

Abstract
Purpose
To report the interim analysis of the phase II single arm noninferiority trial testing the upfront use of dexrazoxane with doxorubicin on progression free survival (PFS) and cardiac function in soft tissue sarcoma (STS).
Patients and Methods
Patients with metastatic or unresectable STS who were candidates for first line treatment with doxorubicin were deemed eligible. An interim analysis was initiated after 33/65 patients were enrolled. Using the historical control of 4.6 months progression free survival (PFS) for doxorubicin in the front-line setting, we tested if the addition of dexrazoxane affected the efficacy of doxorubicin in STS. The study was powered so that a decrease of PFS to 3.7 months would be considered non-inferior. Secondary aims included cardiac related mortality, incidence of heart failure/cardiomyopathy, and expansion of cardiac monitoring parameters including 3D echocardiography. Patients were allowed to continue on doxorubicin beyond 600 mg/m2 if they were deriving benefit and were not demonstrating evidence of symptomatic cardiac dysfunction.
Results
At interim analysis, upfront use of dexrazoxane with doxorubicin demonstrated a PFS of 8.4 months (95% confidence interval: 5.1 – 11.2 months). Only 3 patients were removed from study for cardiotoxicity, all on > 600 mg/m2 doxorubicin. No patients required cardiac hospitalization or had new, persistent cardiac dysfunction with left ventricular ejection fraction remaining below 50%. The median administered doxorubicin dose was 450 mg/m2 (interquartile range 300–750 mg/m2).
Conclusions
At interim analysis, dexrazoxane did not reduce PFS in patients with STS treated with doxorubicin. Involvement of cardio-oncologists is beneficial for the monitoring and safe use of high dose anthracyclines in STS.
Keywords: Soft Tissue Sarcoma, Sarcoma, Doxorubicin, Anthracycline, Dexrazoxane, Heart Failure, Echocardiograph
Statement of Translational Relevance
Anthracyclines are the front-line treatment for STS. Standard of care is to initiate dexrazoxane after a cumulative dose of 300 mg/m2 of doxorubicin or to give doxorubicin as a 72-hour continuous infusion. Based on a theoretical concern from the literature that dexrazoxane will protect tumors from doxorubicin and reduce efficacy, we performed a non-inferiority trial to test this hypothesis. In this first prospective trial in STS of upfront use of dexrazoxane with doxorubicin, we found no evidence of tumor protection and there was, in fact, an increase in PFS from a historical 4.6 months to 8.4 months. Only three patients were removed from study for cardiotoxicity and all patients had recovery of their left ventricular function to normal or their prior baseline by the end of the study. No deaths due to anthracycline-induced heart failure were observed. With careful cardiac monitoring, patients can exceed the 450 mg/m2 lifetime maximum of doxorubicin.
Introduction
While there have been revolutionary advances in the treatment of many cancers over the last decade, soft tissue sarcomas (STS) remain challenging to treat. Anthracyclines, particularly doxorubicin, are the mainstay of therapy (1–3). Unfortunately, the therapeutic potential of doxorubicin is severely limited by its dose-dependent cardiotoxicity, and the median overall survival (OS) for advanced metastatic disease remains poor at approximately 20 months (1). Existing approaches to cardio-protection have provided limited efficacy. These methods include changing drug administration to longer infusions, using a liposomal derivative of doxorubicin, or co-administering cardio-protective drugs such as beta-blockers, especially carvedilol, or inhibitors of the renin-angiotensin-aldosterone system (RAAS) (angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and aldosterone inhibitors) (2–4).
Dexrazoxane was introduced nearly 25 years ago with promising initial findings to limit anthracycline cardiotoxicity (5). However, support for dexrazoxane was diminished when the tumor response rate to doxorubicin was reduced in the dexrazoxane arm in one of two randomized breast cancer trials, although there was no significant difference in time of PFS or OS (6). The original FDA approval permitted introduction of the dexrazoxane only after a cumulative dose of 300 mg/m2 modelling a beneficial study that delayed dexrazoxane administration (7). At a meeting of oncologists and cardiologists to evaluate the use of dexrazoxane, the consensus opinion was that dexrazoxane was underutilized and the data did not support a negative effect of dexrazoxane against tumor efficacy (8). This was corroborated by a recent systematic review and meta-analysis (9). Current practice for treatment of breast cancer still limits doxorubicin to 300 mg/m2 before alternative treatments are begun. However, anthracyclines are the cornerstone of treatment of childhood lymphomas and dexrazoxane is begun at the onset of treatment in order to limit long-term cardiotoxicity (10,11).
We therefore sought to determine if the use of dexrazoxane in adults starting with the first dose of doxorubicin (upfront dexrazoxane) would permit prolonged treatment with doxorubicin and not decrease PFS in metastatic or unresectable STS. Due to concern for timely patient accrual, the study was designed as a non-inferiority trial compared to historical controls (12,13), with 4.6 months PFS from the Phase III study used for statistical purposes (12). For safety, at the study mid-point, this interim analysis was performed.
Methods
The study was conducted at the Siteman Cancer Center/Washington University in St. Louis from May 4, 2016 through the interim analysis date of May 27, 2019. Patients were recruited if they had unresectable or metastatic STS and were planning to initiate treatment with doxorubicin (cycles of 75 mg/m2 every 21 days) with or without olaratumab (starting dose of 15 mg/kg) as routine care. During the time of this trial, olaratumab received FDA approval and subsequent removal of FDA approval as it was deemed to be an inactive drug (1). Patients with a sarcoma measurable according to RECIST 1.1 and histologic grade 2 or 3 were eligible for enrollment. Patients had to be at least 18 years of age, have ECOG performance status of 0 or 1, have adequate organ function, and be able to understand and willing to sign an IRB approved written informed consent. Women of childbearing potential and men had to agree to adequate contraception for the duration of study participation. Prior adjuvant chemotherapy with gemcitabine and/or docetaxel/paclitaxel was allowed.
Sample size was calculated using a significance level (alpha, type I error rate) of .05 and power (1 – β) of .80, so β (type II error rate) = .20. Based on a historical PFS of 4.6 months (12), a sample of 57 patients would be needed to show noninferiority with an allowable difference of 4 weeks, which was classified by the investigators as a clinically meaningful decrease in PFS. The planned total enrollment was 63 patients, including 10% for withdrawal or loss to follow-up.
Patients were excluded if they met any of the following criteria: 1) myocardial infarction with the past 12 months, 2) stable or unstable angina, 2) left ventricular (LV) ejection fraction (EF) ≤ 45%, 3) symptomatic valvular heart disease, 4) prior chemotherapy for advanced or metastatic disease, 5) known brain metastases, 6) a second primary malignancy or a prior malignancy within the last two years (except carcinoma in situ of the cervix, non-metastatic prostate cancer, or basal cell or squamous cell carcinoma of the skin which were treated with local resection only; prior adjuvant androgen deprivation therapy in the case of prostate cancer was permitted, but current adjuvant androgen deprivation therapy was not permitted), 7) current treatment with investigational agents, 8) history of allergic reactions attributed to compounds of similar chemical or biologic composition to dexrazoxane or other agents used in the study, 9) uncontrolled intercurrent illness including, but not limited to, ongoing or active infection, symptomatic congestive heart failure (HF), or psychiatric illness/social situations that would limit compliance with study requirements, 10) pregnancy and/or breast feeding, 11) treatment with combination antiretroviral therapy for HIV due to the potential for pharmacokinetic interactions with dexrazoxane, and 12) prior treatment with anthracyclines.
While on study, patients were treated with doxorubicin at a starting dose of 75 mg/m2 every three weeks. Olaratumab was used at a dose of 15 mg/kg on days 1 and 8 of the 21 day-cycle during its period of FDA approval. Olaratumab was first added to the study protocol after the FDA approved its use in combination with doxorubicin in October 2016. Olaratumab was removed from the protocol on FDA guidance following the negative findings from the Phase III ANNOUNCE trial (1). Dexrazoxane was initiated with cycle 1 and continued through the duration of the study, administered in a 10:1 ratio to doxorubicin (750 mg/m2) over 15 minutes, no more than 30 minutes prior to doxorubicin administration (14). Dosing of dexrazoxane was reduced as needed in patients with moderate to severe renal impairment (creatinine clearance levels < 40 mL/min) by 50%. All treatments were done on an outpatient basis. All clinical decisions were made on the standard of care 2D ECHO. Patients were referred to cardo-oncology when they exceeded 300 mg/m2 of doxorubicin or when there was a change in EF. The study was approved by the Washington University in St. Louis Institutional Review Board.
Patients were followed for a primary endpoint of PFS. Secondary endpoints were measures of cardiac function by echocardiographic left ventricular ejection fraction (LVEF) and global longitudinal strain (GLS), cardiac-related mortality, and incidence of HF or cardiomyopathy. Other secondary objectives were troponin, galectin 3, or BNP levels as early markers of cardiac dysfunction. Patients were defined as having experienced cardiotoxicity during the study if they were removed from the study for cardiac concerns, had a drop in LVEF of >10% to < 50%, or had a HF related hospitalization. Heart failure was defined as positive findings from at least two of the three following categories: symptoms of HF (paroxysmal nocturnal dyspnea, orthopnea, shortness of breath, swelling, fatigue, weight gain), physical findings of HF (increased jugular venous pressure, crackles, edema, S3) and/or positive diagnostic tests (pulmonary edema on CXR, BNP > 100 pg/mL, NT-pro BNP > 125 pg/mL) (15). Heart failure was classified as either systolic (LVEF < 50%) or diastolic (LVEF ≥ 50%). Asymptomatic LVEF dysfunction was defined as a drop in LVEF to <50% without signs or symptoms of HF.
Echocardiographically-determined LVEF function was obtained at pre-treatment baseline and before every other treatment cycle and was interpreted by an accredited expert cardiologist in standard fashion for clinical decision-making. The echo-core laboratory then provided centralized Echo readings by two expert Echo readers. The core laboratory was blinded to all clinical outcome data for uniform analysis, but these readings were not immediately available to the treating physicians. LVEF was determined with contrast enhancement using biplane Simpson’s rule (16). When possible, 3D echocardiograms were obtained, but these were not clinically feasible in a significant number of patients and were not used for clinical decision making. Myocardial strain imaging was obtained from the apical 4-chamber, 2-chamber, and long-axis views to determine global longitudinal strain (GLS), as described previously in detail (GE EchoPac v200, Horton, Norway) (17).
All patients demonstrating a reduction in LVEF up to 50% or with clinical concerns for cardiotoxicity were referred to a cardio-oncologist. Management decisions about cardiac toxicity were made jointly between the oncologist and cardio-oncologist. Use of additional cardio-protective medications, such as beta-blockers and RAAS inhibitors, were encouraged if there was any concern for potential cardiotoxicities.
Patients were followed until the date of interim analysis (May 27, 2019) or until death. Patients were monitored for adverse events (AEs) throughout the study per the NCI Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. The occurrence of HF was further adjudicated retrospectively by agreement of three cardio-oncologists in a manner similar to other cardio-oncology based trials (18).
Baseline demographics and comorbidities, anthracycline dosing, and cancer characteristics were compared between patients with and without cardiotoxicity using Fisher’s exact test for categorical variables and the exact Wilcoxon test for continuous variables. The LVEF was compared between baseline, and time of interim analysis using Kruskal Wallis. Given the sample size, no continuous variables met the definition for normal distribution after reviewing the histograms and Q-Q plots for each variable’s distribution. Statistics were performed using SAS 9.4 (SAS Institute, Cary, NC) using a two-tailed alpha of 0.05.
Results
In total, 42 patients with sarcoma were screened for study eligibility. Eight patients failed screening and one patient withdrew from the study prior to receiving treatment. Thereafter, 33 patients (median age 65, 55% female) were started on study treatment and had at least 90 days of follow-up at the time of interim analysis (27 May 2019).
Over a median of 16.1 months of follow-up (IQR 5.7–25.0 months), PFS was a median of 8.4 months (IQR 4.0 –11.2 months) (Figure 1). At the time of interim analysis, six patients (21.4%) had not yet progressed. There were 13 deaths (12 due to cancer progression and 1 due to myocarditis in a patient who received checkpoint-inhibitor therapy after progressing on study protocol). There were no deaths attributed to doxorubicin.
Figure 1: Median Progression Free Survival.
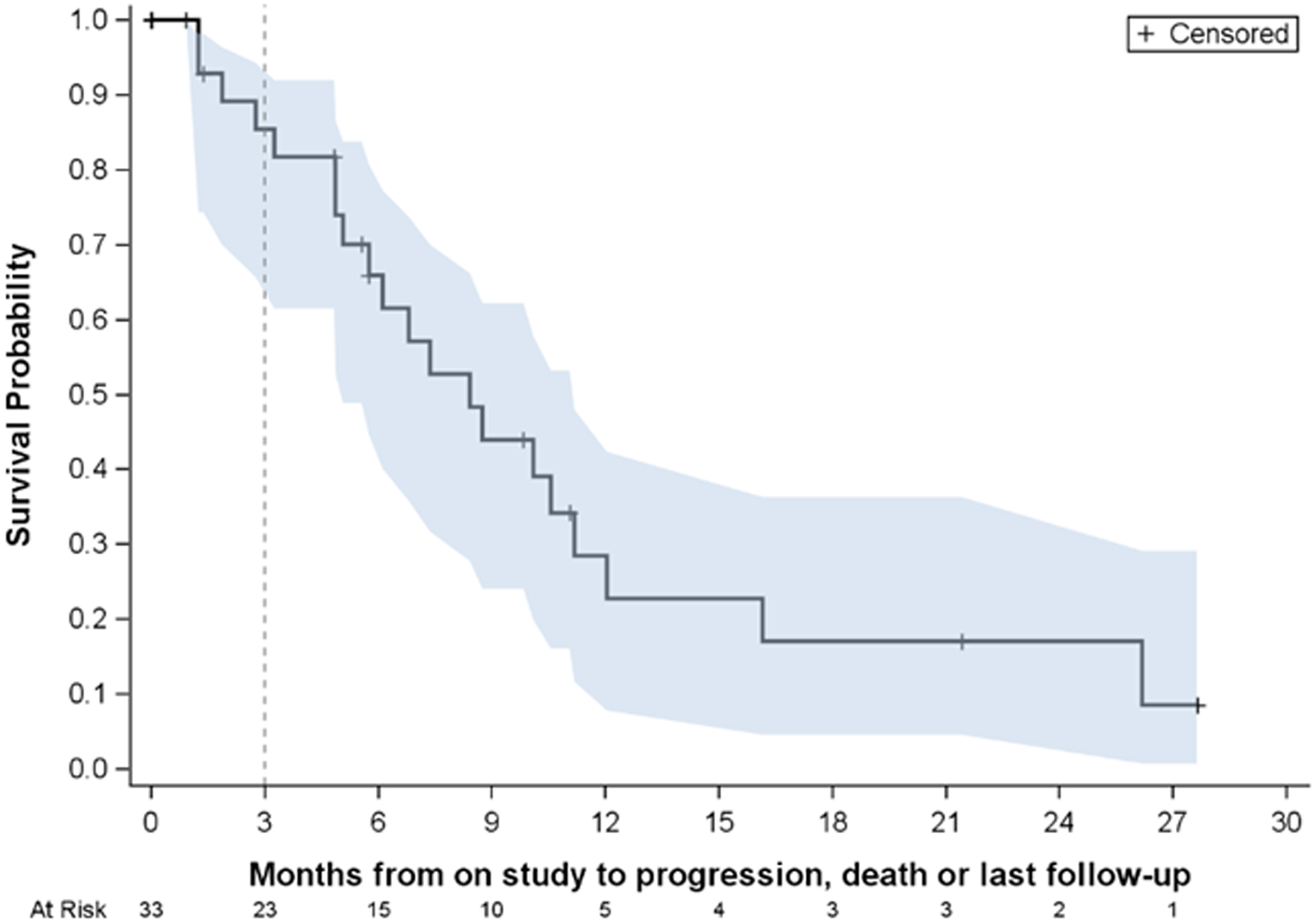
Progression-free survival (PFS) for patients on study. Median PFS is estimated to be 8.4 months with 95% confidence interval (5.1 months, 11.2 months).
Patients received a median of six cycles (450 mg/m2, IQR 300–750 mg/m2) of doxorubicin prior to interim analysis at which time five patients (15.2%) remained on study, three patients (9.1%) were removed for cardiotoxicity, 15 (45.5%) were removed for disease progression, 7 (21.2%) were removed for non-cardiac toxicity, and 3 (9.1%) were removed for patient preference. The number of cycles ranged from one to 38. Three patients only received one cycle (75 mg/m2) prior to being removed for non-cardiac toxicity, and three patients were removed after only two cycles (150 mg/m2) due to disease progression. Conversely, 5 patients received over 1000 mg/m2 of doxorubicin, with 2 patients receiving over 2000 mg/m2 to a maximum of 2850 mg/m2. Of these five patients, only one was taken off study for cardiotoxicity while four were removed due to disease progression.
Overall, eight patients were referred to a cardio-oncologist and three patients were removed from the study due to cardiotoxicity, though two were found not to have cardiotoxicity after Core Lab analysis of the echocardiograms. Baseline comorbidities between patients who did or did not experience cardiotoxicity are shown in Table 1. Cardiac study outcomes are reported in Table 2. LVEF (Figure 2A) and GLS (Figure 2B) determined by Core Lab are demonstrated by patient and stratified by dose for clarity.
Table 1:
Study Characteristics of Patients with or without Cardiotoxicity on Protocol
| Overall N=33 |
No Cardiotoxicity N=30 |
Cardiotoxicity* N=3 |
||
|---|---|---|---|---|
| Demographics | ||||
| Age (yrs), Median (IQR) | 65 (60–68) | 64.5 (58–69) | 65 (62–68) | |
| Age range (yrs) | 35–79 | 35–79 | 62–68 | |
| Female, N (%) | 18 (54.6) | 17 (56.7) | 1 (33.3) | |
| Race, N (%): | ||||
| Black | 5 (15.2) | 4 (13.3) | 1 (33.3) | |
| Caucasian | 28 (84.9) | 26 (86.7) | 2 (66.7) | |
| Cancer History and Anthracycline Dosing | ||||
| Time since Diagnosis (days), Median (IQR) | 115 (43–279) | 141 (46–279) | 43 (28–405) | |
| Prior Chemotherapy | 3 (9.1) | 3 (10.0) | 0 (0) | |
| Prior Chest Radiation | 1 (3.0) | 1 (3.3) | 0 (0) | |
| Doxorubicin Dosing on Study | ||||
| # of cycles, Median (IQR) | 6 (4–10) | 5.5 (4–10) | 9 (8–16) | |
| Total Dose (mg/m2), Median (IQR) | 450 (300–750) | 412.5 (300–750) | 675 (600–1200) | |
| Baseline Cardiac Function | ||||
| Baseline LVEF, Median (IQR) | 65 (60–69) | 65 (60–71) | 68 (48–68) | |
| LVEF < 50%, N (%) | 1 (3.0)** | 0 (0) | 1 (33.3)** | |
| Absolute Baseline strain (GLS), Median (IQR) | 18.8 (16.8–20) | 19.23§ (16.8–20.1) | 17.2 (13.2–18.5) | |
| Baseline Comorbidities | ||||
| Diabetes Mellitus, N (%) | 6 (18.2) | 6 (20.0) | 0 (0.0) | |
| Hyperlipidemia, N (%) | 17 (51.5) | 15 (50.0) | 2 (66.7) | |
| Hypertension, N (%) | 23 (69.7) | 31 (70.0) | 2 (66.7) | |
| Tobacco use, N (%) | 11 (33.3) | 10 (33.3) | 1 (33.3) | |
| Heart Failure (Systolic), N (%) | 2 (6.1) | 1 (3.3) | 1 (33.3) | |
| Arrhythmia, N (%) | 4 (12.1) | 4 (13.3) | 0 (0) | |
| CAD, including coronary calcifications | 9 (27.3) | 8 (26.7) | 1 (33.3) | |
| Obstructive CAD, N (%) | 1 (3.0) | 1 (3.3) | 0 (0.0) | |
| CAD, HF, or any CV risk factor, N (%) | 28 (84.9) | 25 (83.3) | 3 (100) | |
| eGFR (Creatinine), Median (IQR) | 88.4 (68.1–101.9) | 87.6 (68.1–101.9) | 90.0 (52.4–107.2) | |
| Baseline Cardiac Medications | ||||
| Beta-blocker | 6 (18.2) | 5 (16.7) | 1 (33.3) | |
| ACE-I/ARB | 13 (39.4) | 12 (40.0) | 1 (33.3) | |
| Aldosterone Antagonist | 0 (0) | 0 (0) | 0 (0) | - |
| Statin | 11 (33.3) | 10 (33.3) | 1 (33.3) | |
| Aspirin | 12 (36.4) | 11 (36.7) | 1 (33.3) | |
3 patients removed from study for cardiotoxicity including one with baseline LVEF <50% and two with LVEF decline >10% to less than 50%. No patients required HF hospitalization while on study.
The patient with a baseline LVEF of 47% (asymptomatic) is the same one reported to have cardiotoxicity.
3 patients missing baseline strain from No Cardiotoxicity group
Obstructive CAD: prior positive stress, PCI, CABG or MI)
Table 2:
Cumulative Cardiac Based Study Outcomes
| Cardiac Outcomes | On Protocol | Within 1 Year | Within 2 Years |
|---|---|---|---|
| Lowest LVEF, Median (IQR) | 58 (52–64) | 57 (51–63) | 57 (51–63) |
| Final LVEF, Median (IQR)* | 62 (58–67) | 64 (58–68) | 64 (58–68) |
| Final LVEF < 50%, N (%) | 3 (9) | 1 (3) | 1 (3) |
| New LV Dysfunction ≥ 10% decrease to <50%, N (%) | 2 (6) | 3 (9) | 3 (9) |
| Any LV Dysfunction (LVEF < 50%), N (%) | 4 (12) | 6 (18) | 6 (18) |
| Asymptomatic LV Dysfunction, N (%) | 2 (6) | 3 (9) | 2 (9) |
| Systolic HF, Grade II, N (%) | 2 (6) | 3 (9) | 3 (9) |
| Systolic HF, Grade III or more, N (%) | 0 (0) | 0 (0) | 0 (0) |
| Persistent LV Dysfunction, N (%)*** | 1 (3) | 1 (3) | 1 (3) |
| Symptomatic Diastolic HF (LVEF ≥ 50%) | |||
| Grade I or II, N (%) | 0 (0) | 1 (3) | 2 (6) |
| Grade III or more HF, N (%) | 0 (0) | 3 (9) | 4 (12)** |
| Myocarditis (On Checkpoint Inhibitor Therapy) | - | 1 (3) | 2 (6) |
| Cardiac Death**** | 1 (3) | 1 (3) |
All LVEF measurements per clinical echocardiogram. Persistent LV Dysfunction defined as two consecutive echocardiograms with LVEF <50%.
No difference between time points, p = NS.
Includes one patient with acute diastolic HF and segmental PE while on gemcitabine and docetaxel, one patient with admission for pneumonia complicated by acute HF, and two patients with checkpoint inhibitor associated myocarditis.
The only patient with persistent LV dysfunction had baseline LVEF < 50%.
Due to checkpoint inhibitor associated myocarditis.
Figure 2: Cardiac Measurements Stratified by Dose.
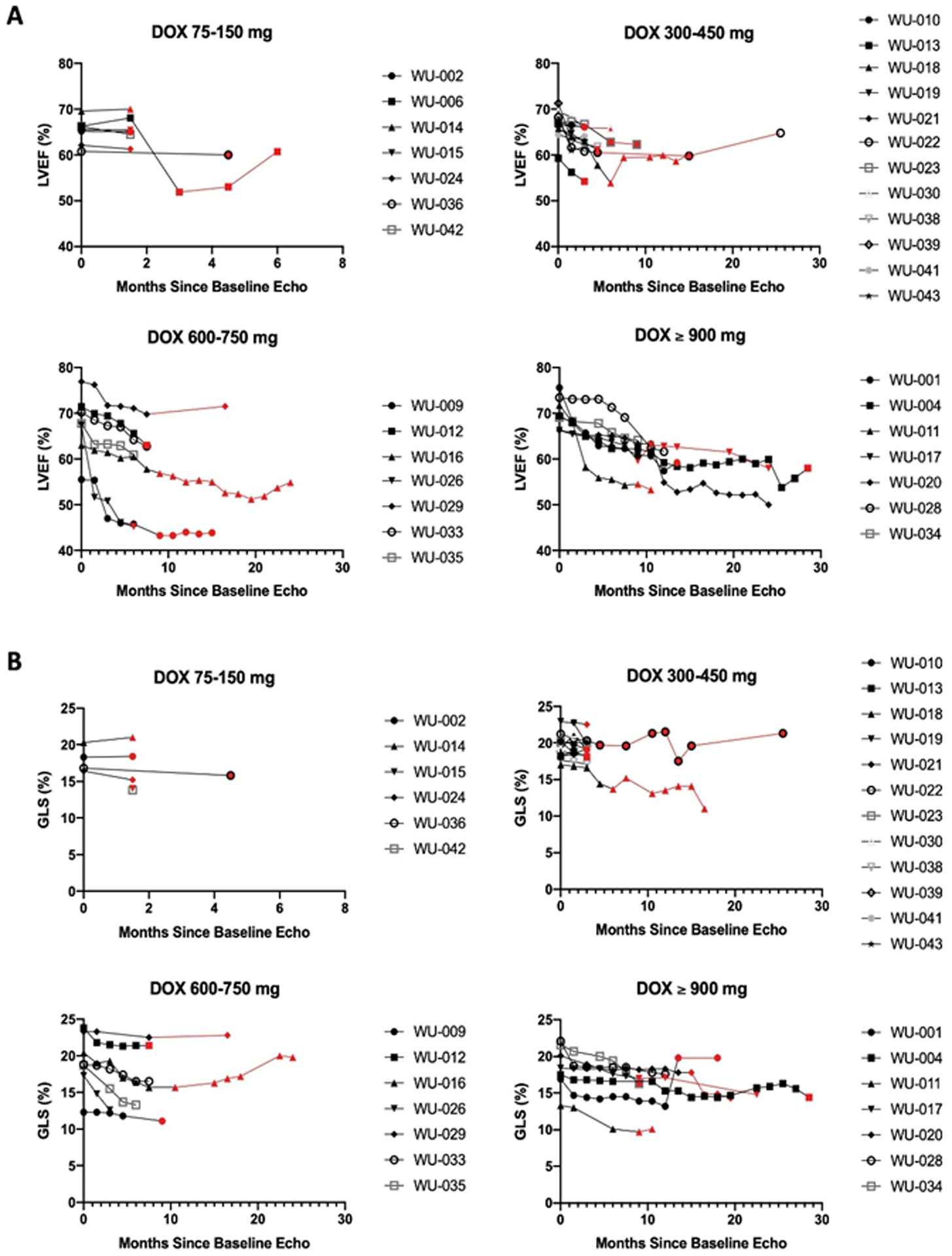
A: The 2D Ejection fractions calculated by the CORE Laboratory presented in patients grouped by dose of doxorubicin. B: The GLS calculated by the CORE laboratory in patients grouped by dose of doxorubicin, presented as absolute value for clarity. In cases where doxorubicin was discontinued, the curves are indicated in red; otherwise, while receiving doxorubicin the curves are black.
The first patient removed for cardiotoxicity was the first patient enrolled. She had a baseline LVEF of 68% that declined to 47% after 16 cycles (1200 mg/m2 doxorubicin) at which time she was removed from the study due to concerns for systolic HF, given some symptoms of mild shortness of breath and fatigue. Subsequent evaluation of the LVEF by the Echo Core Lab found her LVEF to have been 57% at the time of study removal as opposed to the clinically reported LVEF of 47%, so in retrospect, she would not have met criteria to be removed for cardiotoxicity. Her LVEF subsequently improved to 55% on her follow-up clinical echocardiogram. The second patient removed had a baseline LVEF of 48% (Core Lab LVEF 56%). He was removed after 8 cycles (600 mg/m2 doxorubicin) when his clinical LVEF declined to 41% (Core Lab 46%) and he developed a new wall motion abnormality. The LVEF was subsequently 39% on the next clinical echocardiogram (Core Lab 43%) and was measured as 45% (Core Lab 43%) two weeks later. After 2 years, his clinical LVEF was 50% and Core Lab LVEF was 43%. The third patient was removed after 9 cycles (675 mg/m2 doxorubicin). He had a baseline LVEF of 68% which declined to an LVEF of 40% on his clinical echocardiogram. His biomarkers were normal. Subsequent core lab measurement of his ejection fraction was 58% on this echocardiogram, and his LVEF had recovered to 54% on his clinical echocardiogram a month later. All three patients were referred to a cardio-oncologist and placed on cardio-protective therapy at the time of LVEF decline, and all had improvement of their LVEF back to normal or the previous baseline prior to interim analysis.
In long-term follow up, two additional patients were diagnosed with transient systolic dysfunction during the first year post-study. Both patients again had mild reductions in their LVEF with recovery to > 50% on subsequent clinical echocardiograms. One of these patients had been continued on doxorubicin without dexrazoxane after being removed following one cycle of study treatment due to prolonged cessation of therapy. Her LVEF started at 76%, dropped to 48%, and then recovered to 73% (calculated by core lab as 52% with recovery to 61%). The other patient had a drop in his LVEF to 49%, with recovery to 64% at the end of the follow-up period. The core lab never measured an ejection fraction less than 54%. These inconsistencies were seen throughout the study and emphasize the point that echo measurements are, in some respect, operator-dependent, and that decisions regarding treatment should not depend on a single measurement. In the clinical setting, promptness is critical to establish whether a dose should be given and confirmation of decision-critical evaluations needs prompt confirmation.
Among the entire study cohort, there was no difference between LVEF measured at baseline, at the end of the study treatment, and at the end of the follow-up period. GLS and 3D echo were not helpful in identifying patients who developed heart failure early in our study, and with the low rates of cardiotoxicity, the study was not powered to do this.
Discussion
Though upfront dexrazoxane with doxorubicin has been used in selected patients with preexisting cardiac disease where there was special concern about cardiotoxicity (19,20), this is the first prospective study to evaluate the safety and efficacy of using upfront dexrazoxane in combination with doxorubicin for the treatment of metastatic or unresectable STS. We have shown that the combination of dexrazoxane and close collaboration with cardio-oncologists allows for prolonged treatment with doxorubicin and historically longer PFS. OS will be reported at the completion of this trial. There was only minimal reduction of LVEF over the study period with administration of high doses of doxorubicin in these sarcoma patients who are at high risk for the development of HF.
The use of doxorubicin for the treatment of sarcoma is traditionally limited by production of myocardial cell toxicity, which can lead to severe HF. There are many mechanisms by which doxorubicin causes this damage: production of reactive oxygen species, i.e. free radicals, incorporation of iron into the mitochondrial respiratory pathway leading to damage to the cell membrane (21–24), direct interference with DNA metabolism and structure and, emphasized more recently, binding with topoisomerase II (TOP2) leading to double stranded DNA breaks (DSB) (25). While the development of HF is related to the cumulative dose, damage to myocardial cell nuclei can be seen four hours after the very first dose of doxorubicin (26). These processes lead to inhibition of tumor cell growth and cell death. TOP2 has two isoforms, TOP2α mostly in the tumor and TOP2β in the myocardium. There are data suggesting that dexrazoxane depletes and reduces TOP2β levels found in the heart, so the heart is relatively protected from the effects of doxorubicin (27). Levels of TOP2α in the tumor are also reduced, to different levels in different tumors, potentially limiting the efficacy of doxorubicin on the tumor. However, dexrazoxane has been shown to bind with TOP2α leading to DSB and apoptosis (28) in the tumor while TOP2β is depleted in cardiac tissue and not susceptible to doxorubicin (29).
While this trial was originally powered based on the findings of Judson et al (12), where the PFS was 4.6 months, more modern PFS data are available. The PFS for the Palifosfamide trial was 5.2 months (30), 6.3 months in the doxorubicin arm of the Evofosfamide trial (31), and the PFS of the placebo control doxorubicin arm of the Olaratumab Phase 3 clinical trial was 6.8 months. At the time of the interim data analyses for this trial, our finding of a PFS of 8.4 months would still be consistent with non-inferiority regardless of the more modern data.
Due to the importance of aggressive dosing of doxorubicin in the treatment of sarcoma, we have reported the interim analysis of the upfront use of dexrazoxane in combination with doxorubicin. At the time of this report, no reduction in the efficacy of doxorubicin was seen and the Data and Safety Monitoring Board concluded that the full trial should be accrued.
Financial Support
Financial support was provided by a grant from the Richard & Marie Hahn Cancer Medical Research Endowment Fund at the Barnes Jewish Foundation, a gift from Donald and Shirley Sher, divisional funding from the Division of Medical Oncology provided by John D DiPersio, MD, Ph.D, STL Cure Sarcoma 6K, and by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR002345 (JDM). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of health.”
Footnotes
Conflicts of Interest
Brian A. Van Tine: grants from Merck; grants and personal fees from Pfizer; grants from TRACON Pharmaceuticals; grants, personal fees, and other from GlaxoSmithKline; personal fees from Polaris Inc.; personal fees from Lilly; personal fees from Caris Life Sciences; personal fees from Novartis; personal fees from CytRX; personal fees from Plexxikon; personal fees from Epizyme; personal fees from Daiichi Sankyo; personal fees from Adaptimmune; personal fees from Immune Design; personal fees from Bayer; personal fees from Cytokinetics; personal fees from Deciphera; and has a patent issued for the use of ME1 as a biomarker and ACXT3102.
Angela C. Hirbe: consult to Astrazeneca and Springworks
Peter Oppelt: speaking fees/honoraria: Merck, Bristol-Myers, Eisai
Ashley E. Frith: none
Richa Rathore: none
Joshua D. Mitchell: consultant to Pfizer.
Fei Wan: none
Shellie Berry: none
Michele Landeau: none
George A. Heberton: none
John Gorcsan III: research grants for GE, Canon, V-wave and EBR systems
Peter R. Huntjens: none
Yuko Soyama: none
Justin M. Vader: none
Jose A. Alvarez Cardona: none
Kathleen W. Zhang: consultant fees from Eidos Therapeutics
Daniel J. Lenihan: consultant to Roche, Prothena, Lilly Clementia and Reseach Funding from Myocardial Solutions
Ronald J. Krone: none
References:
- 1.Tap WD, Wagner AJ, Schöffski P, Martin-Broto J, Krarup-Hansen A, Ganjoo KN, et al. Effect of Doxorubicin Plus Olaratumab vs Doxorubicin Plus Placebo on Survival in Patients With Advanced Soft Tissue Sarcomas: The ANNOUNCE Randomized Clinical Trial. Jama 2020;323(13):1266–76 doi 10.1001/jama.2020.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gulati G, Heck SL, Ree AH, Hoffmann P, Schulz-Menger J, Fagerland MW, et al. Prevention of cardiac dysfunction during adjuvant breast cancer therapy (PRADA): a 2 × 2 factorial, randomized, placebo-controlled, double-blind clinical trial of candesartan and metoprolol. Eur Heart J 2016;37(21):1671–80 doi 10.1093/eurheartj/ehw022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bosch X, Rovira M, Sitges M, Domenech A, Ortiz-Perez JT, de Caralt TM, et al. Enalapril and carvedilol for preventing chemotherapy-induced left ventricular systolic dysfunction in patients with malignant hemopathies: the OVERCOME trial (preventiOn of left Ventricular dysfunction with Enalapril and caRvedilol in patients submitted to intensive ChemOtherapy for the treatment of Malignant hEmopathies). Journal of the American College of Cardiology 2013;61(23):2355–62 doi 10.1016/j.jacc.2013.02.072. [DOI] [PubMed] [Google Scholar]
- 4.Akpek M, Ozdogru I, Sahin O, Inanc M, Dogan A, Yazici C, et al. Protective effects of spironolactone against anthracycline-induced cardiomyopathy. European journal of heart failure 2015;17(1):81–9 doi 10.1002/ejhf.196. [DOI] [PubMed] [Google Scholar]
- 5.Wiseman LR, Spencer CM. Dexrazoxane. A review of its use as a cardioprotective agent in patients receiving anthracycline-based chemotherapy. Drugs 1998;56(3):385–403 doi 10.2165/00003495-199856030-00009. [DOI] [PubMed] [Google Scholar]
- 6.Swain SM, Whaley FS, Gerber MC, Weisberg S, York M, Spicer D, et al. Cardioprotection with dexrazoxane for doxorubicin-containing therapy in advanced breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 1997;15(4):1318–32. [DOI] [PubMed] [Google Scholar]
- 7.Swain SM, Whaley FS, Gerber MC, Ewer MS, Bianchine JR, Gams RA. Delayed administration of dexrazoxane provides cardioprotection for patients with advanced breast cancer treated with doxorubicin-containing therapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 1997;15(4):1333–40. [DOI] [PubMed] [Google Scholar]
- 8.Swain SM, Vici P. The current and future role of dexrazoxane as a cardioprotectant in anthracycline treatment: expert panel review. J Cancer Res Clin Oncol 2004;130(1):1–7 doi 10.1007/s00432-003-0498-7. [DOI] [PubMed] [Google Scholar]
- 9.Macedo AVS, Hajjar LA, Lyon AR, Nascimento BR, Putzu A, Rossi L, et al. Efficacy of Dexrazoxane in Preventing Anthracycline Cardiotoxicity in Breast Cancer. JACC: CardioOncology 2019;1(1):68–79 doi 10.1016/j.jaccao.2019.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chow EJ, Asselin BL, Schwartz CL, Doody DR, Leisenring WM, Aggarwal S, et al. Late Mortality After Dexrazoxane Treatment: A Report From the Children’s Oncology Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015;33(24):2639–45 doi 10.1200/JCO.2014.59.4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sepe DM, Ginsberg JP, Balis FM. Dexrazoxane as a cardioprotectant in children receiving anthracyclines. Oncologist 2010;15(11):1220–6 doi 10.1634/theoncologist.2010-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Judson I, Verweij J, Gelderblom H, Hartmann JT, Schöffski P, Blay J-Y, et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. The lancet oncology 2014;15(4):415–23 doi 10.1016/S1470-2045(14)70063-4. [DOI] [PubMed] [Google Scholar]
- 13.Tap WD, Jones RL, Van Tine BA, Chmielowski B, Elias AD, Adkins D, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. The Lancet;388(10043):488–97 doi 10.1016/S0140-6736(16)30587-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brier ME, Gaylor SK, McGovren JP, Glue P, Fang A, Aronoff GR. Pharmacokinetics of dexrazoxane in subjects with impaired kidney function. Journal of clinical pharmacology 2011;51(5):731–8 doi 10.1177/0091270010369675. [DOI] [PubMed] [Google Scholar]
- 15.Cornell RF, Ky B, Weiss BM, Dahm CN, Gupta DK, Du L, et al. Prospective Study of Cardiac Events During Proteasome Inhibitor Therapy for Relapsed Multiple Myeloma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2019;37(22):1946–55 doi 10.1200/JCO.19.00231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr 2015;28(1):1–39 e14 doi 10.1016/j.echo.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 17.Bax JJ, Delgado V, Sogaard P, Singh JP, Abraham WT, Borer JS, et al. Prognostic implications of left ventricular global longitudinal strain in heart failure patients with narrow QRS complex treated with cardiac resynchronization therapy: a subanalysis of the randomized EchoCRT trial. Eur Heart J 2017;38(10):720–6 doi 10.1093/eurheartj/ehw506. [DOI] [PubMed] [Google Scholar]
- 18.Voors AA, Dittrich HC, Massie BM, DeLucca P, Mansoor GA, Metra M, et al. Effects of the adenosine A1 receptor antagonist rolofylline on renal function in patients with acute heart failure and renal dysfunction: results from PROTECT (Placebo-Controlled Randomized Study of the Selective Adenosine A1 Receptor Antagonist Rolofylline for Patients Hospitalized with Acute Decompensated Heart Failure and Volume Overload to Assess Treatment Effect on Congestion and Renal Function). Journal of the American College of Cardiology 2011;57(19):1899–907 doi 10.1016/j.jacc.2010.11.057. [DOI] [PubMed] [Google Scholar]
- 19.Ganatra S, Nohria A, Shah S, Groarke JD, Sharma A, Venesy D, et al. Upfront dexrazoxane for the reduction of anthracycline-induced cardiotoxicity in adults with preexisting cardiomyopathy and cancer: a consecutive case series. Cardio-Oncology 2019;5(1):1 doi 10.1186/s40959-019-0036-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schuler MK, Gerdes S, West A, Richter S, Busemann C, Hentschel L, et al. Efficacy and safety of Dexrazoxane (DRZ) in sarcoma patients receiving high cumulative doses of anthracycline therapy - a retrospective study including 32 patients. BMC cancer 2016;16:619 doi 10.1186/s12885-016-2654-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.-Sawyer DB, Peng X, Chen B, Pentassuglia L, Lim CC. Mechanisms of anthracycline cardiac injury: can we identify strategies for cardioprotection? Prog Cardiovasc Dis 2010;53(2):105–13 doi S0033–0620(10)00119–2 [pii] 10.1016/j.pcad.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The Medical Letter. Dexrazoxane for cardiac protection against doxorubicin. Med Lett Drugs Ther 1995;37(962):110–1. [PubMed] [Google Scholar]
- 23.Buss JL, Hasinoff BB. Ferrous ion strongly promotes the ring opening of the hydrolysis intermediates of the antioxidant cardioprotective agent dexrazoxane (ICRF-187). Arch Biochem Biophys 1995;317(1):121–7 doi 10.1006/abbi.1995.1143. [DOI] [PubMed] [Google Scholar]
- 24.Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol 1999;57(7):727–41. [DOI] [PubMed] [Google Scholar]
- 25.Sawyer DB. Anthracyclines and heart failure. N Engl J Med 2013;368(12):1154–6 doi 10.1056/NEJMcibr1214975. [DOI] [PubMed] [Google Scholar]
- 26.Unverferth BJ, Magorien RD, Balcerzak SP, Leier CV, Unverferth DV. Early changes in human myocardial nuclei after doxorubicin. Cancer 1983;52(2):215–21. [DOI] [PubMed] [Google Scholar]
- 27.Deng S, Yan T, Jendrny C, Nemecek A, Vincetic M, Godtel-Armbrust U, et al. Dexrazoxane may prevent doxorubicin-induced DNA damage via depleting both topoisomerase II isoforms. BMC cancer 2014;14:842 doi 10.1186/1471-2407-14-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan T, Deng S, Metzger A, Gödtel-Armbrust U, Porter AC, Wojnowski L. Topoisomerase II{alpha}-dependent and -independent apoptotic effects of dexrazoxane and doxorubicin. Mol Cancer Ther 2009;8(5):1075–85 doi 10.1158/1535-7163.mct-09-0139. [DOI] [PubMed] [Google Scholar]
- 29.Deng S, Yan T, Nikolova T, Fuhrmann D, Nemecek A, Godtel-Armbrust U, et al. The catalytic topoisomerase II inhibitor dexrazoxane induces DNA breaks, ATF3 and the DNA damage response in cancer cells. Br J Pharmacol 2015;172(9):2246–57 doi 10.1111/bph.13046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryan CW, Merimsky O, Agulnik M, Blay JY, Schuetze SM, Van Tine BA, et al. PICASSO III: A Phase III, Placebo-Controlled Study of Doxorubicin With or Without Palifosfamide in Patients With Metastatic Soft Tissue Sarcoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2016;34(32):3898–905 doi 10.1200/JCO.2016.67.6684. [DOI] [PubMed] [Google Scholar]
- 31.Tap WD, Papai Z, Van Tine BA, Attia S, Ganjoo KN, Jones RL, et al. Doxorubicin plus evofosfamide versus doxorubicin alone in locally advanced, unresectable or metastatic soft-tissue sarcoma (TH CR-406/SARC021): an international, multicentre, open-label, randomised phase 3 trial. The lancet oncology 2017;18(8):1089–103 doi 10.1016/S1470-2045(17)30381-9. [DOI] [PMC free article] [PubMed] [Google Scholar]