

Abstract
A pathophysiological consequence of both type 1 and 2 diabetes is remodelling of the myocardium leading to the loss of left ventricular pump function and ultimately heart failure (HF). Abnormal cardiac bioenergetics associated with mitochondrial dysfunction occurs in the early stages of HF. Key factors influencing mitochondrial function are the shape, size and organisation of mitochondria within cardiomyocytes, with reports identifying small, fragmented mitochondria in the myocardium of diabetic patients. Cardiac mitochondria are now known to be dynamic organelles (with various functions beyond energy production); however, the mechanisms that underpin their dynamism are complex and links to motility are yet to be fully understood, particularly within the context of HF. This review will consider how the outer mitochondrial membrane protein Miro1 (Rhot1) mediates mitochondrial movement along microtubules via crosstalk with kinesin motors and explore the evidence for molecular level changes in the setting of diabetic cardiomyopathy. As HF and diabetes are recognised inflammatory conditions, with reports of enhanced activation of the NLRP3 inflammasome, we will also consider evidence linking microtubule organisation, inflammation and the association to mitochondrial motility. Diabetes is a global pandemic but with limited treatment options for diabetic cardiomyopathy, therefore we also discuss potential therapeutic approaches to target the mitochondrial-microtubule-inflammatory axis.
Keywords: diabetic cardiomyopathy, heart failure, Miro1, microtubules, HDAC6, NLRP3, mitochondrial dysfunction, mitochondrial movement
Introduction
Diabetic Mellitus (DM) remains a global epidemic, with an estimated 463 million cases worldwide in 2019, and is associated with marked morbidity and mortality rates (1). Diabetes is a major risk factor for heart failure (HF), with a three-fold higher prevalence for developing coronary artery disease, CAD (2, 3). Although, approximately 50 years ago, it was revealed that myocardial remodelling and dysfunction can also occur in diabetic patients in the absence of CAD, a condition termed diabetic cardiomyopathy (DCM) which commonly advances to HF (4, 5). Recently it has emerged that in addition to HF with reduced ejection fraction (HFrEF) roughly half of all HF cases can be classified as HF with preserved ejection fraction (HFpEF), which is less well-understood in comparison. Further, treatments developed to manage HFrEF have limited efficacy in HFpEF patients; highlighting the need for a more detailed understanding of the disease mechanisms. Significantly, over a third of patients with HFpEF have type 2 diabetes (6, 7) indicating that the diabetic insult leads, at least initially, to the development of a distinct myocardial phenotype.
Mitochondrial dysfunction is a hallmark of HF (8) and more recently identified as a feature of HFpEF (9) and DCM (10). While mitochondrial dysfunction is a broad term a recurring feature of HF both in the presence and absence of diabetes, is morphological remodelling of mitochondria, with reports of both swelling and fragmentation [for a review see (11)]. Mitochondrial size, shape and distribution (factors dictating function) are regulated by mechanisms grouped under the umbrella term “mitochondrial dynamics,” which also encompasses mitochondrial turnover, mitophagy and biogenesis, as reviewed by (12). Mitochondrial movement, although less well-studied, particularly in the heart, is driven by the outer mitochondrial membrane protein Miro1 (also termed RHOT1) and is also believed to be important for regulating and maintaining a healthy mitochondrial network (13).
DCM is a chronic low grade inflammatory condition, with inflammation associated with the pathogenesis of HF and linked to the development of mitochondrial dysfunction [as reviewed recently (14)]. Kaludercic and Di Lisa (15) provided an overview of a number of studies indicating that excessive ROS production due to mitochondrial dysfunction is a causative agent of increased expression and activity of the inflammasome, NLRP3. NLRP3 is a multimeric complex formed from NOD-like receptor 3, the apoptosis-associated speck-like protein containing (ASC) adaptor protein, and caspase-1, (16, 17). Although, details are somewhat sparse (and not within the setting of the heart or DCM) an association between proteins mediating mitochondrial dynamics and NLRP3 inflammasome assembly is also emerging (18, 19).
This review will summarise evidence for the emerging role of dysregulated mitochondrial movement in HF/DCM, the intersection with aberrant mitochondrial dynamics and NLRP3 activity focusing on the involvement of Miro1; as well as considering the putative mechanisms involved.
Mitochondrial Dynamics in Cardiovascular Health and Disease
Previously perceived as “static” organelles within cardiomyocytes, mitochondria are now known to be highly dynamic undergoing restructuring via an equilibrium of fission and fusion (12). In brief, fission, mediated by the GTPase dynamin-related protein 1 (Drp1) and receptors Mitochondrial fission 1 protein (Fis1), Mitochondrial fission factor (MFF), and MiD49/51, leads to the division of a single mitochondrion into two, allowing removal of damaged mitochondria from the network. Whereas fusion, is the amalgamation of two mitochondria into one larger mitochondrion orchestrated by the outer mitochondrial membrane (OMM) proteins Mitofusin 1 (Mfn1) and Mitofusin 2 (Mfn2). Fusion of the inner mitochondrial membrane (IMM) next occurs and is regulated by Optic atrophy 1 (Opa1).
The dynamic nature of mitochondria underpins mitochondrial quality control that is, mitochondrial biogenesis (replacement with healthy mitochondria), maintenance and degradation (mitophagy). Over time mitochondria can accumulate damage as a result of multiple mutations in nuclear-encoded mitochondrial genes or oxidative damage via ROS formation as a by-product of OXPHOS. In brief, mitophagy, the removal of damaged mitochondria, is regulated by PTEN-induced kinase protein 1 (PINK1) which upon activation (via phosphorylation) recruits cytosolic Parkin (20, 21). Parkin selectively phospho-ubiquitinates the OMM proteins (including Mfn1/2) and facilitates the selective binding and extension of autophagosomes around damaged mitochondria, although details of the exact mechanisms involved remain incomplete (22).
Recently another method of “fusion” has been identified in cardiomyocytes via the formation of tubular protrusions known as nanotunnels, visualised by live-cell confocal imaging and electron microscopy (23, 24). Nanotunnels serve as a form of direct intercommunication between cardiac mitochondria over micron distances, which allows the exchange of matrix contents between non-adjacent mitochondria. Whether the frequency of nanotunnels is correlated to cell stress is not yet clear, although links to an imbalance in Ca2+ cycling has been implicated (25).
There is a plethora of studies, both clinical and preclinical, identifying decreased expression of fusion proteins and increased levels of fission proteins in the context of cardiovascular disease (26). For example, depressed levels of Mfn1/2 are a feature of human and rodent hearts with impaired contractility and mitochondrial dysfunction (27, 28). Ablation of Mfn1 and Mfn2 results in dilated cardiomyopathy, impaired mitochondrial respiration and mitochondrial fragmentation (29). Opa1 is also depressed in HF patients (30). Whereas, increased fission, fragmented mitochondria and cell death associated with Drp1 expression has been reported as a feature of HF (31). Consequently, research has focused upon developing pharmacological inhibitors of Drp1, for example, treatment of HL-1 cardiomyocytes with mitochondrial division inhibitor-1 is shown to be cardioprotective against ischemia/reperfusion injury (32). However, the Janus nature of Drp1-mediated pathways should not be ignored as in some conditions promotion of these pathways can be cardioprotective (33). Similarly, reduced levels of PINK1 and Parkin in the heart are also associated with ventricular hypertrophy, mitochondrial swelling (34) and disorganised mitochondria (35). Andres et al. (36) also reported how simvastatin provides cardioprotection by triggering Parkin-dependent mitophagy. A balance between mitochondrial fusion, fission and mitophagy is crucial for maintaining a healthy population of mitochondria.
Miro1 Plays a Central Role Governing Mitochondrial Movement
While Mfn1, Mfn2, Opa1 and Drp1 (and receptors) are essential for regulating mitochondrial size and shape, important for mitochondrial “quality control,” the movement of mitochondria within the cell is also a crucial factor with distribution tightly linked to cellular energy requirements. An elegant study from Bers and colleagues not only captured, using live imaging, fusion and fission events within cardiomyocytes but also tracked mitochondrial movement (37). Interestingly, the study showed that over a 1 h period the net movement of mitochondria between the sarcomeres (interfibrillar, IFM) was <0.3 μm compared to those adjacent to the nucleus (peri-nuclear, PNM) which traversed 2.8 μm; this difference in motility may be due to the IFM being more spatially restricted by the sarcomeric organisation.
Miro1 localised to the OMM, has been firmly established in neurons as central for regulating mitochondrial movement in response to temporal and spatial metabolic demands (13), with impaired mitochondrial trafficking associated with several neurodegenerative diseases (38). Miro1 is also highly expressed in the heart (39) and although less well-studied, knock-down of Miro1 in H9c2 cardiomyoblasts revealed a similar effect upon mitochondrial movement in a Ca2+ dependent manner (40). Interestingly, studies exploring mitochondrial transfer via transplantation of human induced pluripotent mesenchymal stem cells (iPSC-MSCs) for tissue regeneration in models of anthracycline-induced cardiomyopathy identify the intrinsically high Miro1 content of iPSC-MSCs as essential for facilitating mitochondrial relocation and improved cardiac bioenergetics (41). In contrast, a recent study suggested that knockdown of Miro1 in cultured neonatal cardiomyocytes (NRCMs) could be protective against phenylephrine-induced hypertrophy through attenuating mitochondrial fission (42). Whilst different cardiac pathologies likely require different approaches in terms of therapeutic targeting, NRCMs as a model system may not always be directly translatable to the mature cardiomyocyte in which mitochondria show a substrate preference for free fatty acids rather than pyruvate (product of glycolysis); Dorn et al. have proposed that after birth there is cell-wide replacement with “adult” mitochondria (43). It is noteworthy that Miro1 is also decreased in pancreatic cells of patients with type 2 diabetes, with a mouse model of islet Miro1 ablation developing insulin resistance, increased production of ROS, inflammation and dysregulated mitophagy (44). Evidence, mainly from studies of neuronal tissue (38), indicates that loss of Miro1 is a decisive factor leading to “arrested” motility and linked to the accumulation of damaged mitochondria.
A Miro1-Macromolecular Complex Mobilises Mitochondria Along Microtubules
Miro proteins bind to kinesin-1/KIF5 and Milton (also known as trafficking kinesin-binding protein 1, TRAK1 or OIP106) (39); interactions proposed to link mitochondria to the microtubule trafficking apparatus (45, 46). Miro1 also directly interacts with Mfn2 in neuronal cells, an association that is proposed as an essential step mediating mitochondrial movement (47) as shown in Figure 1; although, the molecular basis of this interaction remains unknown as is whether this association occurs in cardiomyocytes. Significantly, as discussed above, cardiac Mfn2 levels are reported to be down-regulated in models of DCM (48). How the loss of Mfn2 (which presumably leads to reduced Miro1-Mfn2 interactions) impacts mitochondrial movement and DCM linked phenotypic changes has yet to be clarified.
Figure 1.

Putative molecular mechanisms of mitochondrial movement. (A) In the absence of cytosolic Ca2+ (nM) Miro1 (bound to the outer mitochondrial membrane) coordinates mitochondrial movement along microtubules via KIF5A, TRAK1 and Mfn2. According to literature Ca2+ binding leads to two possible scenarios; (B) Conformational changes to Miro1, and subsequent detachment from Mfn2 and release of KIF5A from the microtubule trafficking apparatus, or (C) Detachment of Miro1 from Mfn2 and a disruption between TRAK1 and KIF5A interactions. Both scenarios result in halted mitochondrial movement.
Ca2+ binding to Miro1, via two EF-hands within the primary sequence, elicits a conformational change, mediating the association and dissociation of the Miro1-complex assembly from the microtubules, MTs, (with detachment halting mitochondrial motility). This process underpins mitochondrial moment (Figure 1) (49). Aberrant Ca2+ homeostasis is a hallmark of HF in patients (50) and associated with DCM (51). Mitochondria play a central role in regulating cytosolic [Ca2+] and maintaining the cellular redox status (52). Organisation of mitochondria straddling either side of the dyad (formed by t-tubules, specialised regions of the sarcolemma, and junctional sarcoplasmic reticulum) is essential for Ca2+ uptake into the mitochondria for driving bioenergetics (52, 53). While HF is associated with displacement of mitochondria (54), how impaired Ca2+ cycling influences mitochondrial movement along microtubules has yet to be examined in detail. Importantly, the Bers group have shown in isolated cardiomyocytes that under stress conditions, causing mitochondrial damage, there is migration of IFM (fission products) via MTs to the perinuclear region where they undergo mitophagy (37).
Microtubule Organisation and Cardiovascular Function and Disease
The presence of MTs in cardiomyocytes has been known for many years (55). MTs are formed by the polymerisation of α/β-tubulin dimers assembling into rods roughly ~ 25 nm in diameter which can extend up to tens of microns in length (56). Similar to mitochondria, MTs are organised into spatially distinct populations; (i) interfibrillar (55), (ii) those surrounding the nuclear envelope (57), and (iii) cortical MTs perpendicular to myofibrils (58). These differing populations of MTs are commonly believed to explain, in part, the diverse physiological roles of MTs in cardiomyocytes, for example, ion channel trafficking, mechanical signalling pathways fundamental for cardiac contractility and inter-organelle communication, as reviewed by Caporizzo et al. (59).
As also discussed in (59) there are numerous studies linking changes to MT properties to the development and progression of cardiovascular diseases. MT remodelling has been identified as the source of increased mechanical stiffness occurring in the early stages of diastolic dysfunction. Accordingly, there has been interest in the use of reagents that depolymerise MTs. Specifically, post-translational modification (PTM) (detyrosination) of MTs is associated with increased viscoelastic resistance in human failing hearts (60). Stiffening of the myocardium due to MT remodelling in the failing heart is also reported to displace mitochondria adjacent to the sarcolemma (subsarcolemmal mitochondria, SSM) triggering the propagation of abnormal Ca2+ transients, leading to arrhythmogenesis (8). However, since mitochondria have been demonstrated to provide a bridge between calcium cycling (and contractility) and mechano-electric and chemical MT-mediated inter-organelle tethering (61) the impact of disrupting the MT network upon mitochondrial motility and function needs further investigation.
Protein Post-Translational Modifications (PTMs) and Role in Mitochondrial Motility
In addition to Ca2+ homeostasis, regulatory mechanisms mediating mitochondrial motility include the cellular redox balance (62), as well PTMs of mitochondrial (63) and MT proteins (64). Miro1 is also regulated by PTMs being ubiquitinated after phosphorylation via PINK1, triggering Parkin and proteasomal pathways leading to Miro1 degradation (65). Given that the PINK1-Parkin pathway is activated in response to depolarisation of the mitochondrial membrane it is generally considered, in this context, that prevention of mitochondrial movement through Miro1 phosphorylation-degradation is important for preserving mitochondrial quality by segregating those damaged mitochondria for removal from the cell (66). How removal of damaged mitochondria is linked to the transport of mitochondria via Miro1 is not clear, particularly within cardiomyocytes; although the Bers study (37) would suggest that mitochondrial transport of IFM precedes mitophagy. The relationship between mitophagy and Miro1 also appears to be cell-type dependent with Miro1 loss-of-function mutations preventing the induction of mitophagy in neurons (67) but activating mitophagy in fibroblasts (68). Miro1 is also a substrate of the class II histone deacetylase, HDAC6, with acetylation of K105 (murine and rat) corresponding to K92 in human Miro1, involving different lysine residues than those targeted for ubiquitination, indicative of two different processes (69). HDAC6 co-immunopreciptates with Miro1 supporting a direct interaction between the two proteins. Importantly, Kalinski et al. (69) also demonstrated that deactetylation of Miro1 leads to stalled mitochondrial movement with detrimental effects upon axon growth, with pharmacological inhibition or deletion of HDAC6 protecting mitochondria against damage and abnormal mitochondrial clustering.
Notably, one of the main components of MTs, α-tubulin, is also an HDAC6 substrate. Acetylation is important for MT stability, as well-inducing conformationally directed MT organisation for the recruitment of kinesin motor proteins (70). Inhibition of HDAC6 in hippocampal neurons is shown to lead to higher levels of α-tubulin and enhanced mitochondrial motility (71). While the role of MT acetylation remains to be fully understood it is intriguing that HDAC6 separately regulates both Miro1 and MT engagement and disengagement and consequently mitochondrial motility.
Significantly, inhibition of HDAC6 activity is also reported as cardioprotective (72) preventing the development of hypertrophy and fibrosis (73). Further, in the context of DCM, HDAC6 inhibition using tubastatin A (TBA) is shown to be beneficial in a rat model of type 1 diabetes for improving outcomes from ischaemia/reperfusion (I/R) injury (74). Moreover, recently, pan-inhibitors of HDACs have been proposed as novel treatments for treating HFpEF, preserving cardiac function in a small animal model (rat) of hypertension induced LV-dysfunction (75) and a larger animal (feline) model of pressure overload (76).
Increased HDAC6 activity in the failing heart has been known for the past decade (77) but a key question that remains to be fully answered is how is HDAC6 activated? Chen and colleagues demonstrated, using an atrial cell line (HL-1), that mitochondrial dysfunction (impaired OXPHOS) induced by treatment with TNF-α, could be rescued using an inhibitor of HDAC6 (78) indicating a mechanistic link between inflammation, mitochondrial function and HDAC6 activity. Further, HDAC6 inhibition in SH-SY5Y cells (a model for Parkinson's disease) is reported to lead to reduced activation of the inflammasome, NLRP3, inflammatory response concomitant with attenuation of dopaminergic neuronal degeneration (79); although the pathways involved were not described.
Microtubules Play a Critical Role for NLRP3 Activation
Activation of NLRP3 modulates the release of inflammatory cytokines, IL-1β and IL-18, cell death and fibrosis associated with the pathogenesis of DCM (80). The acetylation of α-tubulin, whilst linked to mitochondrial transport, is also shown to play a role in the movement of NLRP3 inflammasome components along microtubules leading to the subsequent apposition of NLRP3 to mitochondrion-associated ASC (81). Multiple studies have established the link between microtubule dynamics and NLRP3 inflammasome activity, for example (82). Significantly NLRP3 activation is a feature of several cardiac pathologies as reviewed in (83). For example, colchicine (a microtubule polymerisation inhibitor) disrupts microtubule/tubulin dynamics suppressing the activation of the NLRP3 inflammasome. Animal models of myocardial infarction treated with colchicine have improved cardiac performance, improved survival rates and attenuated HF development and inflammatory response (84).
Since the acetylation of α-tubulin/NLRP3 is under the control of HDAC6 it is perhaps not surprising that beneficial effects of using HDAC6 inhibitors in preventing IL-1β generation have been demonstrated (85). Although, one macrophage study concluded that HDAC6 is a negative regulator of NLRP3, due to a direct interaction mediated by the ubiquitin binding domains (86); with this mechanism suggested to mediate NLRP3 transport into aggresomes via the microtubule network. More recently, Magupalli et al. also demonstrated in macrophages that specific regions of MTs, the microtubule-organising centre (the centrosome), are the sites for NLRP3 assembly and HDAC6 knockout (and loss of ubiquitin-binding) leads to impaired inflammasome assembly and activation (87). Clearly, there is a complex association between NLRP3 assembly, activation and MTs, which may also be tissue/cell type specific.
An indirect link between Miro1 and NLRP3 has also been identified in a rat pancreatic cells using high-fat and high glucose stressors to mimic T2DM conditions (88). Specifically, cells exhibited dysregulated Ca2+ homeostasis, which was suggested to lead to the dissociation of Miro1 from mitochondria and subsequent impaired mitochondrial movement, stalled mitophagy and accumulation of damaged ROS producing mitochondria that in turn triggered activation of NLRP3.
Concluding Remarks
Here we have highlighted evidence for the physio-pathological role of Miro1-mediated movement of mitochondria along MTs and while the majority of data is from the study of neurons evidence is emerging to support a similar role for Miro1 in the heart (40). Therefore, in addition to studies focussing upon strategies to prevent fission (89, 90) an emerging area for future studies is the delineation of the mechanisms surrounding mitophagy, mitochondrial movement, and role of the Miro1-macromolecular complex. For example, it remains unclear as to whether Miro1 expression and activity influences the processes of fission and fusion and is essential for mitophagy in the heart.
Additionally, we have highlighted a potential link between mitochondrial motility and inflammation and the involvement of deacetylation and HDAC6 (Figure 2). Although technically there remain significant challenges around studying PTMs (91) a better understanding of the functional effects of PTMs on the proteins underpinning mitochondrial motility will provide new avenues for future research. In conclusion, this review article summarises some of the current evidence, and areas where knowledge is lacking, for mitochondrial motility in the heart, and suggests a possible unifying mechanism linking impaired mitophagy, the MT network and the inflammatory response to arrested mitochondrial movement. As the causative agents and mechanisms of mitochondrial dysfunction and impaired motility are discovered, then new promising treatment therapies may emerge for promoting better cardiac outcomes in DCM/HF.
Figure 2.
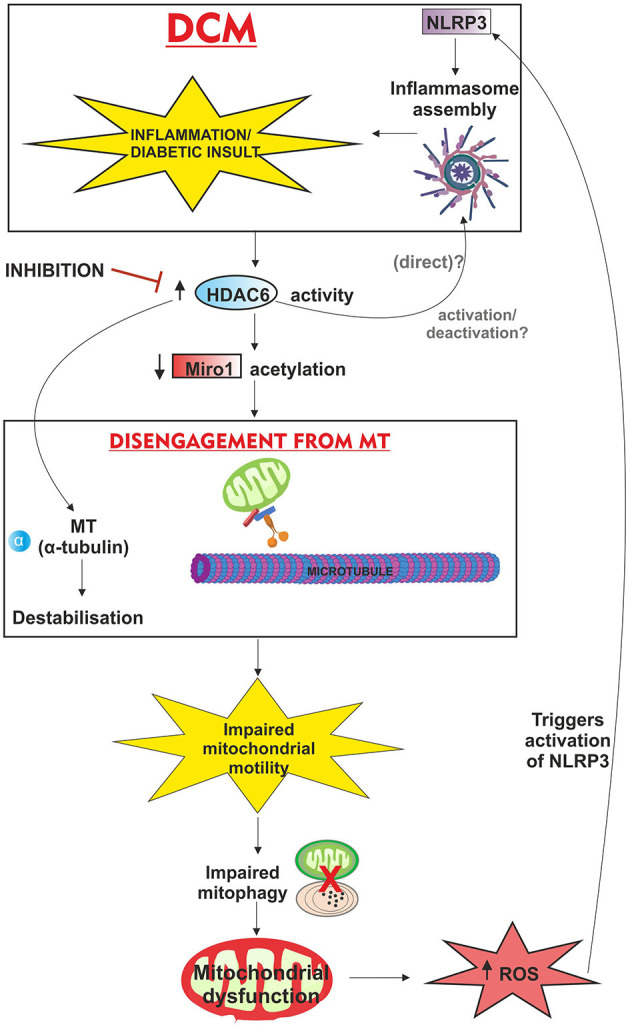
Schematic overview of proposed mechanism of action of HDAC6 and associated deacetylation; linking mitochondrial motility, inflammation and the putative involvement of post-translational modifications in DCM. Inflammation/diabetic insult triggers HDAC6 activity which directly interacts with NLRP3 via ubiquitin binding domains (although it is unknown whether this activates/inactivates NLRP3 and inflammasome assembly in the heart). The combination of microtubule (alpha-tubulin) destabilisation and reduced acetylation (important for microtubule stability), contributes to disengagement of the mitochondria from the microtubule apparatus and halts mitochondrial movement. With mitochondrial motility linked to mitophagy, the removal of damaged mitochondria (potentially ROS producing) is impaired, leading to subsequent mitochondrial dysfunction, and oxidative stress further exacerbating inflammation via NLRP3. Inhibition of HDAC6 activity is also reported as cardioprotective.
Author Contributions
AK and SK wrote and planned the manuscript. ZA and HD contributed to the writing process. SK generated the figures in consultation with AK. All authors contributed and approved the submitted article.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported in part by the British Heart Foundation PG/18/61/33966. HD was supported by King Abdulaziz University and ZA by The Saudi Arabian Cultural Bureau (SACB).
References
- 1.Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res Clin Pract. (2019) 157:107843. 10.1016/j.diabres.2019.107843 [DOI] [PubMed] [Google Scholar]
- 2.Aneja A, Tang WHW, Bansilal S, Garcia MJ, Farkouh ME. Diabetic cardiomyopathy: Insights into pathogenesis, diagnostic challenges, and therapeutic options. Am J Med. (2008) 121:748–57. 10.1016/j.amjmed.2008.03.046 [DOI] [PubMed] [Google Scholar]
- 3.Avogaro A, de Kreutzenberg SV, Negut C, Tiengo A, Scognamiglio R. Diabetic cardiomyopathy: a metabolic perspective. Am J Cardiol. (2004) 93:13–6. 10.1016/j.amjcard.2003.11.003 [DOI] [PubMed] [Google Scholar]
- 4.Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. The Am J Cardiol. (1972) 30:595–602. 10.1016/0002-9149(72)90595-4 [DOI] [PubMed] [Google Scholar]
- 5.Dillmann WH. Diabetic cardiomyopathy. Circ Res. (2019) 124:1160–2. 10.1161/CIRCRESAHA.118.314665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paulus WJ, Dal Canto E. Distinct myocardial targets for diabetes therapy in heart failure with preserved or reduced ejection fraction. JACC Heart Fail. (2018) 6:1–7. 10.1016/j.jchf.2017.07.012 [DOI] [PubMed] [Google Scholar]
- 7.Meagher P, Adam M, Civitarese R, Bugyei-Twum A, Connelly KA. Heart failure with preserved ejection fraction in diabetes: mechanisms and management. Can J Cardiol. (2018) 34:632–43. 10.1016/j.cjca.2018.02.026 [DOI] [PubMed] [Google Scholar]
- 8.Rosca MG, Tandler B, Hoppel CL. Mitochondria in cardiac hypertrophy and heart failure. J Mol Cell Cardiol. (2013) 55:31–41. 10.1016/j.yjmcc.2012.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar AA, Kelly DP, Chirinos JA. Mitochondrial dysfunction in heart failure with preserved ejection fraction. Circulation. (2019) 139:1435–50. 10.1161/CIRCULATIONAHA.118.036259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schilling JD. The mitochondria in diabetic heart failure: from pathogenesis to therapeutic promise. Antioxid Redox Signal. (2015) 22:1515–26. 10.1089/ars.2015.6294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daghistani HM, Rajab BS, Kitmitto A. Three-dimensional electron microscopy techniques for unravelling mitochondrial dysfunction in heart failure and identification of new pharmacological targets. Br J Pharmacol. (2019) 176:4340–59. 10.1111/bph.14499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dorn GW, 2nd. Evolving concepts of mitochondrial dynamics. Annu Rev Physiol. (2019) 81:1–17. 10.1146/annurev-physiol-020518-114358 [DOI] [PubMed] [Google Scholar]
- 13.Burte F, Carelli V, Chinnery PF, Yu-Wai-Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol. (2015) 11:11–24. 10.1038/nrneurol.2014.228 [DOI] [PubMed] [Google Scholar]
- 14.Filard T, Ghinassi B, Di Baldassarre A, Tanzilli G, Morano S, Lenzi A, et al. Cardiomyopathy associated with diabetes: the central role of the cardiomyocyte. Int J Mol Sci. (2019) 20:3299. 10.3390/ijms20133299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaludercic N, Di Lisa F. Mitochondrial ROS formation in the pathogenesis of diabetic cardiomyopathy. Front Cardiovasc Med. (2020) 7:12. 10.3389/fcvm.2020.00012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacDonald JA, Wijekoon CP, Liao KC, Muruve DA. Biochemical and structural aspects of the ATP-binding domain in inflammasome-forming human NLRP proteins. IUBMB Life. (2013) 65:851–62. 10.1002/iub.1210 [DOI] [PubMed] [Google Scholar]
- 17.Li PL. Cardiovascular pathobiology of inflammasomes: inflammatory machinery and beyond. Antioxid Redox Signal. (2015) 22:1079–83. 10.1089/ars.2015.6319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ichinohe T, Yamazaki T, Koshiba T, Yanagi Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc Natl Acad Sci U S A. (2013) 110:17963–8. 10.1073/pnas.1312571110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park S, Won JH, Hwang I, Hong S, Lee HK, Yu JW. Defective mitochondrial fission augments NLRP3 inflammasome activation. Sci Rep. (2015) 5:15489. 10.1038/srep15489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. (2011) 12:9–14. 10.1038/nrm3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nature Comm. (2012) 3:1016. 10.1038/ncomms2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dorn GW, 2nd. Parkin-dependent mitophagy in the heart. J Mol Cell Cardiol. (2016) 95:42–9. 10.1016/j.yjmcc.2015.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang X, Sun L, Ji S, Zhao T, Zhang W, Xu J, et al. Kissing and nanotunneling mediate intermitochondrial communication in the heart. Proc Natl Acad Sci U S A. (2013) 110:2846–51. 10.1073/pnas.1300741110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vincent AE, Ng YS, White K, Davey T, Mannella C, Falkous G, et al. The spectrum of mitochondrial ultrastructural defects in mitochondrial myopathy. Sci Rep. (2016) 6:30610. 10.1038/srep30610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lavorato M, Iyer VR, Dewight W, Cupo RR, Debattisti V, Gomez L, et al. Increased mitochondrial nanotunneling activity, induced by calcium imbalance, affects intermitochondrial matrix exchanges. Proc Natl Acad Sci U S A. (2017) 114:E849–58. 10.1073/pnas.1617788113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vasquez-Trincado C, Garcia-Carvajal I, Pennanen C, Parra V, Hill JA, Rothermel BA, et al. Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol. (2016) 594:509–25. 10.1113/JP271301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation. (2014) 130:554. 10.1161/CIRCULATIONAHA.113.008476 [DOI] [PubMed] [Google Scholar]
- 28.Gao Q, Wang XM, Ye HW, Yu Y, Kang PF, Wang HJ, et al. Changes in the expression of cardiac mitofusin-2 in different stages of diabetes in rats. Mol Med Rep. (2012) 6:811–4. 10.3892/mmr.2012.1002 [DOI] [PubMed] [Google Scholar]
- 29.Chen Y, Liu YQ, Dorn GW. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. (2011) 109:1327–36. 10.1161/CIRCRESAHA.111.258723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen L, Gong Q, Stice JP, Knowlton AA. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovas Res. (2009) 84:91–9. 10.1093/cvr/cvp181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Givvimani S, Pushpakumar S, Veeranki S, Tyagi SC. Dysregulation of Mfn2 and Drp-1 proteins in heart failure. Can J Physiol Pharmacol. (2014) 92:583–91. 10.1139/cjpp-2014-0060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. (2010) 121:2012–22. 10.1161/CIRCULATIONAHA.109.906610 [DOI] [PubMed] [Google Scholar]
- 33.Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, et al. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation. (2016) 133:1249–63. 10.1161/CIRCULATIONAHA.115.020502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci U S A. (2011) 108:9572–7. 10.1073/pnas.1106291108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kubli DA, Quinsay MN, Gustafsson AB. Parkin deficiency results in accumulation of abnormal mitochondria in aging myocytes. Commun Integr Biol. (2013) 6:e24511. 10.4161/cib.24511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andres AM, Hernandez G, Lee P, Huang CQ, Ratliff EP, Sin J, et al. Mitophagy is required for acute cardioprotection by simvastatin. Antioxid Redox Signal. (2014) 21:1960–73. 10.1089/ars.2013.5416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu X, Thai PN, Lu S, Pu J, Bers DM. Intrafibrillar and perinuclear mitochondrial heterogeneity in adult cardiac myocytes. J Mol Cell Cardiol. (2019) 136:72–84. 10.1016/j.yjmcc.2019.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsieh CH, Shaltouki A, Gonzalez AE, Bettencourt da Cruz A, Burbulla LF, St Lawrence E, et al. Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic parkinson's disease. Cell Stem Cell. (2016) 19:709–24. 10.1016/j.stem.2016.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fransson A, Ruusala A, Aspenstrom P. Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem. (2003) 278:6495–502. 10.1074/jbc.M208609200 [DOI] [PubMed] [Google Scholar]
- 40.Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, et al. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A. (2008) 105:20728–33. 10.1073/pnas.0808953105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Yu Z, Jiang D, Liang X, Liao S, Zhang Z, et al. iPSC-MSCs with high intrinsic MIRO1 and sensitivity to TNF-alpha yield efficacious mitochondrial transfer to rescue anthracycline-induced cardiomyopathy. Stem Cell Rep. (2016) 7:749–63. 10.1016/j.stemcr.2016.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conejeros C, Parra V, Sanchez G, Pedrozo Z, Olmedo I. Miro1 as a novel regulator of hypertrophy in neonatal rat cardiomyocytes. J Mol Cell Cardiol. (2020) 141:65–9. 10.1016/j.yjmcc.2020.03.014 [DOI] [PubMed] [Google Scholar]
- 43.Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW, et al. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. (2015) 350:aad2459. 10.1126/science.aad2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Basu K, Lajoie D, Aumentado-Armstrong T, Chen J, Koning RI, Bossy B, et al. Molecular mechanism of DRP1 assembly studied in vitro by cryo-electron microscopy. PLoS ONE. (2017) 12:e0179397. 10.1371/journal.pone.0179397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brickley K, Smith MJ, Beck M, Stephenson FA. GRIF-1 and OIP106, members of a novel gene family of coiled-coil domain proteins: association in vivo and in vitro with kinesin. J Biol Chem. (2005) 280:14723–32. 10.1074/jbc.M409095200 [DOI] [PubMed] [Google Scholar]
- 46.Lopez-Domenech G, Covill-Cooke C, Ivankovic D, Halff EF, Sheehan DF, Norkett R, et al. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. (2018) 37:321–36. 10.15252/embj.201696380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci. (2010) 30:4232–40. 10.1523/JNEUROSCI.6248-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu L, Ding M, Tang D, Gao E, Li C, Wang K, et al. Targeting mitochondrial dynamics by regulating Mfn2 for therapeutic intervention in diabetic cardiomyopathy. Theranostics. (2019) 9:3687–706. 10.7150/thno.33684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, et al. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. (2009) 61:541–55. 10.1016/j.neuron.2009.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bers DM. Altered cardiac myocyte Ca regulation in heart failure Physiology. Bethesda. (2006) 21:380–7. 10.1152/physiol.00019.2006 [DOI] [PubMed] [Google Scholar]
- 51.Pereira L, Matthes J, Schuster I, Valdivia HH, Herzig S, Richard S, et al. Mechanisms of [Ca2+]i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes. (2006) 55:608–15. 10.2337/diabetes.55.03.06.db05-1284 [DOI] [PubMed] [Google Scholar]
- 52.Bertero E, Maack C. Calcium signaling and reactive oxygen species in mitochondria. Circ Res. (2018) 122:1460–78. 10.1161/CIRCRESAHA.118.310082 [DOI] [PubMed] [Google Scholar]
- 53.Kohlhaas M, Maack C. Calcium release microdomains and mitochondria. Cardiovasc Res. (2013) 98:259–68. 10.1093/cvr/cvt032 [DOI] [PubMed] [Google Scholar]
- 54.Belmonte S, Morad M. Shear fluid-induced Ca2+ release and the role of mitochondria in rat cardiac myocytes. Ann N Y Acad Sci. (2008) 1123:58–63. 10.1196/annals.1420.007 [DOI] [PubMed] [Google Scholar]
- 55.Goldstein MA, Entman ML. Microtubules in mammalian heart muscle. J Cell Biol. (1979) 80:183–95. 10.1083/jcb.80.1.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gittes F, Mickey B, Nettleton J, Howard J. Flexural rigidity of microtubules and actin filaments measured from thermal fluctuations in shape. J Cell Biol. (1993) 120:923–34. 10.1083/jcb.120.4.923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stroud MJ. Linker of nucleoskeleton and cytoskeleton complex proteins in cardiomyopathy. Biophys Rev. (2018) 10:1033–51. 10.1007/s12551-018-0431-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Steele DF, Fedida D. Cytoskeletal roles in cardiac ion channel expression. Biochim Biophys Acta. (2014) 1838:665–73. 10.1016/j.bbamem.2013.05.001 [DOI] [PubMed] [Google Scholar]
- 59.Caporizzo MA, Chen CY, Prosser BL. Cardiac microtubules in health and heart disease. Exp Biol Med. (2019) 244:1255–72. 10.1177/1535370219868960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Caporizzo MA, Chen CY, Bedi K, Margulies KB, Prosser BL. Microtubules increase diastolic stiffness in failing human cardiomyocytes and myocardium. Circulation. (2020) 141:902–15. 10.1161/CIRCULATIONAHA.119.043930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Caffarra Malvezzi C, Cabassi A, Miragoli M. Mitochondrial mechanosensor in cardiovascular diseases. Vasc Biol. (2020) 2:R85–92. 10.1530/VB-20-0002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Debattisti V, Gerencser AA, Saotome M, Das S, Hajnoczky G. ROS Control mitochondrial motility through p38 and the motor adaptor miro/trak. Cell Rep. (2017) 21:1667–80. 10.1016/j.celrep.2017.10.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stram AR, Payne RM. Post-translational modifications in mitochondria: protein signaling in the powerhouse. Cell Mol Life Sci. (2016) 73:4063–73. 10.1007/s00018-016-2280-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Magiera MM, Singh P, Gadadhar S, Janke C. Tubulin posttranslational modifications and emerging links to human disease. Cell. (2018) 173:1323–7. 10.1016/j.cell.2018.05.018 [DOI] [PubMed] [Google Scholar]
- 65.Birsa N, Norkett R, Wauer T, Mevissen TE, Wu HC, Foltynie T, et al. Lysine 27 ubiquitination of the mitochondrial transport protein Miro is dependent on serine 65 of the Parkin ubiquitin ligase. J Biol Chem. (2014) 289:14569–82. 10.1074/jbc.M114.563031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. (2011) 147:893–906. 10.1016/j.cell.2011.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Berenguer-Escuder C, Grossmann D, Antony P, Arena G, Wasner K, Massart F, et al. Impaired mitochondrial-endoplasmic reticulum interaction and mitophagy in Miro1-mutant neurons in Parkinson's disease. Hum Mol Genet. (2020) 29:1353–64. 10.1093/hmg/ddaa066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grossmann D, Berenguer-Escuder C, Bellet ME, Scheibner D, Bohler J, Massart F, et al. Mutations in RHOT1 disrupt endoplasmic reticulum-mitochondria contact sites interfering with calcium homeostasis and mitochondrial dynamics in parkinson's disease. Antioxid Redox Signal. (2019) 31:1213–34. 10.1089/ars.2018.7718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kalinski AL, Kar AN, Craver J, Tosolini AP, Sleigh JN, Lee SJ, et al. Deacetylation of Miro1 by HDAC6 blocks mitochondrial transport and mediates axon growth inhibition. J Cell Biol. (2019) 218:1871–90. 10.1083/jcb.201702187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Balabanian L, Berger CL, Hendricks AG. Acetylated microtubules are preferentially bundled leading to enhanced kinesin-1 motility. Biophys J. (2017) 113:1551–60. 10.1016/j.bpj.2017.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen S, Owens GC, Makarenkova H, Edelman DB. HDAC6 regulates mitochondrial transport in hippocampal neurons. PLoS ONE. (2010) 5:e10848. 10.1371/journal.pone.0010848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Demos-Davies KM, Ferguson BS, Cavasin MA, Mahaffey JH, Williams SM, Spiltoir JI, et al. HDAC6 contributes to pathological responses of heart and skeletal muscle to chronic angiotensin-II signaling. Am J Physiol Heart Circ Physiol. (2014) 307:H252–8. 10.1152/ajpheart.00149.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kee HJ, Bae EH, Park S, Lee KE, Suh SH, Kim SW, et al. HDAC inhibition suppresses cardiac hypertrophy and fibrosis in DOCA-salt hypertensive rats via regulation of HDAC6/HDAC8 enzyme activity. Kidney Blood Press Res. (2013) 37:229–39. 10.1159/000350148 [DOI] [PubMed] [Google Scholar]
- 74.Leng Y, Wu Y, Lei S, Zhou B, Qiu Z, Wang K, et al. Inhibition of HDAC6 activity alleviates myocardial ischemia/reperfusion injury in diabetic rats: potential role of peroxiredoxin 1 acetylation and redox regulation. Oxid Med Cell Longev. (2018) 2018:9494052. 10.1155/2018/9494052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jeong MY, Lin YH, Wennersten SA, Demos-Davies KM, Cavasin MA, Mahaffey JH, et al. Histone deacetylase activity governs diastolic dysfunction through a nongenomic mechanism. Sci Transl Med. (2018) 10:eaao0144. 10.1126/scitranslmed.aao0144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wallner M, Eaton DM, Berretta RM, Liesinger L, Schittmayer M, Gindlhuber J, et al. HDAC inhibition improves cardiopulmonary function in a feline model of diastolic dysfunction. Sci Transl Med. (2020) 12:eaay7205. 10.1126/scitranslmed.aay7205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lemon DD, Horn TR, Cavasin MA, Jeong MY, Haubold KW, Long CS, et al. Cardiac HDAC6 catalytic activity is induced in response to chronic hypertension. J Mol Cell Cardiol. (2011) 51:41–50. 10.1016/j.yjmcc.2011.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lkhagva B, Kao YH, Lee TI, Lee TW, Cheng WL, Chen YJ. Activation of Class I histone deacetylases contributes to mitochondrial dysfunction in cardiomyocytes with altered complex activities. Epigenetics. (2018) 13:376–85. 10.1080/15592294.2018.1460032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yan S, Wei X, Jian W, Qin Y, Liu J, Zhu S, et al. Pharmacological Inhibition of HDAC6 attenuates NLRP3 inflammatory response and protects dopaminergic neurons in experimental models of parkinson's disease. Front Aging Neurosci. (2020) 12:78. 10.3389/fnagi.2020.00078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Luo B, Huang F, Liu Y, Liang Y, Wei Z, Ke H, et al. NLRP3 Inflammasome as a molecular marker in diabetic cardiomyopathy. Front Physiol. (2017) 8:519. 10.3389/fphys.2017.00519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. (2013) 14:454–60. 10.1038/ni.2550 [DOI] [PubMed] [Google Scholar]
- 82.Li X, Thome S, Ma X, Amrute-Nayak M, Finigan A, Kitt L, et al. MARK4 regulates NLRP3 positioning and inflammasome activation through a microtubule-dependent mechanism. Nat Commun. (2017) 8:15986. 10.1038/ncomms15986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.An N, Gao Y, Si Z, Zhang H, Wang L, Tian C, et al. Regulatory mechanisms of the NLRP3 inflammasome, a novel immune-inflammatory marker in cardiovascular diseases. Front Immunol. (2019) 10:1592. 10.3389/fimmu.2019.01592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fujisue K, Sugamura K, Kurokawa H, Matsubara J, Ishii M, Izumiya Y, et al. Colchicine improves survival, left ventricular remodeling, and chronic cardiac function after acute myocardial infarction. Circ J. (2017) 81:1174–82. 10.1253/circj.CJ-16-0949 [DOI] [PubMed] [Google Scholar]
- 85.Carta S, Tassi S, Semino C, Fossati G, Mascagni P, Dinarello CA, et al. Histone deacetylase inhibitors prevent exocytosis of interleukin-1beta-containing secretory lysosomes: role of microtubules. Blood. (2006) 108:1618–26. 10.1182/blood-2006-03-014126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hwang I, Lee E, Jeon SA, Yu JW. Histone deacetylase 6 negatively regulates NLRP3 inflammasome activation. Biochem Biophys Res Commun. (2015) 467:973–8. 10.1016/j.bbrc.2015.10.033 [DOI] [PubMed] [Google Scholar]
- 87.Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W, et al. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science. (2020) 369:eaas8995. 10.1126/science.aas8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gao J, Liu RT, Cao S, Cui JZ, Wang A, To E, et al. NLRP3 inflammasome: activation and regulation in age-related macular degeneration. Mediators Inflamm. (2015) 2015:690243. 10.1155/2015/690243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qi X, Qvit N, Su YC, Mochly-Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci. (2013) 126:789–802. 10.1242/jcs.114439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Develop Cell. (2008) 14:193–204. 10.1016/j.devcel.2007.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Smith LE, Rogowska-Wrzesinska A. The challenge of detecting modifications on proteins. Essays Biochem. (2020) 64:135–53. 10.1042/EBC20190055 [DOI] [PubMed] [Google Scholar]