

Abstract
Hyperbranched polyesters with a range of exterior thiol modifications were synthesized through a Michael addition thiol-ene reaction. S-Nitrosothiol nitric oxide (NO) donors were subsequently introduced onto the scaffolds to yield NO-releasing polyesters with total NO storage of ~2.0 μmol mg−1. Multiple decomposition pathways (i.e., use of light, copper ions, and heat) triggered S-nitrosothiol NO donor breakdown and NO release under physiological conditions (37 °C, pH 7.4). The NO-releasing polyesters were characterized as a function of chemical modification and scaffold size or generation. The approaches described herein expand the scope of biodegradable NO-releasing materials with large NO payloads.
Keywords: Nitric oxide; hyperbranched polyesters; thiol-ene reaction; S-nitrosothiol; 2,2-bis(hydroxymethyl)propionic acid (bis-MPA)
Introduction
Nitric oxide (NO) is an endogenously produced diatomic free radical that mediates angiogenesis,1 blood pressure regulation,2 wound healing,3–4 and the immune response.5–6 Due to its relatively short biological half-life (seconds) and reactive nature, the synthesis of scaffolds capable of controlled NO storage and release is important for helping further understand NO’s role in physiology and developing NO-based therapeutics.7–10 To date, N-diazeniumdiolates and S-nitrosothiols (RSNOs) represent the most widely employed NO donors because of their spontaneous NO-release characteristics under physiological conditions (pH 7.4, 37 °C).10 S-Nitrosothiol-based NO donors are attractive as potential therapeutics due to their low toxicity and endogenous nature.11 S-Nitrosothiols are readily synthesized via the reaction of free thiols with nitrosating agents (e.g., NaNO2/HCl). The ensuing NO release is tunable and triggered via several decomposition pathways, including exposure to light, heat, and copper ions.12
Recent research has focused on the synthesis of macromolecular NO-release scaffolds to enhance NO payloads and enable greater tunability of the NO-release kinetics relative to that achieved using small molecule NO donors.13–25 Dendritic polymeric scaffolds exhibit attractive characteristics as macromolecular scaffolds due to their high density of exterior functional groups available for further modifications.16, 17, 22, 24 For example, polyamidoamine (PAMAM) dendrimers functionalized with RSNOs demonstrated large NO storage and tunable NO release kinetics by simply varying the steric structure of the thiol modification. Thiols with a compact structure exhibited extended NO-release kinetics compared to branched thiol structures.22 While these NO-releasing dendrimers displayed antiplatelet activity, the scaffold toxicity against mammalian cells and poor biodegradability may hinder their utility for certain biomedical applications.22
Polyesters represent an alternative polymeric NO-release scaffold with potentially useful biodegradation properties.26–28 Seabra et al. reported the synthesis of S-nitrosothiol-modified polyesters via the condensation of 3-mercapto-1,2-propanediol and mercaptosuccinic acid, followed by a nitrosation reaction.28 The resulting NO donor-modified polyesters were blended with poly(methyl methacrylate) to yield coatings that released ~40 nmol NO cm−2 over 72 h. Yapor et al. subsequently developed citrate-based S-nitrosothiol-modified polyesters capable of storing ~0.45 0μmol NO mg−1 polymer.29 Of practical relevance, the tedious material preparation and relatively low NO storage capacities of these scaffolds limit their utility.
Hyperbranched polyesters are a family of polymers with multivalent structures and diverse exterior functionalities.30 In contrast to their dendrimer counterparts, the synthesis of hyperbranched polyesters is more straightforward as they proceed via one-pot reactions.30 Bis(hydroxymethyl)propionic acid (bis-MPA) has been widely utilized as a building unit in creating hyperbranched polyesters with a high density of modifiable exterior hydroxyl groups and good solubility in organic solvents for subsequent chemical functionalization steps.31–34 Easy synthesis, biodegradability, and solubility in organic solvents have led many research groups to evaluate the potential of these polymers for biomedical application.31, 32, 35–37 For example, Feliu et al. demonstrated that bis-MPA-based polyesters exhibited excellent biodegradability and were non-toxic compared to PAMAM dendrimers.32 Based on these two characteristics, bis-MPA-based hyperbranched polyesters are now being utilized for cancer-targeted drug delivery and non-invasive positron emission tomography (PET) imaging applications.31 The facile chemical modification and excellent biocompatibility of bis-MPA-based hyperbranched polyesters suggest that they may be useful for NO-release applications.
In this study, we report the synthesis of S-nitrosothiol-modified bis-MPA-based hyperbranched polyesters with large NO payloads and controllable NO-release kinetics. The high density of hydroxyl groups on the hyperbranched polyesters scaffold allow for subsequent modification with S-nitrosothiol precursors (i.e., thiol groups) through a Michael addition thiol-ene reaction. A subsequent nitrosation step is then carried out to introduce the S-nitrosothiol NO donors onto the scaffolds. The resulting hyperbranched polyesters NO-release properties were studied as a function of the exterior modification and scaffold size.
Experimental
Materials.
Diethylene triamine pentaacetic acid (DTPA), 5,5’-dithio-bis(2-nitrobenzoic acid) (DTNB), sodium acetate, 2,2-bis(hydroxymethyl)propionic acid (bis-MPA), 2-ethyl-2-(hydroxymethyl)-1,3-propanediol (TMP), 1,2-ethanedithiol (ET), 2,3-butanedithiol (BT), dithiothreitol (DTT), and p-toluenesulfonic acid (p-TSA) were purchased from the Aldrich Chemical Company (Milwaukee, WI). Water was purified using a Millipore Milli-Q UV Gradient A10 System (Bethlehem, PA) to a final resistivity of 18.2 M∙Ωcm and total organic content of ≤6 ppb. Common laboratory salts and solvents were purchased from Fisher Scientific (Pittsburgh, PA). Unless noted otherwise, all materials were used as received without further purification.
Instrumentation.
Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a 400 MHz Bruker instrument. Quantitative carbon nuclear magnetic resonance (13C NMR) was performed on a 600 MHz Bruker instrument following a previously reported method.38 Gel permeation chromatography (GPC) measurements were carried out in tetrahydrofuran (THF) against polystyrene standards using a Waters 2695 system. Elemental analysis was performed on an inductively coupled plasma (ICP) optical emission spectrometer (Prodigy High Dispersion ICP-OES, Teledyne Leeman Labs). The standard addition of a copper reference standard (TraceCERT®, Sigma–Aldrich) and emission intensity at 324.75 nm were utilized to determine the concentration of free copper ion in PBS buffer. Hydrodynamic size and polydispersity index (PDI) were measured by dynamic light scattering (DLS) using a Malvern Zeta Nano-ZS (Westborough, MA).
Synthesis of hyperbranched polyesters.
Hyperbranched polyesters were prepared according to Malmstrom et al.38 Briefly, bis-MPA (5 mmol), TMP (0.555 mmol), and p-TSA (0.02 mmol) were mixed in a three-neck round bottom flask and stirred for 4 h at 140 °C to prepare second generation hyperbranched polyesters (G2-HP). For the synthesis of third generation hyperbranched polyesters (G3-HP), 6.67 mmol bis-MPA and 0.027 mmol p-TSA were subsequently added to this reaction mixture and stirred for an additional 4 h at 140 °C. Synthesis of the fourth generation hyperbranched polyesters (G4-HP) was achieved by adding 13.30 mmol bis-MPA and 0.054 mmol p-TSA to the G3-HP reaction mixture after the prior reaction went to completion, followed by stirring for 4 h at 140 °C. Nitrogen was continuously flowed through the reaction flask to remove water. After reaction completion, acetone was added to the mixture to dissolve the polyesters, and insoluble reactants were removed via filtration. The crude polyesters were then precipitated from acetone by the addition of hexane. This solid was dried under vacuum overnight at room temperature and stored in acetone after resuspension. The 1H NMR data of the hyperbranched polyesters for each generation consisted of the following peaks (400 MHz, acetone-d6, δ): 1.1 – 1.4 (CH3C), 3.68 (CH2OH), and 4.10 – 4.3 (CH2OCO).
Synthesis of acrylate-modified hyperbranched polyesters.
Hyperbranched polyesters (500 mg of G2, G3, or G4) and triethylamine (TEA; 692 μL) were dissolved in 5 mL acetone, followed by the dropwise addition of an acryloyl chloride (AC) solution (0.4 mL AC in 0.6 mL acetone) over a 5-min period. The reaction mixture was stirred for 1 h at room temperature. Precipitate byproducts were removed via filtration. The residual solvent, acryloyl choride, and TEA in the filtrate were then removed under vacuum at room temperature. 1H NMR data of acrylate-modified hyperbranched polyesters consisted of the following peaks (400 MHz, CD2Cl2, δ): 1.1 – 1.4 (CH3C), 3.68 (CH2OH), 4.1 – 4.3 (CH2OCO), 5.84 and 6.27 (CH2=CH), 6.07 (CH2=CH).
Synthesis of 1,2-ethylenedithiol-, 2,3-butanedithiol-, and dithiothreitol-modified hyperbranched polyesters.
The acrylate-modified G2-hyperbranched polyesters (200 mg) were dissolved in 2 mL acetone, followed by the addition of TEA (40 μL) and either 1,2-ethylenedithiol (ET; 815 μL) or 2,3-butanedithiol (BT; 1316 μL). Excess ET or BT (20:1 molar ratio compared to acrylate double bonds) was used to minimize crosslinking of the thiol groups. After stirring at room temperature for 18 h, the product was precipitated in hexane and washed with methanol to remove residual ET or BT. The resulting ET or BT thiol-modified hyperbranched polyesters (G2-HP-ET and G2-HP-BT) were collected by centrifugation and dried under vacuum at room temperature for 2 h. 1H NMR data of G2-HP-ET consisted of the following peaks (400 MHz, CD2Cl2, δ): 1.1 – 1.4 (CH3C), 2.6 – 2.9 (SCH2CH2SH), (SCH2CH2SH), (COCH2CH2SCH2), and (COCH2CH2SCH2), 3.65 (CH2OH), and 4.10 – 4.3 (CH2OCO). 1H NMR data of G2-HP-BT consisted of the following peaks (400 MHz, CD2Cl2, δ): 1.1 – 1.4 (CH3C), (SCHCH3) and (CH3CHSH), 2.6 (COCH2CH2SCHCH3), 2.85 (COCH2CH2SCHCH3), 2.95 (SCHCH3CHCH3SH), 3.2 (SCHCH3CHCH3SH), 3.65 (CH2OH), and 4.1 – 4.3 (CH2OCO).
Dithiothreitol-modified hyperbranched polyesters (G2-HP-DTT, G3-HP-DTT, and G4-HP-DTT) were synthesized using a similar procedure. Dithiothreitol (DTT) (1648 mg) and TEA (40 μL) were mixed with either G2, G3, or G4 hyperbranched polyesters (200 mg) in 2 mL acetone. The resulting thiol-modified polyesters were precipitated in water and collected via centrifugation. The residual solvent was removed under vacuum at room temperature. 1H NMR data of the DTT-modified hyperbranched polyesters consisted of the following peaks. G2-HP-DTT (400 MHz, CD2Cl2, δ): 1.1 – 1.4 (CH3C), 2.6 – 2.9 (COCH2CH2S), (COCH2CH2S), (SCH2CH(OH)), and (CH(OH)CH2SH), 3.65 (CH2OH), 3.70 (SCH2CH(OH)CH(OH)) and (CHCH(OH)CH2SH), 4.10 – 4.3 (CH2OCO). OCO).g thiol-modified polyester e ioned (50nm to 8 mixture, e the NO-release kinetics.netics (half-lives ~208s). We attributed G3-HP-DTT (400 MHz, CD2Cl2, δ): 1.1 – 1.4 (CH3C), 2.6 – 2.9 (COCH2CH2S), (COCH2CH2S), (SCH2CH(OH)), and (CH(OH)CH2SH), 3.65 (CH2OH), 3.70 (SCH2CH(OH)CH(OH)) and (CHCH(OH)CH2SH), 4.10 – 4.3 (CH2OCO). G4-HP-DTT (400 MHz, CD2Cl2, δ): 1.1 – 1.4 (CH3C), 2.6 – 2.9 (COCH2CH2S), (COCH2CH2S), (SCH2CH(OH)), and (CH(OH)CH2SH), 3.65 (CH2OH), 3.70 (SCH2CH(OH)CH(OH)) and (CHCH(OH)CH2SH), 4.1 – 4.3 (CH2OCO).
Nitrosation of thiol-modified hyperbranched polyesters.
The thiol-modified hyperbranched polyesters (10 mg) were dissolved in acetone (1 mL) and 5 M hydrochloric acid (100 μL). An aqueous solution (100 μL) containing sodium nitrite (6.9 mg) and diethylenetriamine pentaacetic acid (DTPA; 500 μM) was then added dropwise to this solution over a 1-min period. After stirring for 1 h at 0℃ (i.e., ice bath) in the dark, the solvent was removed under vacuum. Acetone was used to dissolve the resulting nitrosated polyesters (e.g., G2-HP-ET/NO). The solution was filtered to remove residual salt and then stored at −20 °C in the dark until use.
Ellman’s assay for thiol quantification of thiol-modified hyperbranched polyesters.
The free thiol content was quantified using a modified Ellman’s assay.39, 40 A solution of 2 mM 5,5’-dithio-bis(2-nitrobenzoic acid) (DTNB) in 50 mM sodium acetate was prepared and refrigerated until use. Samples of each polymer were dissolved in dimethyl sulfoxide (DMSO), treated with 50 uL of a 2 mM DTNB solution, and incubated for 5 min at room temperature. The optical densities of these solutions were measured at 414 nm and compared to N-acetyl cysteine standard solutions prepared identically.
Characterization of NO release from S-nitrosothiol-modified hyperbranched polyesters.
S-Nitrosothiol-functionalized hyperbranched polyesters were added to 30 mL deoxygenated 500 μM DTPA-supplemented phosphate buffered saline (PBS) or regular (DTPA-free) PBS at 37 °C. Nitrogen was bubbled through this solution at a flow rate of 70 mL min−1 to carry the liberated NO to a Sievers chemiluminescence nitric oxide analyzer (Boulder, CO). Additional nitrogen flow was supplied to the flask to match the collection rate of the instrument at 200 mL min−1. The real-time NO-release profiles triggered by copper (0.2 mg mL−1 CuBr2), heat (37 °C, shielded from light), or light (37 °C, 200 W, 15 cm above the reaction flask) were recorded until the observed NO levels decreased below 10 ppb mg−1 polyesters.8, 41
Results and discussion
Synthesis of second generation thiol-modified hyperbranched polyesters.
Hyperbranched polyesters (HP) composed of bis-MPA were synthesized as described by Malmstrom et al.38 The core molecule 2-ethyl-2-(hydroxymethyl)-1,3-propanediol (TMP) was reacted with bis-MPA at a molar ratio of 1:9 to yield second generation hyperbranched polyesters (G2-HP). Nuclear magnetic resonance (NMR) data confirmed the branching structure and the chemical composition of the polyesters. A typical hyperbranched polymer consists of three different structural units – dendritic (D), linear (L), and terminal (T). 13C NMR spectra (Figure S1) and a simple calculation (Equation S1) indicated that the degree of branching was ~0.44, corroborating a previous report.42 By integrating the distinct 1H NMR resonance at 3.65 ppm (−CH2OH) versus 4.10 – 4.30 ppm (−CH2O) (Fig. 1A), it was determined that ~8 HP hydroxyl groups on the G2-HP scaffold were available for subsequent chemical modification.
Fig. 1.
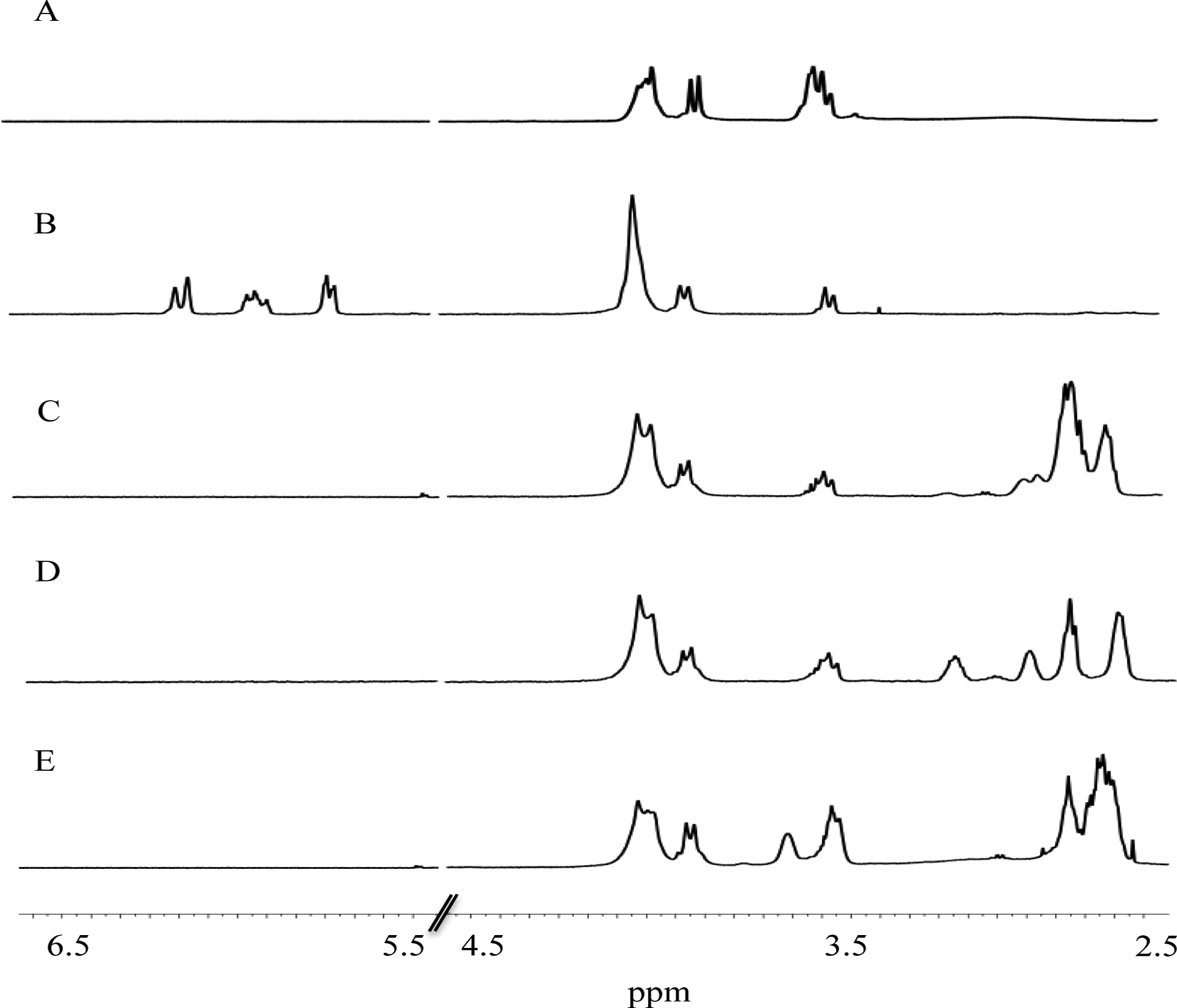
1H NMR spectra for A) G2-HP; B) acrylate-; C) 1,2-ethanedithiol (ET)-; D) 2,3-butanedithiol (BT)-; E) dithiothreitol (DTT)-modified G2-HP.
The exterior hydroxyl groups were next functionalized with acryloyl chloride to introduce alkene groups to the scaffold (Scheme 1A). As expected, resonances for the alkene protons were split into three distinct peaks at 5.84, 6.07, and 6.27 ppm (Fig. 1B).43 The resonance observed at 3.65 ppm corresponded to the methylene protons adjacent to unreacted peripheral hydroxyl groups and was integrated against the alkene resonances (5.84—6.27 ppm) to determine the molar ratio of hydroxyl groups to alkene bonds. The reaction efficiency of G2-HP with acryloyl chloride was roughly 70 ± 7% with an average of 6 unsaturated double bonds per macromolecule. Subsequent reaction of the unsaturated double bonds with 1,2-ethanedithiol (ET), 2,3-butanedithiol (BT), or dithiothreitol (DTT) yielded either two primary (G2-HP-ET and G2-HP-DTT) or one secondary (G2-HP-BT) thiol-modified polyester(s) (Scheme 1B–D). All of the unsaturated double bonds were modified with thiols upon the completion of this reaction, as evidenced by the disappearance of the alkene proton resonances at 5.84, 6.07, and 6.27 ppm (Fig. 1C–E). Dithiol addition was also confirmed by the appearance of peaks at 2.5 to 3 ppm, representing protons on the saturated double bonds and thiol groups (Fig. 1C–E).
Scheme 1.
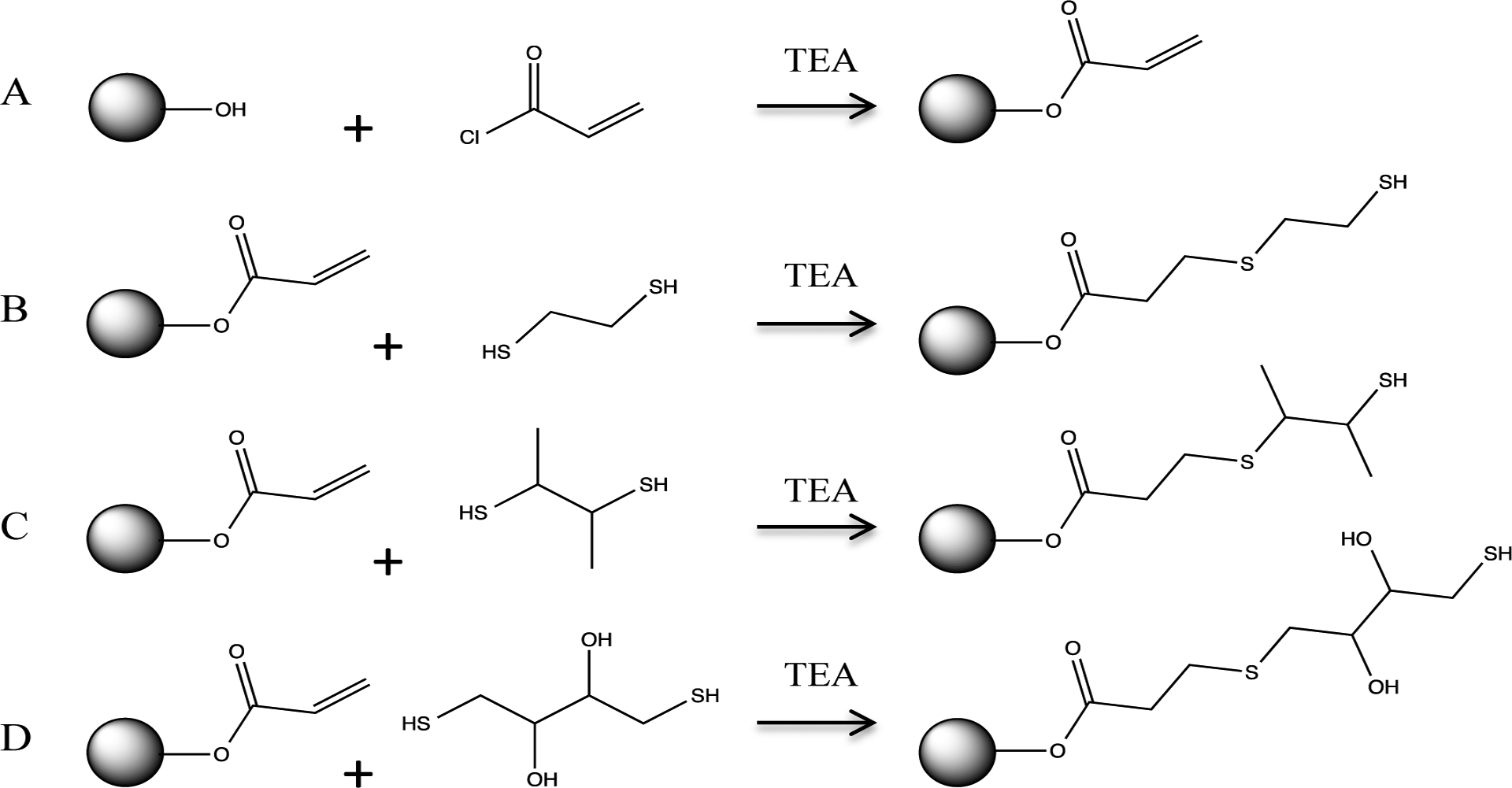
Synthesis of A) acrylate-; B) 1,2-ethanedithiol (ET)-; C) 2,3-butanedithiol (BT)-; D) dithiothreitol (DTT)-modified G2-HP.
Gel permeation chromatography (GPC) was used to determine the molecular weight (MW) of the hyperbranched polyesters. The similarity between the experimental molecular weight of G2-HP-DTT and the theoretical molar mass indicated negligible crosslinking of the DTT thiols during the reaction (Table 1). This result is most likely due to the reducing capacity of DTT.44 A larger experimental molar mass was noted for both the G2-HP-ET and G2-HP-BT scaffolds relative to theoretical values, which may be attributed to crosslinking and/or inter-molecular bridging of the dithiols during the reaction.45 To determine the extent of crosslinking, G2-HP-BT and G2-HP-ET were reacted with the reducing agent DTT overnight (~16 h), respectively. A slight decrease in molar mass was observed only for G2-HP-BT (Mn, 4000 ± 400 g mol−1), indicating potential crosslinking due to disulfide bond formation. Regardless, the still greater experimental molar mass for the reduced G2-HP-BT and G2-HP-ET suggests the formation of inter-molecular bridging between two polyester molecules. Although a 20x excess of dithiol relative to the number of double bonds on the hyperbranched polyester scaffolds was present during reaction, similar observations have been reported for thiol addition reactions.22, 29, 46 Despite the undesirable side reactions, the polyester scaffolds synthesis was reproducible and resulted in robust solubility in most organic solvents.
Table 1.
Molecular weight and PDI for the thiol-modified G2-hyperbranched polyesters.a
| Polyesters | Mn Theoreticalb (g mol−1) | Mn Experimentalc (g mol−1) | PDI |
|---|---|---|---|
| G2-HP | 1179 | 1700 ± 180 | 1.32 ± 0.02 |
| G2-HP-ET | 2783 | 3510 ± 220 | 1.31 ± 0.04 |
| G2-HP-BT | 2867 | 4970 ± 940 | 1.33 ± 0.02 |
| G2-HP-DTT | 2986 | 2530 ± 570 | 1.39 ± 0.13 |
n ≥ 3 separate syntheses;
Theoretical molar mass of thiol-modified G2-HP derived from the experimental molar mass of G2-HP and ~6 dithiols modification for each polyesters.
Molecular weight determined by GPC analysis.
The free thiol content available for subsequent nitrosation and NO storage was determined using the Ellman’s assay (Table 2). Each polyester scaffold exhibited large free thiol content (>2 μmol mg−1), indicating the potential for large NO payloads compared to the polyesters reported by Yapor et al. (~0.9 μmol mg−1).29 Of note, the similar thiol availability for each exterior modification also suggested negligible effects from disulfide and inter-molecular bridge bonding.
Table 2.
Thiol content and total nitric oxide storage for the thiol-modified G2-hyperbranched polyesters.a
| Polyesters | Thiol contentb (μmol mg−1) | t[NO]c (μmol mg−1) | Nitrosation efficiencyd (%) |
|---|---|---|---|
| G2-HP-ET | 1.96 ± 0.04 | 1.60 ± 0.27 | 81.8 ± 13.7 |
| G2-HP-BT | 2.38 ± 0.07 | 1.97 ± 0.41 | 82.8 ± 17.2 |
| G2-HP-DTT | 2.20 ± 0.17 | 1.84 ± 0.21 | 83.6 ± 9.5 |
n ≥ 3 separate syntheses;
Thiol content determined by Ellman’s assay;
NO-release totals measured in 0.2 mg mL−1 CuBr2-supplemented PBS solution.
Nitrosation efficiency calculated from dividing NO totals by thiol content.
S-Nitrosothiol-modified G2-hyperbranched polyesters.
Thiol-modified G2-hyperbranched polyesters were nitrosated using sodium nitrite in acidic solutions to produce NO-releasing polyesters.25, 27, 47 Acetone was selected as the solvent to dissolve G2-HP-ET, G2-HP-BT or G2-HP-DTT before adding the nitrosating agent (i.e., NaNO2/HCl). Due to the susceptibility of ester bonds to hydrolysis under acidic condition, GPC was used to characterize the stability of the polyesters during the nitrosation reaction. All of the polyester systems studied retained similar molecular weight and PDI, indicating negligible hydrolysis, likely the result of our use of an aprotic solvent (acetone) and low reaction temperature (0 °C). Previous reports have also demonstrated that polyesters were resistant to acidic degradation during nitrosation, albeit at a less concentrated hydrochloric acid concentration.25, 27 Of note, a minor peak representing a greater molecular weight species became apparent with time, upon RSNO decomposition. This peak is attributed to binding of discrete polyester polymers via disulfide linkages.
S-nitrosothiol formation on the polyesters (G2-HP-ET/NO, G2-HP-BT/NO, and G2-HP-DTT/NO) was confirmed using UV-vis absorption spectroscopy (Supporting Information). The peaks at ~340 nm (intense) and ~550 nm (broad) correspond to the S-nitrosothiol nO - π* and nN - π* transitions, respectively.28, 46, 48 The deep red color of the nitrosated polyesters along with the UV-vis peak at ~550 nm (Fig. S2) clearly affirmed formation of the NO donor. Hydroxyl groups are also known to react with nitrosating reagents to form alkyl nitrites.49 However, the multiple absorption bands representative of alkyl nitrite formation (in the 300 – 400 nm UV-vis region) were not observed for any of the S-nitrosothiol-modified polyesters or intermediates prior to thiol modification (e.g., G2-HP-AC) indicating that the nitrosation process only produces S-nitrosothiols.
The total NO storage for each S-nitrosothiol modification was measured in a copper bromide (0.2 mg mL−1, 37 °C, pH 7.4) supplemented phosphate buffered saline (PBS) solution under dark conditions. Although blood and tissue contain only trace levels of copper ions (Cu2+),50 the rapid NO release triggered by S-nitrosothiol (RSNO) reaction with Cu2+ is helpful in assessing total NO payloads (Fig. 2a). As shown in Table 2, similar nitrosation efficiencies (~70%) were observed regardless of exterior modification. The total NO storage for each scaffold was ~2 μmol mg−1 and directly correlated with the high free thiol content measured for the thiol-modified G2-HP systems (Table 2). Indeed, these levels are significantly greater than the previously reported NO-releasing polyester-based materials (~0.45 μmol mg−1).29
Fig. 2.
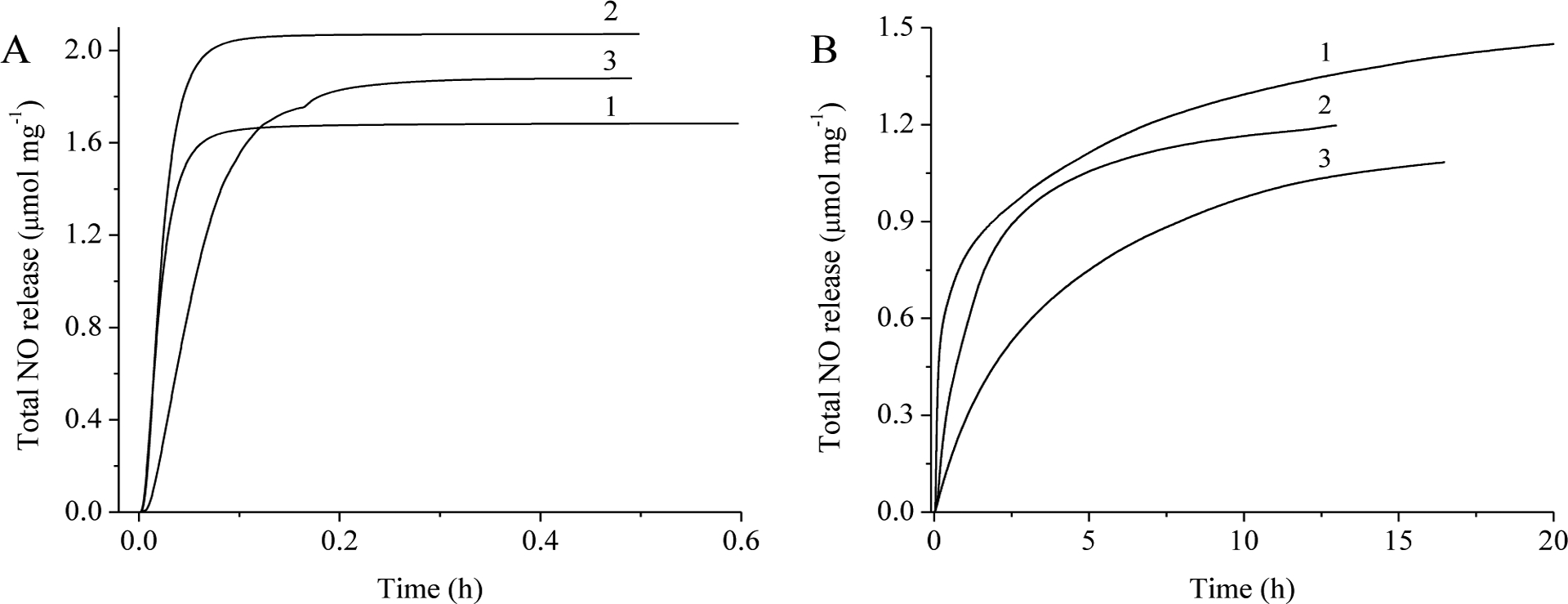
Total nitric oxide release from (1) G2-HP-ET/NO, (2) G2-HP-BT/NO, and (3) G2-HP-DTT/NO in: (A) 0.2 mg mL−1 CuBr2 solution (37 °C, pH 7.4) or (B) DTPA-supplemented PBS solution shielded from light (37 °C, pH 7.4).
The NO-release properties of the hyperbranched polyesters were initially evaluated in PBS solution (37 °C, pH 7.4) without the presence of a copper chelator (e.g. DTPA). Regardless of the S-nitrosothiol modification, each scaffold exhibited rapid NO-release kinetics and comparable NO-release half-lives (~5 min) under light irradiation (Table 3). Similar NO-release kinetics (t1/2 < 10 min) were still observed for each scaffold modification in the absence of light. This rapid NO release was attributed to copper and more specifically, copper ion-mediated S-nitrosothiol breakdown. The relatively high levels of copper present in untreated PBS (21.5 ppm Cu2+ as measured via inductively coupled plasma-optical emission spectrometry) oxidized residual thiolate ions, resulting in Cu+-mediated RSNO decomposition.48
Table 3.
Nitric oxide-release properties of S-nitrosothiol NO donor-modified polyesters in PBS or 500 μM DTPA-supplemented copper free PBS (37 °C, pH 7.4).a
| DTPA | Light | G2-HP-ET/NO | G2-HP-BT/NO | G2-HP-DTT/NO | |||||
|---|---|---|---|---|---|---|---|---|---|
| t[NO]b (μmol mg−1) | t1/2c (min) | t[NO]b (μmol mg−1) | t1/2c (min) | t[NO]b (μmol mg−1) | t1/2c (min) | ||||
| Yes | No | 1.40 ± 0.17 | 84 ± 24 | 0.99 ± 0.20 | 190 ± 46 | 1.47 ± 0.24 | 53 ± 28 | ||
| Yes | Yes | 1.55 ± 0.22 | 36 ± 16 | 1.14 ± 0.24 | 75 ± 19 | 1.43 ± 0.10 | 42 ± 12 | ||
| No | No | 1.40 ± 0.15 | 5 ± 2 | 1.90 ± 0.29 | 10 ± 2 | 1.73 ± 0.17 | 7 ± 2 | ||
| No | Yes | 1.65 ± 0.10 | 5 ± 2 | 1.79 ± 0.12 | 5 ± 3 | 1.69 ± 0.24 | 6 ± 3 | ||
n ≥ 3 separate syntheses;
Total NO storage per milligram polyesters;
Half-life of NO release.
As a result of significantly lower basal Cu2+ levels in the human body, RSNO decomposition under physiological condition will likely be restricted to a thermal RSNO-breakdown mechanism.50, 51 To better understand such behavior, NO release from the polyesters was evaluated in the absence of light and copper by supplementing the PBS with 500 μM diethylene triamine pentaacetic acid (DTPA). Secondary RSNO G2-HP-BT/NO polyesters exhibited both significantly lower NO-release totals and extended NO-release kinetics compared to both primary RSNOs (either G2-HP-ET/NO or G2-HP-DTT/NO; Table 3). Previous studies have demonstrated that methyl groups of secondary thiols act as electron-donating groups, enhancing the stability of the corresponding RSNOs compared to their primary thiol counterparts (e.g., ET and DTT).52 As thermal RSNO decomposition results in homolytic cleavage of the S-N bond (yielding thiyl and NO radicals)48, increased stability from the secondary thiol (BT) may facilitate the recombination of this geminate radical pair and slow the NO donor breakdown (Fig. 2B).53 On the other hand, the more compact structure of primary thiol polyesters scaffold (e.g., G2-HP-ET) should enhance RSNO stability.22, 40 Indeed, a slight increase in half-life was observed for G2-HP-ET/NO (~84 min) compared to G2-HP-DTT/NO (~54 min), corroborating this concept. However, the electron-donating effect of the secondary thiol was found to be more significant in enhancing RSNO stability compared to the compact structure, as evidenced by the extended NO-release kinetics of G2-HP-BT/NO system.
The photolysis mechanism of S-nitrosothiol breakdown was also studied in PBS supplemented with DTPA using a broad-spectrum white light source at a power of 200 W (15 cm above the reaction flask). As expected, light triggered more rapid RSNO decomposition and NO release than thermal decomposition alone for each S-nitrosothiol system, corroborating previous reports of similar behavior for S-nitrosothiol-modified macromolecular scaffolds.22, 40, 54 Despite the more rapid NO release, the secondary RSNO scaffold (G2-HP-BT/NO) still exhibited the slowest NO release, again attributed to enhanced RSNO stability due to favorable electron donating from the methyl groups adjacent to the S-nitrosothiol. Of note, both primary RSNO scaffolds (G2-HP-ET/NO and G2-HP-DTT/NO) were characterized by accelerated RSNO decomposition with light, resulting in similar NO-release kinetics (t1/2 ~40 min).22 In DTPA-supplemented PBS, evaluation of the NO-release characteristics of the G2-HP scaffolds with the different thiol modifications (i.e., ET, BT, and DTT) demonstrated that both the electron-donating groups of secondary thiols and the compact structure of primary thiols enhanced RSNO stability under thermal decomposition conditions, while the NO release under light was only prolonged using NO donors with secondary thiol structures.
Higher generation S-nitrosothiol-modified hyperbranched polyesters.
Larger molecular weight (i.e., higher generation) hyperbanched polyesters modified with S-nitrosothiol NO donors were also synthesized to evaluate the effect of polyesters generation (i.e., size) on NO-release properties. Specifically, G3 and G4-HP were synthesized using TMP to bis-MPA ratios of 1:21 and 1:45, respectively. The theoretical number of hydroxyl groups available for subsequent chemical modification for these polyesters was determined using 1H NMR to be ~18 for G3-HP and ~40 for G4-HP. The 13C NMR data (Fig. S1) indicated a decrease in the degree of branching for the G3-HP (~0.25) and G4-HP (~0.22) scaffolds compared to G2-HP (~0.44). The altered branching may be the result of increased reaction mixture viscosity at higher generations, thus hindering molecule diffusion during the one-pot reaction and reducing linear unit reactivity relative to dendritic units.42
The subsequent acryloyl chloride conversion efficiency was found to be 56 ± 6 and 66 ± 8% for G3-HP and G4-HP, respectively, corresponding to an average of ~10 and 28 double bonds per scaffold. The G3-HP and G4-HP polyesters were then modified with DTT. As expected, any evidence of remaining double bonds was lost following this reaction (Fig. S3), again conveying successful DTT modification. Greater molecular weights were measured by GPC for both the unmodified and DTT-modified scaffolds with each increase in scaffold generation (Table 4). While similar theoretical and experimental molecular weights were determined for the G2-HP and G3-HP scaffolds, the experimental MW for G4-HP was significantly lower than the theoretical value. Indeed, such error is the result of linear polystyrene-based GPC calibration standards, which inherently underestimates the molecular weights of the more globular-like hypebranched polyesters.55 This effect is more pronounced at higher generation or larger molar mass polymers.54 Two distinct peaks were observed in the GPC chromatograms for G3-HP-DTT and G4-HP-DTT (Fig. S4), assignable to the modified polyesters (major peak) and unreactive polyether oligomers (minor peak). The unreactive polyether oligomers (Mn < 2000 g mol−1) were expected from side reactions occurring during the one-pot synthesis of hyperbranched polyesters.42 Relative to their unmodified counterparts, the G3-HP-DTT and G4-HP-DTT were characterized as having lower PDI values as they lacked the byproduct peak from the molecular weight analysis. Resolution of these peaks was not observed for G2-HP-DTT because of similar molar mass to the unreactive oligomers. As such, G2-HP and G2-HP-DTT had similar PDI values.
Table 4.
Molecular weight and PDI for hyperbranched polyester modified with dithiothreitol (DTT) as a function of size (i.e., generation).a
| Polyesters | Mn Theoreticalb (g mol−1) | Mn Experimentalc (g mol−1) | PDI |
|---|---|---|---|
| G2-HP | 1179 | 1700 ± 180 | 1.32 ± 0.02 |
| G2-HP-DTT | 2986 | 2530 ± 580 | 1.39 ± 0.13 |
| G3-HP | 2573 | 2820 ± 270 | 1.44 ± 0.13 |
| G3-HP-DTT | 4950 | 4430 ± 260 | 1.28 ± 0.12 |
| G4-HP | 5359 | 3570 ± 420 | 1.56 ± 0.11 |
| G4-HP-DTT | 9525 | 6240 ± 980 | 1.12 ± 0.00 |
n ≥ 3 separate syntheses.
Theoretical molar mass of thiol-modified HP derived from the experimental molar mass of HP and corresponding amount of dithiols modification for each polyesters.
Molecular weight determined by GPC.
Although G2-HP-DTT and G4-HP-DTT exhibited similar thiol content, the thiol levels for the G3-HP-DTT scaffold was noticeably less (Table 5). This decrease is attributed to lower acryloyl chloride modification efficiency. The success of S-nitrosothiol functional group addition was again examined by UV-vis spectroscopy. The appearance of a characteristic absorption band at ~550 nm confirmed S-nitrosothiol formation. G3-HP-AC (the intermediate prior to thiol incorporation) was also nitrosated to rule out potential alkyl nitrite formation. The UV-vis absorption spectra for G3-HP-AC and G3-HP-AC exposed to nitrosating conditions were nearly identical, showing no evidence of alkyl nitrite formation (Supporting Information). As expected, an additional peak at 1520 cm−1 was observed in the FTIR spectrum of G3-HP-DTT/NO, assignable to the S-N=O stretching vibration (Fig. S5).25, 56 Concomitantly, the sharp but weak S-H stretch mode at ~2550 cm−1 disappeared following the nitrosation reaction. FTIR spectra of G3-HP-AC before and after exposure to the nitrosation conditions were also identical and without alkyl nitrite peaks, demonstrating that free hydroxyl groups are not reactive to the nitrosation conditions.
Table 5.
Thiol content and total nitric oxide storage data for DTT-modified hyperbranched polyesters.a
| Polyesters | Thiol contentb (μmol mg−1) | t[NO]c (μmol mg−1) | Nitrosation efficiencyd (%) |
|---|---|---|---|
| G2-HP-DTT | 2.20 ± 0.17 | 1.84 ± 0.21 | 83.6 ± 9.5 |
| G3-HP-DTT | 1.85 ± 0.16 | 1.48 ± 0.29 | 79.8 ± 15.6 |
| G4-HP-DTT | 2.15 ± 0.13 | 1.84 ± 0.16 | 85.5 ± 7.8 |
n ≥ 3 separate syntheses;
Thiol content determined by Ellman’s assay;
NO-release total amounts measured in 0.2 mg mL−1 CuBr2-supplemented PBS solution.
Nitrosation efficiency calculated from dividing NO totals by thiol content.
Lastly, the NO-release properties of the HP-DTT/NO polyesters were evaluated as a function of scaffold size (Table 5, Table 6). As might be expected based on the smaller thiol levels, G3-HP-DTT/NO exhibited slightly lower NO payloads relative to G4-HP-DTT/NO. Nevertheless, the nitrosation efficiency (capacity to form NO donors) was similar regardless of scaffold size. In PBS without a copper chelator, the NO-release kinetics were similar regardless of size, with light irradiation having little effect on NO release (t1/2 <10 min). However, the NO-release kinetics proved highly dependent on polyester generation in DTPA-supplemented PBS. In the absence of light irradiation, larger sizes led to longer NO-release for the three sizes investigated (i.e., G2, G3, and G4). In this manner, the extended NO-release kinetics for the G3-HP-DTT/NO and G4-HP-DTT/NO scaffolds were attributed to differences in the RSNO microenvironment between the generations. As provided in supplementary information (Table S1), the polyester suspensions exhibited similar hydrodynamic (DLS) sizes regardless of generation. The inherent greater density of RSNO donors (number of RSNO groups per hydrodynamic radius) for G4-HP-DTT/NO cultivated the recombination of geminate radical pairs (i.e., cage effect)25, 53, thus slowing NO release. The faster NO release was observed for the lowest RSNO density system (i.e., G2-HP-DTT/NO). Light irradiation mitigated these differences, particularly for the G2-HP-DTT/NO and G3-HP-DTT/NO systems, due to the acceleration of RSNO decomposition.22 Of potential importance, G4-HP-DTT/NO polyesters still maintained the longest NO-release duration with light, indicating the importance of RSNO donor stability.
Table 6.
Nitric oxide-release properties from S-nitrosothiol NO donor-modified HP-DTT polyesters in PBS or 500 μM DTPA-supplemented PBS (37 °C, pH 7.4).a
| DTPA | Light | G2-HP-DTT/NO | G3-HP-DTT/NO | G4-HP-DTT/NO | |||||
|---|---|---|---|---|---|---|---|---|---|
| t[NO]b (μmol mg−1) | t1/2c (min) | t[NO]b (μmol mg−1) | t1/2c (min) | t[NO]b (μmol mg−1) | t1/2c (min) | ||||
| Yes | No | 1.47 ± 0.24 | 56 ± 24 | 1.26 ± 0.39 | 70 ± 29 | 1.53 ± 0.04 | 171 ± 56 | ||
| Yes | Yes | 1.43 ± 0.10 | 42 ± 12 | 1.05 ± 0.20 | 42 ± 15 | 1.43 ± 0.10 | 132 ± 35 | ||
| No | No | 1.73 ± 0.17 | 7 ± 2 | 1.47 ± 0.32 | 4 ± 2 | 1.83 ± 0.18 | 7 ± 3 | ||
| No | Yes | 1.69 ± 0.24 | 6 ± 3 | 1.50 ± 0.42 | 3 ± 1 | 1.90 ± 0.22 | 9 ± 3 | ||
n ≥ 3 separate syntheses;
Total NO storage per milligram polyesters;
Half-life of NO release.
Conclusion
Herein, we describe the synthesis of S-nitrosothiol NO donor-modified hyperbranched polyesters and the investigation as to how polymer scaffold size (generation) and the NO donor precursor (thiol structure) influence NO release. Extended NO release was observed when increasing the S-nitrosothiol stability. The measured NO payloads (~2 μmol mg−1) represent the largest level for a biodegradable scaffold to date, with potential therapeutic implications.
Supplementary Material
Acknowledgement
Financial support was provided by the National Institutes of Health (DE025207). We acknowledge the Energy Frontier Research Center for Solar Fuels at UNC-Chapel Hill for providing access to FTIR instrumentation.
Footnotes
Disclosure
The corresponding author declares competing financial interest. Mark Schoenfisch is a co-founder and maintains a financial interest in Novan, Inc. Novan is a late-stage pharmaceutical company commercializing macromolecular nitric oxide storage and release vehicles for dermatological indications.
References
- 1.Cooke JP, Atherosclerosis Suppl, 2003, 4, 53–60. [DOI] [PubMed] [Google Scholar]
- 2.Rees DD, Palmer RM and Moncada S, Proc. Natl. Acad. Sci, 1989, 86, 3375–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo J.-d. and Chen AF, Acta Pharmacol. Sin, 2005, 26, 259–264. [DOI] [PubMed] [Google Scholar]
- 4.Schäffer MR, Tantry U, Gross SS, Wasserkrug HL and Barbul A, J. Surg. Res, 1996, 63, 237–240. [DOI] [PubMed] [Google Scholar]
- 5.Bogdan C, Nat. Immunol, 2001, 2, 907–916. [DOI] [PubMed] [Google Scholar]
- 6.MacMicking J, Xie Q.-w. and Nathan C, Annu. Rev. Immunol, 1997, 15, 323–350. [DOI] [PubMed] [Google Scholar]
- 7.Carpenter AW and Schoenfisch MH, Chem. Soc. Rev, 2012, 41, 3742–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coneski PN and Schoenfisch MH, Chem. Soc. Rev, 2012, 41, 3753–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nichols SP, Storm WL, Koh A and Schoenfisch MH, Adv. Drug Delivery Rev, 2012, 64, 1177–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riccio DA and Schoenfisch MH, Chem. Soc. Rev, 2012, 41, 3731–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coneski PN and Schoenfisch MH, Poly. Chem, 2011, 2, 906–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyn H áWilliams D, Chem. Soc. Rev, 1985, 14, 171–196. [Google Scholar]
- 13.Zhang HP, Annich GM, Miskulin J, Stankiewicz K, Osterholzer K, Merz SI, Bartlett RH and Meyerhoff ME, J. Am. Chem. Soc, 2003, 125, 5015–5024. [DOI] [PubMed] [Google Scholar]
- 14.Lu Y, Slomberg DL, Sun B and Schoenfisch MH, Small, 2013, 9, 2189–2198. [DOI] [PubMed] [Google Scholar]
- 15.Slomberg DL, Lu Y, Broadnax AD, Hunter RA, Carpenter AW and Schoenfisch MH, ACS Appl. Mater. Interfaces, 2013, 5, 9322–9329. [DOI] [PubMed] [Google Scholar]
- 16.Lu Y, Slomberg DL, Shah A and Schoenfisch MH, Biomacromolecules, 2013, 14, 3589–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun B, Slomberg DL, Chudasama SL, Lu Y and Schoenfisch MH, Biomacromolecules, 2012, 13, 3343–3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu Y, Sun B, Li C and Schoenfisch MH, Chem. Mater, 2011, 23, 4227–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hetrick EM, Shin JH, Stasko NA, Johnson CB, Wespe DA, Holmuhamedov E and Schoenfisch MH, ACS Nano, 2008, 2, 235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou ZR, Annich GM, Wu YD and Meyerhoff ME, Biomacromolecules, 2006, 7, 2565–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frost MC and Meyerhoff ME, J. Biomed. Mater. Res., Part A, 2005, 72A, 409–419. [DOI] [PubMed] [Google Scholar]
- 22.Stasko NA, Fischer TH and Schoenfisch MH, Biomacromolecules, 2008, 9, 834–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soto RJ, Yang L and Schoenfisch MH, ACS Appl. Mater. Interfaces, 2016, 8, 2220–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Worley BV, Slomberg DL and Schoenfisch MH, Bioconjugate Chem, 2014, 25, 918–927. [DOI] [PubMed] [Google Scholar]
- 25.Liu T, Zhang W, Yang X and Li C, J. Colloid Interface Sci, 2015, 459, 115–122. [DOI] [PubMed] [Google Scholar]
- 26.Coneski PN, Rao KS and Schoenfisch MH, Biomacromolecules, 2010, 11, 3208–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Damodaran VB and Reynolds MM, J. Mater. Chem, 2011, 21, 5870–5872. [Google Scholar]
- 28.Seabra AB, Martins D, Simoes MMSG, da Silva R, Brocchi M and de Oliveira MG, Artif. Organs, 2010, 34, E204–E214. [DOI] [PubMed] [Google Scholar]
- 29.Yapor JP, Lutzke A, Pegalajar-Jurado A, Neufeld BH, Damodaran VB and Reynolds MM, J. Mater. Chem. B, 2015, 3, 9233–9241. [DOI] [PubMed] [Google Scholar]
- 30.Voit BI and Lederer A, Chem. Rev, 2009, 109, 5924–5973. [DOI] [PubMed] [Google Scholar]
- 31.Xiao YL, Hong H, Javadi A, Engle JW, Xu WJ, Yang YA, Zhang Y, Barnhart TE, Cai WB and Gong SQ, Biomaterials, 2012, 33, 3071–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feliu N, Walter MV, Montanez MI, Kunzmann A, Hult A, Nystrom A, Malkoch M and Fadeel B, Biomaterials, 2012, 33, 1970–1981. [DOI] [PubMed] [Google Scholar]
- 33.Carlmark A, Malmstrom E and Malkoch M, Chem. Soc. Rev, 2013, 42, 5858–5879. [DOI] [PubMed] [Google Scholar]
- 34.Malmström E, Johansson M and Hult A, Macromolecules, 1995, 28, 1698–1703. [Google Scholar]
- 35.Prabaharan M, Grailer JJ, Pilla S, Steeber DA and Gong SQ, Biomaterials, 2009, 30, 5757–5766. [DOI] [PubMed] [Google Scholar]
- 36.Zeng XH, Zhang YN and Nystrom AM, Biomacromolecules, 2012, 13, 3814–3822. [DOI] [PubMed] [Google Scholar]
- 37.Karatasos K, J. Phys. Chem. B, 2013, 117, 2564–2575. [DOI] [PubMed] [Google Scholar]
- 38.Malmstrom E, Johansson M and Hult A, Macromolecules, 1995, 28, 1698–1703. [Google Scholar]
- 39.Gabor G and Vincze A, Anal. Chim. Acta, 1977, 92, 429–431. [Google Scholar]
- 40.Lu Y, Shah A, Hunter RA, Soto RJ and Schoenfisch MH, Acta Biomater, 2015, 12, 62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hetrick EM and Schoenfisch MH, Annu. Rev. Anal. Chem, 2009, 2, 409–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Žagar E and Žigon M, Progress in Polymer Science, 2011, 36, 53–88. [Google Scholar]
- 43.Zhang H, Patel A, Gaharwar AK, Mihaila SM, Iviglia G, Mukundan S, Bae H, Yang H and Khademhosseini A, Biomacromolecules, 2013, 14, 1299–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cleland WW, Biochem, 1964, 3, 480–482. [DOI] [PubMed] [Google Scholar]
- 45.Nakamoto H and Bardwell JCA, Biochimica et Biophysica Acta (BBA)-Molecular Cell Research, 2004, 1694, 111–119. [DOI] [PubMed] [Google Scholar]
- 46.Lutzke A, Pegalajar-Jurado A, Neufeld BH and Reynolds MM, J. Mater. Chem. B, 2014, 2, 7449–7458. [DOI] [PubMed] [Google Scholar]
- 47.Riccio DA, Coneski PN, Nichols SP, Broadnax AD and Schoenfisch MH, ACS Appl. Mater. Interfaces, 2012, 4, 796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Williams DLH, Acc. Chem. Res, 1999, 32, 869–876. [Google Scholar]
- 49.Williams DLH, Nitrosation reactions and the chemistry of nitric oxide, Elsevier, 2004. [Google Scholar]
- 50.O’Halloran TV and Culotta VC, J. Biol. Chem, 2000, 275, 25057–25060. [DOI] [PubMed] [Google Scholar]
- 51.Valko M, Morris H and Cronin MTD, Curr. Med. Chem, 2005, 12, 1161–1208. [DOI] [PubMed] [Google Scholar]
- 52.Roy B, Du Moulinet d’Hardemare A and Fontecave M, J. Org. Chem, 1994, 59, 7019–7026. [Google Scholar]
- 53.Shishido SM and Oliveira MG, Photochem. Photobiol, 2000, 71, 273–280. [DOI] [PubMed] [Google Scholar]
- 54.Riccio DA, Nugent JL and Schoenfisch MH, Chem. Mater, 2011, 23, 1727–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhai X, Peleshanko S, Klimenko NS, Genson KL, Vaknin D, Vortman MY, Shevchenko VV and Tsukruk VV, Macromolecules, 2003, 36, 3101–3110. [Google Scholar]
- 56.Zhang X, Mansouri S, Mbeh D, Yahia LH, Sacher E and Veres T, Langmuir, 2012, 28, 12879–12885. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.