

Summary
Dr. John Herrick described the first clinical case of sickle cell anaemia (SCA) in the United States in 1910. Subsequently, four decades later, Ingram and colleagues characterized the A to T substitution in DNA producing the GAG to GTG codon and replacement of glutamic acid with valine in the sixth position of the βS-globin chain. The establishment of Comprehensive Sickle Cell Centers in the United States in the 1970s was an important milestone in the development of treatment strategies and describing the natural history of sickle cell disease (SCD) comprised of genotypes including homozygous haemoglobin SS (HbSS), HbSβ0thalassaemia, HbSC and HbSβ+thalassaemia, among others. Early drug studies demonstrating effective treatments of HbSS and HbSβ0thalassaemia, stimulated clinical trials to develop disease-specific therapies to induce fetal haemoglobin due to its ability to block HbS polymerization. Subsequently, hydroxycarbamide proved efficacious in adults with SCA and was Food and Drug Administration (FDA)-approved in 1998. After two decades of hydroxycarbamide use for SCD, there continues to be limited clinical acceptance of this chemotherapy drug, providing the impetus for investigators and pharmaceutical companies to develop non-chemotherapy agents. Investigative efforts to determine the role of events downstream of deoxy-HbS polymerization, such as endothelial cell activation, cellular adhesion, chronic inflammation, intravascular haemolysis and nitric oxide scavenging, have expanded drug targets which reverse the pathophysiology of SCD. After two decades of slow progress in the field, since 2018 three new drugs were FDA-approved for SCA, but research efforts to develop treatments continue. Currently over 30 treatment intervention trials are in progress to investigate a wide range of agents acting by complementary mechanisms, providing the rationale for ushering in the age of effective and safe combination drug therapy for SCD. Parallel efforts to develop curative therapies using haematopoietic stem cell transplant and gene therapy provide individuals with SCD multiple treatment options. We will discuss progress made towards drug development and potential combination drug therapy for SCD with the standard of care hydroxycarbamide.
Keywords: combination drug therapy, fetal haemoglobin, gene therapy, haematopoietic stem cell transplant, sickle cell anaemia, sickle cell disease
Introduction
Sickle cell disease (SCD) is a global health condition that affects more than 20 million people worldwide with over a billion dollars spent in the United States (US) covering annual healthcare costs.1 In the US, approximately 100,000 people mostly of African descent are affected and 1 in 375 African–American infants are diagnosed with SCD annually by universal newborn screening.2 Compared to the general population, the average life span for individuals with SCD is 48 years of age; however, many adults live to 70 years in well-resourced countries. By contrast, there remains a staggering 50–80% mortality rate in the first five years of life for underdeveloped countries, such as sub-Saharan Africa, providing the impetus for the World Health Organization to declare SCD a global health problem in 2006 and calling for worldwide efforts to develop affordable and efficacious treatment options.3–5
Sickle cell disease is a group of inherited disorders, of which homozygous haemoglobin S (HbSS) and HbSβ0thalassaemia or sickle cell anaemia (SCA) are the most common and clinically severe phenotypes.6 Other compound heterozygote forms of SCD include HbSC, HbSβ0thalassaemia, HbSβ+thalassaemia and other β-globin chain variants combined with HbS. Under hypoxia, acidosis or dehydration conditions, HbS polymerizes and forms linear elongated fibres that distort red blood cells (RBCs), leading to a chronic haemolytic anaemia and acute episodes of pain due to vaso-occlusive (VOC) obstruction of blood flow and tissue ischaemia. The ensuing ischaemia/reperfusion injury leads to the generation of reactive oxygen species and endothelial cell activation with increased adhesion molecule expression, and activation of neutrophils, monocytes and platelets leading to a chronic inflammatory state in SCD. Additionally, intravascular haemolysis of sickle RBCs leads to nitric oxide (NO) depletion by cell-free Hb, contributing to endothelial dysfunction. Moreover, the RBC membrane is damaged by deoxy-HbS polymerization through lipid peroxidation leading to phosphatidylserine exposure, and generation of a hypercoagulable state. These mechanisms contribute to the development of chronic organ damage including sickle nephropathy, pulmonary hypertension, avascular necrosis of bone, chronic lung disease and ultimately to a shortened life expectancy.7–9
Standards of care
Comprehensive Sickle Cell Programmes that implement patient-centred medical home principles early in infancy achieve high uptake of preventative care measures and patient satisfaction. Recently, the National Heart, Lung and Blood Institute released an Expert Panel Report that includes evidence-based guidelines for the treatment of SCD.10 While a full discussion of the current standards of care is beyond the scope of this review, a few major recommendations in childhood made major impact on survival in early childhood. Based on the landmark finding in the Prophylactic Penicillin Study 1,11 universal newborn screening was established in the US to enable early diagnosis and treatment of infants with SCD to reduce morbidity and mortality.11 It is recommended that all infants with HbSS and HbSβ0thalassaemia receive penicillin prophylaxis and PPV23 (23-valent-pneumococcal polysaccharide) vaccine to prevent invasive pneumococcal disease.12 There exists a devastating 10% rate of central nervous system involvement as stroke in the first 20 years of life for individuals with SCA.13–15 The Stroke Prevention Trial in Sickle Cell Anemia demonstrated a 90% relative reduction in risk of stroke in high-risk children receiving regular blood transfusions.16 The standard of care guidelines17 recommend that children with SCA receive annual transcranial Doppler screenings from 2 to 16 years of age. The implementation of early diagnosis, education and recommended treatment and screenings, in a patient-centred medical home setting, is essential to address the medical vulnerabilities of this population.18
FDA-approved treatments for SCD
The human HBB (β-like globin genes) locus is composed of five developmentally regulated globin genes arranged from 5’-HBE1-HBG1-HBG2-HBD-HBB-3’ on chromosome 11, in the order of expression during in utero development. Located ~ 20 kb upstream of HBE1 is a regulatory element called the locus control region (LCR) consisting of five erythroid-specific DNase I hypersensitive sites; each LCR element has core enhancer sequences, that bind ubiquitous and erythroid-specific transcription factors to achieve normal haemoglobin switching. The LCR regulates the expression of the HBB locus by direct interaction with the individual globin gene promoters, facilitated by transcription factors, epigenetic remodelling proteins and DNA looping.19 A complex combination of DNA-binding proteins, such as GATA1, TAL1, E2A, LMO2, LRF/ZBTB7 and LDB1, mediate DNA looping of the LCR with each globin promoter to enhance gene transcription.20–23 Molecular mechanisms that control the switch from fetal haemoglobin (HbF; α2γ2) to adult HbA (α2β2) spawned decades of research to move the field forward to develop novel drugs for treatment of SCD. The regulation of globin gene transcription is tightly controlled by interaction of the LCR,24 and repressive transcription factors that bind to the HBG promoters or HBD/HBB intergenic region such as BCL11A.25 This factor is under intense investigation as a druggable target for effective HbF induction.
In 1948, Dr. Janet Watson observed the delayed onset of clinical symptoms in early infancy due to higher HbF levels during the first year of life.26 Additional evidence for the disease-modifying properties of HbF are supported by the observation that patients with HbS who co-inherited a genetic variant producing hereditary persistence of fetal haemoglobin (HbS-HPFH) are relatively asymptomatic.27,28 The presence of HbF in sickle RBCs delays deoxy-HbS polymerization. Several HPFH variants are produced by inherited deletions in binding sites for cis-acting factors in the HBB locus producing a decrease in HbA production. In addition, HBG promoter mutations are bound by negative transcription factors during developmentally regulated globin gene switching; however, a point mutation in the Xmn1 site leads to elevated HBG expression in adults from different racial/ethnic populations. Genetic mapping and later genome-wide association studies discovered quantitative trait loci in the HBS1L–MYB intergenic region (6q23) and BCL11A (2p16) with potent HbF-modulating abilities.28,29 A frequently asked question is what level of HbF is required for optimal modification of clinical disease severity? Historical data suggest that individuals with HbF levels > 8 6%30 have less VOC episodes, acute chest syndrome and improved survival while individuals with HbF > 20% are protected from stroke31 More recent clinical studies by Estepp et al., support the use of hydroxycarbamide in children, and suggest that the preferred dosing strategy is one that targets a HbF end-point > 20%.32 Another important factor to evaluate in addition to HbF levels is the distribution of HbF in RBCs (F cells) to achieve clinical efficacy. In vitro studies demonstrated the ability of HbF to form haemoglobin hybrids consisting of α2γβ that inhibit HbS polymerization.33 Furthermore, numerous naturally occurring HPFH variants lead to significant HbF levels in the majority of RBCs (pancellular) producing benign clinical phenotypes. Thus, to achieve maximal clinical benefit, it is critical that pharmacologic agents produce a pancellular HbF distribution.
Hydroxycarbamide
The discovery that 5-azacytidine mediated HbF induction in adult baboons, spawned over three decades of research to develop agents for clinical treatment.34 Other cytotoxic agents had similar effects;35–37 however, among them, hydroxycarbamide was most suited for clinical trials.30 Subsequently, the landmark Multicenter Study of Hydroxycarbamide in Patients with Sickle Cell Anemia (MSH) was completed in 1995 demonstrating the clinical benefit of hydroxycarbamide in producing a decrease in VOC pain episodes in 50% of adult patients.38,39 Although hydroxycarbamide has disease-modifying effects unrelated to HbF induction, such as reduction in RBC adhesion and white blood cell counts,40 this agent produces a heterocellular distribution of HbF synthesis. Hydroxycarbamide was approved for use in adult patients with SCA by the US FDA in 1998, and subsequently by the European Medicines Agency in 2007; use in children > 2 years of age with SCA was approved in 2017. Hydroxycarbamide has a tolerable side effect profile when given by daily oral dosing, and proven efficacy.38,41 A recent paediatric clinical trial demonstrated that hydroxycarbamide and phlebotomy were non-inferior to chronic blood transfusions in children with SCA at higher risk for stroke determined by abnormal transcranial Doppler screen.42 Over time, the HbF response to hydroxycarbamide may decline in some individuals, thus creating a need to discover additional pharmacologic agents that induce HbF by novel mechanisms or target downstream effects of HbS polymerization to develop combination drug treatment and prevent chronic pain and progressive organ damage.
Mechanisms of fetal haemoglobin induction by hydroxycarbamide
Hydroxycarbamide is an S-phase-specific cytotoxic, anti-metabolic and anti-neoplastic molecule; it is a potent inhibitor of ribonucleotide diphosphate reductase.43 However, much is still unknown about how hydroxycarbamide induces HbF synthesis. One of the most widely accepted mechanisms is that hydroxycarbamide causes a ‘stress erythropoiesis’ response,44,45 which alters erythroid differentiation kinetics, resulting in a release of reticulocytes and RBCs with high HbF levels (Fig 1). Hydroxycarbamide also reacts with haeme-containing proteins to release nitric oxide (NO).46 Previous studies provide data that HbF induction occurs in response to activation of soluble guanosine monophosphate (sGC) by hydroxycarbamide-derived NO.47,48 Studies in SCD mouse models demonstrated that cell signalling, involving hydroxycarbamide-induced NO-sGC-cyclic guanosine monophosphate (cGMP) and protein kinase signalling path-ways,49 is associated with HBG activation.50 In addition, hydroxycarbamide increases phosphorylation of p38 mitogen-activated protein kinase, and dephosphorylation of extracellular signal-regulated kinase and c-Jun N-terminal kinase,51 effects known to affect erythroid differentiation and induce HBG expression. More recent studies implicate mechanisms involving microRNAs for explaining variances in HbF levels achieved after hydroxycarbamide treatment in SCD patients. The response to hydroxycarbamide was correlated with HbF levels at baseline (miR-494) and maximum tolerated dose (miR-26b and miR-151–3p) microRNA expression.52 Recent data support the ability of hydroxycarbamide to downregulate BCL11A, KLF1 and MYB through miR-15a and miR-16-1 upregulation,53 and TAL 1 levels.54 There is evidence for an inverse correlation of methylated of CpG islands in the HBG2 promoter in SCD patients and HbF levels.52 Based on changes in protein levels of GATA-1, GATA-2 and BCL11A in sickle erythroid progenitors generated in culture, a hydroxycarbamide responsiveness index was proposed.55
Fig 1.
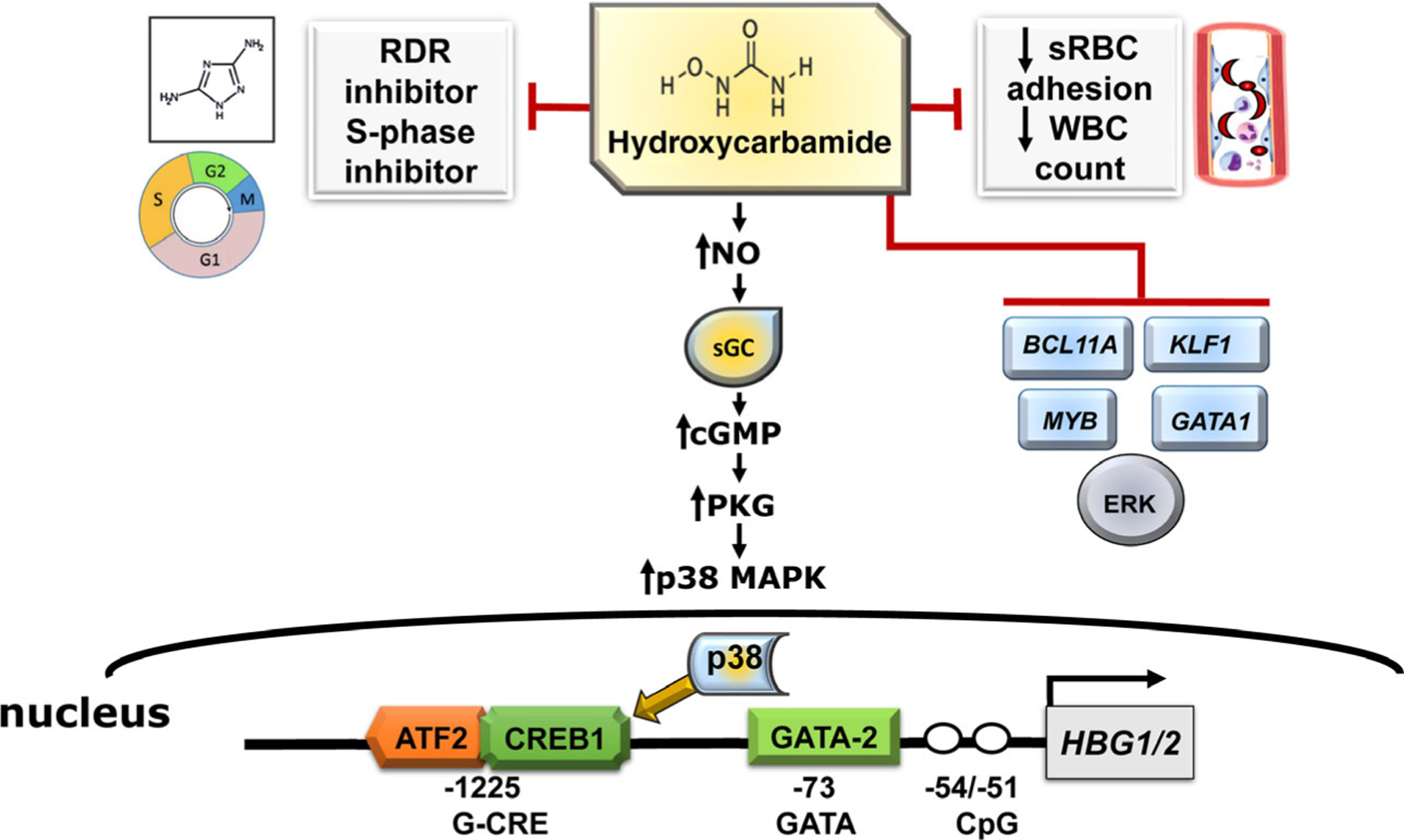
Mechanisms of fetal haemoglobin (HbF) induction by hydroxycarbamide. Shown are the various mechanisms by which hydroxycarbamide impacts the clinical symptoms of sickle cell disease. The generation of nitric oxide (NO) by hydroxycarbamide leads to sGC (soluble guanylyl cyclase) activation and subsequent cGMP-PKG signalling and p38 MAPK (mitogen-activated protein kinase) phosphorylation and repression of ERK MAPK. Once activated, p38 MAPK crosses into the nucleus and activates downstream transcription factors ATF2 and CREB1 to enhance HBG2 expression. Symbols: white circle, unmethylated cytosine. Abbreviations: ATF2, Activating Transcription Factor 2; cGMP-PKG, cyclic guanosine monophosphate-protein kinase g; CREB1, CAMP responsive element binding protein 1; ERK, extracellular signal-regulated kinase; GATA2, GATA-binding factor 2; RDR, ribonucleoside diphosphate reductase; sRBC, sickle red blood cells; WBC, white blood cell.
Recent FDA approval of new therapies
After two decades of clinical use, despite the requirement for regular monitoring of peripheral blood counts and chemistries, hydroxycarbamide has proven to be a safe and effective agent producing mainly reversible bone marrow suppression. Hydroxycarbamide is recommended as the standard of care to be offered to children with SCA starting at nine months of age and adults with severe clinical disease.10 Recently, three additional drugs were FDA-approved including L-glutamine, crizanlizumab and voxelotor.56–59 The phase 3 L-glutamine clinical trial showed a decrease in the median number of VOC pain crises over 48 weeks, regardless of hydroxycarbamide use.60 L-glutamine increases the proportion of reduced nicotinamide adenine dinucleotides in sickle RBCs, which reduces oxidative stress; additional benefits included a decrease in the number of acute chest syndrome episodes. The FDA granted approval of Endari (L-glutamine) for children > 5 years of age and adults with SCA.60 The third disease-modifying drug, voxelotor (Oxbryta) is an HbS polymerization inhibitor with a novel mechanism of action as the first-in-class therapy stabilizing the relaxed (R-state), non-polymerizing confirmation of HbS (shifting the O2 dissociation curve to the left). The net result of treatment is a decrease in the concentration of deoxy-HbS.61 The recently completed phase 3 trial demonstrated a significant increase in total Hb levels and reduction in worsening anaemia and haemolysis in people with SCA.61 The most recently approved crizanlizumab (Adakveo) is a monoclonal antibody against P-selectin that produced a significantly lower frequency of VOC pain episodes regardless of concomitant hydroxycarbamide use.58,62 Now that four FDA-approved drugs are available in the US as disease-modifying therapies in SCA, treatment strategies combining hydroxycarbamide (HbF induction) with drugs targeting other disease pathophysiology are possible. The development of additional novel agents that further ameliorate clinical severity in an additive and/or synergistic manner when combined with hydroxycarbamide, has ushered in an era to develop personalized combination drug regimens for individuals with SCA and/or non-responsive to hydroxycarbamide or with contra-indications for use.
Development of novel clinical therapeutics
After numerous clinical trials failed to produce additional disease-modifying therapies, over the last decade a renewed interest in developing drugs for SCD erupted. It is beyond the scope of this review to discuss the large number of clinical interventions, behaviour and psychosocial trials ongoing for SCD; therefore, we limit our discussion to active interventional trials registered in Clinicaltrials.gov, excluding those related to continued development of hydroxycarbamide, crizanlizumab, L-glutamine and voxelotor (Table I). The agents discussed have made the leap to clinical trials, which is a significant achievement. We organized our discussion into four broad categories including drugs that: (i) enhance metabolic molecules in RBCs; (ii) inhibit HbS polymerization; (iii) targets abnormal vascular physiology; and (iv) have general systemic effects (Fig 2).
Table I.
Summary of active safety and interventional studies for sickle cell disease.*
| Drug | Mechanism of action | Dates | Phase | Subject number | Age | Number of sites | ClinicalTrials.gov Identifier |
|---|---|---|---|---|---|---|---|
| Olinciguat (IW-1701) | Stimulator of soluble guanylate cyclase | 12/2017–2/2020 | 2 | 70 | 16–70 years SCD | 37 | NCT03285178 |
| Riociguat | Stimulator of soluble guanylate cyclase | 11/2017–6/2021 | 2 | 100 | >18 years SCD | 18 | NCT02633397 |
| FT–4202 | PK-R activator | 12/2018–9/20 | 1 | 150 | 12–65 years SCD | 16 | NCT03815695 |
| AG-348 | PK-R activator | 7/2019–12/2020 | 2 | 25 | 18–70 years SCD | 1 | NCT04000165 |
| IMR-687 | PDE9 inhibitor | 1/2018–9/20 | 2 | 70 | 18–55 years SCA | 17 | NCT03401112 |
| Imatinib mesylate | Protein-tyrosine kinase inhibitor | 2/2020–9/2022 | 1 | 20 | 18–25 years SCA | 2 | NCT03997903 |
| Nicotinamide, THU decitabine | Vitamin B3 analogue, cytidine deaminase inhibitor, DNMT1 inhibitor | 1/2020–3/2021 | 1 | 20 | >18 years SCD | 1 | NCT04055818 |
| Benserazide | DOPA decarboxylase inhibitor | 8/2020–9/2023 | 1/2 | 36 | 18–45 years Thal | 2 | NCT04432623 |
| Metformin | Antihyperglycemic agents | 3/2017–11/2023 | 1 | 56 | 10–60 years SCA | 1 | NCT02981329 |
| CSL889 | Haemopexin | 10/2020–5/2022 | 1 | 24 | 18–60 years SCA | NYR | NCT04285827 |
| Ticagrelor | P2Y12-receptor antagonist | 9/2018–12/2020 | 3 | 193 | 2–18 years SCA | 56 | NCT03615924 |
| Ketamine | N-methyl-D-aspartate receptor antagonist | 11/2019–1/2021 | 3 | 40 | 3–25 years SCD | 1 | NCT04150757 |
| Mometasone | Corticosteroids | 11/2018–6/2023 | 2 | 80 | >18 years SCA | 2 | NCT03758950 |
| Ambrisentan | Type-A endothelin receptor antagonist | 9/2015–11/2019 | 1 | 26 | 18–65 years SCA | 1 | NCT02712346 |
| Gamma globulin | Blockade of Fcγ receptors on macrophages | 11/2008–9/2022 | 1/2 | 94 | 12–65 years SCA | 1 | NCT01757418 |
| SCD-101 | Botanical drug with anti-sickling activity | 2/2015–12/2020 | 1 | 60 | 18–55 years SCA | 1 | NCT02380079 |
| SC411 | Omega-3 docosahexaenoic acid | 3/2019–12/2020 | 3 | 210 | 5–17 years SCD | 3 | NCT02604368 |
| HSCT | Matched-unrelated donor/standard care | 11/2016–2/2022 | 2 | 200 | 15–40 years SCD | 35 | NCT02766465 |
| HSCT | Haploidentical T-cell depletion | 5/2020–11/2027 | NA | 38 | 2–25 years SCD | 1 | NCT04207320 |
| HSCT | Matched related ABO-incompatible donor | 7/2017–7/2024 | 2 | 12 | 1–19 years SCD | 1 | NCT03214354 |
| LentiGlobin BB305 | Modified HBB gene | 8/2014–2/2022 | 1/2 | 50 | 12–50 years SCD | 10 | NCT02140554 |
| ARU-1801 | Modified HBG gene | 7/2014–6/2023 | 1/2 | 10 | 18–45 years SCD | 1 | NCT02186418 |
| BCH-BBB694 | Anti-BCL11A RNAi | 2/2018–2/2024 | 1 | 15 | 3–40 years SCD | 1 | NCT03282656 |
| Lenti/G-βAS3-FB | Anti-sickling βAS3 gene | 12/2014–2/2022 | 1/2 | 6 | >18 years SCA | 1 | NCT02247843 |
THU, tetrahydrouridine; HSCT, haematopoietic stem cell transplant; PK-R, pyruvate kinase-R; DNMT1, DNA methyltransferase 1; DOPA, dihydroxyphenylalanine; SCD, sickle cell disease (HbSS, HbSβ0Thal, HbSC and HbSβ+Thal); SCA, sickle cell anaemia (HbSS and HbSβ0Thal); Thal, thalassaemia.
FDA-approved drugs are not included in the table.
Fig 2.
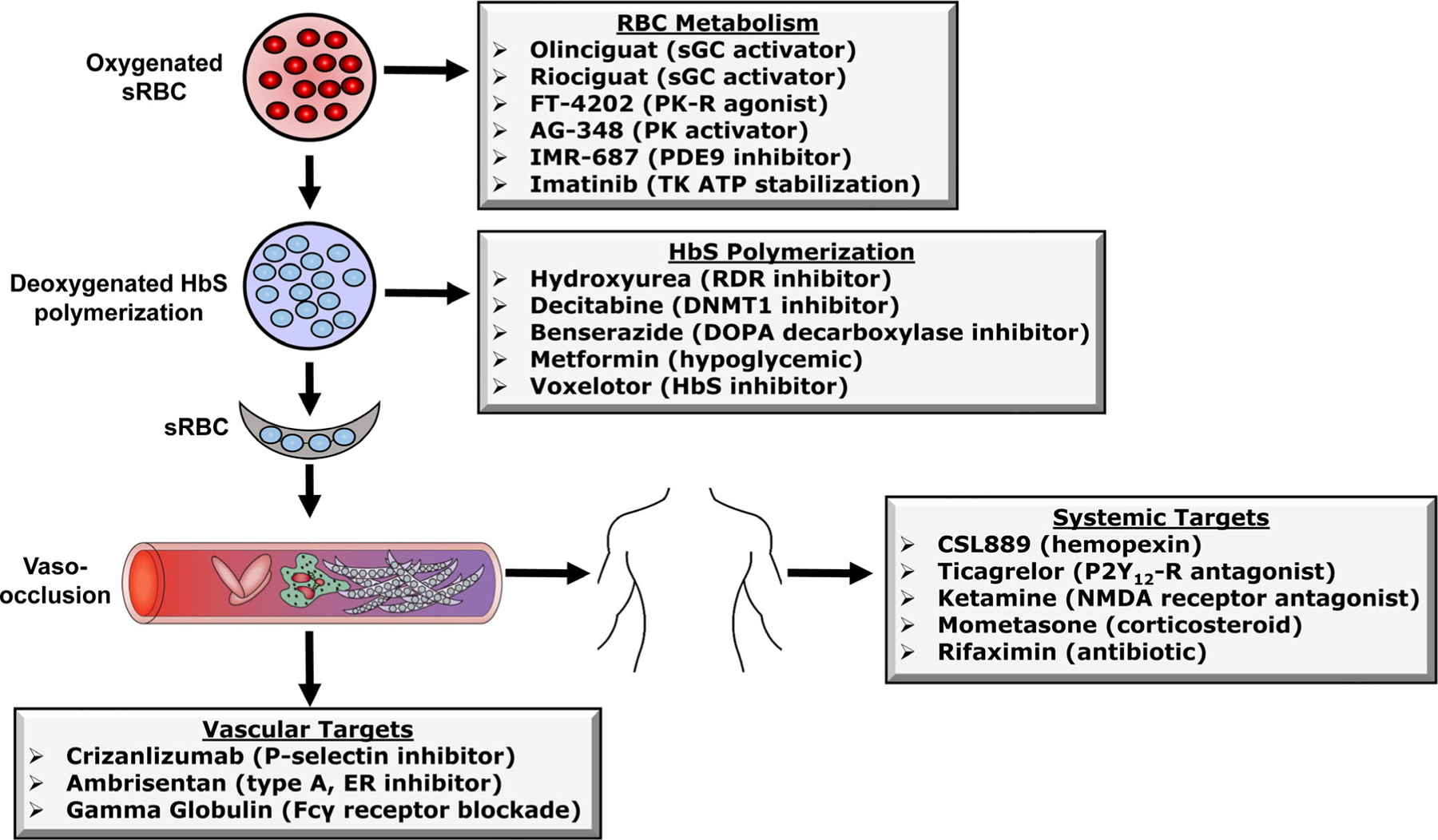
Mechanisms of action of drugs under evaluation in human clinical trials. Shown is a summary of the major cellular, vascular and system mechanisms by which FDA-approved and other drugs improve the clinical severity of sickle cell disease. The effects have been divided into four areas/targets as shown for the sake of discussion in the text. Abbreviations: sRBC, sickle red blood cells; HbS, haemoglobin S; sGC, soluble guanylyl cyclase; PK-R, pyruvate kinase-R; PK; pyruvate kinase; TK, BCR/ABL tyrosine kinase ATP tyrosine kinase, RDR, ribonucleoside diphosphate reductase; DNMT1, DNA methyltransferase 1; DOPA, dihydroxyphenylalanine; ER, endothelial receptor; NMDA, N-Methyl-d-aspartic acid or N-Methyl-d-aspartate; P2YR, purinergic.
Red blood cell metabolism targets
Soluble guanylate cyclase, a key enzyme in the NO signalling pathway, is rapidly attracting interest as a therapeutic target for cardiovascular disease.63–65 Two sGC agonists, riociguat and olinciguat, are currently in clinical development for SCD. After NO binds a haeme group in sGC, the synthesis of cGMP is stimulated to produce vaso-relaxation and inhibition of smooth muscle proliferation, leukocyte activation and platelet aggregation.66 In addition, Cokic and others47,48 provided data that HbF induction by hydroxycarbamide involves activation of sGC through the generation of NO. Early in vitro work67–69 showed that treatment of normal and thalassaemia primary erythroid progenitors with protoporphyrin IX (PPIX) (sGC activator) and bromo-sGC-activated HBG expression. Therefore, clinical and experimental data provide the rationale for current clinical trials to develop combination treatment with hydroxycarbamide and sGC activators.
Riociguat is the only FDA-approved medical therapy for chronic thromboembolic pulmonary hypertension. A case series of six SCD patients demonstrated the safety and tolerability of riociguat use in adults with SCD.70 Riociguat is a suitable vasodilator for pulmonary hypertension without increasing VOC crises in SCD; a phase 2 trial at the University of Pittsburgh is under way to develop riociguat (Table I). A second sGC activator, olinciguat, was investigated in the phase 2-STRONG-SCD trial (Cyclerion, Boston, MA, USA). Enrollment was recently completed, and results are expected to become available in late 2020.
Pyruvate kinase mediates the transfer of a phosphate group from phosphoenolpyruvate to adenosine diphosphate to produce pyruvate and ATP during glycolysis. Decreased concentrations of 2,3-diphosphoglycerate causes a leftward shift in the oxygen dissociation curve thus facilitating oxygen availability to tissues. Activation of pyruvate kinase is predicted to increase ATP in sickle RBCs, decrease 2,3-diphosphoglycerate and HbS polymerization. Another agent, FT-4202 is a novel, small-molecule allosteric activator of erythrocyte pyruvate kinase-R predicted to decrease 2,3-diphosphoglycerate and HbS polymerization and RBC sickling; the increase in ATP should promote RBC repair and reduce chronic haemolysis. FT-4202 was well tolerated in healthy volunteers providing rationale for a phase 1 clinical trial initiation in 2018. A second agent, AG-348 (mitapivat sulfate), previously developed for pyruvate kinase deficiency launched in 2020 as a phase 2 trial to determine efficacy in SCD.
IMR-687 is a small-molecule inhibitor of phosphodiesterase 9A that acts primarily on RBCs with the potential to act on white blood cells and adhesion mediators implicated in the pathophysiology of SCD. This agent is currently being studied in a phase 2 clinical trial in adults with SCA. Another agent, imatinib mesylate (Gleevec) is an oral protein tyrosine kinase inhibitor that targets the BCR-ABL tyrosine kinase, the constitutive active abnormal molecule created by the Philadelphia chromosome in chronic myeloid leukaemia (CML).71 Preclinical data in mouse models suggest that imatinib inhibited c-kit-mediated mast cell activation, and resultant neurogenic inflammation causing chronic pain.72 This agent was used in two SCA patients who experienced reduced frequency of VOC episodes and pain during imatinib treatment for Ph1-positive CML; a phase 1 trial to determine its safety for SCA launched in 2020.
Haemoglobin F inducers
Direct modification of DNA by methylation of its cytidine residues is associated with transcriptional repression or gene silencing during development.73 The CpG nucleotides in the γ-globin promoters of adults (with lower HbF) are methylated by a group of enzymes known as the DNA methyltransferases (DNMT). This process is completed over the first year of life in normal infants.74 The development of DNMT inhibition, through gene silencing in human erythroid precursors, using chemicals or DNMT1 knockout in transgenic mice, results in reactivation of HBG transcription.75,76 These observations led to investigations of DNMT1 inhibitors including cytidine 5-azacytidine and its analogue 2′deoxy-5-azacytidine (decitabine) for HbF induction.77–81 Decitabine incorporates into DNA82 and is a potent DNMT1 inhibitor with a favourable safety profile. Parenteral decitabine has been shown to be effective in increasing HbF in patients with SCD, including those refractory to hydroxycarbamide therapy.83 However, oral administration of decitabine is not effective, due to rapid degradation by intestinal cytidine deaminase. Newer regimens incorporating oral administration of tetrahydrouridine before decitabine resulted in a significant increase in F cells, HbF and total Hb level in people with SCD.84 In the US, the FDA recently approved the combination of oral tetrahydrouridine and decitabine (INQOVI) for use in myelodysplastic syndrome.85
Benserazide is an L-Dopa decarboxylase inhibitor, approved in Europe for Parkinson’s disease for over two decades.86 To expand safe and orally bioavailable γ-globin-inducing agents for SCD and β-thalassaemia, a high-throughput drug screen was complete that showed the ability of benserazide to increase γ-globin promoter activity. Three lead drugs were subsequently evaluated in erythroid progenitors, anaemic baboons and transgenic mice.87 Benserazide produced > 20-fold induction of γ-globin mRNA expression in anaemic baboons and increased F-cell numbers by 3 5-fold in transgenic mice. Recently, a phase 1 clinical trial was opened, to test benserazide for safety and toxicity in β-thalassaemia patients. Another agent, metformin, currently used in type 2 diabetes was shown to induce HbF levels in primary erythroid cells through FOXO3 silencing.88 Subsequently, a phase 1 clinical trial was initiated in patients with SCA in 2017; the results of the trial outcome have not been published.
Vascular targets
Pulmonary arterial hypertension is a progressive disease defined by an elevation in pulmonary arterial pressure that can lead to right heart failure and death. Ambrisentan is a selective endothelin receptor antagonist approved for the treatment of idiopathic, heritable pulmonary arterial hypertension. This agent was tested in SCA in a phase 1, clinical trial (Table I), which is under analysis at this time.89 Intravenous immunoglobulin decreases neutrophil adhesion to endothelium and red blood cell-neutrophil interactions in sickle cell mice during VOC. In a phase I clinical trial SCA patients were treated with a single dose of intravenous immunoglobulin, which showed a decrease in membrane-activated complex-1 (Mac-1) from baseline in low dose (200–400 mg/kg) with limited adverse events. These data provide evidence that intravenous immunoglobulin can decrease neutrophil adhesion through Mac-1 inhibition.90 A phase 1, clinical trial is currently under way (Table I).
Systemic targets
After intravascular haemolysis the Hb released in plasma will be oxidized into met-Hb, which further disassociates into free haeme and globin chains. After the oxidation of free haeme, it is bound and detoxified by haemopexin, a plasma protein with high binding affinity.91 Haeme is a toxic and pro-oxidant molecule released in excessive amounts in SCA and thalassaemia patients.92,93 When free haeme overwhelms the ability of haemopexin to detoxify, haeme-iron loading of reticuloendothelial system macrophages and chronic inflammation occurs.94,95 To test the safety and tolerability of haemopexin (CSL8) a single-dose exploratory trial in patients with SCA is scheduled to open in late 2020. A chronic inflammatory state contributes to the most common clinical symptom of SCD, which is VOC pain. Many opioid-based therapies, such as morphine, hydrocodone, codeine and dilaudid, have addiction potential when used long-term on a daily basis. In addition, there is a high risk of developing morphine-induced tolerance and hyperalgesia, creating a need to develop non-opioid agents for pain treatment. Several clinical trials are in progress with the primary end-point to decrease VOC pain episodes in SCD (Table I). Ticagrelor (Hestia3) is an anti-platelet drug that will be evaluated for efficacy, safety and tolerability in children with severe SCA. Ticagrelor blocks platelets, which may reduce symptoms of SCA caused by ischaemia during VOC episodes. Ketamine is a non-opioid N-methyl-d-aspartic acid (NMDA) receptor antagonist commonly used for anaesthesia, sedation and pain.96,97 While the identity of the active substances is unknown in SCD-101 (botanical drug), this agent has a direct anti-sickling effect on RBCs; a multi-site phase 1 study in SCA patients is ongoing to test this agent (Table I). Mometasone is a topical corticosteroid used to treat skin conditions, such as eczema and psoriasis, and inflammation.98 A phase 2 trial will test the ability of inhaled mometasone to reduce vascular cell adhesion molecule levels in SCA patients. Studies have shown that SCA patients have a deficiency of essential omega-3 fatty acids and supplementation supports a decrease in VOC pain.99 The primary objective of the SC411 (omega-3 docosahexaenoic acid) phase 3 trial in SCD patients is to evaluate the safety and tolerability of three different doses of SC411 and the primary end-point, number of VOC episodes.
Opportunities for precision medicine using combination therapy
The elucidation of the downstream effects of deoxy-HbS polymerization (ischaemia/reperfusion injury, chronic inflammation, NO scavenging, endothelial cell activation, increased expression of adhesion molecules, etc.), on one hand, and a better understanding of the mechanisms of fetal-to-adult Hb switching over the past three decades have provided the rationale to conceptualize combination therapies for SCD. These can be combination anti-switching therapies, and/or combination of anti-switching and anti-sickling targeting one or more downstream effects.9 The proof of concept for combined therapies was provided in two previous studies including Atweh et al.100 demonstrating hydroxycarbamide and butyrate resulted in higher HbF levels for individuals failing treatment with butyrate alone. Moreover, Xu et al.101 demonstrated that erythroid-specific knockout of BCL11A in SCD mice results in higher HbF expression, which was enhanced further with epigenetic modifiers, such as the histone deacetylase inhibitor suberoylanilide hydroxamic acid, and the DNMT1 inhibitor 5-azacytidine. The challenge for the future is how to design/conduct rational combination therapeutic regimens. These approaches will likely be guided by severity of clinical phenotype, principles of combination chemotherapy for neoplastic diseases (such as non-overlapping toxicities) and testing in preclinical animal models. Combination of orally available, safe and well-tolerated agents will likely have a huge impact on the global burden of SCD, especially in low-resource, developing countries.
Haematopoietic stem cell transplant cure
Several clinical trials are ongoing to expand haematopoietic stem cell transplant options for patient with SCD. The Bone Marrow Transplantation vs. Standard of Care in Patients with Severe Sickle Cell Disease (BMT CTN 1503) (STRIDE2) clinical trial will compare survival and sickle-related outcomes in adolescents and young adults (age 15–40) with severe SCD. Patients will be evaluated after bone marrow transplantation or standard of care (Table I) with the primary outcome of two-year overall survival. This is a prospective phase II multicentre trial of haematopoietic stem cell transplantation based on availability of human leukocyte antigen (HLA)-matched related or unrelated donor after confirmation of clinical eligibility. The goal of the study is to establish that the difference in the proportion of patients surviving is not significantly more than 15% lower in the donor arm at two years after assignment to treatment arm. The study has so far enrolled 127 patients (64% of projected 200) and will be closed at the end of November 2020.
The overwhelming majority of haematopoietic stem cell transplantations have been performed in children with HLA-identical sibling donors and have resulted in excellent rates of survival and cure of SCD. Increasingly, the use of alternate donors, such as non-sibling HLA-matched donors, unrelated donors and haplo-identical donors, has the potential to expand the applicability of haematopoietic stem cell transplantation. In view of the excellent results with matching sibling donor stem cell transplantation for SCA, patient with cerebral vasculopathy, silent infarcts, stenosis, or abnormal transcranial Doppler screen (with familial HLA typing) should be offered this option. A more recent trial seeks to expand the donor pool further. For example, the NCT04207320 trial (Table I) will investigate a safe and curative approach to treating SCD using haploidentical haematopoietic stem cell transplantation using αβ+ T-cell depletion for children and adolescents with severe SCD.102 A second study, NCT03214354, will evaluate the safety and efficacy of a non-myeloablative conditioning regimen for allogeneic haematopoietic stem cell transplantation in paediatric patients with SCD who have a matched related major ABO-incompatible donor. The non-myeloablative regimen used is alemtuzumab, total body irradiation and sirolimus for immune suppression. This study will expand the access of haematopoietic stem cell transplantation for patients with SCA who are currently not eligible because of donor restrictions. The remaining 25 active haematopoietic stem cell transplantation protocols in Clinicaltrials.gov for SCA are beyond the scope of this review.
Gene therapy cure
Gene therapy using ex vivo autologous haematopoietic stem cell transplantation removes the limitation of matched related donor availability, decreases the need for immuno-suppressive drugs, and trhe risk of graft-versus-host disease. There are several possible gene-editing strategies to treat individuals with HbSS disease by gene therapy including correction of the A to T point mutation in the HBB locus or induction of HbF through: (i) HBG gene addition; (ii) downregulation of BCL11A in an erythroid-specific manner targeting the exon 2 enhancer; or (iii) introduction of HPFH deletions. Creating HPFH mutations using CRISPR–Cas9 technology was established as an alternative approach using in vitro tissue culture systems.103 Several HPFH mutations occur in the −118 to −114 region, upstream of the HBG1 and HBG2 transcription start sites that disrupt a cognate-binding element (TGACC) for the repressor protein BCL11A. Proof-of-principal studies using CRISPR–Cas9 technology demonstrated feasibility of this approach to induce HbF and recapitulate naturally occurring HPFH mutations.104–109 Studies using the same approach to disrupt expression of KLF1 (exons 2 and 3), a major transcription factor involved in HBG silencing during haemoglobin switching resulted in reactivation of HbF expression.110,111
Correction of the HBB or high-level pancellular HbF expression could be curative, therefore we will discuss gene therapy drugs currently in clinical trials to achieve both approaches.112,113 In 2014, the first SCA patient was successfully treated with gene therapy as part of a phase 1/2 clinical trial (HGB-205). This is a lentivirus-based vector consisting of a modified human β-globin gene that inhibits HbS polymerization.114 Fifteen months after treatment, the level of anti-sickling β-globin remained ~ 50% of total β-like globin chains without recurrence of VOC crises and correction of other biologic markers of severe disease.115 Subsequently, the first clinical trial for SCD was opened in the US with the LentiGlobin BB305 drug product. To date, over 20 patients have been treated with variations of BB305 and/or preparatory regimens combined with autologous transplant of ex vivo transduced haematopoietic CD34+ stem cells.115 Most of the experience and safety data collected using the LentiGlobin BB305 drug product comes from the non-randomized, open-label, multi-site, single-dose, phase 1/2 study (NCT02140554) for adults and adolescents with severe SCD.
Two methods to increase HbF expression have reached in-patient treatment. The phase 1/2 clinical trial NCT02186418 determined the feasibility and safety of a lentiviral vector-comprised HBG was initiated in 2014. Subsequently in 2018, the FDA-approved ARU-1801 as an orphan drug with gene therapy potential for SCD. ARU-1801 is expected to increase HbF expression to prevent HbS polymerization and this drug is novel since engraftment occurs with a reduced-intensity-conditioning regimen. Two adult HbS-β0thalassaemia patients were treated and early results showed excellent safety, feasibility and rapid bone marrow recovery. The drug also produced a significant amelioration of clinical symptoms with 20% vector-derived HbF positive RBCs. Recent progress was also made to develop a lentiviral vector carrying the short hairpin miR233 that mediates downregulation of BCL11A via RNA interference selectively in erythroid cells using a microRNA adapted short hairpin RNA.116 After remarkable in vitro data, supporting high-level HbF induction and preclinical data demonstrating the production of the clinical-grade drug BCH-bbb694, a pilot and feasibility clinical trial for the treatment of SCD was initiated (NCT03282656). Other more experimental approaches to develop gene therapy for SCD include gene editing using CRISR–Cas9 technology are under way. Recent studies showed a ~40% reduction in BCL11A mRNA with gene knockout with a corresponding two-fold increase in HBG mRNA transcripts and 30–40% HbF induction.117,118 Similar decreases in BCL11A expression and increases in HBG transcription were observed in SCD-derived erythroid progenitors. Subsequent preclinical studies with HbSS mice using CRISPR–Cas9 approaches increase the number of RBCs carrying HbF by 30–40%. In March 2020, the US FDA cleared the start of a phase 1/2 clinical trial testing the CRISPR–Cas9-based drug, OTQ923, in adults with severe SCA.
Future directions
Where do we go from here after five decades of laboratory, animal and clinical trials data explicating the pathophysiology of SCD, and successful treatment of patients with broad cytotoxic agents such as hydroxycarbamide and newer mechanism-based therapies? With expanded knowledge of the pathophysiology of SCD, drugs that directly target HbS polymerization in RBCs have launched new avenues for investigation. For example, voxelotor emerging as a first-in-class agent was recently shown to be safe and effective to use in SCA. Recently, the American Society of Hematology, Research Collaborative, Clinical Trials Network was established in 2018 (https://www.ashresearchcollaborative.org/clinical-trials-network) with the goal ‘to improve the lives of people affected by blood diseases and to accelerate progress in hematology’. This network will facilitate the efficient identification of clinical trial sites and allow testing of new drug candidates in the patient populations most likely to benefit and will facilitate more rapid drug approval. This initiative is a transformative, multi-faceted, patient-centric effort to improve outcomes for individuals with SCD, both in the US and worldwide, by bringing together all medical and support staff personal dedicated to improving the lives of people with SCD.
The race is not over — we have a lot more work to do. Now that we have preclinical HbSS mouse models, which facilitate drug screening before human trials, we can rapidly test the effects of drug combination therapies with hydroxycarbamide and other agents that work by different mechanisms, such as L-glutamine and crizanlizumab that target downstream effects of VOC episodes. Alternatively, combining hydroxycarbamide with other agents such as decitabine, or other agents under development could produce additive HbF induction or be offered to patients non-responsive to hydroxycarbamide (Table I). Potential development of novel therapies such as microRNAs, haemoglobin substitutes for acute anaemia, etc. are still ongoing. Whether there is a class of other agents to be discovered using high-throughput drug screens, computer modelling and/or artificial intelligence are new frontiers to be explored.
Bone marrow transplantation and gene therapy efforts have been steady over the last 40 years, with haematopoietic stem cell transplantation (HSCT) moving forward, so that families with sibling donors have the choice for safe treatment in medical centres with the expertise to carry out these complex procedures. With the establishment of centres of excellence and gene therapy efforts by many approaches, the stage is set to provide options for cure for every child or adult using their own haematopoietic stems cells. We applaud Dr. Francis Collins, Director of the National Institutes of Health, for supporting the Cure Sickle Cell Disease Initiative (CURESCI) launched in September 2018 through the leadership of the National Heart Lung and Blood Institute. This initiative will fund gene research therapy efforts for academic faculty combined with the laudable efforts of pharmaceutical companies, to bring gene therapy to a reality. We are all labouring (primary doctors, haematologists, nurses, research staff, drug companies, American Society of Hematology, National Institutes of Health and beyond), on behalf of families, to improve the quality of care delivered and improved access to care, such that all patients benefit from these wonderful advances.
Acknowledgements
This work was supported in part by HL149365-01A1 to BSP, U24HL13339 to AK and HL144641 to AS-D.
References
- 1.Kauf TL, Coates TD, Huazhi L, Mody-Patel N, Hartzema AG. The cost of health care for children and adults with sickle cell disease. Am J Hematol. 2009;84:323–7. [DOI] [PubMed] [Google Scholar]
- 2.Ashley-Koch A, Yang Q, Olney RS. Sickle hemoglobin (HbS) allele and sickle cell disease: a HuGE review. Am J Epidemiol. 2000;151:839–45. [DOI] [PubMed] [Google Scholar]
- 3.Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood. 2010;115:3447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gardner K, Douiri A, Drasar E, Allman M, Mwirigi A, Awogbade M, et al. Survival in adults with sickle cell disease in a high-income setting. Blood. 2016;128:1436–8. [DOI] [PubMed] [Google Scholar]
- 5.Thein SL, Howard J. How I treat the older adult with sickle cell disease. Blood. 2018;132:1750–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weatherall DJ. The role of the inherited disorders of hemoglobin, the first “molecular diseases,” in the future of human genetics. Annu Rev Genomics Hum Genet. 2013;14:1–24. [DOI] [PubMed] [Google Scholar]
- 7.Hebbel RP, Boogaerts MA, Eaton JW, Steinberg MH. Erythrocyte adherence to endothelium in sickle-cell anemia. A possible determinant of disease severity. N Engl J Med. 1980;302:992–5. [DOI] [PubMed] [Google Scholar]
- 8.Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350:886–95. [DOI] [PubMed] [Google Scholar]
- 9.Hebbel RP, Hedlund BE. Sickle hemoglobin oxygen affinity-shifting strategies have unequal cerebrovascular risks. Am J Hematol. 2018;93:321–5. [DOI] [PubMed] [Google Scholar]
- 10.Lung Heart N., and Blood Institute. Evidence-Based Management of Sickle Cell Disease: Expert Panel Report. 2014;2014:1–48. [Google Scholar]
- 11.Gaston MH, Verter JI, Woods G, Pegelow C, Kelleher J, Presbury G, et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N Engl J Med. 1986;314:1593–9. [DOI] [PubMed] [Google Scholar]
- 12.Farrall M, Scambler P, Klinger KW, Davies K, Worrall C, Williamson R, et al. Cystic fibrosis carrier detection using a linked gene probe. J Med Genet. 1986;23:295–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicklas TA, Kuvibidila S, Gatewood LC, Metzinger AB, Frempong KO. Prevalence of anaemia and iron deficiency in urban Haitian children two to five years of age. J Trop Pediatr. 1998;44:133–8. [DOI] [PubMed] [Google Scholar]
- 14.Adams JG 3rd, Coleman MB. Simian switching suggests solution to the symptoms of the sickle cell syndromes. Exp Hematol. 1992;20:1151–3. [PubMed] [Google Scholar]
- 15.Adams RJ, McKie VC, Brambilla D, Carl E, Gallagher D, Nichols FT, et al. Stroke prevention trial in sickle cell anemia. Control Clin Trials. 1998;19:110–29. [DOI] [PubMed] [Google Scholar]
- 16.Barriteau CM, Murdoch A, Gallagher SJ, Thompson AA. A patient-centered medical home model for comprehensive sickle cell care in infants and young children. Pediatr Blood Cancer. 2020;67:e28275. [DOI] [PubMed] [Google Scholar]
- 17.Reeves SL, Madden B, Freed GL, Dombkowski KJ. Transcranial Doppler Screening Among Children and Adolescents With Sickle Cell Anemia. JAMA Pediatr. 2016;170:550–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wutzen J, Lewicki Z. The effect of immobilization on ultrastructural changes in the myocardium of rats on a low-magnesium diet. Mater Med Pol. 1985;17:221–6. [PubMed] [Google Scholar]
- 19.Kim A, Dean A. Chromatin loop formation in the beta-globin locus and its role in globin gene transcription. Mol Cells. 2012;34:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Breda L, Motta I, Lourenco S, Gemmo C, Deng W, Rupon JW, et al. Forced chromatin looping raises fetal hemoglobin in adult sickle cells to higher levels than pharmacologic inducers. Blood. 2016;128:1139–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Love PE, Warzecha C, Li L. Ldb1 complexes: the new master regulators of erythroid gene transcription. Trends Genet. 2014;30:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li L, Freudenberg J, Cui K, Dale R, Song SH, Dean A, et al. Ldb1-nucleated transcription complexes function as primary mediators of global erythroid gene activation. Blood. 2013;121:4575–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Z, Huang S, Chang LS, Agulnick AD, Brandt SJ. Identification of a TAL1 target gene reveals a positive role for the LIM domain-binding protein Ldb1 in erythroid gene expression and differentiation. Mol Cell Biol. 2003;23:7585–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Enver T, Raich N, Ebens AJ, Papayannopoulou T, Costantini F, Stamatoyannopoulos G. Developmental regulation of human fetal-to-adult globin gene switching in transgenic mice. Nature. 1990;344:309–13. [DOI] [PubMed] [Google Scholar]
- 25.Sankaran VG, Xu J, Byron R, Greisman HA, Fisher C, Weatherall DJ, et al. A functional element necessary for fetal hemoglobin silencing. N Engl J Med. 2011;365:807–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watson J The significance of the paucity of sickle cells in newborn Negro infants. Am J Med Sci. 1948;215:419–23. [DOI] [PubMed] [Google Scholar]
- 27.Shaukat I, Pudal A, Yassin S, Hoti N, Mustafa S. Blessing in disguise; a case of Hereditary Persistence of Fetal Hemoglobin. J Community Hosp Intern Med Perspect. 2018;8:380–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akinsheye I, Alsultan A, Solovieff N, Ngo D, Baldwin CT, Sebastiani P, et al. Fetal hemoglobin in sickle cell anemia. Blood. 2011;118:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galarneau G, Palmer CD, Sankaran VG, Orkin SH, Hirschhorn JN, Lettre G. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet. 2010;42:1049–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Platt OS, Orkin SH, Dover G, Beardsley GP, Miller B, Nathan DG. Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. J Clin Invest. 1984;74:652–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Powars DR, Weiss JN, Chan LS, Schroeder WA. Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood. 1984;63:921–6. [PubMed] [Google Scholar]
- 32.Estepp JH, Smeltzer MP, Kang G, Li C, Wang WC, Abrams C, et al. A clinically meaningful fetal hemoglobin threshold for children with sickle cell anemia during hydroxyurea therapy. Am J Hematol. 2017;92:1333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Noguchi CT, Rodgers GP, Schechter AN. Intracellular polymerization of sickle hemoglobin: disease severity and therapeutic goals. Prog Clin Biol Res. 1987;240:381–91. [PubMed] [Google Scholar]
- 34.DeSimone J, Heller P, Hall L, Zwiers D. 5-Azacytidine stimulates fetal hemoglobin synthesis in anemic baboons. Proc Natl Acad Sci U S A. 1982;79:4428–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papayannopoulou T, Torrealba de Ron A, Veith R, Knitter G, Stamatoyannopoulos G. Arabinosylcytosine induces fetal hemoglobin in baboons by perturbing erythroid cell differentiation kinetics. Science. 1984;224:617–9. [DOI] [PubMed] [Google Scholar]
- 36.Veith R, Papayannopoulou T, Kurachi S, Stamatoyannopoulos G. Treatment of baboon with vinblastine: insights into the mechanisms of pharmacologic stimulation of Hb F in the adult. Blood. 1985;66:456–9. [PubMed] [Google Scholar]
- 37.Liu DP, Liang CC, Ao ZH, Jia PC, Chen SS, Wang RX, et al. Treatment of severe beta-thalassemia (patients) with myleran. Am J Hematol. 1990;33:50–5. [DOI] [PubMed] [Google Scholar]
- 38.Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995;332:1317–22. [DOI] [PubMed] [Google Scholar]
- 39.Steinberg MH, McCarthy WF, Castro O, Ballas SK, Armstrong FD, Smith W, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am J Hematol. 2010;85:403–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferster A, Vermylen C, Cornu G, Buyse M, Corazza F, Devalck C, et al. Hydroxyurea for treatment of severe sickle cell anemia: a pediatric clinical trial. Blood. 1996;88:1960–4. [PubMed] [Google Scholar]
- 41.Kinney TR, Sleeper LA, Wang WC, Zimmerman RA, Pegelow CH, Ohene-Frempong K, et al. Silent cerebral infarcts in sickle cell anemia: a risk factor analysis. The Cooperative Study of Sickle Cell Disease. Pediatrics. 1999;103:640–5. [DOI] [PubMed] [Google Scholar]
- 42.Ware RE, Davis BR, Schultz WH, Brown RC, Aygun B, Sarnaik S, et al. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial Doppler flow velocities in children with sickle cell anaemia-TCD With Transfusions Changing to Hydroxyurea (TWiTCH): a multicentre, open-label, phase 3, non-inferiority trial. Lancet. 2016;387:661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elford HL. Effect of hydroxyurea on ribonucleotide reductase. Biochem Biophys Res Commun. 1968;33:129–35. [DOI] [PubMed] [Google Scholar]
- 44.Galanello R, Veith R, Papayannopoulou T, Stamatoyannopoulos G. Pharmacologic stimulation of Hb F in patients with sickle cell anemia. Prog Clin Biol Res. 1985;191:433–45. [PubMed] [Google Scholar]
- 45.Stamatoyannopoulos G, Veith R, Galanello R, Papayannopoulou T. Hb F production in stressed erythropoiesis: observations and kinetic models. Ann N Y Acad Sci. 1985;445:188–97. [DOI] [PubMed] [Google Scholar]
- 46.Huang J, Sommers EM, Kim-Shapiro DB, King SB. Horseradish peroxidase catalyzed nitric oxide formation from hydroxyurea. J Am Chem Soc. 2002;124:3473–80. [DOI] [PubMed] [Google Scholar]
- 47.Cokic VP, Smith RD, Beleslin-Cokic BB, Njoroge JM, Miller JL, Gladwin MT, et al. Hydroxyurea induces fetal hemoglobin by the nitric oxide-dependent activation of soluble guanylyl cyclase. J Clin Invest. 2003;111:231–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lou TF, Singh M, Mackie A, Li W, Pace BS. Hydroxyurea generates nitric oxide in human erythroid cells: mechanisms for gamma-globin gene activation. Exp Biol Med (Maywood). 2009;234:1374–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Almeida CB, Scheiermann C, Jang JE, Prophete C, Costa FF, Conran N, et al. Hydroxyurea and a cGMP-amplifying agent have immediate benefits on acute vaso-occlusive events in sickle cell disease mice. Blood. 2012;120:2879–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ikuta T, Thatte HS, Tang JX, Mukerji I, Knee K, Bridges KR, et al. Nitric oxide reduces sickle hemoglobin polymerization: potential role of nitric oxide-induced charge alteration in depolymerization. Arch Biochem Biophys. 2011;510:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park JI, Choi HS, Jeong JS, Han JY, Kim IH. Involvement of p38 kinase in hydroxyurea-induced differentiation of K562 cells. Cell Growth Differ. 2001;12:481–6. [PubMed] [Google Scholar]
- 52.Walker AL, Steward S, Howard TA, Mortier N, Smeltzer M, Wang YD, et al. Epigenetic and molecular profiles of erythroid cells after hydroxyurea treatment in sickle cell anemia. Blood. 2011;118:5664–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pule GD, Mowla S, Novitzky N, Wonkam A. Hydroxyurea down-regulates BCL11A, KLF-1 and MYB through miRNA-mediated actions to induce gamma-globin expression: implications for new therapeutic approaches of sickle cell disease. Clin Transl Med. 2016;5:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grieco AJ, Billett HH, Green NS, Driscoll MC, Bouhassira EE. Variation in Gamma-Globin Expression before and after Induction with Hydroxyurea Associated with BCL11A, KLF1 and TAL1. PLoS One. 2015;10: e0129431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu X, Li B, Pace BS. NRF2 mediates gamma-globin gene regulation and fetal hemoglobin induction in human erythroid progenitors. Haematologica. 2017;102:e285–e288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Emmaus Medical Inc. ENDARI (L-glutamine oral powder) [Package insert] Torrance CA. 2017. [Google Scholar]
- 57.Ballas SK. The Evolving Pharmacotherapeutic Landscape for the Treatment of Sickle Cell Disease. Mediterr J Hematol Infect Dis. 2020;12: e2020010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017;376:429–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Global blood therapeutics. OXBRYTA (voxelotor) tablets [Package insert] San Francisco, CA: 2019. [Google Scholar]
- 60.Niihara Y, Miller ST, Kanter J, Lanzkron S, Smith WR, Hsu LL, et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N Engl J Med. 2018;379:226–35. [DOI] [PubMed] [Google Scholar]
- 61.Vichinsky E, Hoppe CC, Ataga KI, Ware RE, Nduba V, El-Beshlawy A, et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N Engl J Med. 2019;381:509–19. [DOI] [PubMed] [Google Scholar]
- 62.Kutlar A, Kanter J, Liles DK, Alvarez OA, Cancado RD, Friedrisch JR, et al. Effect of crizanlizumab on pain crises in subgroups of patients with sickle cell disease: A SUSTAIN study analysis. Am J Hematol. 2019;94:55–61. [DOI] [PubMed] [Google Scholar]
- 63.Tobin JV, Zimmer DP, Shea C, Germano P, Bernier SG, Liu G, et al. Pharmacological Characterization of IW-1973, a Novel Soluble Guanylate Cyclase Stimulator with Extensive Tissue Distribution, Antihypertensive, Anti-Inflammatory, and Antifibrotic Effects in Preclinical Models of Disease. J Pharmacol Exp Ther. 2018;365:664–75. [DOI] [PubMed] [Google Scholar]
- 64.Zhao Y, Vanhoutte PM, Leung SW. Vascular nitric oxide: Beyond eNOS. J Pharmacol Sci. 2015;129:83–94. [DOI] [PubMed] [Google Scholar]
- 65.Breitenstein S, Roessig L, Sandner P, Lewis KS. Novel sGC stimulators and sGC activators for the treatment of heart failure. Handb Exp Pharmacol. 2017;243:225–47. [DOI] [PubMed] [Google Scholar]
- 66.Stasch JP, Pacher P, Evgenov OV. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation. 2011;123:2263–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ikuta T, Ausenda S, Cappellini MD. Mechanism for fetal globin gene expression: role of the soluble guanylate cyclase-cGMP-dependent protein kinase pathway. Proc Natl Acad Sci U S A. 2001;98:1847–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kuroyanagi Y, Kaneko Y, Muta K, Park BS, Moi P, Ausenda S, et al. cAMP differentially regulates gamma-globin gene expression in erythroleukemic cells and primary erythroblasts through c-Myb expression. Biochem Biophys Res Commun. 2006;344:1038–47. [DOI] [PubMed] [Google Scholar]
- 69.Inoue A, Kuroyanagi Y, Terui K, Moi P, Ikuta T. Negative regulation of gamma-globin gene expression by cyclic AMP-dependent pathway in erythroid cells. Exp Hematol. 2004;32:244–53. [DOI] [PubMed] [Google Scholar]
- 70.Weir NA, Conrey A, Lewis D, Mehari A. Riociguat use in sickle cell related chronic thromboembolic pulmonary hypertension: A case series. Pulm Circ. 2018;8:2045894018791802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Waller CF. Imatinib mesylate. Recent Results Cancer Res. 2018;212:1–27. [DOI] [PubMed] [Google Scholar]
- 72.Vincent L, Vang D, Nguyen J, Gupta M, Luk K, Ericson ME, et al. Mast cell activation contributes to sickle cell pathobiology and pain in mice. Blood. 2013;122:1853–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van der Ploeg LH, Flavell RA. DNA methylation in the human gamma delta beta-globin locus in erythroid and nonerythroid tissues. Cell. 1980;19:947–58. [DOI] [PubMed] [Google Scholar]
- 74.Mabaera R, Richardson CA, Johnson K, Hsu M, Fiering S, Lowrey CH. Developmental- and differentiation-specific patterns of human gamma- and beta-globin promoter DNA methylation. Blood. 2007;110:1343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Charache S, Dover G, Smith K, Talbot CC Jr, Moyer M, Boyer S. Treatment of sickle cell anemia with 5-azacytidine results in increased fetal hemoglobin production and is associated with nonrandom hypomethylation of DNA around the gamma-delta-beta-globin gene complex. Proc Natl Acad Sci U S A. 1983;80:4842–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu J, Bauer DE, Kerenyi MA, Vo TD, Hou S, Hsu YJ, et al. Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc Natl Acad Sci U S A. 2013;110:6518–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gilmartin AG, Groy A, Gore ER, Atkins C, Long ER 3rd, Montoute MN, et al. In vitro and in vivo induction of fetal hemoglobin with a reversible and selective DNMT1 inhibitor. Haematologica. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fathallah H, Atweh GF. Induction of fetal hemoglobin in the treatment of sickle cell disease. Hematology Am Soc Hematol Educ Program. 2006;58–62. [DOI] [PubMed] [Google Scholar]
- 79.Kalantri SA, Ray R, Chattopadhyay A, Bhattacharjee S, Biswas A, Bhattacharyya M. Efficacy of decitabine as hemoglobin F inducer in HbE/beta-thalassemia. Ann Hematol. 2018;97:1689–94. [DOI] [PubMed] [Google Scholar]
- 80.Koshy M, Dorn L, Bressler L, Molokie R, Lavelle D, Talischy N, et al. 2-deoxy 5-azacytidine and fetal hemoglobin induction in sickle cell anemia. Blood. 2000;96:2379–84. [PubMed] [Google Scholar]
- 81.Lavelle D, Engel JD, Saunthararajah Y. Fetal Hemoglobin Induction by Epigenetic Drugs. Semin Hematol. 2018;55:60–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lavelle DE. The molecular mechanism of fetal hemoglobin reactivation. Semin Hematol. 2004;41:3–10. [DOI] [PubMed] [Google Scholar]
- 83.Saunthararajah Y, Molokie R, Saraf S, Sidhwani S, Gowhari M, Vara S, et al. Clinical effectiveness of decitabine in severe sickle cell disease. Br J Haematol. 2008;141:126–9. [DOI] [PubMed] [Google Scholar]
- 84.Molokie R, Lavelle D, Gowhari M, Pacini M, Krauz L, Hassan J, et al. Oral tetrahydrouridine and decitabine for non-cytotoxic epigenetic gene regulation in sickle cell disease: A randomized phase 1 study. PLoS Medicine. 2017;14:e1002382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Food and Drug Administration. FDA approves oral combination of decitabine and cedazuridine for myelodysplastic syndromes. July 7, 2020.
- 86.Alborghetti M, Nicoletti F. Different Generations of Type-B Monoamine Oxidase Inhibitors in Parkinson’s Disease: From Bench to Bedside. Curr Neuropharmacol. 2019;17:861–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Boosalis MS, Sangerman JI, White GL, Wolf RF, Shen L, Dai Y, et al. Novel inducers of fetal globin identified through high throughput screening (HTS) are active in vivo in anemic baboons and transgenic mice. PLoS One. 2015;10:e0144660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang Y, Paikari A, Sumazin P, Ginter Summarell CC, Crosby JR, Boerwinkle E, et al. Metformin induces FOXO3-dependent fetal hemoglobin production in human primary erythroid cells. Blood. 2018;132:321–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rivera-Lebron BN, Risbano MG. Ambrisentan: a review of its use in pulmonary arterial hypertension. Ther Adv Respir Dis. 2017;11:233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Manwani D, Chen G, Carullo V, Serban S, Olowokure O, Jang J, et al. Single-dose intravenous gammaglobulin can stabilize neutrophil Mac-1 activation in sickle cell pain crisis. Am J Hematol. 2015;90:381–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fiorito V, Chiabrando D, Petrillo S, Bertino F, Tolosano E. The multi-faceted role of heme in cancer. Front Oncol. 2019;9:1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tolosano E, Fagoonee S, Morello N, Vinchi F, Fiorito V. Heme scavenging and the other facets of hemopexin. Antioxid Redox Signal. 2010;12:305–20. [DOI] [PubMed] [Google Scholar]
- 93.Schaer DJ, Buehler PW, Alayash AI, Belcher JD, Vercellotti GM. Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood. 2013;121:1276–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vinchi F, Costa da Silva M, Ingoglia G, Petrillo S, Brinkman N, Zuercher A, et al. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood. 2016;127:473–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ghosh S, Adisa OA, Chappa P, Tan F, Jackson KA, Archer DR, et al. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J Clin Invest. 2013;123:4809–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Low YH, Gan TJ. NMDA Receptor Antagonists, Gabapentinoids, alpha-2 Agonists, and Dexamethasone and Other Non-Opioid Adjuvants: Do They Have a Role in Plastic Surgery? Plast Reconstr Surg. 2014;134:69S–82S. [DOI] [PubMed] [Google Scholar]
- 97.Dimitrov IV, Harvey MG, Voss LJ, Sleigh JW, Bickerdike MJ, Denny WA. Ketamine esters and amides as short-acting anaesthetics: Structure-activity relationships for the side-chain. Bioorg Med Chem. 2019;27:1226–31. [DOI] [PubMed] [Google Scholar]
- 98.Gupta AK, Versteeg SG. Topical Treatment of Facial Seborrheic Dermatitis: A Systematic Review. Am J Clin Dermatol. 2017;18:193–213. [DOI] [PubMed] [Google Scholar]
- 99.Pollack S Omega-3 fatty acids in sickle cell anemia. Blood. 2013;121:2366–7. [DOI] [PubMed] [Google Scholar]
- 100.Atweh GF, Sutton M, Nassif I, Boosalis V, Dover GJ, Wallenstein S, et al. Sustained induction of fetal hemoglobin by pulse butyrate therapy in sickle cell disease. Blood. 1999;93:1790–7. [PMC free article] [PubMed] [Google Scholar]
- 101.Xu J, Peng C, Sankaran VG, Shao Z, Esrick EB, Chong BG, et al. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science. 2011;334:993–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Leonard A, Tisdale J, Abraham A. Curative options for sickle cell disease: haploidentical stem cell transplantation or gene therapy? Br J Haematol. 2020;189:408–23. [DOI] [PubMed] [Google Scholar]
- 103.Traxler EA, Yao Y, Wang YD, Woodard KJ, Kurita R, Nakamura Y, et al. A genome-editing strategy to treat beta-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med. 2016;22:987–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Metais JY, Doerfler PA, Mayuranathan T, Bauer DE, Fowler SC, Hsieh MM, et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. 2019;3:3379–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Demirci S, Leonard A, Haro-Mora JJ, Uchida N, Tisdale JF. CRISPR/Cas9 for Sickle Cell Disease: Applications, Future Possibilities, and Challenges. Adv Exp Med Biol. 2019;1144:37–52. [DOI] [PubMed] [Google Scholar]
- 106.Khosravi MA, Abbasalipour M, Concordet JP, Berg JV, Zeinali S, Arashkia A, et al. Targeted deletion of BCL11A gene by CRISPR-Cas9 system for fetal hemoglobin reactivation: A promising approach for gene therapy of beta thalassemia disease. Eur J Pharmacol. 2019;854:398–405. [DOI] [PubMed] [Google Scholar]
- 107.Humbert O, Radtke S, Samuelson C, Carrillo RR, Perez AM, Reddy SS, et al. Therapeutically relevant engraftment of a CRISPR-Cas9-edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci Transl Med. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Weber L, Frati G, Felix T, Hardouin G, Casini A, Wollenschlaeger C, et al. Editing a gamma-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci Adv. 2020;6:eaay9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ye L, Wang J, Tan Y, Beyer AI, Xie F, Muench MO, et al. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and beta-thalassemia. Proc Natl Acad Sci U S A. 2016;113:10661–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lamsfus-Calle A, Daniel-Moreno A, Antony JS, Epting T, Heumos L, Baskaran P, et al. Comparative targeting analysis of KLF1, BCL11A, and HBG1/2 in CD34(+) HSPCs by CRISPR/Cas9 for the induction of fetal hemoglobin. Sci Rep. 2020;10:10133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shariati L, Khanahmad H, Salehi M, Hejazi Z, Rahimmanesh I, Tabatabaiefar MA, et al. Genetic disruption of the KLF1 gene to overexpress the gamma-globin gene using the CRISPR/Cas9 system. J Gene Med. 2016;18:294–301. [DOI] [PubMed] [Google Scholar]
- 112.Renneville A, Van Galen P, Canver MC, McConkey M, Krill-Burger JM, Dorfman DM, et al. EHMT1 and EHMT2 inhibition induces fetal hemoglobin expression. Blood. 2015;126:1930–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Masuda T, Wang X, Maeda M, Canver MC, Sher F, Funnell AP, et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science. 2016;351:285–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pawliuk R, Westerman KA, Fabry ME, Payen E, Tighe R, Bouhassira EE, et al. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science. 2001;294:2368–71. [DOI] [PubMed] [Google Scholar]
- 115.Ribeil JA, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E, et al. Gene Therapy in a Patient with Sickle Cell Disease. N Engl J Med. 2017;376:848–55. [DOI] [PubMed] [Google Scholar]
- 116.Brendel C, Negre O, Rothe M, Guda S, Parsons G, Harris C, et al. Preclinical Evaluation of a Novel Lentiviral Vector Driving Lineage-Specific BCL11A Knockdown for Sickle Cell Gene Therapy. Mol Ther Methods Clin Dev. 2020;17:589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Basak A, Munschauer M, Lareau CA, Montbleau KE, Ulirsch JC, Hartigan CR, et al. Control of human hemoglobin switching by LIN28B-mediated regulation of BCL11A translation. Nat Genet. 2020;52:138–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Taghavi SA, Hosseini KM, Tamaddon G, Kasraian L. Inhibition of gamma/beta Globin Gene Switching in CD 34(+) Derived Erythroid Cells by BCL11A RNA Silencing. Indian J Hematol Blood Transfus. 2019;35:758–64. [DOI] [PMC free article] [PubMed] [Google Scholar]