

Abstract
Background and objective
Synthetic ginsenoside compounds G-Rp (1,3, and 4) and natural ginsenosides in Panax ginseng 20(S)-Rg3, Rg6, F4 and Ro have inhibitory actions on human platelets. However, the inhibitory mechanism of ginsenoside Rk1 (G-Rk1) is still unclear thus, we initiated investigation of the anti-platelet mechanism by G-Rk1 from Panax ginseng.
Methodology
Our study focused to investigate the action of G-Rk1 on agonist-stimulated human platelet aggregation, inhibition of platelet signaling molecules such as fibrinogen binding with integrin αIIbβ3 using flow cytometry, intracellular calcium mobilization, fibronectin adhesion, dense granule secretion, and thromboxane B2 secretion. Thrombin-induced clot retraction was also observed in human platelets.
Key Results
Collagen, thrombin, and U46619-stimulated human platelet aggregation were dose-dependently inhibited by G-Rk1, while it demonstrated a more effective suppression on collagen-stimulated platelet aggregation using human platelets. Moreover, G-Rk1 suppressed collagen-induced elevation of Ca2+ release from endoplasmic reticulum, granule release, and αIIbβ3 activity without any cytotoxicity.
Conclusions and implications
These results indicate that G-Rk1 possess strong anti-platelet effect, proposing a new drug candidate for treatment and prevention of platelet-mediated thrombosis in cardiovascular disease.
Keywords: Anti-platelet, Ginsenoside-Rk1, Integrin αIIbβ3, Thrombosis, Clot retraction
Graphical abstract
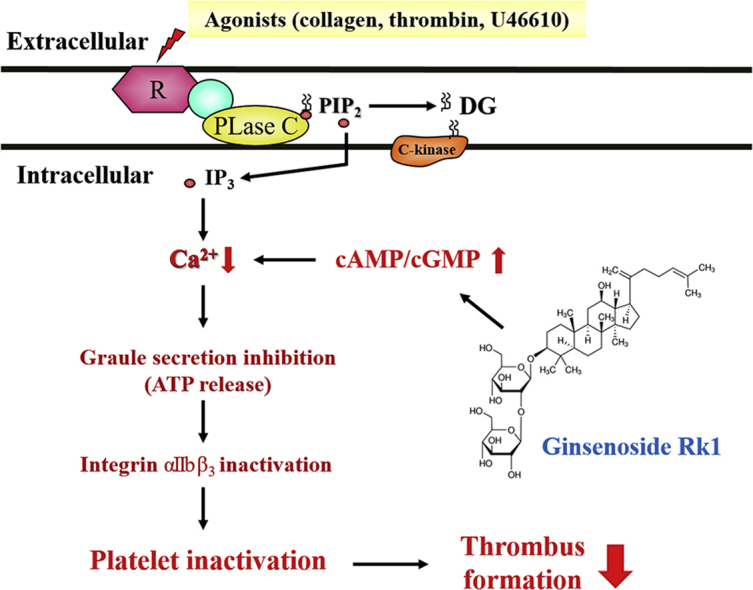
1. Introduction
Circulatory platelets are essential factor for hemostasis, while simultaneously posing a risk for thrombosis. One of the underlying causes of cardiovascular diseases (CVDs) is pathophysiologic over-activation of platelets leading to thrombosis and other platelet related cardiovascular ailmets [1]. Therefore, drugs that suppress platelets activity are necessary. CVDs are considered one of the leading causes of mortality globally [2] and several potential factors are involved in CVDs. Among the factors, platelet-mediated hemostatic plug is most important factor leading thrombotic complications [3]. Pharmacological suppression of platelets has been proven effective in prevention of CVDs, but, despite availability of several antiplatelet and antithrombotic drugs with higher efficacy, survival rate is low. For example, some common antiplatelet drugs have been reported for their side effects, i.e., aspirin causes gastric ulcers and clopidogrel causes aplastic anemia and thrombocytopenic purpura [4], thus, it necessitates the discovery of various drug candidates for treatment and prevention of thrombotic complications that lead to development of CVDs.
Collagens are concealed by endothelial cell layers and support blood vessel wall. In normal circulation of blood, collagen is not exposed to the outside of blood vessels. However, when endothelial wall is injured, collagen can bind to integrin α2β1 and glycoprotein VI leading upregulation of mitogen-activated protein kinases (MAPKs), PI3K/Akt phosphorylation [5,6]. Activated platelets are accumulated at the damaged site and interact using glycoprotein IIb/IIIa (integrin αIIb/β3), activating outside-in signaling pathway affecting Ca2+ mobilization, ATP secretion, thrombus formation and platelet-mediated clot retraction. During activation of platelet, diacylglycerol lipase and monoacylglycerol lipase sequentially hydrolyze diacylglycerol produced from the degradation of phosphatidylinositol 4,5-bisphosphate located on the platelet membrane producing arachidonic acid, which is converted to thromboxane A2 (TXA2) [7]. TXA2 has been found to induce secretion and morphological changes of intracellular granules through the activation of platelets [8].
Ginsenosides from Panax ginseng are known for induction of various biological activities. Major constituents of red and white ginseng are ginsenoside-Rb1, -Rc, -Rd, -Re, and -Rg1 [9], while minor ginsenoside constituents are ginsenoside-Rg5, -Rk1, -Rg6 and -F4 are found in red ginseng [10]. Our recent study has reported anti-platelet effect of F4 and Rg6, unique fraction of ginseng [11,12]. Ginsenoside-Rk1 is reported for its inhibitory effects on human hepatocellular carcinoma [13], and it can suppress pro-inflammatory responses in RAW264.7 [14]. Moreover, it has also been reported that G-Rk1 suppresses platelet aggregation [15], however, its inhibitory mechanism on platelet functions is yet to be explored. Thus, we investigated the regulatory action of G-Rk1 on human platelets.
2. Materials and Methods
2.1. Chemicals and reagents sources
Ambo Institute (Daejeon, Korea) supplied ginsenoside Rk1 (G-Rk1). Chrono-Log (Havertown, PA, USA) Corporation supplied platelet agonists (collagen and thrombin). Invitrogen (Eugene, OR, USA) provided Fura 2-AM (2-acetoxymethyl) and Alexa Fluor 488 conjugated fibrinogen. Cayman Chemical (Ann Arbor, MI, USA) supplied U46619, ATP luminescent assay kit. Cell Signaling (Beverly, MA, USA) supplied the lysis buffer and antibodies against phospho-VASP (Ser157), phospho-inositol-3-phosphate receptor type I (Ser1756), phospho-cPLA2 (Ser505), phospho-p38, phospho-Akt (Ser473), β-actin, and anti-rabbit secondary antibody. Fibronectin-Coated cell adhesion assay kit was procured from Cell Biolabs (San Diego, CA, USA).
2.2. Preparation of human platelets suspension
The human platelet-rich plasma (PRP) from normal healthy human volunteers with informed consent was obtained from the Korean Red Cross Blood Center (Suwon, Korea), and study protocols were approved by the approval of Namseoul University Institutional Review Board (1041479-HR-201803-003). The PRP was centrifuged for 10 min at 1,300g at room temperature, and pellet was washed twice using washing buffer (pH 6.5), and re-suspended pellet with suspension buffer (pH 6.9) according to the previous research [11]. All procedures were performed at room temperature i.e., 25°C. The suspension of platelets was adjusted to 5 × 108/mL concentration [16].
2.3. Platelet aggregation
For platelet aggregation, washed platelets (108/mL) were pre-incubated for 3 min in presence or absence of G-Rk1 along with 2 mM CaCl2 at 37°C, then collagen (2.5 μg/mL), U46619 (200 nM), and thrombin (0.05 U/mL) were added for stimulation, and the total volume became 400 μl. The aggregation assay was conducted for 5 minutes under continuous stirring condition. An increase in light transmission can convert into platelet aggregation rate (%). 0.1% dimethyl sulfoxide solution was used to dissolve G-Rk1, and the same amount of dimethyl sulfoxide solution was added in all experiments.
2.4. Cytotoxicity assay
G-Rk1 was examined for any cytotoxic effects via lactate dehydrogenase (LDH) leakage from cytosol of platelets. Washed human platelets (108/mL) were incubated with different concentrations of G-Rk1 for 2 hours and centrifuged for 2 min at 12,000 g. The supernatant was used to detect the cytotoxic effects using LDH assay kit. The plate was read at a wavelength of 490 nm using Synergy HT Microplate Reader (BioTek Instruments, Winoosku, VT., USA).
2.5. Intracellular calcium concentration
The Fura 2-AM (5 μM) and PRP mixture was pre-incubated at 37°C for 60 min and then platelets (108/mL) were washed with washing buffer as aforementioned. After washing step, platelets were suspended using suspending buffer and pre-incubated with or without G-Rk1 for 3 min at 37°C. The platelets were stimulated with collagen (2.5 μg/mL) in the presence of 2 mM CaCl2. A spectro-fluorometer (SFM-25; BioTek Instruments, Italy) was used to measure Fura 2-AM fluorescence according to the Grynkiewicz method [17] to calculate the [Ca2+]i values.
2.6. Measurement of thromboxane B2
Since thromboxane A2 (TXA2) is unstable and transforms into thromboxane B2 (TXB2) quickly, therefore, TXA2 generation was measured by detecting TXB2 production [7]. After platelet activation, the reaction was stopped by adding indomethacin (0.2 mM) with EDTA (5 mM). The amounts of TXB2 were determined using a TXB2 EIA kit Cayman Chemical (Ann Arbor, MI, USA). The plate was read at a wavelength of 420 nm using a Synergy HT Microplate Reader (BioTek Instruments, Winoosku, VT., USA).
2.7. Measurement of ATP
Platelets (108/mL) were pre-incubated for 3 min at 37°C with G-Rk1, then stimulated with collagen (2.5 μg/mL) in the presence of 2 mM CaCl2 to terminate ATP release, followed by centrifugation. The supernatant was used for detection of ATP release. ATP luminescent assay kit (Cayman Chemical, Ann Arbor, MI, USA) was performed with luminescence detection area using a Synergy HT Microplate Reader (BioTek Instruments, Winoosku, VT., USA).
2.8. Immunoblotting
Platelet aggregation was performed for 5 min and stopped by addition of lysis buffer and lysates of platelet were calculated using a bicinchoninic acid protein assay kit (Pierce Biotechnology, IL, USA). For Western blotting, proteins (15 μg) from platelets, pre-incubated with G-Rk1, were treated by SDS-PAGE (8%), and transferred onto PVDF membranes which were then probed with the primary (1:1,000) and secondary antibodies (1:10,000). Membranes were visualized by enhanced chemiluminescence. The blots were analyzed using the Quantity One, Ver. 4.5 (BioRad, Hercules, CA, USA).
2.9. Measurement of fibrinogen binding to αIIbβ3
Platelets, pre-incubated with G-Rk1, were treated with fibrinogen (30 μg/mL, Alexa Flour 488-conjugated) at 37°C for 5 mins. 0.5% paraformaldehyde in cold PBS was added to fix the interaction between platelet integrin and Alexa Flour 488-conjugated human fibrinogen. All procedures were conducted in the absence of light. The fibrinogen binding to integrin αIIbβ3 was conducted by the fluorescence of fibrinogen using flow cytometry (BD Biosciences, San Jose, CA, USA), and data were analyzed by the CellQuest software (BD Biosciences).
2.10. Fibronectin adhesion
Platelets (108/mL) were pre-incubated with G-Rk1 and CaCl2 (2 mM) for 1 h at 37°C in the presence of collagen (2.5 μg/mL) and washed five times with PBS followed by addition of cell stain solution and was placed for 10 min to ensure proper staining to measure absorbance. Extraction solution was added after a washing step to detach the adhered platelet plaque from fibronectin coated well. The plate was read at a wavelength of 560 nm using Synergy HT Microplate Reader (BioTek Instruments, Winooski, VT, USA).
2.11. Platelet-mediated fibrin clot retraction
Human PRP (500 μL) was poured into a polyethylene tube and samples were pre-incubated in presence or absence of various concentration of G-Rk1 for 15 min at 37°C, and clot retraction was triggered by adding thrombin (0.05 U/mL). Pictures of fibrin clot were taken using a digital camera at 15 min interval. Image J Software was used to calculate the clot area (v1.46, National Institutes of Health, USA).
2.12. Animals
Seven-week-old male C57BL/6 mice weighing 20–22 g were purchased from Orient Co. (Seoul, Korea). Mice were acclimatized for 1 week in an air-conditioned room with a 12 h/12 h light/dark cycle at a temperature and humidity of 22°C ± 2°C and 55% ± 10%, respectively. Animal experiment was conducted following IACUC guidelines, and the protocol was approved by the Ethics Committee of Kyungpook National University, Daegu, Republic of Korea (KNU2020-91).
2.13. Acute pulmonary thromboembolism in mice
A collagen- and epinephrine-induced acute pulmonary thromboembolism experiment was carried out in mice as previously described [18]. Briefly, mice were administered saline, G-Rk1 (30 mg/kg i.p.), or ASA (100 mg/kg i.p.) once a day for three days (10 mice in each group). Then, 1 h after the final injection, the mice were challenged with 0.1 mL of a mixture containing collagen (500 μg/mL) and epinephrine (60 μg/mL) by smooth injection into one of the tail veins. The mortality of mice in each group was monitored for 10 min, and the data are presented as the percentage of surviving animals in each treatment group. At the end of each experiment, surviving animals were euthanized with an overdose of anesthesia.
2.14. Statistical analyses
Data is analyzed by one-way analysis of variance was performed followed by Tukey-Kramer method. SPSS 21.0.0.0 software (SPSS, Chicago, IL, USA). All data is presented as the mean ± standard deviation. A p-value of 0.05 or less was considered statistically significant.
3. Results
3.1. Effects of G-Rk1 on human platelets aggregation and cytotoxicity
To determine anti-platelet effect by Rk1, three agonists were used. Collagen at 2.5 μg/mL, thrombin at 0.05 U/mL, and U46619 at 200 nM were used for optimum aggregation of human platelets (Fig. 1A, B, 1C). However, collagen induced platelets treated with G-Rk1 (3.75, 7.5, 15, and 30 μM) were most significantly reduced (28.3, 67.7, 89.2, and 98.9%, respectively) as compared to thrombin and U46619 induced platelet aggregation without cytotoxicity (Fig. 1D). DMSO 0.1% seemed to have no affect for agonist-induced platelet aggregation [19].
Fig. 1.

Effects of G-Rk1 on platelet aggregation and cytotoxicity. (A) Effect of G-Rk1 on collagen-induced human platelet aggregation. (B) Effect of G-Rk1 on thrombin-induced human platelet aggregation. (A) Effect of G-Rk1 on U46619-induced human platelet aggregation. (D) Effect of G-Rk1 on cytotoxicity. Platelet aggregation and cytotoxicity were carried out as described in “Materials and Methods” section. The data are expressed as the mean ± standard deviation (n = 4). ∗p < 0.05, ∗∗p < 0.01 versus each agonist-stimulated human platelets. NS, not significant.
3.2. Inhibitory effects of G-Rk1 on [Ca2+]i mobilization, dense granule secretion, and IP3RI phosphorylation
Considering the fact that intracellular ion concentration ([Ca2+]i) plays a vital role and is essential for platelet activation, we focused our investigation to explore the effect of G-Rk1 on antagonistic activity of Ca2+. As shown in Fig. 3A, [Ca2+]i levels were elevated from 101.2 ± 0.6 nM to 694.6 ± 8.4 nM by collagen (2.5 μg/mL) treatment in vehicle treated platelets. However, G-Rk1 dose-dependently (3.75 to 30 μM) reduced the collagen-stimulated increased [Ca2+]i levels (Fig. 2A). Notably, the inhibition rate of G-Rk1 (60 μM) was estimated as 83.2% may inhibit. Therefore, we expected that G-Rk1 may inhibits calcium release from endoplasmic reticulum and investigated the phosphorylation of calcium mobilization signaling molecule. As shown in Fig. 2B, G-Rk1 (3.75 to 30 μM) increased inositol 1, 4, 5-triphosphate receptor type I (IP3RI) phosphorylation (Ser1756) in collagen-stimulated human platelet aggregation in a dose-dependent manner. This shows that the decline of cytosolic calcium concentration by G-Rk1 resulted from IP3RI (Ser1756) phosphorylation. Because G-Rk1 inhibited collagen-stimulated [Ca2+]i mobilization (Fig. 2A), we explored whether G-Rk1 is involved in inhibition of dense granule secretion and examined ATP release. It is known that high [Ca2+]i levels facilitates the phosphorylation of myosin light chain and pleckstrin to facilitate granule release (i.e., dense body, α-granule), and platelet aggregation [20]. As shown in Fig. 2C, G-Rk1 dose dependently inhibited collagen-stimulated ATP secretion.
Fig. 3.

Effects of G-Rk1 on TXA2 generation and cPLA2 and p38 MAPK phosphorylation. (A) Effects of G-Rk1 on collagen-induced TXA2 generation. (B) Effect of G-Rk1 on collagen-induced cPLA2 (Ser505) phosphorylation. (C) Effect of G-Rk1 on collagen-induced p38 MAPK phosphorylation. Measurement of TXA2 generation and Western blot was performed as described in “Materials and Methods” section. The data are expressed as the mean ± standard deviation (n = 4). ∗p < 0.05, ∗∗p < 0.01 versus the collagen-stimulated human platelets.
Fig. 2.

Effects of G-Rk1 on [Ca2+]i mobilization and IP3RI phosphorylation. (A) Effect of G-Rk1 on collagen-induced [Ca2+]i mobilization. (B) Effect of G-Rk1 on collagen-induced IP3RI (Ser1756) phosphorylation. (C) Effects of G-Rk1 on collagen-induced ATP release. [Ca2+]i mobilization, ATP release and Western blot were performed as described in “Materials and Methods” section. The data are expressed as the mean ± standard deviation (n = 4). ∗p < 0.05, ∗∗p < 0.01 versus the collagen-stimulated human platelets.
3.3. Measurement of TXB2 and cPLA2 and p38 MAPK dephosphorylation
As shown in Fig. 3A, TXA2 (determined as TXB2) levels in intact platelets were 1.8 ± 0.3 ng/108 platelets, but the collagen (2.5 μg/mL) markedly increased TXA2 level to 227.3 ± 6.4 ng/108 platelets. However, G-Rk1 potently abridged TXA2 generation to 2.4 ± 0.7 ng/108 platelets (98.9% inhibition at 30 μM) (Fig. 3A). For identification of inhibitory effect of G-Rk1 on TXA2 production, we investigated cPLA2 phosphorylation. The cPLA2 has been reported to play key role in regulating the AA release in human platelets. As shown in Fig. 3B, the cPLA2 was phosphorylated at Ser505 by collagen in vehicle treated control platelets, but G-Rk1 significantly inhibited cPLA2 phosphorylation in a dose-dependent manner. Further, previous studies have shown that the cPLA2 activity is achieved by p38 MAPK (p38) and p38 also being activated through phosphorylation. As shown in Fig. 3C, collagen elevated p38 phosphorylation, but G-Rk1 inhibited its phosphorylation dose-dependently.
3.4. Inhibitory effects of G-Rk1 on fibrinogen binding to integrin αIIbβ3 and fibronectin adhesion
Next, the inhibitory effects of G-Rk1 on fibrinogen binding to αIIbβ3 in human platelets were investigated. Agonist-stimulated platelet triggers inside-out signaling pathway through conformational change in glycoprotein IIb/IIIa (also called αIIbβ3) structure which increases its binding affinity towards fibrinogen. Collagen elevated the binding of fibrinogen to αIIbβ3 in vehicle treated control platelets (Fig. 4A and b, 4B), with a rate of 86.7 ± 0.8%. However, G-Rk1 significantly attenuated the αIIbβ3 and fibrinogen interaction in a dose-dependently manner (Fig. 4A-c-f, 4B). The inhibition rate by G-Rk1 (30 μM) was 77.2%. Moreover, αIIbβ3 also serves as a binding molecule of fibronectin which is crucial for platelet adhesion to vascular endothelium. Therefore, we examined whether G-Rk1 affect fibronectin adhesion. As shown in Fig. 4C, G-Rk1 suppressed collagen-stimulated fibronectin adhesion. Bovine serum albumin coated well is used as negative control.
Fig. 4.

Effects of G-Rk1 on fibrinogen binding to αIIb/β3 and Fibronectin adhesion. (A) The flow cytometry histograms on fibrinogen binding. a, Intact platelets (base); b, Collagen (2.5 μg/mL); c, Collagen (2.5 μg/mL) + G-Rk1 (3.75 μM); d, Collagen (2.5 μg/mL) + G-Rk1 (7.5 μM); e, Collagen (2.5 μg/mL) + G-Rk1 (15 μM); f, Collagen (2.5 μg/mL) + G-Rk1 (30 μM). (B) Effects of G-Rk1 on collagen-induced fibrinogen binding (%). (C) Effects of G-Rk1 on collagen-induced fibronectin adhesion. Measurement of fibrinogen binding and fibronectin adhesion was carried out as described in “Materials and Methods” section. The data are expressed as the mean ± standard deviation (n = 4). ∗p < 0.05, ∗∗p < 0.01 versus the collagen-stimulated human platelets.
3.5. Effects of G-Rk1 on regulation of VASP and Akt
Phosphorylated vasodilator-stimulated phosphoprotein (VASP) inhibits Actin Dynamics which activates αIIb/β3 [21,22], while phosphatidylinositol 3-kinase (PI3K)/Akt phosphorylation associated with αIIbβ3 activation [23]. As G-Rk1 showed the inhibitory action on collagen-induced αIIb/β3 activation (Fig. 4A and C), we investigated the effect of G-Rk1 on VASP Ser157 phosphorylation and Akt Ser473 dephosphorylation in collagen-stimulated platelets. G-Rk1 upregulated VASP-Ser157 phosphorylation significantly (Fig. 5A) and decreased Akt-Ser473 phosphorylation (Fig. 5B) in a dose-dependent manner.
Fig. 5.

Effects of G-Rk1 on VASP and Akt phosphorylation. (A) Effect of G-Rk1 on collagen-induced VASP (Ser157) phosphorylation. (B) Effect of G-Rk1 on collagen-induced Akt (Ser473) phosphorylation. Western blot was performed as described in “Materials and Methods” section. The data are expressed as the mean ± standard deviation (n = 4). ∗p < 0.05, ∗∗p < 0.01 versus the collagen-stimulated human platelets.
3.6. G-Rk1 modulates outside-in signaling by inhibiting clot retraction
Activated integrin αIIbβ3 by platelet agonists and fibrinogen binding, transduces signals into the cell which primes various actions in platelets such as platelet spreading, granule secretion, stable adhesion, protein phosphorylation, platelet contraction, ultimately lead to stable thrombus formation and clot retraction (termed as outside-in signaling). Thus, we finally examined the inhibitory effects of G-Rk1 on thrombin-stimulated fibrin clot retraction. Fig. 6A shows thrombin-induced fibrin clot build up and contraction with an inhibition rate of 96.9%. However, the phenomenon was effectively suppressed by G-Rk1 (3.75, 7.5, 15, 30 μM) dose-dependently, with inhibitory degrees of 62.3, 46.9, 41.7, 30.5, 16.0%, respectively, compared to thrombin-induced clot retraction (Fig. 6B). Y27632 was used as positive control.
Fig. 6.

Effects of G-Rk1 on fibrin clot retraction. (A) Photographs of fibrin clot (B) Effects of G-Rk1 on thrombin-retracted fibrin clot (%). Quantification of fibrin clot retraction was performed as describe in “Materials and Methods” section. The data are expressed as the mean ± standard deviation (n = 4). ∗p < 0.05, ∗∗p < 0.01 versus the thrombin-stimulated human PRP.
3.7. G-Rk1 protects mice from pulmonary thromboembolism
Collagen combined with epinephrine induced pulmonary thromboembolism model has been commonly used to test the in vivo effects of antithrombotic agents [24]. We injected mice with a mixture of collagen and epinephrine to evaluate in vivo effects of G-Rk1 on thrombus formation. We observed a significant decrease in mortality for the G-Rk1-treated group, highlighting the protective role of G-Rk1 against thrombosis in mice (p < 0.001; Fig. 7).
Fig. 7.

G-Rk1 inhibits pulmonary thromboembolism in mice. Mice were pre-treated with saline, G-Rk1 (30 mg/kg i.p.), or ASA (100 mg/kg i.p.) once a day for 3 days (n = 10 in each group). One hour after the final administration, mice were challenged with a collagen + epinephrine mixture and checked for survival at 10 min. ∗∗∗p < 0.001 compared to the control group.
4. Discussion
Our previous ginseng study reported that 20(S)-Rg3, Ro, and synthetic compounds such as G-Rp1, G-Rp3, G-Rp4, and minor fraction ginsenoside, F4, Rg6, have anti-platelet effects. These physiological effects of ginseng suggest new directions and approaches in CVDs [25]. In the present study, we evaluated that whether G-Rk1 leads anti-platelets and anti-thrombotic potential on collagen induced human platelets. Of several platelet activating molecules, collagen, thrombin, TXA2 and intracellular calcium are known to be crucial for activation of platelets. G-Rk1 significantly blocked various agonists-elevated human platelet aggregation. Among agonists, we confirmed that G-Rk1 potent inhibitory action in collagen-induced platelet aggregation. Therefore, we checked collagen-stimulated Ca2+ mobilization, ATP release, fibrinogen binding, fibronectin adhesion, and thrombin-induced clot retraction and associated signaling molecules. Physiological agonists such as collagen, ADP, thrombin and U46619 increase [Ca2+]i influencing on phosphorylation of calcium-calmodulin-dependent myosin light chain, leading to ATP release and calpain activation. Calpain in platelets consists of calpain-1 and calpain-2 and acts Ca2+ dependent cysteine protease. It has been well known that calpain modulates outside-in signaling pathway [26]. G-Rk1 suppressed collagen-induced [Ca2+]i level (Fig. 2A), ATP release (Fig. 2C) and elevated IP3RI (Ser1756) phosphorylation (Fig. 2B). The IP3RI is found on endoplasmic reticulum surface, which induces Ca2+ mobilization but, the action of IP3RI is inhibited by its phosphorylation (Ser1756). In addition, G-Rk1 suppressed collagen-induced TXA2 generation. The TXA2 is a strong autacoid molecule, affecting circulating platelet and stimulates platelet-mediated hemostasis mechanism. Intracellular Ca2+ can activate on endogenous enzyme, cPLA2. The cPLA2 is a crucial mediator for TXA2 production. Upon stimulation by agonists, cPLA2 is translocated from cytosol to membranes in the presence of intracellular Ca2+ and mitogen-activated protein kinases p38 phosphorylates at Ser505 for full catalytic activity [27,28]. G-Rk1 inhibited the phosphorylation of p38 and cPLA2 dose-dependently, which influence on TXA2 generation (Fig. 3A).
G-Rk1 downregulated αIIb/β3 activity affecting fibrinogen binding and fibronectin adhesion (Fig. 4A and C) through upregulation of phosphorylation of VASP (Fig. 5A) and downregulation of Akt phosphorylation (Fig. 5B). The binding between αIIb/β3 and fibrin derived from fibrinogen is a key mediator for the clot build up and clot retraction. The αIIb/β3 stimulated outside in signaling pathway leading the platelet cytoskeleton modification, platelet spreading and clot retraction. Our results showed that G-Rk1 suppressed collagen-induced fibrinogen biding and fibronectin adhesion and clot retraction together with VASP (Ser157) phosphorylation and Akt (Ser473) dephosphorylation. VASP in platelets is a key mediator for actin dynamics and focal adhesions leading αIIb/β3 activation. The VASP also has a phosphorylation site at Ser157, and its phosphorylation leads inhibition of αIIb/β3 affinity. As shown in Fig. 6A, G-Rk1 inhibited clot retraction dose-dependently. These data mean that downregulation of Ca2+ by phosphorylation of IP3RI (Ser1756) and suppression of αIIb/β3 affinity by phosphorylation VASP (Ser157) facilitates delay of clot retraction.
It is established that a mixture of collagen and epinephrine, a standard agonist for induction of platelet activation, has been used to study defects in platelet function as well as pulmonary thromboembolism in vivo [29]. As expected, we observed that collagen and epinephrine injection rapidly induced fatal pulmonary thromboembolism in majority of the control mice; however, mice treated with G-Rk1 were protected against thrombosis, which indicated that G-Rk1 modulated platelet function and inhibited thrombus formation.
In conclusion, we confirm that G-Rk1 decreases calcium mobilization, fibrinogen-binding to αIIb/β3, fibronectin adhesion and thrombin-stimulated clot retraction, which are achieved by the phosphorylation of IP3RI (Ser1756), VASP (Ser157), and dephosphorylation of Akt (Ser473) and decreased of TXA2 production through downregulation of cPLA2 (Ser505) and p38 MAPK. Therefore, we suggest that G-Rk1 is an effective compound for prevention of platelet-mediated CVDs.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
Funding for this paper was provided by Namseoul University.
References
- 1.Andrews R.K., Berndt M.C. Platelet physiology and thrombosis. Thromb Res. 2004;114:447–453. doi: 10.1016/j.thromres.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 2.Mozaffarian D., Benjamin E.J., Go A.S., Arnett D.K., Blaha M.J., Cushman M., etal Executive summary: heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation. 2016;133:447–454. doi: 10.1161/CIR.0000000000000366. [DOI] [PubMed] [Google Scholar]
- 3.Jennings L.K. Role of platelets in atherothrombosis. Am J Cardiol. 2009;103:4A–10A. doi: 10.1016/j.amjcard.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 4.Barrett N.E., Holbrook L., Jones S., Kaiser W.J., Moraes L.A., Rana R., etal Future innovations in anti-platelet therapies. Br J Pharmacol. 2008;154:918–939. doi: 10.1038/bjp.2008.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farndale R.W. Collagen-induced platelet activation. Blood Cell Mol Dis. 2006;36:162–165. doi: 10.1016/j.bcmd.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 6.Chen H., Kahn M.L. Reciprocal signaling by integrin and nonintegrin receptors during collagen activation of platelets. Mol Cell Biol. 2003;23:4764–4777. doi: 10.1128/MCB.23.14.4764-4777.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohkubo S. Thromboxane A2-mediated shape change: independent of Gq-phospholipase C-Ca2+ pathway in rabbit platelets. Br J Pharmacol. 1996;117:1095–1104. doi: 10.1111/j.1476-5381.1996.tb16702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamberg M., Svensson J., Samuelsson B. Thromboxanes: a new group of biologically active compounds derived from prostaglandin endoperoxides. P Natl Acad Sci. 1975;72:2994–2998. doi: 10.1073/pnas.72.8.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nam K.Y. The comparative understanding between red ginseng and white ginsengs, processed ginsengs (Panax ginseng CA Meyer) Journal of Ginseng Research. 2005;29(1):1–18. [Google Scholar]
- 10.Ha Y.W., Lim S.S., Ha I.J., Na Y.C., Seo J.J., Shin H., etal Preparative isolation of four ginsenosides from Korean red ginseng (steam-treated Panax ginseng CA Meyer), by high-speed counter-current chromatography coupled with evaporative light scattering detection. J Chromatogr A. 2007;1151:37–44. doi: 10.1016/j.chroma.2007.01.038. [DOI] [PubMed] [Google Scholar]
- 11.Shin J.H., Kwon H.W., Lee D.H. Ginsenoside F4 inhibits platelet aggregation and thrombus formation by dephosphorylation of IP3RI and VASP. J Appl Biol Chem. 2019;62:93–100. [Google Scholar]
- 12.Kwon H.W. Ginsenoside Rg6 modulates platelet function and thrombus formation via inhibition of phosphoproteins. J Korean Soc Food Sci Nutr. 2020;49:229–235. [Google Scholar]
- 13.Kim Y.J., Kwon H.C., Ko H., Park J.H., Kim H.Y., Yoo J.H., etal Anti-tumor activity of the ginsenoside Rk1 in human hepatocellular carcinoma cells through inhibition of telomerase activity and induction of apoptosis. Biol Pharm Bull. 2008;31:826–830. doi: 10.1248/bpb.31.826. [DOI] [PubMed] [Google Scholar]
- 14.Qian Y.U., Ke-Wu Z.E.N.G., Xiao-Li M.A., Jiang Y., Peng-Fei T.U., Wang Xue-Mei. Ginsenoside Rk1 suppresses pro-inflammatory responses in lipopolysaccharide-stimulated RAW264. 7 cells by inhibiting the Jak2/Stat3 pathway. Chin J Nat Medicines. 2017;15:751–757. doi: 10.1016/S1875-5364(17)30106-1. [DOI] [PubMed] [Google Scholar]
- 15.Ju H.K., Lee J.G., Park M.K., Park S.J., Lee C.H., Park J.H., etal Metabolomic investigation of the anti-platelet aggregation activity of ginsenoside Rk1 reveals attenuated 12-HETE production. J Proteome Res. 2012;11:4939–4946. doi: 10.1021/pr300454f. [DOI] [PubMed] [Google Scholar]
- 16.Born G.V.R., Hume M. Effects of the numbers and sizes of platelet aggregates on the optical density of plasma. Nature. 1967;215:1027–1029. doi: 10.1038/2151027a0. [DOI] [PubMed] [Google Scholar]
- 17.Grynkiewicz G., Poenie M., Tsien R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 18.Canobbio I., Cipolla L., Consonni A., Momi S., Guidetti G., Oliviero B., Falasca M., Okigaki M., Balduini C., Gresele P. Impaired thrombin-induced platelet activation and thrombus formation in mice lacking the Ca(2+)-dependent tyrosine kinase Pyk2. Blood. 2013;121:648–657. doi: 10.1182/blood-2012-06-438762. [DOI] [PubMed] [Google Scholar]
- 19.Kwon H.W. Inhibitory effects of PD98059, SB203580, and SP600125 on α-and δ-granule release and intracellular Ca2+ levels in human platelets. Biomed Sci Lett. 2018;24:253–262. [Google Scholar]
- 20.Kaibuchi K., Sano K., Hoshijima M., Takai Y., Nishizuka Y. Phosphatidylinositol turnover in platelet activation; calcium mobilization and protein phosphorylation. Cell Calcium. 1982;3:323–335. doi: 10.1016/0143-4160(82)90020-3. [DOI] [PubMed] [Google Scholar]
- 21.Laurent V., Loisel T.P., Harbeck B., Wehman A., Gröbe L., Jockusch B.M. Role of proteins of the Ena/VASP family in actin-based motility of Listeria monocytogenes. J Cell Biol. 1999;144:1245–1258. doi: 10.1083/jcb.144.6.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sudo T., Ito H., Kimura Y. Phosphorylation of the vasodilator-stimulated phosphoprotein (VASP) by the anti-platelet drug, cilostazol, in platelets. Platelets. 2003;14:381–390. doi: 10.1080/09537100310001598819. [DOI] [PubMed] [Google Scholar]
- 23.Morello F., Perino A., Hirsch E. Phosphoinositide 3-kinase signalling in the vascular system. Cardiovasc Res. 2009;82:261–271. doi: 10.1093/cvr/cvn325. [DOI] [PubMed] [Google Scholar]
- 24.Lhermusier T., Severin S., Van Rothem J., Garcia C., Bertrand-Michel J., Le Faouder P., Hechler B., Broccardo C., Couvert P., Chimini G. ATP-binding cassette transporter 1 (ABCA1) deficiency decreases platelet reactivity and reduces thromboxane A2 production independently of hematopoietic ABCA1. Journal of Thrombosis and Haemostasis. 2016;14:585–595. doi: 10.1111/jth.13247. [DOI] [PubMed] [Google Scholar]
- 25.Irfan M., Kim M., Rhee M.H. Anti-platelet role of Korean ginseng and ginsenosides in cardiovascular diseases. J Ginseng Res. 2020;44:24–32. doi: 10.1016/j.jgr.2019.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Azam M., Andrabi S.S., Sahr K.E., Kamath L., Kuliopulos A., Chishti A.H. Disruption of the mouse μ-calpain gene reveals an essential role in platelet function. Mol Cell Biol. 2001;21:2213–2220. doi: 10.1128/MCB.21.6.2213-2220.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin L.L., Wartmann M., Lin A.Y., Knopf J.L., Seth A., Davis R.J. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72:269–278. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- 28.Adam F., Kauskot A., Rosa J.P., Bryckaert M. Mitogen-activated protein kinases in hemostasis and thrombosis. J Thromb Haemost. 2008;6:2007–2016. doi: 10.1111/j.1538-7836.2008.03169.x. [DOI] [PubMed] [Google Scholar]
- 29.Angelillo-Scherrer A., de Frutos P., Aparicio C., Melis E., Savi P., Lupu F., Arnout J., Dewerchin M., Hoylaerts M., Herbert J. Deficiency or inhibition of Gas6 causes platelet dysfunction and protects mice against thrombosis. Nat Med. 2001;7:215–221. doi: 10.1038/84667. [DOI] [PubMed] [Google Scholar]