

Keywords: machine learning, Phox2B, random forest, supervised learning, unsupervised learning
Abstract
Modern neurophysiology research requires the interrogation of high-dimensionality data sets. Machine learning and artificial intelligence (ML/AI) workflows have permeated into nearly all aspects of daily life in the developed world but have not been implemented routinely in neurophysiological analyses. The power of these workflows includes the speed at which they can be deployed, their availability of open-source programming languages, and the objectivity permitted in their data analysis. We used classification-based algorithms, including random forest, gradient boosted machines, support vector machines, and neural networks, to test the hypothesis that the animal genotypes could be separated into their genotype based on interpretation of neurophysiological recordings. We then interrogate the models to identify what were the major features utilized by the algorithms to designate genotype classification. By using raw EEG and respiratory plethysmography data, we were able to predict which recordings came from genotype class with accuracies that were significantly improved relative to the no information rate, although EEG analyses showed more overlap between groups than respiratory plethysmography. In comparison, conventional methods where single features between animal classes were analyzed, differences between the genotypes tested using baseline neurophysiology measurements showed no statistical difference. However, ML/AI workflows successfully were capable of providing successful classification, indicating that interactions between features were different in these genotypes. ML/AI workflows provide new methodologies to interrogate neurophysiology data. However, their implementation must be done with care so as to provide high rigor and reproducibility between laboratories. We provide a series of recommendations on how to report the utilization of ML/AI workflows for the neurophysiology community.
NEW & NOTEWORTHY ML/AI classification workflows are capable of providing insight into differences between genotypes for neurophysiology research. Analytical techniques utilized in the neurophysiology community can be augmented by implementing ML/AI workflows. Random forest is a robust classification algorithm for respiratory plethysmography data. Utilization of ML/AI workflows in neurophysiology research requires heightened transparency and improved community research standards.
INTRODUCTION
Neurophysiology data sets show high dimensionality and suffer in many instances high signal to noise ratios. These challenges make it difficult to identify baseline differences between animal groups, and often neurophysiologists have to resort to a spectrum of physiological maneuvers to highlight dysfunctions in animals. Additional techniques to identify novel machine learning and artificial intelligence (ML/AI) methodologies include a collection of mathematical and computer science tools with broad problem-solving capabilities for multiple fields. Of particular relevance to investigative neurophysiology is the capacity to mine pre-existing data sets to refine and/or crystalize hypotheses before executing wet-laboratory experiments. Furthermore, the capacity of ML/AI tools to identify distinctions in groups with significantly overlapping features is unmatched [reviewed by Vu et al. (1)]. Thus, neurophysiological studies represent an ideal implementation of ML/AI. For instance, unrestrained murine respiratory plethysmography captures over a dozen features, often averaged in 2-s bins of information over long recordings periods. This leads to large data sets without a clear consensus on how best to evaluate the data. A commonly utilized approach is to reduce the data by evaluating, subjectively, the recording and isolating six 10-s periods with stable breath patterns during a day-time recording (for instance, see Refs. 2–5). Sleep studies only use a subset of the EEG recording frequency steps in the designation of vigilance states and frequently discard all other data located in high frequency bands (e.g., γ: 30–80 Hz; for instance, Ref. 6). In addition to the subjectivity of these approaches, discarding a majority of the data captured on the animal represents additional drawback, as instructive features hidden within the data may not be identified by human investigators, and its implementation in biomedical research workflows has become a necessity for modern neuroscientists (7).
A second challenge in neurophysiology research represents the interpretation of gene expression studies obtained from fixed brain tissue. Research on murine brainstem gene expression patterns has lagged that of forebrain and spinal cord research. For instance, querying the GEO (Gene Expression Omnibus) database at the time of writing this manuscript showed less than 8,000 GEO submissions associated with the terms “medulla” or “pons,” a stark contrast to the over 37,000 results obtained from GEO search results for “hippocampus.” The advent of Nanostring technology, which uses predesignated codesets of small oligonucleotides, has the capacity to transform neurophysiology research. This methodology 1) requires significantly lower levels of RNA than other methods such as microarray or RNAseq, 2) Nanostring does not require RNA amplification, and 3) these expression studies can be carried out on formalin-fixed paraffin or OCT embedded tissues, enabling retrospective analysis of banked tissue data sets (8). This technology opens to us the possibility of performing gene expression studies on banked tissues from human, nonhuman primate, and other experimental systems. However, no consensus in the neurophysiology/neuroscience community exists as to the best methodology with which to normalize such Nanostring data sets. For instance, within the Nanostring normalization package, there are over 80 potential permutations of how to normalize these data sets.
In this technical report, we apply ML/AI approaches to interpreting high dimensionality physiology data. To achieve this, we performed a secondary analysis of plethysmographic and electroencephalographic data previously published by our group, performed on animals that underwent genetic ablation of Phox2b-derived astrocytes (9). Adult mice suffering Phox2b-derived astrocyte ablation show a deficient hypoxic chemoreflex during the roll-off/phase II of the acute hypoxic chemoreflex and show a mildly fractured sleep architecture. These animals also showed neurodegenerative ultrastructural features in the axonal terminals of the ventrolateral medulla, insinuating that neurons projecting into the ventrolateral medulla may be undergoing changes in line with apoptotic activation. First, we make the case that discarding the majority of the data from respiratory physiology or EEG results in important feature loss by demonstrating that even at baseline respiratory physiology patterns between control and mutant animals can be observed when traditional techniques are unable to do so. We then propose a workflow with which to study and analyze Nanostring brainstem gene expression studies for neurophysiology research. We also provide in the Supplemental Materials the prototype code we used to perform these studies in the R programming language, an open source statistical language, in the Supplemental Information (all Supplemental Material is available at https://figshare.com/articles/figure/code4manuscript_pdf/13491351).
MATERIALS AND METHODS
Animal Husbandry
Procedures utilizing mice were performed in accordance with The Ohio State University Institutional Animal Care and Use Committee guidelines (protocol number: 2012A00000162-R1). Animal use was in accordance with the NIH Guide for the Care and Use of Laboratory Animals and was approved by The Ohio State University (protocol number 2012A00000162-R2). ALDH1L1loxp-GFP-STOP-loxp-DTA mice (10) and PHOX2Bcre mice (11) were interbreeding to perform the astrocyte ablation technique described by Tsai et al. (10). Animals were housed under standard 12-h light/dark cycle with ad libitum access to food and water.
Whole Body Plethysmography in Conscious Mice
Breathing patterns were measured in conscious mice using barometric, unrestrained whole body plethysmography captured by DSI Buxco system and Finepoint Software. Mice were acclimatized to the plethysmography chambers 2–3 days before the experiments. On the experimental day, freely moving mice were kept in a 500-mL plethysmography chamber with room air for 45–60 min before the ventilatory parameters were recorded. The methods are presented in details in Ref. 9.
Physiological Experiments under Sleep State
Following biotransmitter implantation surgery (F20-EET, DSI), mice were singly housed and their cages were placed on top of receiver boards (RPC-1; DSI) in ventilated cabinets. These boards relay telemetered data to a data exchange matrix (DSI) and a computer running Ponemah software (v. 6.1; DSI, St. Paul, MN). Mice were allowed to recover from the surgery for 2 wk before beginning 2 days of baseline sleep recording. For sleep architecture and spectral analyses methods, see Ref. 9. The data structure of the EEG data was tabular, with each row representing a 10-s bin of data. Each feature contained spectral recordings, which were class numeric, vigilance state, sex, and gender, coded as factors. The levels of genotype were mutant and control. The levels of sleep were rapid eye movement (REM) sleep, wake, and non-rapid eye movement (NREM) sleep. As described in Czeisler et al. (9), each mouse was recorded over a 2-day interval in light and wake cycles, with each row composed of a 10-s bin of data. The feature Time therefore was coded as a sequence of integers from the startpoint of the recording to the end of the recording. The pooled data contained 55 features of a total of 276,480 rows of data.
RNA Extraction and Nanostring Assay
We compared the gene expression profile in the ventrolateral (VLM) and dorsal medulla [nucleus of tractus solitarius (NTS), area postrema (AP), dorsal motor nucleus of vagus (DMNV)] of PHOX2Bcre, ALDH1L1loxp-GFP-STOP-loxp-DTA, and control ALDH1L1loxp-GFP-STOP-loxp-DTA animals. The mice were perfused through the ascending aorta first with 20 mL of phosphate buffered saline and then with 50 mL of 4% paraformaldehyde. The brains were extracted, cryoprotected, and sectioned in the coronal plane at 40 μm. The samples were then microdissected under a dissecting microscope. The dorsal medulla (NTS, AP, and DMNV) was dissected approximately at the bregma level of −7.32 to −7.76 mm and 0.75 mm lateral to the midline. The ventrolateral medulla samples were dissected at bregma level of −6.36 mm to −6.96 mm. The area below the nucleus ambiguus was dissected between 1.0 mm and 1.55 lateral to the midline. At bregma level 6.36 to 6.48 mm, only the ventral surface was dissected (12). RNA was extracted by the FFPE RNA extraction kit (Norgen, Cat. No. 25300). RNA was then subjected to NanoString mRNA analysis (NanoString Technologies, Seattle, WA) neuropathology panel. The RNA was extracted and purified using a FFPE RNA Purification Kit (Cat. No. 25300; Norgen Kit). Briefly, 100 ng of total RNA was hybridized overnight with nCounter NeuroPath Reporter (8 μL) probes in hybridization buffer and in excess of nCounter NeuroPath Capture probes (2 μL) at 65°C for 17 h. After overnight hybridization, probe excess was removed using two-step magnetic beads-based purification on an automated fluidic handling system (nCounter Prep Station). Biotinylated capture probe-bound samples were immobilized and recovered on a streptavidin-coated cartridge. The abundance of specific target molecules was then quantified using the nCounter digital analyzer. Individual fluorescent barcodes and target molecules present in each sample were recorded with a CCD camera by performing a high-density scan (325 fields of view). Images were processed internally into a digital format and exported as Reporter Code Count (RCC).
Machine Learning/Artificial Intelligence Methods and Workflows
All code has been uploaded to a public repository in compliance with this Journal’s Editorial policy (https://figshare.com/articles/figure/code4manuscript_pdf/13491351). Baseline raw respiratory data, EEG recordings, and gene expression were analyzed using R statistics, machine learning toolbox. These parameters were used to generate linear models of mice breathing frequency (f) and tidal volume (TVb) as factors of both inspiratory and expiratory breathing. Predictive features generated from our linear models, data frames were built with each row representing individual animals and each column representing a feature. Data frames were then centered and scaled before analysis utilizing the “scale” function in R’s base package. Model validation was performed with tools from the caret package. All hyperparameters are detailed in the supplementary materials which contains our code. The supplementary materials contain the code we used to generate the predictive modeling tools. Principal component analysis (PCA) and linear discriminant analysis (LDA) were performed utilizing custom R-scripts and the MASS package (13), and visualized using ggplot2 packages within R to identify data clustering (14). With regard to hyperparameter tuning, we utilized the tune function available in R’s caret package. We empirically changed the tuning parameters and evaluating the results on validation sets using the keras package for the neural networks. Hyperparameters utilized in the random forest are delineated in the supplemental file containing the custom R scripts. This was compared against kmeans cluster analysis found with the “kmeans” function in R’s base package and visualized using the factoextra package (15). Feature selection was done utilizing the Boruta package (16). We utilized the keras implementation for neural networks [reviewed by Hackenberger (17)], the random forest R library for random forest (18), the Joust package for adaboost, and the e1071 package for implementation for support vector machines (19). Random forest algorithms were built by randomly subsetting 70% of the data set as training and 30% as test data within the random Forest package. Two sets of analyses were performed: one where all breaths were split 70% training/30% testing, and another where all the breaths from 70% of the animals were placed into testing and the remaining 30% of the animals were placed into testing. For the latter, we generated a for loop that resulted in multiple permutations of the animals that were split into testing and training and collecting the outcome of the accuracy on the test-set prediction in each iteration. Similar approaches were utilized for the EEG prediction analyses. The CARET package was used in R to generate confusion matrixes and compared the overall accuracy of the model to the no information rate as a primary metric (20, 21). Pertinent evaluation metrics for each test is clarified in the legends of each figure or table.
To obtain the approximate number of transitions in Phox2b-ablated mice during the light cycle, we utilized a markov modeling approach using similar methodology described by Perez-Atencio and colleagues (22). We modeled three transitions for each EEG cycle: REM sleep, NREM sleep, and Wake. We utilized the markovchain package and its dependencies in R to construct the model (23). The transition probabilities of each transition were calculated across all animals in the control and mutant groups to construct a transition matrix of a 12-h light cycle. The markov chain was then commenced with the first state of set to Wake for both control and mutant groups. This model was capable of quickly calculating the amount of transitions in each state over a light cycle period.
RESULTS
ML/AI Techniques Can Identify Group Differences Using Unrestrained Whole Body Plethysmography Even When Significant Overlap Exists between Groups
To develop these workflows, we performed a secondary analysis of baseline physiological data from a study set previously published by our group (9). Figure 1 shows histograms of frequency of distribution in the key parameters obtained from unrestrained whole-body plethysmography. Note that by traditional techniques commonly used in our community, no significant difference in the arithmetic mean is noted between control and mutant mice when evaluated at baseline room air (Fig. 1, A–I). These classical statistical analyses might suggest that no difference exists between groups when in fact differences may manifest only when evaluating all features simultaneously. To identify subtle differences between groups, we then pooled all of the data from each measurement and then assigned class labels (either control or mutant) to each measurement based on animal genotypes. Based on these analyses, we predicted that at baseline the control and mutant animals would show significant overlap. We tested this by first performing a linear discriminant analysis (LDA), which generates a linear model that separates groups by maximizing distance between groups while minimizing variance within groups (for review, see Ref. 24). The data set we are analyzing represents typical respiratory plethysmography data, which includes over 10 features with thousands of measurements. LDA is a supervised linear dimensionality reduction technique that permits us to visualize how distinct the control and mutant groups are. We performed an LDA analysis utilizing frequency, tidal volume, minute ventilation, inspiratory time, expiratory time, EF50, EIP, and EEP as predictors of genotype. This resulted in a data matrix composed of 14 rows and 12,213 columns, with each column representing a 10-s bin of data (3,103 rows representing mutant data points across 6 animals, and 9,110 rows obtained from 19 animals). As shown in Fig. 2A, the mutant and control genotypes demonstrated significant overlap between groups when evaluated using a linear analysis. We then attempted to identify if there were different clusters of breath measurements. To achieve this, we determined across all of the features what the optimal number of data clusters could be generated by performing the silhouette test across multiple different center values for the kmeans cluster algorithm using the same parameters as used for the LDA analysis. The silhouette analysis is used to study the separation distance between resulting clusters, and thereby identifies the optimal number of clusters for kmeans cluster analysis. After pooling all the data from control and mutant animals, we found that the optimal number of clusters for baseline room air respiratory data was 2 (Fig. 2B), and we then plotted the distribution of the two clusters between control and mutant animals (Fig. 2C, note that the control and mutant animals had a similar distribution in the quantity of measurements that were present in cluster 1 or cluster 2 based on a kmeans separation).
Figure 1.
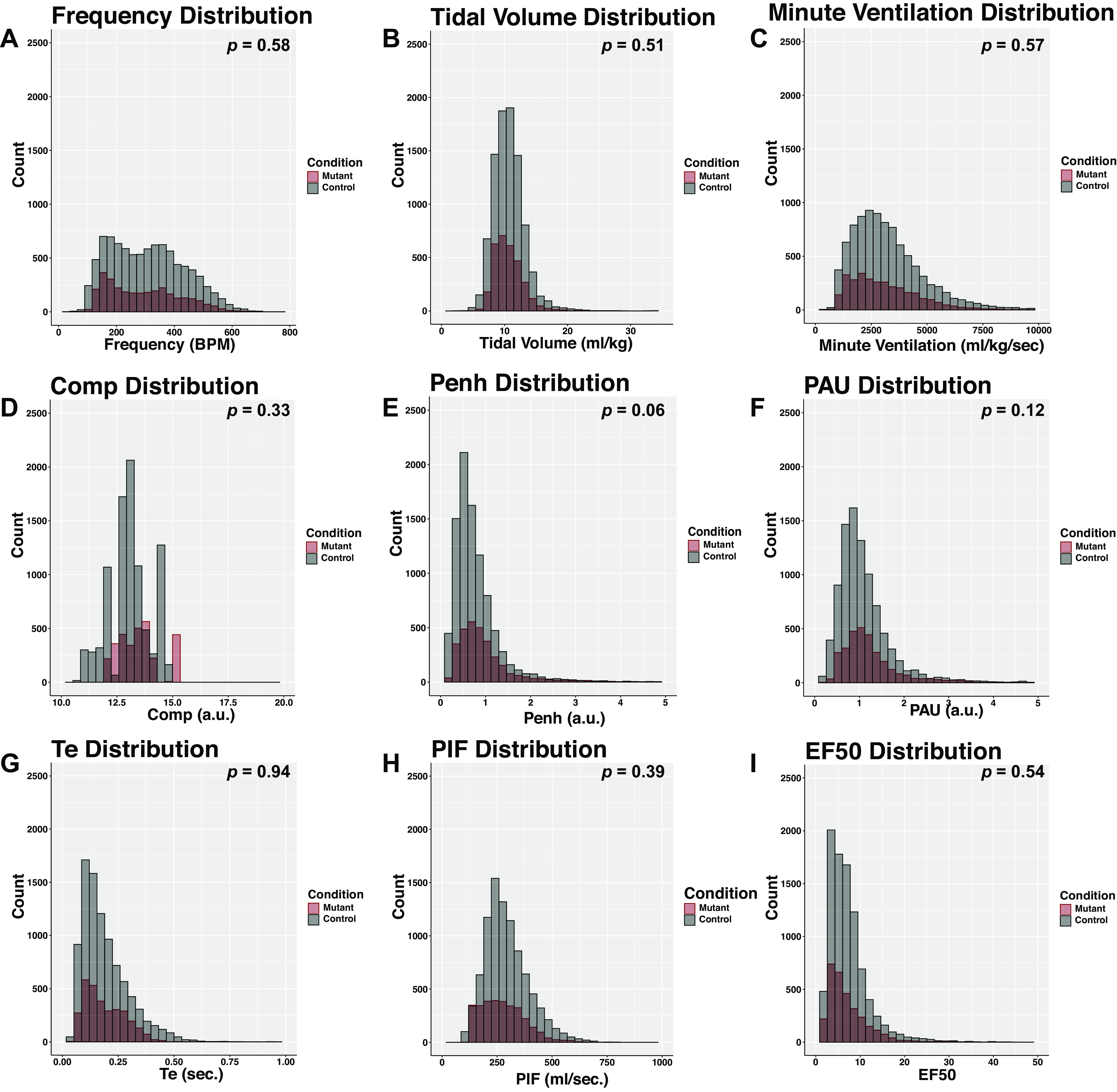
Plethysmography recordings show significant overlap between Phox2bcre, Aldh1l1DTA mice and littermate controls. Data from Phox2bcre, Aldh1l1DTA mice and littermate controls obtained from unrestrained plethysmography recordings of baseline room air are illustrated as density plots. Gray bars represent littermate control mice, and red bars denote Phox2bcre, Aldh1l1DTA mice (labeled as mutant in the figure). Data were recorded as 10-s bins of averaged data from a Buxco plethysmograph system, averaged for each animal, and differences between the two groups was performed by t test. The t test result value is indicated in the top right corner. n = 6 Phox2bcre, Aldh1l1DTA mice, n = 19 negative littermate controls. Each graph shows the value of each parameter, with the y-axis showing the quantity of measurements identified in designated bin. A: f, frequency (breaths per min). B: TVb, tidal volume (mL/kg). C: MVb, minute ventilation (mL/kg/min). D: Comp, compensation factor, unitless number calculated by Buxco and related to ambient humidity. E: Penh, enhanced pause (index of constriction; unitless numbers). F: PAU, pause (index of constriction; unitless numbers). G: Te, expiratory time (s). H: PIFb, peak inspiratory flow (mL/kg/s). I: EF50, peak expiratory flow at 50th percentile (mL/kg/s).
Figure 2.
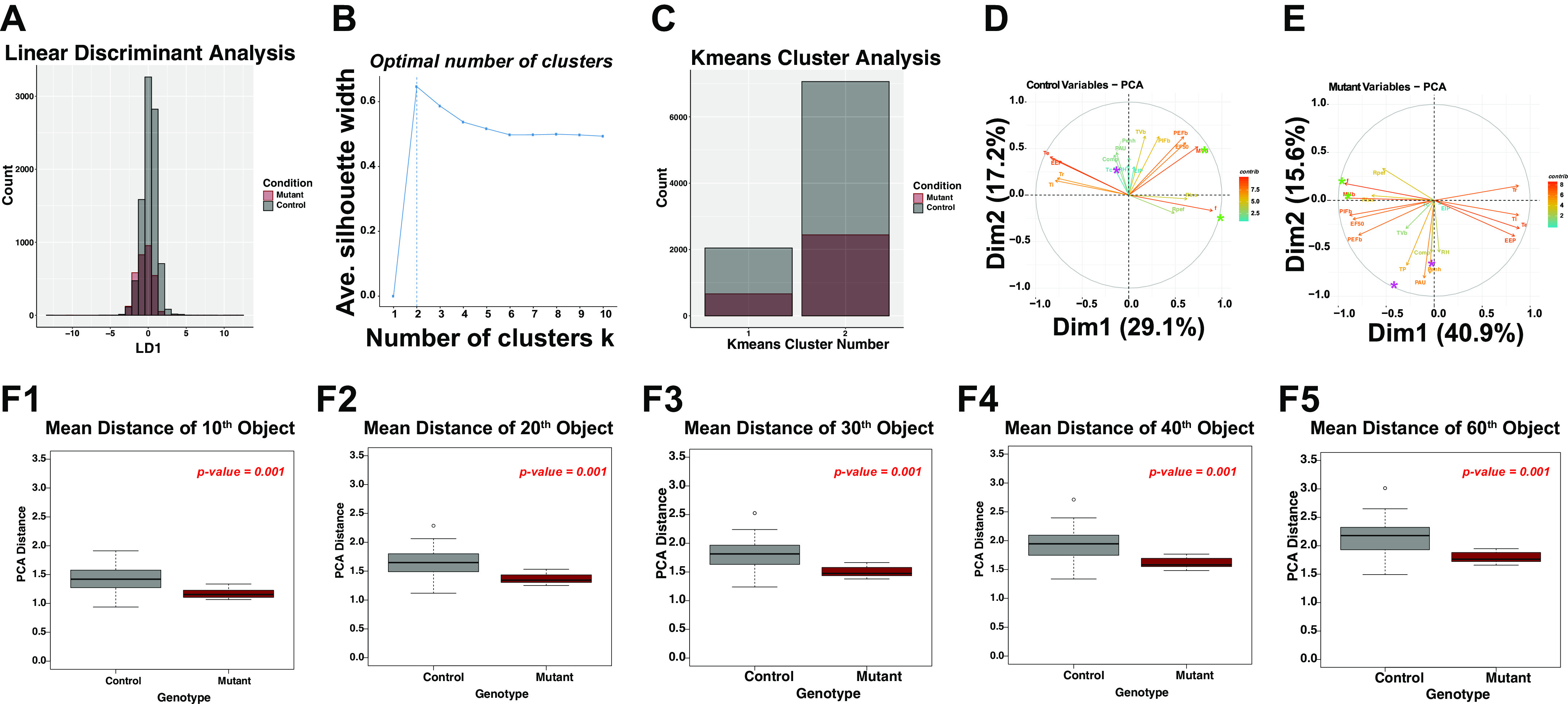
Baseline room air measurements show significant overlap. A: linear discriminant analysis results of all measured data. Red is mutant, and black is control. Note that there is significant overlap between the LDA graphs. LDA has only one dimension, as there are only two class labels (i.e., control and mutant). The y-axis is total count of measurements in the respective x-axis bin. B: optimal k-means analysis. The y-axis is average silhouette width; x-axis is the number of centers k used in the k-means algorithm. Two clusters were determined to be optimal for clustering in this data set. C: density distribution of mutant and control measurements of each cluster. Similar distributions exist in cluster between control and mutants. Bar chart shows kmeans cluster number and total count of measurements in the respective cluster. D: eigenvector plots of principal component analysis. E: eigenvector plots of principal component analysis of mutant data. F shows the PCA Euclidean distance (PCA distance) of each data point/object. The t test probability is labeled in top right, red. Comp, factor applied to box flow to estimate animal’s flow; EEP, end expiratory pause; EF50, expiratory flow at 50% expired volume; EIP, end inspiratory pause; f, respiratory rate; MVb, minute ventilation; PAU, pause (index of constriction); Penh, enhanced pause (index of constriction); PEFb, estimated peak expiratory flow; PIFb, estimated peak inspiratory flow; RH, relative humidity; Rinx, rejection index (percentage of breaths before a breath is accepted); Rpef, the location into expiration where the peak occurs as a fraction of Te; TB, duration of breaking; Tbody, body temperature; Tc, chamber temperature; Te, expiratory time; Ti, inspiratory time; TP, duration of pause before inspiration; Tr, relaxation time; TVb, tidal volume. F: density calculations, Euclidian distance in the PCA for the 10th distant (F1), 20th distant (F2), 30th distant (F3), 40th distant (F4), and 60th distant (F5) data point.
We next evaluated the effect of each measured feature on variance of the data set for control and mutant animals. To achieve this, we performed a principal components analysis (PCA) for the control and mutant animals and plotted the eigenvectors/eigenvalues for each feature on a two-dimensional graph containing principal component 1 on the x-axis and principal component 2 on the y-axis. This analysis yielded some insightful results that raised the possibility that the relationships between features may be distinct between the groups (Fig. 2, D and E). For instance, note that the respiratory compensation factor (Comp), relative humidity (RH), and time of pause at end of expiration (TP) showed more significant contributions to measurement variance in the mutant data set relative to the control data set (purple asterisk). Furthermore, the mutants had stronger correlations between the features respiratory frequency (f) and minute volume (MVb; Fig. 2, D and E, green asterisk, note that in mutant animals the f and MVb eigenvectors are in the same direction and magnitude, whereas in the control animals f and MVb are much more separated). These data indicate that in the mutants variances in MVb are due more to variance in f than TVb relative to the control.
Having demonstrated significant overlap between features in the baseline plethysmographic measurements, we next sought to evaluate data variance in each animal through data density estimations. Specifically, we pooled all data from control and mutant animals and performed a principal component analysis of the pooled data for the following features: f, TVb, MVb, Comp, compensation factor (unitless number calculated by Buxco and related to ambient humidity); EEP, end expiratory pause; EIP, end inspiratory pause; EF50, peak expiratory flow at 50th percentile; MVb, minute ventilation; PAU, pause (unitless index of constriction); Penh, enhanced pause (unitless index of constriction); PEFb, estimated peak expiratory flow; PIFb, peak inspiratory flow; Rpef, the location into expiration where the peak occurs as a fraction of Te; RH, relative humidity; Tc, chamber temperature; Te, expiratory time; Ti, inspiratory time; Tr, relaxation time; TVb, tidal volume. We then obtained a distance map of the data so that the Euclidean distance of each data point to all other data in the animal’s recordings was obtained (25). To obtain a measure of data density within each animal, we obtained the mean Euclidian distance in the PCA (which we term PCA Distance) for the 10th distant, 20th distant, 30th distant, 40th distant, and 60th distant data point (referred to as object in Figure 2, F1–F5). We noted significant differences between control and mutant groups. Specifically, on average, the PCA distance of each data point/object was smaller in the mutant animals relative to the control animals. These data indicate that each plethysmographic data point in each mutant animal is more compact, a finding suggesting lesser variance in respiratory parameters.
Having discovered that the mutant and control animals showed distinct correlations and distinct density of respiratory parameter measurements, we tested the extent to which we could predict class labels using common nonlinear machine learning predictive methodologies. As a control, we generated a randomized data set where the class labels were scrambled. For this scrambled data set, we then took all pooled measurements and randomly separated 70% of the data for training and 30% of the data for testing. These separate training and test data sets were randomly sampled and split from the pooled data. We then trained three algorithms to identify class label. These models included random forest, support vector machines, and a neural net (see methods). The confusion matrix of these results are illustrated in Table 1. Note that the Random Forest algorithm was capable of accurately labeling the test set class with over 98% accuracy. The support vector machines, using linear and radial kernels, were able to detect at ∼ 95% accuracy. Random Forest and Support Vector Machine models trained on scrambled data were unable to show improved accuracy relative to the no information rate. Our neural network model showed the weakest capacity for classification in these experiments but was still capable of beating the no information rate.
Table 1.
Outcomes of class predictions obtained from baseline room air plethysmography data
| Parameter | Random Forest Model | Scrambled Random Forest Model | SVM-Linear Model | Scrambled SVM-Linear Model | SVM-Radial Model | Scrambled SVM-Radial Model | Neural Network | Scrambled Neural Network |
|---|---|---|---|---|---|---|---|---|
| Accuracy | 0.9876 | 0.7384 | 0.9595 | 0.7369 | 0.9503 | 0.7415 | 0.7545 | 0.7396 |
| 95% CI | (0.9833, 0.991) | (0.7234, 0.7529) | (0.9524, 0.9659) | (0.7219, 0.7515) | (0.9425, 0.9573) | (0.7266, 0.7561) | (0.7398, 0.7689) | (0.7245, 0.7544) |
| No information rate | 0.7457 | 0.7415 | 0.7457 | 0.7415 | 0.732 | 0.7415 | 0.7259 | 0.7396 |
| P-value (Acc> NIR) | < 2.2e-16 | 0.6734 | < 2.2e-16 | 0.7399 | < 2.2e-16 | 0.509 | 7.96E-05 | 0.5091 |
| Kappa | 0.967 | −9.00E-04 | 0.8919 | −0.0059 | 0.868 | 0 | 0.1534 | 0 |
| McNemar’s test P value | 0.0007937 | <2e-16 | 0.005287 | <2e-16 | < 2.2e-16 | <2e-16 | < 2.2e-16 | <2e-16 |
| Sensitivity | 0.9625 | 0.0056 | 0.9011 | 0.003 | 0.8438 | 0 | 0.1162 | 0 |
| Specificity | 0.9961 | 0.993 | 0.9795 | 0.992 | 0.9893 | 1 | 0.99557 | 1 |
| Positive predictive value | 0.9883 | 0.238 | 0.9374 | 0.14 | 0.9665 | Undefined | 0.91 | Undefined |
| Negative predictive value | 0.9873 | 0.74 | 0.9667 | 0.74 | 0.9454 | 0.74 | 0.748 | 0.74 |
| Positive class | Mutant | Mutant | Mutant | Mutant | Mutant | Mutant | Mutant | Mutant |
Results from a confusion matrix were generated from the caret package in R and summarized above. Acc, accuracy; CI, confidence interval; NIR, no information rate; SVM, support vector machine.
We next set out to identify which features in the baseline room air experiments were most important in the analysis. We utilized two approaches to this end. First, we identified the random forest model’s Mean Decrease Gini values for each parameter and graphed these data as a pie chart (Fig. 3A; Refs. 16, 26, 27). Note that the feature Comp, RH, and chamber temperature (Tc) showed the highest importance in classification by the random forest model. We next performed more precise feature selection by passing each factor into the Boruta algorithm. In the Boruta workflow, a shadow feature is generated for each feature by scrambling all of the data on that feature (16, 26). It then compares the efficiency of classification from the original feature with unscrambled data and the shadow feature with the scrambled data. By comparing the impact on random forest-mediated classification of the original feature to the shadow feature, the Boruta algorithm learns which of these features should be rejected. We plotted our results of the Boruta analysis in Fig. 3B. We note that all plethysmography features were important in classification, as they all beat their shadow features. We also conclude that the relationships of Comp, RH, and Tc were major drivers in separating class labels between these mutant and control mice. However, all features in the data set were important in classification since each feature defeated its shadow feature in the Boruta Algorithm test. To evaluate the extent to which a random forest would be able to detect individual animal differences without information from genotype, we generated a random forest and tested the capacity to accurately classify the breaths of each animal. The random forest was remarkably efficient at identifying each animal with a peak accuracy of 75% compared with 88% accuracy in identifying genotype (Fig. 3C). These data in Fig. 3C raise the possibility that some of the distinctions identified in the random forest model may be derived from the algorithm identifying which individual animals were in each group rather than identifying treatment differences. To address this problem, we implemented a permutation approach by repeating a classifier 1,000 times using a for loop as follows. First, to computationally facilitate this workflow, we took a pooled data frame from all of our measurements that included f, TVb, MVb, Penh, PAU, Comp, PIFb, PEFb, Ti, Tr, TP, RH, Te, EF50, EIP, and EEP. After we performed a principal components analysis, we found that 90% of the variance was captured in principal components 1 through 6. We then generated a data frame that included PC1 through 6 as well as the distance to each measurements’ 20th nearest neighbor in the PCA space for that animal. To address this problem, we selected one mouse out of the mutant data set (representing ∼ 15% of the total mutant data) and three mice from the control set (representing ∼16% of the total control data) as a test set. We then generated a new training data set that contained the remaining five mutant animals and five randomly selected control animals that were not included in the test set. This specific design was performed so as to prevent a random forest classifier from seeing data from an animal in the training set. This training set had dimensions of 4,800 rows and nine columns. Once the random forest was trained on the five mutant and five control animals, they were tested on the mutant test and control test set. In each permutation, a different set of five control animals were randomly selected for the training set, which we performed so as to prevent a class imbalance training problem. As a control, we included in each permutation scrambled data in each permutation. We repeated this permutation 1,000 times using a for loop in R and collected the accuracies of the predictions. These are plotted in Fig. 3D, where each black-gray circle represents the accuracy of one permutation. We note that the mean accuracy in detecting mutant mice was 65%, but that the distribution was bimodal, with some of the permutations failing to accurately predict that mice were mutant (notice that a cluster of 31% of the permutations showed inaccuracies with only 21% of their measurements being deemed mutant, whereas 69% of the permutations demonstrated very high accuracies showing an arithmetic mean of 84% of their measurements being deemed as derived from a mutant). We conclude that at baseline, respiratory parameters in control and mutant mice show nonlinear differences. We also conclude that in ML implementations of respiratory plethysmography, algorithms utilized for class label predictions must show accuracies at least higher than the algorithm’s capacity to identify measurements must in the minimum be higher over their capacity to detect individual subject differences.
Figure 3.
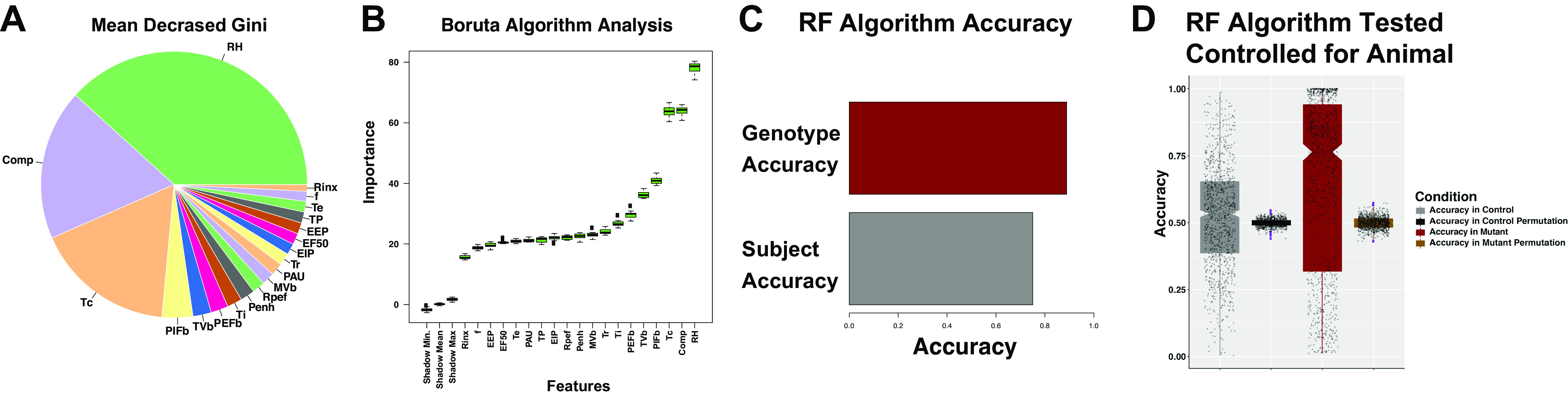
Importance measures from the Random Forest algorithm for genotype classification. The Random Forest algorithm was queried for importance measures. The mean decrease Gini (MDG) for each parameter is graphed in a pie chart in A. The MDG is calculated by the Random Forest package, where the decrease in node impurity is summed and averaged across all trees. In B, the boruta algorithm results are plotted. Green box-whisker plots denote original features, and red denotes the minimum, mean, and maximum shadow feature results (denoted as Shadow Min., Shadow Mean, Shadow Max.). The y-axis is the ranger normalized permutation importance. Note that all features beat the shadow features, indicating that each feature was important for classification. Using two techniques, RH, Comp, and Tc showed the most importance. C: accuracy of a 500 tree random forest in detecting class label (genotype = control vs. mutant in red, subject = animal number in gray). Note that the genotype algorithm shows more accuracy than class label problems to detect animal. D: accuracies of the predictions, each black-gray circle represents the accuracy of one permutation. Comp, factor applied to box flow to estimate animal’s flow; EEP, end expiratory pause; EF50, expiratory flow at 50% expired volume; EIP, end inspiratory pause; f, respiratory rate; MVb, minute ventilation; PAU, pause (index of constriction); Penh, enhanced pause (index of constriction); PEFb, estimated peak expiratory flow; PIFb, estimated peak inspiratory flow; RH, relative humidity; Rinx, rejection index (percentage of breaths before a breath is accepted); Rpef, the location into expiration where the peak occurs as a fraction of Te; TB, duration of breaking; Tbody, body temperature; Tc, chamber temperature; Te, expiratory time; Ti, inspiratory time; TP, duration of pause before inspiration; Tr, relaxation time; TVb, tidal volume. Hyperparameters were tuned using caret package.
Identification of Group Differences in Electroencephalographic Studies with Machine Learning Methodologies
Broadly speaking, utilization of machine learning algorithms in EEG interpretation has focused on correlating specific EEG findings to transcriptional data sets (28), or in utilization of algorithms to identify specific EEG features in an automated way (29). To our knowledge, no approaches attempting to use ML/AI approaches to identify EEG patterns in genotypes exist in the scientific literature. We therefore re-evaluated the EEG data set previously published by our group where Phox2b-derived astrocytes were experimentally ablated. In these studies, we found subtle differences in sleep patterns between male and female mutant mice, as well as an overall fractured sleep pattern (9). We also were interested in determining the extent to which EEG patterns could distinguish genotype. However, in our prior study, we only evaluated a subset of frequency bands (0.5–25 Hz) between animals that were focused on vigilance state data and discarded all remaining data (in other words, all data from recordings obtained over >25 Hz were ignored in the prior analysis). We first pooled the data in the mutant and control mice, performed a central scaling, and evaluated the variance by PCA (Fig. 4, A and B). For this PCA, all of the frequency steps were included in the analysis, as well as the features Sleep, Time, and Sex. Note that plotting the eigenvectors/eigenvalues demonstrates two distinct patterns in mutant versus control mice. First, although the Sleep feature contributed significantly to the variance in both groups, note that Sex shows an opposite covariance to Sleep in the control versus the mutant mice, indicating that in these groups the relationships between Sleep and Sex are distinct between control and mutant mice, a finding consistent with our previous analyses (9). We also note that different frequencies show distinct hierarchical clustering patterns between control and mutant mice (Fig. 4, C and D). We conclude that evaluating all of the data (rather than a subset of frequency components) reveals distinct trends between groups in an unbiased fashion.
Figure 4.
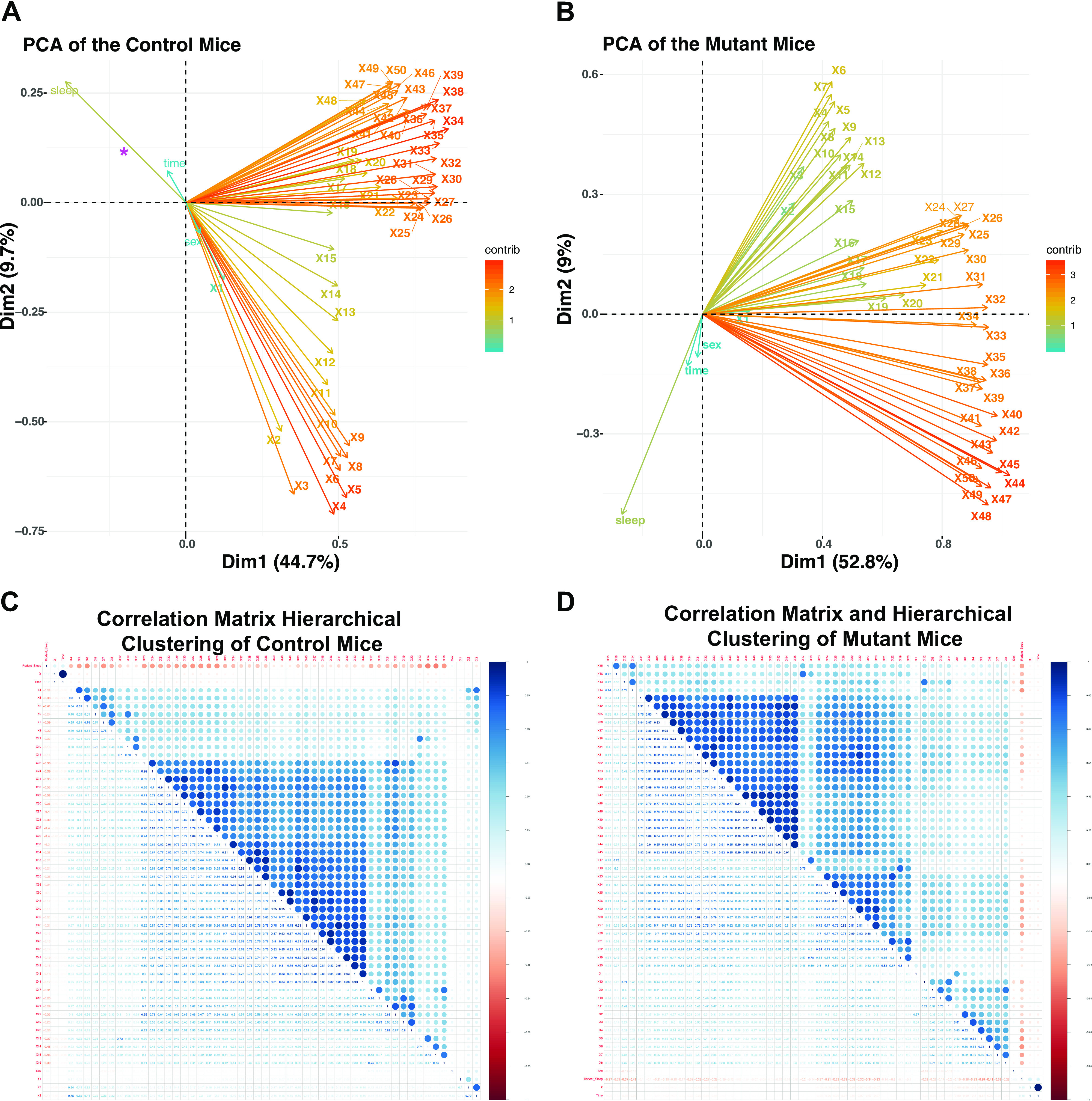
EEG analysis by machine learning workflows in Phox2bcre, Aldh1l1DTA mice. Raw EEG data from all 50 frequency steps, as well as data from sleep, sex, and time underwent principal component analysis (PCA), and the eigenvectors/eigenvalues are plotted for PC1 and PC2 for the control (A) and mutant (B) animals. Correlation matrices for the control (C) and the mutant mice (D). Pierson’s correlation coefficient is color coded with blue showing positive (+1) and red showing negative (−1) correlation. Note that the correlation matrices in the control and mutant mice are ordered in different ways based on the result of a hierarchical clustering assay. In the control, three main clusters appear, whereas in the mutant four discernable groups can be identified by hierarchical clustering.
We next set out to determine if we could develop a machine learning prediction scheme capable of determining if a recording came from a mutant or control animal. Each recording represents a 10-s bin of information, containing 50 frequency steps, sex, and sleep status. We randomly separated the data into a training set (70%) and test set (30%). We utilized three prediction algorithms: adaboost [a gradient-boosted machine model, (30)], random forest, and multilayer perceptron (a neural network model). We noted that training these models required significantly more computation time for both the adaboost and random forest models relative to the neural network model, yet both adaboost and random forest outperformed the neural network (Table 2). Although the accuracy was relatively low at ∼71–73% accuracy for the tree-based prediction models, the probability that this prediction would be this accurate by chance was extremely low (P < 10−16). Due to the method in which we separated the train and test data, concerns arose regarding the extent to which the model was merely detecting animals relative to detecting genotype. To address this, we utilized similar approaches to the plethysmography data where we performed repetitive loops that left out animal data from the experiment, and we tested the extent to which the EEG patters could perform this prediction. To our surprise, we found the accuracy to be incredibly low and below the accuracy that could be identified by chance. Specifically, the mean accuracy of predicting that a recording came from a mutant was 37% (SD = 14.4), whereas the mean accuracy of predicting that recording came from a control was 43.9% (SD = 14.5) after 366 iterations of the loop. We conclude that evaluation of the raw EEG recordings is capable of identifying features that are important in predicting the animals but incapable separating animals into mutant and control groups.
Table 2.
Predictions obtained from baseline EEG data
| Parameter | Adaboost | Random Forest | Neural Network |
|---|---|---|---|
| Accuracy | 0.7323 | 0.7182 | 0.6123 |
| 95% CI | 0.7292, 0.7353 | 0.7151, 0.7213 | 0.6089, 0.6156 |
| No information rate | o.5005 | 0.5007 | 0.5013 |
| P value (Acc> NIR) | < 2.2 X 10−16 | < 2.2 X 10−16 | |
| Kappa | 0.4645 | 0.4365 | 0.2254 |
| McNemar’s test P value | 5.16 × 10−8 | < 2.2 × 10−16 | < 2.2 × 10−16 |
| Sensitivity | 0.7423 | 0.7425 | 0.39 |
| Specificity | 0.7222 | 0.6939 | 0.83 |
| Positive predictive value | 0.7281 | 0.7076 | 0.7017 |
| Negative predictive value | 0.7366 | 0.7299 | 0.5772 |
| Positive class | Mutant | Mutant | Control |
Results from a confusion matrix were generated from the caret package in R and summarized above. Acc, accuracy; CI, confidence interval; NIR, no information rate.
A finding that we identified in our prior study was the tendency for a fractured sleep pattern. This finding raised the possibility that EEG findings may be distinct within the mutant relative to the control animals. Figure 5A demonstrates the potential transitions that could be detected in our EEG/EMG recordings. To test this hypothesis, we pooled all of the data together and performed a principal component analysis. We found that PC1 through PC15 successfully reduced the dimensions to 90% of the variance in the data set. We then determined for each 10-s bin which transition delineated in Fig. 5A would occur in the next 10-s recording period. Our analysis was focused on hours 1–12 of the recording period, corresponding to day time. Since the self-transitions from Wake to Wake (A), REM to REM (C), and NREM to NREM (F) were most common, modeling these transitions would pose a significant class imbalance problem for a random forest. We therefore trained a random forest algorithm to identify the non–self-transitions (transitions B, D, H, G, E in Fig. 5A colored in blue font) as a function of Sleep State (Wake, NREM, or REM), PC1 through PC15 from the EEG data, genotype, and sex. We then found that the model showed an accuracy of over 84% (no information rate of 27%, P < 10−16, Table 3 shows the confusion matrix). Figure 5B shows the mean decrease Gini importance in the trained RF model. Note that genotype and sex showed very little importance for the random forest classification model. Rather, current vigilance state and PC3 and PC15 showed the most important impact in classification accuracy. We conclude that males and females, as well as controls and mutants, do not have a significantly distinct relationship between EEG pattern and vigilance state transition. We then sought to utilize markov chain modeling of phase transitions to estimate across all mutant and control animals the average number of transitions that could be detected in control and mutant animals. These data are shown in Fig. 5C (control) and Fig. 5D (mutant). We found that calculating Markov Chain models could tell us the relative time that the animal would spend in wake, NREM, or REM per day cycle, which was then multiplied times the proportion of the respective transitions. We conclude that performing a Markov chain model is capable of easily quantifying the average vigilance state transitions in each genotype.
Figure 5.
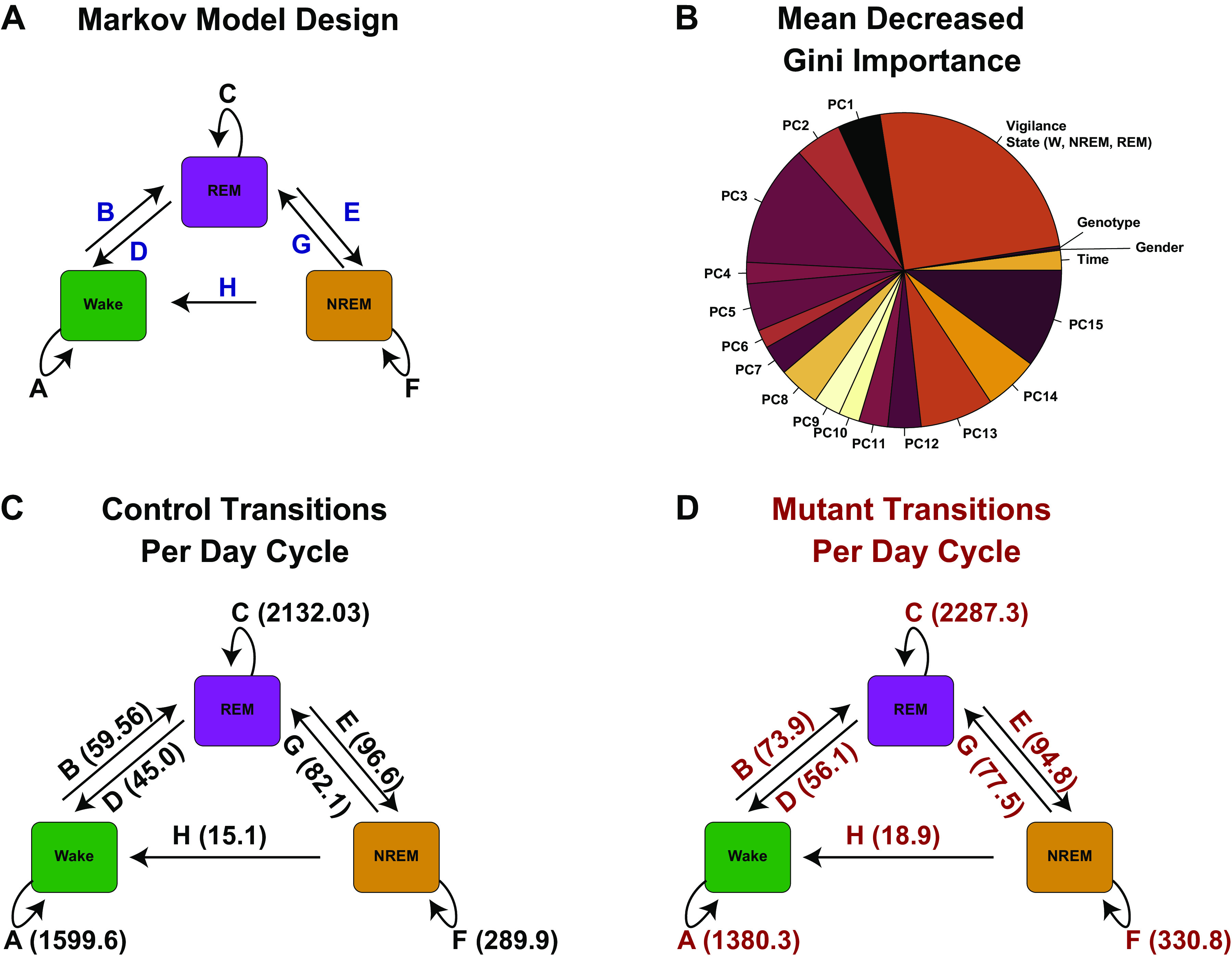
Vigilance state transition analysis. A: model of transition state analysis. For purposes of modeling, we assumed the Markov property in vigilance state transition. B: pie chart of mean decreased Gini importance of random forest prediction model where class label was non–self-transition (blue transitions in A) and predictor variables were current vigilance state, gender, genotype, and principal components 1–15 of the EEG data taken during hours 1–12. C: average transition events determined from the Markov model for control (C) and mutant (D). NREM, non-rapid eye movement sleep; REM, rapid eye movement sleep; W, wake.
Table 3.
Confusion matrix of random forest prediction algorithm for predicting next step vigilance state in time 1–12 hours
| B | D | E | G | H | |
|---|---|---|---|---|---|
| B | 826 | 0 | 0 | 12 | 0 |
| D | 0 | 505 | 88 | 0 | 0 |
| E | 0 | 153 | 815 | 0 | 0 |
| G | 0 | 0 | 0 | 701 | 168 |
| H | 0 | 0 | 0 | 4 | 3 |
Self-transitions were excluded, as these resulted in significant class imbalance predictions as they far outnumbered the vigilance state transitions.
Ablation of Phox2b-Derived Astrocytes Results in Activation of Apoptosis-Related Pathways
Prior ultrastructural studies demonstrated findings consistent with neuro-axonal dystrophic morphologies of axon terminals projecting into the VLM of mice harboring ablation of Phox2b-derived astrocytes (9). Therefore, we decided to interrogate pathway activation in these regions and propose a usable workflow from which to interpret the gene expression data. To achieve this, we performed microdissection of the VLM and dorsal medulla following paraformaldehyde fixation and extracted RNA from these regions for Nanostring analysis. However, several statistical issues are raised in the interpretation of Nanostring data. First, the available “off the shelf codesets” (such as the neurodegenerative codeset package utilized in this study) represent genes that show some relationship to each other. This raises issues regarding how best to correct for multiple comparisons. Indeed, manufacturer guidelines suggest that P value adjustments may not even be required for interpretation of Nanostring codeset data. Second, we note that the Nanostring Norm package in R offers 89 distinct normalization methodologies, and no consensus in the scientific community exists regarding which normalization methodology is best. We therefore performed a for-loop to utilize all normalization methodologies in R and plotted relative log expression (RLE) plots of each analysis. RLE plots represent a quick and easy way in which to evaluate data variation in high dimensionality data sets such as microarray and Nanostring data sets and permits evaluating normalization procedure efficiency (31). We identified the four best RLE plots by visual inspection to identify where the median of the RLE plot was closest to value = 0 (red line in Fig. 6, left). We then plotted volcano plots of differential gene expression in the ventral lateral medulla or dorsal medulla of control and mutant animals (Fig. 6, second and third columns). Such interactive analyses with data take advantages of visual capabilities and is thoroughly used in data analytics applications (for review, see Refs. 32, 33). Depending on the normalization methodologies utilized, we obtained slight differences in the absolute values of the log2-fold activation and P value of the comparisons. We conclude that when evaluating Nanostring data, evaluation of differentially expressed genes should be performed after reviewing data from a spectrum of normalization methods that meet the RLE plot criteria described above. Table 4 tabulates the differentially expressed genes in the RTN and NTS obtained from this analysis.
Figure 6.

Evaluation of normalization efficiency in Nanostring platform. Relative log expression (RLE) plots were performed in all 89 possible normalization methodologies available in the NanostringNorm R package. Based on the proximity of the RLE =0 line to the median of the RLE for each condition, we chose four normalization methods, all of which gave slightly different results (A1, B1, C1, and D1). In each graph, the normalization method implemented is shown at head of each graph. Volcano plots showing the differential gene expression for the ventral lateral medulla microdissection (mutant/control) are shown in A2, B2, C2, and D2, with similar data analysis for the dorsal medulla microdissections graphed in A3, B3, C3, and D3.
Table 4.
Differentially expressed genes
| Gene_Name | RTN_Control_Mean | RTN_Mutant_Mean | RTN_P value | NTS_Control_Mean | NTS_Mutant_Mean | NTS_P value | RTN_log2 |
|---|---|---|---|---|---|---|---|
| Lrrc4 | 199.79 | 181.57 | 0.52 | 197.35 | 164.83 | 0 | −0.14 |
| Stambpl1 | 160.94 | 153.2 | 0.52 | 233.8 | 189.29 | 0.01 | −0.07 |
| Comt | 481.1 | 499.68 | 0.6 | 412.01 | 442.63 | 0.02 | 0.05 |
| Drd4 | 19.5 | 23.97 | 0.62 | 15.65 | 6.32 | 0.02 | 0.3 |
| Lamp1 | 3,147.08 | 2,761.9 | 0.06 | 2,399.43 | 2,044.2 | 0.02 | −0.19 |
| Atrn | 675.7 | 663.94 | 0.85 | 620.49 | 572.81 | 0.03 | −0.03 |
| Efna1 | 55.6 | 46.6 | 0.31 | 27.65 | 22.52 | 0.03 | −0.25 |
| Casp3 | 60.76 | 47.02 | 0.11 | 56 | 68.34 | 0.03 | −0.37 |
| Npc1 | 1,614.06 | 1,731.31 | 0.52 | 848.85 | 945.18 | 0.04 | 0.1 |
| Snrpa | 200.26 | 227.33 | 0.24 | 227.39 | 183.25 | 0.04 | 0.18 |
| Rhoa | 1,037.04 | 986.98 | 0.29 | 1,038.29 | 839.35 | 0.04 | -0.07 |
| F2 | 5.82 | 9.02 | 0.48 | 7.4 | 0.35 | 0.04 | 0.63 |
| Aldh1l1 | 477.29 | 510.28 | 0.56 | 472.72 | 524.13 | 0.05 | 0.1 |
| Erlec1 | 292.87 | 317.9 | 0.27 | 358.05 | 321.52 | 0.05 | 0.12 |
| Camk2d | 613.97 | 598.21 | 0.8 | 1,062.78 | 910.89 | 0.05 | −0.04 |
| Ran | 790.37 | 716.48 | 0.22 | 1,150.13 | 962.48 | 0.05 | −0.14 |
| Mmp2 | 30.59 | 35.35 | 0.47 | 26.66 | 20.54 | 0.06 | 0.21 |
| Gata2 | 112.4 | 151.41 | 0.02 | 41.03 | 46.78 | 0.06 | 0.43 |
| Osmr | 70.76 | 74.65 | 0.84 | 29.08 | 36.17 | 0.06 | 0.08 |
| Hpgds | 66.92 | 68.5 | 0.92 | 47 | 55.47 | 0.06 | 0.03 |
NTS, nucleus of tractus solitarius; RTN, retrotrapezoid nucleus.
The aforementioned analysis of differentially expressed genes does not include potential interactors between genotype and neuroanatomical location. To address this further, we next performed linear discriminant analyses (LDA) and linear regression (LR) on all of the genes. These analyses are graphed in Fig. 7 for data that underwent normalization using “geo.mean” as the code count, “mean” as the background correction, and a “housekeeping.geo.mean” as the sample content correction. We focused on six genes that were commonly regulated by mutant as factor or mutant-factor interacting with the dorsal brainstem as the factor. For the LDA, we used gene expression from the Nanostring assay to predict class. Class was a factor composed of the following levels: NTS-Control (n = 3), NTS-Mutant (n = 3), VLM-Control (n = 3), and VLM-Mutant (n = 3). The advantage of the LDA in this context is that it provided a different method of evaluating the statistical significance of the gene expression relative to the volcano plot assay. Specifically, the LDA provided coefficients, which are plotted as histograms in Fig. 6, A1, B1, C1, D2, and F2, where the x-axis represents the value of the linear discriminant (LD1). LD thus provides a separate methodology to evaluate statistical significance relative to evaluation of differentially expressed genes as performed above. To evaluate interactions between genotype and neuroanatomical location, we generated LR models for each gene where genotype and neuroanatomical location predicted gene count. In this model, we generated an interaction between genotype and neuroanatomical location. Thus, for each gene its count was calculated by four parameters: an intercept, genotype as a dummy variable (0 = control, 1 = mutant), neuroanatomical location as a dummy variable (0 = RTN, 1 = Dorsal Medulla, or DM), and the interaction of genotype and neuroanatomical location. These coefficients are shown as bar graphs in Fig. 7, A2, B2, C2, D2, E2, and F2. After having identified these genes, we then evaluated the wikipathways to interrogate the pathways most commonly associate with such differentially expressed genes. These wikipathways were then condense into a word cloud which was presented as the graphical abstract. The main pathways that were identified in the differentially expressed genes were implicated in apoptosis. We conclude that ablation of Phox2b-derived astrocytes results in subtle changes in gene transcription that modulate apoptosis, a finding concordant with our prior ultrastructural data.
Figure 7.

Linear discriminant analysis (LDA) and linear regression analysis of the gene codesets. Linear discriminant analysis and linear regression analysis were performed on every gene evaluated in the Nanostring neurodegeneration platform. We chose to illustrate six genes that showed interesting separation on the linear discriminant analysis and/or linear regression. Evaluation of all of the genes are present as supplemental data. A1, B1, C1, D1, E1, and F1 represent histograms of the distribution of the LDA scores for each group. The bar graphs (A2, B2, C2, D2, E2, and F2) show for each linear regression the intercept (orange), mutant class coefficient (green), mutant class-dorsal medulla interaction (turquois), and the dorsal medulla (purple). The P value for each coefficient is shown in red on top of each bar graph.
DISCUSSION
In this study, we tested the hypothesis that animals showing Phox2b-derived astrocyte ablation had different characteristics at baseline compared with littermate control mice. To test this hypothesis, we performed a reanalysis of a previously published data set whose baseline physiological data had been analyzed using traditional techniques. These traditional techniques had only identified findings following physiological challenges/maneuvers, but the extent to which these animals show differences at baseline physiological conditions had remained elusive. Our results indicated that even at baseline, physiological differences could be identified between mutant and littermate control mice using machine learning/artificial intelligence (ML/AI) approaches. Additionally, data coming from mutant and control mice could be classified as belonging to each group in an unbiased way with ∼95% (plethysmography) and ∼72% (EEG) accuracy using ML/AI workflows. These differences inspired us to attempt to identify transcriptional pathways that were distinct in the mutants and the controls. Through microdissection of the dorsal brainstem structures, we were able to discover dysregulation of apoptotic pathway genes.
ML/AI Models Interpreted in a Physiological Context
Given the sensitivity of ML/AI methods, a significant question that remains in these types of studies is the extent to which statistically significant changes detected in ML/AI represent changes that show biological significance. Neurophysiologists often debate the extent to which such changes detected in experimental paradigms are relevant to physiological outcomes. In the case of mice with Phox2b-ablated astrocytes, we find that baseline breathing shows significances, in particular the data distribution is more compact in mutant mice, and these mice show larger drivers of variance in specific parameters, which are distinct from the control mice. The whole body plethysmography (WBP) and barometric method offers a unique opportunity to record the breathing pattern in conscious animals. The precision of this technique relies on the accuracy of the humidity, body and ambient temperatures, and barometric pressure measurements for the tidal volume computation from the pressure oscillations (34). The system used to record the ventilatory parameters presented has chambers fitted with pneumotachs, which allow air to pass in and out of the chamber. The air resistance created by the pneumotach screens cause small pressure changes in the chamber relative to the ambient air (35). A sensitive pressure transducer measures these pressure oscillations inside the chamber that are synchronous with breathing and from which flow can be derived. The airways usually have humidity and temperature higher than the air inside them, therefore the mouse humidifies and heats the inhaled air during inspiration causing air expansion, and contraction when mouse exhales the cooled air (35).
Humidity is one of the key variables for the correct measurement and conversion of pressure oscillations into tidal volume (36). Our data show using PCA for the control and mutant animals that relative humidity (measured using a digital humidity sensor in the chamber) showed more significant contributions to measurement variance in the mutant data set relative to the control data set. The same was observed with Comp and TP. In the whole body plethysmography used, Comp is calculated by Drorbaugh and Fenn or Epstein equations (37, 38). For the data analyzed, default setting was selected to apply Drorbaugh and Fenn and Comp was applied to the flow and volume data to approximate the actual flows and volumes from the chamber to more physiological values from the mouse (35). The tidal volume estimated from the flow signal is equal to the product of the volume measured and Comp. The plethysmography system also uses Comp to estimate de MVb (product of the volume measured, f, and Comp).
The TP represents the percentage of the breath occupied by transition from expiration to inspiration and changes of TP is an indicator of pulmonary irritation (35).
The analysis of the breathing pattern is important and necessary for many clinical or research purposes. The measurement of the quantity of air passing into the lungs over a period of time, represented by the MVb, is the product of the air inhaled with each breath (TVb) and of the number of breaths per minute (f). The principal components analysis performed also shows that mutants had stronger correlations between the features f and MVb when compared with controls, indicating that mutant variances in MVb are due more to variance in f than TVb. The present data confirm the utility of ML/AI-based workflows for the analysis plethysmography data generating novel hypotheses for investigators in an efficient manner.
These data led us to then test the hypothesis that distinct gene activation may have arisen in mice in medulla regions implicated in respiratory control. For these reasons, we interrogated pathway activation in the VLM and dorsal medulla regions. However, these workflows represent novel methodologies for neurophysiologists, and so we propose a usable workflow from which to interpret the gene expression data obtained by Nanostring. Previous data showed that ablation of Phox2b-derived astrocytes results in axon terminals in the mutants average larger in area and perimeter. Furthermore, absence of Phox2b-derived astrocytes also increased the percentage of axon terminals showing autophagic vacuoles/phagosomes (9). Here we identified using linear regression and linear discriminant analyses, six genes that were commonly regulated by mutant as factor, or mutant-factor interacting with the dorsal brainstem as the factor. Interrogating the pathways most commonly associate with such differentially expressed genes, we noticed a relation with apoptotic pathways, a finding concordant with our prior ultrastructural data.
As the VLM and dorsal medulla of mutant mice used for Nanostring analysis lack Phox2b-derived astrocytes, it is important to emphasize that these cells exerts structural, metabolic, and functional effects on neurons, reinforcing that interactions between astrocytes and neurons play crucial roles in central nervous system homeostasis (39, 40).
Evidences shows that astrocytes provide powerfully control of the neuronal processes involved with circuit formation, supporting evidences that these cells are integral to neural circuit function and that their impairment and axon degeneration have been found to contribute to neuronal dysfunction in several neurodegenerative conditions (41–44). Thus, we conclude that ablation of Phox2b-derived astrocytes results in changes in gene transcription that modulate neurodegeneration and apoptosis, a finding concordant with our prior ultrastructural data.
Data Analysis by ML/AI Approaches
The rise of machine learning and artificial intelligence techniques has enabled us to evaluate high dimensionality data sets for physiology research using open source software platforms [such as Rstudio (https://rstudio.com/) and Python (https://www.python.org/)]. ML/AI are highly powerful tools, but their implementation is not without challenges and requires special considerations, both technical and ethical, in biomedical research. Although the neuroscience and neurophysiology community has embraced machine learning approaches for the interrogation of genomics studies, application of these computational techniques to other areas of physiology research such as respiratory plethysmography measurements or interpretation of EEG data has been more slowly adopted. In our approach, we begin first by interacting with the data by attempting linear dimensionality reduction techniques such as LDA (supervised) and PCA (unsupervised). These initial studies give us a sense of how the features interact in the groups to drive distance between groups or variance between observations within the groups. We next determine if the data contains sufficient features to render any capacity of accurate group classification as either mutant or a control mouse. If we were successful in classification, model interrogation techniques (such as mean decreased Gini values and Boruta algorithm for random forest) inform us as to which features are most important for classification. In our data sets, we obtained good prediction techniques using random forest, a technique that shows benefits of model interpretation relative to neural networks. We note that a significant problem with ML/AI techniques is that not all are amenable to facile interpretation. Lack of model interpretability represents a significant concern for “black box” algorithms such as neural networks and support vector machines. We advocate for choosing ML/AI algorithms that balance high accuracy, computational efficiency, and model interpretability. We also note that this sentiment has been echoed by other neuroscientists (1, 45).
In addition to the technical considerations for choosing ML/AI algorithms, we strongly advocate for the inclusion of proper research ethics standards. Specifically, the ethical standards delineated by the National Institutes of Health policy statement “Principles and Guidelines for Reporting Preclinical Research” (https://www.nih.gov/research-training/rigor-reproducibility/principles-guidelines-reporting-preclinical-research) clearly states that preclinical research requires “Rigorous Statistical Analyses” and “Transparency in Reporting.” Both of these tenets of ethical preclinical research require significant thought in the ML/AI era since the statistical testing is essentially outsourced to computational platforms. For these reasons, we advocate inclusion of all code utilized to generate the data as supplementary materials and also to openly include alternate analyses where the community has not reached consensus on an appropriate “Rigorous Statistical Analysis.” This is highly relevant to Nanostring data analysis. In this manuscript, we illustrate a workflow that we believe is required for proper interrogation of Nanostring data sets. We note that this technology requires small quantities of input RNA, making it amenable to perform Nanostring gene expression assays on tissues following fixation. However, data normalization significantly alters the output of the Nanostring analysis. To be compliant with “Transparency of Reporting,” we included what we felt were the most optimally normalized Nanostring analyses based off of relative log expression (RLE) graphs. From observations of the RLE plots, we then chose to evaluate by LDA and linear regression genes that were commonly differentially regulated between control and mutant mice. These analyses yielded differential gene expression of genes involved in apoptosis, a finding supporting our prior observations of dystrophic axon terminals in the ventral lateral medulla of Phox2b-astrocyte ablated mice.
GRANTS
This work was supported by NIH/National Heart, Lung, and Blood Institute (R01HL132355 for C. M. Czeisler and J. J. Otero) and NIH/National Cancer Institute P30 CA016058. This research was also supported by public funding from São Paulo Research Foundation (FAPESP Grants: 2019/01236-4 to A. C. Takakura and 2015/23376-1 to T. S. Moreira). T. M. Silva acknowledges FAPESP 2017/12678‐2 and 2018/03994-0 funding from São Paulo Research Foundation. This study was also supported by Serrapilheira Institute (Grant: Serra-1812–26431).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.M.S., C.M.C., and J.J.O. conceived and designed research; T.M.S., C.M.C., J.C.B., M.J.A., D.A.C., P.F., and A.E.T. performed experiments; T.M.S., C.M.C., J.J.O., J.C.B., M.J.A., D.A.C., J.Z., P.F., A.E.T., A.C.T., and T.S.M. analyzed data; T.M.S., C.M.C., J.J.O., J.C.B., M.J.A., D.A.C., J.Z., P.F., A.E.T., A.C.T., and T.S.M. interpreted results of experiments; T.M.S., C.M.C., J.J.O., and M.J.A. prepared figures; T.M.S., C.M.C., and J.J.O. drafted manuscript; T.M.S., C.M.C., J.J.O., J.C.B., M.J.A., D.A.C., J.Z., P.F., A.E.T., A.C.T., and T.S.M. edited and revised manuscript; T.M.S., C.M.C., J.J.O., J.C.B., M.J.A., D.A.C., J.Z., P.F., A.E.T., A.C.T., and T.S.M. approved final version of manuscript.
REFERENCES
- 1.Vu M-AT, Adali T, Ba D, Buzsaki G, Carlson D, Heller K, Liston C, Rudin C, Sohal VS, Widge AS, Mayberg HS, Sapiro G, Dzirasa K. A shared vision for machine learning in neuroscience. J Neurosci 38: 1601–1607, 2018. doi: 10.1523/JNEUROSCI.0508-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crone SA, Viemari J-C, Droho S, Mrejeru A, Ramirez J-M, Sharma K. Irregular breathing in mice following genetic ablation of V2a neurons. J Neurosci 32: 7895–7906, 2012. doi: 10.1523/JNEUROSCI.0445-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ray RS, Corcoran AE, Brust RD, Kim JC, Richerson GB, Nattie E, Dymecki SM. Impaired respiratory and body temperature control upon acute serotonergic neuron inhibition. Science 333: 637–642, 2011. doi: 10.1126/science.1205295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ray RS, Corcoran AE, Brust RD, Soriano LP, Nattie EE, Dymecki SM. Egr2-neurons control the adult respiratory response to hypercapnia. Brain Res 1511: 115–125, 2013. doi: 10.1016/j.brainres.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun JJ, Huang T-W, Neul JL, Ray RS. Embryonic hindbrain patterning genes delineate distinct cardio-respiratory and metabolic homeostatic populations in the adult. Sci Rep 7: 9117, 2017. doi: 10.1038/s41598-017-08810-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borniger JC, Don RF, Zhang N, Boyd RT, Nelson RJ. Enduring effects of perinatal nicotine exposure on murine sleep in adulthood. Am J Physiol Regul Integr Comp Physiol 313: R280–R289, 2017. doi: 10.1152/ajpregu.00156.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glaser JI, Kording KP. The development and analysis of integrated neuroscience data. Front Comput Neurosci 10: 11, 2016. doi: 10.3389/fncom.2016.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warren S. Simultaneous, multiplexed detection of RNA and protein on the nanostring ®nCounter® platform. Methods Mol Biol 1783: 105–120, 2018. doi: 10.1007/978-1-4939-7834-2_5. [DOI] [PubMed] [Google Scholar]
- 9.Czeisler CM, Silva TM, Fair SR, Liu J, Tupal S, Kaya B, Cowgill A, Mahajan S, Silva PE, Wang Y, Blissett AR, Goksel M, Borniger JC, Zhang N, Fernandes-Junior SA, Catacutan F, Alves MJ, Nelson RJ, Sundaresean V, Rekling J, Takakura AC, Moreira TS, Otero JJ. The role of PHOX2B-derived astrocytes in chemosensory control of breathing and sleep homeostasis. J Physiol 597: 2225–2251, 2019. doi: 10.1113/JP277082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsai H-H, Li H, Fuentealba LC, Molofsky AV, Taveira-Marques R, Zhuang H, Tenney A, Murnen AT, Fancy SPJ, Merkle F, Kessaris N, Alvarez-Buylla A, Richardson WD, Rowitch DH. Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science 337: 358–362, 2012. doi: 10.1126/science.1222381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scott MM, Williams KW, Rossi J, Lee CE, Elmquist JK. Leptin receptor expression in hindbrain Glp-1 neurons regulates food intake and energy balance in mice. J Clin Invest 121: 2413–2421, 2011. doi: 10.1172/JCI43703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paxinos G, Franklin KBJ. The Mouse Brain in Sterotaxic Coordinates (2nd ed.). San Diego, CA: Academic, 2001. [Google Scholar]
- 13.Venables WN, Ripley BD. Modern Applied Statistics with S (4th ed.). New York: Springer, 2002. [Google Scholar]
- 14.Wang W, Alzate-Correa D, Alves MJ, Jones M, Garcia AJ, Zhao J, Czeisler CM, Otero JJ. Machine learning-based data analytic approaches for evaluating post-natal mouse respiratory physiological evolution. Respir Physiol Neurobiol 283: 103558, 2021. doi: 10.1016/j.resp.2020.103558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kassambara A, Mundt F. factoextra: Extract and visualize the results of multivariate data analyses. https://rpkgs.datanovia.com/factoextra/index.html.
- 16.Kursa M, Rudnicki W. Feature selection with the Boruta package. J Stat Softw 36: 1–13, 2010. doi: 10.18637/jss.v036.i11. [DOI] [Google Scholar]
- 17.Hackenberger BK. Tensors all around us. Croat Med J 60: 369–374, 2019. doi: 10.3325/cmj.2019.60.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liaw A, Wiener M. Classification and regression by randomForest. R News 2: 18–22, 2002. [Google Scholar]
- 19.Meyer DM, Dimitriadou E, Hornik K, Weingessel A, Leisch F. e1071: Misc Functions of the Department of Statistics, Probability Theory Group (Formerly: E1071), TU Wien 2019.
- 20.Kuhn M. Building predictive models in R using the caret package. J Stat Softw 28, 2008. doi: 10.18637/jss.v028.i05. [DOI] [Google Scholar]
- 21.Kuhn M. caret: Classification and regression training. 2019. https://cran.r-project.org/web/packages/caret/caret.pdf.
- 22.Perez-Atencio L, Garcia-Aracil N, Fernandez E, Barrio LC, Barios JA. A four-state Markov model of sleep-wakefulness dynamics along light/dark cycle in mice. PLoS One 13: e0189931, 2018. doi: 10.1371/journal.pone.0189931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spedicato GA, Kang TS, Yalamanchi SB, Yadav D, Cordon I. The markovchain package: a package for easily handling discrete Markov chains in R. 2016. https://cran.r-project.org/web/packages/markovchain/vignettes/an_introduction_to_markovchain_package.pdf.
- 24.Mai Q. A review of discriminant analysis in high dimensions. Wires Computational Statistics 5: 190–197, 2013. doi: 10.1002/wics.1257. [DOI] [Google Scholar]
- 25.Alzate-Correa D, Liu JM-L, Jones M, Silva TM, Alves MJ, Burke E, Zuñiga J, Kaya B, Zaza G, Aslan MT, Blackburn J, Shimada MY, Fernandes-Junior SA, Baer LA, Stanford KI, Kempton A, Smith S, Szujewski CC, Silbaugh A, Viemari J-C, Takakura AC, Garcia AJ, Moreira TS, Czeisler CM, Otero JJ. Neonatal apneic phenotype in a murine congenital central hypoventilation syndrome model is induced through non-cell autonomous developmental mechanisms. Brain Pathol 31: 84–102, 2020. doi: 10.1111/bpa.12877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kursa MB, Jankowski A, Rudnicki WR. Boruta - a system for feature selection. Fund Inform 101: 271–286, 2010. doi: 10.3233/FI-2010-288. [DOI] [Google Scholar]
- 27.Menze BH, Kelm BM, Masuch R, Himmelreich U, Bachert P, Petrich W, Hamprecht FA. A comparison of random forest and its Gini importance with standard chemometric methods for the feature selection and classification of spectral data. BMC Bioinformatics 10: 213, 2009. doi: 10.1186/1471-2105-10-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nollet M, Hicks H, McCarthy AP, Wu H, Moller-Levet CS, Laing EE, Malki K, Lawless N, Wafford KA, Dijk D-J, Winsky-Sommerer R. REM sleep's unique associations with corticosterone regulation, apoptotic pathways, and behavior in chronic stress in mice. Proc Natl Acad Sci USA 116: 2733–2742, 2019. doi: 10.1073/pnas.1816456116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pfammatter JA, Maganti RK, Jones MV. An automated, machine learning-based detection algorithm for spike-wave discharges (SWDs) in a mouse model of absence epilepsy. Epilepsia Open 4: 110–122, 2019. doi: 10.1002/epi4.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morra JH, Tu ZW, Apostolova LG, Green AE, Toga AW, Thompson PM. Comparison of AdaBoost and support vector machines for detecting Alzheimer’s disease through automated hippocampal segmentation. IEEE Trans Med Imaging 29: 30–43, 2010. doi: 10.1109/TMI.2009.2021941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gandolfo LC, Speed TP. RLE plots: visualizing unwanted variation in high dimensional data. PLoS One 13: e0191629, 2018. doi: 10.1371/journal.pone.0191629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aung T, Niyeha D, Heidkamp R. Leveraging data visualization to improve the use of data for global health decision-making. J Glob Health 9: 020319, 2019. 020319. doi: 10.7189/jogh.09.020319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malik WA, Unlu A. Interactive graphics: exemplified with real data applications. Front Psychol 2: 11, 2011. doi: 10.3389/fpsyg.2011.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mortola JP, Frappell PB. On the barometric method for measurements of ventilation, and its use in small animals. Can J Physiol Pharmacol 76: 937–944, 1998. doi: 10.1139/cjpp-76-10-11-937. [DOI] [PubMed] [Google Scholar]
- 35.DSI. FinePointe WBP Manual. St. Paul, MN: Data Sciences International, 2017. [Google Scholar]
- 36.Mortola JP, Frappell PB. Measurements of air ventilation in small vertebrates. Respir Physiol Neurobiol 186: 197–205, 2013. doi: 10.1016/j.resp.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 37.Drorbaugh JE, Fenn WO. A barometric method for measuring ventilation in newborn infants. Pediatrics 16: 81–87, 1955. [PubMed] [Google Scholar]
- 38.Epstein MA, Epstein RA. A theoretical analysis of the barometric method for measurement of tidal volume. Respir Physiol 32: 105–120, 1978. doi: 10.1016/0034-5687(78)90103-2. [DOI] [PubMed] [Google Scholar]
- 39.Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60: 430–440, 2008. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 40.Turnquist C, Horikawa I, Foran E, Major EO, Vojtesek B, Lane DP, Lu X, Harris BT, Harris CC. p53 isoforms regulate astrocyte-mediated neuroprotection and neurodegeneration. Cell Death Differ 23: 1515–1528, 2016. doi: 10.1038/cdd.2016.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clarke LE, Barres BA. Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci 14: 311–321, 2013[Erratum inNat Rev Neurosci14: 451, 2013]. doi: 10.1038/nrn3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poskanzer KE, Molofsky AV. Dynamism of an astrocyte in vivo: perspectives on identity and function. Annu Rev Physiol 80: 143–157, 2018. doi: 10.1146/annurev-physiol-021317-121125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmidt RE, Parvin CA, Green KG. Synaptic ultrastructural alterations anticipate the development of neuroaxonal dystrophy in sympathetic ganglia of aged and diabetic mice. J Neuropathol Exp Neurol 67: 1166–1186, 2008. doi: 10.1097/NEN.0b013e318190d6db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11: 251–253, 2008. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fellous JM, Sapiro G, Rossi A, Mayberg H, Ferrante M. Explainable artificial intelligence for neuroscience: behavioral neurostimulation. Front Neurosci 13: 1346, 2019. doi: 10.3389/fnins.2019.01346. [DOI] [PMC free article] [PubMed] [Google Scholar]