

Keywords: breathing, motor neuron, plasticity, respiratory, spinal cord
Abstract
Moderate acute intermittent hypoxia (mAIH; 35–55 mmHg PaO2) elicits phrenic long-term facilitation (pLTF) by a mechanism that requires activation of Gq protein-coupled serotonin type 2 receptors, MEK/ERK MAP kinase, and NADPH oxidase activity and is constrained by cAMP-PKA signaling. In contrast, severe AIH (sAIH; 25–35 mmHg PaO2) elicits Gs protein-coupled adenosine type 2 A receptor-dependent pLTF. Another Gs protein-coupled receptor, serotonin 7 receptors, elicits phrenic motor facilitation (pMF) by a mechanism that requires exchange protein activated by cyclic AMP (EPAC) and phosphatidylinositol 3-kinase/Akt (PI3K/Akt) activation and is constrained by NADPH oxidase activity. Here, we tested the hypothesis that the same downstream signaling mechanisms giving rise to serotonin 7 (vs. serotonin 2) receptor-induced pMF underlie sAIH-induced pLTF. In anesthetized rats, sAIH-induced pLTF was compared after pretreatment with intrathecal (C4) injections of inhibitors for: 1) EPAC (ESI-05); 2) MEK/ERK (UO126); 3) PKA (KT-5720); 4) PI3K/Akt (PI828); and 5) NADPH oxidase (apocynin). In partial agreement with our hypothesis, sAIH-induced pLTF was abolished by ESI-05 and PI828 and marginally enhanced by apocynin but, surprisingly, was abolished by UO126 and attenuated by KT-5720. Mechanisms of sAIH-induced pLTF reflect elements of both Gq and Gs pathways to pMF, likely as a consequence of the complex, cross-talk interactions between them.
NEW & NOTEWORTHY Distinct mechanisms give rise to pLTF induced by moderate and severe AIH. We demonstrate that, unlike moderate AIH, severe AIH-induced pLTF requires EPAC and PI3K/Akt and is marginally constrained by NADPH oxidase activity. Surprisingly, sAIH-induced pLTF requires MEK/ERK activity similar to moderate AIH-induced pLTF and is reduced by PKA inhibition. We suggest sAIH-induced pLTF arises from complex interactions between dominant mechanisms characteristic of moderate versus severe AIH-induced pLTF.
INTRODUCTION
Plasticity is a prominent feature of neuromotor systems, including the neural system controlling breathing. A general term for plasticity in the phrenic motor system is phrenic motor facilitation (pMF), which can be elicited pharmacologically (1–5) or by exposure to acute intermittent hypoxia (AIH) (6–10). AIH elicits a specialized form of pMF known as phrenic long-term facilitation (pLTF) (7, 9, 11–15). pLTF is characterized by a persistent (and progressive) increase in the amplitude of integrated phrenic bursts lasting many hours post-AIH.
Phrenic motor neurons exhibit pMF in response to spinally injected drugs targeting serotonin (2, 4, 5), adenosine (1), or adrenergic receptors (3). However, Gq (e.g., 5-HT2A, 5-HT2B, and α-1) versus Gs (e.g., A2A and 5-HT7) protein-coupled receptors elicit pMF by distinct, but interacting, cellular mechanisms (1–3, 5, 16–21). Similar pMF is revealed with intrathecal injections of downstream kinase activators, including activators of protein kinase C (PKC; 22), exchange protein activated by cAMP (EPAC; 18, 23), or protein kinase A (PKA; 23). These pathways are named for the G protein canonically coupled to the initiating neurotransmitter receptor, particularly Gq (Q pathway) and Gs (S pathway) coupled receptors (6).
AIH-induced pLTF also results from distinct cellular mechanisms, and the predominant mechanism depends on the severity of hypoxemia within each hypoxic episode of the AIH protocol. Whereas spinal 5-HT2 receptors and the Q pathway predominate with moderate AIH (21, 24), spinal A2A receptors and the S pathway predominate when AIH consists of more severe hypoxic episodes (sAIH; 15).
The S and Q pathways are not independent but interact via powerful cross-talk inhibition (17–19, 25, 26). Subthreshold activation of the S pathway constrains moderate AIH-induced pLTF via A2A (27) and/or 5-HT7 receptor activation (28). An essential mediator of such S to Q cross-talk inhibition is cyclic AMP, acting via PKA (not EPAC) (18, 19, 26). Conversely, subthreshold Q pathway activation constrains sAIH-induced pLTF via 5-HT2A (26) or 5-HT2B receptor activation (19). Spinal 5-HT2A receptor activation inhibits 5-HT7 receptor-induced pMF (i.e., S pathway) and sAIH-induced pLTF by a mechanism that requires spinal PKCδ activity (26), whereas 5-HT2B receptors inhibit S pathway-induced pMF via NADPH oxidase activity (19). Although moderate AIH-induced pLTF requires NADPH oxidase activity and reactive oxygen species formation (29–31), the impact of NADPH oxidase inhibition on sAIH-induced pLTF has not been explored.
To date, most studies of the S pathway concern pMF induced via intrathecal drug injections (6, 19, 26), with little direct evidence concerning mechanisms contributing to sAIH-induced versus moderate AIH-induced pLTF. The major goal of this study was to investigate the role of enzymes known to play critical roles in S versus Q pathways to pMF, or their interactions. Specifically, we tested the hypothesis that spinal enzyme activities known to regulate the S pathway, including EPAC (18, 23) and phosphatidylinositol 3-kinase/Akt (PI3K/Akt) (1, 32), are necessary for sAIH-induced pLTF. We also tested the importance of an enzyme mediating S to Q pathway cross-talk inhibition: PKA (18, 28). We compared these results with outcomes following inhibition of an enzyme required for 5-HT2A receptor-dependent, moderate AIH-induced pLTF, MEK/ERK MAP kinases (21, 32), and another mediating Q to S pathway cross-talk inhibition, NADPH oxidase (26). Our results reveal a complex picture but do confirm S pathway predominance in sAIH-induced pLTF, while leaving other questions unanswered.
MATERIALS AND METHODS
Experimental Groups
Experiments were performed using adult (3–4 mo) male Sprague Dawley rats (Harlan colony 211, Houston, TX). The Animal Care and Use Committee at the University of Wisconsin-Madison approved all experimental procedures. Each rat group received one of two treatments: 1) severe acute intermittent hypoxia (sAIH; PaO2: 25–30 mmHg), or 2) sham normoxia (i.e., time controls without AIH). Further, all rat groups received intrathecal drug delivery as described in detail below (Neurophysiology Protocol).
Neurophysiological Preparation
Experimental procedures have been described previously in detail in multiple publications (e.g., 15, 32). In brief, rats were anesthetized with isoflurane, tracheotomized, and ventilated (Rodent Ventilator, Model 683; Harvard Apparatus, Holliston, MA; tidal volume ∼2.5 mL, frequency ∼70–80). Isoflurane anesthesia was maintained (3.5% in 50% O2, balance N2) throughout surgical procedures; rats were converted to urethane anesthesia over 15–20 min (1.8 g kg−1, i.v.) while slowly withdrawing isoflurane. Adequacy of anesthesia was verified before protocols commenced and immediately after completion by the lack of pressor responses or obvious respiratory neural responses to toe pinch with a hemostat. After conversion to urethane anesthesia, rats were given a continuous intravenous infusions to maintain body fluid and acid-base balance; infusions (1.5–6 mL/kg/h) consisted of a 1:2:0.13 mixture of 6% Hetastarch (in 0.9% sodium chloride), lactated Ringer’s solution, and 8.4% sodium bicarbonate.
Body temperature was assessed with a rectal thermometer (Fisher Scientific, Pittsburgh, PA) and maintained (37.5 ± 1°C) with a heated surgical table. To monitor end-tidal PaCO2 (PetCO2), a flow-through carbon dioxide analyzer was used with sufficient response time to measure PetCO2 in rats (Capnogard, Novametrix, Wallingford, CT). PetCO2 was maintained at ∼45 mmHg throughout the surgical preparation.
Rats were vagotomized bilaterally, and a polyethylene catheter (PE50 ID: 0.58 mm, OD: 0.965 mm; Intramedic, Maryland) was inserted into the right femoral artery to monitor blood pressure (Gould Pressure Transducer, P23ID) and draw samples for arterial blood gas analysis. These arterial blood samples were analyzed for partial pressures of O2 (PaO2) and CO2 (PaCO2) with a blood gas analyzer (ABL 800, Radiometer, Westlake, OH); blood gases were assessed during baseline, all three hypoxic episodes, and at 15, 30, 60, and 90 min post-AIH. PaCO2 was maintained within ± 1.5 mmHg of baseline levels by adjusting inspired CO2 and/or ventilator rate throughout the experiment. After the third hypoxic episode, baseline O2 levels were restored and then maintained for the duration of experiments.
The left phrenic nerves were isolated (dorsal approach), cut distally, desheathed, and covered with a saline-soaked cotton ball until placing the nerves on bipolar silver electrodes (see Neurophysiology Protocol below). Laminectomy was performed at cervical level 2 (C2) to enable intrathecal delivery of drugs. Once rats were converted to urethane anesthesia, a minimum of 1 h was allowed before protocols commenced.
Neurophysiology Protocol
To enable intrathecal drug delivery, a small incision was made in the dura at C2, and a soft silicone catheter (2 Fr; Access Technologies, Skokie, IL) was inserted caudally 3–4 mm until the tip rested approximately over the C4 segment to deliver drugs near the phrenic motor nucleus before sAIH [three 5-min episodes of isocapnic hypoxia (25–30 mmHg) separated by 5-min intervals of baseline oxygen levels (PaO2 ≥ 150 mmHg)]. The intrathecal catheter was attached to a 50-µL Hamilton syringe filled with drug or vehicle solutions as described below. The left phrenic nerve was submerged in mineral oil and placed on bipolar silver electrodes to record nerve activity. Neural signals were amplified (×10,000), band-pass filtered (300–10,000 Hz, Model 1800, A-M Systems, Carlsborg, WA), full-wave rectified, and integrated (Paynter filter, time constant, 50 ms, MA-821, CWE Inc., Ardmore, PA). Integrated nerve bursts were digitized (8 kHz) and analyzed using WINDAQ data acquisition system (DATAQ Instruments, Akron, OH). Rats were then paralyzed using pancuronium bromide for neuromuscular blockade (2.5 mg kg−1, i.v.). To begin protocols, the apneic CO2 threshold was determined by lowering PetCO2 until nerve activity ceased for approximately 1 min. The recruitment threshold was then determined by slowly increasing PetCO2 until nerve activity resumed (11). PetCO2 was raised ∼2 mmHg above the recruitment threshold, and ∼15 min were allowed to establish a stable baseline. This procedure assures comparable baseline phrenic nerve activities across individual rats, at least relative to the CO2 apneic/recruitment thresholds. Further, this procedure assures comparability with other studies concerning molecules contributing to or regulating phrenic motor plasticity.
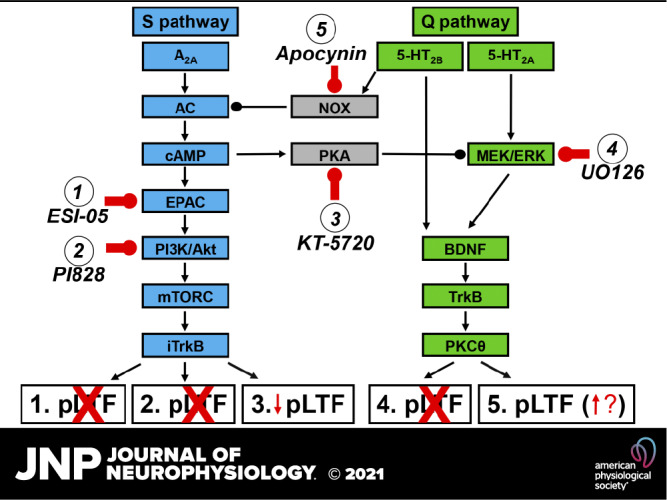
Rats were divided into groups to test the hypotheses that enzymes regulating the S pathway to pMF or mediating S to Q pathway cross-talk inhibition also regulate sAIH-induced pLTF (i.e., EPAC, PI3K/Akt, and PKA). As a contrast, we also tested an enzyme known to regulate the Q (not S) pathway to pMF (i.e., MEK/ERK and MAPK), and an enzyme mediating Q to S pathway cross-talk inhibition (i.e., NADPH oxidase or NOX). All drug or vehicle injections took place after a 15-min baseline was established, and were given in 2–3 µL boluses; all drug doses used were determined from previous studies (18, 28, 30, 32). Rat groups included: 1) vehicle for ESI-05 and PI828 [20% DMSO dissolved in saline, 10–12 µL, 20 min before sAIH; (18, 28) n = 3]; 2) ESI-05 [EPACi; 10 µL, 20 min before sAIH, 2 mM; BioLog Life Science Institute; (18) n = 5]; 3) ESI-05 time control (TC or no hypoxia; n = 2); 4) PI828 [2-(4-Morpholinyl)-8-(4-aminopheny)l-4H-1-benzopyran-4-one, PI3K/Akti; 12 µL, 20 min before sAIH, 100 µM; Tocris Bioscience; (32) n = 5]; 5) PI828 TC (n = 2); 6) vehicle for KT-5720 [20% DMSO in saline, 15 µL, 5 min before sAIH; (28) n = 7]; 7) KT-5720 [PKAi; 15 µL, 5 min before sAIH, 100 µM; Tocris Bioscience; (28) n = 8]; 8) vehicle TC (n = 3); 9) KT-5720 TC (n = 3); 10) vehicle for UO126 [20% DMSO dissolved in saline, 12 µL, 20 min before sAIH; (32) n = 5]; 11) UO126 [(1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene, MEK/ERKi; 12 µL, 20 min before sAIH, 100 µM; Promega; (32) n = 5]; 12) vehicle TC (n = 1); 13) UO126 TC (n = 2); 14) vehicle for apocynin [10% DMSO dissolved in artificial cerebral spinal fluid (aCSF), 12 µL, 20 min before sAIH; (30) n = 5]; 15) apocynin [NADPH oxidase inhibitor (NOXi); 12 µL, 20 min before sAIH, 600 µM; Sigma; (30) n = 5]; 16) vehicle TC (n = 2); and 17) apocynin TC (n = 2). Since there were no apparent differences in vehicle (with sAIH) or TC groups (no AIH), and multiple previous studies demonstrating minimal experimental drift in time control experiments, all vehicle and TC rats were grouped together in S pathway or Q pathway studies for statistical analyses (S: vehicle n = 10, TC n = 10; Q: vehicle n = 10, TC n = 7). In this way, we were able to assure that drugs or vehicle did not elicit unexpected pMF (i.e., a time-dependent drift in time control experiments) while minimizing animal use consistent with national standards for animal research. The targets for the respective drugs are depicted in a working (hypothetical) cellular model of AIH-induced phrenic motor plasticity in Fig. 1.
Figure 1.

Working model of sAIH-induced pLTF. pLTF can be regulated by Gq (5-HT2A/B) or Gs (A2A) protein-coupled metabotropic receptors (the Q and S pathways, respectively). Following severe AIH, we hypothesize that activation of A2A receptors activates adenyl cyclase (AC) and cAMP, with subsequent activation of EPAC, PI3K/Akt, mTORC, and iTrkB. Activation of EPAC drives PI3K activation, phosphorylating Akt to cause pLTF. In contrast, following moderate AIH, both 5-HT2 isoforms are activated, with 5-HT2A receptor activation increasing MEK/ERK activity. Both 5-HT2 isoforms then utilize downstream signaling via BDNF, TrkB and PKCθ, although this convergence has not been verified. We further hypothesized that the S pathway is constrained by NADPH oxidase (NOX), whereas the Q pathway is constrained by PKA activity. We intrathecally delivered the inhibitors to key enzymes in this working cellular model, including: 1) ESI-05 (targeting EPAC), 2) PI828 (targeting PI3K/Akt), 3) KT-5720 (targeting PKA), 4) UO126 (targeting MEK/ERK), and 5) apocynin (targeting NADPH oxidase). AIH, acute intermittent hypoxia; BDNF, brain-derived neurotrophic factor; EPAC, exchange protein activated by cyclic AMP; mTORC, mammalian target of rapamycin; PI3K/Akt, phosphatidylinositol 3-kinase/Akt; pLTF, phrenic long-term facilitation; sAIH, severe AIH.
Data Analyses
Integrated phrenic nerve burst amplitude was averaged over 1-min bins at each time (baseline, all three hypoxic episodes, and 15, 30, 60, and 90 min post-AIH) and normalized as a percent change from baseline. Respiratory frequency was expressed as a change from baseline (bursts per min). All statistical comparisons between treatment groups for nerve burst amplitude and frequency were made via two-way ANOVA with a repeated measures design (time post-AIH: 15, 30, 60, and 90 min; or hypoxic episodes for the short-term hypoxic response). A one-way ANOVA was used to compare nerve burst amplitude and frequency at 90 min posthypoxia. With statistically significant ANOVAs, individual comparisons were made using Tukey post hoc test (SigmaPlot v. 14.0; Systat Software Inc., San Jose, CA). Differences were considered significant if P < 0.05. Values are expressed as means ± 1 SE.
RESULTS
Blood Gases and Mean Arterial Pressures
During hypoxic episodes, arterial PCO2 (PaCO2) was successfully maintained within 1.5 mm Hg of its baseline value at all times in all groups (Table 1). Therefore, changes in integrated phrenic nerve burst amplitude following AIH cannot be attributed to differences in chemoreceptor feedback, which can influence the apparent magnitude of pLTF (11, 14, 33). PaO2 was successfully maintained within the target range for severe AIH (25–30 mmHg), and mean arterial pressure (MAP) did not differ within groups during hypoxic episodes (Table 1). As expected, PaO2 and mean arterial pressure differed among groups when AIH versus TCs were compared during hypoxic episodes (Table 1). In addition, significant differences were detected during hypoxic episodes for MAP when comparing rats treated with KT-5720 (PKA inhibitor) or apocynin (NADPH oxidase inhibitor) to rats treated with ESI-05 (EPAC inhibitor) or PI828 (PI3K/Akt inhibitor; HX1: P = 0.026 for apocynin vs. ESI-05, P = 0.035 for apocynin vs. PI828, P = 0.017 for KT-5720 vs. ESI-05, and P = 0.024 for KT-5720 vs. PI828; HX2: P = 0.043 for apocynin vs. ESI-05, P = 0.033 for apocynin vs. PI828, P = 0.020 for KT-5720 vs. ESI-05, and P = 0.014 for KT-5720 vs. PI828; HX3: P = 0.048 for apocynin vs. ESI-05, P = 0.022 for apocynin vs. PI828, P = 0.041 for KT-5720 vs. ESI-05, and P = 0.017 for KT-5720 vs. PI828; Table 1).
Table 1.
Arterial Pco2, Po2, and MAP during hypoxic episodes
| PaCO2, mmHg |
PaO2, mmHg |
MAP, mmHg |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| HX1 | HX2 | HX3 | HX1 | HX2 | HX3 | HX1 | HX2 | HX3 | |
| Spinal groups | |||||||||
| Vehicle 1 | 47.6 ± 1.5 | 48.0 ± 1.3 | 47.6 ± 1.3 | 30 ± 1a | 26 ± 1a | 27 ± 1a | 56 ± 7a | 51 ± 7a | 52 ± 7a |
| ESI-05 | 48.5 ± 0.9 | 50.6 ± 1.3 | 51.1 ± 0.8 | 28 ± 1a | 25 ± 1a | 27 ± 1a | 73 ± 5 | 67 ± 3 | 64 ± 4 |
| PI828 | 48.3 ± 2.9 | 47.8 ± 3.5 | 49.0 ± 2.5 | 27 ± 2a | 27 ± 1a | 28 ± 1a | 72 ± 4 | 68 ± 3 | 66 ± 3 |
| KT-5720 | 47.0 ± 0.9 | 45.0 ± 1.4 | 46.3 ± 1.5 | 29 ± 1a | 28 ± 1a | 27 ± 1a | 45 ± 3a,b,c | 39 ± 3a,b,c | 38 ± 3a,b,c |
| (–AIH) 1 | 47.7 ± 1.0 | 48.2 ± 0.8 | 47.7 ± 1.0 | 332 ± 9 | 338 ± 9 | 333 ± 8 | 90 ± 5 | 90 ± 4 | 90 ± 4 |
| Vehicle 2 | 45.9 ± 1.5 | 44.2 ± 1.4 | 44.1 ± 1.4 | 28 ± 1a | 27 ± 1a | 29 ± 1a | 54 ± 8a | 47 ± 6a | 47 ± 6a |
| UO126 | 49.3 ± 1.1 | 48.4 ± 1.3 | 48.3 ± 2.4 | 28 ± 2a | 27 ± 1a | 29 ± 2a | 61 ± 6 | 52 ± 6a | 52 ± 6a |
| Apocynin | 44.9 ± 2.1 | 46.1 ± 5.3 | 45.4 ± 2.1 | 27 ± 1a | 26 ± 1a | 25 ± 1a | 43 ± 2a,b,c | 39 ± 2a,b,c | 36 ± 1a,b,c |
| (–AIH) 2 | 44.9 ± 1.6 | 45.3 ± 1.6 | 46.1 ± 1.7 | 283 ± 35 | 285 ± 33 | 294 ± 33 | 86 ± 2 | 85 ± 2 | 85 ± 2 |
All values are expressed as means ± SE. HX1, HX2, and HX3 designate hypoxic episodes 1, 2, and 3, respectively. AIH, acute intermittent hypoxia; –AIH, without AIH; EPAC, exchange protein activated by cyclic AMP; ESI-05, EPAC inhibitor; KT-5720, PKA inhibitor; MAP, mean arterial pressure; PI3K/Akt, phosphatidylinositol 3-kinase/Akt; PI828, PI3K/Akt inhibitor; UO126, MEK/ERK inhibitor. Significant differences from: aappropriate time control group (-AIH), bESI-05, and cPI828. A two-way ANOVA with repeated measures design followed by a Tukey post hoc test was utilized. Differences were considered significant if P < 0.05. The n for each group is as follows: vehicle 1, n = 10; ESI-05, n = 5; PI828, n = 5; KT-5720, n = 8; (−AIH) 1, n = 10; vehicle 2, n = 10; UO126, n = 5; apocynin, n = 5; (−AIH) 2, n = 7.
No differences were detected when comparing PaCO2 during baseline or 90-min post-hypoxia; PaO2 remained above 150 mmHg at all times posthypoxia, and the only significant difference we observed was when comparing the second time control group at baseline versus 90-min post-AIH (P = 0.006; Table 2). For mean arterial pressure during baseline and at 90-min posthypoxia, we observed slight but significant differences within (both vehicle groups; P = 0.019 for vehicle 1, and P = 0.037 for vehicle 2 comparing baseline with 90-min post-AIH) and across groups [baseline: ESI-05 vs. apocynin, P = 0.047; 90-min posthypoxia: ESI-05 and PI828 vs. vehicle (P = 0.043 vs. ESI-05, and P = 0.004 vs. PI828), apocynin (P = 0.032 vs. ESI-05, and P = 0.004 vs. PI828), and KT-5720 (P = 0.045 vs. ESI-05, and P = 0.005 vs. PI828); Table 2]; changes in mean arterial pressure of <20 mmHg from baseline have minimal reported effect on respiratory activity in rats (11, 34). Thus, differential pLTF expression observed following drug treatments was not affected by differences in PaCO2, PaO2, or blood pressure regulation.
Table 2.
Arterial Pco2, Po2, and MAP during baseline and 90-min posthypoxia
| PaCO2, mmHg |
PaO2, mmHg |
MAP, mmHg |
||||
|---|---|---|---|---|---|---|
| Baseline | 90 Min | Baseline | 90 Min | Baseline | 90 Min | |
| Spinal groups | ||||||
| Vehicle 1 | 48.4 ± 1.1 | 48.7 ± 1.1 | 359 ± 10 | 329 ± 13 | 92 ± 5 | 80 ± 6a |
| ESI-05 | 49.0 ± 0.9 | 49.2 ± 1.0 | 338 ± 5 | 328 ± 6 | 110 ± 2c | 105 ± 5b,c,d |
| PI828 | 48.2 ± 2.7 | 48.1 ± 2.6 | 309 ± 39 | 312 ± 20 | 105 ± 4 | 110 ± 5b,c,d |
| KT-5720 | 47.3 ± 0.9 | 47.3 ± 0.9 | 353 ± 10 | 324 ± 6 | 88 ± 3 | 79 ± 4 |
| (–AIH) 1 | 48.2 ± 1.0 | 48.7 ± 1.0 | 329 ± 9 | 330 ± 12 | 90 ± 5 | 88 ± 4 |
| Vehicle 2 | 46.4 ± 1.1 | 46.7 ± 1.1 | 313 ± 20 | 323 ± 9 | 87 ± 4 | 76 ± 5a |
| UO126 | 48.3 ± 0.9 | 48.1 ± 1.2 | 294 ± 25 | 350 ± 14 | 93 ± 2 | 84 ± 4 |
| Apocynin | 45.2 ± 2.3 | 45.5 ± 2.3 | 284 ± 40 | 332 ± 26 | 82 ± 6 | 75 ± 8 |
| (–AIH) 2 | 46.4 ± 1.2 | 46.7 ± 1.0 | 283 ± 36 | 347 ± 23a | 87 ± 2 | 83 ± 4 |
Values are expressed as means ± SE. AIH, acute intermittent hypoxia; –AIH, without AIH; EPAC, exchange protein activated by cyclic AMP; ESI-05, EPAC inhibitor; KT-5720, PKA inhibitor; MAP, mean arterial pressure; PI3K/Akt, phosphatidylinositol 3-kinase/Akt; PI828, PI3K/Akt inhibitor; UO126, MEK/ERK inhibitor. Significant differences from: abaseline, bappropriate vehicle group, capocynin, and dKT-5720. A two-way ANOVA with repeated measures design followed by a Tukey post hoc test was utilized. Differences were considered significant if P < 0.05. The n for each group is as follows: vehicle 1, n = 10; ESI-05, n = 5; PI828, n = 5; KT-5720, n = 8; (−AIH) 1, n = 10; vehicle 2, n = 10; UO126, n = 5; apocynin, n = 5; (−AIH) 2, n = 7.
sAIH-Induced pLTF Is EPAC and PI3K/Akt Dependent
To test the hypothesis that enzymes required for S pathway-induced pMF are also required for sAIH-induced pLTF, we first studied sAIH-induced pLTF following spinal inhibition of EPAC (with ESI-05) or PI3K/Akt (with PI828). Representative phrenic neurograms during protocols for each treatment group are illustrated in Fig. 2, A–D. Whereas sAIH-induced pLTF was similar to previously reported values in vehicle-treated rats (15), significantly greater versus baseline (P < 0.001 at all times post-AIH), and versus time-controls (P = 0.003 at 15 min, and P < 0.001 at 30-, 60-, and 90-min post-AIH), pLTF was abolished with EPACi at 30 (P = 0.003), 60 (P < 0.001), and 90 min (P < 0.001) post-AIH, as well as PI3K/Akt inhibition at all times post-AIH (P = 0.008 at 15 min, P < 0.001 at 30-, 60-, and 90-min post-AIH) versus vehicle-treated rats (Fig. 2, A–C and E).
Figure 2.

pLTF is abolished following ESI-05 and PI828 treatment. A–D: representative traces of compressed, integrated phrenic nerve activity before and after sAIH following pretreatment with vehicle (A), ESI-05 (EPAC inhibitor, B), PI828 (PI3K/Akt inhibitor, C), and KT-5720 (PKA inhibitor, D). The dashed line in each trace represents baseline. E: phrenic burst amplitude (percent change from baseline) in vehicle (open diamonds)-, PKA inhibitor (filled squares)-, EPAC inhibitor (filled triangles)-, and PI3K/Akt inhibitor (filled circles)-treated rats after AIH, and time control (without AIH, gray diamonds)-treated rats. pLTF is significant in vehicle-treated rats versus EPACi-treated rats at 30-, 60-, and 90-min posthypoxia ($), and at all time-points post-AIH versus time control-treated rats (*), baseline (+), PKAi (#), and PI3K/Akti (&) (all P < 0.05). PKAi-treated rats showed significant pLTF versus baseline at 30-, 60-, and 90-min posthypoxia, and time control-treated rats at 90-min posthypoxia (all P < 0.05). F: frequency (change from baseline; bursts/min) in vehicle-, PKA inhibitor-, EPAC inhibitor-, and PI3K/Akt inhibitor-treated rats after AIH, and time control-treated rats. Small frequency LTF in vehicle-treated rats is observed at 30- and 60-min post-AIH versus baseline (+) (P < 0.05). PI3K/Akti-treated rats had decreased frequency LTF compared with baseline (+) at 15-min post-AIH (P < 0.05). A two-way ANOVA with repeated measures design was utilized, followed by a Tukey post hoc test. Values are means ± SE. AIH, acute intermittent hypoxia; EPAC, exchange protein activated by cyclic AMP; LTF, long-term facilitation; PI3K/Akt, phosphatidylinositol 3-kinase/Akt; pLTF, phrenic long-term facilitation; sAIH, severe AIH. The n for each group is as follows: vehicle 1, n = 10; ESI-05, n = 5; PI828, n = 5; KT-5720, n = 8; time control, n = 10.
In vehicle-treated rats, pLTF was 51 ± 15%, 58 ± 14%, 76 ± 9%, and 78 ± 9% at 15, 30, 60, and 90 min, respectively (Fig. 2E). Following EPACi, pLTF was 16 ± 6%, 11 ± 6%, 15 ± 6%, and 10 ± 5% at 15, 30, 60, and 90 min, respectively (Fig. 2E). PI3K/Akt inhibition decreased pLTF to 7 ± 5%, 1 ± 5%, 7 ± 7%, and 3 ± 7% at 15, 30, 60, and 90 min, respectively (Fig. 2E).
Frequency LTF was statistically significant, but small, as reported before (15, 35). Phrenic burst frequency in vehicle treated rats was significantly different from baseline at 30- (P = 0.006) and 60 (P = 0.003)-min post-AIH (4 ± 2 bursts/min at 30 and 60 min; Fig. 2F). Rats treated with PI3K/Akt inhibitor exhibited less frequency change from baseline at 15 min post-AIH only (−5 ± 3 bursts/min; P = 0.017; Fig. 2F) and was unchanged at any time in rats after EPACi (P > 0.05; Fig. 2F).
PKA Inhibition Attenuates sAIH-Induced pLTF
To test the hypothesis that PKA, a protein activated by cAMP but known for its role in S to Q cross-talk inhibition (18, 23), is not necessary for sAIH-induced pLTF, we studied sAIH-induced pLTF following spinal PKA inhibition (with KT-5720). A typical phrenic neurogram during an experimental protocol is illustrated in Fig. 2. To our surprise, sAIH-induced pLTF was decreased but not abolished by PKA inhibition [20 ± 10%, 26 ± 12%, 33 ± 11%, and 34 ± 9% at 15, 30, 60, and 90 min, respectively; vs. vehicle-treated rats at all times post-AIH (P = 0.049 at 15, P = 0.033 at 30, P = 0.002 at 60, and P = 0.001 at 90 min); vs. baseline at 30 (P = 0.004), 60 (P < 0.001), and 90 (P < 0.001) min; and vs. time control rats at 90 min (P = 0.028); Fig. 2, D and E]. PKAi had not effect on frequency at any time (Fig. 2E).
sAIH-Induced pLTF Is MEK/ERK Dependent
To test the hypothesis an enzyme required for Q (not S) pathway pMF is unnecessary for sAIH-induced pLTF, we studied sAIH-induced pLTF following spinal MEK/ERK inhibition (with UO126). Contrary to our hypothesis, sAIH-induced pLTF was abolished by MEK/ERK inhibition [10 ± 3%, 8 ± 10%, 15 ± 8%, and 17 ± 4% at 15, 30, 60, and 90 min, respectively; vs. vehicle-treated rats at 60 (P = 0.006) and 90 min (P = 0.005); Fig. 3A, B, and D]. After MEK/ERK inhibition, sAIH-induced frequency LTF was −8 ± 7, −5 ± 6, −1 ± 2, and −1 ± 1 at 15, 30, 60, and 90 min, respectively, in rats treated with the MEK/ERK inhibitor (Fig. 3E). Frequency changes after MEK/ERK inhibition were significantly different from baseline and time control rats at 15-min post-AIH only (P = 0.007 vs. baseline; P = 0.008 vs. time control), and versus vehicle-treated rats at 15- (P = 0.016) and 30 (P = 0.048)-min post-AIH (Fig. 3E).
Figure 3.

pLTF is abolished following UO126, but marginally constrained by apocynin treatment. A–C: representative traces of compressed, integrated phrenic nerve activity before and after sAIH following pretreatment with vehicle (A), UO126 (MEK/ERK inhibitor, B), and apocynin (NADPH oxidase or NOX inhibitor, C). The dashed line in each trace represents baseline. D: phrenic burst amplitude (percent change from baseline) in vehicle (open diamonds)-, NOX inhibitor (filled squares)-, and MEK/ERK inhibitor (filled triangles)-treated rats after AIH, and time control (without AIH, gray diamonds)-treated rats. pLTF is significant in vehicle-treated rats at all time-points post-AIH versus time control-treated rats (*) and baseline (+), and versus MEK/ERKi-treated rats at 60- and 90-min posthypoxia ($) (all P < 0.05). NOXi-treated rats showed significant pLTF versus baseline, time control-treated rats, and MEK/ERKi-treated rats at all time-points post-AIH (all P < 0.05). E: Frequency (change from baseline; bursts/min) in vehicle-, NOX inhibitor-, and MEK/ERK inhibitor-treated rats after AIH, and time control-treated rats. Small frequency LTF in NOXi-treated rats is observed at all time points post-AIH versus MEK/ERKi-treated rats ($), but only at 30- and 90-min post-AIH versus baseline (+) and time control-treated rats (*) (P < 0.05). MEK/ERKi-treated rats had decreased frequency LTF compared with time control-treated rats (*) and baseline (+) at 15-min post-AIH and compared with vehicle-treated rats at 15- and 30-min post-AIH (#) (all P < 0.05). A two-way ANOVA with repeated measures design followed by a Tukey post hoc test was utilized. Values are means ± SE. AIH, acute intermittent hypoxia; LTF, long-term facilitation; PI3K/Akt, phosphatidylinositol 3-kinase/Akt; pLTF, phrenic long-term facilitation; sAIH, severe AIH. The n for each group is as follows: vehicle 2, n = 10; UO126, n = 5; apocynin, n = 5; time control, n = 7.
sAIH-Induced pLTF Is Marginally Constrained by NADPH Oxidase Activity
To test the hypothesis an enzyme shown to inhibit S pathway-induced pMF also inhibits sAIH-induced pLTF, we studied sAIH-induced pLTF following spinal NADPH oxidase inhibition with apocynin. Typical phrenic neurograms during experimental protocols for each treatment group are illustrated in Fig. 3, A–C. Vehicle-treated rats exhibited similar sAIH-induced pLTF as in the first vehicle-treated group of this study (Fig. 2), and as observed in vehicle-treated rats in a previous publication (15); pLTF was 51 ± 10%, 53 ± 8%, 76 ± 9%, and 80 ± 8% at 15, 30, 60, and 90 min, respectively [P < 0.001 at all time points vs. baseline, and rats treated as time-controls at all time-points post-AIH (P = 0.017 at 15, P = 0.007 at 30, and P < 0.001 at 60 and 90 min); Fig. 3, A and D]. As predicted, sAIH-induced pLTF was present and increased following NADPH oxidase inhibition (pLTF was 94 ± 49%, 85 ± 35%, 105 ± 31%, and 118 ± 32% at 15, 30, 60, and 90 min, respectively; P < 0.001 at all time points vs. baseline and rats treated as time controls; P = 0.001 at 15, P = 0.003 at 30, and P < 0.001 at 60 and 90 min vs. rats treated with the MEK/ERK inhibitor; and P = 0.089 at 15, P = 0.271 at 30, P = 0.386 at 60, and P = 0.156 at 90 min vs. vehicle-treated rats; Fig. 3, A-D).
Again, frequency LTF was small and, consistent with the first set of studies (Fig. 2); frequency in NADPH oxidase inhibitor treated rats was significantly elevated from rats treated with the MEK/ERK inhibitor at all time points (P < 0.001 at 15-min and 30-min, P = 0.040 at 60-min, and P = 0.014 at 90-min posthypoxia), from baseline as well as versus time control rats at 30- (P = 0.022 vs. baseline; P = 0.036 vs. time control) and 90 (P = 0.023 vs. baseline; P = 0.006 vs. time control)-min post-AIH (Fig. 3E). Frequency was −1 ± 1, 2 ± 1, 1 ± 1, and 3 ± 1 bursts/min at 15, 30, 60, and 90 min, respectively in vehicle-treated rats, and 4 ± 2, 8 ± 2, 7 ± 2, and 8 ± 1 at 15, 30, 60 and 90 min, respectively in rats treated with the NOXi (Fig. 3E).
Summary of Effects on sAIH-Induced pLTF
In summary, at 90-min post-AIH, apocynin-treated rats had greater nerve burst amplitude LTF versus rats treated with inhibitors for PI3K/Akt, EPAC, PKA, and MEK/ERK [all P < 0.001; marginally significant for apocynin vs. vehicle-treated rats (P = 0.057; Fig. 4A), and greater nerve burst frequency LTF vs. rats treated with inhibitors for PI3K/Akt (P = 0.009), PKA (P = 0.022), and MEK/ERK (P = 0.004) (Fig. 4B)]. Combined vehicle-treated rats exposed to sAIH (Figs. 2 and 3, n = 20) had significantly larger nerve burst amplitudes versus rats treated as time controls (i.e., no AIH; combined group from Figs. 2 and 3, n = 17; P < 0.001), or rats pretreated with inhibitors of EPAC (P < 0.001), PI3K/Akt (P < 0.001), PKA (P = 0.002), or MEK/ERK MAPK (P < 0.001) (P < 0.05; Fig. 4A).
Figure 4.

Direct comparison of the change in burst amplitude (percent baseline) and burst frequency (bursts/min) of the phrenic nerve following sAIH at 90-min posthypoxia. A and B: change in phrenic burst amplitude (A) or burst frequency (B) following pretreatment with vehicle, EPAC inhibitor (EPACi), PI3K/Akt inhibitor (PI3K/Akti), PKA inhibitor (PKAi), MEK/ERK inhibitor (MEK/ERKi), NADPH oxidase inhibitor (NOXi), or rats treated as time controls. A: phrenic amplitude after AIH at 90-min posthypoxia is significantly higher in vehicle-treated rats and NOXi rats compared with all other groups B: phrenic burst frequency after AIH at 90-min posthypoxia is significantly higher in NOXi rats compared time control-treated rats, PKAi-treated rats, MEK/ERKi-treated rats, and PI3K/Akti-treated rats. * vs. time control-treated rats, < vs. PKAi-treated rats, # vs. vehicle-treated rats, @ vs. MEK/ERKi-treated rats, $ vs. EPACi-treated rats, and & vs. PI3K/Akti-treated rats; all P < 0.05. A one-way ANOVA followed by a Tukey post hoc test was utilized; values are means ± SE. AIH, acute intermittent hypoxia; EPAC, exchange protein activated by cyclic AMP; PI3K/Akt, phosphatidylinositol 3-kinase/Akt; sAIH, severe AIH; TC, time control.
Drug Effects on Progressive Augmentation of the Short-Term Hypoxic Phrenic Response
Progressive augmentation is a progressive increase in the magnitude of the short-term hypoxic ventilatory (or phrenic) response from the initial to the final episode of an intermittent hypoxia protocol and has been previously described following sAIH (15, 36, 37). Here, we verified that sAIH-treated rats exhibit progressive augmentation (Fig. 5). Specifically, progressive augmentation was present in both vehicle-treated rat groups (vehicle 1: HX1 vs. HX2 and HX3, P < 0.001, and HX2 vs. HX3, P = 0.025; vehicle 2: HX1 vs. HX2 and HX3, P < 0.001, and HX2 vs. HX3, P = 0.001), PKA inhibitor-treated rats (HX1 vs. HX2, P = 0.012, and HX1 vs. HX3, P < 0.001), PI3K/Akt inhibitor-treated rats (HX1 vs. HX2, P = 0.017, and HX1 vs. HX3, P < 0.001), EPAC inhibitor-treated rats (HX1 vs. HX2, P = 0.004, and HX1 vs. HX3, P < 0.001), NADPH oxidase inhibitor-treated rats (HX1 vs. HX3, P = 0.038), and MEK/ERK inhibitor-treated rats (HX1 vs. HX3, P = 0.007; Fig. 5). All episodes were significantly different from time control rats (HX1: vehicle 1, P < 0.001; PKA inhibitor, P < 0.001; PI3K/Akt inhibitor, P = 0.008; EPAC inhibitor, P = 0.009; vehicle 2, P = 0.030; NADPH oxidase inhibitor, P = 0.004; MEK/ERK inhibitor, P = 0.049; HX2: vehicle 1, P < 0.001; PKA inhibitor, P < 0.001; PI3K/Akti, P = 0.001; EPAC inhibitor, P < 0.001; vehicle 2, P = 0.027; NADPH oxidase inhibitor, P = 0.003; MEK/ERK inhibitor, P = 0.048; HX3: vehicle 1, P < 0.001; PKA inhibitor, P < 0.001; PI3K/Akt inhibitor, P < 0.001; EPAC inhibitor, P < 0.001; vehicle 2, P = 0.012; NADPH oxidase inhibitor, P = 0.003; MEK/ERK inhibitor, P = 0.039; Fig. 5). In time control rats without AIH, there were no detectable increases in phrenic activity at times corresponding to hypoxic episodes (P > 0.05; Fig. 5).
Figure 5.

Short-term hypoxic phrenic response. A and B: progressive augmentation of the short-term hypoxic phrenic response was observed in all sAIH-treated rats including vehicle-, EPAC inhibitor (EPACi)-, PI3K/Akt inhibitor (PI3K/Akti)-, PKA inhibitor-, MEK/ERK inhibitor (MEK/ERKi)-, and NADPH oxidase inhibitor (NOXi)-treated rats. 1, 2, and 3 represent hypoxic episodes for sAIH-treated rats or normoxic episodes for time control-treated rats. In A and B, the hypoxic response progressively increases with each episode (* indicates significantly different than 1; and + indicated significantly different from 3; both P < 0.05). There were no changes in time control-treated rats, and all treatment groups were significantly different from time control-treated rats (P < 0.05). A two-way ANOVA with repeated measures design followed by a Tukey post hoc test was utilized; values are means ± SE. AIH, acute intermittent hypoxia; EPAC, exchange protein activated by cyclic AMP; PI3K/Akt, phosphatidylinositol 3-kinase/Akt; sAIH, severe AIH; TC, time control.
DISCUSSION
We demonstrate that enzymes necessary for the S pathway to pMF (defined by spinal receptor agonist or enzyme activators) also underlie sAIH-induced pLTF. Specifically, spinal EPAC and PI3K/Akt inhibition blocked sAIH-induced pLTF. Further, inhibition of NADPH oxidase, an enzyme regulating the S pathway via cross-talk inhibition, augments sAIH-induced pLTF as predicted. On the other hand, the picture is complex, since inhibition of a divergent signaling pathway (PKA), or an enzyme required for the Q pathway, but not the S pathway (MEK/ERK MAPK), either impaired (PKAi) or abolished (MEK/ERKi) sAIH-induced pLTF. We cannot explain these surprising findings at this time. However, AIH elicits multiple mechanisms that compete with one another in regulating phrenic motor plasticity (6, 16, 25, 38). The data presented here point out that we have a lot to learn concerning the regulation of phrenic motor plasticity but confirm that the severity of hypoxia during AIH is a key factor in its regulation.
Progressive Augmentation Was Not Affected by Any Drug Treatment
Unlike moderate AIH, sAIH elicits consistent progressive augmentation of the short-term hypoxic phrenic response [(15); Fig. 5, this study]. The mechanism of progressive augmentation remains unknown, but it is not dependent on serotonergic receptor activation (37, 39). We now show that sAIH-induced progressive augmentation does not require EPAC, PI3K/Akt, PKA, MEK/ERK, or NADPH oxidase activity (Fig. 5). The still unknown mechanism(s) of progressive augmentation during sAIH requires further investigation.
sAIH-Induced pLTF Requires Spinal EPAC and PI3K/Akt Activity
Unlike moderate AIH, which is serotonin-dependent and adenosine constrained (27), sAIH-induced pLTF is A2A receptor dependent (15). However, signaling mechanisms downstream from A2A receptors have only been inferred from studies of pMF following intraspinal injections of pharmacological agents and/or siRNAs following A2A (e.g., 1, 20) or 5-HT7 receptor agonists (2, 18; see Fig. 1). This is the first study verifying that two key downstream signaling kinases necessary for the S pathway to pMF, EPAC and PI3K/Akt, are also required for sAIH-induced pLTF (Figs. 2 and 4). The requirement for EPAC signaling is consistent with reports that EPAC, not PKA inhibition, abolishes 5-HT7 receptor-induced pMF (18), whereas downstream EPAC signaling likely requires PI3K/Akt (1) and mammalian target of rapamycin (mTOR), which is indirectly activated by PI3K/Akt (40–43) and is known to be required for sAIH-induced pLTF (44).
PKA Regulates sAIH-Induced pLTF
PKA is not required for 5HT7 receptor-induced pMF (18) but does constrain moderate AIH-induced pLTF (23, 28). On the other hand, paradoxically, PKA activation alone is sufficient to elicit pMF by an unknown mechanism (23). Here, we report the unanticipated finding that spinal PKA inhibition reduces, but does not abolish, sAIH-induced pLTF (Figs. 2 and 4). Thus, 1) PKA may play an unknown role in enabling sAIH-induced pLTF, but not 5-HT2A receptor-induced pMF (23); 2) A2A and 5-HT7 receptors differ in their downstream signaling, similar to 5-HT2A versus 5-HT2B receptors (21); and/or 3) PKA-dependent effects are “dose-dependent,” with higher PKA activity capable of eliciting pMF independently from the S or Q pathways (23).
NADPH Oxidase Activity Marginally Constrains sAIH-Induced pLTF
Moderate AIH-induced pLTF (i.e., predominant Q pathway) requires NADPH oxidase activity and reactive oxygen species formation (30, 31). Similarly, 5-HT2B, but not 5-HT2A receptor-induced pMF requires NADPH oxidase activity (5). On the flip side, 5-HT2B receptors constrain 5-HT7 receptor-induced pMF via NADPH oxidase activity (19). It has not been shown that similar, NADPH oxidase activity-dependent cross-talk inhibition constrains sAIH-induced pLTF.
Complex relationships such as the seeming dual role of NADPH oxidase in regulating phrenic motor plasticity must be interpreted in the context of the interactive, cellular network regulating phrenic motor plasticity. Our observation that NOX inhibition marginally increases sAIH-induced pLTF is reminiscent of our prior report that NADPH-dependent ROS formation underlies cross-talk inhibition from 5-HT2B to 5-HT7 receptors (19); similar S to Q cross-talk inhibition between A2A receptor-induced pMF could account for the tendency of apocynin to increase pLTF in this study (Figs. 2 and 4). On the other hand, NADPH oxidase activity is necessary for 5-HT2B receptor-induced pMF. The parsimonious explanation for this bidirectional (and seemingly paradoxical relationship) is that 5-HT2B receptors stimulate NADPH oxidase activity, suppressing adenyl cyclase activity (45, 46), thereby minimizing cAMP formation and PKA activity. It follows that NOX inhibition would reverse these effects, increasing PKA activity and suppressing the Q pathway to pMF. In this way, NOX inhibition or ROS scavenging would block pMF elicited by intraspinal serotonin (29), 5-HT2B receptor activation (5), and moderate AIH (30).
sAIH-Induced pLTF Requires Spinal MEK/ERK MAPK Activity
Although spinal MEK/ERK inhibition with UO126 blocks moderate AIH-induced pLTF (32), we did not anticipate that it would also block sAIH-induced pLTF (Figs. 3 and 4). We can only speculate concerning mechanisms of MEK/ERK involvement in sAIH-induced pLTF at this time. However, EPAC enhances hippocampal long-term potentiation by a mechanism that also requires ERK MAPK activity (47). Our data suggest that MEK/ERK activity is necessary for both Q and S pathway-dependent pMF and pLTF.
Possible MEK/ERK involvement in the S pathway is suggested by reports that ERK both activates and inactivates different phosphodiesterase-4 isoforms (48). Thus, it is possible that ERK enables sAIH-induced pLTF by indirect regulation of cAMP formation by, for example, attenuating phosphodiesterase-4 activity. ERK phosphorylation inactivates long phosphodiesterase-4 isoforms, which would increase cAMP levels (48–51), driving PKA and EPAC activation. In the context of this study, ERK inactivation of long phosphodiesterase 4 isoforms would drive the S pathway to pMF and sAIH-induced pLTF. Consequently, MEK/ERK inhibition would reduce cAMP levels, EPAC activation, and the driving force for S pathway-dependent pMF and pLTF. Even reduced cAMP levels may still be sufficient to activate PKA and suppress 5-HT2 receptor-induced pLTF expected with sAIH since cAMP levels needed to activate PKA (and cross-talk inhibition) are well below those required for EPAC and S pathway activation [see (23)]. Possibilities concerning ERK-phosphodiesterase-4 interactions in regulating sAIH-induced pLTF warrant further investigation.
Significance and Conclusions
In conclusion, the S pathway to pMF is the predominant driver of sAIH-induced pLTF since this A2A receptor-dependent form of phrenic motor plasticity requires EPAC and PI3K/Akt activity, similar to 5-HT7 receptor-induced pMF. Further, NADPH oxidase, a cross-talk inhibitor of 5-HT7 receptor-induced pMF, marginally constrains sAIH-induced pLTF, presumably via ROS formation. On the other hand, surprising observations that MEK/ERK activity is necessary for, and PKA activity modulates, sAIH-induced pLTF suggest there is more to learn about this important model of phrenic motor plasticity. Although spinal PKA activity is sufficient to elicit pMF in rats (23), mechanisms of this plasticity are unknown. We speculate that the MEK/ERK pathway suppresses phosphodiesterases, enabling adequate cAMP formation and accumulation to activate the high-threshold cAMP-activated kinase, EPAC. Thus, MEK/ERK inhibition would undermine the ability to initiate the S pathway to phrenic motor facilitation.
Although the complex, interacting mechanisms of phrenic motor plasticity are difficult to explain, they may offer physiological advantages, such as enabling appropriate responses to: 1) physiological challenges that differ in severity (e.g., mild to severe hypoxia) or duration (acute through lifelong), and/or 2) the ability to express emergent properties such as pattern sensitivity (25) and/or accumulation of functional benefits (i.e., metaplasticity; 17, 52). This complexity also offers potential molecular targets, as we attempt to optimize the therapeutic potential of intermittent hypoxia to elicit meaning functional recovery of breathing ability and nonrespiratory motor behaviors in clinical disorders such as spinal cord injury and motor neuron diseases (8, 53–55).
GRANTS
This work was supported by the Francis Families Foundation (N. L. Nichols), NIH K99/R00 HL119606 (N. L. Nichols), NIH R01 HL080209 (G. S. Mitchell) and R01 HL148030 (G. S. Mitchell).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.L.N. and G.S.M. conceived and designed research; N.L.N. performed experiments; N.L.N. analyzed data; N.L.N. and G.S.M. interpreted results of experiments; N.L.N. prepared figures; N.L.N. drafted manuscript; N.L.N. and G.S.M. edited and revised manuscript; N.L.N. and G.S.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Kalen Nichols for assistance with blood-gas analysis and Safraaz Mahammed for custom-designed software used for data analysis.
Present addresses: N. L. Nichols, Department of Biomedical Sciences, University of Missouri, Columbia, Missouri; G. S. Mitchell, Breathing Research and Therapeutics Center, Department of Physical Therapy and McKnight Brain Institute, University of Florida, Gainesville, Florida.
REFERENCES
- 1.Golder FJ, Ranganathan L, Satriotomo I, Hoffman M, Lovett-Barr MR, Watters JJ, Baker-Herman TL, Mitchell GS. Spinal adenosine A2a receptor activation elicits long-lasting phrenic motor facilitation. J Neurosci 28: 2033–2042, 2008. doi: 10.1523/JNEUROSCI.3570-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoffman MS, Mitchell GS. Spinal 5-HT7 receptor activation induces long-lasting phrenic motor facilitation. J Physiol 589: 1397–1407, 2011. doi: 10.1113/jphysiol.2010.201657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huxtable AG, MacFarlane PM, Vinit S, Nichols NL, Dale EA, Mitchell GS. Adrenergic alpha1 receptor activation is sufficient, but not necessary for phrenic long-term facilitation. J Appl Physiol (1985) 116: 1345–1352, 2014. doi: 10.1152/japplphysiol.00904.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacFarlane PM, Mitchell GS. Episodic spinal serotonin receptor activation elicits long-lasting phrenic motor facilitation by an NADPH oxidase-dependent mechanism. J Physiol 587: 5469–5481, 2009. doi: 10.1113/jphysiol.2009.176982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacFarlane PM, Vinit S, Mitchell GS. Serotonin 2A and 2B receptor-induced phrenic motor facilitation: differential requirement for spinal NADPH oxidase activity. Neuroscience 178: 45–55, 2011. doi: 10.1016/j.neuroscience.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dale-Nagle EA, Hoffman MS, MacFarlane PM, Mitchell GS. Multiple pathways to long-lasting phrenic motor facilitation. Adv Exp Med Biol 669: 225–230, 2010. doi: 10.1007/978-1-4419-5692-7_45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci 26: 239–266, 2003. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitchell GS. Respiratory plasticity following intermittent hypoxia: a guide for novel therapeutic approaches to ventilatory control disorders. In: Genetic Basis for Respiratory Control Disorders, edited by Gaultier C.New York: Springer, 2007, p. 291–306. [Google Scholar]
- 9.Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB Jr.. Invited review: intermittent hypoxia and respiratory plasticity. J Appl Physiol (1985) 90: 2466–2475, 2001. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- 10.Mitchell GS, Johnson SM. Neuroplasticity in respiratory motor control. J Appl Physiol (1985) 94: 358–374, 2003. doi: 10.1152/japplphysiol.00523.2002. [DOI] [PubMed] [Google Scholar]
- 11.Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol 104: 251–260, 1996. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- 12.Baker TL, Mitchell GS. Episodic but not continuous hypoxia elicits long-term facilitation of phrenic motor output in rats. J Physiol 529: 215–219, 2000. doi: 10.1111/j.1469-7793.2000.00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuller DD, Bach KB, Baker TL, Kinkead R, Mitchell GS. Long term facilitation of phrenic motor output. Respir Physiol 121: 135–146, 2000. doi: 10.1016/s0034-5687(00)00124-9. [DOI] [PubMed] [Google Scholar]
- 14.Hayashi F, Coles SK, Bach KB, Mitchell GS, McCrimmon DR. Time-dependent phrenic nerve responses to carotid afferent activation: intact vs. decerebellate rats. Am J Physiol Regul Integr Comp Physiol 265: R811–R819, 1993. doi: 10.1152/ajpregu.1993.265.4.R811. [DOI] [PubMed] [Google Scholar]
- 15.Nichols NL, Dale EA, Mitchell GS. Severe acute intermittent hypoxia elicits phrenic long-term facilitation by a novel adenosine-dependent mechanism. J Appl Physiol (1985) 112: 1678–1688, 2012. doi: 10.1152/japplphysiol.00060.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Devinney MJ, Huxtable AG, Nichols NL, Mitchell GS. Hypoxia-induced phrenic long-term facilitation: emergent properties. Ann N Y Acad Sci 1279: 143–153, 2013. doi: 10.1111/nyas.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fields DP, Mitchell GS. Spinal metaplasticity in respiratory motor control. Front Neural Circuits 9: 2, 2015. doi: 10.3389/fncir.2015.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fields DP, Springborn SR, Mitchell GS. Spinal 5-HT7 receptors induce phrenic motor facilitation via EPAC-mTORC1 signaling. J Neurophysiol 114: 2015–2022, 2015. doi: 10.1152/jn.00374.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perim RR, Fields DP, Mitchell GS. Cross-talk inhibition between 5-HT2B and 5-HT7 receptors in phrenic motor facilitation via NADPH oxidase and PKA. Am J Physiol Regul Integr Comp Physiol 314: R709–R715, 2018. doi: 10.1152/ajpregu.00393.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seven YB, Perim RR, Hobson OR, Simon AK, Tadjalli A, Mitchell GS. Phrenic motor neuron adenosine 2A receptors elicit phrenic motor facilitation. J Physiol 596: 1501–1512, 2018. doi: 10.1113/JP275462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tadjalli A, Mitchell GS. Cervical spinal 5-HT2A and 5-HT2B receptors are both necessary for moderate acute intermittent hypoxia-induced phrenic long-term facilitation. J Appl Physiol (1985) 127: 432–443, 2019. doi: 10.1152/japplphysiol.01113.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devinney MJ, Fields DP, Huxtable AG, Peterson TJ, Dale EA, Mitchell GS. Phrenic long-term facilitation requires PKCtheta activity within phrenic motor neurons. J Neurosci 35: 8107–8117, 2015. doi: 10.1523/JNEUROSCI.5086-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fields DP, Mitchell GS. Divergent cAMP signaling differentially regulates serotonin-induced spinal motor plasticity. Neuropharmacology 113: 82–88, 2017. doi: 10.1016/j.neuropharm.2016.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuller DD, Zabka AG, Baker TL, Mitchell GS. Phrenic long-term facilitation requires 5-HT receptor activation during but not following episodic hypoxia. J Appl Physiol (1985) 90: 2001–2006, 2001. doi: 10.1152/jappl.2001.90.5.2001. [DOI] [PubMed] [Google Scholar]
- 25.Devinney MJ, Nichols NL, Mitchell GS. Sustained hypoxia elicits competing spinal mechanisms of phrenic motor facilitation. J Neurosci 36: 7877–7885, 2016. doi: 10.1523/JNEUROSCI.4122-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perim RR, Fields DP, Mitchell GS. Protein kinase Cdelta constrains the S-pathway to phrenic motor facilitation elicited by spinal 5-HT7 receptors or severe acute intermittent hypoxia. J Physiol 597: 481–498, 2019. doi: 10.1113/JP276731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffman MS, Golder FJ, Mahamed S, Mitchell GS. Spinal adenosine A2(A) receptor inhibition enhances phrenic long term facilitation following acute intermittent hypoxia. J Physiol 588: 255–266, 2010. doi: 10.1113/jphysiol.2009.180075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoffman MS, Mitchell GS. Spinal 5-HT7 receptors and protein kinase A constrain intermittent hypoxia-induced phrenic long-term facilitation. Neuroscience 250: 632–643, 2013. doi: 10.1016/j.neuroscience.2013.06.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacFarlane PM, Mitchell GS. Respiratory long-term facilitation following intermittent hypoxia requires reactive oxygen species formation. Neuroscience 152: 189–197, 2008. doi: 10.1016/j.neuroscience.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacFarlane PM, Satriotomo I, Windelborn JA, Mitchell GS. NADPH oxidase activity is necessary for acute intermittent hypoxia-induced phrenic long-term facilitation. J Physiol 587: 1931–1942, 2009. doi: 10.1113/jphysiol.2008.165597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacFarlane PM, Wilkerson JE, Lovett-Barr MR, Mitchell GS. Reactive oxygen species and respiratory plasticity following intermittent hypoxia. Respir Physiol Neurobiol 164: 263–271, 2008. doi: 10.1016/j.resp.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoffman MS, Nichols NL, Macfarlane PM, Mitchell GS. Phrenic long-term facilitation after acute intermittent hypoxia requires spinal ERK activation but not TrkB synthesis. J Appl Physiol (1985) 113: 1184–1193, 2012. doi: 10.1152/japplphysiol.00098.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olson EB Jr, Bohne CJ, Dwinell MR, Podolsky A, Vidruk EH, Fuller DD, Powell FL, Mitchel GS. Ventilatory long-term facilitation in unanesthetized rats. J Appl Physiol (1985) 91: 709–716, 2001. doi: 10.1152/jappl.2001.91.2.709. [DOI] [PubMed] [Google Scholar]
- 34.Walker JKL, Jennings DB. Metabolic change induced by phenylephrine infusion stimulates ventilation, despite increased blood pressure, in conscious rats. FASEB J 9: A565, 1995. [Google Scholar]
- 35.Baker-Herman TL, Mitchell GS. Determinants of frequency long-term facilitation following acute intermittent hypoxia in vagotomized rats. Respir Physiol Neurobiol 162: 8–17, 2008. doi: 10.1016/j.resp.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mateika JH, Narwani G. Intermittent hypoxia and respiratory plasticity in humans and other animals: does exposure to intermittent hypoxia promote or mitigate sleep apnoea? Exp Physiol 94: 279–296, 2009. doi: 10.1113/expphysiol.2008.045153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Powell FL, Milsom WK, Mitchell GS. Time domains of the hypoxic ventilatory response. Respir Physiol 112: 123–134, 1998. doi: 10.1016/s0034-5687(98)00026-7. [DOI] [PubMed] [Google Scholar]
- 38.Perim RR, Mitchell GS. Circulatory control of phrenic motor plasticity. Respir Physiol Neurobiol 265: 19–23, 2019. doi: 10.1016/j.resp.2019.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Millhorn DE, Eldridge FL, Waldrop TG. Prolonged stimulation of respiration by a new central neural mechanism. Respir Physiol 41: 87–103, 1980. doi: 10.1016/0034-5687(80)90025-0. [DOI] [PubMed] [Google Scholar]
- 40.Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, Thomas G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell 11: 1457–1466, 2003. doi: 10.1016/S1097-2765(03)00220-X. [DOI] [PubMed] [Google Scholar]
- 41.Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci 33: 67–75, 2010. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jaworski J, Sheng M. The growing role of mTOR in neuronal development and plasticity. Mol Neurobiol 34: 205–219, 2006. doi: 10.1385/MN:34:3:205. [DOI] [PubMed] [Google Scholar]
- 43.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 10: 151–162, 2002. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 44.Dougherty BJ, Fields DP, Mitchell GS. Mammalian target of rapamycin is required for phrenic long-term facilitation following severe but not moderate acute intermittent hypoxia. J Neurophysiol 114: 1784–1791, 2015. doi: 10.1152/jn.00539.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haenen GR, Plug HJ, Vermeulen NP, Timmerman H, Bast A. Contribution of 4-hydroxy-2,3-trans-nonenal to the reduction of beta-adrenoceptor function in the heart by oxidative stress. Life Sci 45: 71–76, 1989. doi: 10.1016/0024-3205(89)90437-2. [DOI] [PubMed] [Google Scholar]
- 46.Haenen GR, Veerman M, Bast A. Reduction of beta-adrenoceptor function by oxidative stress in the heart. Free Radic Biol Med 9: 279–288, 1990. doi: 10.1016/0891-5849(90)90002-z. [DOI] [PubMed] [Google Scholar]
- 47.Gelinas JN, Banko JL, Peters MM, Klann E, Weeber EJ, Nguyen PV. Activation of exchange protein activated by cyclic-AMP enhances long-lasting synaptic potentiation in the hippocampus. Learn Mem 15: 403–411, 2008. doi: 10.1101/lm.830008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dumaz N, Marais R. Integrating signals between cAMP and the RAS/RAF/MEK/ERK signalling pathways. Based on the anniversary prize of the Gesellschaft fur Biochemie und Molekularbiologie Lecture delivered on 5 July 2003 at the Special FEBS Meeting in Brussels. FEBS J 272: 3491–3504, 2005. doi: 10.1111/j.1742-4658.2005.04763.x. [DOI] [PubMed] [Google Scholar]
- 49.Gao Y, Nikulina E, Mellado W, Filbin MT. Neurotrophins elevate cAMP to reach a threshold required to overcome inhibition by MAG through extracellular signal-regulated kinase-dependent inhibition of phosphodiesterase. J Neurosci 23: 11770–11777, 2003. doi: 10.1523/JNEUROSCI.23-37-11770.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoffmann R, Baillie GS, MacKenzie SJ, Yarwood SJ, Houslay MD. The MAP kinase ERK2 inhibits the cyclic AMP-specific phosphodiesterase HSPDE4D3 by phosphorylating it at Ser579. EMBO J 18: 893–903, 1999. doi: 10.1093/emboj/18.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang HT, Zhao Y, Huang Y, Dorairaj NR, Chandler LJ, O'Donnell JM. Inhibition of the phosphodiesterase 4 (PDE4) enzyme reverses memory deficits produced by infusion of the MEK inhibitor U0126 into the CA1 subregion of the rat hippocampus. Neuropsychopharmacology 29: 1432–1439, 2004. doi: 10.1038/sj.npp.1300440. [DOI] [PubMed] [Google Scholar]
- 52.MacFarlane PM, Vinit S, Mitchell GS. Enhancement of phrenic long-term facilitation following repetitive acute intermittent hypoxia is blocked by the glycolytic inhibitor 2-deoxyglucose. Am J Physiol Regul Integr Comp Physiol 314: R135–R144, 2018. doi: 10.1152/ajpregu.00306.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dale EA, Ben Mabrouk F, Mitchell GS. Unexpected benefits of intermittent hypoxia: enhanced respiratory and nonrespiratory motor function. Physiology (Bethesda) 29: 39–48, 2014. doi: 10.1152/physiol.00012.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gonzalez-Rothi EJ, Lee KZ, Dale EA, Reier PJ, Mitchell GS, Fuller DD. Intermittent hypoxia and neurorehabilitation. J Appl Physiol (1985) 119: 1455–1465, 2015. doi: 10.1152/japplphysiol.00235.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Navarrete-Opazo A, Mitchell GS. Therapeutic potential of intermittent hypoxia: a matter of dose. Am J Physiol Regul Integr Comp Physiol 307: R1181–R1197, 2014. doi: 10.1152/ajpregu.00208.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]