

Keywords: epilepsy, KCNQ3, neurological disorders, seizures, retigabine
Abstract
Retigabine is a first-in-class potassium channel opener approved for patients with epilepsy. Unfortunately, several side effects have limited its use in clinical practice, overshadowing its beneficial effects. Multiple studies have shown that retigabine acts by enhancing the activity of members of the voltage-gated KCNQ (Kv7) potassium channel family, particularly the neuronal KCNQ channels KCNQ2-KCNQ5. However, it is currently unknown whether retigabine’s action in neurons is mediated by all KCNQ neuronal channels or by only a subset. This knowledge is necessary to elucidate retigabine’s mechanism of action in the central nervous system and its adverse effects and to design more effective and selective retigabine analogs. In this study, we show that the action of retigabine in excitatory neurons strongly depends on the presence of KCNQ3 channels. Deletion of Kcnq3 severely limited the ability of retigabine to reduce neuronal excitability in mouse CA1 and subiculum excitatory neurons. In addition, we report that in the absence of KCNQ3 channels, retigabine can enhance CA1 pyramidal neuron activity, leading to a greater number of action potentials and reduced spike frequency adaptation; this finding further supports a key role of KCNQ3 channels in mediating the action of retigabine. Our work provides new insight into the action of retigabine in forebrain neurons, clarifying retigabine’s action in the nervous system.
NEW & NOTEWORTHY Retigabine has risen to prominence as a first-in-class potassium channel opener approved by the Food and Drug Administration, with potential for treating multiple neurological disorders. Here, we demonstrate that KCNQ3 channels are the primary target of retigabine in excitatory neurons, as deleting these channels greatly diminishes the effect of retigabine in pyramidal neurons. Our data provide the first indication that retigabine controls neuronal firing properties primarily through KCNQ3 channels.
INTRODUCTION
Retigabine {N-[2-amino-4-(4-fluorobenzylamino)-phenyl] carbamic acid ethyl ester}, also known as ezogabine, is a first-in-class compound approved by the Food and Drug Administration for treating partial adult seizures with or without secondary generalized seizures (1). In addition to epilepsy, researchers have also suggested retigabine as a possible treatment for tinnitus (2), amyotrophic lateral sclerosis (3), migraines (4), chronic and neuropathic pain (5), stroke (6), traumatic brain injury (7), and Huntington’s disease (8).
It is widely accepted that the primary molecular targets of retigabine are members of the voltage-gated KCNQ (Kv7) potassium channel family (9, 10), although some evidence suggests GABAA receptor involvement as well (11, 12). It is unclear whether retigabine exerts its effects in neurons through all neuronal KCNQ channels or through a select few. The KCNQ potassium family has five members (KCNQ1–KCNQ5), four of which (KCNQ2–KCNQ5) are neuronal (13, 14). Similar to other voltage-gated potassium channels, functional KCNQ channels are tetramers of identical (homomer) or compatible (heteromer) subunits (15). Retigabine primarily acts on neuronal KCNQ channels by shifting their activation to hyperpolarized membrane potentials and increasing their probability of opening (16, 17). The extent of this effect depends on the KCNQ channel subtype. For instance, the predominantly nonneuronal KCNQ1 channel is insensitive to retigabine, whereas KCNQ3 channels show the highest affinity and efficacy for retigabine among all KCNQ channels (1, 17, 18). KCNQ2 and KCNQ5 respond well to retigabine, but to a lesser extent than KCNQ3 channels (1). Because of its preference for KCNQ2-5 channels, researchers have proposed retigabine as a treatment for children carrying pathogenic loss-of-function variants of KCNQ2 and KCNQ3 channels (19). The current thinking is that retigabine may boost the activity of the remaining functional KCNQ2/3 channels, possibly alleviating the symptoms of KCNQ2/3-related disorders. However, retigabine administration to patients with KCNQ2 variants has shown mixed results (19).
Despite growing interest in the anticonvulsant properties of retigabine (19) and its proposed use for treating various conditions caused by neuronal hyperexcitability, this agent is associated with several drawbacks. For instance, phase-II enzymes in the liver rapidly metabolize retigabine, requiring a thrice-daily dosing regimen (15). An additional concern is the chemical instability of retigabine, which may be related to the photosensitivity of retigabine under prolonged ultraviolet exposure (20), potentially resulting in retinal and mucocutaneous blue-gray discoloration. Lastly, urinary retention is a common side effect associated with retigabine (21). Although it has been widely assumed that urinary retention occurs as retigabine activates KCNQ4 channels residing in bladder detrusor smooth muscles (22, 23), recent work has shown that retigabine directly targets neuronal fibers that sense bladder fullness rather than detrusor smooth muscles (24). These findings suggest that the beneficial and some (but not all) of the adverse effects of retigabine may arise through neuronal channels. However, it is currently unknown which KCNQ channels primarily mediate retigabine’s action in the nervous system. Identifying the primary target of retigabine is not only important for the design of compounds with greater specificity, but also for determining which patients with KCNQ2 and KCNQ3 loss-of-function pathogenic variants might benefit the most from retigabine administration. Based on the robust response of KCNQ3 channels to retigabine in heterologous cell lines, we investigated whether retigabine’s action in neurons requires KCNQ3 channels.
MATERIALS AND METHODS
All experiments were performed according to the guidelines described in the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Connecticut, Storrs.
Animals and Genotyping
For all experiments, we did not discriminate between male and female mice. Mice were housed under a 14-h to 10-h light and dark cycle and food was provided ad libitum. Kcnq3 germline knockout mice were generated previously (25) by crossing Kcnq3 flox (Kcnq3fl/fl) mice to Hprt-cre mice (https://www.jax.org/strain/004302). In a subset of experiments, we used wild-type C57BL/6J (https://www.jax.org/strain/000664) or neurotensin (NTS) Cre mice crossed with enhanced green fluorescent protein, as NTS can act as a marker for subiculum neurons (https://www.jax.org/strain/017525). We found no difference between NTS Cre mice and wild-type Kcnq3 knockout littermates with respect to retigabine effects; thus, we pooled the data together. PCR genotyping of mouse-tail prep DNA was performed using the following primers: Kcnq3, 5′- TCCACTTCCATGTTCAATGC-3′, 5′- CAGCACTCCCATGACAAATG-3′, and 5′- TCTCCCATGGCAAGTATTCC-3′. The primers amplified a 255 bp wild-type fragment and/or a 429 bp mutant fragment.
Slice Preparation and Electrophysiology
Postnatal day 14–20 (P14–P20) mice were anesthetized using isoflurane (Baxter Healthcare, Deerfield, IL) and euthanized by decapitation. Brains were quickly removed and placed in ice-cold cutting solution consisting of the following: 25 mM NaHCO3, 200 mM sucrose, 10 mM glucose, 2.5 mM KCl, 1.3 mM NaH2PO4, 0.5 mM CaCl2, and 7 mM MgCl2. Cerebella were removed, and 300-μm coronal or horizontal slices were cut using a vibratome (Leica VT1200S). Slices were then transferred to a holding chamber containing artificial cerebrospinal fluid (aCSF) consisting of the following: 125 mM NaCl, 26 mM NaHCO3, 2.5 mM KCl, 1 mM NaH2PO4, 1.3 mM MgCl2, 1.5 mM CaCl2, and 12 mM d-glucose. We used 1.5 mM CaCl2, as this concentration allows for better approximation of neuronal firing properties (26). Slices were recovered at 35°C for 30 min, and then left at room temperature (∼22°C) for at least 1 h before electrophysiological recording. Both cutting and aCSF solutions were saturated with 95% O2-5% CO2.
Whole cell recordings were obtained using electrodes pulled from thin-walled borosilicate glass capillaries (World Precision Instruments, Sarasota, FL) that had resistances of 2–4 MΩ when filled with recording solution. Electrodes were pulled using a Sutter Instruments puller (P-1000, Sutter instruments, CA). The internal recording solution for whole cell recordings of subiculum neurons consisted of the following: 115 mM potassium gluconate, 20 mM KCl, 4 mM Mg·ATP, 0.3 mM Na4·GTP, 10 mM HEPES, and 10 mM Na2-phosphocreatine (osmolarity ∼300 mosmol/kgH2O) (27). For hippocampal CA1 pyramidal neurons, the internal solution consisted of the following: 130 mM potassium methylsulfate, 10 mM KCl, 4 NaCl, 4 mM Mg·ATP, 0.4 mM Na4·GTP, 10 mM HEPES, and 5 mM Tris-phosphocreatine (osmolarity ∼300–310 mosmol/kgH2O). The pH was adjusted to 7.2–7.3 with KOH. We recorded from neurons that had an initial resting membrane potential of −65 to −70 mV. All recordings were performed at high temperature (28°C–32°C) using a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA), filtered at 2 kHz and sampled at 50 kHz. Bridge balancing was engaged throughout the recordings. The recordings were not adjusted for the junction potential. Retigabine (10 µM) was bath applied in the current clamp experiments. In a subset of experiments, 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX; 50 µM), d-2-amino-5-phosphonovaleric acid (D-AP5; 25 µM), and picrotoxin (100 µM) (Abcam) were added to the aCSF to block α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-mediated, N-methyl-d-aspartate (NMDA)-mediated, and GABA-mediated synaptic transmission, respectively.
Analysis and Quantification
Data were analyzed offline using Clampfit (Molecular Devices, Sunnyvale, CA) and Prism 8 (GraphPad, La Jolla, CA) software. Statistical significance was determined using unpaired or paired Student’s t tests, nonparametric Mann–Whitney or paired Wilcoxon tests, or a two-way repeated-measures (RM) or mixed effects analysis of variance (ANOVA). We used nonparametric tests for data that did not pass normality tests. In the figure legends, we report both n values that indicate the number of cells and the number of animals used to obtain these cells. This work was not blinded, as application of retigabine distinguished wild-type cells from Kcnq3-null neurons from the first recording. The number of cells was based on previously published work (28).
RESULTS
Previous work in expression systems has shown that KCNQ3 channels respond robustly to retigabine (17, 18), a KCNQ2-5 activator. To examine whether KCNQ3 provides neurons with heightened retigabine sensitivity, we compared the effects of retigabine in control and Kcnq3-knockout mice. First, we confirmed that retigabine primarily acts through KCNQ channels in control animals. For this, we applied retigabine (10 µM) followed by the pan-KCNQ channel blocker ML-252 (10 µM) in CA1 pyramidal neurons of the hippocampus. We used ML-252 as it has very high affinity and potency across neuronal KCNQ channels (29). Indeed, application of 10 µM ML-252 readily reversed the effect of retigabine on dampening the excitability of CA1 pyramidal neurons (Fig. 1, A and B). In cells not incubated with retigabine, application of ML-252 alone increased CA1 activity (control+75pA = 16 ± 3 AP, +ML252+75pA = 34 ± 6 AP, n = 4; P = 0.017 t = 4.87, df = 3, paired Student’s t test; data not shown; AP refers to action potentials), confirming that blocking KCNQ channels elevates neuronal excitability.
Figure 1.
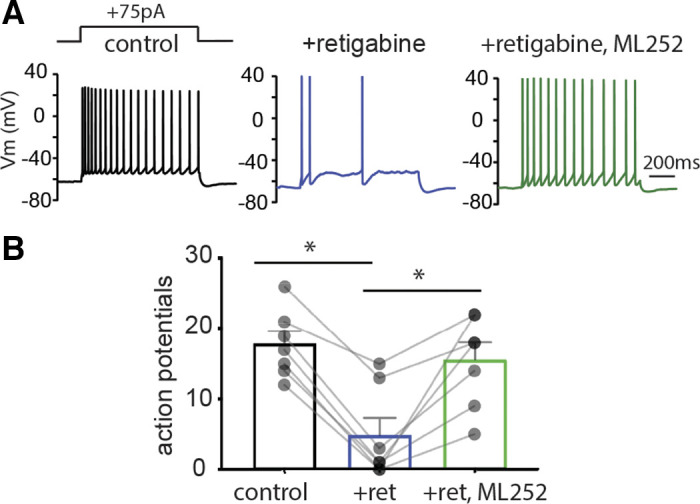
Retigabine (ret) effects on CA1 pyramidal neurons are reversed by the nonselective pan-KCNQ inhibitor ML-252. A: representative voltage responses to a +75 pA current injection step (1 s) in CA1 pyramidal neurons from wild-type neurons and neurons in the presence of retigabine (10 µM) or retigabine (10 μM) + ML252 (10 µM). Membrane potential was kept constant by injecting a small direct current (DC) through the recording pipette. B: summary graphs (n = 7 from 4 mice) showing the effect ML-252 on retigabine-induced responses. *Significance of less than 0.05. One-way repeated-measures (RM) ANOVA P = 0.0001, F(2, 12) = 21.8. Tukey’s multiple comparisons test: control vs. retigabine P = 0.0001; control vs. retigabine+ML252 = 0.5395; retigabine vs. retigabine+ML252 P = 0.0007. For statistical analysis we used the number of cells as n.
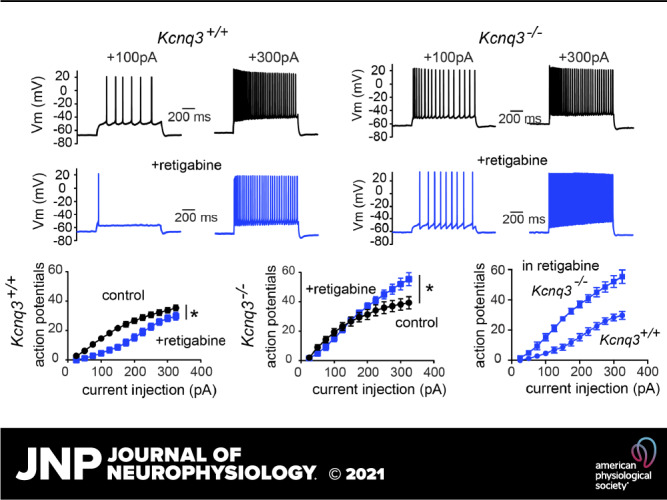
Next, we examined whether retigabine would have similar impacts on CA1 pyramidal neuronal activity in control and Kcnq3-knockout mice. In the presence of 10 µM retigabine, Kcnq3+/+ neurons required a much greater depolarization to induce a train of action potentials (Fig. 2A). In particular, we found that small depolarizing current injections (range: +25 pA to +100 pA) led to one or a few action potentials in the presence of retigabine (+50 pA; control: 5.9 ± 1.3 AP; +retigabine: 0.8 ± 0.34 AP, n = 18; P = 0.0005, t = 4.321, df = 17, paired Student’s t test). However, as we increased the depolarizing currents to increase excitability, the pyramidal neurons gradually overcame some of the retigabine-induced hypoexcitability (Fig. 2A). This behavior was apparent for current injections at or greater than +200 pA (i.e., at +325 pA; control: 35.4 ± 1.9 AP; +retigabine: 29.8 ± 2.6 AP, n = 18; P = 0.0005, t = 4.321, df = 17, paired Student’s t test). Thus, the reduction in action potential number caused by retigabine shifted from ∼80%–90% to ∼15%, depending on the depolarization level.
Figure 2.

Retigabine dampening of CA1 pyramidal neuron excitability depends on KCNQ3 channels. A: representative voltage responses to two current injection steps (1 s) in CA1 pyramidal neurons from control neurons before and after bath application of retigabine (10 µM). Membrane potential was kept constant by injecting a small direct current (DC) through the recording pipette. B; representative voltage responses to two current injection steps (1 s) in CA1 pyramidal neurons from Kcnq3-null neurons before and after bath application of retigabine (10 µM). Membrane potential was kept constant by injecting a small DC through the recording pipette. C: summary graphs showing the effect of 10 µM retigabine on action potential number in Kcnq3+/+ [two-way repeated-measures (RM) ANOVA P < 0.0001, F(12, 204) = 11.02, n = 18 from 10 mice] and Kcnq3−/− [two-way RM ANOVA P < 0.0001, F(12,144) = 14.74, n = 13 from 6 mice] neurons, and the comparison between Kcnq3+/+ and Kcnq3−/− neurons in the presence of 10 µM retigabine. D: summary graphs showing the effect of 10 µM retigabine on the input resistance (Rin) in Kcnq3+/+ [two-way RM ANOVA P < 0.0001, F(3,27) = 138.9, n = 16 from 9 mice] and Kcnq3−/− [two-way RM ANOVA P = 0.1597, F(3, 18) = 1.938, n = 13 from 6 mice]. *Significance of less than 0.05. Data are represented as means ± SE. For statistical analysis we used the number of cells as n.
After confirming that retigabine blunts CA1 pyramidal neuron firing activity under our recording conditions, we recorded the response of Kcnq3-null neurons to retigabine. We found that retigabine was ineffective in reducing excitability in Kcnq3-deficient neurons (Fig. 2, A and B). Instead, with retigabine, strong step depolarizing currents (> +250 pA) led to a greater number of action potentials and a higher final firing frequency (i.e., the frequency of the last two action potentials) (control Ff+325pA: 32.8 ± 4.6; +retigabine Ff +325pA: 45.7 ± 4.1, n = 13; P = 0.009, t = 4.353 df = 12, paired Student’s t test). As a result, Kcnq3-null CA1 pyramidal neurons fired twice as many action potentials as their wild-type counterparts in the presence of retigabine (Fig. 2C).
In addition to regulating the number of action potentials, retigabine significantly reduced the input resistance of CA1 pyramidal neurons. In particular, retigabine led to a decrease in the pyramidal neuron input resistance, with the most pronounced effects observed for modest current injections (Fig. 2D, left; −25 pA; control: 148 ± 10 MΩ; +retigabine: 27 ± 2.0 MΩ, n = 16). This effect was absent in Kcnq3-null neurons (Fig. 2D, right), suggesting that retigabine exerts its effect on input resistance through KCNQ3-containing channels.
We also examined the effect of retigabine in CA1 pyramidal neurons by using a ramp protocol. KCNQ channels have slow activation kinetics; thus, slow ramp protocols reveal different aspects of the roles of KCNQ channels in regulating neuronal excitability. As observed with our step protocols, retigabine application reduced the number of action potentials during the slow ramp protocol in control neurons (control: 29 ± 2 AP; +retigabine: 11 ± 2 AP, n = 16; P < 0.0001, t = 7.69, df = 15, paired Student’s t test). We did not observe this effect in Kcnq3-null neurons (Fig. 3; control: 35 ± 4.5 AP; +retigabine: 34 ± 2 AP, n = 12; P < 0.86, t = 0.17, df = 11, paired Student’s t test). Importantly, a closer examination of our recordings showed that the number of action potentials increased in the presence of retigabine in 7 of the 12 recordings for Kcnq3-null neurons. We did not observe such an increase in the action potential number due to retigabine in ramp recordings from any of the cells recorded from wild-type slices.
Figure 3.

Retigabine’s inability to reduce excitability in Kcnq3-null neurons is independent of stimulation protocol. A: representative voltage responses to a ramp protocol (120 pA/s) in CA1 pyramidal neurons from Kcnq3+/+ and Kcnq3−/− slices before and after bath application of retigabine (10 µM). Membrane potential was kept constant by injecting a small direct current (DC) through the recording pipette. B: summary graphs showing the effect of 10 µM retigabine (ret) on action potential number. *Significance of less than 0.05. (Kcnq3+/+ paired Student’s t test P < 0.0001, t = 7.687, df = 15, n = 16 from 9 mice; Kcnq3−/− paired Student’s t test P = 0.8648, t = 0.1743, df = 11, n = 12 from 6 mice). For statistical analysis we used the number of cells as n.
What contributes to the increased firing caused by retigabine in Kcnq3-null neurons during strong depolarizations? Previous work has shown that deletion of Kcnq3 increases the magnitude of the slow afterhypepolarization (sAHP) current in pyramidal neurons (25). Consistent with this, we found that the sAHP was larger in Kcnq3-null neurons (sAHP+325pA control: −2.8 ± 0.3 mV, n = 16; Kcnq3−/−: −5.0 ± 0.4 mV, n = 13; P = 0.0007, t = 3.822, df = 27 unpaired Student’s t test), whereas there was no difference in the fast afterhyperpolarization (fAHP control: −7.887 ± 1.327 mV, n = 13; Kcnq3−/−: −6.306 ± 0.9766 mV, n = 12; P = 0.3540, t = 0.946 df = 23 unpaired Student’s t test). Thus, it is possible that retigabine drives the activity of the remaining KCNQ channels and, in conjunction with a larger sAHP, leads to a hyperpolarization that is sufficient for a quicker recovery of sodium channels from inactivation. In turn, this would lead to a greater firing frequency but would be insufficient to shunt neuronal excitability.
In addition, boosting the retigabine-induced hyperpolarization may be an ongoing action of synaptic receptors, which were not blocked in our experiments. Thus, we repeated our experiments in the presence of synaptic blockers for GABAA, AMPA, and NMDA receptors. In the presence of synaptic blockers, retigabine application modestly increased the number of action potentials (Fig. 4, A and B; action number at +325pA went from 34 ± 1.6 to 40 ± 2.1, n = 13, P = 0.0004, t = 4.858, df = 12 paired Student’s t test), and the final firing frequency (frequency of the last two action potentials), which significantly increased (control Ff+325pA = 24.7 ± 1.5 Hz, +retigabine Ff +325pA = 33.8 ± 2.1, n = 13; P < 0.0001 t = 6.35, df = 12; paired Student’s t test). As mentioned earlier, we observed a similar increase in the final frequency for Kcnq3-null cell recordings obtained in the absence of synaptic blockers. In contrast to the final frequency, retigabine led to a decrease in the initial frequency (frequency of the first two action potentials) (Fig. 4B), which diminished the spike frequency adaptation measured as the ratio of the final frequency to the initial frequency (Ff+325pA/Fin+325pA = 0.22 ± 0.02 to 0.41 ± 0.05, n = 13; P = 0.0001 t = 5.49 df = 12 paired Student’s t test). This result suggests that the increased excitability caused by retigabine is influenced by, but does not require, ongoing synaptic activity.
Figure 4.

Retigabine increases excitability in Kcnq3-null neurons independent of fast synaptic activity. A: representative voltage responses of +300 pA current injection step (1 s) in CA1 pyramidal neurons from Kcnq3−/−- neurons before and after bath application of retigabine (10 µM) in the presence of the synaptic blockers D-AP5, NBQX, and picrotoxin. Membrane potential was kept constant by injecting a small direct current (DC) through the recording pipette. B: summary graphs showing the effect of 10 µM retigabine on action potential number [two-way repeated-measures (RM) ANOVA < 0.0001, F(12, 144) = 8.482, n = 13 from 5 mice], initial firing frequency [mixed effects two-way ANOVA P = 0.0024, F(12, 144) = 2.717, n = 13 from 5 mice], and final firing frequency [mixed effects two-way ANOVA P < 0.0001, F(12, 144) = 9.449, n = 13 from 5 mice) in Kcnq3−/− CA1 pyramidal neurons. *Significance of less than 0.05. Data are represented as means ± SE. For statistical analysis we used the number of cells as n.
CA1 pyramidal neurons express all of the major neuronal KCNQ channels: KCNQ2, KCNQ3, and KCNQ5. Thus, the absence of KCNQ3, KCNQ2, KCNQ5, or both KCNQ2 and KCNQ5 could be responsible for the effect of retigabine in pyramidal neurons. Therefore, we examined the effect of retigabine in a cell population with a different set of KCNQ channels. We focused on subiculum pyramidal neurons (30). Earlier studies have shown that KCNQ3 channels, but not KCNQ5 channels, are highly expressed in the subiculum (31), suggesting that retigabine may regulate the properties of subiculum neurons in a manner distinct from that of CA1 pyramidal neurons.
The subiculum is divided into two broad areas, proximal and distal, with each region projecting to different target areas (32). Importantly, neurons in the two regions exhibit different firing properties, classified as regular spiking and burst spiking neurons (32). We distinguished the two groups of neurons (regular versus burst spiking) based on their input resistance and their initial firing frequency following strong depolarization currents (33). We note that bursting neurons can be further subdivided into weak and strong bursters (27, 34); the majority of our cells were weak bursters, possibly due to the extracellular Ca2+ concentration (1.5 mM), as strength of bursting is driven by T-type calcium channels (34).
Consistent with previous work, subiculum burst spiking neurons (defined as neurons with more than 100 Hz final frequency at +200pA) had a twofold higher initial frequency, compared with regular spiking neurons (Fig. 5, A and B; regular spiking Fin+325pA = 92 ± 8 Hz, n = 14; burst spiking Fin+325pA = 202 ± 8 Hz, n = 20). The burst spiking neurons also exhibited a lower input resistance (burst spiking Rin−25pA = 108 ± 13 MΩ, n = 20) than regular spiking neurons (Rin−25pA = 182 ± 15 MΩ, n = 14). Moreover, we found that the number of action potentials fired by burst spiking neurons was slightly higher than that of regular spiking neurons (burst spiking: AP+325pA = 43 ± 2.0, n = 20; regular spiking: AP+325pA = 37 ± 3, n = 14).
Figure 5.

Kcnq3 deletion differentially affects the firing properties of subiculum burst and regular spiking neurons. A: representative voltage responses from a +200 pA current injection step (1 s) in regular (left) and burst (right) firing spiking neurons. Insets in burst spiking neurons show the first few action potentials, highlighting the difference in their initial firing frequency. Membrane potential was kept constant by injecting a small direct current (DC) through the recording pipette. B: summary graphs showing the effect of Kcnq3 channel loss on the action potential number and initial firing frequency in either regular or burst spiking neurons. Initial frequency decreased in the absence of KCNQ3 channels (Kcnq3+/+ n = 20 from 9 mice; Kcnq3−/− n = 9 from 4 mice). Statistics for bottom right panel: for 50 pA P = 0.105, t = 1.767 df = 11; 75 pA P = 0.034, t = 2.265 df = 22; 100 pA P = 0.02, t = 2.48 df = 26; 125 pA P = 0.0175, t = 2.53 df = 27; 150 pA P = 0.0155, t = 2.583 df = 27; 175 pA P = 0.024, t = 2.398 df = 27; 200 pA P = 0.0205, t = 2.461 df = 27; 225 pA P = 0.0383, t = 2.178, df=27, 250 pA P = 0.0165, t = 2.557, df = 27; 275 pA P = 0.0206, t = 2.459 df = 27; 300 pA P = 0.0447 t = 2.105 df = 27; 325 pA P = 0.0321 t = 2.26 df = 27, unpaired Student’s t test. *Statistical significance of less than 0.05. Data are represented as means ± SE. For statistical analysis we used the number of cells as n.
KCNQ3 deletion moderately altered the firing properties of burst spiking neurons, but it did not affect the firing behavior of regular spiking neurons (Fig. 5B). The most pronounced effects we observed for burst spiking neurons upon Kcnq3 deletion were 1) a decrease in the initial action potential frequency for all depolarizing current stimuli (Fig. 5) and 2) a reduction in the sAHP (sAHP+325pA control: −1.8 ± 0.3 mV, n = 20; Kcnq3−/− −0.6 ± 0.2 mV, n = 6; P = 0.033 Mann–Whitney test). Beyond this impact, we did not observe any effects on the total number of action potentials fired (Fig. 5) or the final firing frequency (Kcnq3+/+ Ff+325pA = 35.2 ± 1.6 Hz, n = 20; Kcnq3−/− Ff+325pA = 38.2 ± 3.4 Hz, n = 6; P = 0.398, t = 0.8606, df = 24 unpaired Student’s t test). This result suggests that the loss of KCNQ3 channels in subiculum neurons likely unmasked another conductance that can decelerate firing in burst spiking neurons, as the loss of KCNQ3 should have caused an increase in the firing frequency, not a decrease. We did not investigate this issue further.
Next, we examined the response of burst spiking neurons to retigabine. Similar to other cell types, 10 µM retigabine caused a strong reduction in burst spiking neuron excitability (Fig. 6). In particular, we found that retigabine reduced both the number of action potentials (Fig. 6, A and B) and the final frequency (Kcnq3+/+ Ff+325 = 34.2 ± 1.7 Hz, n = 10; +retigabine Ff+325 = 17 ± 2.5 Hz, n = 11; P = 0.0005, t = 5.294, df = 9 paired Student’s t test). In contrast to the recordings of CA1 pyramidal neurons, we did not observe a concomitant change in input resistance due to retigabine (burst spiking Rin−25pA = 107.6 ± 14.6 MΩ, +retigabine = 111.3 ± 12.4, n = 11). This result suggested that retigabine could reduce excitability without necessarily decreasing the neuron input resistance measured in the soma. Despite the presence of some differences between CA1 and subiculum neurons in response to retigabine, Kcnq3-null burst spiking neurons were also less responsive to retigabine at small (<+100 pA), intermediate (between +100 pA and +200 pA), and large (>200 pA) current injections, with stronger effects occurring at intermediate current injections (Fig. 6B, middle and right panels). Unlike CA1 pyramidal neurons, these cells did not exhibit an increase in firing frequency (Kcnq3−/− Ff+325 = 34 ± 3.1 Hz, +retigabine Ff+325 = 30 ± 5 Hz, n = 9; P = 0.13 t = 1.6 df = 8 paired Student’s t test). To ensure that this result was not due to our use of differing internal solutions between CA1 and subicular neurons, we repeated our experiments using a K-MeSO4-based CA1 internal solution. Similar to our earlier experiments (Fig. 6), retigabine did not increase firing in Kcnq3-null subicular neurons (control: 39 ± 3 AP; +retigabine: 34 ± 3 AP, n = 3; P = 0.25 Wilcoxon paired test).
Figure 6.

Retigabine dampening of subiculum burst spiking excitability depends on KCNQ3 channels. A: left, representative voltage responses to two current injection steps (1 s) in burst spiking neurons from Kcnq3+/+ neurons before and after bath application of retigabine (10 µM). Membrane potential was kept constant by injecting a small direct current (DC) through the recording pipette. Right, representative voltage responses to two current injection steps (1 s) in burst spiking neurons from Kcnq3−/− neurons before and after bath application of retigabine (10 µM). Membrane potential was kept constant by injecting a small DC through the recording pipette. B: left, summary graph showing the effect of 10 µM retigabine on action potential number in Kcnq3+/+ neurons [two-way repeated-measures (RM) ANOVA P < 0.0001 F(12, 120) = 10.79, n = 11 from 7 mice]. Middle, summary graph showing the effect of 10 µM retigabine on action potential number in Kcnq3−/− neurons [two-way RM ANOVA P = 0.0006, F(12, 96) = 3.247, n = 9 from 4 mice]. Data are represented as means ± SE. Right, percentage inhibition of retigabine in Kcnq3+/+ and Kcnq3−/− burst spiking neurons (statistics for right panel: for 100 pA P = 0.0219, 125 pA P = 0.0188,150pA P = 0.0043, 250 pA P = 0.0023, 275 pA P = 0.0004, 300 pA P = 0.0074, 325 pA P = 0.0016, Mann–Whitney test), (for 175 pA P = 0.0035, t = 3.356 df = 18; 200 pA P = 0.0042, t = 3.279 df = 18; 225 pA P = 0.0026, t = 3.498 df = 18; Student’s unpaired t tests). For statistical analysis we used the number of cells as n. *Statistical significance of less than 0.05.
Together, these results suggest that KCNQ3 channels are the primary targets of retigabine in some excitatory neurons; moreover, depending on the neuron type, retigabine can potentially increase neuronal activity in the absence of KCNQ3 channels.
DISCUSSION
Retigabine, a KCNQ channel activator, has received much attention over the years as a possible treatment for multiple neurological disorders. Importantly, retigabine has also been suggested as a therapeutic intervention for patients carrying KCNQ2 and KCNQ3 pathogenic loss-of-function variants, as retigabine potentially could boost the activity of the remaining functional KCNQ2/3 channels (19, 35). Here, using Kcnq3-knockout mice we demonstrated that KCNQ3 channels are the main molecular target of retigabine in excitatory neurons of the hippocampus and subiculum, two brain areas implicated in spatial navigation and learning and memory (32). Our work is also consistent with the recent observation that retigabine’s ability to decrease neuronal excitability in the neocortex closely follows the distribution of Kcnq3 mRNA in the different cortical layers (36), further supporting our suggestion that KCNQ3 channels are the principal targets of retigabine in the brain.
Our data are consistent with several previous reports in heterologous systems. First, expression of KCNQ2-KCNQ4 channels in heterologous cells showed that application of retigabine leads to ∼−40 mV shift in the half-activation voltage (V0.5) of KCNQ3 channels. A much smaller effect was observed for KCNQ2 (ΔV0.5 ∼−17 mV to −24 mV), KCNQ2/3 (ΔV0.5 ∼−30 mV), and KCNQ4 channels (ΔV0.5 ∼−14 mV). Second, similar experiments demonstrated that the retigabine EC50 is 0.6 µM for KCNQ3, fourfold lower than that for KCNQ2 channels (EC50 ∼2.5 µM) (18). KCNQ5 has an EC50 of 2–6 µM, and KCNQ2/3 and KCNQ3/5 heteromers have a comparable EC50 of ∼1.4–1.6 µM (1). Third, under voltage clamp, application of retigabine in Kcnq3-null CA1 pyramidal neurons did not affect the holding current, unlike in wild-type neurons (37). Building on these findings, we showed that retigabine requires KCNQ3 channels to exert robust effects on the firing properties of CA1 pyramidal neurons as well as a subclass of subiculum neurons.
Our study also revealed several unexpected findings. For instance, in the absence of synaptic blockers, retigabine increased the excitability of CA1 pyramidal neurons following strong depolarization, an effect that was maintained even in the absence of synaptic activity. A likely scenario is that retigabine exerts its effects via activating the remaining KCNQ2 and KCNQ5 channels in pyramidal neurons. Previous work has shown that Kcnq3 deletion, in contrast to Kcnq2 deletion, does not affect KCNQ2 and KCNQ5 channel levels (28). As a result, in the presence of ongoing background synaptic events, increasing KCNQ2 and KCNQ5 activity might provide sufficient amounts of hyperpolarization to relieve sodium channel inactivation when neurons are strongly depolarized, but not enough to shunt and dampen neuronal excitability. Consistent with this possibility, we previously found that, in the presence of high extracellular potassium to drive KCNQ2 and KCNQ5 channel activation in Kcnq3-null neurons, retigabine could still dampen the neuronal excitability of CA1 pyramidal neurons, an effect that was reversed by the application of the pan-KCNQ blocker XE991 (28). Alternatively, in parallel to activating KCNQ channels, retigabine might also inhibit potassium channels such as Kv2.1 (38) and/or increase the activity of channels mediating the fast AHP (39). Such effects might increase the firing frequency of pyramidal neurons, an effect that might become even more pronounced in the absence of KCNQ3 channels. Independent of the precise mechanism, loss of KCNQ3 channels might unmask an increased excitatory effect of retigabine under basal conditions.
In contrast to CA1 pyramidal neurons, the activity of subiculum neurons was not potentiated by retigabine in the absence of KCNQ3 channels. In addition, retigabine decreased the firing properties of these neurons without changing their input resistance, measured in the soma. Currently, we do not know the precise mechanism for the lack of change in the input resistance by retigabine, but it might relate to the absence of KCNQ5 channels in subiculum neurons (31). KCNQ5 channels are found throughout the hippocampal formation but might be absent from the subiculum region, unlike KCNQ3 channels. KCNQ5 and KCNQ3 could form heteromers, the properties of which differ from the properties of KCNQ2/3 channels (40). Thus, it is possible that the changes in the input resistance found in CA1 pyramidal neurons were due to activation of KCNQ3/5 channels, a complex that might not be present in subiculum neurons. The absence of KCNQ5-specific pharmacological blockers prevents us from testing this hypothesis directly. Alternatively, subiculum and CA1 excitatory neurons might simply respond differently to the application of retigabine due to the overall expression of different ion channel clades.
Our work has implications for the administration of retigabine to patients with KCNQ2/3 pathogenic variants as well. Considering that retigabine primarily targets KCNQ3 channels, retigabine’s effect will be greatly diminished in patients carrying KCNQ3 loss-of-function variants and possibly in patients with KCNQ2 loss-of-function variants that exert a strong defect in KCNQ2/3 channels in the brain. Thus, patients that have primarily intact KCNQ3 channels would show stronger benefits from retigabine. In summary, our study establishes KCNQ3 as the major target of retigabine in some forebrain neurons, an important step toward developing more specific analogs for retigabine that are potentially effective against a wide array of brain hyperexcitability diseases.
GRANTS
This work was supported by National Institutes of Health Grants NS101596, HL137094, and NS108874 (to A.V.T.) and a grant from the Italian Ministry for University and Research, PRIN 2017ALCR7C (to M.T.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.T. and A.V.T. conceived and designed research; N.V. and A.L. performed experiments; N.V. and A.L. analyzed data; N.V., A.L., and A.V.T. interpreted results of experiments; N.V. and A.L. prepared figures; N.V. and A.L. drafted manuscript; M.T. and A.V.T. edited and revised manuscript; N.V., A.L., M.T., and A.V.T. approved final version of manuscript.
REFERENCES
- 1.Gunthorpe MJ, Large CH, Sankar R. The mechanism of action of retigabine (ezogabine), a first‐in‐class K+ channel opener for the treatment of epilepsy. Epilepsia 53: 412–424, 2012. doi: 10.1111/j.1528-1167.2011.03365.x. [DOI] [PubMed] [Google Scholar]
- 2.Kalappa BI, Soh H, Duignan KM, Furuya T, Edwards S, Tzingounis AV, Tzounopoulos T. Potent KCNQ2/3-specific channel activator suppresses in vivo epileptic activity and prevents the development of tinnitus. J Neurosci 35: 8829–8842, 2015. doi: 10.1523/JNEUROSCI.5176-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kovalchuk MO, Heuberger JA, Sleutjes BT, Ziagkos D, van den Berg LH, Ferguson TA, Franssen H, Groeneveld GJ. Acute effects of riluzole and retigabine on axonal excitability in patients with amyotrophic lateral sclerosis: a randomized, double‐blind, placebo‐controlled, crossover trial. Clin Pharmacol Ther 104: 1136–1145, 2018. doi: 10.1002/cpt.1096. [DOI] [PubMed] [Google Scholar]
- 4.Zhang X, Jakubowski M, Buettner C, Kainz V, Gold M, Burstein R. Ezogabine (KCNQ2/3 channel opener) prevents delayed activation of meningeal nociceptors if given before but not after the occurrence of cortical spreading depression. Epilepsy Behav 28: 243–248, 2013. doi: 10.1016/j.yebeh.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abd-Elsayed A, Jackson M, Gu SL, Fiala K, Gu J. Neuropathic pain and Kv7 voltage-gated potassium channels: the potential role of Kv7 activators in the treatment of neuropathic pain. Mol Pain 15: 1744806919864256, 2019. doi: 10.1177/1744806919864256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bierbower SM, Choveau FS, Lechleiter JD, Shapiro MS. Augmentation of M-type (KCNQ) potassium channels as a novel strategy to reduce stroke-induced brain injury. J Neurosci 35: 2101–2111, 2015. doi: 10.1523/JNEUROSCI.3805-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vigil FA, Bozdemir E, Bugay V, Chun SH, Hobbs M, Sanchez I, Hastings SD, Veraza RJ, Holstein DM, Sprague SM, Carver CM, Cavazos JE, Brenner R, Lechleiter JD, Shapiro MS. Prevention of brain damage after traumatic brain injury by pharmacological enhancement of KCNQ (Kv7,“M-type”) K+ currents in neurons. J Cereb Blood Flow Metab 40: 1256–1273, 2020. doi: 10.1177/0271678X19857818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao Y, Bartolomé-Martín D, Rotem N, Rozas C, Dellal SS, Chacon MA, Kadriu B, Gulinello M, Khodakhah K, Faber DS. Rescue of homeostatic regulation of striatal excitability and locomotor activity in a mouse model of Huntington’s disease. Proc Natl Acad Sci USA 112: 2239–2244, 2015. doi: 10.1073/pnas.1405748112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rundfeldt C. The new anticonvulsant retigabine (D-23129) acts as an opener of K+ channels in neuronal cells. Eur J Pharmacol 336: 243–249, 1997. doi: 10.1016/S0014-2999(97)01249-1. [DOI] [PubMed] [Google Scholar]
- 10.Sills GJ, Rogawski MA. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 168: 107966, 2020. doi: 10.1016/j.neuropharm.2020.107966. [DOI] [PubMed] [Google Scholar]
- 11.Treven M, Koenig X, Assadpour E, Gantumur E, Meyer C, Hilber K, Boehm S, Kubista H. The anticonvulsant retigabine is a subtype selective modulator of GABA A receptors. Epilepsia 56: 647–657, 2015. doi: 10.1111/epi.12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Rijn CM, Willems-van Bree E. Synergy between retigabine and GABA in modulating the convulsant site of the GABAA receptor complex. Eur J Pharmacol 464: 95–100, 2003. doi: 10.1016/S0014-2999(03)01426-2. [DOI] [PubMed] [Google Scholar]
- 13.Greene DL, Hoshi N. Modulation of Kv7 channels and excitability in the brain. Cell Mol Life Sci 74: 495–508, 2017. doi: 10.1007/s00018-016-2359-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci 1: 21–30, 2000. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- 15.Barrese V, Stott JB, Greenwood IA. KCNQ-encoded potassium channels as therapeutic targets. Annu Rev Pharmacol Toxicol 58: 625–648, 2018. doi: 10.1146/annurev-pharmtox-010617-052912. [DOI] [PubMed] [Google Scholar]
- 16.Tatulian L, Brown DA. Effect of the KCNQ potassium channel opener retigabine on single KCNQ2/3 channels expressed in CHO cells. J Physiol 549: 57–63, 2003. doi: 10.1113/jphysiol.2003.039842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tatulian L, Delmas P, Abogadie FC, Brown DA. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci 21: 5535–5545, 2001. doi: 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schenzer A, Friedrich T, Pusch M, Saftig P, Jentsch TJ, Grötzinger J, Schwake M. Molecular determinants of KCNQ (Kv7) K+ channel sensitivity to the anticonvulsant retigabine. J Neurosci 25: 5051–5060, 2005. doi: 10.1523/JNEUROSCI.0128-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Millichap JJ, Park KL, Tsuchida T, Ben-Zeev B, Carmant L, Flamini R, Joshi N, Levisohn PM, Marsh E, Nangia S, Narayanan V, Ortiz-Gonzalez XR, Patterson MC, Pearl PL, Porter B, Ramsey K, McGinnis EL, Taglialatela M, Tracy M, Tran B, Venkatesan C, Weckhuysen S, Cooper EC. KCNQ2 encephalopathy: features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol Genet 2: e96, 2016. doi: 10.1212/NXG.0000000000000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar M, Reed N, Liu R, Aizenman E, Wipf P, Tzounopoulos T. Synthesis and evaluation of potent KCNQ2/3-specific channel activators. Mol Pharmacol 89: 667–677, 2016. doi: 10.1124/mol.115.103200. [DOI] [PubMed] [Google Scholar]
- 21.Brickel N, Gandhi P, VanLandingham K, Hammond J, DeRossett S. The urinary safety profile and secondary renal effects of retigabine (ezogabine): a first‐in‐class antiepileptic drug that targets KCNQ (Kv7) potassium channels. Epilepsia 53: 606–612, 2012. doi: 10.1111/j.1528-1167.2012.03441.x. [DOI] [PubMed] [Google Scholar]
- 22.Bientinesi R, Mancuso C, Martire M, Bassi PF, Sacco E, Currò D. Kv7 channels in the human detrusor: channel modulator effects and gene and protein expression. Naunyn Schmiedebergs Arch Pharmacol 390: 127–137, 2017. doi: 10.1007/s00210-016-1312-9. [DOI] [PubMed] [Google Scholar]
- 23.Gopalakrishnan M, Shieh CC. Potassium channel subtypes as molecular targets for overactive bladder and other urological disorders. Expert Opin Ther Targets 8: 437–458, 2004. doi: 10.1517/14728222.8.5.437. [DOI] [PubMed] [Google Scholar]
- 24.Tykocki NR, Heppner TJ, Dalsgaard T, Bonev AD, Nelson MT. The Kv7 channel activator retigabine suppresses mouse urinary bladder afferent nerve activity without affecting detrusor smooth muscle K+ channel currents. J Physiol 597: 935–950, 2019. doi: 10.1113/JP277021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim KS, Duignan KM, Hawryluk JM, Soh H, Tzingounis AV. The voltage activation of cortical KCNQ channels Ddepends on global PIP2 levels. Biophys J 110: 1089–1098, 2016. doi: 10.1016/j.bpj.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forsberg M, Seth H, Björefeldt A, Lyckenvik T, Andersson M, Wasling P, Zetterberg H, Hanse E. Ionized calcium in human cerebrospinal fluid and its influence on intrinsic and synaptic excitability of hippocampal pyramidal neurons in the rat. J Neurochem 149: 452–470, 2019. doi: 10.1111/jnc.14693. [DOI] [PubMed] [Google Scholar]
- 27.Staff NP, Jung H-Y, Thiagarajan T, Yao M, Spruston N. Resting and active properties of pyramidal neurons in subiculum and CA1 of rat hippocampus. J Neurophysiol 84: 2398–2408, 2000. doi: 10.1152/jn.2000.84.5.2398. [DOI] [PubMed] [Google Scholar]
- 28.Soh H, Pant R, LoTurco JJ, Tzingounis AV. Conditional deletions of epilepsy-associated KCNQ2 and KCNQ3 channels from cerebral cortex cause differential effects on neuronal excitability. J Neurosci 34: 5311–5321, 2014. doi: 10.1523/JNEUROSCI.3919-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheung Y-Y, Yu H, Xu K, Zou B, Wu M, McManus OB, Li M, Lindsley CW, Hopskins CR. Discovery of a series of 2-phenyl-N-(2-(pyrrolidin-1-yl) phenyl) acetamides as novel molecular switches that modulate modes of Kv7. 2 (KCNQ2) channel pharmacology: Identification of (S)-2-phenyl-N-(2-(pyrrolidin-1-yl) phenyl) butanamide (ML252) as a potent, brain penetrant Kv7. 2 channel inhibitor. J Med Chem 55: 6975–6979, 2012. doi: 10.1021/jm300700v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cembrowski MS, Wang L, Lemire AL, Copeland M, DiLisio SF, Clements J, Spruston N. The subiculum is a patchwork of discrete subregions. eLife 7: e37701, 2018. doi: 10.7554/eLife.37701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tzingounis AV, Heidenreich M, Kharkovets T, Spitzmaul G, Jensen HS, Nicoll RA, Jentsch TJ. The KCNQ5 potassium channel mediates a component of the afterhyperpolarization current in mouse hippocampus. Proc Natl Acad Sci USA 107: 10232–10237, 2010. doi: 10.1073/pnas.1004644107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cembrowski MS, Spruston N. Heterogeneity within classical cell types is the rule: lessons from hippocampal pyramidal neurons. Nat Rev Neurosci 20: 193–204, 2019. doi: 10.1038/s41583-019-0125-5. [DOI] [PubMed] [Google Scholar]
- 33.Graves AR, Moore SJ, Bloss EB, Mensh BD, Kath WL, Spruston N. Hippocampal pyramidal neurons comprise two distinct cell types that are countermodulated by metabotropic receptors. Neuron 76: 776–789, 2012. doi: 10.1016/j.neuron.2012.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joksimovic SM, Eggan P, Izumi Y, Joksimovic SL, Tesic V, Dietz RM, Orfila JE, DiGruccio MR, Herson PS, Jevtovic-Todorovic V, Zorumski CF, Todorovic SM. The role of T‐type calcium channels in the subiculum: to burst or not to burst? J Physiol 595: 6327–6348, 2017. doi: 10.1113/JP274565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lauritano A, Moutton S, Longobardi E, Tran Mau-Them F, Laudati G, Nappi P, Soldovieri MV, Ambrosino P, Cataldi M, Jouan T, Lehalle D, Maurey H, Philippe C, Miceli F, Vitobello A, Taglialatela M. A novel homozygous KCNQ3 loss-of-function variant causes non-syndromic intellectual disability and neonatal-onset pharmacodependent epilepsy. Epilepsia Open 4: 464–475, 2019. doi: 10.1002/epi4.12353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Farhi SL, Parot VJ, Grama A, Yamagata M, Abdelfattah AS, Adam Y, Lou S, Kim JJ, Campbell RE, Cox DD, Cohen AE. Wide-area all-optical neurophysiology in acute brain slices. J Neurosci 39: 4889–4908, 2019. doi: 10.1523/JNEUROSCI.0168-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tzingounis AV, Nicoll RA. Contribution of KCNQ2 and KCNQ3 to the medium and slow afterhyperpolarization currents. Proc Natl Acad Sci USA 105: 19974–19979, 2008. doi: 10.1073/pnas.0810535105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stas JI, Bocksteins E, Jensen CS, Schmitt N, Snyders DJ. The anticonvulsant retigabine suppresses neuronal KV2-mediated currents. Sci Rep 6: 35080, 2016. doi: 10.1038/srep35080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peng H, Bian X-L, Ma F-C, Wang K-W. Pharmacological modulation of the voltage-gated neuronal Kv7/KCNQ/M-channel alters the intrinsic excitability and synaptic responses of pyramidal neurons in rat prefrontal cortex slices. Acta Pharmacol Sin 38: 1248–1256, 2017. doi: 10.1038/aps.2017.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lerche C, Scherer CR, Seebohm G, Derst C, Wei AD, Busch AE, Steinmeyer K. Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J Biol Chem 275: 22395–22400, 2000. doi: 10.1074/jbc.M002378200. [DOI] [PubMed] [Google Scholar]