrocytes
(A and B) Diagram of 2 different modes of TNF-α stimulation on chondrocytes. The expression of angiogenic factors (VEGF, FGF2, IGF-1, EGF, TGF-β, and IL-6) was measured in chondrocytes cultured with TNF-α for 1 day (A) or 7 days (B).
(C) qRT-PCR analysis of VEGF and FGF2 expression in chondrocytes cultured with IFN-γ or IL-17 for 7 days, followed by the removal of the stimulus for 0 days or 7 days, respectively.
(D and E) FGF2 expression was measured by WB (D) and ELISA (E) in chondrocytes cultured with TNF-α, IFN-γ, or IL-17 for 7 days.
(F) IHC analysis of FGF2 protein in the spine joint, sacroiliac joint, knee joint and foot joint of SKG mice. Scale bar, 50 μm.
(G) Quantification of the proportions of FGF2-positive chondrocytes in SKG mice (n = 10 mice per group).
(H) IHC analysis of FGF2 protein in femur heads of healthy donors (HD), ankylosing spondylitis patients (AS) and osteoarthritis patients (OA). Scale bar, 100 μm.
(I) Quantification of the proportions of FGF2-positive chondrocytes in femur heads. (n = 10 people per group). Bars show the means ± SD.
∗p < 0.05 and ∗∗p < 0.01, 2-tailed Student's t-test (A, B, C, D, E, and G) and one-way ANOVA followed by Dunnett's post hoc test (I). d0 = day 0; d7 = day 7; HD = healthy donors; AS = ankylosing spondylitis; OA = osteoarthritis.
Previous studies showed that curdlan-treated SKG mice develop arthritis in peripheral joints and in axial joints (Ruutu et al., 2012; Sakaguchi et al., 2003). Eight weeks after treatment with curdlan, SKG mice exhibited signs of inflammation. We then sacrificed the animals and performed immunohistochemistry (IHC) staining for FGF2 in the axial joints (spine, sacroiliac) and peripheral joints (knee, foot). FGF2 was expressed at higher levels in cartilage from curdlan-treated SKG mice than in cartilage from untreated SKG mice (Figures 1F and 1G). Because cartilage is always replaced by bone in AS patients after surgery, it is difficult to obtain enough cartilage tissue for chondrocyte extraction from AS patients. Alternatively, in patients with AS, we performed hematoxylin and eosin (H&E) and IHC staining on the femur head, which contains a small amount of residual cartilage. The remaining cartilage was invaded by granulation tissue and expressed FGF2 at higher levels than the femur head in non-AS individuals (Figures 1H,1I, and S3). The characteristics of healthy donors and AS patients have been previously reported (Liu et al., 2019). These results suggest that extended inflammation induces FGF2 expression in the chondrocytes of patients with AS.
Chondrocytes secrete FGF2 during extended inflammation to induce angiogenesis
To explore whether FGF2 is an important factor for angiogenesis during the 7-day stimulation of chondrocytes, we performed tube-formation and transwell migration assays. The results demonstrated that the angiogenic activities of human umbilical vein endothelial cells (HUVECs) were enhanced by conditioned media (CM) from chondrocytes following 7 days of stimulation with TNF-α, IFN-γ, or IL-17. In contrast, treatment of HUVECs with an FGF-2 antibody (Ab) significantly abolished chondrocyte CM-induced HUVEC tube formation and migration (Figures 2A–2D). To further characterize the angiogenic function of FGF-2 expression in chondrocytes, an in vivo Matrigel plug assay was performed. The results showed that plugs containing CM from TNF-α-, IFN-γ- or IL-17-stimulated chondrocytes exhibited a notable increase in blood vessels containing red blood cells, which was inhibited dramatically by an FGF2 Ab (Figures 2E and 2F). Therefore, proinflammatory factor-induced FGF-2 expression in chondrocytes subsequently promotes angiogenesis in vitro and in vivo.
Figure 2.

Chondrocytes secrete FGF2 to induce angiogenesis
The conditioned media (CM) of chondrocytes cultured with TNF-α, IFN-γ, or IL-17 for 7 days were collected separately and then applied to HUVECs.
(A–D) Tube-formation (A) and transwell migration (C) assays were performed on HUVECs cultured in CM. The numbers of branches (B) and migratory cells (D) were calculated and quantified using ImageJ software. Scale bar, 100 μm.
(E and F) Matrigel plugs containing chondrocyte CM, TNF-α-treated chondrocyte CM, INF-γ-treated chondrocyte CM, and IL-17-treated chondrocyte CM with or without an FGF-2-neutralizing antibody were subcutaneously injected into nude mice. The plugs were collected on day 7. Paraffin sections of Matrigel plugs were stained with H&E (E), and vessel structures containing red blood cells were quantified (F) (n = 6 mice per group). Scale bar, 100 μm.
Bars show the means ± SD. ∗p < 0.05 and ∗∗p < 0.01, 2-tailed Student's t-test.
Extended inflammation triggers ERS and enhances the angiogenic ability of chondrocytes
Because inflammation is an important cause of ERS (Grootjans et al., 2016), we examined the expression of ERS-related genes in chondrocytes. The expression of GRP78 and GRP94 in chondrocytes was significantly increased after 7 days of stimulation with inflammatory factors (Figures 3A, 3C, and 3D). The UPR is the major protective mechanism by which ERS is relieved and involves 3 pathways (IRE1a, PERK, and ATF6) (Hetz and Papa, 2018). Further detection of IRE1a, PERK, and ATF6 revealed that their expression in stimulated chondrocytes was higher than that in control chondrocytes (Figure 3B). ATF6 was expressed at the highest levels among proteins involved in the 3 key pathways (Figure 3B). We also examined chondrocyte ERS levels in vivo by IHC staining of cartilage samples collected from AS patients. Both GRP78 and ATF6 were expressed at high levels in the chondrocytes of patients with AS (Figure 3E).
Figure 3.

Seven-day stimulation with inflammatory factors triggers ERS in chondrocytes and enhances angiogenesis
(A and B) The expression of ER stress (ERS) genes (GRP78, GRP94; (A) and unfolded protein response (UPR) pathway genes (IRE1a, XBP-1, PERK, CHOP, ATF6, ERO1B; (B) was measured in chondrocytes cultured with TNF-α, IFN-γ or IL-17 for 7 days.
(C and D) Protein levels of GRP78, GRP94, and ATF6 were measured with WB and quantified using ImageJ software.
(E) IHC analysis of GRP78 and ATF6 expression on femur heads of healthy donors (HD), ankylosing spondylitis patients (AS) and osteoarthritis patients (OA). Scale bar, 100 μm. Quantification of the proportions of GRP78- and ATF6-positive chondrocytes in femur heads (n = 10 people per group).
(F) FGF2 expression was measured by ELISA in chondrocytes cultured with 4-PBA.
(G) Transwell migration assays were performed on HUVECs cultured in chondrocyte CM. The number of migratory cells was calculated and quantified using ImageJ software. Scale bar, 100 μm. Bars show the means ± SD.
∗p < 0.05 and ∗∗p < 0.01, 2-tailed Student's t-test (A, B, D, F, and G) and one-way ANOVA followed by Dunnett's post hoc test (E).
Previous studies have shown that ERS induces angiogenic activity (Binet and Sapieha, 2015). We thus explored whether the angiogenic effect of stimulated chondrocytes was related to ERS. The ERS alleviator 4-phenyl butyric acid (4-PBA) significantly reduced FGF2 expression in stimulated chondrocytes (Figure 3F) and abolished the ability of stimulated chondrocytes to promote the migration of HUVECs (Figure 3G) and tube formation (Figures S4 and S5). All of these results indicate that proinflammatory factors in AS stimulate chondrocytes and promote angiogenic activity by increasing ERS levels.
ERS induces FGF2 expression in chondrocytes through the ATF6 pathway
While many studies have focused on the relationship between IRE1a or PERK and angiogenesis, few have proven the angiogenic role of ATF6. As the expression of ATF6 was the highest in proinflammatory factor-stimulated chondrocytes, we explored the role of ATF6 in the angiogenic effect of stimulated chondrocytes. First, chondrocytes were transfected with plasmids encoding an shRNA targeting ATF6 (Figure S6). We then examined FGF2 and GRP78 (a known target gene of ATF6) expression in control and proinflammatory factor-stimulated chondrocytes. FGF2 and GRP78 expression was significantly reduced by ATF6 knockdown in stimulated chondrocytes but remained unchanged in control chondrocytes (Figures 4A–4D). Furthermore, tube-formation and transwell migration assays showed that the knockdown of ATF6 significantly reduced the angiogenic effect of stimulated chondrocytes but not control chondrocytes (Figure S7).
Figure 4.

Activation of the ATF6 pathway promoted FGF2 expression in chondrocytes
(A) FGF2 mRNA levels were measured in chondrocytes with ATF6 knockdown.
(B and C) Protein levels of ATF6, GRP78, and FGF2 were measured with WB and quantified using ImageJ software.
(D) FGF2 expression was measured by ELISA in chondrocytes with ATF6 knockdown.
(E) Confocal images show that ATF6 translocated to the nucleus in chondrocytes after TNF-α, IFN-γ, and IL-17 stimulation, which was blocked by the ATF6 activation inhibitor Ceapin-A7, Scale bar, 20 μm.
(F) Mean fluorescence intensity (MFI) was quantified by ImageJ software.
(G) ELISA analysis of FGF2 expression on chondrocytes treated with or without Ceapin-A7.
(H) Tube formation assays were performed with CM derived from chondrocytes treated with or without Ceapin-A7.
The number of branches was calculated and quantified using ImageJ software (I). Scale bar, 100 μm. Bars show the means ± SD.
∗p < 0.05 and ∗∗p < 0.01, 2-tailed Student's t-test (A, C, D, G, and I) and one-way ANOVA followed by Dunnett's post hoc test (F). NC = negative control (control shRNA).
ATF6 activation includes ATF6 cleavage by S1P and S2P in the Golgi apparatus and thus translocation to the nucleus (Hillary and FitzGerald, 2018). An analysis of confocal images showed that ATF6 translocated to the nucleus of chondrocytes after stimulation with TNF-α (Figures 4E and 4F). Ceapin-A7 is a specific inhibitor of ATF6 activation that functions by inhibiting the transport of ATF6 from the ER to the Golgi apparatus (Gallagher and Walter, 2016). To explore whether FGF2 expression and the angiogenic effect of stimulated chondrocytes were related to activated or inactivated ATF6, we treated stimulated chondrocytes with Ceapin-A7. The inhibition of ATF6 activation by Ceapin-A7 decreased FGF2 expression (Figures 4G, S8A, and S8B) and the ability of stimulated chondrocytes to promote the migration of HUVECs and tube formation (Figures 4H, 4I, and S8C). Taken together, these results indicate that proinflammatory factors induce FGF2 expression in chondrocytes at least partially by activating the ATF6 pathway.
ATF6 directly promotes FGF2 transcription
Activated ATF6 mediates gene expression by recognizing ERSE I (CCAAT-N9-CCAC(G)) and ERSE II (ATTGG-N1-CCAC(G)) (Kokame et al., 2001). To explore whether ATF6 promoted FGF2 expression in chondrocytes in a direct or an indirect manner, we scanned FGF2 promoter regions spanning from −10,000 bp to 0 bp of the transcription start site (TSS) for a potential ERSE in various species. The analysis revealed ERSE sequences in the FGF2 promoters of various species, including humans, mice, rabbits, cattle, and sheep (Figure 5A). We also identified 4 potential ERSEs in the promoters of hFGF2, among which 2 potential ERSEs, namely, M1 (−1420 to −1402) and M2 (−1784 to −1766), were located at -2000-0 bp (Figure 5B). These results support that ATF6 directly binds to the FGF2 promoter and mediates its transcription.
Figure 5.

ATF6 directly binds to the FGF2 promoter and promotes its transcription
(A) Schematic representation of the FGF2 promoters from humans (Homo sapiens, hFGF2), mice (Mus musculus, mFGF2), rabbits (Oryctolagus cuniculus, oFGF2), cattle (Bos taurus, bFGF2), and sheep (Ovis aries, ovFGF2). Different squares represent the possible ATF6-binding sites (ERSE I or ERSE II) of different species. TSS, transcriptional start site.
(B) Diagram showing the noncanonical ERSE I and ERSE II in the hFGF2 promoter. NF-γ is a transcription factor that binds to the CCAAT sequence.
(C and D) Both the mRNA (C) and protein (D) levels of FGF2 in chondrocytes were measured.
(E) After transfection of ATF6 (1–373) in chondrocytes, the DNA binding ability of ATF6 to the hFGF2 and GRP78 promoters was analyzed using ChIP assays.
(F) Diagram of the locations of ERSE I (M1, M2) in the hFGF2 5′-flanking region, their sequences (WT), and the mutations to those sequences (mt1, mt2, mt3).
(G and H) Chondrocytes were transfected with plasmids encoding hFGF2(−2000/-1173)-Luc WT, mt1, mt2, or mt3. Then, chondrocytes were transfected with ATF6-expressing vectors (G) or treated with ERS inducers (Tg and Tm; H). Forty-eight hours later, luciferase levels were measured in extracts.
Bars show the means ± SD. ∗p < 0.05 and ∗∗p < 0.01, 2-tailed Student's t-test (C, D, and E) and one-way ANOVA followed by Dunnett's post hoc test (G and H).
To explore whether ATF6 directly binds to the FGF2 promoter and mediates transcription, we overexpressed full-length ATF6, the active form of ATF6 (1-373), or the negative mutation ATF6 (1-373) m1 in chondrocytes. FGF2 expression in chondrocytes was promoted by ATF6 and ATF6 (1-373) but not ATF6 (1-373) m1 (Figures 5C and 5D). We then cloned a truncated version of the hFGF2 5′-flanking sequence (−2000 to −1173) in front of firefly luciferase (referred to as WT-Luc). M1 and M2 were mutated either singly or together (m1-Luc, m2-Luc, and m3-Luc) (Figure 5F). As shown in Figure 5G, transfection with ATF6 (1-373) significantly increased luciferase activity in WT-Luc-, m1-Luc- and m2-Luc-transfected 293T cells but not m3-Luc-transfected 293T cells, indicating that ATF6 binds to WT-Luc, m1-Luc and m2-Luc but not m3-Luc. We further used thapsigargin (Tg) and tunicamycin (Tm) to induce endogenous ATF6 expression. Tg and Tm significantly increased luciferase activity in WT-Luc-, m1-Luc- and m2-Luc-transfected 293T cells but not m3-Luc-transfected 293T cells (Figure 5H). Finally, we performed a ChIP assay to explore the DNA-binding ability of ATF6 to the FGF2 promoter. The results revealed that ATF6 bound to the M1 and M2 sequences in the FGF2 promoter, as well as the GRP78 gene promoters, in chondrocytes. Both exogenous, active ATF6 and the expression of endogenous ATF6 increased ATF6 binding to the human and the mouse FGF2 promoter, which was inhibited by Ceapin-A7 (Figures S9 and S10). These results suggest that ATF6 directly binds to the FGF2 promoter and promotes its transcription.
ATF6 aggravates angiogenesis-osteogenesis coupling in mice
To observe the effect of ERS on angiogenesis and osteogenesis, we performed an intra-articular injection of Tg to induce ERS in mouse chondrocytes (Figure S11A). IHC staining was also performed on the knee joints of experimental mice. Tg treatment significantly increased the expression of ATF6, GRP78, and FGF2. However, after treatment with the ATF6 inhibitor Ceapin-A7, ATF6 expression was not changed, while the expression of GRP78 and FGF2 was significantly decreased (Figure S11B), which suggested that FGF2 and GRP78 act downstream of ATF6 in mouse chondrocytes. To explore the changes in angiogenesis and osteogenic activity in the knee joint, we performed IF staining of the endothelial cell marker CD31 and the osteoblast marker Osteocalcin (OSX) in experimental mice. As shown in Figure S11C and D, Tg treatment significantly increased CD31+ vessels and OSX+ cells near the endosteum in bone marrow, suggesting that ERS leads to increased angiogenesis and osteogenic activity. However, after treatment with the ATF6 inhibitor Ceapin-A7, CD31+ vessels and OSX+ cells were significantly reduced, suggesting that the enhancement of angiogenesis and osteogenic activity is achieved mainly through the ATF6 pathway.
We further treated SKG mice with Ceapin-A7. One week after curdlan induction, SKG mice were treated with drinking water containing DMSO or 1 μM Ceapin-A7 (Figure 6A). Because a previous study showed that ATF6 antagonized chronic liver injury (Cinaroglu et al., 2011; Hillary and FitzGerald, 2018), we evaluated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels in the sera of SKG mice. Neither ALT nor AST levels were elevated in Ceapin-A7-treated mice (Figure S12), indicating that Ceapin-A7 did not cause liver damage in mice. To determine whether Ceapin-A7 influences the inflammatory state in SKG mice, we also examined the severity of arthritis (Figure 6B) and the local expression of TNF-α, IFN-γ and IL-17 in hind paws (Figure 6C). TNF-α, IFN-γ and IL-17 levels did not change in Ceapin-A7-treated mice, indicating that Ceapin-A7 does not influence the inflammatory state in SKG mice. Twelve weeks after curdlan induction, SKG mice developed severe arthritis. IF staining of CD31 and OSX in the vertebral body showed that the numbers of CD31+ vessels and OSX+ cells were increased in untreated SKG mice compared with Ceapin-A7-treated SKG mice (Figures 6D and 6E). We also performed a micro-CT scan and found bony bridging between vertebrae (spinal ankylosis) (6/10, n = 10). Following treatment with Ceapin-A7, the occurrence of spinal ankylosis decreased (1/10, n = 10) (Figure 6F). Similar osteogenic changes were observed in the hind paws, and new bone formed in the hind paws (16/20, n = 10). Following treatment with Ceapin-A7, the incidence of osteophyte formation was significantly reduced (4/20, n = 10) (Figure 6G). Moreover, IHC staining showed low FGF2 expression in the cartilage of SKG mice treated with Ceapin-A7 (Figures 6F and 6G). Furthermore, we also obtained the same results using CIA models (Figure S13). These results indicate that the ATF6 inhibitor Ceapin-A7 slows the pathological progression of osteogenesis by inhibiting angiogenesis-osteogenesis coupling. Taken together, these data suggest that chondrocytes stimulated by extended inflammation undergo ERS via activation of the ATF6 pathway and thus secrete more FGF2 to aggravate angiogenesis-osteogenesis coupling in AS.
Figure 6.

ATF6 regulates angiogenesis-osteogenesis coupling through FGF2 in vivo
(A) Representative SKG mice 12 weeks after curdlan injection. Insets show a healthy, untreated SKG mouse (left), a curdlan-treated SKG mouse with arthritis (middle) and a curdlan-treated SKG mouse treated with Ceapin-A7.
(B) Clinical scores in SKG mice were measured (n = 10 mice per group).
(C) Local mRNA expression of TNF-α, IFN-γ and IL-17 in the hind paws of SKG mice. (n = 10 mice per group).
(D) IF staining of vertebral bodies from SKG mice showing endothelial cells (CD31+) and osteoblasts (OSX+). Scale bar, 50 μm.
(E) Quantification of the proportions of CD31 + vessels and OSX + cells (n = 10 mice per group).
(F and G) Representative micro-CT images of the spine (F) and ankle (G) obtained from SKG mice at baseline, 12 weeks after induction, and 12 weeks after induction and treated with Ceapin-A7. The red arrows point to bony bridging between vertebrae in (F) and osteophytes in (G). IHC staining of FGF2 in spine and ankle. Scale bar, 50 μm.
Bars show the means ± SD. ∗p < 0.05 and ∗∗p < 0.01, one-way ANOVA followed by Dunnett's post hoc test.
Discussion
Although inflammation is an important pathologic process in AS, the association between inflammation and pathologic bone formation has not yet been illuminated. More than 10 years ago, some researchers hypothesized that enthesitis was uncoupled from pathologic bone formation in patients with AS (Poddubnyy and Sieper, 2017). In vitro experiments showed that proinflammatory factors, such as TNF-α and IL-1, accelerated osteoclast activity while inhibiting osteoblast differentiation (McInnes and Schett, 2007). Furthermore, many mechanisms other than inflammation, such as BMP, Wnt, Hh signaling and mechanical loading, can explain pathologic bone formation in AS (Haynes et al., 2012; Lories et al., 2005; Ruiz-Heiland et al., 2012). We previously showed that mesenchymal stem cells from patients with AS secreted more BMP-2 but less Noggin coupled with abnormal osteogenic differentiation than cells from healthy individuals (Xie et al., 2016). However, whether these signaling pathways act downstream of inflammation or independent of inflammation remains unknown. In our study, we showed that proinflammatory factor stimulation promoted the extended angiogenesis effect of chondrocytes, even after the removal of proinflammatory factors. This result indicates that we should take into account the interval between two inflammatory periods during which there might be abnormal osteogenic activity.
The structural destruction and replacement of cartilage by granulation tissue is an important pathological change observed in patients with AS (Lories, 2018). Actually, an abnormal cartilage structure might be a common change before osteophyte formation. In 2009, Benjamin et al. (2009) applied an IHC assay to spinal osteophytes from elderly individuals and found a mixture of bone, cartilage, blood vessels, and other tissues. The authors also observed active osteogenesis and osteolysis activity among osteophytes. This discovery indicated that the formation of osteophytes is the result of cartilage calcification, during which blood vessels play an important role. Additionally, IHC staining of the hip joints and facet joints from AS patients revealed an abnormal cartilage structure, inside of which blood vessels and granulation tissue, accompanied by active osteogenesis and osteolysis activity, were observed (Bleil et al., 2014, 2016). Taken together, these results suggest that blood vessel invasion is closely related to cartilage destruction and calcification (Lories, 2018). However, the mechanism of blood vessel invasion remains unknown. As we showed above, chondrocytes treated with proinflammatory factors secrete a variety of angiogenic factors and promote endothelial cell migration and lumen formation. Similarly, in hip joints from patients with AS, chondrocytes express high levels of FGF2. Therefore, we hypothesize that inflammation is a major cause of the structural destruction of cartilage by promoting vascular invasion. Further study is needed to clarify the role of inflammation in the structural destruction of cartilage and pathologic bone formation.
Currently, whether ERS plays a role in AS development remains controversial (Pedersen and Maksymowych, 2019). Although several lines of evidence have demonstrated high ERS in HLA-B27 transgenic animals, data from patients with AS are lacking. Dong et al. (2008) revealed that macrophages in peripheral joints from patients with AS expressed high levels of GRP78, which indicated high levels of ERS. However, some studies showed that the ERS level in several tissues, including the peripheral blood, synovia, and intestine, from patients with AS did not differ from that in controls (Ciccia et al., 2014). We found that the levels of GRP78 and ATF6 were higher in chondrocytes in the femur heads of AS patients than in those of controls, prompting chondrocytes in patients with AS to have high levels of ERS. Similarly, high levels of GRP78 and ATF6 were found in the chondrocytes of SKG mice. In addition, we treated chondrocytes with proinflammatory factors for 7 days and successfully induced ERS. All of these results indicate that certain cell types in patients with AS have a high level of ERS and might be induced by extended inflammation.
ERS is a key regulator of angiogenesis (Binet and Sapieha, 2015). Many studies have focused on the mechanisms by which ERS regulates angiogenic activities. A previous study showed that the expression of the key angiogenic factor VEGF was under the control of Xbp-1, ATF4 and ATF6 (De Palma et al., 2017). Ghosh et al. (2010) found that ATF6 promoted VEGF transcription by directly binding to its gene promoter in the liver cancer cell line HepG2. However, in our study, we did not observe a change in VEGF mRNA levels in chondrocytes after ATF6 knockdown. This result suggested that ATF6 was not a key transcription factor by which VEGF transcription was regulated in chondrocytes. However, we noted that FGF2 mRNA levels decreased after ATF6 knockdown or ATF6 inhibition in chondrocytes. We also demonstrated that ATF6 directly bound to the FGF2 promoter region in human chondrocytes. Furthermore, we found that the ATF6 α-specific inhibitor Ceapin-A7 slowed the pathological progression of osteogenesis by inhibiting angiogenesis-osteogenesis coupling. However, ATF6 α and ATF6 β are regulated in different manners, which needs further exploration (Mehrbod et al., 2019; Wu et al., 2011). These results indicate that ATF6-mediated FGF2 transcription is an important pathway by which ERS regulates angiogenic activities.
FGF2 is a well-studied polypeptide that functions in angiogenesis, tissue regeneration, and neuroprotection (Akl et al., 2016; Bossé and Rola-Pleszczynski, 2008; Woodbury and Ikezu, 2014). Several lines of evidence suggest that FGF2 promotes cartilage repair and bone growth (Huang et al., 2018; Poudel et al., 2019; Yang et al., 2018) and plays an important role in the process of osteoarthritis (Wang et al., 2019; Yu et al., 2019). However, the direct regulatory effect of FGF2 on osteoblasts is still under debate. Some studies have suggested that the FGF2 signaling pathway enhances the transcriptional activity of Runx2 and promotes osteoblast differentiation (Kawane et al., 2018), while others have indicated that FGF2 inhibits osteoblast differentiation (Lee et al., 2016). Nevertheless, it is hypothesized that the effect of FGF2 on cartilage repair and bone growth depends on its angiogenesis promotion effect (Poudel et al., 2019). By performing IHC staining on sections of femoral heads from patients with AS, we found that FGF2 was highly expressed in femoral head chondrocytes of patients with AS and that there was an increase in vascular structures in cartilage tissues. Based on the results described above, we conclude that FGF2 plays an important role in the processes of pathologic osteogenesis, and it is likely to be achieved by promoting vascular growth.
In summary, we identified ATF6 as a critical positive regulator of angiogenesis-osteogenesis coupling in AS. Activated by extended inflammation, ATF6 increases FGF2 expression by mediating FGF2 transcription in chondrocytes. This study also demonstrated that ATF6 in chondrocytes may represent a promising therapeutic target for AS.
Limitations of the study
Several study limitations are acknowledged in this work. Our findings are restricted to 7-day stimulation with inflammatory factors. Future studies performing longer stimulation with inflammatory factors are needed to validate the long-term effects of inflammatory factors on chondrocytes. Our mouse model of AS is SKG mice. Because a specific animal model of AS is currently unavailable, a discussion of the specificity of AS treatment in animal models is not accurate. In addition, because the pathogenesis of AS remains unclear, no disease model perfectly mimics AS. SKG mice are a model of arthritis that can only be used to explain the effects of inflammation on joints and cartilage. However, because of its characteristics that can lead to spinal fusion, it is also used as an AS model. Finally, since cartilage is always replaced by bone in patients undergoing surgery for AS, we failed to obtain sufficient cartilage tissue from patients with AS for chondrocyte extraction.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-FGF2 | Signalway Antibody | #36769 |
| Rabbit polyclonal anti-ATF6 | Signalway Antibody | #32008 |
| Rabbit monoclonal anti-ATF6 | Abcam | #ab227830 |
| Rabbit monoclonal anti-GRP78 | Cell Signaling Technology | AB_2119845 |
| Rabbit monoclonal anti-GRP94 | Cell Signaling Technology | AB_2722657 |
| Rabbit monoclonal anti-GAPDH | Cell Signaling Technology | AB_10622025 |
| Rabbit polyclonal anti-CD31 | Abcam | AB_726362 |
| Rabbit polyclonal anti-OSX | Abcam | AB_10675660 |
| Goat polyclonal anti-FGF2 | R&D systems | AB_2104324 |
| Biological samples | ||
| Patient-derived articular cartilage | Sun Yat-sen Memorial Hospital (Sun Yat-sen University, Guangzhou, China) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Curdlan | InvivoGen | tlrl-curd |
| Thapsigargin | Sigma-Aldrich | 67526-95-8 |
| Tunicamycin | Sigma-Aldrich | 11089-65-9 |
| 4-phenyl butyric acid | Sigma-Aldrich | 15118-60-2 |
| Ceapin-A7 | Sigma-Aldrich | 2323027-38-7 |
| Recombinant human TNF-α | PeproTech | 300-01A |
| Recombinant human IFN-γ | PeproTech | 300-02 |
| Recombinant human IL-17 | PeproTech | 200-17 |
| Critical commercial assays | ||
| Human FGF basic DuoSet ELISA | R&D systems | DY233 |
| SimpleChIP Enzymatic ChIP Kit | Cell Signaling Technology | #9002 |
| Experimental models: cell lines | ||
| human umbilical vein endothelial cells | iCell Bioscience | iCell-0045a |
| human embryonic kidney cell line 293T | iCell Bioscience | iCell-h237 |
| Experimental models: organisms/strains | ||
| Mouse: C57BL/6N | The Guangdong Medical Laboratory Animal Center | N/A |
| Mouse: BALB/c | The Guangdong Medical Laboratory Animal Center | N/A |
| Mouse: SKG | S. Sakaguchi (University of Kyoto, Kyoto, Japan) | N/A |
| Mouse: C57BL/6J | the National Laboratory Animal Center (Taipei, Taiwan) | RMRC11005 |
| Oligonucleotides | ||
| Primers used in qRT-PCR for VEGF, FGF2, IGF-1, EGF, TGF-β, IL-6, GAPDH, GRP78, GRP94, IRE1, PERK, and ATF6, see Table S2 | This paper | N/A |
| Primer used in CHIP: GRP78 Forward: 5’-GCG GAG CAG TGA CGT TTA TT-3’ Reverse: 5’-GAC CTC ACC GTC GCC TAC T-3’ | This paper | N/A |
| Primer used in CHIP: FGF2 M1 Forward: 5’-TCTGAGCAAATAGGCCTTGCT-3’ Reverse: 5’-GGCTGAAGCCCCTGTAACAAA-3’ | This paper | N/A |
| Primer used in CHIP: FGF2 M2 Forward: 5’-ATATGCCTGGTTTTGGGCCT-3’ Reverse: 5’-CAGCCTACCGAATAGCTGGG-3’ | This paper | N/A |
| Plasmid: Pcgn-ATF6 | Addgene | #11974 |
| Plasmid: Pcgn-ATF6 (1-373) | Addgene | #27173 |
| Plasmid: Pcgn-ATF6 (1-373) m1 | Addgene | #27174 |
| Software and algorithms | ||
| ImageJ software | National Institutes of Health | imagej.nih.gov/ij/ |
| MicroCT Ray software V3.0 | Scanco Medical | http://www.scanco.ch/ |
| Other | ||
| angiogenesis μ-slides | Ibidi | 81506 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Huiyong Shen (shenhuiy@mail.sysu.edu.cn).
Material availability
This study did not generate new, unique reagents.
Data and code availability
This study did not generate datasets.
Experimental model and subject details
Patient recruitment
This study was approved by the Ethics Committee of The Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, China. Twenty trauma patients and twenty AS patients who underwent hip replacement surgery were recruited, and femur heads were collected. The characteristics of the study subjects are listed in Table S1. All AS patients met the modified New York criteria. Written informed consent was obtained from all participants.
Mice
All animal care protocols and experiments were reviewed and approved by the Ethics Committee of Sun Yat-sen University. C57BL/6N male mice and BALB/c male nude mice (4 weeks old) were purchased from the Guangdong Medical Laboratory Animal Center (Guangdong, China). SKG mice (4 weeks old) were obtained from S. Sakaguchi (University of Kyoto, Kyoto, Japan). C57BL/6J male mice (8–10 weeks old) were purchased from the National Laboratory Animal Center (Taipei, Taiwan). These mice were maintained according to the guidelines established by the Animal Care Committee of China Medical University.
For the curdlan induced model, disease was induced in C67BL/6N, BALA and SKG mice between 6 and 10 weeks of age using 3 mg of curdlan (InvivoGen, USA) administered by intraperitoneal injection. Clinical features in the mice were monitored weekly and scored by the same observer, who was blinded to treatment, as follows: 0, no swelling or redness; 0.1, swelling or redness of digits; 0.5, mild swelling and/or redness of wrists or ankle joints; and 1, severe swelling of the larger joints. The scores of the affected joints were summed; the maximum possible score was 6. All mice were housed in a specific pathogen-free facility.
The CIA mouse model was established in C57BL/6J mice between 8 and 10 weeks of age using a previously reported protocol (Cinaroglu et al., 2011; Mehrbod et al., 2019). Briefly, in the primary immunization with bCII/CFA emulsion, bCII (100 μg) was dissolved in dilute acetic acid (0.1 M) to the desired concentration (2 mg/ml), emulsified in 0.25 ml CFA (1:1) and then injected intradermally into the base of the tail. At 3 weeks after primary immunization, a collagen/IFA emulsion was used to ensure the induction of the incidence of CIA. bCII (100 μg) was emulsified in IFA (1:1) and then injected into the hind leg as a booster. The incidence of arthritis can be observed within 6 weeks after primary immunization, and overall, 95% of the mice will develop severe arthritis.
Cell culture and treatment
Primary human articular chondrocytes were collected from the articular cartilage through enzymatic digestion. Briefly, cartilage slices were minced and washed clean with 1% PBS, followed by digestion in a mixture of 0.25% collagenase II (Gibco, USA) in FBS (Hangzhou Sijiqing Biological Engineering Material Co. Ltd., China) free DMEM (glucose 4500 mg/L; Gibco) supplemented with Pen-Strep (Guangzhou Jingxin Biotechnology Co., Ltd., China) at 37°C for 3-5 h in a spinner flask in an incubator with a 5% CO2 atmosphere. Then, the cell suspension was used to establish cultures in a T75 flask. At 3-4 days after harvesting, primary chondrocytes were replated at 80% to 85% confluence in 6-well plates and used for further studies.
HUVECs and the human embryonic kidney cell line 293T were obtained from iCell Bioscience (Shanghai, China). HUVECs were cultured in endothelial growth medium 2 (EGM-2; Lonza, Switzerland) containing 10% FBS and the supplied growth factors, and cells at the third to fourth passages were used in all experiments. 293T cells were cultured in high-glucose DMEM supplemented with 10% FBS.
The ERS inducers Tm (1 μg/ml) and Tg (1 μM), the ERS alleviator 4-phenyl butyric acid (4-PBA, 5 mM) and the ATF6 inhibitor Ceapin-A7 (500 nM) were purchased from Sigma-Aldrich. Chondrocytes were cultured in DMEM supplemented with Tm or Tg for 6 h. Chondrocytes were cultured in DMEM supplemented with 4-PBA or Ceapin-A7 for 24 h. The inflammatory cytokines TNF-α (10 ng/ml), IFN-γ (500 U/L) and IL-17 (10 ng/ml) were obtained from PeproTech. For 1 day of stimulation, chondrocytes were incubated with TNF-α for 1 day. For 7 days of stimulation, chondrocytes were incubated with TNF-α, IFN-γ or IL-17 for 7 days, and the medium was changed every other day. Then, the stimulus was removed, and the samples were cultured in DMEM and harvested after an additional 7 days. After the stimulus was removed, the samples were cultured in EGM-2 for 24 h to produce CM for further experiments (Figure 1A).
Method details
Plasmid transfection
Plasmid transfections were performed using Lipofectamine 3000 (Invitrogen, USA) according to the manufacturer’s instructions; shRNAs and shATF6 were purchased from GenePharma (Shanghai, China). pCGN-ATF6, pCGN-ATF6 (1-373) and pCGN-ATF6 (1-373) m1 were gifts from Professor Ron Prywes (Addgene plasmid 11974, 27173, 27174).
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from cultured cells or tissues using TRIzol Reagent (Invitrogen, USA) and transcribed into cDNA using a PrimeScriptTM RT Reagent Kit (Takara Bio, Mountain View, CA, USA) according to the manufacturer’s instructions. qRT-qPCR was performed with a SYBR PrimeScript RT-qPCR Kit (Takara, USA) for 40 cycles at 95°C for 5 s and 60°C for 30 s on a Light Cycler 480 Real-Time PCR System (Roche, Switzerland). All primer sequences used in this study are listed in the supplemental information (Table S2).
Western blotting (WB)
Cells were harvested and lysed in RIPA buffer containing a protease inhibitor cocktail (1:100). Total protein was extracted, and the concentration was determined by the BCA method (Thermo Fisher Scientific, USA). Then, equal amounts of total protein were separated by 10% SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore). The membranes were blocked and incubated with primary antibodies against FGF2 (36769, SAB), ATF6 (32008, SAB), GRP78 (3177S, CST), GRP94 (20292, CST), and GAPDH (5174, CST). Protein bands were then incubated with secondary anti-mouse or anti-rabbit peroxidase-linked antibodies (Beyotime). We then used enhanced chemiluminescent (ECL) detection reagents (Beyotime) to obtain the final result. Three replicates were performed for all WB analyses. WB results were quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Enzyme-linked immunosorbent assay (ELISA)
To measure FGF-2 production by chondrocytes in CM, cells were seeded into 6-well plates (6×104 cells/well). Then, the cells were treated with TNF-α (10 ng/ml), IFN-γ (500 U/L) and IL-17 (10 ng/ml) and incubated in a humidified incubator at 37°C for 7 days. After incubation, the supernatant CM was collected and stored at -80°C until the assay was performed. FGF-2 in the CM was assayed using a human FGF basic DuoSet ELISA Development Kit (R&D Systems) according to the manufacturer’s procedure.
Immunohistochemistry (IHC) and immunofluorescence (IF)
Paraffin-embedded sections were prepared, mounted onto slides, deparaffinized in xylene, rehydrated in a graded alcohol series and washed in deionized water. After antigen retrieval (sections were heated at 95-100°C on a hotplate for 30 min in 10 mM sodium citrate, pH 6.0), intrinsic peroxidase activity was blocked by incubation with 3% H2O2. Nonspecific antibody-binding sites were blocked using 3% BSA in PBS. Sections were then incubated with appropriately diluted primary antibodies specific for human or mouse CD31 (ab28364, Abcam), OSX (ab93876, Abcam), FGF2 (36,769, SAB), ATF6 (32,008, SAB) or GRP78 (3177S, CST) at 4°C overnight. Then, we followed the instructions of the SP Rabbit & Mouse HRP Kit (CWBio) for IHC. Alternatively, we incubated the slides with a fluorescence-labeled secondary antibody (CST). Slides were observed under a light microscope or a fluorescence microscope.
Transwell migration assay
The transwell migration assay was performed using transwell inserts (8.0-μm pore size; Costar) in 24-well plates. HUVECs (104 cells in 200 μL of EGM-2 with 10% FBS) were then seeded into the upper chamber, and 300 μL of chondrocyte CM was placed in the lower chamber. Cells on the lower side of the transwell membrane were examined and counted under a microscope after crystal violet staining.
Tube-formation assay
Matrigel (BD Biosciences) was melted at 4°C, added to angiogenesis μ-slides (81,506, Ibidi, Germany) at 10 μL/well, and then incubated at 37°C for 30 min. HUVECs (2×103 cells) were resuspended in a 1:1 mixture of EGM-2 serum-free medium and chondrocyte CM (total 50 μL) and added to the wells. After 12 hr of incubation at 37°C, HUVEC tube formation was assessed by microscopy, and each well was imaged under a light microscope. The numbers of branches were calculated and quantified using ImageJ software.
In vivo Matrigel plug assay
Four-week-old male nude mice were divided into three groups (n = 10 for each group) and subcutaneously injected with 150 μL of Matrigel containing chondrocyte CM, control CM, TNF-α-treated CM, and TNF-α-treated CM with FGF-2 neutralizing antibody (Ab; AF-233, R&D Systems). On day 7, the Matrigel plugs were harvested: some were fixed with 3.7% paraformaldehyde for at least 2 days and then embedded in paraffin and subsequently processed for IHC and IF staining, whereas others were evaluated by Drabkin's method (Drabkin's Reagent Kit, Sigma-Aldrich) to quantify the hemoglobin concentration.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed according to the manufacturer's instructions provided in the SimpleChIP Enzymatic ChIP Kit (CST). Briefly, the ChIP assay was performed using protein A/G agarose (Thermo Fisher Scientific) and an anti-ATF6 antibody (ab227830, Abcam). The immunoprecipitated DNA was used to amplify DNA fragments via PCR with specific primers. The primer sequences for GRP78 were 5′-GCGGAGCAGTGACGTTTATT-3′ (forward) and 5′-GACCTCACCGTCGCCTACT-3′ (reverse). The primer sequences for FGF2 M1 were 5′-TCTGAGCAAATAGGCCTTGCT-3′ (forward) and 5′-GGCTGAAGCCCCTGTAACAAA-3′ (reverse). The primer sequences for FGF2 M2 were 5′-ATATGCCTGGTTTTGGGCCT-3′ (forward) and 5′-CAGCCTACCGAATAGCTGGG-3′ (reverse).
Construction of reporter plasmids and luciferase assays
FGF2-ERSE (ERSE-like site) was cloned into the pGL3-basic luciferase reporter plasmid. Chondrocytes (2.5×104 cells per well) were seeded in triplicate into 24-well plates (Corning). After incubation for 24 hr, the cells with either ATF6 plasmids or ATF6-mutant plasmids were transfected with 200 ng of FGF2-ERSE-luciferase-reporter plasmids using Lipofectamine 3000 (Invitrogen) according to the manufacturer's recommendations. Each transfection included the same amount of Renilla, which was used to standardize transfection efficiency. Then, the cells were allowed to recover in medium containing 10% FBS for 24 hr. Forty-eight hours posttransfection, firefly and Renilla signals were measured using a Dual Luciferase Reporter Assay Kit (Promega) and are presented as the increase in activation over the reporter alone.
Micro-CT analysis
Lumbar spine (spinal segment including intervertebral disc and adjacent endplates) and hind paw specimens were obtained from mice postmortem and fixed with 4% paraformaldehyde. For micro-CT scans, specimens were fitted in a cylindrical sample holder and scanned using a Scanco lCT40 scanner set to 55 kVp and 70 lA. For visualization, the segmented data were imported and reconstructed as 3-dimensional images using MicroCT Ray software V3.0 (Scanco Medical).
Quantification and statistical analysis
Statistical analyses were performed with SPSS 13.0 (SPSS, Inc., Chicago, IL, USA). The data are shown as the means ± SD. For comparisons of 2 groups, a 2-tailed Student's t-test was used. Comparisons of multiple groups were performed by using one-way analysis of variance (ANOVA) followed by Dunnett's post hoc test. All experiments were repeated at least 3 times. Statistical significance was presumed at ∗P < 0.05 and ∗∗P < 0.01. Detailed information is provided in the figure legends.
Acknowledgments
This study was financially supported by grants from the National Natural Science Foundation of China of China (81971518), the National Natural Science Foundation of China of China (81871750), the Natural Science Foundation of Guangdong Province (2018A030313232), the Shenzhen Key Medical Discipline Construction Fund (ZDSYS20190902092851024), and the Health Welfare Fund Project of Futian District (FTWS2020078).
The authors thank Prof. Pheier Saw (Medical Research Center, Sun Yat-sen Memorial Hospital, SYSU, China) for her guidance.
Author contributions
M.Ma, H.Li, and P.Wang planned the project, performed the experiments, interpreted the data, and drafted the article. W.Yang and R.Mi designed the research and conducted data analysis. J.Zhuang and Y.Jiang provided technical support and revised the article. X.Shen and Y.Lu collected human samples and isolated and cultured chondrocytes. Y.Wu and H.Shen supervised the study.
Declaration of interests
The authors declare no competing interests.
Published: July 23, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102791.
Contributor Information
Yanfeng Wu, Email: wuyf@mail.sysu.edu.cn.
Huiyong Shen, Email: shenhuiy@mail.sysu.edu.cn.
Supplemental information
References
- Akl M.R., Nagpal P., Ayoub N.M., Tai B., Prabhu S.A., Capac C.M., Gliksman M., Goy A., Suh K.S. Molecular and clinical significance of fibroblast growth factor 2 (FGF2/bFGF) in malignancies of solid and hematological cancers for personalized therapies. Oncotarget. 2016;7:44735–44762. doi: 10.18632/oncotarget.8203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraliakos X., Haibel H., Listing J., Sieper J., Braun J. Continuous long-term anti-TNF therapy does not lead to an increase in the rate of new bone formation over 8 years in patients with ankylosing spondylitis. Ann. Rheum. Dis. 2014;73:710–715. doi: 10.1136/annrheumdis-2012-202698. [DOI] [PubMed] [Google Scholar]
- Benjamin M., Toumi H., Suzuki D., Hayashi K., McGonagle D. Evidence for a distinctive pattern of bone formation in enthesophytes. Ann. Rheum. Dis. 2009;68:1003–1010. doi: 10.1136/ard.2008.091074. [DOI] [PubMed] [Google Scholar]
- Bettigole S.E., Glimcher L.H. Endoplasmic reticulum stress in immunity. Annu. Rev. Immunol. 2015;33:107–138. doi: 10.1146/annurev-immunol-032414-112116. [DOI] [PubMed] [Google Scholar]
- Binet F., Sapieha P. ER stress and angiogenesis. Cell Metab. 2015;22:560–575. doi: 10.1016/j.cmet.2015.07.010. [DOI] [PubMed] [Google Scholar]
- Bleil J., Maier R., Hempfing A., Schlichting U., Appel H., Sieper J., Syrbe U. Histomorphologic and histomorphometric characteristics of zygapophyseal joint remodeling in ankylosing spondylitis. Arthritis Rheumatol. 2014;66:1745–1754. doi: 10.1002/art.38404. [DOI] [PubMed] [Google Scholar]
- Bleil J., Maier R., Hempfing A., Sieper J., Appel H., Syrbe U. Granulation tissue eroding the subchondral bone also promotes new bone formation in ankylosing spondylitis. Arthritis Rheumatol. 2016;68:2456–2465. doi: 10.1002/art.39715. [DOI] [PubMed] [Google Scholar]
- Bleil J., Sieper J., Maier R., Schlichting U., Hempfing A., Syrbe U., Appel H. Cartilage in facet joints of patients with ankylosing spondylitis (AS) shows signs of cartilage degeneration rather than chondrocyte hypertrophy: implications for joint remodeling in AS. Arthritis Res. Ther. 2015;17:170. doi: 10.1186/s13075-015-0675-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosse Y., Rola-Pleszczynski M. FGF2 in asthmatic airway-smooth-muscle-cell hyperplasia. Trends Mol. Med. 2008;14:3–11. doi: 10.1016/j.molmed.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Ciccia F., Accardo-Palumbo A., Rizzo A., Guggino G., Raimondo S., Giardina A., Cannizzaro A., Colbert R.A., Alessandro R., Triolo G. Evidence that autophagy, but not the unfolded protein response, regulates the expression of IL-23 in the gut of patients with ankylosing spondylitis and subclinical gut inflammation. Ann. Rheum. Dis. 2014;73:1566–1574. doi: 10.1136/annrheumdis-2012-202925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinaroglu A., Gao C., Imrie D., Sadler K.C. Activating transcription factor 6 plays protective and pathological roles in steatosis due to endoplasmic reticulum stress in zebrafish. Hepatology. 2011;54:495–508. doi: 10.1002/hep.24396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Palma M., Biziato D., Petrova T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer. 2017;17:457–474. doi: 10.1038/nrc.2017.51. [DOI] [PubMed] [Google Scholar]
- Dong W., Zhang Y., Yan M., Liu H., Chen Z., Zhu P. Upregulation of 78-kDa glucose-regulated protein in macrophages in peripheral joints of active ankylosing spondylitis. Scand. J. Rheumatol. 2008;37:427–434. doi: 10.1080/03009740802213310. [DOI] [PubMed] [Google Scholar]
- Gallagher C.M., Walter P. Ceapins inhibit ATF6α signaling by selectively preventing transport of ATF6α to the Golgi apparatus during ER stress. Elife. 2016;5:e11880. doi: 10.7554/eLife.11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh R., Lipson K.L., Sargent K.E., Mercurio A.M., Hunt J.S., Ron D., Urano F. Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PLoS One. 2010;5:e9575. doi: 10.1371/journal.pone.0009575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grootjans J., Kaser A., Kaufman R.J., Blumberg R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016;16:469–484. doi: 10.1038/nri.2016.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F., Han X., Wu Z., Cheng Z., Hu Q., Zhao Y., Wang Y., Liu C. ATF6a, a Runx2-activable transcription factor, is a new regulator of chondrocyte hypertrophy. J. Cell Sci. 2016;129:717–728. doi: 10.1242/jcs.169623. [DOI] [PubMed] [Google Scholar]
- Haynes K.R., Pettit A.R., Duan R., Tseng H.W., Glant T.T., Brown M.A., Thomas G.P. Excessive bone formation in a mouse model of ankylosing spondylitis is associated with decreases in Wnt pathway inhibitors. Arthritis Res. Ther. 2012;14:R253. doi: 10.1186/ar4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C., Papa F.R. The unfolded protein response and cell fate control. Mol. Cell. 2018;69:169–181. doi: 10.1016/j.molcel.2017.06.017. [DOI] [PubMed] [Google Scholar]
- Hillary R.F., FitzGerald U. A lifetime of stress: ATF6 in development and homeostasis. J. Biomed. Sci. 2018;25:48. doi: 10.1186/s12929-018-0453-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G., Zhao G., Xia J., Wei Y., Chen F., Chen J., Shi J. FGF2 and FAM201A affect the development of osteonecrosis of the femoral head after femoral neck fracture. Gene. 2018;652:39–47. doi: 10.1016/j.gene.2018.01.090. [DOI] [PubMed] [Google Scholar]
- Kawane T., Qin X., Jiang Q., Miyazaki T., Komori H., Yoshida C.A., Matsuura-Kawata V., Sakane C., Matsuo Y., Nagai K. Runx2 is required for the proliferation of osteoblast progenitors and induces proliferation by regulating Fgfr2 and Fgfr3. Sci. Rep. 2018;8:13551. doi: 10.1038/s41598-018-31853-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokame K., Kato H., Miyata T. Identification of ERSE-II, a new cis-acting element responsible for the ATF6-dependent mammalian unfolded protein response. J. Biol. Chem. 2001;276:9199–9205. doi: 10.1074/jbc.M010486200. [DOI] [PubMed] [Google Scholar]
- Krishnan Y., Grodzinsky A.J. Cartilage diseases. Matrix Biol. 2018;71-72:51–69. doi: 10.1016/j.matbio.2018.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landewe R., Dougados M., Mielants H., van der Tempel H., van der Heijde D. Physical function in ankylosing spondylitis is independently determined by both disease activity and radiographic damage of the spine. Ann. Rheum. Dis. 2009;68:863–867. doi: 10.1136/ard.2008.091793. [DOI] [PubMed] [Google Scholar]
- Lee M.N., Kim J.W., Oh S.H., Jeong B.C., Hwang Y.C., Koh J.T. FGF2 stimulates COUP-TFII expression via the MEK1/2 pathway to inhibit osteoblast differentiation in C3H10T1/2 cells. PLoS One. 2016;11:e0159234. doi: 10.1371/journal.pone.0159234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Wang P., Xie Z., Wang S., Ma M., Li J., Li M., Cen S., Tang S.A., Zheng G. Abnormal inhibition of osteoclastogenesis by mesenchymal stem cells through the miR-4284/CXCL5 axis in ankylosing spondylitis. Cell Death Dis. 2019;10:188. doi: 10.1038/s41419-019-1448-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lories R.J. Advances in understanding the pathophysiology of spondyloarthritis. Best Pract. Res. Clin. Rheumatol. 2018;32:331–341. doi: 10.1016/j.berh.2018.12.001. [DOI] [PubMed] [Google Scholar]
- Lories R.J., Derese I., Luyten F.P. Modulation of bone morphogenetic protein signaling inhibits the onset and progression of ankylosing enthesitis. J. Clin. Invest. 2005;115:1571–1579. doi: 10.1172/JCI23738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lories R.J., Haroon N. Evolving concepts of new bone formation in axial spondyloarthritis: insights from animal models and human studies. Best Pract. Res. Clin. Rheumatol. 2017;31:877–886. doi: 10.1016/j.berh.2018.07.007. [DOI] [PubMed] [Google Scholar]
- McInnes I.B., Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- Mehrbod P., Ande S.R., Alizadeh J., Rahimizadeh S., Shariati A., Malek H., Hashemi M., Glover K.K.M., Sher A.A., Coombs K.M. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence. 2019;10:376–413. doi: 10.1080/21505594.2019.1605803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar C., Scherer A., Baraliakos X., de Hooge M., Micheroli R., Exer P., Kissling R.O., Tamborrini G., Wildi L.M., Nissen M.J. TNF blockers inhibit spinal radiographic progression in ankylosing spondylitis by reducing disease activity: results from the Swiss Clinical Quality Management cohort. Ann. Rheum. Dis. 2018;77:63–69. doi: 10.1136/annrheumdis-2017-211544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen S.J., Maksymowych W.P. The pathogenesis of ankylosing spondylitis: an update. Curr. Rheumatol. Rep. 2019;21:58. doi: 10.1007/s11926-019-0856-3. [DOI] [PubMed] [Google Scholar]
- Poddubnyy D., Sieper J. Mechanism of new bone formation in axial spondyloarthritis. Curr. Rheumatol. Rep. 2017;19:55. doi: 10.1007/s11926-017-0681-5. [DOI] [PubMed] [Google Scholar]
- Poudel S.B., Min C.K., Lee J.H., Shin Y.J., Kwon T.H., Jeon Y.M., Lee J.C. Local supplementation with plant-derived recombinant human FGF2 protein enhances bone formation in critical-sized calvarial defects. J. Bone Miner. Metab. 2019;37:900–912. doi: 10.1007/s00774-019-00993-2. [DOI] [PubMed] [Google Scholar]
- Ranganathan V., Gracey E., Brown M.A., Inman R.D., Haroon N. Pathogenesis of ankylosing spondylitis - recent advances and future directions. Nat. Rev. Rheumatol. 2017;13:359–367. doi: 10.1038/nrrheum.2017.56. [DOI] [PubMed] [Google Scholar]
- Ruiz-Heiland G., Horn A., Zerr P., Hofstetter W., Baum W., Stock M., Distler J.H., Nimmerjahn F., Schett G., Zwerina J. Blockade of the hedgehog pathway inhibits osteophyte formation in arthritis. Ann. Rheum. Dis. 2012;71:400–407. doi: 10.1136/ard.2010.148262. [DOI] [PubMed] [Google Scholar]
- Ruutu M., Thomas G., Steck R., Degli-Esposti M.A., Zinkernagel M.S., Alexander K., Velasco J., Strutton G., Tran A., Benham H. β-glucan triggers spondylarthritis and Crohn's disease-like ileitis in SKG mice. Arthritis Rheum. 2012;64:2211–2222. doi: 10.1002/art.34423. [DOI] [PubMed] [Google Scholar]
- Sakaguchi N., Takahashi T., Hata H., Nomura T., Tagami T., Yamazaki S., Sakihama T., Matsutani T., Negishi I., Nakatsuru S. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature. 2003;426:454–460. doi: 10.1038/nature02119. [DOI] [PubMed] [Google Scholar]
- Sieper J., Poddubnyy D. Axial spondyloarthritis. Lancet. 2017;390:73–84. doi: 10.1016/S0140-6736(16)31591-4. [DOI] [PubMed] [Google Scholar]
- Van der Heijde D., Landewé R., Baraliakos X., Houben H., Van Tubergen A., Williamson P., Xu W., Baker D., Goldstein N., Braun J. Radiographic findings following two years of infliximab therapy in patients with ankylosing spondylitis. Arthritis Rheum. 2008;58:3063–3070. doi: 10.1002/art.23901. [DOI] [PubMed] [Google Scholar]
- Van der Heijde D., Landewé R., Einstein S., Ory P., Vosse D., Ni L., Lin S.L., Tsuji W., Davis J.C., Jr. Radiographic progression of ankylosing spondylitis after up to two years of treatment with etanercept. Arthritis Rheum. 2008;58:1324–1331. doi: 10.1002/art.23471. [DOI] [PubMed] [Google Scholar]
- Van der Heijde D., Salonen D., Weissman B.N., Landewé R., Maksymowych W.P., Kupper H., Ballal S., Gibson E., Wong R. Assessment of radiographic progression in the spines of patients with ankylosing spondylitis treated with adalimumab for up to 2 years. Arthritis Res. Ther. 2009;11:R127. doi: 10.1186/ar2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Liu S., Li J., Yi Z. The role of the fibroblast growth factor family in bone-related diseases. Chem. Biol. Drug Des. 2019;94:1740–1749. doi: 10.1111/cbdd.13588. [DOI] [PubMed] [Google Scholar]
- Wendling D., Verhoeven F., Prati C. Anti-IL-17 monoclonal antibodies for the treatment of ankylosing spondylitis. Expert Opin. Biol. Ther. 2019;19:55–64. doi: 10.1080/14712598.2019.1554053. [DOI] [PubMed] [Google Scholar]
- Woodbury M.E., Ikezu T. Fibroblast growth factor-2 signaling in neurogenesis and neurodegeneration. J. Neuroimmune Pharmacol. 2014;9:92–101. doi: 10.1007/s11481-013-9501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Ruas J.L., Estall J.L., Rasbach K.A., Choi J.H., Ye L., Boström P., Tyra H.M., Crawford R.W., Campbell K.P. The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1α/ATF6α complex. Cell Metab. 2011;13:160–169. doi: 10.1016/j.cmet.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z., Wang P., Li Y., Deng W., Zhang X., Su H., Li D., Wu Y., Shen H. Imbalance between bone morphogenetic protein 2 and noggin induces abnormal osteogenic differentiation of mesenchymal stem cells in ankylosing spondylitis. Arthritis Rheumatol. 2016;68:430–440. doi: 10.1002/art.39433. [DOI] [PubMed] [Google Scholar]
- Yang W., Cao Y., Zhang Z., Du F., Shi Y., Li X., Zhang Q. Targeted delivery of FGF2 to subchondral bone enhanced the repair of articular cartilage defect. Acta Biomater. 2018;69:170–182. doi: 10.1016/j.actbio.2018.01.039. [DOI] [PubMed] [Google Scholar]
- Yu X., Qi Y., Zhao T., Fang J., Liu X., Xu T., Yang Q., Dai X. NGF increases FGF2 expression and promotes endothelial cell migration and tube formation through PI3K/Akt and ERK/MAPK pathways in human chondrocytes. Osteoarthr. Cartil. 2019;27:526–534. doi: 10.1016/j.joca.2018.12.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets.