

Summary
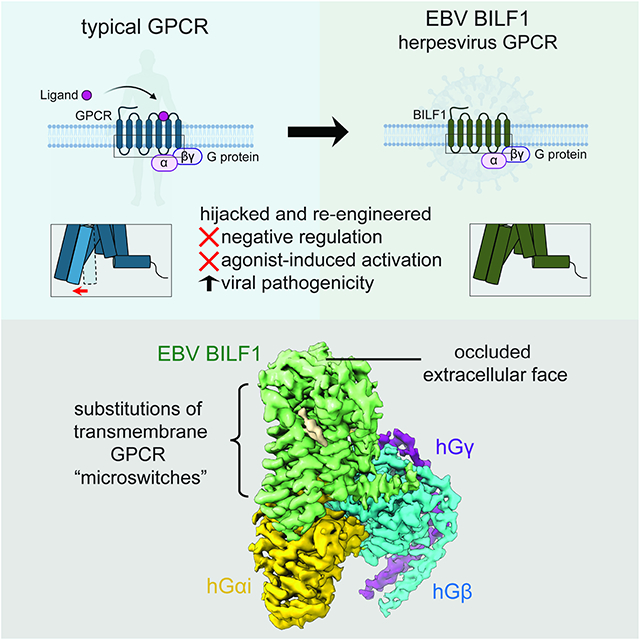
Epstein-Barr virus (EBV) encodes a G protein-coupled receptor (GPCR) termed BILF1 that is essential for EBV-mediated immunosuppression and oncogenesis. BILF1 couples with inhibitory G protein (Gi), the major intracellular signaling effector for human chemokine receptors, and exhibits constitutive signaling activity; the ligand(s) for BILF1 are unknown. We studied the origins of BILF1’s constitutive activity through structure determination of BILF1 bound to the inhibitory G protein (Gi) heterotrimer. The 3.2 Å resolution cryo-electron microscopy structure revealed an extracellular loop within BILF1 that blocked the typical chemokine binding site, suggesting ligand-autonomous receptor activation. Rather, amino acid substitutions within BILF1 transmembrane regions at hallmark ligand-activated class A GPCR “microswitches” stabilized a constitutively active BILF1 conformation for Gi coupling in a ligand-independent fashion. Thus, the constitutive activity of BILF1 promotes immunosuppression and virulence independent of ligand availability, with implications for the function of GPCRs encoded by related viruses and for therapeutic targeting of EBV.
Graphical Abstract
eTOC blurb
Herpesviruses hijack endogenous G protein-coupled receptors (GPCRs) signaling to evade host immunity. Tsutsumi et al. report the cryo-EM structure of BILF, an Epstein-Barr virus encoded GPCR, in complex with human Gi heterotrimer and reveal how a class A GPCR evolved constitutive activities to disrupt host chemokine signaling.
Introduction
Epstein-Barr virus (EBV), also known as human herpesvirus 4, is a ubiquitous virus that primarily targets human B cells and endothelial cells, and is estimated to infect almost 90% of the human population (Young et al., 2016). During EBV infection there is an interplay between EBV and the host that promotes viral persistence, including a variety of strategies to manipulate the host immune response. While most EBV infections are asymptomatic, EBV causes widespread immunosuppression, and is known to be the causative agent for mononucleosis. EBV has oncogenic activity on B cells and is implicated in endemic Burkitt’s lymphoma, as well as other cancers (Farrell, 2019; Mundo et al., 2020). The EBV genome encodes various immune modulators, among them, BILF1 is a putatively ‘orphan’ G protein-coupled receptor (GPCR) (Beisser et al., 2005; Paulsen et al., 2005). BILF1 is expressed predominantly in the early lytic phase contributing to immune evasion of EBV-infected cells during viral replication (Nijmeijer et al., 2010; Zuo et al., 2009), but also detected during viral latency (Tierney et al., 2015). The EBV lytic replication cycle and periodic reactivation allow the virus to produce infectious particles and spread. BILF1 physically associates with major histocompatibility complex class I (MHC-I) molecules to enhance internalization and depletion of MHC-I from the host cell surface to escape surveillance by cytotoxic CD8+ T cells (Fares et al., 2019; Griffin et al., 2013; Zuo et al., 2009).
An important feature of BILF1 is that it appears to exhibit constitutive GPCR activities despite the fact that the existence of a natural ligand has not been ascertained (De Groof et al., 2021). Thus, mere expression of BILF1 on the infected cell surface is enough to initiate a cascade of downstream events to enhance its virulence and pathogenesis in an unregulated fashion. BILF1 is coupled with inhibitory G protein (Gi) (Beisser et al., 2005; Lyngaa et al., 2010; Paulsen et al., 2005), which is the major intracellular signaling effector for human chemokine receptors (Flock et al., 2017; Murphy, 1994). One effect of BILF1-mediated Gi activation is to attenuate Gi signaling of other endogenous chemokine receptors expressed on the infected cell by hijacking the host signal transduction pathway (Nijmeijer et al., 2010). In this scenario, BILF1 exhausts the intracellular Gi pool via its constitutive Gi coupling, leading to impaired signaling by the human CXC chemokine receptor (CXCR) 4 upon stimulation by the CXC chemokine ligand 12 (CXCL12). The ‘cis repression’ phenomenon is also observed with endogenous human GPCRs, where prolonged activity of one GPCR reduces signaling from another GPCR that shares the same intracellular signaling pathway (Canals and Milligan, 2008; Ziffert et al., 2020). In addition to its role in immune evasion, BILF1’s constitutive Gi coupling likely enhances its neoplastic activity and tumorigenesis (Lyngaa et al., 2010). Collectively, BILF’s role in promoting EBV infection and oncogenesis is enabled by its constitutive activities and makes the BILF1-Gi signaling axis an attractive target for therapeutic intervention (De Groof et al., 2021).
Despite their importance in viral pathogenesis, immune evasion and cancer, structural information about viral GPCRs (vGPCRs) (Montaner et al., 2013) encoded by β- and γ-herpesviruses is limited compared to mammalian GPCRs. We previously showed how US28, a chemokine receptor encoded by human cytomegalovirus (HCMV), acts as a “chemokine trap” that binds to and is activated by human CX3C chemokine ligand 1 (CX3CL1, fractalkine) in order to subvert the host immune system (Burg et al., 2015). In contrast, EBV-encoded BILF1 immunosuppresses the host through its constitutive activity, but the identity of a putative ligand and its role(s) in the immunomodulatory functions of BILF1 remain undefined despite considerable efforts (Beisser et al., 2005) - BILF1 is assumed to possess a chemokine-type ligand. The absence of a known ligand as also prevented a full understanding of the mechanisms underlying the robust constitutive BILF1-Gi signaling.
We took a structural approach to examine the basis of constitutive BILF1-Gi signaling. Through cryo-electron microscopy (cryo-EM), we solved the structure of BILF1 in complex with Gi heterotrimer at 3.2 Å resolution. Structural and functional analysis revealed that BILF1 has occluded access to the typical soluble GPCR ligand-binding site and assumes an atypical ligand-independent “on state” compared to canonical class A GPCRs. The BILF1-dependent Gi activities were highly tolerant to mutagenesis, suggesting that interactions throughout the entire protein are important to retain robust constitutive activity and negate the need for a host ligand. Thus, the constitutive activity of BILF1, which enables its major immunoevasive and oncogenic activities, results from evolved ability to signal in the absence of a natural host or viral ligand. This constitutive activity results in unregulated host immune suppression and oncogenic transformation.
Results
Overall structure of the signaling BILF1-Gi holocomplex
Wild-type full-length BILF1, a vGPCR (Figure 1A), was expressed in human embryonic kidney (HEK) 293S GnTI− cells and extracted in a buffer containing the detergent lauryl maltose neopentyl glycol (LMNG) with cholesterol hemisuccinate (CHS). The purified receptor was assembled with Gi heterotrimer in the presence of a G protein stabilizing antibody scFv16 (Maeda et al., 2018) in a buffer containing LMNG, CHS, and glyco-diosgenin (GDN), yielding the monodisperse holocomplex (Figure S1A and S1B). We performed single-particle cryo-EM for the sample, which yielded a 3D reconstruction of the BILF1 signaling complex at a global indicated resolution of 3.2 Å (Figure 1B, S1C-S1F, and S2; Table S1). The receptor has the typical seven-transmembrane (7TM) GPCR topology with an α-helix 8 (H8) that runs along the cell membrane plane (Figures 1C). The BILF1-Gi complex adopts the single canonical nucleotide-free state (Figure S1E), with the overall docking mode similar to mammalian GPCR-G protein complexes (Wang et al., 2020). BILF1 interacts exclusively with the Gαi1 subunit and has limited contact with the Gβ1γ2 subunits as seen in other GPCR-Gi/o complexes, with interface areas of 1180 Å2 and 50 Å2, respectively.
Figure 1. Cryo-EM structure of the transmembrane EBV BILF1 in complex with human Gi heterotrimer.

(A) A cartoon representation of the phylogenetic relationships between vGPCRs encoded by human herpesviruses (HHV) and human chemokine receptors. The herpesvirus strains are indicated at the bottom of the vGPCR names. The figure was modified from (Montaner et al., 2013). EBV: Epstein-Barr virus. HCMV: Human cytomegalovirus. KSHV: Kaposi's sarcoma-associated herpesvirus. CCR, CXCR, CX3CR, XCR, and ACKR: CC, CXC, CX3C, XC, and atypical chemokine receptors, respectively. (B) 3.2 Å cryo-EM 3D reconstruction from the full-length EBV BILF1 in complex with human Gαi1Gβ1γ2 heterotrimer and the G protein complex stabilizing antibody scFv16. (C) Cartoon representations of the BILF1-Gαi1Gβiγ2-scFv16 model from top-down and side views. Disulfide bond-forming cysteines are displayed as sticks. In the cryo-EM map and the cartoon model, the components are colored in green (BILF1), yellow (Gαi1), cyan (Gβ1), purple (Gγ2), and gray (scFv16). The cryo-EM map is depicted around a 3 Å distance from the model built, and the contour level is set at 4.7σ. An unassigned density observed between TM6 and TM7 is not modeled but highlighted in wheat color in the map.
Extracellular structure of BILF1 in comparison with human and viral chemokine receptors
A striking feature of BILF1 is that extracellular loop (ECL) 2 forms a lid that caps the extracellular vestibule (Figures 2A and B). This feature differs from the chemokine-binding GPCRs CXCR2, CXCR4, and US28 (Figures 2C and S3), where the extracellular vestibule is open and accessible for chemokine recognition. BILF1-ECL3 also undergoes an inward movement, collectively blocking the typical orthosteric ligand binding pocket and restricting access of soluble ligands to the extracellular pocket. BILF1 has not been functionally paired with chemokines, or any ligand, for either signaling or ligand sequestration (De Groof et al., 2021), so the occluded pocket supports the hypothesis that BILF1 is likely not an ‘orphan’ but rather has evolved to not require binding to chemokines or other ligands. Comparison of the current active-state structure of BILF1 with the inactive state of the closest human homolog, CXCR4, shows that the TM helices are structurally different, yielding a root-mean-square deviation (RMSD) of ~8.1 Å (Figure S3B).
Figure 2. BILF1 shows the occluded extracellular ligand-binding pocket in comparison with chemokine receptors.

(A) A top view of the BILF1 structure. N-terminal loop (blue), ECL1 (pale green), ECL2 (red), and ECL3 (wheat) are shown as transparent surface presentation and overlaid on the cartoon model of BILF1 (green). (B) The extracellular cartoon view of BILF1 showing the ECL2 structure capping the extracellular pocket, with ECL2 highlighted with red color. ECL2a and ECL2b indicate the loops composed of amino acid residues between L167 and M173, and between R173 and K180, respectively. Disulfide bond-forming cysteines are displayed as sticks. (C) Extracellular structures of CXCR2 (light orange, PDB ID: 6LFO), CXCR4 (cyan, PDB ID: 4RWS), and US28 (pink, PDB ID: 4XT1). The ECL2s are highlighted with red color and dashed circles. Disulfide bondforming cysteines are displayed as sticks. The extracellular pocket of BILF1 is blocked by ECL2, whereas it is largely open in the cases of chemokine receptors (CXCR2, CXCR4, and US28). (D) Gi signaling of the wild-type and ECL2 modified BILF1. ECLGGG, ECLCXCR4a, and ECLCXCR4b donate the BILF1 ECL2 mutants harboring L167G-N168G-R169G mutation with G170-P171-N172 deletion, L167E-N168A-R169D-G170D-P171R-N172Y-M173I mutation, and R175D-E176R-G177F-P178Y-T179P-K180N mutation, respectively. ECLCXCR4ab is a combination mutant of ECLCXCR4a and ECLCXCR4b. See also Figure S5A. IP3 accumulation was measured in HEK293 cells expressing GαΔ6qi4myr and transfected with 0, 0.3, 0.6, 1.3, 2.5, 5, 10, 30, or 50 ng plasmid DNA/well of each BILF1 construct along with the same concentrations of the empty vector (negative control). The left panel shows the gene dose-dependent signaling as an overlaid line graph, and the right panel shows the values at 10 ng DNA/well condition as a bar graph subjected to statistical tests. Normalization is performed at 10 ng wild-type BILF1 DNA/well as 100% Gi signaling. (E) Total protein expression of wild-type and the ECL2 mutant BILF1. ELISA assay was performed on permeabilized HEK293 cells using 0, 10, 30, or 50 ng DNA/well of each FLAG-tagged BILF1 construct and the empty vector (negative control). The left panel shows the gene dose-dependent protein expression as an overlaid line graph, and the right panel shows the values at 50 ng DNA/well condition as a bar graph for statistical tests. Normalization is performed at 50 ng wild-type BILF1 DNA/well as 100% total protein expression. Mutant labeling on x-axis is colored in black: wild-type-like expression, red: higher expression, blue: lower expression. The experiments were performed at least three times in duplicates. The squares in the line graphs and the bars in the bar graphs indicate mean of the relative experimental values. The error bars represent standard errors of mean (SEM), and statistical significance was calculated using Student t-test; ** p < 0.01.
The disposition of ECL2 in viral and chemokine GPCRs may provide clues about function. Among the viral GPCR family, BILF1 belongs to a branch that is distinct from HCMV-encoded US28, and is closer in evolutionary relatedness to Kaposi's sarcoma herpesvirus (KSHV) chemokine receptor ORF74 and orphan HCMV GPCRs UL33 and UL78 (Montaner et al., 2013). In both the human (eg. CXCR2 and CXCR4) and viral chemokine receptors, ECL2 forms a β-hairpin (Figure 2C) and plays key roles in their ligand-binding functions (Burg et al., 2015; Liu et al., 2020; Qin et al., 2015). In human class A GPCRs, ECL2 is connected to transmembrane helix (TM) 3 by a conserved disulfide bond in order to maintain the architecture of the ligand binding pocket and contributes to agonist recognition. In the structure of the CX3CL1-US28 complex, the agonist:ECL2 interaction is mediated via the chemokine’s β1–β2 turn (the 30s loop) (Burg et al., 2015; Miles et al., 2018). In the BILF1 structure, ECL2 (G162-P178), which includes three glycine-proline (GP) repeats, sits on top of the extracellular ligand binding pocket, acting like a self ‘ligand’ that is closely packed by ECL1 and ECL3 (Figure 2B). The extracellular architecture is further stabilized by the extra disulfide bond between the N-terminal loop and ECL3 via the C28-C258 bridge, which is a signature of chemokine receptors. The ECL2 sequence is highly conserved among BILF1 sequences encoded by nonhuman primate lymphocryptoviruses (Spiess et al., 2015) (Figure S4), strongly implying that these BILF1 homologs also have occluded ligand binding pockets.
We initially hypothesized ECL2 may play a crucial role in its constitutive activity, perhaps acting as a “self-agonist” as has been seen in the GPR52 structure (Lin et al., 2020). However, radical substitutions of BILF1 ECL2 residues did not abolish the constitutive Gi signaling (Figures 2D and S5), although they substantially reduced the protein expression levels (Figure 2E).
The Gi signaling activity of BILF1 mutants was assessed by a membrane-proximal cell-based assay in HEK293 cells expressing BILF1 constructs, together with a chimeric Gi protein (GαΔ6qi4myr) that recognizes Gi-coupled GPCRs, but elicits Gq-dependent phospholipase C activation. We measured the accumulation of inositol phosphate (IP3) as a readout of this chimeric Gi signaling (Figure 2D). To validate the system, we first compared the signaling between the wild-type BILF1 and the K122A3.50 mutant. K1223.50 locates at the DR3.50Y-like EK3.50T motif (superscripts donate generic numbering for class A GPCRs (Isberg et al., 2015); for BILF1, the numbering is based on the structural comparison with CXCR4 (Figures S3 and S4)), which is typically important for both the conformational activation of class A GPCRs and the direct interaction with G protein (Zhou et al., 2019). The alanine substitution of this residue is known to silence BILF1’s Gi-cAMP signaling and reduce its ability to transform mammalian cells (Lyngaa et al., 2010). Consistent with this, the K122A3.50 mutant showed significantly lower Gi signaling (~20% activities), with even ~50% higher protein expression than the wild-type BILF1. We note that both the wild-type and mutant BILF1 showed gene-dose dependent increase of the signaling up to 10 ng DNA/well, and decrease at the higher gene concentrations. As similar behaviors were observed for most other mutants tested in this study regardless of their expression levels, we chose the 10 ng DNA/well condition for normalization and analysis of all cell-based signaling experiments. We also noticed a few mutants showed a trait to reach the maximum signaling at lower gene does, where we performed an additional analysis at 5 ng DNA/well. For comparison of protein expression levels, we used the 50 ng DNA/well conditions due to limited detection sensitivity at lower concentrations (Figure 2E).
We next assessed the ECL2 chimeras using the same system. A replacement of residues L167-N172 with a triglycine linker (ECLGGG) maintained ~60% of Gi signaling, but with ~30% of protein expression compared to the wild-type. The same outcome was observed by exchanging either 7 amino acid residues before or 6 amino acids after Cys174 in ECL2, termed ECL2a or ECL2b (Figure S5A), respectively, with the corresponding CXCR4 residues, as well as for the combined exchange of both ECL2a and ECL2b with those of CXCR4, substantiating that the conserved residues in ECL2 are not essential for Gi signaling. We also determined inhibition of the forskolin-induced cAMP response element (CRE) activity (Figure S5), where the differences amongst mutants were smaller than in the membrane-proximal assay. Compared to the CRE inhibition by the wild-type BILF1 at 10 ng DNA/well, K122A3.50 showed ~75% inhibition, giving only small range to rank the activities of mutants. Nevertheless, the result was analogous to the IP3 assay, and these findings indicate ECL2 has only supportive effects for BILF1’s Gi signaling, but is more critical for its protein expression and stability. This is in contrast to the orphan GPR52, whose ECL2 is deeply plugged into the major ligand binding pocket, and works as a built-in ‘agonist’ and is indispensable for receptor coupling to Gs protein (Lin et al., 2020).
BILF1 is a structural analog of lipid GPCRs
Although thought to be a chemokine GPCR homolog (de Munnik et al., 2015), the overall structural topology of BILF1 is more similar to lipid GPCRs (prostaglandin receptors and thromboxane A2 receptor) (Audet et al., 2019; Fan et al., 2019; Morimoto et al., 2019; Toyoda et al., 2019) (Figure 3A). Prostanoid receptors share a similar ECL2 structure capping the TM helical bundle that blocks access of soluble ligands, yet they accommodate lipidic agonists and antagonists into the major ligand binding groove from the gap between TM1 and TM7 at the side of the receptors buried in the plasma membrane. Although BILF1 shows a tight TM1-TM7 packing, we observed a gap between TM6 and TM7 that might be large enough to accommodate aliphatic small molecules or lipids. Indeed, we observed a prominent EM density embedded in the gap (Figures 3B and S2E). As this shape of the density does not resemble any components used during the purification, we surmised that it could be an endogenous lipidic compound co-purified with BILF1 from the HEK293 cell membrane instead, although we do not have biochemical evidence for the identity of this density. Additionally, we observed weak cryo-EM density at the tip of the Y2677.37 sidechain which could be a sulfate or phosphate modification site (Figure 3B right). To probe the potential importance of this density occupying the TM6-TM7 gap, a selection of residues in TM6 (M2406.50, L2416.51 and L2436.53) and TM7 (Y2677.37 and A2717.42) that make contact with the unassigned density were selected for substitution with either alanine or phenylalanine (Figures 3C-3F and S5). However, the effects of these mutations were either small or insignificant on Gi signaling. The single point “bulky” mutant A271F7.42 reduced expression by 30~40% likely due to its effect on protein stabilities, while M240F6.50 and L241F6.51 maintained protein expression at the level of the wild-type BILF1. M240F6.50 and L241F6.51 mutants achieved wild-type-like signaling with its maximum at 5 ng DNA/well. The A271F single point mutant as well as quadruple (TM6x7-4A) and quintuple (TM6x7-4A1F) mutants showed slightly lower signaling compared to the wild-type BILF1. Nevertheless, the differences observed with both IP3 and CRE signaling assay were not substantial compared to ECL2 chimeras, and we conclude that the unassigned molecule occupying this site is likely not critical to BILF1’s constitutive Gi activity. However, we do not rule out that there could be a physiological molecule that stabilizes the pre-formed active conformation.
Figure 3. BILF1 has topological similarities to lipid GPCRs but shows a ligand-independent Gi activity.

(A) Structural comparison between BILF1 (green), prostaglandin E2 receptor 4 (EP4, gray, PDB ID: 5YWY), and thromboxane receptor (TP, yellow, PDB ID: 6IIU). The cartoon models are displayed side-by-side from the side view with disulfide bond-forming cysteines shown as sticks. The ECL2s are colored in red. (B) An assigned density observed between TM6 and TM7. The overall cryo-EM map is shown at the left panel with the red dashed-square closed-up in the right panel, where the surface representation of the unassigned density (wheat) and the surrounding BILF1 structure are display. The cartoon BILF1 model (dark green) and the amino acid residues contacting the unassigned density are shown as sticks with the overlaid cryo-EM density (white). The red dashed circle indicates an extra cryo-EM density continuous from the Y267 sidechain. The map contour level is set to 3.8σ. (C, E) Gi signaling of wild-type and the TM-gap mutant BILF1. IP3 accumulation was measured in HEK293 cells expressing GαΔ6qi4myr, with 0, 0.3, 0.6, 1.3, 2.5, 5, or 10 ng/well of each construct transfected as indicated in the figures. TM6x7-4A and TM6x7-4A1F donate the BILF1 variants harboring M240A-L241A-L243A-Y267A and M240A-L241A-L243A-Y267A-A271F mutations, respectively. The left line graphs show the gene dose-dependent signaling, and the right bar graphs show the relative values at the 5 ng or 10 ng DNA/well conditions subjected to statistical tests. Normalization is performed at 10 ng wild-type BILF1 DNA/well as 100% Gi signaling. (D, F) Total expression of wild-type or the mutant BILF1. The left line graphs show the gene dose-dependent protein expression at 0, 10, 30, or 50 ng DNA/well measured by anti-FLAG ELISA, and the right bar graphs show the relative values at the 50 ng DNA/well condition for statistical tests. Normalization is performed at 50 ng wild-type BILF1 DNA/well as 100% total expression. Mutant labeling on x-axis is colored in black: wild-type-like expression, red: higher expression, blue: lower expression. The experiments were performed at least three times in duplicates. The squares in the line graphs and the bars in the bar graphs indicate mean of the relative experimental values. The error bars represent standard errors of mean (SEM), and statistical significance was calculated using Student t-test; ** p < 0.01, * p < 0.05.
Remodeled structure of BILF1 transmembrane region stabilizes constitutive Gi activities
Given the small effect by the putative aliphatic ligand, and that ECL2 is not acting as a self-agonist, an important question is how BILF1 has structurally evolved into a robust constitutively active GPCR. In short, a structure-wide, global evolutionary modification of the typical class A GPCR structural motifs appears to underlie BILF1 constitutive activity. A striking feature of the BILF1-7TM structure is conversion of several conserved GPCR “microswitches” that are necessary for agonist-induced conformational changes and allosteric modulation in ligand-activated GPCRs (Zhou et al., 2019). Examples include the structural motifs: [NP7.50xxY7.53], [DR3.50Y], [CW6.48xP6.50], and [p5.50-I3.40-F6.44], the ionic lock and the sodium-binding pocket. Interestingly, analogous sequence conversions of these microswitches have been synthetically generated by directed mutagenesis of canonical ligand-activated GPCRs to create hyperactivated or constitutive forms, such as Rhodopsin (Rho) (Fritze et al., 2003), β2-adrenergic receptor (Chen et al., 2002), A2A adenosine receptor (Zhou et al., 2019), and a chemokine receptor CXCR2 (Burger et al., 1999). BILF1 appears to have naturally evolved mutations in these microswitches to transform them into constitutively active motifs.
As one example, in class A GPCRs, P7.50 is 93.7% conserved (Gómez Tamayo et al., 2018) and positioned at the NP7.50xxY7.53 motif of TM7 (Figures 4A and 4B), where the proline residue mediates its transition from the inactive to active conformation for G protein binding, upon agonist stimulation. Generally, in an inactive state, P7.50 kinks the intracellular end of TM7, with Y7.53 facing the TM1-TM2 side, and interacting with F8.50 on C-terminal helix 8 (H8) (Figure 4A). In an active state, the kink is relaxed and TM7 adopts a straighter conformation, where Y7.53 is turned inward to form the G protein binding interface with the DR3.50Y motif at TM3. BILF1, on the other hand, has the LI7.50ILY7.53 sequence at the corresponding position, resulting in a linear TM7 and an active receptor conformation (Figures 4A and 4B). A second microswitch is on TM3, where the canonical DR3.50Y motif, found in ligand-activated GPCRs, is replaced by an EK3.50T motif in BILF1 (Figures 4A and 4B). Another microswitch, the CW6.48xP6.50 motif important for agonist recognition in class A GPCRs, is converted to SW6.48xM6.50 in BILF1 (Figure 4C). In ligand-activated GPCRs, P6.50 of this motif creates a hinge on TM6 that plays a pivotal role in moving the cytoplasmic half of TM6 outward upon ligand-mediated activation of the receptor to create an open cavity for G protein binding. In this scenario, C6.47 works as a rotamer switch to rearrange the TM6-TM7 interface upon activation (Olivella et al., 2013).
Figure 4. BILF1 possesses remodeled conformational switches that result in its constitutive Gi activity.

(A) Comparison of the TM7 conformations among active BILF1 (green), active human Rhodopsin (Rho, pink, PDB ID: 6CMO), and inactive bovine Rho (gray, PDB ID: 1F88). BILF1 has the linear helical structure at the bottom of TM7 that interacts with the EKT motif at TM3. (B) A close-up view around the bottom region of BILF1-TM7 (right) indicated as the red dashed square in the overall BILF1 structure (left), showing the intramolecular interaction network. E121 at the D(E)RY-like EKT motif shows extensive interaction with the TM2 residues to stabilize the structure. Polar and aromatic interactions with <4 Å distance are highlighted with red dashed lines. (C) A close-up view around the top region of BILF1-TM6 (right) indicated as the red dashed-square in the overall BILF1 structure (left), showing the region containing the CWxP motif-like SWxM. (D, F) Effect on Gi signaling by single point mutations or the motif reversions that would disrupt the intrahelical packing of BILF1. NPxxY, CWxM, CXxP, and SWxP indicate BILF1 variants harboring L278N-I279P, S237C, S237C-M240P, and M240P mutations, respectively. The left line graphs show BILF1 gene-dose IP3 accumulation measured in HEK293 cells expressing GαΔ6qi4myr and transfected with the constructs indicated in the figures. The right bar graphs indicate the relative signaling amplitude at 10 ng DNA/well conditions for statistical analyses. Data were normalized to 10 ng wild-type BILF DNA/well as 100% singling. (E, G) Total expression of wild-type and the BILF1 mutants. The left line graphs show gene-dose BILF1 expression measured by anti-FLAG ELISA on permeabilized HEK293 cells transfected with the same constructs as the signaling assays, but normalization to the 50 ng wild-type BILF1 DNA/well condition. All cell-based assay were performed at least three times in duplicates. The squares in the line graphs and the bars in the bar graphs indicate mean of the normalized experimental values. The error bars represent standard errors of mean (SEM), and statistical significance was calculated using Student t-test; ** p < 0.01, * p < 0.05.
In order to assess the functionalities of these altered motifs in BILF1, we made single point mutation and reverted the motifs to canonical class A GPCR residues at the corresponding positions (Figures 4D-G and S5). Y282A7.53 mutation had analogous, but less substantial effects compared to the alanine substitution of the partner residue K1223.50, resulting in ~60% reduction of the signaling with ~20% higher protein expression than the wild-type BILF1 (Figures 4A, 4D and 4E). Similarly, the LI7.50ILY to NP7.50ILY conversion showed ~40% attenuation in signaling with ~20% lower expression (Figures 4A, 4F and 4G). Another mutation on the EK3.50T motif, E121A3.49, (Figures 4B, 4D and 4E), showed ~50% expression and signaling compared to the wild-type, consistent with the sidechain mediating the extensive intramolecular hydrogen-bonding network to maintain the receptor structure. The functional pressure for this sequence conversion is evident in that EK3.50 T to ER3.50 T mutation maintains comparable or slightly reduced activity to wild-type BILF1, whereas the EKT to DRY conversion significantly reduces signaling (Lyngaa et al., 2010), suggesting that EKT has been evolutionarily optimized for BILF1. We also tested single point mutants L75A2.47 and H115A3.43 around this region that appear to mediate important inter-helical packing with the sidechains facing the receptor core, and observed lower signaling and expression. The CWxP6.50 motif-like SWxM6.50 was also targeted for mutagenesis. In TM6, the introduction of the kink residue P6.50 didn’t change the pattern of the signaling amplitudes, whereas the S6.47 to C6.47 conversion slightly weakened the signaling by ~20%.
While we identified a few sequence modifications that attenuated the BILF1 expression and signaling, in general, the receptor activity was very robust to the mutational disruption. These results indicate BILF1 has evolutionary accumulated mutations that favor constitutive activity at multiple locations within the TM bundle, making it highly tolerant to mutational pressure that may alter its pathogenic functions.
Structural comparison of the Gi interface between BILF1 and endogenous GPCRs
The overall assembly and binding mode between BILF1 and Gi bear close similarities to mammalian GPCR-G protein complexes (Figures 5A and S6) with K1223.50 at the center of the BILF1:Gαi interface (Figure 5B). However, we observed a notable difference that appeared to define a higher specificity between BILF1 and Gi for more efficient disruption of chemokine-induced Gi signaling. The Gαi/o family of G proteins has an aromatic residue at the α5 C-terminal end, which is recognized by BILF1 in a manner different from other Gi/o-coupled GPCRs. In BILF1-Gi, the C-terminal F(−1) of Gαi is trapped by the aromatic and cationic cluster formed by H66 at ICL1 and F2867.57 and H2888.49 at the TM7-H8 turn (Figures 5B, S2D and S4). This conformation is further stabilized by the ionic interaction between C-terminal carboxylate of Gαi and R2226.32. Most other GPCR-Gi/o complexes place the F(−1) sidechain at the TM6 side, while F(−1) of μ-opioid receptor (μOR) is at the intermediate position between BILF1 and other GPCR-Gi/o complexes (Figure 5C) (Kang et al., 2018; Kato et al., 2019; Koehl et al., 2018; Krishna Kumar et al., 2019; Maeda et al., 2019). We initially hypothesized this unique interface could be a tunable site, because the intracellular cavity between ICL2 and TM7-H8 is generally considered to be the pharmacological hotspot for human chemokine receptors (Jaeger et al., 2019; Nicholls et al., 2008; Oswald et al., 2016; Salchow et al., 2010; Zheng et al., 2016). However, the alanine substitutions of this site did not make substantial changes (Figures 5D, 5E and S5B). We also examined a bulky mutant at the cytoplasmic end of TM3, A125F3.53 (Figures 5D, 5E and S5B), which may mimic the Gi inhibition by a small molecule. We chose this residue as it appeared to be the only appropriate small amino acid on the interface, but one aromatic ring at this site was not enough to make a critical effect on the signaling. These results indicate the BILF1 activity is also robust to Gi interface disruption by mutagenesis, except for the core interface residue K1223.50.
Figure 5. The BILF1-Gαi interface shows overall similarities and local differences to endogenous GPCR-Gi complexes.

(A) Structural comparison among the Gi complexes with either viral or mammalian GPCRs. Cartoon models of BILF1 (green), Rho (gray, PDB ID: 6CMO), μ-opioid receptor (μOR, purple, PDB ID: 6DDE) and cannabinoid receptor-1 (CB1, dark green, PDB ID: 6N4B) complexed with Gi heterotrimer (Gαi1: yellow, Gβ1: cyan, Gγ2: purple) are shown side-by-side on the left panel. ScFv16 is omitted for clarity. Orange lines indicate approximate cell membrane boundaries. A top-down view of their structural superimposition is displayed on the right with all the GPCRs shown as the transparent wheat surface representation. The Gβ1γ2 subunit is omitted for clarity, and the Gαi subunit is colored in yellow (BILF1), gray (Rho), purple (μOR), and dark green (CB1). For the superimposition, structural alignment is based on α carbons (Cα) of residue 3.37 - 3.51 (generic GPCR numbering). (B) Interaction between the intracellular pocket of BILF1 (green) and the C-terminal region of Gαi1 (yellow). The close-up view of the red dashed box on the overall BILF1-Gαi1 model is displayed in the black square. C-terminal F(−1) is trapped by the aromatic cluster around the TM7-H8 turn and ICL2, and the C-terminal carboxylate is interacting with R2226.32. Polar and aromatic interactions with <4 Å distance are highlighted with red dashed lines. (C) Recognition of C-terminal α5 helix of Gαi/o by viral or mammalian GPCRs in their canonical binding mode. In addition to the four structures superimposed in (A), the neurotensin receptor-1 (NTSR1)-Gi (light purple, PDB ID: 6OS9) and muscarinic acetylcholine receptor M2 (M2R)-Go (cyan, PDB ID: 6OIK) are superimposed. The helices in GPCRs are shown as transparent cylinders, and Gα chains are shown as thin lines. The structures are aligned using Cα of Gαi/o proteins. (D) Effect on Gi signaling by the BILF1 mutations at the BILF1-Gαi interface. The left line graphs show BILF1 gene-dose IP3 accumulation measured in HEK293 cells expressing GαΔ6qi4myr and transfected with the constructs indicated in the figures. Data were normalized to the 10 ng wild-type BILF1 DNA/well condition. The right bar graphs show the relative values at the 5 ng or 10 ng DNA/well conditions subjected to statistical tests as indicated in the figures. (E) Total expression of wild-type and the mutant BILF1. The left line graphs show gene-dose BILF1 expression measured by anti-FLAG ELISA on permeabilized HEK293 cells transfected with the same constructs as the signaling assay, but normalized to the 50 ng wild-type BILF1 DNA/well condition. All cell-based assay were performed at least three times in duplicates. The squares in the line graphs and the bars in the bar graphs indicate mean of the normalized experimental values. The error bars represent standard errors of mean (SEM), and statistical significance was calculated using Student t-test; ** p < 0.01, * p < 0.05.
Discussion
By visualizing the structure of EBV BILF1 in complex with a human Gi protein, and revealing the complex structural origins of its high constitutive GPCR signaling underlying its pathogenic activities, we now provide a template for antagonist drug design. Unlike other viral GPCRs, BILF1 does not appear to be a human chemokine GPCR homolog, but topologically rather more resembles an evolved version of lipid GPCRs. It is noteworthy that human genome encodes an atypical chemokine receptor ACKR3, also known as CXCR7, that is evolutionarily intermediate between chemokine and opioid receptors and works as a broad-spectrum scavenger for opioid peptides (Meyrath et al., 2020). Based upon an unknown ancestral host GPCR, BILF1 has undergone an overall global evolutionary remodeling of its TM helices to favor maintenance of stable constitutive activity. The etiology of the ligand-independent activity appears to derive from the collective action of many mutations to canonical structural microswitches known to mediate ligand-dependent conformational changes in typical class A GPCRs. For example, BILF1 does not possess the ionic lock and sodium-binding pocket motifs, which are negative regulators of typical class A GPCRs. BILF1 might have lost this regulatory switch as an outcome of host-pathogen co-evolution, thereby disrupting G protein signaling homeostasis of infected cells. The “constitutively active motifs” observed in EBV BILF1 are broadly conserved in BILF1 orthologs from other lymphocryptoviruses, indicating it is a shared immune evasion mechanism among the broad species (Spiess et al., 2015).
One important function of BILF1 is downregulation of host cell surface MHC-I molecules, which is commonly employed by many types of viruses such as herpesviruses, lentiviruses and coronavirus SARS-CoV-2 (Schuren et al., 2016; Zhang et al., 2020). Those include ER resident type-I transmembrane proteins US2, US3, US6, and US11 encoded by HCMV (Gewurz et al., 2001; Wiertz et al., 1996), membrane-bound ubiquitin E3 ligases K3 and K5 encoded by KSHV (Ishido et al., 2000), a multifunction intracellular protein Nef encoded by lentiviruses such as HIV (Schwartz et al., 1996). As a ubiquitous human virus, EBV itself encodes multiple immunomodulatory proteins to disrupt several aspects of host immune reactions, including a membrane protein BNLF2A, a DNase BGLF5, and BILF1 that downregulate MHC-I during the lytic phase of infection. Among them, BILF1 plays a major role in interfering with CD8+ T cell recognition in cooperation with BNLF2A (Quinn et al., 2014). Unlike these other viral proteins, BILF1 is a GPCR that serves dual functions of MHC-I downregulation and disruption of the chemokine-Gi signaling. The correlation between BILF1’s Gi activity and MHC downregulation is controversial (Fares et al., 2019; Zuo et al., 2011), but the two functions appear to contribute to persistence of EBV infection independently. Endocytosis of the BILF1-MHC-I complex drives depletion of MHC-I from the cell surface; however, it is still unclear if the process is β-arrestin dependent as observed in many class A GPCRs.
BILF1 does not act alone. Thus, inhibition of BILF1 activities may be compensated to some extent by the presence of alternative pathways for host subversion by the virus. EBV persistence is achieved by concerted actions of a variety of EBV-encoded proteins that subvert host immunity, including latent membrane protein LMP2A (B cell receptor homolog), and lytic proteins BCRF1 (interleukin-10 homolog) and BILF1. EBV-encoded immunomodulatory proteins alter the complex B cell signaling network to enhance viral pathogenesis, facilitating immune evasion during viral latency and replication, yet the reprogrammed B cells are also more prone to be transformed. The BILF1-Gi axis disrupts host G protein signaling homeostasis (Nijmeijer et al., 2010), to impair normal activation and trafficking of EBV-infected B cells induced by chemokine stimulation. On the other hand, the BILF1-Gi signaling has the ability to induce tumorigenesis in vitro and in mice (Lyngaa et al., 2010). In fact, BILF1 expression was detected in Burkitt’s lymphoma cell lines even during latency, and EBV-transformed B cell (Quinn et al., 2014; Tierney et al., 2015). It is also expressed ubiquitously in a variety of EBV-positive cancers (Chakravorty et al., 2019).
Given BILF1’s important role in viral pathogenesis, immune suppression and neoplastic transformation, it is a prime target for inhibition of signaling after EBV infection (De Groof et al., 2021). Although BILF1 forms a structurally active conformation in a ligand-independent fashion to be compatible with constitutive and pathogenic G protein binding, the energetic basis of BILF1 constitutive activity is distributed throughout the structure, indicating it will be a challenging target for a conventional inverse agonist given the lack of localized targetable structural “hotspots” that would uncouple its activity. Indeed, our mutational analysis failed to significantly impair signaling, suggesting that small molecules that targeting the BILF1-Gi interface may be a more productive path than targeting the traditional ‘orthosteric’ ligand binding sites accessible from the extracellular face of the receptor.
Limitations of Study
Although the structure we report suggests that BILF1 is a structural analog of lipid GPCRs, and may have a lipid-like modulator based on observation of unassigned electron density. However, as BILF1 has features commonly seen in both lipid GPCRs and chemokine receptors, the evolutionary origin of BILF1 still remains elusive. Also, the assignment of the lipid-like density observed between TM6 and TM7, that would require precise analytical, biophysical, biochemical and pharmacological characterizations, has not been performed, therefore we cannot rule out the existence of an endogenous modulator of BILF1 function.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, K. Christopher Garcia (kcgarcia@stanford.edu).
Materials availability
All unique and stable reagents generated in this study are available via the Lead Contact upon a reasonable request.
Data and code availability
The cryo-EM map has been deposited in the Electron Microscopy Data Bank (EMDB) under accession code EMD-22338 and the model coordinate has been deposited in the Protein Data Bank (PDB) under accession number 7JHJ.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mammalian cell lines and culture conditions
To perform the signaling and protein expression assays for BILF1 variants, adhesion HEK293 cells (ATCC CRL-1573) were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Thermo Fisher) with 10% (v/v) fetal bovine serum (FBS, Sigma-Aldrich), 180 units/ml penicillin and 45 μg/ml streptomycin (Substrate Department, Faculty of Health and Medical Sciences, University of Copenhagen) at 37°C with 10% CO2. For recombinant BILF1 production, suspension HEK293S GnTI− cells (ATCC CRL-3022) were grown in FreeStyle 293 Expression Medium (Thermo Fisher) supplemented with 1% (v/v) FBS (Sigma-Aldrich) and maintained at 37°C with 5% CO2 and gentle agitation.
Insect cell lines and culture conditions
For recombinant protein expression, baculovirus was produced in Spodoptera frugiperda (Sf9) ovarian cells (ATCC CRL-1711) maintained in Sf-900 III medium (Thermo Fisher) with 10% (v/v) FBS (Sigma-Aldrich) and GlutaMAX (Thermo Fisher). G protein and scFV16 were expressed by baculoviral infection of Trichoplusia ni (Hi5) ovarian cells (Expression Systems, Cat# 94-002) maintained in ESF 921 Insect Cell Culture Medium (Expression Systems). Insect cells were grown at 27°C with ambient CO2 and gentle agitation.
METHOD DETAILS
Preparation of recombinant BILF1
The coding sequence of full-length EBV BILF1 (UniProt ID: P03208, residues 1-312) was cloned into the BacMam vector pVLAD6 (Dukkipati et al., 2008), which includes a C-terminal 3C protease site followed by a protein C epitope and an octahistidine tag. P0 virus was then prepared in SF9 by co-transfecting the expression vector and BestBac Linearized Baculovirus DNA (Expression Systems, CA, USA) using FuGENE HD Transfection Reagent (Promega, WI, USA), and the virus was amplified from P0 to P2. BILF1 was expressed in HEK293S GnTI− cells with BacMam baculovirus transduction. 10% (v/v) P2 virus and 10 mM sodium butyrate (Sigma-Aldrich, MO, USA) were added to cells at a density of 2-3 x 106 cells/ml and culture flasks were shaken at 5% CO2 and 37°C for 24 hrs. After centrifugation, cells were washed with phosphate-buffered saline (PBS) supplemented with 5 mM EDTA and 1:1000 protease inhibitor cocktail (PIC, Sigma), weighed and stored at −20°C. Approximately 30 grams of cell pellet was thawed and lysed with a Dounce homogenizer in a lysis buffer composed of 20 mM Tris-HCl pH 8.0, 5 mM EDTA, 2 mg/ml iodoacetamide, and 1:1000 PIC. The lysate was centrifuged at 48,000 x g for 1 hr and the membrane pellet was resuspended and nutated for 2 hrs in 400 ml of a solubilization buffer consisting of 10 mM HEPES-Na pH 7.2, 150 mM NaCl (HBS), 1% (w/v) Lauryl Maltose Neopentyl Glycol (LMNG, Anatrace; OH, USA), 0.2% (w/v) cholesterol hemisuccinate (CHS, Anatrace), 10% (v/v) glycerol, 2 mg/ml iodoacetamide, and cOmplete PIC (Roche, Basel, Switzerland).
After centrifugation at 48,000 x g for 1 hr, 10 ml Ni-NTA resin (Qiagen, Hilden, Germany) was added to the supernatant, and the mixture was stirred at 4°C overnight. The resin was then collected in a column; washed with HBS with 0.1% (w/v) LMNG, 0.01% (w/v) CHS, 10% (v/v) glycerol, and 20 mM imidazole (wash buffer); and eluted in wash buffer supplemented to 230 mM imidazole. The eluate was then supplemented with 2 mM CaCl2 and further purified over an in-house anti-Protein C affinity sepharose column. The receptor was eluted in HBS with 0.01% (w/v) LMNG, 0.001% (w/v) CHS, 0.2 mg/ml Protein C peptide and 5 mM EDTA, and further purified by size-exclusion chromatography (SEC) equilibrated with HBS, 0.01% (w/v) LMNG and 0.001% (w/v) CHS. The yield of the SEC purified BILF1 was ~5 mg from one preparation.
Preparation of Gi heterotrimer and scFv16
Gαi1Gβ1γ2 and scFv16 were each prepared separately as described (Koehl et al., 2018; Maeda et al., 2018). Gαi1, Gβ1 with an N-terminal 3C protease-cleavable His tag, Gγ2 were co-expressed in Hi5 cells with the baculovirus system. The membrane fraction was isolated, and solubilized with a buffer containing 20 mM HEPES-Na pH 7.4, 100 mM NaCl, 1% (w/v) sodium cholate, 0.05% (w/v) DDM, 5 mM MgCl2, 2 μL Calf Intestinal Alkaline Phosphatase (CIP, NEB), 5 mM 2-mercaptoethanol, 10 μM GDP, 5 mM imidazole, and PIC (Sigma). The detergent-solubilized Gαi1Gβ1γ2 heterotrimer was loaded over Ni-NTA resin, and the buffer was gradually exchanged to 20 mM HEPES-Na pH 7.4, 100 mM NaCl, 0.05% DDM, 1 mM MgCl2, 0.1 mM TCEP, 10 μM GDP, 20 mM imidazole (wash buffer). The protein was eluted with increased imidazole concentration to 200 mM, and dialyzed against the wash buffer supplemented with 1 mM MnCl2 in the presence of in-house 3C protease prepared with Rosetta 2 (DE3) and protein phosphatases (Antarctic Phosphatase, CIP, and Lambda Protein Phosphatase from NEB) overnight. The next day, the reaction was applied onto fresh Ni-NTA resin to remove the cleaved tag and His-tagged 3C protease. The flow-through was concentrated to ~20 mg/ml for aliquoting and flash-freezing with 10% (v/v) glycerol.
ScFv16 was expressed from Hi5 cells with a C-terminal 3C protease-cleavable His tag using the baculovirus system and purified from culture supernatant using Ni-NTA column. 3C tag was cleaved and removed, and the scFv16 solution was purified over a size exclusion chromatography column Superdex 75 10/300 GL equilibrated with HBS. The peak fraction was concentrated to ~20 mg/ml and flash-frozen with 10% (v/v) glycerol. Protein aliquots were kept at −80°C until future use.
Preparation of BILF1-Gαiβ1γ2-scFv16
1.2 and 1.4 molar excess of purified Gαi1β1γ2 and scFv16, respectively, were added to the SEC elution containing BILF1. The HBS-based buffer was then adjusted to contain 1% (w/v) LMNG, 0.1% (w/v) CHS, 0.1% (w/v) GDN, 0.1 mM TCEP and 1 mM MnCl2 (G protein coupling buffer), and incubated at room temperature for 1 hr, followed by addition of 5 μL Lambda Protein Phosphatase (NEB) to the complexing reaction. After incubation on ice for another 1 hr, 5 μL apyrase (NEB) was added, and the reaction mixture was incubated at 4°C overnight to hydrolyze GDP in the reaction.
Final pull-down was carried out on agarose bound Concanavalin A (Con A, Vector Laboratories, CA, USA) that binds to glycosylated N-terminus of BILF1 to remove excess G protein, scFv16, and impurities. The reaction was 10-fold diluted with the G protein coupling buffer, mixed with 4 mL of Con A resin, and nutated at 4°C for 2 hrs. The resin was then washed to reduce the detergent concentration to 0.01% (w/v) LMNG, 0.01% (w/v) GDN, 0.001% (w/v) CHS. The BILF1-Gαi1β1γ2-scFv16 complex was eluted in HBS containing 0.001% (w/v) LMNG, 0.001 (w/v) GDN, 0.0001% (w/v) CHS, 0.1 mM TCEP, 250 mM methyl α-D-mannopyranoside and 250 mM methyl α-D-glucopyranoside.
The Con A elution was concentrated down to 500 μL and purified over Superdex200 10/300 GL size-exclusion column equilibrated with HBS containing 0.001% (w/v) LMNG, 0.001 (w/v) GDN, 0.0001% (w/v) CHS and 0.1 mM TCEP. The final yield of the complex was ~3 mg from one preparation. Peak fractions were concentrated to approximately 30 mg/ml for cryo-EM study. The quality of the BILF1-Gi-scFv16 complex was checked with negative-stain EM with uranyl formate (VWR) (Figure 1B) before cryo-EM specimen preparation.
Cryo-EM experiment and analysis
3.5 μL of the BILF1-Gαi1β1γ2-scFv16 sample was applied to glow-discharged 200 mesh gold grids (Quantifoil R1.2/1.3) with or without the addition of 0.1% (w/v) n-octyl-β-D-glucopyranoside (βOG). Excess sample was blotted away by filter paper for 1 sec before plunge-freezing using a Vitrobot Mark IV (Thermo Fisher Scientific) at 18°C and 100% humidity.
For initial trials, we imaged the specimens prepared either with or without βOG on an FEI Titan Krios operated at 300 kV equipped with a Gatan K3 camera but without an energy filter. However, the resolutions of the refined 3D reconstructions were limited to 4.35 Å. To generate the final 3.2 Å map, movies were collected using the same specimen preparation batch as above but only with βOG on a Titan Krios operated at 300 kV equipped with K2 Summit camera and a Gatan imaging filter at a nominal magnification of 130,000x in super-resolution counting mode, corresponding to a physical pixel size of 1.06 Å. Each movie was recorded for a total of 8 sec with 0.2 sec exposure per frame at an exposure rate of ~7 electrons/pixel/sec and the defocus range between −1.0 and −2.0 μm, using SerialEM (Mastronarde, 2005) with beam-image shift to collect 9 images from 9 holes per stage shift and focus.
A total of 2,237 movies were subjected to beam-induced motion correction using MotionCor2 (Zheng et al., 2017) on the GUI of Relion version 3.1 (Zivanov et al., 2018). The contrast transfer function parameters of the motion-corrected micrographs were estimated from the dose-weighted averages of all frames using Gctf v1.06 (Zhang, 2016). A reference-based auto-picking was performed to extract 1,652,930 particles for classification. After iterative rounds of 2D classifications, 710,554 selected particles were used to generate the 3D initial model and subjected to 3D classification. The final 253,960 particles were iteratively refined using Relion 3D auto-refine followed by CTF refinement and Bayesian polishing, which yielded 3.2 Å map as determined by gold-standard Fourier shell correlation (FSC) using the 0.143 cutoff. Local map resolutions were estimated using Bsoft (Heymann, 2018).
Model building and refinement
A homology model for BILF1 was generated on the SWISS-MODEL server (swissmodel.expasy.org) that automatically chose Neuropeptide Y receptor Y1 (PDB ID: 5ZBQ, 15.16% sequence identity to BILF1) (Yang et al., 2018) as a template. The model and Gαi1β1γ2-scFv16 (from PDB ID: 6DDE) (Koehl et al., 2018) structure were roughly placed into the initial cryo-EM density map at a resolution of 4.6 Å using UCSF Chimera (Pettersen et al., 2004) and Coot (Emsley and Cowtan, 2004) to fit individual TMs, real-space refined using Phenix (Liebschner et al., 2019) with global minimization and rigid body constraints. The model was modified and built on Coot and refined on 4.6 Å and then on 4.35 Å maps from FEI Titan Krios equipped with Gatan K3 detector. To create the final model, the 4.35 Å model was fitted into the 3.2 Å map and further fixed and refined iteratively using Coot for manual model building, inspection and correction, ISOLDE (Croll, 2018) on UCSF ChimeraX (Goddard et al., 2018) for MD-based model idealization, and Phenix for real-space refinement. The structure and map were visualized using UCSF ChimeraX and PyMol (Schrödinger, LLC). Unless stated in figures or figure legends, the map contour levels are set to 4.7σ. This value appears to be reasonable to visualize most part of protein density, except for peripheral regions of the 3D volume. The secondary structures in the figures were assigned using DSSP (Joosten et al., 2011).
BILF1 constructs for functional analysis
Wild-type and mutant BILF1 variants were each cloned into the pcDNA3.1+ vector with an N-terminal FLAG-tag. The constructs were synthesized and verified by GenScript. HEK293 cells were transfected with expression vectors using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s recommendation.
IP3 assay
HEK293 cells were seeded in 96 well plates at 35,000 cells/well, and one day after seeding, the cells were transfected with plasmid DNA concentrations of 0, 0.3, 0.6, 1.3, 2.5, 5, 10, 30, or 50 ng/well. All cells were co-transfected with 30 ng of the chimeric G protein GαΔ6qi4myr that converts Gi signaling to Gq signaling for output reads (Heydorn et al., 2004). One day after transfection, the culture media was exchanged to the media containing myo-[3H]inositol (5 μl/ml, 2 μCi/ml) in 0.1 ml and the cells were incubated overnight. On the following day, the cells were washed twice in PBS and incubated in 0.1 ml of Hanks’ balanced salt solution (Invitrogen) supplemented with 10 mM LiCl at 37°C for 90 min. The assay medium was then removed, and the cells were extracted by the addition of 50 μl of 10 mM formic acid to each well, followed by incubation on ice for 30-60 min. The [3H]inositol phosphates in the formic acid cell lysates were thereafter quantified by adding yttrium silicate-poly-D-Lys-coated SPA beads. 35 μl of cell extract were mixed with 60 μl of SPA bead suspension in water (12.5 μg/μl) in a white 96-well plate. Plates were sealed and shaken on a table shaker for at least 30 min. SPA beads were allowed to settle and react with the extract for at least 8 hrs before radioactivity was determined using a Packard Top Count NXT scintillation counter (PerkinElmer Life Sciences). The experiments were performed at least three times in duplicates. Normalization was performed with the wild-type max signaling at 10 ng DNA/well as 100%. At the higher receptor concentrations, the signals declined. Thus, we displayed the results with the receptor concentrations up to 10 ng/well.
CRE-transcription factor luciferase assay
HEK293 cells were seeded at 35,000 cells per/well in white 96-well plates pre-treated with poly-D-lysine. On the following day, the cells were transiently transfected with receptor constructs at 0, 0.3, 0.5, 1, 2.5, or 10 ng DNA/well. All cells were co-transfected with transcription reporter plasmids CRE-Luc at 30 ng/well. After 24 hrs, the cells were incubated with 10 μM forskolin (Sigma, US) for 5 hrs, washed with PBS and incubated for 30 min with a mixture of SteadyLite (50 μL/well, PerkinElmer) and PBS (50 μL/well). The plates were read on EnVision Multilabel Plate Reader (PerkinElmer) using the luciferase program. The experiments were performed at least three times in triplicates. Normalization was performed with the wild-type max signaling at 10 ng DNA/well as 100%.
Detection of BILF1 expression by ELISA
HEK293 cells were seeded in transparent Poly-D-lysine pre-coated 96-well plates in the same way as the cell signaling assays. On the following day, the cells were transiently transfected with 0, 10, 30, or 50 ng/well of plasmid DNA using Lipofectamine 2000 (Invitrogen). 24 hrs after transfection, cells were fixed with 3.7% paraformaldehyde in PBS-CaCl2 (pH 7.3) for 10 min, blocked with 2% (w/v) bovine serum albumin (BSA) in PBS-CaCl2 for 30 min, and subsequently permeabilized with 0.1% (w/v) saponin (Sigma-Aldrich) in 2% (w/v) BSA-PBS-CaCl2 for 20 min. Receptors were detected by incubation with M2 anti-FLAG antibody (Sigma-Aldrich) at 1:2250 in 1% (w/v) BSA-PBS-CaCl2 for 1 hr at RT. After washing, cells were incubated with secondary goat anti-mouse horseradish peroxidase-conjugated IgG antibody (Sigma-Aldrich) at 1:1000 in PBS-CaCl2 for 1 hr at RT. Peroxidase activity was detected by 3,3’−5,5’-tetramethylbenzidine substrate (TMB) (Sigma-Aldrich) for 5 min and the reaction was terminated with 0.2M H2SO4. Absorbance was measured at 450 nm using EnVision Multilabel Plate Reader (PerkinElmer). Normalization was performed with the wild-type max expression at 50 ng DNA/well as 100%.
QUANTIFICATION AND STATISTICAL ANALYSIS
For all cell-based signaling and protein expression assays, at least three independent experiments were performed in duplicates. The data were expressed as mean ± standard error of means (SEM), and data analysis was performed in GraphPad Prism version 8.3.0 using unpaired non-parametric student t-tests. P values < 0.05 was considered statistically significant.
Supplementary Material
Table 1.
Summary of the relative Gi signaling and total expression of BILF1 variants
| BILF1 variant | relative Gi signaling (GαΔ6qi4myr-IP3, 10 ng DNA/well) |
relative expression (ELISA, 50 ng DNA/well) |
|---|---|---|
| wild-type | 100% | 100% |
| K122A | 16.0 ± 4.5 % (**) | 151.3 ± 16.4% (**) |
| ECL2GGG | 63.8 ± 2.8% (**) | 25.2 ± 7.3% (**) |
| ECL2CXCR4a | 63.9 ± 6.4% (**) | 32.1 ± 2.0% (**) |
| ECL2CXCR4b | 58.5 ± 5.7% (**) | 37.3 ± 0.7% (**) |
| ECL2CxCR4ab | 59.5 ± 4.4% (**) | 27.1 ± 4.1% (**) |
| TM6x7-4A | 89.1 ± 8.8% (*) | 111.1 ± 1.4% (*) |
| TM6x7-4A1F | 78.0 ± 5.1% (**) | 108.5 ± 2.7% (ns) |
| A271F | 84.3 ± 13.2% (**) | 66.4 ± 13.5% (**) |
| M240F | 112.6 ± 7.5% (*) | 92.9 ± 9.0% (ns) |
| L241F | 91.5 ± 2.8% (*) | 91.7 ± 9.0% (ns) |
| Y282A | 41.8 ± 4.2% (**) | 129.7 ± 7.1% (**) |
| E121A | 55.1 ± 2.0% (**) | 58.7 ± 3.2% (**) |
| H115A | 58.1 ± 4.9% (**) | 139.4 ± 7.2% (**) |
| L75A | 50.4 ± 10.4% (**) | 97.8 ± 7.5% (ns) |
| NPxxY | 59.1 ± 3.8% (**) | 84.8 ± 7.8 (*) |
| CWxM | 76.8 ± 3.2% (**) | 100.7 ± 4.9% (ns) |
| CWxP | 77.8 ± 3.7% (**) | 98.2 ± 10.6% (ns) |
| SWxP | 92.0 ± 8.6% (ns) | 90.5 ± 3.6% (ns) |
| A125F | 76.8 ± 0.8% (**) | 88.2 ± 3.2% (*) |
| H288A | 98.9 ± 5.7% (ns) | 91.5 ± 7.1% (ns) |
| F286-H288A | 87.1 ± 4.2% (**) | 85.0 ± 9.3% (*) |
| H66A-F286A-H288A | 76.0 ± 2.3% (**) | 79.7 ± 6.1% (**) |
ns: not significant
p < 0.05
p < 0.01
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-FLAG M2 antibody | Sigma-Aldrich | Cat# F1804, RRID: AB_262044 |
| Goat polyclonal anti-Mouse IgG (H+L), HRP conjugated | Invitrogen | Cat# 62-6520, RRID: AB_2533947 |
| Bacterial and Virus Strains | ||
| Rosetta 2 (DE3) | Sigma-Aldrich | Cat# 71400-M |
| Chemicals, Peptides, and Recombinant Proteins | ||
| C-terminal protein C-6xHis tagged full-length EBV BILF1 | This paper | N/A |
| Gαi1-Gβ1-Gγ2 (GDP-bound) | Produced in house | N/A |
| scFv16 | Produced in house | N/A |
| 3C protease | Produced in house | N/A |
| Uranyl formate | VWR | Cat# 101410-83 |
| Lauryl Maltose Neopentyl Glycol (LMNG) | Anatrace | Cat# NG310 |
| n-Dodecyl-β-D-Maltoside (DDM) | Anatrace | Cat# D310 |
| glyco-diosgenin (GDN) | Anatrace | Cat# GDN101 |
| n-Octyl-β-D-Glucopyranoside (βOG) | Anatrace | Cat# O311 |
| Cholesteryl Hemisuccinate (CHS) | Anatrace | Cat# CH210 |
| Sodium cholate | Sigma-Aldrich | Cat# C6445 |
| Protease Inhibitor Cocktail | Sigma-Aldrich | Cat# P8849 |
| cOmplete Protease Inhibitor Cocktail | Roche | Cat# 05056489001 |
| HEPES | Fisher Scientific | Cat# BP310 |
| Tris Base | Fisher Scientific | Cat# BP152 |
| Tris Hydrochloride | Fisher Scientific | Cat# BP153 |
| Glycerol | Fisher Scientific | Cat# BP229 |
| Iodoacetamide | Sigma-Aldrich | Cat# I1149 |
| Sodium Chloride | Fisher Scientific | Cat# S271 |
| Calcium chloride dihydrate | Sigma-Aldrich | Cat# C7902 |
| Magnesium chloride hexahydrate | Sigma-Aldrich | Cat# M2393 |
| Manganese(II) chloride tetrahydrate | Sigma-Aldrich | Cat# M3634 |
| TCEP solution | Thermo Scientific | Cat# 77720 |
| EDTA | Fisher Scientific | Cat# BP120 |
| Protein C peptide | GenScript | N/A |
| Methyl α-D-mannopyranoside | Sigma-Aldrich | Cat# M6882 |
| Methyl α-D-glucopyranoside | Sigma-Aldrich | Cat# M9376 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat# M3148 |
| Imidazole | ACROS Organics | Cat# 122025000 |
| GDP | Sigma Aldrich | Cat# G7127 |
| Apyrase | New England Biolabs | Cat# M0398S |
| Antarctic Phosphatase | New England Biolabs | Cat# M0289S |
| Alkaline Phosphatase, Calf Intestinal | New England Biolabs | Cat# M0290S |
| Lambda Protein Phosphatase | New England Biolabs | Cat# P0753S |
| FreeStyle 293 Expression Medium | Thermo Fisher | Cat# 12338026 |
| ESF 921 Insect Cell Culture Medium, Protein Free | Expression Systems | Cat# 96-001-01 |
| Sf-900 III SFM (Serum-Free Media) | Thermo Fisher | Cat# 12658019 |
| GlutaMAX | Thermo Fisher | Cat# 35050079 |
| Fetal Bovine Serum (FBS) | Sigma-Aldrich | Cat# F7524 |
| Sodium butyrate | Sigma-Aldrich | Cat# 303410 |
| FuGENE HD Transfection Reagent | Promega | Cat# E2312 |
| Lipofectamine 2000 transfection reagent | Invitrogen | Cat# 11668019 |
| Dulbecco’s Modified Eagle Medium (DMEM) | Thermo Fisher | Cat# 10569010 |
| Penicillin solution | Immunology and Microbiology, Substrate Department, Faculty of Health and Medical Sciences, University of Copenhagen | N/A |
| Streptomycin solution | Immunology and Microbiology, Substrate Department, Faculty of Health and Medical Sciences, University of Copenhagen | N/A |
| Inositol, Myo, -[2-3H(N)]-, Specific Activity: 10-25Ci (370-925GBq)/mmol | Perkin Elmer | Cat# NET114A005MC |
| Hanks’ balanced salt solution (HBSS) | Thermo Fisher | Cat# 14025100 |
| YSi-poly-d-Lys-coated SPA beads | Perkin Elmer | Cat# RPNQ0010 |
| Poly-D-lysine | Thermo Fisher | Cat# A3890401 |
| Forskolin | Sigma-Aldrich | Cat# F6886 |
| Formaldehyde solution | Sigma-Aldrich | Cat# 252549 |
| Saponin from quillaja bark | Sigma-Aldrich | Cat# S7900 |
| 3, 3', 5, 5'-tetramethylbenzidine (TMB) substrate | Eco-tek | Cat# 4395A |
| Critical Commercial Assays | ||
| Steadylite plus reporter gene assay system | Perkin Elmer | Cat# 6066759 |
| Deposited Data | ||
| Cryo-EM structure of BILF1-Gi-scFv16 | RCSB PDB | PDB ID: 7JHJ |
| Cryo-EM map of BILF1-Gi-scFv16 | EMDB | EMD-22338 |
| Experimental Models: Cell Lines | ||
| Homo Sapiens: HEK293 cells | ATCC | CRL-1573 |
| Homo Sapiens: HEK293S GnTI− cells | Reeves, P.J. et al., Proc Natl Acad Sci USA. (2002), 99(21), 13419-13424. | CRL-3022 |
| Mus musculus: HPC-4 MOUSE HYBRIDOMA | ATCC | HB-9892 |
| Spodoptera frugiperda: Sf9 cells | ATCC | CRL-1711 |
| Trichuplusia ni: Tni or Hi5 cells | Expression Systems | Cat# 94-002 |
| Recombinant DNA | ||
| pVLAD6 full-length EBV BILF1 | This paper | N/A |
| pVL1392 human Gαi1 | Maeda, S. et al. Nat Commun. (2018), 9, 3712. | N/A |
| pVL1392duet human Gβ1-3C protease site-6xHis Gγ2 | Maeda, S. et al. Nat Commun. (2018), 9, 3712. | N/A |
| pVL1392 gp67 signal peptide-scFv16-3C protease site-6xHis | Maeda, S. et al. Nat Commun. (2018), 9, 3712. | N/A |
| BestBac 1.0 Linearized Baculovirus DNA | Expression Systems | Cat# 91-001 |
| pcDNA3.1+ | Genscript | Cat# SC1317 |
| pcDNA3.1+ GαΔ6qi4myr | Kostenis, E. et al. J Pharmacol Exp Ther. (2005). 313, 78-87. | N/A |
| pLuc-MCS CRE-cis reporter plasmid | Agilent-Stratagene | Cat# 219075 |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 | Paulsen, S. et al. J Virol (2005). 79, 536–46. | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring K122A mutation (K122A) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring L167G N168G R169G mutation and G170 P171 N172 deletion (ECLGGG) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring L167E N168A R169D G170D P171R N172Y M173I mutation (ECLCXCR4a) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring R175D E176R G177F P178Y T179P K180N mutation (ECLCXCR4b) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring L167E N168A R169D G170D P171R N172Y M173I R175D E176R G177F P178Y T179P K180N mutation (ECLCXCR4ab) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring M240A L241A L243A Y267A mutation (TM6x7-4A) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring M240A L241A L243A Y267A A271F mutation (TM6x7-4A1F) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring A271F mutation (A271F) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring M240F mutation (M240F) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring L241F mutation (L241F) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring Y282A mutation (Y282A) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring E121A mutation (E121A) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring H115A mutation (H115A) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring L75A mutation (L75A) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring L278N I279P mutation (NPxxY) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring S237C mutation (CWxM) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring S237C M240P mutation (CWxP) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring M240P mutation (SWxP) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring A125F mutation (A125F) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring H288A mutation (H288A) | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring F286A H288A mutation | This paper | N/A |
| pcDNA3.1+ N-terminal FLAG tagged full-length EBV BILF1 harboring H66A F286A H288A mutation | This paper | N/A |
| Software and Algorithms | ||
| SWISS-MODEL | Waterhouse, A. et al., Nucleic Acids Res. (2018). 46(W1), W296-W303 | https://swissmodel.expasy.org/ |
| SerialEM | Mastronarde, D.N. J. Struct. Biol. (2005). 152, 36-51. | http://bio3d.colorado.edu/SerialEM/ |
| MotionCor2 | Zheng S.Q. et al., Nat. Methods (2017). 14, 331-332. | http://msg.ucsf.edu/em/software/motioncor2.html |
| Relion 3.1 | Zivanov, J. et al. eLife (2018). e42166. | https://github.com/3dem/relion |
| Bsoft | Heymann, J.B. et al. Protein Sci. (2018). 27, 159-171. | https://lsbr.niams.nih.gov/bsoft/ |
| Coot 0.9 | Emsley P., Cowtan K. Acta Cryst. (2004). D60, 2126-2132. | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix suite | Adams, P.V. et al., Acta Cryst. (2010). D66, 213-221. | https://www.phenix-online.org/ |
| Isolde | Croll, T.I. Acta Cryst. (2018). D74, 519-530. | http://isoldehttps//isolde.cimr.cam.ac.uk/ |
| UCSF Chimera 1.14 | Pettersen, E.F. et al., J Comput Chem. (2004) 1605-1612. | https://www.cgl.ucsf.edu/chimera/ |
| UCSF ChimeraX 1.0 | Pettersen, E.F. et al., Protein Sci. (2020). | https://www.rbvi.ucsf.edu/chimerax/ |
| PyMol 2.3.0 | Schrödinger | http://www.pymol.org/;RRID: SCR_000305 |
| Prism 8 | GraphPad | https://www.graphpad.com/ |
| Other | ||
| Anti-Protein C affinity agarose | in-house | N/A |
| Agarose bound Concanavalin A | Vector Laboratories | Cat# AL-1003 |
| Ni-NTA Agarose | QIAGEN | Cat# 30250 |
Highlights.
3.2Å cryo-EM structure of the Epstein-Barr virus GPCR BILF1 in complex with human Gi
Global remodeling of class A GPCR microswitches leads to constitutive Gi signaling
Unexpected dissimilarity to chemokine receptors with an occluded extracellular face
Topological similarity to lipid GPCRs with a density observed between TM helices
Acknowledgements
Cryo-EM data were collected at the Stanford-SLAC Cryo-EM facility; We thank Dr. Elizabeth Montabana for user support. We thank Andrew Velasco for his technical assistance and Drs. Kevin Jude and Michelle Yen for scientific discussion. This work was supported by the NIH (R01AI125320) to K.C.G., and the Kirsten and Freddy Johansens Foundation (2017-112697), the Lundbeck foundation (R242-2017-409) and the NovoNordisk Foundation (NNF17OC0029222) to M.M.R. K.C.G. is an investigator of the Howard Hughes Medical Institute and the Younger Family Chair, and is a member of the Ludwig Institute. N.T. was supported by the Long-Term Fellowships from the Human Science Frontier Program Organization (LT000011/2016-L). M.M. was supported by the Slovenian Research Agency (Young Researcher PhD program and P4-0053). A part of the graphical abstract was created with biorender.com.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Audet M, White KL, Breton B, Zarzycka B, Han GW, Lu Y, Gati C, Batyuk A, Popov P, Velasquez J, et al. (2019). Crystal structure of misoprostol bound to the labor inducer prostaglandin E 2 receptor. Nat. Chem. Biol 15, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisser PS, Verzijl D, Gruijthuijsen YK, Beuken E, Smit MJ, Leurs R, Bruggeman CA, and Vink C (2005). The Epstein-Barr Virus BILF1 Gene Encodes a G Protein-Coupled Receptor That Inhibits Phosphorylation of RNA-Dependent Protein Kinase. J. Virol 79, 441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burg JS, Ingram JR, Venkatakrishnan AJ, Jude KM, Dukkipati A, Feinberg EN, Angelini A, Waghray D, Dror RO, Ploegh HL, et al. (2015). Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science (80-. ). 347, 1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger M, Burger JA, Hoch RC, Oades Z, Takamori H, and Schraufstatter IU (1999). Point mutation causing constitutive signaling of CXCR2 leads to transforming activity similar to Kaposi’s sarcoma herpesvirus-G protein-coupled receptor. J. Immunol 163, 2017–2022. [PubMed] [Google Scholar]
- Canals M, and Milligan G (2008). Constitutive activity of the cannabinoid CB1 receptor regulates the function of co-expressed Mu opioid receptors. J. Biol. Chem 283, 11424–11434. [DOI] [PubMed] [Google Scholar]
- Chakravorty S, Yan B, Wang C, Wang L, Quaid JT, Lin CF, Briggs SD, Majumder J, Canaria DA, Chauss D, et al. (2019). Integrated pan-cancer map of EBV-associated neoplasms reveals functional host–virus interactions. Cancer Res. 79, 6010–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Lin FF, Xu M, Riek RP, Novotny J, and Graham RM (2002). Mutation of a single TMVI residue, Phe282, in the β2-adrenergic receptor results in structurally distinct activated receptor conformations. Biochemistry 41, 6045–6053. [DOI] [PubMed] [Google Scholar]
- Croll TI (2018). ISOLDE: A physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. Sect. D Struct. Biol 74, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukkipati A, Park HH, Waghray D, Fischer S, and Garcia KC (2008). BacMam system for high-level expression of recombinant soluble and membrane glycoproteins for structural studies. Protein Expr. Purif 62, 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Fan H, Chen S, Yuan X, Han S, Zhang H, Xia W, Xu Y, Zhao Q, and Wu B (2019). Structural basis for ligand recognition of the human thromboxane A 2 receptor. Nat. Chem. Biol 15, 27–33. [DOI] [PubMed] [Google Scholar]
- Fares S, Spiess K, Olesen ETB, Zuo J, Jackson S, Kledal TN, Wills MR, and Rosenkilde MM (2019). Distinct Roles of Extracellular Domains in the Epstein-Barr Virus-Encoded BILF1 Receptor for Signaling and Major Histocompatibility Complex Class I Downregulation. MBio 10, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell PJ (2019). Epstein–Barr Virus and Cancer. Annu. Rev. Pathol. Mech. Dis 14, 29–53. [DOI] [PubMed] [Google Scholar]
- Flock T, Hauser AS, Lund N, Gloriam DE, Balaji S, and Babu MM (2017). Selectivity determinants of GPCR-G-protein binding. Nature 545, 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritze O, Filipek S, Kuksa V, Palczewski K, Hofmann KP, and Ernst OP (2003). Role of the conserved NPxxY(x)5,6F motif in the rhodopsin ground state and during activation. Proc. Natl. Acad. Sci. U. S. A 100, 2290–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewurz BE, Gaudet R, Tortorella D, Wang EW, Ploegh HL, and Wiley DC (2001). Antigen presentation subverted: Structure of the human cytomegalovirus protein US2 bound to the class I molecule HLA-A2. Proc. Natl. Acad. Sci. U. S. A 98, 6794–6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, and Ferrin TE (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez Tamayo JC, Olivella M, Ríos S, Hoogstraat M, Gonzalez A, Mayol E, Deupi X, Campillo M, and Cordomí A (2018). GPCR-SAS: A web application for statistical analyses on G protein-coupled receptors sequences. PLoS One 13, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin BD, Gram AM, Mulder A, Van Leeuwen D, Claas FHJ, Wang F, Ressing ME, and Wiertz E (2013). EBV BILF1 Evolved To Downregulate Cell Surface Display of a Wide Range of HLA Class I Molecules through Their Cytoplasmic Tail. J. Immunol 190, 1672–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groof TWM, Elder EG, Siderius M, Heukers R, Sinclair JH, and Smit MJ (2021). Viral G Protein–Coupled Receptors: Attractive Targets for Herpesvirus-Associated Diseases. Pharmacol. Rev 73, 828–846. [DOI] [PubMed] [Google Scholar]
- Heydorn A, Ward RJ, Jorgensen R, Rosenkilde MM, Frimurer TM, Milligan G, and Kostenis E (2004). Identification of a novel site within G protein α subunits important for specificity of receptor-G protein interaction. Mol. Pharmacol 66, 250–259. [DOI] [PubMed] [Google Scholar]
- Heymann JB (2018). Guidelines for using Bsoft for high resolution reconstruction and validation of biomolecular structures from electron micrographs. Protein Sci. 27, 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isberg V, De Graaf C, Bortolato A, Cherezov V, Katritch V, Marshall FH, Mordalski S, Pin JP, Stevens RC, Vriend G, et al. (2015). Generic GPCR residue numbers - Aligning topology maps while minding the gaps. Trends Pharmacol. Sci 36, 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishido S, Wang C, Lee B-S, Cohen GB, and Jung JU (2000). Downregulation of Major Histocompatibility Complex Class I Molecules by Kaposi’s Sarcoma-Associated Herpesvirus K3 and K5 Proteins. J. Virol 74, 5300–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger K, Bruenle S, Weinert T, Guba W, Muehle J, Miyazaki T, Weber M, Furrer A, Haenggi N, Tetaz T, et al. (2019). Structural Basis for Allosteric Ligand Recognition in the Human CC Chemokine Receptor 7. Cell 178, 1222–1230.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosten RP, te Beek T. a H., Krieger E, Hekkelman ML, Hooft RWW, Schneider R, Sander C, and Vriend G (2011). A series of PDB related databases for everyday needs. Nucleic Acids Res. 39, D411–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Kuybeda O, De Waal PW, Mukherjee S, Van Eps N, Dutka P, Zhou XE, Bartesaghi A, Erramilli S, Morizumi T, et al. (2018). Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature 558, 553–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato HE, Zhang Y, Hu H, Suomivuori CM, Kadji FMN, Aoki J, Krishna Kumar K, Fonseca R, Hilger D, Huang W, et al. (2019). Conformational transitions of a neurotensin receptor 1–Gi1 complex. Nature 572, 80–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehl A, Hu H, Maeda S, Zhang Y, Qu Q, Paggi JM, Latorraca NR, Hilger D, Dawson R, Matile H, et al. (2018). Structure of the μ-opioid receptor–Gi protein complex. Nature 558, 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna Kumar K, Shalev-Benami M, Robertson MJ, Hu H, Banister SD, Hollingsworth SA, Latorraca NR, Kato HE, Hilger D, Maeda S, et al. (2019). Structure of a Signaling Cannabinoid Receptor 1-G Protein Complex. Cell 176, 448–458.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, et al. (2019). Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. Sect. D Struct. Biol 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Li M, Wang N, Wu Y, Luo Z, Guo S, Han G, Li S, Yue Y, Wei X, et al. (2020). Structural basis of ligand recognition and self-activation of orphan GPR52. Nature. [DOI] [PubMed] [Google Scholar]
- Liu K, Wu L, Yuan S, Wu M, Xu Y, Sun Q, Li S, Zhao S, Hua T, and Liu ZJ (2020). Structural basis of CXC chemokine receptor 2 activation and signalling. Nature 585, 135–140. [DOI] [PubMed] [Google Scholar]
- Lyngaa R, Nørregaard K, Kristensen M, Kubale V, Rosenkilde MM, and Kledal TN (2010). Cell transformation mediated by the Epstein-Barr virus G protein-coupled receptor BILF1 is dependent on constitutive signaling. Oncogene 29, 4388–4398. [DOI] [PubMed] [Google Scholar]
- Maeda S, Koehl A, Matile H, Hu H, Hilger D, Schertler GFX, Manglik A, Skiniotis G, Dawson RJP, and Kobilka BK (2018). Development of an antibody fragment that stabilizes GPCR/G-protein complexes. Nat. Commun 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Qu Q, Robertson MJ, Skiniotis G, and Kobilka BK (2019). Structures of the M1 and M2 muscarinic acetylcholine receptor/G-protein complexes. Science (80-. ). 364, 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Meyrath M, Szpakowska M, Zeiner J, Massotte L, Merz MP, Benkel T, Simon K, Ohnmacht J, Turner JD, Krüger R, et al. (2020). The atypical chemokine receptor ACKR3/CXCR7 is a broad-spectrum scavenger for opioid peptides. Nat. Commun 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles TF, Spiess K, Jude KM, Tsutsumi N, Burg JS, Ingram JR, Waghray D, Hjorto GM, Larsen O, Ploegh HL, et al. (2018). Viral GPCR US28 can signal in response to chemokine agonists of nearly unlimited structural degeneracy. Elife 7, e35850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaner S, Kufareva I, Abagyan R, and Gutkind JS (2013). Molecular Mechanisms Deployed by Virally Encoded G Protein–Coupled Receptors in Human Diseases. Annu. Rev. Pharmacol. Toxicol 53, 331–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto K, Suno R, Hotta Y, Yamashita K, Hirata K, Yamamoto M, Narumiya S, Iwata S, and Kobayashi T (2019). Crystal structure of the endogenous agonist-bound prostanoid receptor EP3. Nat. Chem. Biol 15, 8–10. [DOI] [PubMed] [Google Scholar]
- Mundo L, Del Porro L, Granai M, Siciliano MC, Mancini V, Santi R, Marcar L, Vrzalikova K, Vergoni F, Di Stefano G, et al. (2020). Frequent traces of EBV infection in Hodgkin and non-Hodgkin lymphomas classified as EBV-negative by routine methods: expanding the landscape of EBV-related lymphomas. Mod. Pathol 33, 2407–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Munnik SM, Smit MJ, Leurs R, and Vischer HF (2015). Modulation of cellular signaling by herpesvirus-encoded G protein-coupled receptors. Front. Pharmacol 6, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy PM (1994). The molecular biology of leukocyte chemoattractant receptors. Annu. Rev. Immunol 12, 593–633. [DOI] [PubMed] [Google Scholar]
- Nicholls DJ, Tomkinson NP, Wiley KE, Brammall A, Bowers L, Grahames C, Gaw A, Meghani P, Shelton P, Wright TJ, et al. (2008). Identification of a putative intracellular allosteric antagonist binding-site in the CXC chemokine receptors 1 and 2. Mol. Pharmacol 74, 1193–1202. [DOI] [PubMed] [Google Scholar]
- Nijmeijer S, Leurs R, Smit MJ, and Vischer HF (2010). The Epstein-Barr virus-encoded G protein-coupled receptor BILF1 hetero-oligomerizes with human CXCR4, scavenges Gαi proteins, and constitutively impairs CXCR4 functioning. J. Biol. Chem 285, 29632–29641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivella M, Caltabiano G, and Cordomí A (2013). The role of Cysteine 6.47 in class A GPCRs. BMC Struct. Biol 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald C, Rappas M, Kean J, Doré AS, Errey JC, Bennett K, Deflorian F, Christopher JA, Jazayeri A, Mason JS, et al. (2016). Intracellular allosteric antagonism of the CCR9 receptor. Nature 540, 462–465. [DOI] [PubMed] [Google Scholar]
- Paulsen SJ, Rosenkilde MA, Eugen-Olsen J, and Kledal TN (2005). Epstein-Barr virus-encoded BILF1 is a constitutively active G protein-coupled receptor. J. Virol 79, 536–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Qin L, Kufareva I, Holden LG, Wang C, Zheng Y, Zhao C, Fenalti G, Wu H, Han GW, Cherezov V, et al. (2015). Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science (80-. ). 347, 1117–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn LL, Zuo J, Abbott RJM, Shannon-Lowe C, Tierney RJ, Hislop AD, and Rowe M (2014). Cooperation between Epstein-Barr Virus Immune Evasion Proteins Spreads Protection from CD8+ T Cell Recognition across All Three Phases of the Lytic Cycle. PLoS Pathog. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salchow K, Bond M, Evans S, Press N, Charlton S, Hunt P, and Bradley M (2010). A common intracellular allosteric binding site for antagonists of the CXCR2 receptor: Research paper. Br. J. Pharmacol 159, 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuren ABC, Costa AI, and Wiertz EJHJ (2016). Recent advances in viral evasion of the MHC Class I processing pathway. Curr. Opin. Immunol 40, 43–50. [DOI] [PubMed] [Google Scholar]
- Schwartz O, Maréchal V, Le Gall S, Lemonnier F, and Heard JM (1996). Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med 2, 338–342. [DOI] [PubMed] [Google Scholar]
- Spiess K, Fares S, Sparre-Ulrich AH, Hilgenberg E, Jarvis MA, Ehlers B, and Rosenkilde MM (2015). Identification and Functional Comparison of Seven-Transmembrane G-Protein-Coupled BILF1 Receptors in Recently Discovered Nonhuman Primate Lymphocryptoviruses. J. Virol 89, 2253–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tierney RJ, Shannon-Lowe CD, Fitzsimmons L, Bell AI, and Rowe M (2015). Unexpected patterns of Epstein-Barr virus transcription revealed by a High throughput PCR array for absolute quantification of viral mRNA. Virology 474, 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda Y, Morimoto K, Suno R, Horita S, Yamashita K, Hirata K, Sekiguchi Y, Yasuda S, Shiroishi M, Shimizu T, et al. (2019). Ligand binding to human prostaglandin E receptor EP4 at the lipid-bilayer interface. Nat. Chem. Biol 15, 18–26. [DOI] [PubMed] [Google Scholar]
- Wang J, Hua T, and Liu ZJ (2020). Structural features of activated GPCR signaling complexes. Curr. Opin. Struct. Biol 63, 82–89. [DOI] [PubMed] [Google Scholar]
- Wiertz EJHJ, Jones TR, Sun L, Bogyo M, Geuze HJ, and Ploegh HL (1996). The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 84, 769–779. [DOI] [PubMed] [Google Scholar]
- Yang Z, Han S, Keller M, Kaiser A, Bender BJ, Bosse M, Burkert K, Kögler LM, Wifling D, Bernhardt G, et al. (2018). Structural basis of ligand binding modes at the neuropeptide y Y1 receptor. Nature 556, 520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young LS, Yap LF, and Murray PG (2016). Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 16, 789–802. [DOI] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang J, Chen Y, Luo B, Yuan Y, Huang F, Yang T, Yu F, Liu J, Liu B, et al. (2020). The ORF8 Protein of SARS-CoV-2 Mediates Immune Evasion through Potently Downregulating MHC-I. BioRxiv doi: 10.1101/2020.05.24.111823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Qin L, Zacarías NVO, De Vries H, Han GW, Gustavsson M, Dabros M, Zhao C, Cherney RJ, Carter P, et al. (2016). Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 540, 458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Yang D-H, Wu M, Guo Y, Guo W, Zhong L, Cai X, Dai A, Jang W, Shakhnovich EI, et al. (2019). Common activation mechanism of class A GPCRs. Elife 8, e50279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziffert I, Kaiser A, Babilon S, Mörl K, and Beck-Sickinger AG (2020). Unusually persistent Gαi-signaling of the neuropeptide Y2 receptor depletes cellular Gi/o pools and leads to a Gi-refractory state. Cell Commun. Signal 18, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJH, Lindahl E, and Scheres SHW (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, e42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo J, Currin A, Griffin BD, Shannon-Lowe C, Thomas WA, Ressing ME, Wiertz EJHJ, and Rowe M (2009). The Epstein-Barr virus G-protein-coupled receptor contributes txo immune evasion by targeting MHC class I molecules for degradation. PLoS Pathog. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo J, Quinn LL, Tamblyn J, Thomas WA, Feederle R, Delecluse H-J, Hislop AD, and Rowe M (2011). The Epstein-Barr Virus-Encoded BILF1 Protein Modulates Immune Recognition of Endogenously Processed Antigen by Targeting Major Histocompatibility Complex Class I Molecules Trafficking on both the Exocytic and Endocytic Pathways. J. Virol 85, 1604–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM map has been deposited in the Electron Microscopy Data Bank (EMDB) under accession code EMD-22338 and the model coordinate has been deposited in the Protein Data Bank (PDB) under accession number 7JHJ.