

Abstract
Background:
Advancements in sequencing capabilities have enhanced the study of the human microbiome. There are limited studies focused on the gastro-intestinal (gut) microbiome of infants, particularly the impact of diet between breast-fed (BF) versus formula-fed (FF). It is unclear what effect, if any, early feeding has on short-term or long-term composition and function of the gut microbiome.
Results:
Using a shotgun metagenomics approach, differences in the gut microbiome between BF (n = 10) and FF (n = 5) infants were detected. A Jaccard distance principle coordinate analysis was able to cluster BF versus FF infants based on the presence or absence of species identified in their gut microbiome. Thirty-two genera were identified as statistically different in the gut microbiome sequenced between BF and FF infants. Furthermore, the computational workflow identified 371 bacterial genes that were statistically different between the BF and FF cohorts in abundance. Only seven genes were lower in abundance (or absent) in the FF cohort compared to the BF cohort, including CRISPR/Cas9; whereas, the remaining candidates, including autotransporter adhesins, were higher in abundance in the FF cohort compared to BF cohort.
Conclusions:
These studies demonstrated that FF infants have, at an early age, a significantly different gut microbiome with potential implications for function of the fecal microbiota. Interactions between the fecal microbiota and host hinted at here have been linked to numerous diseases. Determining whether these non-abundant or more abundant genes have biological consequence related to infant feeding may aid in understanding the adult gut microbiome, and the pathogenesis of obesity.
Keywords: Metagenomics, Next generation sequencing, Gut microbiome, Whole genome, Breast-feeding, Infants
1. Background
Obesity is a national epidemic, with one in three adolescents overweight, [1] and one in five considered clinically obese. Adolescents with obesity are at high risk for “adult” morbidities in their youth: cardiovascular disease, type-2 diabetes, joint injury, sleep apnea, and non-alcoholic fatty liver disease (NAFLD) [2]. Early dietary content is critical to the long-term development of obesity in children and adolescents [3]. Infants who are breast-fed have lower risk for childhood and adult obesity compared to formula-fed infants [4]. Different feeding regimens in infancy have been shown to contribute to differences in weight gain [5] and to alter the gastrointestinal microbial environment [6]. The multitude of organisms that live in the human digestive tract, the fecal microbiota, and their genomes, the gut microbiome, in turn influence gastrointestinal satiety hormone secretion and signaling, primarily through short-chain fatty acids [7]. Formula-feeding increases the bacterial phyla Firmicutes and Proteobacteria and decreases Actinobacteria as compared to breast-feeding [8,9]. While the fecal microbiota differences between breast- and formula-fed infants typically converge by the end of the second or third year of life [10], there is evidence that the effect of the fecal microbiota’s divergence early in the first year of life on metabolic, immunologic, and cardiovascular diseases is significant in the long term [11]. Very early disruptions in the gut microbiome of infants by antibiotics, delivery mode, or altered environments seem to play a lasting effect on its population and function [4,12].
Two studies have demonstrated differences in the fecal microbiota of infants who are breast-fed versus those who are formula-fed [8,9]. These groups used fecal collection from infants to assess the metagenomics (genomes of genetic material isolated from environmental samples) of the two populations of infants. The sample sizes were small, but key differences were noted. As stated, it is unknown whether the differences in early fecal microbiota, and its corresponding gut microbiome, change the gut environment irrevocably and confer altered satiety regulation to children based on their different initial nutrition. The study hypothesizes that a protective or an ameliorative effect of breastfeeding on later risk for obesity occurs via fecal microbiota modulation of satiety hormone expression and regulation.
Prior work comparing the gut microbiome of breast-fed and formula-fed infants used a targeted approach (16S rRNA sequencing) [9] or metagenomic and metatranscriptomic sequencing [8] based on Roche (Nutley, NJ) 454 technology. The present study utilized a whole-genome approach coupled with Illumina HiSeq technology to assess taxa diversity between groups. Computational bioinformatics allowed characterization of key functional differences by way of relative fecal microbiota gene abundance comparisons. The study presented here aims to strictly and rigorously characterize differences in the very early gut microbiome of breast-fed vs. formula-fed infants; the primary goal is to determine if diet-related changes in the gut microbiome early in infancy initiate long-term cascading consequences even if abundance or taxonomic population traits normalize with food introduction and with early childhood growth. This cross-sectional pilot study sets the groundwork for a longitudinal mapping of the gut microbiome trajectory in these infants in the first two years of life.
2. Results
2.1. Subjects
Fifteen subjects were enrolled, and duplicate fecal samples were processed for each subject. Table 1 details demographic information about subjects. Ten infants were exclusively breast-fed (BF) and five were exclusively formula-fed (FF). Infants were of similar age at enrollment/collection of sample (BF, 45–95 days vs. FF 46–100 days), similar weight at birth (BF, mean 3.23 kg vs. FF, mean 3.37 kg) and enrollment (5.10 kg vs. 5.06 kg), with similar maternal age (33 years vs. 33 years), paternal age (35 years vs. 34 years), BMI (27.9 kg/m2 vs. 26.7 kg/m2), and (maternal) pre-pregnancy BMI (26.5 kg/m2 vs. 24.7 kg/m2).
Table 1.
Subject Characteristics/Demographics.
| Breast-fed (n = 10) | Formula-fed (n = 5) | |
|---|---|---|
| Sex, F | 50% | 0% |
| Age, days (mean, STD) | 79, 15 | 65,22 |
| (median, IQR) | 82, 14 | 59,22 |
| Race, Caucasian | 80% | 60% |
| Ethnicity, Hispanic | 20% | 20% |
| Delivery, SVD | 90% | 60% |
| Birth weight, g (mean, STD) | 3.23, 0.46 | 3.37, 0.3 |
| Enrollment weight, g (mean, STD) | 5.10, 0.75 | 5.06, 0.40 |
| Maternal age, years (mean, STD) | 33, 4.0 | 33, 6.1 |
| Paternal age, years (mean, STD) | 35, 6.5 | 34, 6.6 |
| Maternal BMI, kg/m2 (mean, STD) | 27.9, 7.4 | 26.7, 3.4 |
| Maternal pre-pregnancy BMI, kg/m2 (mean, STD) | 26.5, 6.9 | 24.7, 4.2 |
| Paternal BMI, kg/m2 (mean, STD) | 29.8, 9.6 | 26.5, 2.2 |
Descriptive statistics for subject characteristics; two-tailed Student’s T-test for numerical values, Fisher’s exact test for categorical values. There were no statistically significant differences between the two cohorts for any characteristic measured except for maternal height (not shown), which was lower in the formula-fed cohort (63 in. vs. 66.3 in., p < 0.01).
2.2. Metagenomic sequencing Beta-diversity
Each subject provided adequate duplicate fecal samples for shotgun metagenomics analysis (Fig. 1). Library sequencing via Illumina HiSeq technology resulted in over 376 million raw paired end reads with each library having an average of over 12.5 million read pairs. Even after adapter trimming and the decontamination of human and PhiX the average number of read pairs per library remained over 12 million (Supplementary Table 1). Taxonomic analysis was completed via the Sunbeam pipeline with Kraken1 and yielded successful classification of over 269 million reads with 253 million classified to at least the genus level, over 81% of which were attributable to a species. Results between biological replicates were consistent with R2 values ranging from 0.86 to 0.93 (Supplementary Fig. 1).
Fig. 1.
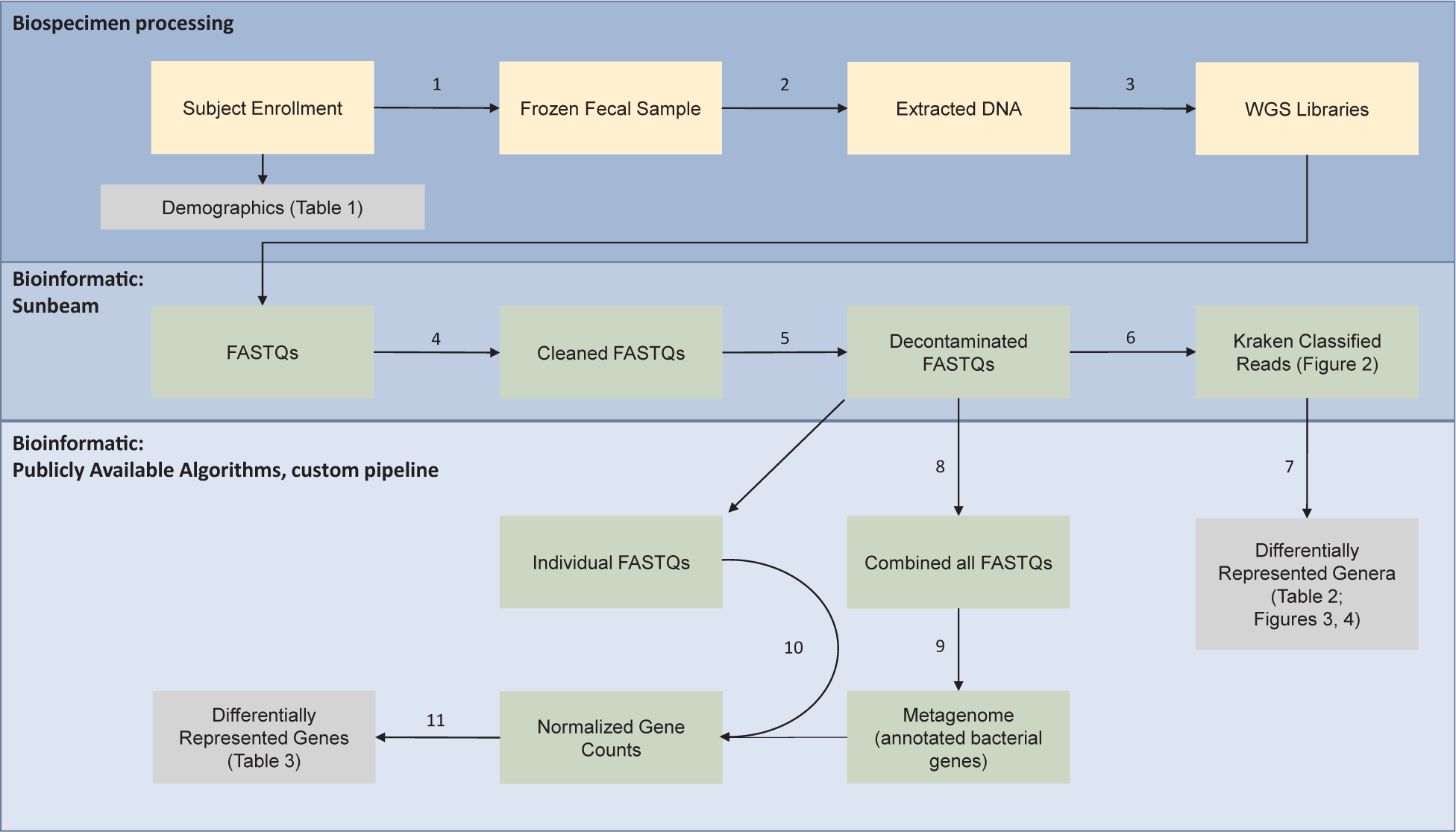
Metagenomic workflow for comparing breast-fed versus formula-fed infants. Bio specimen processing (top box): Subjects were enrolled into two groups; breast-fed (BF) and formula-fed (FF) and demographics are summarized in Table 1. (1) A fresh fecal sample was collected and flash frozen per subject. (2) DNA was extracted from the fecal sample and (3) used to prepare shotgun metagenomic libraries for next generation sequencing (Illumina platform). Bioinformatics: Sunbeam (middle box): Raw reads (FASTQ) were (4) quality trimmed to remove adapter sequences and low-quality bases. The cleaned FASTQ files (5) were mapped to the human genome and PhiX to remove contaminating (un-specific) or control reads. The reads from the decontaminated FASTQ (6) were classified using the Kraken database. Bioinformatics: Publicly available algorithms, custom pipeline (bottom box): The Kraken classified reads were (7) analyzed via edgeR to determine differentially represented genera, summarized in Table 2 and Figs. 2–4. The decontaminated FASTQ files were pooled (8) to create one large library for de novo assembly (MEGAHIT) of a metagenome (9), annotated with prodigal and NCBI COGs. Reads from the individual FastQs were aligned, using STAR and RSEM, to the metagenome (10). Normalized gene counts were calculated via edgeR and results are displayed in Table 2.
Breast-fed and formula-fed cohorts were examined using both Bray-Curtis dissimilarity and Jaccard distance principal coordinate analysis. Technical replicates at the individual level clustered together; however, there was no clear pattern of beta-diversity by abundance at the cohort level (Fig. 2A). On the other hand, presence or absence of species did demonstrate clustering at the cohort level (Fig. 2B). Based on these results, the difference in the fecal microbiota between early infancy formula-fed and breast-fed infants at the species level is dependent on presence or absence, rather than abundance, of taxa.
Fig. 2.
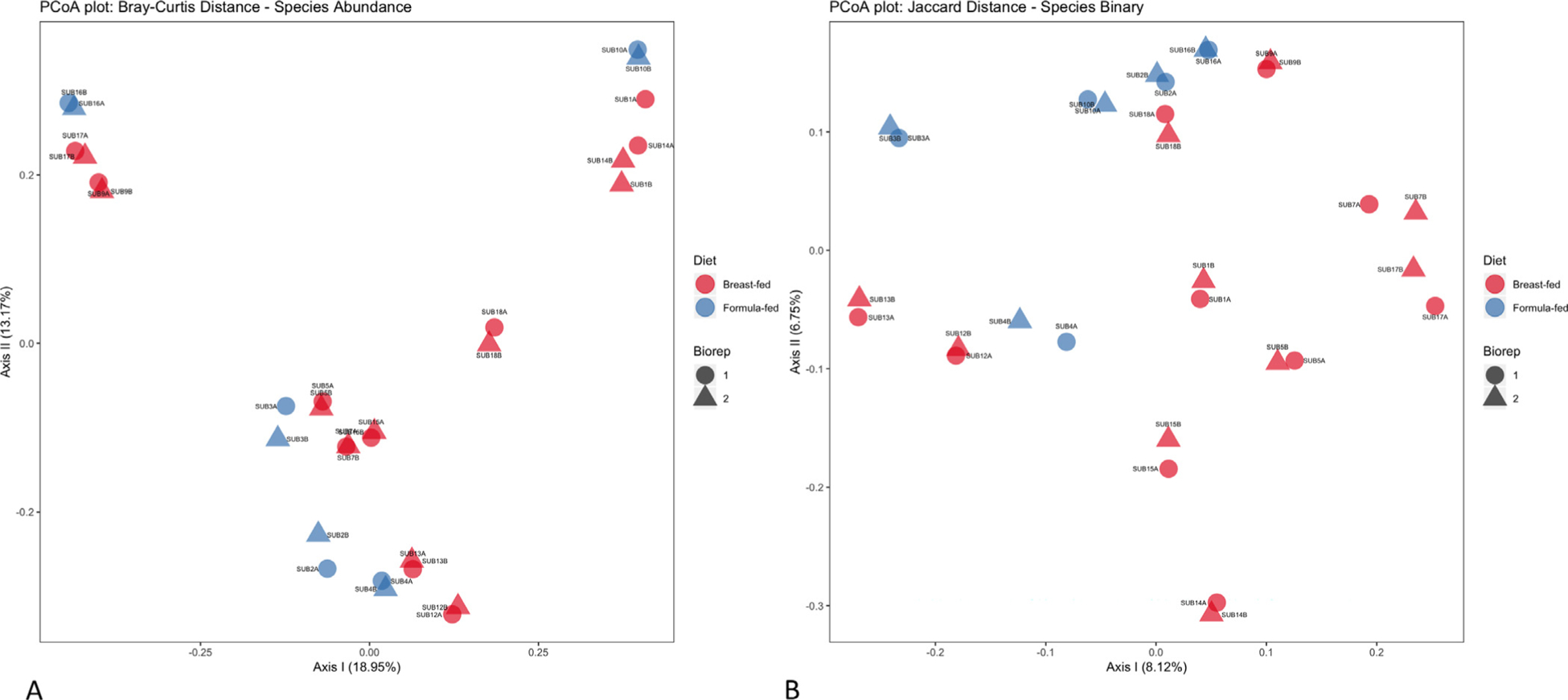
Principle coordinates analysis based on species level data. (A) Bray-Curtis distance plot based on species abundance per subject. Each data point represents either a breast-fed (red) or formula-fed (blue) subject. The shape of the data points represents either technical replicate 1 (circle) or technical replicate 2 (triangle). Axis 1 has a variance of 17.25% and axis 2 has a variance of 12.04%. (B) Jaccard Distance plot based on presence or absence of species per subject. Each data point represents either a breast-fed (red) or formula-fed (blue) subject. The shape of the data points represents either technical replicate 1 (circle) or technical replicate 2 (triangle). Axis 1 has a variance of 7.62% and axis 2 has a variance of 6.46%.
2.3. Phylogenetic abundance
While principal coordinate analysis clustering by species demonstrated differences between the cohorts based on presence or absence, we also assessed phylogenetic abundance in each cohort at the genus level. The top twenty most abundant genera were determined and plotted via box-whisker plots to examine distribution differences between the cohorts (Fig. 3), and statistical testing via edgeR [13] with an FDR cutoff of 0.01 revealed five out of those twenty genera were statistically different between breast-fed and formula-fed infants (Fig. 3, asterisks). In total, there were thirty-two genera with statistically significant differences in abundance between breast-fed and formula-fed infants (Supplementary Table 2). Twelve genera were decreased in abundance in the formula-fed infants, including Haemophilus, Parabacteroides, Serratia, and Lactobacillus, while twenty genera were increased in the formula-fed infants, including Clostridioides, Enterococcus, Stenotrophomonas, and Akkermansia. Relative abundance of the top 20 genera in each sample is represented in Supplementary Fig. 3.
Fig. 3.
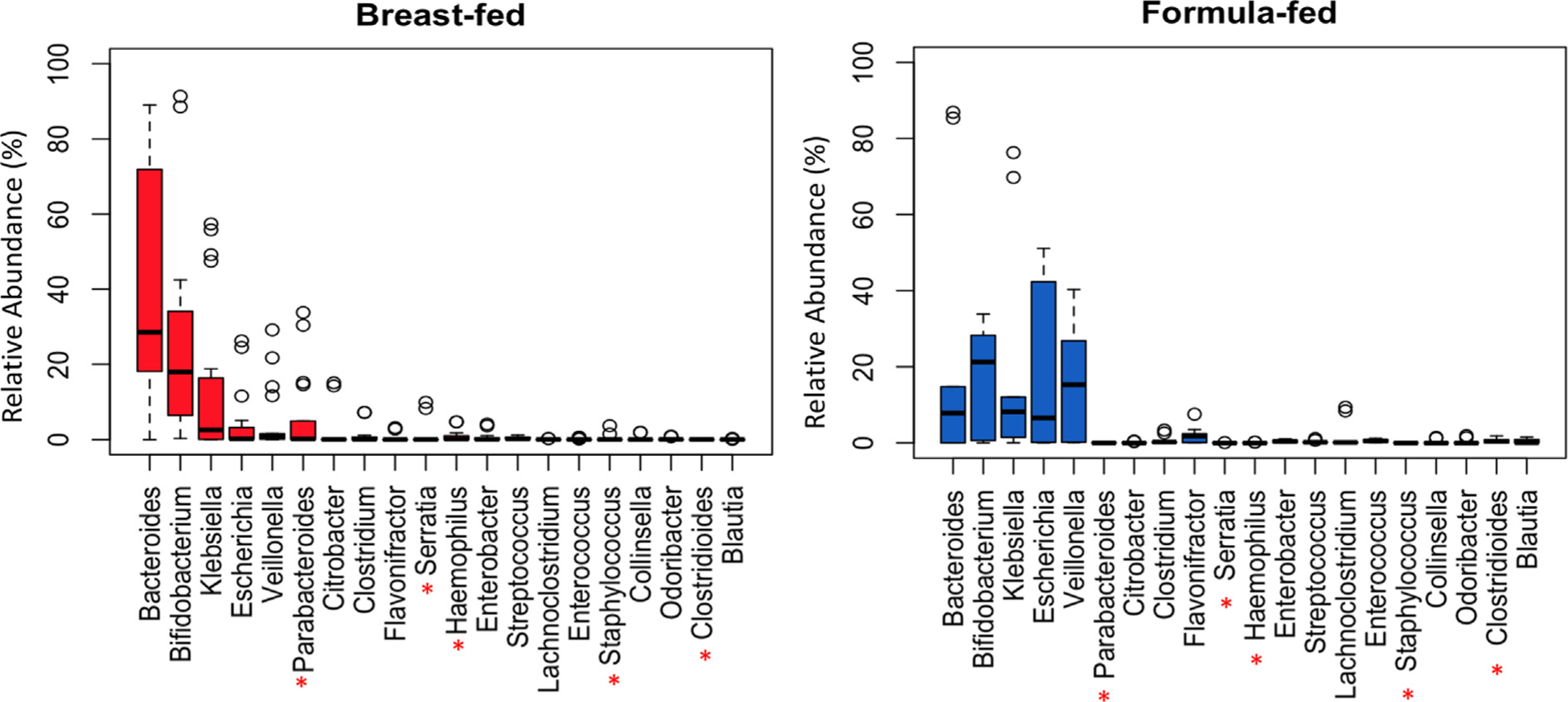
Distribution of genera identified in the gut microbiome of breast-fed and formula-fed infants. Left Panel: Box-plot of the top most abundant genera in breast-fed infants (red boxes). Right Panel: Box-plot of the top most abundant genera in formula-fed infants (blue boxes). The red asterisks represent the genera that were statistically different between the breast-fed and formula-fed cohorts. The y-axis represents phylogenetic abundance (percentage), and each genus is represented on the x-axis.
2.4. Differential gene counts and annotation
A co-assembly of all sequencing reads across all subjects was created and had a total length of 435,829,348 base pairs (bp) and 305,432 total contigs. The N50 was 3,422 bp and the mean was 1,426 bp. Maximum contig length was 378,421 bp. Over 32,000 contigs were greater than 2,500 bp in length and were used for gene prediction. Reads from individual samples were then mapped back to the combined metagenome, gene abundances were calculated, and statistical testing was performed using edgeR. The computational workflow identified 371 genes that statistically different abundances between breast-fed and formula-fed infant samples using an FDR cutoff of 0.01. Of note, only seven of these genes had low abundance in formula-fed compared to breast-fed while the remaining 364 had high abundance (Table 2).
Table 2A.
Differentially Abundant Genes in Formula-fed Infants.
| Gene ID | logFC | logCPM | P Value | FDR | Accession | Function |
|---|---|---|---|---|---|---|
| 167627 | −12.88 | 6.78 | 3.6E-04 | 0.009 | COG3513 | CRISPR/Cas system Type II associated protein, contains McrA/HNH and RuvC-like nuclease domains |
| 162597 | −12.13 | 8.40 | 3.2E-05 | 0.002 | COG0474 | Magnesium-transporting ATPase (P-type) |
| 158392 | −10.71 | 7.83 | 1.0E-04 | 0.004 | COG1196, COG1511, COG3941 | Chromosome segregation ATPase; Uncharacterized membrane protein YhgE, phage infection protein (PIP) family; Phage tail tape-measure protein, controls tail length |
| 203652 | −9.94 | 9.24 | 3.8E-04 | 0.010 | COG0085 | DNA-directed RNA polymerase, beta subunit/140 kD subunit |
| 236312 | −8.72 | 10.61 | 3.3E-04 | 0.009 | COG0787 | Alanine racemase |
2.5. Validation of Cas9 identity and abundance
The aforementioned bioinformatic analysis identified a Cas9 gene as being completely absent in all formula-fed samples. While there are multiple Cas9 genes originating from various bacteria, the potential impact of this finding made its validation paramount. To confirm that the gene in question was a Cas9, the Arg rich region was identified in the peptide sequence manually and the canonical HNH and RuvC domains [14] were identified via InterPro (Fig. 4A). A number of other Cas9 specific domains were also found, a few of which, such as the Cas9 topo homology domain, are specific to Actinobacteria.
Fig. 4.
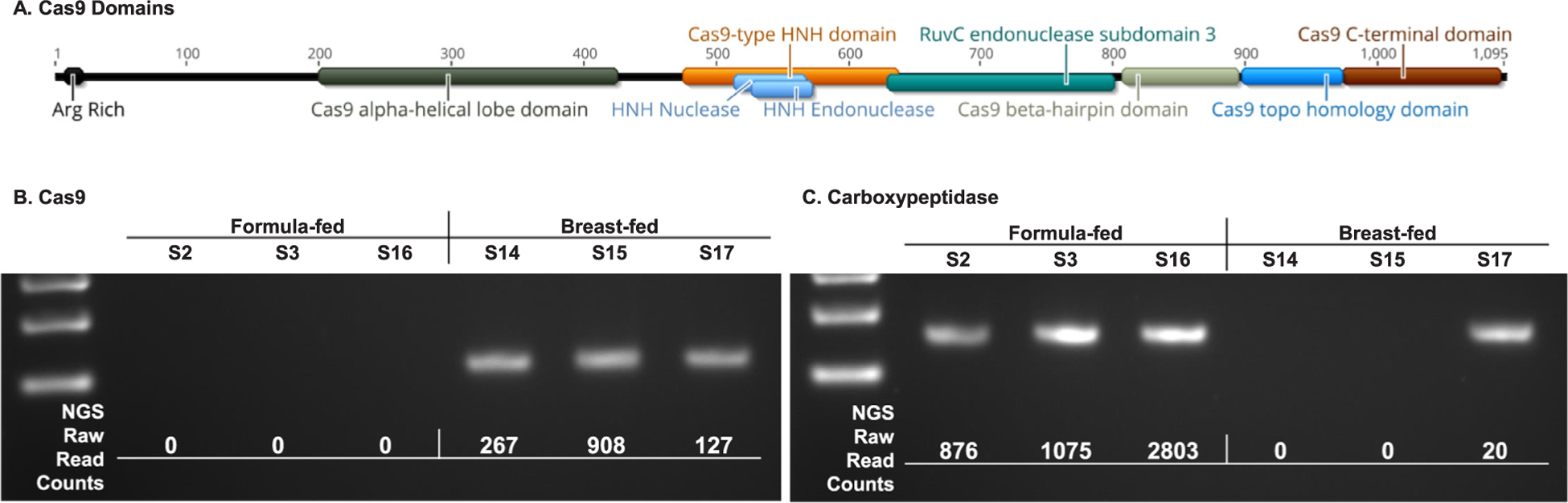
Cas9 Validations. InterPro was used to analyze the amino acid sequence coded for by the Cas9 gene to validate its identity (A). Non-quantitative PCR was used to validate the results of the bioinformatic analysis for Cas9 (B) as well as a Carboxypeptidase (C).
Since the shotgun sequencing and subsequent bioinformatic analysis indicated that this Cas9 gene was completely absent from formula-fed samples, non-quantitative PCR was used to validate these results in 6 samples. A PCR product for Cas9 was observed in all breast-fed but none of the formula-fed samples (Fig. 4B). A carboxypeptidase was also examined and yielded PCR products in all formula-fed, but only one breast-fed sample. The presence or absence of PCR products for both genes correlated perfectly with the presence or absence of raw read counts which mapped to these genes in the bioinformatic analysis.
3. Discussion
Improved understanding of the gut microbiome and its components in the context of the gut-brain-adipose axis [15], create opportunities to develop novel therapeutic interventions for myriad medical conditions [16], including obesity [17]. The intestinal milieu is comprised of both intrinsic (host origin) and extrinsic (non-host origin) factors including epithelial enterocytes, cytokines, paracrine hormones, microbes, and inflammatory mediators, directly impact human health.
Recognizing the debate about the effect of breast-feeding vs. formula feeding on long-term gut health (including the health and function of the microenvironment) [18], this study sought to explore the role of exclusive single-source feeding in the gut microbiome early in life. Ongoing studies will collect feeding logs, additional anthropo-metric data, and fecal samples; this paper summarizes the results of the baseline gut microbiome analysis.
The hypothesis was that the fecal microbiota would differ in formula-fed and breast-fed infants, with specific increases in microbial diversity and relative microbial gene abundance as early as the first 2 months. These changes early in life would impact long-term cellular processes locally and throughout the host by way of interaction with the gut epithelium in an entero-endocrine manner [7]. A recent study demonstrated the susceptibility of specialized epithelial cells in the gut to metabolically active compounds generated by the fecal microbiota and these cells’ importance in neural pathways [19]. Future longitudinal studies will help determine if the changes are permanent or not.
In this study, the BF and FF cohorts demonstrated clustering / similarity based on the presence/absence of species (Fig. 2B). The global abundance of bacteria is similar between the two groups. Greater variation in species diversity implied an early divergence in infants who varied by feeding source. Bacteroides genus predominated in BF, as expected, while the prevalence of Bifidobacterium genus was comparable in the two groups. Notably, the potentially beneficial genus Lactobacillus [20] was more than four-fold lower in the FF group perhaps related to the mode of delivery. A more striking difference in the presence, absence, and abundance of Klebsiella, Escherichia, and Veillonella between BF and FF is observed (Fig. 3) implying early divergence. On average, Escherichia genus was more abundant in FF samples; in other studies have noted associations with patient disease later in life [21]. Overall, the abundance of 12 genera decreased significantly in FF and 20 genera increased significantly in FF as compared to BF (Supplementary Table 2). These observations show that formula feeding dramatically influences the diversity of the gut microbiome early in infancy. Over time, feeding sources typically converge with introduction of solids and transition to table foods. The presence, absence, and relative abundance of fecal microbiota species stabilize and become more “adult-like” beginning at age three years [22]. Individual differences may persist. It is not known whether the early differences in species/genera presence or absence fundamentally alters the function of the gut microenvironment long-term.
The count differences of the bacterial genes detected in the cohorts revealed that FF infants, compared to BF infants, had seven genes that were significantly lower in relative abundance including a CRISPR associated protein 9 (Cas9), Magnesium-transporting ATPase (MgATPase), DNA-directed RNA polymerase (DNA-RNAPol), Chromosome segregation ATPase (ChromATPase), Uncharacterized membrane protein YhgE (YhgE), Phage infection protein family (PIP), and an Alanine racemase (AlaRace). In contrast, nine genes had higher relative abundance in the FF infants (Table 2B). Of note, Autotransporter adhesin (ATAdhes) was the greatest increased abundance gene for bacteria in FF infants. ATAdhes is a family of molecules involved in microbe adherence to cellular structures and in forming biofilms [23]. Biofilms can both contribute to disease and form barriers important for immune function [24]. In the cohort analyzed, 3 out of 5 FF infants had a high abundance of ATAdhes, 2 out of 5 had no or nominal abundance, and all 10 BF infants had no or nominal abundance. Cas9 was the greatest decreased abundance gene for bacteria in FF infants. The lack of CRISPR/Cas or altered alleles of the gene is associated with pathogenic strains and drug-resistance in Escherichia [25]. In the cohort analyzed, this gene was not detected in any of the five FF samples. The foothold for more pathogenic-potential bacteria created in the FF environment merits further examination in light of the absent Cas9 gene abundance. Whether the lack of Cas9 gene changes over time, or persists, may help to explain the divergence in non-BF infants’ gut microbiome. If more association with a shift to Escherichia or Veillonella in FF infants becomes clear with subsequent temporal sampling, Cas9 gene absence is one plausible explanation.
Table 2B.
Differentially Abundant Genes in Formula-fed Infants.
| Gene ID | logFC | logCPM | P Value | FDR | Accession | Function |
|---|---|---|---|---|---|---|
| 231306 | 12.043 | 9.672 | 2.75E-09 | 1.92E-05 | COG5295 | Autotransporter adhesin |
| 141164 | 11.605 | 10.333 | 9.35E-10 | 1.36E-05 | COG5295 | Autotransporter adhesin |
| 174057 | 11.142 | 8.900 | 6.09E-09 | 1.92E-05 | COG1032 | Radical SAM superfamily enzyme YgiQ, UPF0313 family |
| 179341 | 10.627 | 9.914 | 6.69E-08 | 1.22E-04 | COG1239, COG1240 | Mg-chelatase subunit ChlD |
| 238386 | 10.485 | 9.778 | 6.73E-09 | 1.92E-05 | COG0744 | Membrane carboxypeptidase (penicillin-binding protein) |
| 241599 | 10.202 | 7.317 | 2.34E-07 | 1.80E-04 | COG0119 | Isopropylmalate/homocitrate/citramalate synthases |
| 179344 | 10.180 | 8.850 | 8.87E-09 | 1.92E-05 | COG0609 | ABC-type Fe3 + -siderophore transport system, permease component |
| 145985 | 10.078 | 8.892 | 1.88E-07 | 1.52E-04 | COG1984 | Allophanate hydrolase subunit 2 |
| 175186 | 9.846 | 8.624 | 1.04E-07 | 1.34E-04 | COG2271 | Sugar phosphate permease |
| 193265 | 9.601 | 8.654 | 5.34E-07 | 3.63E-04 | COG0795 | Lipopolysaccharide export LptBFGC system, permease protein LptF |
The top seven, and top ten, genes that mapped as the most significantly decreased abundance (A) or increased abundance (B) in the formula-fed infants compared to breast-fed infants.
This study is limited by the size of the cohort and the cross-sectional examination of the gut microbiome under the selection criteria. While the small size limits generalizability, it mirrors other studies on infant feeding and fecal microbiota make-up [8,9]. Notably, fecal samples from an additional four formula-fed infants, collected in an earlier study under the same ethics approval and consent, were processed in a different manner but included in a principal coordinate analysis (Supplementary Fig. 2, Supplementary Table 3). Clustering seen with the original 15 subject cohort was again noted when the four additional samples were added to the analysis, supporting the conclusions reached. The whole genome metagenomics and the bioinformatics computational pipeline in this study yields a detailed examination of the two groups. More data is needed to obtain a longitudinal picture of the gut microenvironment and will help to determine if observed trends do indeed persist beyond early infancy. The present study did not account for any concomitant urinary microbiome [26].
4. Methods
4.1. Aim, design, and setting
The study intended to confirm differences in the gut microbiome resulting from early infant nutrition as determined by whole genome untargeted (shotgun) metagenomic sequencing of fecal samples from breast-fed and formula-fed infants to determine β-diversity, relative abundance, and functional profiles. Study design is described graphically in Fig. 1. The setting was a tertiary children’s academic hospital center serving a population of infants from four surrounding states. The Nemours Institutional Review Board approved the study. The study was registered at ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03751137.
4.2. Recruitment
Fifteen healthy, term infants between 6 weeks 0 days and 14 weeks 6 days of age who were exclusively breast-fed (BF) or formula-fed (FF) were recruited. Infants were excluded if they had any other sources of nutrition, dietary restrictions (e.g. hypoallergenic formula), consumed higher density formula (greater than20 calories/ounce), had exposure to antibiotics, or had any gastrointestinal infection or disease that affected the integrity of the intestinal mucosa. Fecal samples and clinical data on infants were collected, including demographic information, maternal and paternal age (years) at infant’s birth, maternal and paternal height and weight, delivery method, maternal antibiotic use (breast-feeding mothers only), and maternal over-the-counter or prescription medications taken during pregnancy.
4.3. Sample collection
Soiled diapers were sampled within 6 h of defecation for 10 subjects; the remainder of subjects’ fecal samples were collected within 12 to 24 h. Stool was collected by application of two duplicate swabs (Copan Diagnostics, Murrieta, CA) for metagenomics sequencing. The containers were placed immediately into a dry ice ethanol bath and then transferred to a −80° C freezer until processing. Processing was completed at the Microbiome Center at the Children’s Hospital of Philadelphia within 6 months of freezing.
4.4. DNA Extraction and sequencing
DNA was extracted from samples using the DNeasy PowerSoil kit using the manufacturer’s instructions (Qiagen, Germantown, MD). Libraries were generated from 1 ng of DNA using the NexteraXT kit (Illumina, San Diego, CA, USA) and sequenced on the Illumina HiSeq 2500 using 2×125bp chemistry in High Output mode. Extraction controls (no template) and DNA free water were included to empirically assess environmental and reagent contamination. Laboratory-generated mock communities consisting of DNA from Vibrio campbellii, Cryptococcus diffluens, and Lambda phage were included as positive controls.
4.5. Bioinformatics analysis
FASTQ files were analyzed using the “QC” and “Classify” portions of the Sunbeam pipeline (https://github.com/sunbeam-labs/sunbeam). Trimmomatic [27] was configured for adapter removal and quality trimming using “leading” and “trailing” settings of 3 with a sliding window size of 4 bp and a required quality of 15. The resulting cleaned FASTQ files were mapped to the GRCh38 assembly of the human genome and the PhiX genome using BWA MEM (Li H. 2013, https://arxiv.org/abs/1303.3997) with default settings; unmapped reads were compiled into “decontaminated” FASTQ files for downstream analysis. Kraken1 [28] was used to classify the decontaminated reads via a full Kraken database built on 2018.10.23. Raw read counts, which were classified down to the genus level, were analyzed using edgeR with TMM normalization [29] to calculate statistical significance.
4.6. Contig Assembly, Annotation, and functional analysis
Decontaminated FASTQ files from all samples in the previously discussed analysis were concatenated together and contig assembly was performed using MEGAHIT [30]. Contigs which were ≥ 1,500 bp in length were kept for further analysis. Gene prediction was performed using Prodigal [31], and functional annotations were added using NCBI COGs [32]. The decontaminated reads for each sample were then mapped to the annotated contigs using STAR [33] and ENCODE’s standard settings. RSEM [34] was used to produce gene counts which were analyzed using edgeR with TMM normalization to calculate statistical significance. Only those genes with an FDR less than or equal to 0.01 as well as an average of 150 TMM normalized counts in either group were considered statistically significant.
4.7. Data upload to NIH sequence read archive
Data files used for the study are available via the National Institutes of Health Sequence Read Archive, accession # PRJNA542703.
Supplementary Material
Acknowledgements
Lisa Mattei, CHOP Microbiome Center; Kyle Bittenger, CHOP Bioinformatics; Rachel Marine, CDC; Adebowale Adeyemi, Nemours Gastroenterology; Alan Robbins, Nemours Biomedical Research.
Funding
This work was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number U54-GM104941.
Abbreviations:
- BF
Breast-fed
- FF
Formula-fed
- BMI
Body-mass index
- N50
The minimum contig length needed to cover 50% of the metagenome.
- ENCODE
Encyclopedia of DNA Elements
- FDR
False Discovery Rate
- CRISPR
Clustered Regularly Interspaced Short Palindromic Repeats
- PCoA
Principal Coordinates Analysis
- TMM
Trimmed Mean of M-values
Footnotes
Ethics approval and consent to participate
All subjects were given permission to participate by a parent or guardian via a signed parental permission form that was approved by the Nemours Institutional Review Board.
Availability of data and material: The datasets generated and/or analyzed during the current study are being uploaded to the NIH Sequence Read Archive (SRA).
Financial disclosures
None.
Declaration of Competing Interest
None of the authors has a conflict of interest to report.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.humic.2019.100057.
References
- [1].Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA - J Am Med Assoc 2014;311:806–14. 10.1001/jama.2014.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].McCrindle BW. Cardiovascular Consequences of Childhood Obesity. Can J Cardiol 2015;31:124–30. 10.1016/j.cjca.2014.08.017. [DOI] [PubMed] [Google Scholar]
- [3].Singhal A, Lanigan J. Breastfeeding, early growth and later obesity. Obes Rev 2007;8:51–4. 10.1111/j.1467-789X.2007.00318.x. [DOI] [PubMed] [Google Scholar]
- [4].Cox LM, Yamanishi S, Sohn J, Alekseyenko AV, Leung JM, Cho I, et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 2014;158:705–21. 10.1016/j.cell.2014.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Harder T, Bergmann R, Kallischnigg G, Plagemann A. Duration of breastfeeding and risk of overweight: a meta-analysis. Am J Epidemiol 2005;162:397–403. 10.1093/aje/kwi222. [DOI] [PubMed] [Google Scholar]
- [6].De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci 2010;107:14691–6. 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cani PD, Everard A, Duparc T. Gut microbiota, enteroendocrine functions and metabolism. Curr Opin Pharmacol 2013;13:935–40. 10.1016/j.coph.2013.09.008. [DOI] [PubMed] [Google Scholar]
- [8].Schwartz S, Friedberg I, Ivanov IV, Davidson LA, Goldsby JS, Dahl DB, et al. A metagenomic study of diet-dependent interaction between gut microbiota and host in infants reveals differences in immune response. Genome Biol 2012;13.. 10.1186/gb-2012-13-4-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lee SA, Lim JY, Kim BS, Cho SJ, Kim NY, Bin Kim O, et al. Comparison of the gut microbiota profile in breast-fed and formula-fed Korean infants using pyrosequencing. Nutr Res Pract 2015;9:242–8. 10.4162/nrp.2015.9.3.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature 2012;486:222–7. 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ross MC, Lernmark A, Hagopian W, Gibbs RA, Xavier RJ, Hutchinson DS, et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 2018;562:583–8. 10.1038/s41586-018-0617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cox LM, Blaser MJ. Antibiotics in early life and obesity. Nat Rev Endocrinol 2015;11:182–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009;26:139–40. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 2014;343:1247997. 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Beumer J, Clevers H. How the Gut Feels, Smells, and Talks. Cell 2017;170:10–1. 10.1016/j.cell.2017.06.023. [DOI] [PubMed] [Google Scholar]
- [16].Lynch SV, Pedersen O. The Human Intestinal Microbiome in Health and Disease. N Engl J Med 2016;375:2369–79. 10.1056/NEJMra1600266. [DOI] [PubMed] [Google Scholar]
- [17].Bouter KE, van Raalte DH, Groen AK, Nieuwdorp M. Role of the Gut Microbiome in the Pathogenesis of Obesity and Obesity-Related Metabolic Dysfunction. Gastroenterology 2017;152:1671–8. 10.1053/j.gastro.2016.12.048. [DOI] [PubMed] [Google Scholar]
- [18].Pannaraj PS, Li F, Cerini C, Bender JM, Yang S, Rollie A, et al. Association between breast milk bacterial communities and establishment and development of the infant gut microbiome. JAMA Pediatr 2017;171:647–54. 10.1001/jamapediatrics.2017.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bellono NW, Bayrer JR, Leitch DB, Castro J, Zhang C, O’Donnell TA, et al. Enterochromaffin Cells Are Gut Chemosensors that Couple to Sensory Neural Pathways. Cell 2017;170(185–198):e16 10.1016/j.cell.2017.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Montoya-Williams D, Lemas DJ, Spiryda L, Patel K, Carney OO, Neu J, et al. The Neonatal Microbiome and Its Partial Role in Mediating the Association between Birth by Cesarean Section and Adverse Pediatric Outcomes. Neonatology 2018;114:103–11. 10.1159/000487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Singh P, Teal TK, Marsh TL, Tiedje JM, Mosci R, Jernigan K, et al. Intestinal microbial communities associated with acute enteric infections and disease recovery. Microbiome 2015;3:45. 10.1186/s40168-015-0109-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rodríguez JM, Murphy K, Stanton C, Ross RP, Kober OI, Juge N, et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb Ecol Heal Dis 2015;26.. 10.3402/mehd.v26.26050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vo JL, Martínez Ortiz GC, Subedi P, Keerthikumar S, Mathivanan S, Paxman JJ, et al. Autotransporter Adhesins in Escherichia coli Pathogenesis. Proteomics 2017;17.. 10.1002/pmic.201600431. [DOI] [PubMed] [Google Scholar]
- [24].Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science (80- 2018;)(359):592–7. 10.1126/science.aah3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Koonin EV, Makarova KS, Zhang F. Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol 2017;37:67–78. 10.1016/j.mib.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Morand A, Cornu F, Dufour J-C, Tsimaratos M, Lagier J-C, Human Raoult D. Bacterial Repertoire of the Urinary Tract: a Potential Paradigm. Shift. J Clin Microbiol 2019;57.. 10.1128/JCM.00675-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114–20. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wood DE, Kraken Salzberg SL. Ultrafast metagenomic sequence classification using exact alignments. Genome Biol 2014;15.. 10.1186/gb-2014-15-3-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pereira MB, Wallroth M, Jonsson V, Kristiansson E. Comparison of normalization methods for the analysis of metagenomic gene abundance data. BMC Genomics 2018;19:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015;31:1674–6. 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- [31].Hyatt D, Locascio PF, Hauser LJ, Uberbacher EC. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 2012;28:2223–30. 10.1093/bioinformatics/bts429. [DOI] [PubMed] [Google Scholar]
- [32].Tatusov RL, Koonin EV, Lipman DJ. A RTICLES A Genomic Perspective on Protein Families. Science 2012;631:631–7. 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- [33].Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li B, Dewey CN. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. Bioinforma Impact Accurate Quantif Proteomic Genet Anal Res 2014:41–74. 10.1201/b16589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.