

Abstract
A quantitative structure-activity relationship (QSAR) study is performed on 48 novel 4,5,6,7-tetrahydrobenzo[D]-thiazol-2 derivatives as anticancer agents capable of inhibiting c-Met receptor tyrosine kinase. The present study is conducted using multiple linear regression, multiple nonlinear regression and artificial neural networks. Three QSAR models are developed after partitioning the database into two sets (training and test) via the k-means method. The obtained values of the correlation coefficients by the three developed QSAR models are 0.90, 0.91 and 0.92, respectively. The resulting models are validated by using the external validation, leave-one-out cross-validation, Y-randomization test, and applicability domain methods. Moreover, we evaluated the drug-likeness properties of seven selected molecules based on their observed high activity to inhibit the c-Met receptor. The results of the evaluation showed that three of the seven compounds present drug-like characteristics.
In order to identify the important active sites for the inhibition of the c-Met receptor responsible for the development of cancer cell lines, the crystallized form of the Crizotinib-c-Met complex (PDB code: 2WGJ) is used. These sites are used as references in the molecular docking test of the three selected molecules to identify the most suitable molecule for use as a new c-Met inhibitor. A comparative study is conducted based on the evaluation of the predicted properties of ADMET in silico between the candidate molecule and the Crizotinib inhibitor. The comparison results show that the selected molecule can be used as new anticancer drug candidates.
Keywords: C-Met inhibitors, QSAR, ADMET, Molecular docking, Crizotinib
C-Met inhibitors, QSAR, ADMET, Molecular docking, Crizotinib
1. Introduction
Recently, aromatic heterocyclic thiazole compounds have received increasing attention in the medicinal chemistry domain [1,2]. Thiazoles and their derivatives have shown significant biological activities due to their anti-inflammatory properties [3], anticonvulsifs [4], insecticides [5], antioxydants, anti-tumor, antihypertensives, pesticides [6], and antidiabetic potential [7]. In particular, 1,3-thiazole structures have been successfully used as effective anti-cancer agents. For example, the 1,3-thiazole formulations S3U937 Figure 1 (A) [8], and S8A375 Figure 1 (B) [9], showed anticancer activities against different types of cancer [10] (Figure 1).
Figure 1.

(A) and (B) represent successively the structures of Zopolrestat and N-(6-substituted-1,3-benzothiazol-2-yl)-benzenesulfonamide molecules.
The c-Met receptor tyrosine kinase represents an interesting anti-cancer target [11]. Optimization of the series of 2-amino-5-aryl-3-benzyloxypyridine molecules used in the development of a new clinical candidate, namely Crizotinib (PF-02341066) (Figure 2) [12], which demonstrated strong inhibition of c-Met kinase tyrosine in Vitro and in Vivo tests, and shows excellent pharmaceutical properties and strong inhibition of tumor growth [13]. Crizotinib inhibits the c-Met kinase and perturbs the c-Met signaling pathway [14]. Overall, this agent inhibits the growth of cancer cells.
Figure 2.

Molecular structure of the Crizotinib (PF-02341066), c-Met Cell IC50 8 nM inhibitor [13].
To develop a new molecule for use in a drug, a very long synthesis process must be followed.
For this reason, pharmaceutical industries are moving towards innovation and new research methods, including the prediction of molecule activities before their synthesis. The use of molecular modeling techniques such as QSAR and molecular docking have become very important methods [15]. In the context of the development of new molecules inhibiting c-Met, a molecular modeling study of 4,5,6,7-tetrahydrobenzo [D]-Thiazol-2-Y derivatives is performed in this work using the molecular series of these derivatives synthesized by Mohareb et al. [16] since these derivatives showed high biological activity and a high capacity to inhibit the c-Met protein more effectively than the Crizotinib inhibitor.
According to the studies conducted by James et al. [13] and Christensen et al. [14], there is a significant relationship between c-Met kinase activity and tumor cell growth. It is therefore necessary to find effective inhibitors of c-Met to remove the tumor cells generated by the increased enzymatic activity of the c-Met protein. In this work, candidate inhibitors of the studied series are determined to be used in the inhibition of c-Met as novel anticancer agents. These are determined by QSAR and molecular docking analysis. In addition, the evaluation of ADMET properties in silico is performed to confirm the use of the best selected inhibitor that can be used as anticancer drug.
The rest of this work is organized as follows: the second section presents materials and methods. The third section includes the simulation results and discussions. As well as the final section concludes the performed work.
2. Materials and methods
Quantitative structure-activity relationships (QSAR) are powerful methods used in drug discovery [15, 28]. We calculated multidimensional molecular descriptors (Constitutional, Topological, Physico-chemical, Geometrical, and Quantum) in order to identify the regions of space related to the inhibition of the enzymatic activity of the c-Met protein. To this aim, we construct QSAR models by using statistical methods [17]. In this study, we used a set of 48 molecules of 4,5,6,7-tetrahydrobenzo[d]-thiazol-2-yl derivatives to construct QSAR models. To develop these models, we used multiple linear regression (MLR) analysis, multiple nonlinear regression (MNLR) and artificial neural networks (ANN). The predictive capacity of the developed models is tested by several validation techniques that are: internal and external validations and Y-randomization methods. In addition, we also examined the applicability (AD) of the QSAR model obtained by MLR based on William's plot to detect outliers and external compounds [18]. In addition, we evaluated the drug-likeness properties of a set of molecules that achieved high biological activity against cancer cell growth [19]. The evaluation of drug-likeness properties is performed by respecting a set of basic rules that are: Lipinski [20], Veber [21] and Egan rules [22]. After the selection of the candidate molecules, we perform the molecular docking of the selected compounds with the c-Met receptor. This is to identify the different types of interactions between the selected molecules and the active sites of c-Met, as well as to determine the positions and orientations of these molecules in the c-Met receptor pocket.
2.1. The studied compounds
To perform the molecular modeling, we use the experimental IC50 values of anti-cancer enzymatic activity data of a series of 48 compounds of novel 4,5,6,7-Tetrahydrobenzo[D]-Thiazol-2-Yl previously synthesized and presented by Mohareb et al. [16]. The observed activities IC50 (nM) are converted to pIC50 level (pIC50 = -logIC50), which are presented in Table 1.
Table 1.
Studied compounds and their observed pIC50 [16].
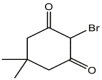 |
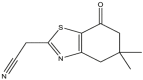 |
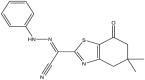 |
 |
||||
| 01 | pIC50 = 8.87 | 02 | pIC50 = 4.64 | 03 | pIC50 = 8.28 | 04 | pIC50 = 8.28 |
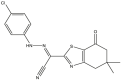 |
 |
 |
 |
||||
| 05 | pIC50 = 9.49 | 06 | pIC50 = 8.90 | 07 | pIC50 = 8.33 | 08 | pIC50 = 8.11 |
 |
 |
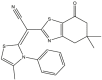 |
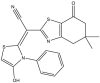 |
||||
| 09 | pIC50 = 10.10 | 10 | pIC50 = 8.90 | 11 | pIC50 = 8.44 | 12 | pIC50 = 9.38 |
 |
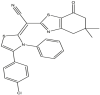 |
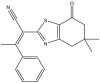 |
 |
||||
| 13 | pIC50 = 8.88 | 14 | pIC50 = 9.64 | 15 | pIC50 = 8.20 | 16 | pIC50 = 9.59 |
 |
 |
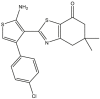 |
 |
||||
| 17 | pIC50 = 8.62 | 18 | pIC50 = 8.08 | 19 | pIC50 = 9.32 | 20 | pIC50 = 8.62 |
 |
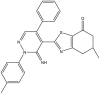 |
 |
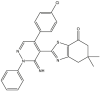 |
||||
| 21 | pIC50 = 8.28 | 22 | pIC50 = 8.46 | 23 | pIC50 = 9.34 | 24 | pIC50 = 8.49 |
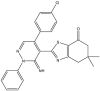 |
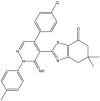 |
 |
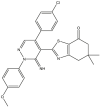 |
||||
| 25 | pIC50 = 8.94 | 26 | pIC50 = 8.46 | 27 | pIC50 = 9.55 | 28 | pIC50 = 8.86 |
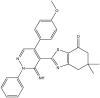 |
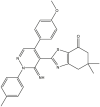 |
 |
 |
||||
| 29 | pIC50 = 8.37 | 30 | pIC50 = 8.08 | 31 | pIC50 = 8.88 | 32 | pIC50 = 8.42 |
 |
 |
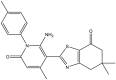 |
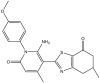 |
||||
| 33 | pIC50 = 8.27 | 34 | pIC50 = 8.97 | 35 | pIC50 = 8.25 | 36 | pIC50 = 8.73 |
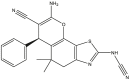 |
 |
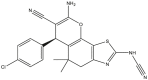 |
 |
||||
| 37 | pIC50 = 8.14 | 38 | pIC50 = 8.55 | 39 | pIC50 = 8.99 | 40 | pIC50 = 9.55 |
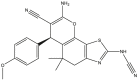 |
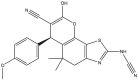 |
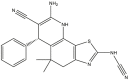 |
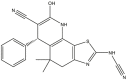 |
||||
| 41 | pIC50 = 8.47 | 42 | pIC50 = 8.85 | 43 | pIC50 = 8.20 | 44 | pIC50 = 9.09 |
 |
 |
 |
 |
||||
| 45 | pIC50 = 9.24 | 46 | pIC50 = 9.49 | 47 | pIC50 = 8.19 | 48 | pIC50 = 8.68 |
2.2. Calculation of molecular descriptors
In the present study, we have based on the computation of 15 different molecular descriptors of the studied series that belong to different classes (1D, 2D, 3D) to develop linear and non-linear mathematical models.
Tables 2 and 3 show the computed descriptors by using Chem3D V16 [23], ChemSketch12 [24] and Gaussian 09 software [25]. The topological, physico-chemical and geometrical descriptors are calculated after optimizing the energy of each compound using the MM2 method (force field method with gradient for root mean square (RMS) of 0.01 kcal/mol) [26]. The geometrical structure of the studied compounds is also optimized by the Becke's three-parameter hybrid method and the Lee-Yang-Parr B3LYP function using the 6-31G(d) basis to calculate the quantum chemical descriptors using the Gaussian 09W software [25,26]. We calculate the molecular descriptors to be used in the development of 2D-QSAR models, as these descriptors represent the most important structural properties of the studied molecules. After calculating these descriptors for all the molecules of the studied series, we determine the quantitative relationship between these descriptors and the biological inhibitory activity of the c-Met enzyme. The quantitative relationship is constructed via statistical methods (MLR, MNLR and ANN) for representing QSAR models.
Table 2.
List of descriptors used in this work.
| Descriptors | Symbol | Class |
|---|---|---|
| Molecular weight | MW | Constitutional |
| Balaban's index | IB | Topological |
| Wiener Index | WI | |
| Coefficient of partition Octanol/Water | LogP | Physico-chemical |
| Polarizability | αe(cm3) | |
| Parachor | Pc(cm3) | |
| Energy of Van der Waals | EVDW (ev) | Geometrical |
| Molecular volume | MV (cm3) | |
| Energy gap | EGap(ev) | Quantum (Electronic) |
| Energy HOMO | EHOMO(ev) | |
| Energy LUMO | ELUMO(ev) | |
| Index of electrophilicity | w (ev) | |
| Electronegativity | x (ev) | |
| Chemical potential | μ (ev) | |
| Chemical hardness | ɳ (ev) |
Table 3.
Calculated descriptors values and observed activity.
| EVDW | LogP | αe | IB | WI | Pc | MW | MV | EGap | ω | EHOMO | ELUMO | χ | μ | η | pIC50 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.23 | 1.37 | 17.85 | 16816 | 140 | 378.20 | 217.99 | 152.2 | 4.32 | 5.73 | -7.13 | -2.82 | 4.97 | -4.97 | 3.90 | 8.87 |
| 2 | 0.32 | 1.31 | 23.16 | 55529 | 351 | 482.30 | 220.07 | 180 | 4.61 | 4.86 | -7.04 | -2.43 | 4.74 | -4.74 | 3.58 | 8.64 |
| 3 | 0.72 | 2.75 | 36.92 | 357864 | 1257 | 667.70 | 324.10 | 247 | 3.15 | 6.51 | -6.10 | -2.95 | 4.53 | -4.53 | 3.74 | 8.28 |
| 4 | 0.74 | 3.25 | 38.68 | 443941 | 1437 | 698.80 | 338.12 | 262.2 | 3.07 | 6.39 | -5.97 | -2.89 | 4.43 | -4.43 | 3.66 | 8.28 |
| 5 | 0.73 | 3.46 | 38.75 | 1048196 | 1437 | 696.60 | 358.07 | 256.3 | 3.15 | 7.01 | -6.27 | -3.12 | 4.70 | -4.70 | 3.91 | 9.49 |
| 6 | 0.83 | 2.67 | 39.23 | 548184 | 1641 | 718.00 | 354.12 | 268.7 | 2.90 | 6.39 | -5.75 | -2.85 | 4.30 | -4.30 | 3.58 | 8.90 |
| 7 | 1.57 | 4.90 | 52.52 | 1025259 | 2696 | 905.70 | 459.12 | 327.9 | 3.06 | 6.10 | -5.85 | -2.79 | 4.32 | -4.32 | 3.55 | 8.33 |
| 8 | 1.60 | 5.40 | 54.27 | 1189694 | 2953 | 936.80 | 473.13 | 343 | 3.02 | 6.08 | -5.80 | -2.77 | 4.28 | -4.28 | 3.53 | 8.11 |
| 9 | 1.59 | 5.61 | 54.34 | 1189694 | 2953 | 934.60 | 493.08 | 337.1 | 3.09 | 6.41 | -6.00 | -2.91 | 4.45 | -4.45 | 3.68 | 10.10 |
| 10 | 1.69 | 4.82 | 54.83 | 1381564 | 3243 | 956.00 | 489.13 | 349.5 | 2.98 | 6.03 | -5.73 | -2.75 | 4.24 | -4.24 | 3.49 | 8.90 |
| 11 | 0.92 | 5.43 | 43.94 | 565181 | 1760 | 829.70 | 393.10 | 300.1 | 3.50 | 3.78 | -5.39 | -1.89 | 3.64 | -3.64 | 2.76 | 8.44 |
| 12 | 0.83 | 4.67 | 42.71 | 565181 | 1760 | 806.60 | 395.08 | 277.1 | 3.48 | 3.93 | -5.44 | -1.96 | 3.70 | -3.70 | 2.83 | 9.38 |
| 13 | 1.23 | 6.80 | 51.77 | 1048196 | 2756 | 963.50 | 455.11 | 344.3 | 3.48 | 3.83 | -5.39 | -1.91 | 3.65 | -3.65 | 2.78 | 8.88 |
| 14 | 1.24 | 7.51 | 53.71 | 1217615 | 3022 | 1000.60 | 489.07 | 356.3 | 3.50 | 4.05 | -5.51 | -2.01 | 3.76 | -3.76 | 2.89 | 9.64 |
| 15 | 0.82 | 3.61 | 36.79 | 332448 | 1167 | 716.40 | 322.11 | 270.1 | 3.84 | 5.39 | -6.47 | -2.63 | 4.55 | -4.55 | 3.59 | 8.20 |
| 16 | 0.84 | 4.32 | 38.73 | 411602 | 1332 | 753.50 | 356.08 | 282.1 | 3.83 | 5.80 | -6.63 | -2.80 | 4.71 | -4.71 | 3.76 | 9.59 |
| 17 | 0.93 | 3.52 | 39.44 | 508042 | 1521 | 775.00 | 352.13 | 294.1 | 3.51 | 5.36 | -6.09 | -2.58 | 4.34 | -4.34 | 3.46 | 8.62 |
| 18 | 0.67 | 3.99 | 39.94 | 326194 | 1270 | 751.50 | 354.09 | 276.2 | 3.52 | 4.06 | -5.54 | -2.02 | 3.78 | -3.78 | 2.90 | 8.08 |
| 19 | 0.68 | 4.70 | 41.88 | 399857 | 1442 | 788.60 | 388.05 | 288.1 | 3.57 | 4.30 | -5.70 | -2.13 | 3.92 | -3.92 | 3.03 | 9.32 |
| 20 | 0.78 | 3.93 | 42.59 | 489129 | 1639 | 810.10 | 384.10 | 300.2 | 3.45 | 4.04 | -5.46 | -2.01 | 3.73 | -3.73 | 2.87 | 8.62 |
| 21 | 1.42 | 4.28 | 49.88 | 890240 | 2487 | 881.40 | 426.15 | 324.5 | 3.73 | 4.23 | -5.84 | -2.11 | 3.97 | -3.97 | 3.04 | 8.28 |
| 22 | 1.45 | 4.78 | 51.63 | 1041253 | 2741 | 912.50 | 440.17 | 339.7 | 3.70 | 4.14 | -5.77 | -2.06 | 3.91 | -3.91 | 2.99 | 8.46 |
| 23 | 1.44 | 5.00 | 51.70 | 1041253 | 2741 | 910.30 | 460.11 | 333.8 | 3.74 | 4.63 | -6.03 | -2.29 | 4.16 | -4.16 | 3.23 | 9.34 |
| 24 | 1.54 | 4.20 | 52.18 | 1217953 | 3027 | 931.70 | 456.16 | 346.1 | 3.64 | 4.10 | -5.69 | -2.04 | 3.86 | -3.86 | 2.95 | 8.49 |
| 25 | 1.44 | 5.00 | 51.70 | 1037088 | 2730 | 910.30 | 460.11 | 333.8 | 3.68 | 4.65 | -5.98 | -2.30 | 4.14 | -4.14 | 3.22 | 8.94 |
| 26 | 1.47 | 5.49 | 53.45 | 1206190 | 2997 | 941.30 | 474.13 | 349 | 3.65 | 4.57 | -5.90 | -2.26 | 4.08 | -4.08 | 3.17 | 8.46 |
| 27 | 1.46 | 5.71 | 53.52 | 1206190 | 2997 | 939.10 | 494.07 | 343.1 | 3.69 | 5.05 | -6.17 | -2.47 | 4.32 | -4.32 | 3.40 | 9.55 |
| 28 | 1.56 | 4.91 | 54.00 | 1403224 | 3297 | 960.50 | 490.12 | 355.4 | 3.59 | 4.53 | -5.82 | -2.24 | 4.03 | -4.03 | 3.13 | 8.86 |
| 29 | 1.54 | 4.20 | 52.18 | 1403224 | 3005 | 931.70 | 456.16 | 346.1 | 3.73 | 4.06 | -5.76 | -2.03 | 3.89 | -3.89 | 2.96 | 8.37 |
| 30 | 1.56 | 4.70 | 53.93 | 1209134 | 3286 | 962.80 | 470.18 | 361.3 | 3.71 | 3.96 | -5.69 | -1.98 | 3.83 | -3.83 | 2.90 | 8.08 |
| 31 | 1.56 | 4.91 | 54.00 | 1398566 | 3286 | 960.50 | 490.12 | 355.4 | 3.72 | 4.45 | -5.93 | -2.21 | 4.07 | -4.07 | 3.14 | 8.88 |
| 32 | 1.66 | 4.12 | 54.48 | 1398566 | 3601 | 981.90 | 486.17 | 367.8 | 3.66 | 3.91 | -5.62 | -1.95 | 3.78 | -3.78 | 2.87 | 8.42 |
| 33 | 1.01 | 3.38 | 41.94 | 1618347 | 1739 | 802.80 | 379.14 | 294.9 | 3.31 | 4.40 | -5.47 | -2.16 | 3.81 | -3.81 | 2.99 | 8.27 |
| 34 | 1.03 | 4.14 | 43.88 | 558287 | 1946 | 839.90 | 413.10 | 306.9 | 3.34 | 4.65 | -5.61 | -2.27 | 3.94 | -3.94 | 3.11 | 8.97 |
| 35 | 1.04 | 3.88 | 43.86 | 669086 | 1946 | 841.10 | 393.15 | 311.2 | 3.30 | 4.33 | -5.43 | -2.13 | 3.78 | -3.78 | 2.96 | 8.25 |
| 36 | 1.13 | 3.38 | 44.59 | 669086 | 2181 | 861.40 | 409.15 | 318.9 | 3.29 | 4.32 | -5.42 | -2.13 | 3.77 | -3.77 | 2.95 | 8.73 |
| 37 | 0.76 | 1.80 | 40.83 | 801064 | 1613 | 799.50 | 374.12 | 274.6 | 4.01 | 3.80 | -5.90 | -1.90 | 3.90 | -3.90 | 2.90 | 8.14 |
| 38 | 0.64 | 1.66 | 40.01 | 516572 | 1613 | 786.70 | 375.10 | 268.4 | 4.17 | 4.16 | -6.25 | -2.08 | 4.17 | -4.17 | 3.12 | 8.55 |
| 39 | 0.78 | 2.51 | 42.75 | 516572 | 1803 | 836.60 | 408.08 | 285.4 | 4.05 | 4.05 | -6.07 | -2.02 | 4.05 | -4.05 | 3.04 | 8.99 |
| 40 | 0.66 | 2.37 | 41.92 | 618482 | 1803 | 823.90 | 409.07 | 279.2 | 4.20 | 4.41 | -6.41 | -2.20 | 4.31 | -4.31 | 3.25 | 9.55 |
| 41 | 0.88 | 1.72 | 43.36 | 618482 | 2021 | 858.10 | 404.13 | 296.3 | 3.92 | 3.75 | -5.79 | -1.87 | 3.83 | -3.83 | 2.85 | 8.47 |
| 42 | 0.76 | 1.57 | 42.53 | 740649 | 2021 | 845.40 | 405.12 | 290.1 | 4.03 | 4.10 | -6.08 | -2.05 | 4.06 | -4.06 | 3.06 | 8.85 |
| 43 | 0.73 | 2.55 | 41.59 | 740649 | 1613 | 807.40 | 373.14 | 276.1 | 3.56 | 3.73 | -5.42 | -1.86 | 3.64 | -3.64 | 2.75 | 8.20 |
| 44 | 0.68 | 2.25 | 40.76 | 516572 | 1613 | 794.70 | 374.12 | 269.9 | 3.72 | 3.93 | -5.68 | -1.96 | 3.82 | -3.82 | 2.89 | 9.09 |
| 45 | 0.75 | 3.26 | 43.50 | 516572 | 1803 | 844.50 | 407.10 | 286.9 | 3.60 | 3.99 | -5.59 | -1.99 | 3.79 | -3.79 | 2.89 | 9.24 |
| 46 | 0.70 | 2.96 | 42.68 | 618482 | 1803 | 831.80 | 408.08 | 280.7 | 3.76 | 4.19 | -5.85 | -2.09 | 3.97 | -3.97 | 3.03 | 9.49 |
| 47 | 0.85 | 2.47 | 44.11 | 618482 | 2021 | 866.00 | 403.15 | 297.8 | 3.55 | 3.68 | -5.39 | -1.84 | 3.61 | -3.61 | 2.73 | 8.19 |
| 48 | 0.79 | 2.17 | 43.29 | 740649 | 2021 | 853.30 | 404.13 | 291.6 | 3.71 | 3.87 | -5.65 | -1.93 | 3.79 | -3.79 | 2.86 | 8.68 |
2.3. Statistical methods
To construct QSAR models, we studied a series of 48 molecules obtained by in vitro synthesis [16]. All the studied molecules showed a strong enzymatic inhibitory activity of c-Met (pIC50 > 8). To develop the QSAR models in this study, we use the statistical methods presented below.
2.3.1. Principal component analysis (PCA)
PCA is a very efficient method for assembling the information encoded in components, and is widely used to understand the distribution of components and the links between them [29]. The PCA method is based on descriptive statistics, and the main objective of using this method is to extract as much information as possible from the database [30]. In this work, we use the PCA method to identify the different molecular descriptors that will contribute to the development of the QSAR models. The PCA method is applied using fifteen descriptors calculated for each molecule in the series of 4,5,6,7-Tetrahydrobenzo[D]-Thiazol-2-Yl derivatives.
2.3.2. K-means method
After selecting the most important molecular descriptors that are uncorrelated through the PCA method, we divide the database into two sets (training and test). Thus, the training and test sets include 80% and 20% of the total data, respectively [31]. The training set is used to develop QSAR models, while the test set is used to evaluate the effectiveness of each developed model. The procedure of dividing the data set into training and test sets is performed by the k-means classification method [32]. In each cluster obtained after the K-means division method, one compound from each cluster is randomly selected as part of the test set, while the remaining compounds are selected as the training set. After this division, we obtain ten compounds for the test set and thirty-eight compounds for the training set.
2.4. Analysis of the structure-activity relationship
To carry out the analysis of the relationship between the structure and activity of the studied compounds, the most widely used methods in the development of QSAR models are used, which are: MLR [33], MNLR [30], and ANN [34]. This is to build QSAR models capable of predicting the biological efficacy of molecules in inhibiting the enzymatic activity of the c-Met protein. Through the obtained QSAR models, the biological activity is related to the molecular descriptors. The descriptors of the obtained model represent very important parameters that can influence the inhibition of the enzymatic activity of the c-Met kinase protein. In our work, the MNLR and MLR models are developed with XLSTAT V. 2019 software [35], and the ANN model is developed with MatlabV.2015a software [36]. In the statistical analysis of QSAR models, we rely on classical analytical approaches [37], where the main parameters used in this approaches are: the determination coefficient (R2) (Eq. 1), correlation coefficient, adjusted coefficient () (Eq. 2), the mean squared error (MSE) (Eq. 3), high F-value (F > 0.33) and the level of signification (p-value) is traditionally between 1% and 5% [38].
| (1) |
| (2) |
| (3) |
where is the value of the observed response, is the value of the predicted response, is the average value of observed/predicted responses, p is the number of explicative variables in the model, and n is the number of individuals.
After partitioning the dataset, we use the descriptors obtained by the PCA method to develop an accurate and statistically acceptable QSAR model using the MLR method. This is done in order to define the most important descriptors to be used as inputs in the development of other QSAR models via the MNLR and ANN techniques.
2.4.1. Multiple linear regression (MLR)
The MLR method is widely used in QSAR studies for molecular descriptor selection due to its simplicity and robustness [39]. MLR is also used to identify the descriptors used as input parameters in the development of QSAR models by the MNLR and ANN methods [40]. MLR is based on the hypothesis that the dependent variable is linked linearly to certain independent variables according to the following relationship given by Eq. (4).
| (4) |
Where is the dependent variable (biological activity to be predicted), are the independent variables (molecular descriptors), n is the number of molecular descriptors, is the constant in Eq. (4), represent the coefficients of the descriptors.
2.4.2. Multiple nonlinear regression (MNLR)
The MNLR method is a non-linear approach (exponential, logarithmic, polynomial...), which consists in determining the mathematical model that best describes the non-linear variation of a molecular property or biological activity (Y) as it relates to molecular descriptors (Xi) [41]. In this context, we use the polynomial model of the second order to build the QSAR model via the MNLR technique, based on the descriptors that are determined by the MLR model. The nonlinear relationship between molecular descriptors and biological activity is done according to Eq. (5).
| (5) |
Where: is the dependent variable (biological activity to be predicted), are the independent variables (the molecular descriptors), n is the number of molecular descriptors, is the constant in the equation of the model, and represent the coefficients of the descriptors in the equation of the model.
2.4.3. Artificial neural network (ANN)
Artificial neural networks are used to augment the probability of characterization of the compound and to generate a predictive model linking all quantitative molecular descriptors obtained from the MLR model and the values of observed biological activities [42]. We develop the QSAR model based on the ANN method to confirm the accuracy of the selected molecular descriptors that are obtained by the MLR model. Moreover, the ANN model allows us to obtain the biological activity predictions for each molecule with high accuracy. The ANN model that we develop in this work is of the feed-forward type [34]. This method is based on the sigmoid transport function in the hidden layer and the linear transfer function in the output layer. The ANN architecture in this work is composed of three layers of neurons, called the input layer, hidden layer, and output layer, as shown in Figure 3.
Figure 3.

The architecture of the ANN model used in this study.
The input layer contains a number of neurons equal to or less than the number of descriptors obtained using the multiple linear regression model, and the output layer contains the predicted activity values. In order to determine the number of hidden neurons within the hidden layer, it is necessary to calculate the parameter ρ, this parameter being calculated according to the relation = (number of weights)/(number of connections) [43]. According to the recommendations of some authors, the value of the parameter ρ should be between 1 and 3 to ensure that the ANN model is statistically acceptable, and that the forecasts obtained through this model are made in a way that ensures the contribution of all the elements of the database used [44,45].
2.5. Statistical testing and validation of the QSAR models proposed
After developing the QSAR models, it is necessary to perform certain statistical tests to confirm the validity of the proposed models. In this work, the developed QSAR models are validated by an internal and an external validation. Also, we perform a Y-randomization test to evaluate the efficiency of the original model obtained by the MLR method, and then we determine the applicability domain of the MLR model.
2.5.1. Internal validation
To validate the QSAR models developed by MLR, MNLR and ANN, we use the internal validation procedure named leave-one-out cross-validation (LOOCV) [46]. This validation is based on the calculation of the coefficient value by using Eq. (6). According to [46] the value of should be more than 0.5. This indicates that the developed model is robust in the internal prediction.
| (6) |
Where is the value of the observed response, is the value of the response predicted by Loo-cv, is the mean value of the observed/predicted responses.
2.5.2. External validation
In this test, we apply the QSAR models developed to predict the activities of the compounds of the test set. The test set contains compounds from the series of molecules studied in this work, but these compounds did not contribute to the development of the QSAR models. We assessed the external ability of the QSAR models to predict the activity of the test set molecules by calculating the coefficient between the observed pIC50 values and the predicted pIC50 values after the inclusion of the test set. The importance of evaluating the value of in the external validation of QSAR models has been described by Globarikh and Tropsha [46]. Accordingly, it has been described that when the value of is greater than 0.5, the model is statistically acceptable in prediction and can be applied to new external data [47].
2.5.3. Y-randomization test
The Y-randomization test is used to avoid the possibility of random correlation between descriptors and their corresponding biological activities in the model that was initially obtained by the MLR technique. Therefore, any random correlation between X values (molecular descriptors) and Y values (biological activity) will affect the efficiency and validity of the MLR model as well as the MNLR and ANN models. the Y-randomization test is distributing randomly the experimental properties/activity values on the descriptors of the original model, and thanks to this distribution, new models are generated [48]. The QSAR model is considered acceptable and was not obtained by chance through the Y-randomization test, when the average random correlation coefficient () of the randomly constructed models is less than the correlation coefficient () of the original non-random model [49].
2.6. Applicability domain (AD)
The applicability domain of the original QSAR model is obtained by MLR. This step is the last step in the validation of the developed QSAR models in the present work. The QSAR model cannot be considered as a universal model, because it is developed on a limited number of compounds that do not cover the total space chemical [50]. The applicability domain of the QSAR model is defined as a space that includes the chemical space of the molecules in the training set. This space contains the molecules with correctly predicted activities [50]. Thus, QSAR models cannot accurately predict the characteristics of all compounds involved. Therefore, determining the applicability domain of QSAR models is of great importance to identify molecules that are not correctly predicted in terms of activities. Hence, molecules that are outside the applicability domain of the QSAR model will not be considered for molecular modeling. In addition, the verification and validation process through the definition of the applicability domain is very important according to the Organization for Economic Cooperation and Development (OECD) [51]. In the absence of the required AD analyses, any QSAR model can predict the activity of any compound, even with a completely different structure than the molecules under study. There are several methods for defining the AD models [52], but the most frequently used method is the determination of the leverage values effect () with () for each compound, is the vector descriptive of the compound to be found, is matrix of the descriptor values of the model for compounds of the training set, and the exponent refers to the transposition of the matrix/vector [51]. The Williams plot is used to determine the AD within a square zone [53] and the level of leverage () [50] with is the number of compounds in the training set and is the number of selected descriptors in the model. When the leverage effect h for the compound is higher than the alert leverage of the same compound, indicates that this compound negatively affects the constructed model, so it is considered outside the applicability domain [18].
In this work, we define the applicability domain of the Williams plot type by using MatlabV2015a software.
2.7. Drug likeness and in silico pharmacokinetics ADMET prediction
We note that many potential therapeutic agents fail to reach clinical trials because of their unfavorable parameters of ADMET (absorption, distribution, metabolism, elimination and toxicity) [50,51]. The drug-likeness is the most recent method proposed to identify compounds that are recommended for use in drugs that must respect certain rules that are important: Lipinski's [20], Veber's [21] and Igan's [22] rules. In this work, we predict the drug-likeness properties of compounds that have experimentally and predictively demonstrated excellent activity, so that these molecules are within the applicability of the previously determined QSAR model. The Lipinski, Veber and Igan rules are based on the evaluation of ADME properties of human drugs. These rules are very useful to discover drugs based on the 2D structure of small molecules, and on the bioavailability of these molecules by the oral administration [56]. Compounds whose physical and chemical properties do not meet at least two of the Lipinski, Veber and Igan rules are subject to a variety of problems in their pharmacokinetic properties related to ADMET. Less than 10% of drugs that reach the clinical trial phase do not meet any of these rules (Lipinski, Veber and Igan). We also evaluate two other factors, which are the number of Rotatable bonds (n-ROTB) and Topological polar surface area (TPSA) [57]. The prediction of these factors allows us to know if the molecule interacts with the receptor in a flexible mode or an inflexible one [58]. In this study, we evaluate the drug-likeness and pharmacokinetic in silico properties of the molecules to be selected as inhibitory agents of c-Met enzyme activity, using the online SwissADME [59] and pkCSM [60] servers, respectively.
2.8. Molecular docking
Molecular docking has recently become an essential tool in drug discovery [61], because of its ability to predict the conformation and mode of binding of the ligand to the receptor binding site. In this study, we dock the selected molecules by evaluating the drug-likeness properties with the c-Met receptor in order to identify the most appropriate candidate molecule for the inhibition of c-Met enzymatic activity. c-Met protein is also called tyrosine kinase Met protein or hepatocyte growth factor receptor (HGFR) [13]. Similar to most receptor tyrosine kinases (RTKs), hepatocyte growth factor (HGF) c-Met is also a regulator of many critical cellular processes, including embryonic development, cell growth, differentiation, vasodilation and tissue regeneration [62]. High enzymatic expression of HGF/c-Met also leads to the growth of different types of solid tumors in humans [63]. Inhibition of the high enzymatic activity of c-Met protein leads to inhibition of various cancer cell lines [13]. A large number of small molecules have been mentioned recently in the inhibition of c-Met enzymatic activity. However, the FDA has approved only one molecule as the lead anti-tumor drug in the c-Met inhibitor class, called Crizotinib (PF-02341066) [13]. The crystalline structure of c-Met (code PDB: 2WGJ) is obtained from the RCSB protein database [64], The 2WGJ crystal complex is formed by the co-crystallized ligand of Crizotinib bound to the c-Met receptor.
Firstly, before performing the molecular docking, the c-Met protein is prepared by removing water molecules, the associated ligand (crizotinib) and all non-protein elements. Then, polar hydrogen atoms are added to the c-Met receptor structure. Next, the preparation of ligands that will be docked to c-Met consists of adding hydrogen atoms to these ligands and optimizing their structure. The site of binding is defined as the volume occupied by the co-crystallized ligand Crizotinib in the c-Met receptor pocket with a radius of 5 Å.
The protein and ligands as well as the ligand entry site into the c-Met protein pocket are prepared in the present work by using Discovery Studio 2016 software [65]. Also, AutoDock software (ADT) MGLTools 1.5.6 packages [66] are used in the re-docking of the Crizotinib ligand with the c-Met receptor and in the docking of molecules (5, 16, and 46) with c-Met receptor. The 3D grid is constructed using the AUTOGRID algorithm which determines the box grid where the total binding energy of the ligands with the receptor are measured [67]. The grid is defined for x = 60, y = 60, z = 60 with a distance of 0.375 Å between the grid points. We then identified the coordinates x = 21.70 Å, y = 83.73 Å, z = 4.28 Å as the docking site of the selected ligands in the c-Met protein pocket. Next, we dock the ligands with the c-Met protein using Lamarckian Genetic Algorithms (LGA) to obtain the best molecular docking match [67].
This work explores molecular docking for two purposes. The first one consists in displaying the ligand visualization profiles in the c-Met receptor pocket, and comparing the realized binding energies between the ligands and the c-Met receptor. The second aim is the identification of the most important active sites of the c-Met protein, which are responsible for its enzymatic activity at the source of cancer, and to analyze the interactions that occur between the docked molecules and the identified active sites. Next, the candidate molecules for inhibition of c-Met enzyme activity are selected and their docking is studied. This is achieved based on the results of the drug-like evaluation of the compounds that show high biological activity predicted by the QSAR models. The binding modes of Crizotinib inhibitor with c-Met receptor and the active sites with which Crizotinib inhibitor interacts are identified according to the study reported by J. Jean Cui et al [13]. The active sites interacted with the Crizotinib are used as reference to predict the inhibition of c-Met enzymatic activity.
To validate the molecular docking procedure, we re-docked the crystallized ligand of Crizotinib to the c-Met receptor to determine the predicted binding energy between Crizotinib and the c-Met receptor, as well as to predict the reference active sites involved in the inhibition of c-Met activity. To ensure that the docking procedure is acceptable and valid, the range of the RMSD must be less than 2 Å according to Y. Westermaier et al. [68].
3. Results and discussion
After performing the PCA analyses, the following descriptors are selected: EVDW, LogP, αe, Pc, MW, MV, and ɳ as inputs for developing the QSAR model through the MLR technique. The seven aforementioned descriptors are selected among fifteen ones based on the correlation coefficient values. In fact, the descriptors of lowest correlation coefficients between them are selected as shown in the correlation matrix presented in Table 4. Then, the values of these descriptors are attributed to all the 48 molecules studied in the form of a matrix of 7 columns and 48 rows. Then, the database obtained is divided into two sets (training and test). This division is done using the K-means technique. The outcomes of this division are presented in Table 5. From these results, the following molecules (2, 6, 8, 14, 22, 23, 24, 26, 38 and 42) are selected for the test set, and the next molecules (1, 3, 4, 5, 7, 9, 10, 11, 12, 13, 15, 16, 17, 18, 19, 20, 21, 25, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 39, 40, 41, 43, 44, 45, 46, 47, and 48) are selected for the training set.
Table 4.
Matrix of the correlation between different descriptors obtained.
| 1 | ||||||||||||||||
| -0.04 | 1 | |||||||||||||||
| 0.23 | 0.71 | 1 | ||||||||||||||
| 0.05 | 0.81 | 0.72 | 1 | |||||||||||||
| 0.01 | 0.85 | 0.57 | 0.83 | 1 | ||||||||||||
| 0.04 | 0.94 | 0.66 | 0.76 | 0.97 | 1 | |||||||||||
| 0.06 | 0.82 | 0.64 | 0.77 | 0.92 | 0.92 | 1 | ||||||||||
| 0.17 | 0.87 | 0.68 | 0.88 | 0.82 | 0.96 | 0.96 | 1 | |||||||||
| 0.00 | 0.81 | 0.73 | 0.87 | 0.81 | 0.95 | 0.96 | 0.95 | 1 | ||||||||
| 0.04 | -0.38 | -0.45 | -0.39 | -0.32 | -0.29 | -0.30 | -0.32 | -0.36 | 1 | |||||||
| 0.19 | 0.03 | 0.06 | -0.16 | -0.04 | -0.15 | -0.33 | -0.19 | -0.22 | -0.47 | 1 | ||||||
| -0.22 | 0.33 | 0.36 | 0.54 | 0.36 | 0.43 | 0.62 | 0.50 | 0.56 | -0.94 | -0.47 | 1 | |||||
| -0.21 | -0.01 | -0.04 | 0.20 | 0.07 | 0.18 | 0.37 | 0.23 | 0.25 | 0.39 | -0.99 | 0.55 | 1 | ||||
| 0.24 | -0.19 | -0.19 | -0.43 | -0.25 | -0.35 | -0.57 | -0.42 | -0.47 | 0.11 | 0.81 | -0.92 | -0.86 | 1 | |||
| -0.24 | 0.19 | 0.19 | 0.43 | 0.25 | 0.35 | 0.57 | 0.42 | 0.47 | -0.11 | -0.81 | 0.92 | 0.86 | -1 | 1 | ||
| 0.23 | -0.08 | -0.07 | -0.32 | -0.16 | -0.27 | -0.48 | -0.33 | -0.37 | -0.15 | 0.94 | -0.741 | -0.96 | 0.96 | -0.96 | 1 |
Table 5.
Classification k-means.
| k-means division results | |
|---|---|
| 1 | 1 2 3 4 7 15 16 17 18 19 20 39 44 45 |
| 2 | 5 6 13 21 25 |
| 3 | 8 9 10 11 27 28 29 30 31 32 33 |
| 4 | 14 12 40 41 46 |
| 5 | 22 |
| 6 | 23 34 37 |
| 7 | 24 35 36 |
| 8 | 26 |
| 9 | 38 43 48 |
| 10 | 42 47 |
3.1. Multiple linear regression
The resulting QSAR model via the MLR technique is given by Eq. (7) below.
| (7) |
where
From Eq. (7) it is clear that the seven selected descriptors by the PCA technique are linearly correlated with the values of the biological inhibitory activity of c-Met (pIC50). We evaluate the performance of the QSAR model obtained by the MLR technique by the values of the following parameters: R2, F, MSE, P-value and . The higher value of the coefficient of determination (R2 = 0.81), the lower value of the mean square error (MSE = 0.06) and the high value of the statistical confidence degree (F = 18.23), indicate that the QSAR model shown in Eq. (7) is statistically acceptable. In addition, the achieved p-value that is less than 0.05 (Pr < 0.0001) indicates that the QSAR model equation is statistically significant with level greater than 95%. In addition, the value of the cross-validation correlation coefficient (), greater than 0.5, indicates the accuracy of the obtained QSAR model through MLR technique. The value of less than R2 value indicates the fragility and weakness of the model when excluding any element of the training set. Figure 4 shows the relationship between the observed activity values and the predicted ones of (pIC50). The latter are obtained by QSAR model based on the MLR technique for the molecules in both test and the training sets.
Figure 4.

Correlations between the observed activity values and the predicted ones via the MLR model.
From Figure 4, we notice that the distribution of observed and predicted pIC50 values are significantly correlated, which is due to the low obtained MSE value. Thus, it is clear that the experimentally obtained values and the predicted ones from the QSAR model are correlated. Therefore, it is apparent that the seven descriptors in Eq. (7) show a strong linear correlation with the biological activity of pIC50 that inhibits the enzymatic activity of the carcinogenic protein c-Met. In order to improve the relationship between the predicted activities obtained by the developed QSAR model via the MLR technique and the seven molecular descriptors, new QSAR models are developed using two different nonlinear techniques, namely the MNLR and ANN techniques. The following descriptors: EVDW, LogP, αe, Pc, MW, MV and ɳ are used as input parameters in these two techniques.
3.2. Multiple nonlinear regression (MNLR)
The nonlinear QSAR model obtained by the MNLR technique is presented in Eq. (8) below.
| (8) |
where
From the following performance parameters of the obtained nonlinear QSAR model: (R = 0.91), (R2 = 0.82) and (MSE = 0.07), it is clear that this model is statistically acceptable. Similarly, the value of the cross-validation coefficient () indicates that the nonlinear model is internally validated, and that the efficiency and reliability of this model is related to the contribution of all the elements in the training set (N = 38) to the construction of this model.
The uniform distribution of the observed experimental pIC50 values against the predicted ones obtained by the MNLR technique (Figure 5) confirms the greater efficiency of the developed QSAR model.
Figure 5.

Correlations between the observed activity values and the predicted activity values via the MNLR model.
3.3. Artificial neural networks (ANN)
When developing a QSAR model by using the ANN technique, the following architecture 7-4-1 with is used. With a value of it is apparent that the number 4 in the hidden layer is proportional to the number of descriptors 7 in the input layer in order to predict the pIC50 values expressed as 1 in the output layer. The developed QSAR model via the ANN technique shows a high value of the determination coefficient (R2 = 0.84) and a low value of the mean square error (MSE = 0.04), as well as the value of the cross-validation coefficient () is lower than the R2 value. These results confirm the efficiency of the QSAR model in the prediction of the anticancer biological activity of the studied molecules. Therefore, the selection of the seven descriptors EVDW, LogP, αe, Pc, MW, MV and ɳ in predicting pIC50 values is a successful. From Figure 6, the even distribution of candidate pIC50 values in the training and the test sets ensures that the pIC50 values obtained by the ANN model predictions are very close to the experimentally observed values.
Figure 6.

Correlations between the observed activity values and the predicted activity values via the ANN model.
We present in Table 6 the pIC50 values predicted by the MLR, MNLR and ANN models that are developed in the present study. The three QSAR models developed are successfully internally validated. In order to test the accuracy of the predictive power of obtained QSAR models, we perform an external validation. In the following paragraph, we present the results of the performed test.
Table 6.
The observed and predicted values of anticancer biological activities by the QSAR models developed based on the training set and testing set.
| Training Set | |||||||
|---|---|---|---|---|---|---|---|
| N |
|
|
|
||||
| 1 | 8.866 | 8.97 | 9.895 | 8.84 | 8.103 | 8.76 | 8.97 |
| 3 | 8.279 | 8.38 | 8.430 | 8.37 | 8.414 | 8.29 | 8.61 |
| 4 | 8.277 | 8.44 | 8.477 | 8.43 | 8.495 | 8.40 | 8.65 |
| 5 | 9.495 | 9.27 | 9.220 | 9.28 | 9.233 | 9.33 | 9.35 |
| 7 | 8.326 | 8.80 | 8.990 | 8.86 | 9.031 | 8.73 | 8.72 |
| 9 | 10.097 | 9.66 | 9.537 | 9.72 | 9.521 | 9.66 | 9.37 |
| 10 | 8.903 | 9.01 | 9.051 | 9.10 | 9.190 | 8.90 | 8.68 |
| 11 | 8.442 | 8.66 | 8.751 | 8.73 | 8.802 | 8.66 | 8.66 |
| 12 | 9.377 | 9.01 | 8.879 | 9.15 | 9.042 | 9.19 | 9.08 |
| 13 | 8.883 | 9.08 | 9.232 | 8.81 | 8.826 | 8.89 | 8.94 |
| 15 | 8.204 | 8.36 | 8.439 | 8.44 | 8.564 | 8.31 | 8.32 |
| 16 | 9.585 | 9.25 | 9.114 | 9.31 | 9.144 | 9.36 | 9.20 |
| 17 | 8.618 | 8.45 | 8.413 | 8.49 | 8.438 | 8.33 | 8.47 |
| 18 | 8.080 | 8.29 | 8.374 | 8.40 | 8.527 | 8.49 | 8.42 |
| 19 | 9.319 | 9.15 | 9.111 | 9.22 | 9.156 | 9.39 | 9.17 |
| 20 | 8.618 | 8.49 | 8.472 | 8.51 | 8.477 | 8.51 | 8.67 |
| 21 | 8.281 | 7.90 | 7.825 | 7.91 | 7.786 | 8.13 | 8.11 |
| 25 | 8.936 | 8.81 | 8.816 | 8.80 | 8.772 | 8.73 | 8.87 |
| 27 | 9.553 | 9.71 | 9.778 | 9.69 | 9.785 | 9.66 | 9.62 |
| 28 | 8.860 | 8.98 | 9.023 | 9.00 | 9.038 | 8.82 | 8.83 |
| 29 | 8.372 | 8.08 | 8.057 | 8.11 | 8.055 | 8.15 | 8.12 |
| 30 | 8.077 | 8.17 | 8.212 | 8.09 | 8.093 | 8.16 | 8.12 |
| 31 | 8.879 | 8.99 | 9.034 | 9.01 | 9.035 | 8.84 | 8.84 |
| 32 | 8.417 | 8.25 | 8.230 | 8.30 | 8.275 | 8.16 | 8.19 |
| 33 | 8.269 | 8.24 | 8.253 | 8.34 | 8.361 | 8.22 | 8.41 |
| 34 | 8.967 | 9.10 | 9.132 | 9.19 | 9.248 | 9.06 | 9.35 |
| 35 | 8.253 | 8.34 | 8.372 | 8.38 | 8.404 | 8.27 | 8.45 |
| 36 | 8.733 | 8.44 | 8.412 | 8.50 | 8.433 | 8.27 | 8.59 |
| 37 | 8.138 | 8.29 | 8.341 | 8.28 | 8.297 | 8.31 | 8.47 |
| 39 | 8.987 | 9.17 | 9.221 | 9.19 | 9.222 | 9.21 | 9.21 |
| 40 | 9.553 | 9.64 | 9.702 | 9.65 | 9.688 | 9.64 | 9.38 |
| 41 | 8.470 | 8.51 | 8.544 | 8.45 | 8.434 | 8.34 | 8.53 |
| 43 | 8.203 | 8.22 | 8.240 | 8.25 | 8.259 | 8.36 | 8.34 |
| 44 | 9.086 | 8.51 | 8.452 | 8.54 | 8.426 | 8.62 | 8.67 |
| 45 | 9.244 | 9.10 | 9.101 | 9.14 | 9.114 | 9.25 | 9.15 |
| 46 | 9.495 | 9.39 | 9.399 | 9.44 | 9.428 | 9.54 | 9.34 |
| 47 | 8.194 | 8.45 | 8.507 | 8.43 | 8.511 | 8.39 | 8.47 |
| 48 |
8.682 |
8.74 |
8.775 |
8.71 |
8.717 |
8.68 |
8.77 |
|
Test set | |||||||
| N |
|
|
ANN |
||||
| 2 | 8.636 | 7.90 | 7.96 | 8.18 | |||
| 6 | 8.900 | 8.48 | 8.47 | 8.33 | |||
| 8 | 8.107 | 8.92 | 8.86 | 8.94 | |||
| 14 | 9.638 | 9.92 | 9.55 | 9.42 | |||
| 22 | 8.457 | 7.99 | 7.90 | 8.15 | |||
| 23 | 9.337 | 8.81 | 8.80 | 8.74 | |||
| 24 | 8.487 | 8.07 | 8.10 | 8.14 | |||
| 26 | 8.458 | 8.90 | 8.78 | 8.84 | |||
| 38 | 8.553 | 8.77 | 8.72 | 8.74 | |||
| 42 | 8.848 | 8.97 | 8.88 | 8.82 | |||
3.4. External validation
We perform the external validation test by evaluating the power of the QSAR models to predict the pIC50 activity values of the molecules from the test set, by computing the coefficient of correlation with represents an important criterion in evaluating the performance of externally validated models in predicting the activities of molecules not involved in the development of the models. The achieved values of are 0.67, 0.69 and 0.64 for the MLR, MNLR and ANN models, respectively. The values of the three models are close to each other, and these values are also greater than 0.5. Hence, the external validation of the QSAR models ensures the strong power of these models to predict pIC50 values.
3.5. Performance comparison of QSAR models
In Table 7, we present a summary of the results of internal and external validation on the QSAR models obtained in this study.
Table 7.
Comparison of MLR, MNLR and ANN models performance.
| coefficients | Training set |
Test set |
|||||
|---|---|---|---|---|---|---|---|
| MSE | |||||||
| MLR | 0.90 | 0.81 | 0.62 | 0.06 | 0.82 | 0.67 | 0.07 |
| MNLR | 0.91 | 0.82 | 0.64 | 0.07 | 0.83 | 0.69 | 0.13 |
| ANN | 0.92 | 0.84 | 0.78 | 0.062 | 0.80 | 0.64 | 0.11 |
The comparison between the performance of MLR, MNLR and ANN models in terms of the coefficients (R, R2, MSE) indicates that all the developed models are statistically significant, and show high internal and external predictive ability.
Hence, MLR, MNLR, and ANN models are capable to correlate a strong quantitative relationship between molecular descriptors (LogP, Polarizability, Parachor, Molecular Weight, Molecular Volume and Chemical Hardness) and biological activity (pIC50) that inhibits the enzymatic activity of c-Met protein. Thus, the QSAR models developed via MLR, MNLR and ANN techniques can be exploited to predict the activity values of other molecules that can be designed by making modifications to the structure of 4,5,6,7-Tetrahydrobenzo[D]-Thiazol-2-Yl derivatives in order to obtain new molecules with stronger biological activities than those observed. In this work, instead of designing new molecules and predicting their activities, we select the most apt molecules to inhibit the enzymatic activity of the c-Met protein among the series of 4,5,6,7-tetrahydrobenzo[D]-Thiazol-2-Yl derivatives that we study. For this selection, we rely on the high activity values obtained by the predictions of the QSAR models. Before selecting the molecules with the highest predicted biological activity, we perform two essential tests to validate the efficiency of the predicted pIC50 values. The performed tests are: Y-randomization and applicability domain tests. Both tests are used to avoid the selection of one or more molecules whose activities are not correctly predicted in this study.
3.6. Y-randomization test
In order to ensure the quality of the QSAR models that select the candidate molecules to inhibit c-Met activity, a Y-randomization test is performed on the original QSAR model that is obtained by the MLR technique. This test is performed to reduce the possibility of randomly obtaining a strong correlation between the seven descriptors in (Eq. 7) and the biological inhibitory activity of c-Met.
We perform the Y-randomization test by randomly distributing the Y values (pIC50) fifty times without changing the seven X descriptors. We perform the Y randomization test by randomly distributing the Y values (pIC50) fifty times without changing the seven X descriptors. The random distribution of Y values to X descriptors enabled the generation of 50 new QSAR models. Each new model has new values of (). The results of the Y-randomization test for the first 50 iterations are presented in Table 8. Through these results, we find that the values of , , and obtained by the randomly constructed models are lower than the values of obtained by the original model. These results confirm that the original model obtained in Eq. (7) is robust and that the correlation between the seven descriptors and biological activity is not due to chance. Thus, we can confirm that the pIC50 values predicted by the QSAR models based on the seven descriptors presented in the original model (Eq. 7) are not due to chance.
Table 8.
Y-Randomization test results.
| Model | |||
|---|---|---|---|
| Original | 0.90 | 0.81 | 0.62 |
| Random 1 | 0.42 | 0.18 | -1.59 |
| Random 2 | 0.38 | 0.14 | -0.55 |
| Random 3 | 0.33 | 0.11 | -0.33 |
| Random 4 | 0.34 | 0.12 | -0.54 |
| Random 5 | 0.34 | 0.11 | -0.55 |
| Random 6 | 0.40 | 0.16 | -0.31 |
| Random 7 | 0.38 | 0.14 | -0.31 |
| Random 8 | 0.28 | 0.08 | -0.47 |
| Random 9 | 0.27 | 0.07 | -0.74 |
| Random 10 | 0.49 | 0.24 | -0.21 |
| Random 11 | 0.40 | 0.16 | -0.40 |
| Random 12 | 0.59 | 0.34 | -0.21 |
| Random 13 | 0.29 | 0.09 | -0.33 |
| Random 14 | 0.46 | 0.21 | -0.57 |
| Random 15 | 0.43 | 0.19 | -0.44 |
| Random 16 | 0.57 | 0.32 | -0.37 |
| Random 17 | 0.36 | 0.13 | -0.77 |
| Random 18 | 0.43 | 0.18 | -0.24 |
| Random 19 | 0.55 | 0.30 | -0.24 |
| Random 20 | 0.44 | 0.20 | -0.40 |
| Random 21 | 0.58 | 0.33 | -0.56 |
| Random 22 | 0.41 | 0.17 | -0.80 |
| Random 23 | 0.38 | 0.14 | -0.36 |
| Random 24 | 0.47 | 0.22 | -0.37 |
| Random 25 | 0.42 | 0.17 | -0.34 |
| Random 26 | 0.44 | 0.20 | -0.30 |
| Random 27 | 0.46 | 0.21 | -0.21 |
| Random 28 | 0.44 | 0.20 | -1.10 |
| Random 29 | 0.40 | 0.16 | -0.90 |
| Random 30 | 0.41 | 0.17 | -0.51 |
| Random 31 | 0.24 | 0.06 | -0.58 |
| Random 32 | 0.36 | 0.13 | -0.44 |
| Random 33 | 0.41 | 0.17 | -0.41 |
| Random 34 | 0.31 | 0.09 | -1.28 |
| Random 35 | 0.26 | 0.07 | -0.57 |
| Random 36 | 0.48 | 0.23 | -0.25 |
| Random 37 | 0.48 | 0.23 | -0.28 |
| Random 38 | 0.59 | 0.35 | 0.03 |
| Random 39 | 0.52 | 0.27 | -1.79 |
| Random 40 | 0.39 | 0.15 | -0.56 |
| Random 41 | 0.58 | 0.33 | -0.05 |
| Random 42 | 0.22 | 0.05 | -1.05 |
| Random 43 | 0.43 | 0.19 | -0.33 |
| Random 44 | 0.64 | 0.41 | 0.06 |
| Random 45 | 0.36 | 0.13 | -2.17 |
| Random 46 | 0.21 | 0.04 | -0.47 |
| Random 47 | 0.59 | 0.35 | -0.04 |
| Random 48 | 0.33 | 0.11 | -0.72 |
| Random 49 | 0.55 | 0.31 | -0.12 |
| Random 50 | 0.58 | 0.34 | -0.05 |
3.7. Applicability domain (AD)
In the present test we determine the applicability domain of the original QSAR model obtained by MLR technique by analyzing the relationship between residual values and the leverage effect. Figure 7 shows the applicability domain obtained by the Williams diagram. The leverage effect threshold value is h∗ = 0.52 and the distribution of normalized residual values and leverage level values were calculated and determined in Figure 7 of the Williams plot. The pIC50 values predicted by the QSAR model are correct and valid only for compounds within the applicability domain located to the left of the leverage threshold h∗ = 0.52, while molecules outside the applicability domain are not predicted correctly.
Figure 7.

William plot for normalized residuals and leverage of the original QSAR model obtained by using MLR technique.
In Figure 7, we note that the molecules (2 and 8) in the test set have a standard deviation outside the ± x range (x = 2.5), but even so, these molecules remain in the applicability domain adjacent to the other molecules. We note that only one molecule is outside the applicability domain and that is molecule 1 of the training set. It could be that the activity value of this compound was not correctly predicted due to the availability of incorrect experimental data for this molecule. For this reason, we will remove molecule 1 from the list of molecules we propose to use as novel inhibitors of c-Met enzymatic activity, and also exclude this molecule from molecular modeling for the rest of this study. Thus, we use only the molecules within the applicability domain situated to the left of the h∗ threshold as candidate group members for c-Met inhibition. From these molecules only those with the highest predicted pIC50 values will be selected.
3.8. Selection of new anticancer molecules for pharmaceutical applications
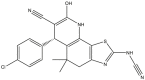
Based on the predicted pIC50 values shown in Table 9 in the attached file, as well as the information obtained by the applicability domain, we selected seven compounds that presented high pIC50 values in terms of c-Met enzymatic inhibitory activity. All of these seven molecules present higher predicted pIC50 values than those experimentally observed in comparison with the drug Crizotinib. We report the predicted pIC50 values for the selected molecules, as well as the experimental pIC50 value for the Crizotinib drug in Table 9.
Table 9.
Predicted values of pIC50 obtained by the QSAR models developed and observed pIC50 for Crizotinib.
| Compounds | pred (pIC50) c-Met |
IC50 (nM) c-Met |
|||||
|---|---|---|---|---|---|---|---|
| MLR | MNLR | ANN | MLR | MNLR | ANN | ||
| 5 | 9.27 | 9.28 | 9.33 | 0.537 | 0.190 | 0.467 | |
| 9 | 9.66 | 9.72 | 9.66 | 0.212 | 0.281 | 0.218 | |
| 14 | 9.92 | 9.55 | 9.42 | 0.120 | 0.490 | 0.380 | |
| 16 | 9.25 | 9.31 | 9.36 | 0.562 | 0.204 | 0.436 | |
| 27 | 9.71 | 9.69 | 9.66 | 0.194 | 0.223 | 0.218 | |
| 40 | 9.64 | 9.65 | 9.64 | 0.230 | 0.363 | 0.230 | |
| 46 |
9.39 |
9.44 |
9.54 |
0.407 |
0.190 |
0.288 |
|
| Crizotinib | |||||||
| Observed (pIC50 = 8.09) | Observed (IC50 =8 nM) | ||||||
The prediction results of pIC50 values presented in Table 9 that are obtained by the three models MLR, MNLR and ANN show that these values are very close to each other. Therefore, it is difficult to favor one molecule over another and select it as the best inhibitor of c-Met enzymatic activity based only on the predicted pIC50 values. Therefore, to select the most candidate molecules for inhibition of c-Met kinase enzymatic activity among the seven molecules, we perform an in-silico study in which we predict the drug-like ADME properties of these molecules. Based on the results of the evaluation of the ADME properties of each molecule, we will select only the molecules that have drug-like properties.
3.9. Evaluation of drug-likeness properties
The first aim of this study is to predict the drug-like properties of the seven molecules (5, 9, 14, 16, 27, 40, 46) in order to describe the biological activity of these compounds and to investigate their beneficial or toxic effects on the organism if they are used in pharmaceutical applications. The second aim is to identify molecules with drug-like properties in order to study their docking with the c-Met receptor. In Table 10, we present the evaluation results of the drug-like properties obtained by using the SwissADME online server.
Table 10.
The properties ADME of the seven best compounds selected.
| Entry |
ABS |
TPSA (A2) |
n-ROTB |
MW |
LogP |
n-OHN acceptors |
n-OHNH donors |
Lipinski's violations |
Veber Violations |
Egan Violation |
S.A |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Rule | - | - | - | <500 | ≤5 | <10 | <5 | ≤1 | ≤1 | ≤1 | 0 < S.A<10 |
| 5 | High | 106.38 | 3 | 358.85 | 3.46 | 4 | 1 | 0 | 0 | 0 | 3.47 |
| 9 | Low | 136.89 | 3 | 494.03 | 5.61 | 4 | 1 | 1 | 1 | 1 | 4.18 |
| 14 | Low | 115.16 | 3 | 490.04 | 7.51 | 3 | 0 | 1 | 1 | 1 | 4.14 |
| 16 | High | 81.99 | 2 | 356.87 | 4.32 | 3 | 0 | 0 | 0 | 0 | 3.46 |
| 27 | Low | 99.87 | 3 | 495.42 | 5.71 | 4 | 1 | 1 | 1 | 1 | 3.97 |
| 40 | Low | 118.17 | 2 | 409.89 | 2.37 | 5 | 1 | 0 | 0 | 0 | 4.26 |
| 46 | High | 120.97 | 2 | 408.90 | 2.96 | 4 | 2 | 0 | 0 | 0 | 4.32 |
Abbreviations: ABS: Absorption, TPSA: Topological Polar Surface Area, n-ROTB: Number of Rotatable Bonds, MW: Molecular Weight, Log P: logarithm of partition coefficient of compound between n-octanol and water, n-ONHN acceptors: Number of hydrogen bond acceptors, n-OHNH donors: Number of hydrogen bonds donors, S.A: Synthetic accessibility.
The results presented in Table 10 indicate that the compounds (5, 16, 40 and 46) meet all the rules of Lipinski, Veber and Egan, which indicates the absence of oral bioavailability problems for these compounds. Where the compounds 5, 16 and 46 demonstrated a high absorption capacity. In contrast compound 40 showed a low absorption capacity. While it is noted that compounds 9, 14 and 27 showed a deviation on the high lipid affinity level (LogP >5) in Lipinski, Veber and Egan rules. This deviation results in higher metabolic turnover, lower solubility and poor oral absorption. Therefore, we will not consider the compounds (9, 14 and 27) as candidates for pharmacological use. Based on these results, we can select the compounds (5, 16 and 46) that do not cause any oral bioavailability problems and have drug-like properties. However, for the TPSA when it is less than 140 Å2 and the number of rotatable bonds is less than 10, the compounds becomes more flexible and is more able to interact with the target receptor [69]. We note from Table 10 that the TPSA values for all seven compounds are less than 140 Å2 and also have the n-ROTB value less than 10, so all seven compounds can interact flexibly with the c-Met receptor. We also predict the synthetic accessibility of the compounds presented in Table 10. The evaluation of the synthetic accessibility of the molecule ranges from 1 (easy to synthesize) to 10 (very difficult to synthesize) [70]. We find that the S.A values for the three selected drug-like molecules (5, 16 and 46) are between 3.46 and 4.32, which means that the S.A values are far from 10 and close to 1, therefore the possibility to synthesize these molecules is very easy. Based on these results, we select the three compounds (5, 16 and 46) as the most potent candidates to inhibit the c-Met receptor and the best in terms of bioavailability in the human body, also in terms of the flexibility of these molecules when interacting with the c-Met receptor kinase. Moreover, the selected molecules are easy synthesis. We illustrate in Figure 8 the 3D structures of the three selected compounds based on the predicted drug-likeness properties.
Figure 8.

3D structures of the selected molecules by evaluating the drug-likeness properties.
In the next step, we test the docking of the three selected molecules with the c-Met receptor kinase to determine the most stable molecule in the c-Met receptor pocket. To do this, we compare the number and types of interactions that take place between the ligands (L5, L16, L46) and the active sites of the c-Met receptor, also we compare the binding energies of the ligands with the same receptor. Then, we select the most appropriate ligand among the three ligands to replace the inhibitor Crizotinib to treat cancer by inhibiting the enzymatic activity of c-Met.
3.10. Molecular docking
Before docking the ligands (L5, L16 and L46) with the c-Met receptor, a first step is to identify the active sites with which the co-crystallized ligand (Crizotinib) interacted within the c-Met receptor pocket. In this step, we visualize the structure of the 2WGJ crystal complex to identify the active sites. Figure 9 shows the most important active sites of the c-Met protein to which the Crizotinib inhibitor was bound (Met1160A, Pro1158A and Tyr1230A), which were reported by 2D PoseView interaction diagrams via the ProteinsPlus online server [71].
Figure 9.

2D diagram of the interactions between the co-crystallized ligand (Crizotinib) and the active sites in the 2WGJ crystal complex.
Figure 10 shows the active sites (Tyr1159, Met1160, Ala1108, Pro1158, Met1211, Val1092, Leu1157, Ala1226, Ala1221, Tyr1230, Asp1222, Leu1140 and Ile1084) in the c-Met protein with which the Crizotinib inhibitor co-crystallized was reacted. We find these interactions by analyzing 2D and 3D visualizations of the 2WGJ complex using Discovery Studio 2016 software.
Figure 10.

(a) 2D and (b) 3D interactions of the co-crystallized ligand (Crizotinib) with the active sites of C-Met in the complex crystal form 2WGJ.
The 2D and 3D visualizations in Figure 10 of the types of interactions that have occurred between the active sites in the c-Met protein and the achieved co-crystallized ligand (Crizotinib) by analyzing the complex (2WGJ) via the Discovery Studio 2016 program are much clearer than the 2D visualization in Figure 9 that is obtained by using ProteinsPlus server. From these results, we consider the following active sites (Tyr1159, Met1160, Ala1108, Pro1158, Met1211, Val1092, Leu1157, Ala1226, Ala1221, Tyr1230, Asp1222, Leu1140, and Ile1084) shown in Figure 10 as the most important sites that conduct the inhibition of c- Met enzyme activity.
In the next step, we perform a re-docking of the Crizotinib with the c-Met receptor to confirm and select the most important reference sites active in the inhibition of c-Met activity, and to verify the ability of the AutoDock (ADT) software for implementing the protocol of molecular docking. Figure 11 shows the superimposed view of the conformation between the docked ligand and the native crystalline ligand in the c-Met receptor pocket, such that the RMSD value between the ligands is 1.428 Å.
Figure 11.

(a) and (b): Re-docking pose with RMSD value it 1.428 Å (Green = Original, Brown = Docked).
After performing the molecular re-docking, we observe in Figure 11 that there is an almost perfect superposition between the original and re-docked ligand in the c-Met receptor pocket. Furthermore, the value of RMSD (1.428 Å) less than 2 Å, which indicates the efficiency of the AutoDock software (ADT) in achieving excellent molecular docking, and therefore we perform the docking test of the selected ligands (5, 16 and 46) inside the c-Met receptor pocket depending on the program AutoDock (ADT) based on the LGA. Also, it appears from Figure 12 that the Crizotinib ligand interacts with the active sites (Ala1108, Met1160, Pro1158, Ala1226, ALa1221, Leu1159, Leu1140, Asp1222, Tyr1230, Val1092, Met1211 and Ile1084) It can be seen from the re-docking results that the interactions of Crizotinib occurred with the same active sites identified by visualizing the docking position in the crystal complex (2WGJ) in Figure 10.
Figure 12.

2D interactions of the Crizotinib with the active sites in the C-Met receptor predicted by re-docking.
Through the identification of the most active sites (Ala1108, Met1160, Pro1158, Ala226, ALa1221, Leu1159, Leu1140, Asp1222, Tyr1230, Val1092, Met1211 and Ile1084) involved in the inhibition of the enzymatic activity of the c-Met receptor by Crizotinib inhibitor. We will take these sites as references that explain the enzymatic activity of c-Met, and thus any interaction between the ligands (5, 16 and 46) with any of the reference sites can cause an inhibitory response of c-Met. Inhibition of c-Met results in the blockage of cancer cell growth. Also, the values of binding energy that occur between the ligands (L5, L16 and L46) will be used to determine the most stable ligand in the c-Met receptor pocket.
We present the visualizations of the interaction profiles obtained by molecular docking of the ligands 5, 16, and 46 with the c-Met receptor in Figure 13, as well as the binding energy values that are obtained between these ligands and the c-Met receptor. Based on the molecular docking results shown in Figure 13, the ligands (5, 16, and 46) are docked into the c-Met receptor pocket through the interactions with the following residues: Tyr 1159, Met1160, Met1211, Tyr1230, Tyr1159, Ile1084, Val1092, Ala1108, Ala1221, Gly1163 and His1162.
Figure 13.

2D and 3D docking poses interactions between (a): ligand 5, (b): ligand 16 and (c): ligand 46 with C-Met active sites.
Table 11 summarizes the results of the interactions predictions between the ligands 5, 16, 46 and Lref with active sites in the c-Met receptor as well as the binding energies of these ligands with the same receptor.
Table 11.
Docking results of ligands L5, L16, L46 and ligand reference Lref (Crizotinib) at receptor c-Met sites.
| Ligands | Complex | Binding Energy (kcal/mol) | Hydrogen-Binding Interaction |
Hydrophobic Interaction |
|---|---|---|---|---|
| Lref | Lref-A | -7.02 | Met1160, Pro1158, Asp1222, Ile1084 | Ala1108, Ala1221, Ala1226, Leu1157, Leu1140, Tyr1230, Val1092, Met1211 |
| L5 | X-A | -7.08 | Tyr1159, Met1160, Ile1084 | Met1211, Tyr1230 |
| L16 | X–B | -8.27 | Gly1163, Met1160, Tyr1159 | Ile1084, Ala1108, Val1092, Tyr1230 |
| L46 | X–C | -7.40 | Met1160, Tyr1159, Lys1161, His1162 | Ile1084, His1162 |
From the complex X-A we can notice that the ligand 5, binds to four reference sites important in the inhibition of the enzymatic activity c-Met kinase, these sites are the following active residues: Met1160 (3.39 Ǻ), Ile1084 (2.24 Ǻ), Met1211 (3.97 Ǻ) and Tyr1230 (4.80 Ǻ) the interaction of ligand 5 with these residues is as follows: hydrogen bonding with Ile1084 and Met1160, by Pi-Sigma interaction with Met1211, and by T-shaped Pi-Pi and Pi-alkyl interactions with Tyr1230. Ligand 5 also binds to residue Tyr1159 (2.60 Ǻ), which we did not identify as an important site in the inhibition of c-Met enzymatic activity (see Figure 12). We further note that the value of the binding energy of ligand 5 with the c-Met receptor is equal to -7.08 kcal/mol.
From the complex X–B in Figure 13 we can observe that the ligand 16, binds to five important sites of reference which are important in the inhibition of c-Met kinase enzyme activity, these sites are: Met1160 (2.94), Ile1084 (4.12), Ile1084 (4.5 Ǻ), Ala1108 (3.63 Ǻ) and Val1092 (4.13 Ǻ), Val1092 (5.23 Ǻ) and Tyr1230 (5.05 Ǻ), Tyr1230 (5.35 Ǻ). The binding of ligand 16 to these residues is performed as follows: a hydrogen bonding interaction with Met1160, the interactions Pi-Pi Stacked and Pi-T-Shaped with Tyr1230, and the reactions Pi-alkyl with Ile1084, Ala1108 and Val1092. Ligand 16 also binds to residues Gly1163 (3.28 Ǻ) and Tyr1159 (4.01 Ǻ), we did not report these amino acids as important sites in the inhibition of c-Met enzyme activity, as previously shown in Figure 12. We also note that the binding energy value of ligand 16 with the c-Met receptor is equal -8.27 Kcal/mol.
From the X–C complex, it is remarkable that the ligand 46, binds only with two reference sites important in the inhibition of c-Met kinase activity, these sites are: Met1160 (1.85 Ǻ), Ile1084 (5.24 Ǻ). The binding of ligand 46 to these residues is performed as follows: hydrogen bonding interaction with Met1160, Pi-alkyl interaction with Ile1084. Ligand 46 also binds to residues, Tyr1159 (2.41 Ǻ), Lys1161 (2.04 Ǻ), His1162 (4.11 Ǻ) and His1162 (3.24 Ǻ), we have not reported these amino acids as important sites in the inhibition of c-Met enzyme activity, as previously shown in Figure 12. The binding energy value of ligand 46 with the c-Met receptor is equal -7.40 kcal/mol.
The comparison of the number of important sites involved in c-Met inhibition with which the ligands L5, L16, L46 interacted indicates that the ligand (L16) interacted with the greatest number of sites important (five sites) in c-Met inhibition compared to the number of sites with which L5 (four sites) and L46 (two sites) interacted. We also find that the binding energy of the ligand (L16) with the c-Met receptor (-8.27 kcal/mol) is less than the binding energies of the ligands L5 (-7.08 kcal/mol) and L46 (-7.4 kcal/mol) with c-Met, which means that the ligand L16 is more stable and more localized in the pocket of the c-Met receptor than the ligands L5 and L46. Thus, the X–B complex shows better stability than the complexes X-A and X–C. Based on the comparison of the molecular docking results, the number of important sites with which the ligand L16 is bound, as well as its most stable binding energy permits to select the compound L16 as the best candidate for the inhibition of the enzymatic activity of the c-Met receptor among the three ligands (L5, 16 and L46). Therefore, based on these results, we can explain the difference in the inhibition mode of c-Met enzymatic activity by the molecules (5, 16 and 46) due to the placement of the root and the orientation of the structure to identified active sites as important references in c-Met inhibition. Moreover, we can conclude that the location and the characteristics of the roots in the structure of the compounds (5, 16 and 46) can influence the biological activity pIC50. This influence being reflected in the number of bonds that can be created between the compound and the active sites of the future target and also in the binding energy that is produced between the compound and the target receptor. We confirm this hypothesis by the 3D visualization of the complex (X–B). In Figure 14, we notice that the phenyl ring level in ligand 16 is bound to the active site (5.35 Ǻ) Tyr1230 with a Pi-Pi Stacked interaction, and this reaction is identical to the interaction of the phenyl ring in the Crizontib ligand with the same active site Tyr1230 (3.92 Ǻ) in the Lref-A complex. In addition, the reaction is carried out by hydrogen bonding between the nitrogen atom of ligand 16 and the amino acid Met1160 (2.94 Ǻ). The same hydrogen bonding is carried out between the nitrogen atom of the Crizotinib molecule with the amino acid Met1160 (2.07 Ǻ).
Figure 14.

Comparison between 3D interactions of the complexes (a): X–B and (b): Lref-A.
In terms of binding energy values, the drug is most potent when it has the lowest binding energy value (more negative value) [72]. This binding results in the formation of more interactions between the receptor and the ligand due to more free energy being liberated. As result, the desired target is more accessible compared to ligands with lower negative values. By comparing the values of the binding energies of the ligands (L5, L16 and L46) with the c-Met receptor obtained by the molecular docking protocol, we can interpret the lowest experimental IC50 value for the molecule 16 (0.26 nM) compared to the experimental IC50 values of the molecules 5 (0.32 nM) and 46 (0.32 nM). Therefore, ligand 16 has the lowest binding energy (-8.27 Kcal/mol) compared to ligands 5 (-7.08 Kcal/mol) and 46 (-7.40 kcal/mol). Thus, the low IC50 value of molecule 16 reflects the high activity of this molecule in the therapeutic use. Also, we note that the binding energy value of Crizotinib ligand (-7.04 Kcal/mol) is higher than the binding energy values of the ligand (5, 16 and 46), which confirms the stronger biological activity of these molecules in inhibiting the enzymatic activity of c-Met protein compared to Crizotinib molecule.
Based on the achieved results of the molecular docking predictions, it is clear that the structure of ligand 16 that is interacted with the largest number of active sites important in inhibiting the enzymatic activity of c-Met. In addition, ligand 16 is well inserted into the pocket of c-Met and produced better stability binding energy compared to L5 and L46 ligands. Hence, the structure of molecule 16 can be used to improve the inhibition of the enzymatic activity of c-Met protein. In addition, the proposal of other molecules based on the structure of molecule 16 can be achieved by identifying additional information about the properties of each region of the structure of compound 16. Also, we can modify the structure of molecule 16 and evaluating the effect of these modifications on the pIC50 values in order to design new molecules based on the performed modifications. For this purpose, it is preferable to combine the study of 2D-QSAR with molecular docking and 3D-QSAR. In a future work, we will combine these studies to design new molecules that can be added to the 4,5,6,7-Tetrahydrobenzo[D]-Thiazol-2-Yl derivatives and we will investigate their anticancer drug kinetics.
Based on the present study results, we confirm the selection of compound 16 as a novel candidate agent for pharmacological use in the treatment of cancerous tumors resulting from the activity of the c-Met kinase enzyme. In order to confirm the validity of using the molecule 16 in the treatment of cancer by inhibiting the enzymatic activity of the c-Met protein, we perform in the following section an evaluation of the pharmacokinetic parameters in silico of this molecule. Also, a comparison will be performed between the Crizotinib and compound 16 in terms of ADMET properties.
3.11. In silico pharmacokinetics ADMET predictions
Both compound 16 and Crizotinib are undergoing ADMET in the present in silico studies by using the pkCSM tool [73]. The letter is used to predict in silico ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) properties of the selected compound 16, as well as the properties of the Crizotinib compound that is previously used as a drug. The results of the ADMET properties prediction are computed and then presented in Table 12.
Table 12.
Predicted properties ADMET in silico for compounds 16 and Crizotinib.
| Properties |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Absorption | Distribution | Metabolism | Excretion | Toxicity | |||||||||
| models | Intestinal absorption (human) | VDss (human) | BBB permeability | CNS permeability | CYP | Total clearance | AMES toxicity | ||||||
| Substrate | Inhibitor | ||||||||||||
| 2D6 | 3A4 | 1A2 | 2C19 | 2C9 | 2D6 | 3A4 | |||||||
| Unity |
Numeric (%absorbed) |
Numeric (Log L kg−1) |
Numeric (Log BB) |
Numeric (Log PS) |
Categorical (yes/no) |
Numeric (log mL min−1 kg−1) |
Categorical (yes/no) |
||||||
| Predicted values | |||||||||||||
| Comp.16 | 94.70 | 0.474 | 0.396 | -1.438 | No | Yes | Yes | Yes | Yes | No | Yes | 0.113 | No |
| Crizotinib | 91.38 | 1.315 | -1.016 | -3.005 | No | Yes | No | Yes | No | No | Yes | 0.583 | No |
Based on the obtained results in Table 12, we can conclude that:
-
-
In terms of the percentage of absorption by the human intestines, a value less than 30% indicates that the absorption is low [74]. Compound 16 presented a absorption value higher than 94%, which guarantees a good absorption by the human intestine best than the Crizotinib compound (91.38%).
-
-
In terms of distribution indicators, the size of the distribution (VDss) is considered high if its value is greater than 0.45 [75]. The standard value for blood-brain barrier (BBB) permeability is good if its value is more than 0.3 and poor if LogBB < -1 [76]. For the CNS index, compounds with LogPS > -2 are considered capable of penetrating the CNS, whereas compounds with LogPS < -3 are considered incapable of penetrating the CNS [77]. The distribution indices reported by molecule 16 indicated a better distribution capacity than Crizotinib.
-
-
In terms of metabolism, cytochrome P450 (CYP) is an important enzyme for detoxification. CYP enzymes are present in all tissues of the body [70]. This enzyme oxidizes foreign microorganisms to facilitate their excretion. Many drugs are inhibited by cytochrome CYP, and some can also be activated by it. Inhibitors of this enzyme may affect the metabolism of the drug, and the drug may have a reverse effect [78]. Therefore, it is indispensable to evaluate the ability of compounds to inhibit cytochromes (CYP). Up to now, 17 categories of CYPs have been identified in humans. Although only CYP1, CYP2, CYP3 and CYP4 are responsible in the metabolism of drugs, Thus, only the types (1A2, 2C9, 2C19, 2D6 and 3A4) are responsible for biotransformation for more than 90% of drugs pass the first step of metabolism [79]. The two isoforms 2D6 and 3A4 are mainly responsible for drug metabolism [80]. A study performed by A. Puccini et al. [81] indicates that the metabolism of Crizotinib in the liver is affected by an increase in CYP3A4 enzyme activity, which inhibited Crizotinib activity. For this reason, we are relying on the evaluation the effect of the compound 16 on the CYP3A4 enzyme (inhibitor or substrate) in order to predict the metabolic effect of CYP3A4 on the activity of the compound 16 that is proposed to use as drug. From the obtained results of the molecule 16 properties metabolism, we can see that this molecule can be a CYP3A4 substrate as well as an inhibitor of CYP3A4. This indicates that the metabolism of the compound 16 as drug is acceptable, and therefore molecule 16 can successfully reach the therapeutic target before being oxidized and excreted.
-
-
The drug clearance index in excretion properties is important for determining drug dosage ratios to achieve stable drug concentrations [75]. Where clearance is done in the liver and excretion in the kidneys. Therefore, lower value of the clearance index indicates that the higher the persistence of drugs in the body. We evaluate the excretion property in this study to determine the level of stability of molecule 16 as a drug in the body before its excretion. The predictive values of this index showed that the total clearance index of molecule 16 is 0.113 that is lower than the total clearance index of crizotinib (0.583), and therefore molecule 16 may persist in the body better than Crizotinib, it can be explained that the stability of molecule 16 for a longer period in the body compared to Crizotinib led to an increase in the activity of molecule 16 in the inhibition of the enzyme c-Met at a dose lower than the dose used by the Crizotinib molecule.
-
-
In terms of the toxicity indicator, it is necessary to check whether the predicted compounds are non-toxic. The letter indicator is important in the selection of drugs. The AMES test is widely used to evaluate the toxicity of compounds [82]. Therefore, in this work, we evaluate the toxicity of the molecules (16 and Crizotinib) based on AMES test predictions. According to the study presented in [75], all compounds in the database are toxic, although we fortunately found in the in silico evaluation of the toxicity characteristic of molecule 16 that this molecule is not toxic.
Based on the obtained results in silico ADMET properties evaluation for both compounds 16 and Crizotinib, we find that compound 16 meets all the pharmacokinetic conditions that are evaluated in this study. Therefore, molecule 16 can be used in the future as a drug to treat cancer by inhibiting the enzymatic activity of the c-Met protein. Molecule 16 can also be used in the design of new compounds with stronger biological activities, with other properties and new uses.
4. Conclusion
This study contributed to the development of mathematical models that were able to determine the quantitative relationship between the biological anticancer activity and the molecular structure of a series of 4,5,6,7-tetrahydrobenzo[D]-thiazol-2-Yl derivatives. Also in this work, the molecular properties necessary for robust activity to inhibit the C-Met receptor tyrosine kinase and thus achieve an anti-cancer response were detected. The resulting QSAR models were analyzed and validated for statistical significance and predictive power through internal and external validations, as well as via a Y-randomization test and domain of applicability. The predictive ability of QSAR models that is obtained by three analysis methods (MLR, MNLR, ANN) has been shown that the proposed models are very strong for all these methods. The analysis of the developed QSAR model equations showed that the following seven important descriptors influence the biological inhibitory activity of the c-Met receptor: LogP, polarization, Parachor, molecular weight, molecular volume and chemical hardness. The descriptors identified in this work by QSAR models can be used effectively to predict the anticancer activity values of new compounds that can be designed based on the structure of 4,5,6,7-Tetrahydrobenzo[D]-Thiazol-2-Yl derivatives. This will allow to significantly reduce the drug development process and the cost of synthesis at the pharmaceutical chemistry laboratories. In this work, seven molecules with the highest c-Met inhibition activity were selected based on the predictions of the 2D-QSAR models that were developed. After performing an evaluation of the drug-likeness properties of the seven molecules, the results showed that the molecules (5, 16 and 46) had acceptable drug-likeness properties.
A molecular docking study that was performed for the molecules (5, 16 and 46) with the c-Met receptor, showed that the molecule 16 is the best candidate for the inhibition of c-Met carcinogenic activity. Where the molecule 16 established more interactions compared to the molecules (5 and 46) with the reference active sites in the c-Met receptor. Furthermore, molecular docking results showed that the binding energy of molecule 16 in the c-Met receptor pocket is higher than the binding energies of molecules 5 and 46 with the same receptor, indicating that molecule 16 is well stable with the c-Met receptor. In addition, the pharmacokinetic evaluation of ADMET properties in silico between molecule 16 and Crizotinib indicates that molecule 16 has better pharmacokinetic properties than Crizotinib. This could explain the higher biological activity observed for molecule 16 (pIC50 = 9.59) compared to the biological activity observed for Crizotinib (pIC50 = 8.0). Thus, molecule 16 could be proposed as a novel agent useful in the treatment of cancer by inhibiting the enzymatic activity of the c-Met protein. These details and information provided in this work can offer many opportunities to medicinal chemists to develop new anticancer drugs, through the adoption of the structure of molecule 16 in the design of new compounds with stronger biological activities anti-cancer. Thus, the structure of 4,5,6,7-Tetrahydrobenzo[D]-Thiazol-2-Yl derivatives may be a new pathway in the development of new drugs for the treatment of cancer and other diseases.
In future work, we will develop 3D-QSAR models based on the series of 4,5,6,7-Tetrahydrobenzo [D]-Thiazol-2-Yl derivatives, and we will adopt molecule 16 as a reference molecule in the design of novel inhibitors of c-Met enzymatic activity in order to propose new derivatives of 4,5,6,7-Tetrahydrobenzo [D]-Thiazol-2-Yl structure and also to predict its biological activities against the growth of non-small cell lung cancer cells as a model of cancer treatment.
Declarations
Author contribution statement
Ossama Daoui: Conceived and designed the experiments; Performed the experiments; Contributed reagents, materials, analysis tools or data; Wrote the paper.
Souad Elkhattabi, Samir Chtita: Conceived and designed the experiments; Analyzed and interpreted the data; Contributed reagents, materials, analysis tools or data; Wrote the paper.
Rachida Elkhalabi, Hsaine Zgou, Adil Touimi Benjelloun: Conceived and designed the experiments.
Funding statement
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Data availability statement
Data will be made available on request.
Declaration of interests statement
The authors declare no conflict of interest.
Additional information
No additional information is available for this paper.
References
- 1.Turan-Zitouni G., Altıntop M.D., Özdemir A., Kaplancıklı Z.A., Çiftçi G.A., Temel H.E. Synthesis and evaluation of bis-thiazole derivatives as new anticancer agents. Eur. J. Med. Chem. 2016;107:288–294. doi: 10.1016/j.ejmech.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Fouedjou R.T. Cameroonian medicinal plants as potential candidates of SARS-CoV-2 inhibitors. J. Biomol. Struct. Dyn. avr. 2021:1–15. doi: 10.1080/07391102.2021.1914170. vol. 0, no 0. [DOI] [PubMed] [Google Scholar]
- 3.Pignatello R. Synthesis and biological evaluation of thiazolo-triazole derivatives. Eur. J. Med. Chem. 1991;26(9):929–938. [Google Scholar]
- 4.Trapani G. Synthesis and anticonvulsant activity of some 1,2,3,3a-tetrahydropyrrolo[2,1-b]benzothiazol-1-ones and pyrrolo[2,1-b]thiazole analogues. Eur. J. Med. Chem. janv. 1994;29(3):197–204. [Google Scholar]
- 5.Yu H. Synthesis and insecticidal activity of N-substituted (1,3-thiazole)alkyl sulfoximine derivatives. J. Agric. Food Chem. déc. 2008;56(23):11356–11360. doi: 10.1021/jf802802g. [DOI] [PubMed] [Google Scholar]
- 6.El-Gazzar A.B.A., Youssef M.M., Youssef A.M.S., Abu-Hashem A.A., Badria F.A. Design and synthesis of azolopyrimidoquinolines, pyrimidoquinazolines as anti-oxidant, anti-inflammatory and analgesic activities. Eur. J. Med. Chem. févr. 2009;44(2):609–624. doi: 10.1016/j.ejmech.2008.03.022. [DOI] [PubMed] [Google Scholar]
- 7.Dahmani R., Manachou M., Belaidi S., Chtita S., Boughdiri S. Structural characterization and QSAR modeling of 1,2,4-triazole derivatives as α-glucosidase inhibitors. New J. Chem. janv. 2021;45(3):1253–1261. [Google Scholar]
- 8.Qiu X.-L. Synthesis and biological evaluation of a series of novel inhibitor of Nek2/Hec1 analogues. J. Med. Chem. mars 2009;52(6):1757–1767. doi: 10.1021/jm8015969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsou H.-R. Discovery and optimization of 2-(4-substituted-pyrrolo [2, 3-b] pyridin-3-yl) methylene-4-hydroxybenzofuran-3 (2H)-ones as potent and selective ATP-competitive inhibitors of the mammalian target of rapamycin (mTOR) Bioorg. Med. Chem. Lett. 2010;20(7):2321–2325. doi: 10.1016/j.bmcl.2010.01.135. [DOI] [PubMed] [Google Scholar]
- 10.Dawood K.M., Eldebss T.M.A., El-Zahabi H.S.A., Yousef M.H., Metz P. Synthesis of some new pyrazole-based 1,3-thiazoles and 1,3,4-thiadiazoles as anticancer agents. Eur. J. Med. Chem. déc. 2013;70:740–749. doi: 10.1016/j.ejmech.2013.10.042. [DOI] [PubMed] [Google Scholar]
- 11.Cañadas I., Rojo F., Arumí-Uría M., Rovira A., Albanell J., Arriola E. C-MET as a new therapeutic target for the development of novel anticancer drugs. Clin. Transl. Oncol. avr. 2010;12(4):253–260. doi: 10.1007/s12094-010-0501-0. [DOI] [PubMed] [Google Scholar]
- 12.Tanizaki J. MET tyrosine kinase inhibitor crizotinib (PF-02341066) shows differential antitumor effects in non-small cell lung cancer according to MET alterations. J. Thorac. Oncol. oct. 2011;6(10):1624–1631. doi: 10.1097/JTO.0b013e31822591e9. [DOI] [PubMed] [Google Scholar]
- 13.Cui J.J. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal–epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK) J. Med. Chem. sept. 2011;54(18):6342–6363. doi: 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- 14.Rodig S.J., Shapiro G.I. Crizotinib, a small-molecule dual inhibitor of the c-Met and ALK receptor tyrosine kinases. Curr. Opin. Investig. Drugs Lond. Engl. 2000. déc. 2010;11(12):1477–1490. [PubMed] [Google Scholar]
- 15.Vilar S., Cozza G., Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. déc. 2008;8(18):1555–1572. doi: 10.2174/156802608786786624. [DOI] [PubMed] [Google Scholar]
- 16.Mohareb R.M., Abouzied A.S., Abbas N.S. Synthesis and biological evaluation of novel 4, 5, 6, 7-tetrahydrobenzo [D]-Thiazol-2-Yl derivatives derived from dimedone with anti-tumor, c-met, tyrosine kinase and Pim-1 inhibitions. Anti-Canc Agent Med. Chem. Former. Curr. Med. Chem.-Anti-Canc Agent. 2019;19(12):1438–1453. doi: 10.2174/1871520619666190416102144. [DOI] [PubMed] [Google Scholar]
- 17.Tropsha A. Best practices for QSAR model development, validation, and exploitation. Mol. Inform. 2010;29(6-7):476–488. doi: 10.1002/minf.201000061. [DOI] [PubMed] [Google Scholar]
- 18.Roy K., Kar S., Ambure P. On a simple approach for determining applicability domain of QSAR models. Chemometr. Intell. Lab. Syst. 2015;145:22–29. [Google Scholar]
- 19.Butina D., Segall M.D., Frankcombe K. Predicting ADME properties in silico: methods and models. Drug Discov. Today. 2002;7(11):S83–S88. doi: 10.1016/s1359-6446(02)02288-2. [DOI] [PubMed] [Google Scholar]
- 20.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997;23(1-3):3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 21.Veber D.F., Johnson S.R., Cheng H.-Y., Smith B.R., Ward K.W., Kopple K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002;45(12):2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 22.Egan W.J., Merz K.M., Baldwin J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000;43(21):3867–3877. doi: 10.1021/jm000292e. [DOI] [PubMed] [Google Scholar]
- 23.ChemOffice Download ChemDraw and Chem3D. http://www.chem.ox.ac.uk/software/chemoffice.html (consulté le mars 22, 2021)
- 24.Structure Drawing Software for Academic and Personal Use ACD/ChemSketch. https://www.acdlabs.com/resources/freeware/chemsketch/index.php (consulté le mars 22, 2021)
- 25.Gaussian.com Expanding the limits of computational chemistry. http://gaussian.com/
- 26.Allinger N.L. Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J. Am. Chem. Soc. 1977;99(25):8127–8134. [Google Scholar]
- 28.Chtita S., Ghamali M., Ousaa A., Aouidate A., Belhassan A., Taourati A.I., Masand V.H., Bouachrine M., Lakhlifi T. QSAR study of anti-Human African Trypanosomiasis activity for 2-phenylimidazopyridines derivatives using DFT and Lipinski’s descriptors. Heliyon. 2019;5(3) doi: 10.1016/j.heliyon.2019.e01304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wold S., Esbensen K., Geladi P. Principal component analysis. Chemometr. Intell. Lab. Syst. 1987;2(1):37–52. [Google Scholar]
- 30.David C.C., Jacobs D.J. Protein Dynamics. Springer; 2014. Principal component analysis: a method for determining the essential dynamics of proteins; pp. 193–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lagunin A., Zakharov A., Filimonov D., Poroikov V. QSAR modelling of rat acute toxicity on the basis of PASS prediction. Mol. Inform. 2011;30(2-3):241–250. doi: 10.1002/minf.201000151. [DOI] [PubMed] [Google Scholar]
- 32.Leonard J.T., Roy K. On selection of training and test sets for the development of predictive QSAR models. QSAR Comb. Sci. 2006;25(3):235–251. [Google Scholar]
- 33.Papa E., Dearden J.C., Gramatica P. Linear QSAR regression models for the prediction of bioconcentration factors by physicochemical properties and structural theoretical molecular descriptors. Chemosphere. 2007;67(2):351–358. doi: 10.1016/j.chemosphere.2006.09.079. [DOI] [PubMed] [Google Scholar]
- 34.Salt D.W., Yildiz N., Livingstone D.J., Tinsley C.J. The use of artificial neural networks in QSAR. Pestic. Sci. 1992;36(2):161–170. [Google Scholar]
- 35.XLSTAT version 2019.1 XLSTAT, Your data analysis solution. https://www.xlstat.com/fr/articles/xlstat-version-2019-1
- 36.Download Matlab Best software & apps. https://en.softonic.com/downloads/matlab
- 37.Veerasamy R., Rajak H., Jain A., Sivadasan S., Varghese C.P., Agrawal R.K. Validation of QSAR models-strategies and importance. Int. J. Drug Discov. 2011;3:511–519. [Google Scholar]
- 38.Kiralj R., Ferreira M. Basic validation procedures for regression models in QSAR and QSPR studies: theory and application. J. Braz. Chem. Soc. 2009;20(4):770–787. [Google Scholar]
- 39.Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment. Elsevier; 2015. [Google Scholar]
- 40.Chtita S. Investigation of antileishmanial activities of acridines derivatives against promastigotes and amastigotes form of parasites using quantitative structure activity relationship analysis. Adv. Phys. Chem. nov. 2016;2016 [Google Scholar]
- 41.Chtita S., Hmamouchi R., Larif M., Ghamali M., Bouachrine M., Lakhlifi T. QSPR studies of 9-aniliioacridine derivatives for their DNA drug binding properties based on density functional theory using statistical methods: model, validation and influencing factors. J. Taibah Univ. Sci. nov. 2016;10(6):868–876. [Google Scholar]
- 42.Prado-Prado F.J., García-Mera X., González-Díaz H. Multi-target spectral moment QSAR versus ANN for antiparasitic drugs against different parasite species. Bioorg. Med. Chem. 2010;18(6):2225–2231. doi: 10.1016/j.bmc.2010.01.068. [DOI] [PubMed] [Google Scholar]
- 43.Kuurková V. Kolmogorov’s theorem and multilayer neural networks. Neural Network. 1992;5(3):501–506. [Google Scholar]
- 44.So et S.S., Richards W.G. Application of neural networks: quantitative structure-activity relationships of the derivatives of 2, 4-diamino-5-(substituted-benzyl) pyrimidines as DHFR inhibitors. J. Med. Chem. 1992;35(17):3201–3207. doi: 10.1021/jm00095a016. [DOI] [PubMed] [Google Scholar]
- 45.Andrea T.A., Kalayeh H. mai 2002. Applications of Neural Networks in Quantitative Structure-Activity Relationships of Dihydrofolate Reductase Inhibitors.https://pubs.acs.org/doi/pdf/10.1021/jm00113a022 (consulté le mars 22, 2021) [DOI] [PubMed] [Google Scholar]
- 46.Golbraikh A., Tropsha A. Beware of q2! J. Mol. Graph. Model. janv. 2002;20(4):269–276. doi: 10.1016/s1093-3263(01)00123-1. [DOI] [PubMed] [Google Scholar]
- 47.Chtita S. QSAR study of unsymmetrical aromatic disulfides as potent avian SARS-CoV main protease inhibitors using quantum chemical descriptors and statistical methods. Chemometr. Intell. Lab. Syst. 2021;210:104266. doi: 10.1016/j.chemolab.2021.104266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rücker C., Rücker G., Meringer M. Y-randomization and its variants in QSPR/QSAR. J. Chem. Inf. Model. nov. 2007;47(6):2345–2357. doi: 10.1021/ci700157b. [DOI] [PubMed] [Google Scholar]
- 49.Roy K., Mitra I. On various metrics used for validation of predictive QSAR models with applications in virtual screening and focused library design. Comb. Chem. High Throughput Screen. 2011;14(6):450–474. doi: 10.2174/138620711795767893. [DOI] [PubMed] [Google Scholar]
- 50.Netzeva T.I. Current status of methods for defining the applicability domain of (quantitative) structure-activity relationships: the report and recommendations of ecvam workshop 52. Altern. Lab. Anim. 2005;33(2):155–173. doi: 10.1177/026119290503300209. [DOI] [PubMed] [Google Scholar]
- 51.Gramatica P. Principles of QSAR models validation: internal and external. QSAR Comb. Sci. 2007;26(5):694–701. [Google Scholar]
- 52.Eriksson L., Jaworska J., Worth A.P., Cronin M.T., McDowell R.M., Gramatica P. Methods for reliability and uncertainty assessment and for applicability evaluations of classification-and regression-based QSARs. Environ. Health Perspect. 2003;111(10):1361–1375. doi: 10.1289/ehp.5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frey A., Di Canzio J., Zurakowski D. A statistically defined endpoint titer determination method for immunoassays. J. Immunol. Methods. 1998;221(1-2):35–41. doi: 10.1016/s0022-1759(98)00170-7. [DOI] [PubMed] [Google Scholar]
- 56.Hansch C., Leo A., Mekapati S.B., Kurup A. QSAR and ADME. Bioorg. Med. Chem. juin 2004;12(12):3391–3400. doi: 10.1016/j.bmc.2003.11.037. [DOI] [PubMed] [Google Scholar]
- 57.Jin Z. Structure-based virtual screening of influenza virus RNA polymerase inhibitors from natural compounds: molecular dynamics simulation and MM-GBSA calculation. Comput. Biol. Chem. 2020;85:107241. doi: 10.1016/j.compbiolchem.2020.107241. [DOI] [PubMed] [Google Scholar]
- 58.Chtita S. QSAR study of N-substituted oseltamivir derivatives as potent avian influenza virus H5N1 inhibitors using quantum chemical descriptors and statistical methods. New J. Chem. févr. 2020;44(5):1747–1760. [Google Scholar]
- 59.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7(1):1–13. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pires D.E.V., Blundell T.L., Ascher D.B. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. Mai 2015;58(9):4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chtita S., Belhassan A., Aouidate A., Belaidi S., Bouachrine M., Lakhlifi T. Discovery of potent SARS-CoV-2 inhibitors from approved antiviral drugs via docking and virtual screening. Comb. Chem. High Throughput Screen. févr. 2021;24(3):441–454. doi: 10.2174/1386207323999200730205447. [DOI] [PubMed] [Google Scholar]
- 62.Christensen J.G. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol. Canc. Therapeut. 2007;6(12):3314–3322. doi: 10.1158/1535-7163.MCT-07-0365. [DOI] [PubMed] [Google Scholar]
- 63.Hu H., Chen F., Dong Y., Liu Y., Gong P. Discovery of novel dual c-Met/HDAC inhibitors as a promising strategy for cancer therapy. Bioorg. Chem. 2020;101:103970. doi: 10.1016/j.bioorg.2020.103970. [DOI] [PubMed] [Google Scholar]
- 64.Bank R.P.D. RCSB PDB - 2WGJ: X-ray Structure of PF-02341066 bound to the kinase domain of c-Met. https://www.rcsb.org/structure/2WGJ (consulté le mars 22, 2021)
- 65.Discovery Systèmes Free download: BIOVIA discovery Studio visualizer. Dassault Systèm. mars 20, 2020 https://discover.3ds.com/discovery-studio-visualizer-download [Google Scholar]
- 66.MGLTools 1.5.6 RC3 release announcement — MGLTools. http://mgltools.scripps.edu/News/mgltools-1-5-6-release-announcement (consulté le mars 22, 2021)
- 67.Morris G.M. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998;19(14):1639–1662. [Google Scholar]
- 68.Westermaier Y., Barril X., Scapozza L. Virtual screening: an in silico tool for interlacing the chemical universe with the proteome. Methods. janv. 2015;71:44–57. doi: 10.1016/j.ymeth.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 69.Sayed A.M. Nature as a treasure trove of potential anti-SARS-CoV drug leads: a structural/mechanistic rationale. RSC Adv. 2020;10(34):19790–19802. doi: 10.1039/d0ra04199h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ouassaf M., Belaidi S., Khamouli S., Belaidi H., Chtita S. Combined 3D-QSAR and molecular docking analysis of thienopyrimidine derivatives as Staphylococcus aureus inhibitors. Acta Chim. Slov. 2021 [PubMed] [Google Scholar]
- 71.Zentrum für Bioinformatik Universität hamburg - proteins plus server. https://proteins.plus/ (consulté le mai 27, 2021)
- 72.Sattari A., Ramazani A., Aghahosseini H. Repositioning therapeutics for COVID-19: virtual screening of the potent synthetic and natural compounds as SARS-CoV-2 3CLpro inhibitors. J. Iran. Chem. Soc. mars 2021 [Google Scholar]
- 73.Ouassaf M., Belaidi S., Mogren Al Mogren M., Chtita S., Ullah Khan S., Thet Htar T. Combined docking methods and molecular dynamics to identify effective antiviral 2, 5-diaminobenzophenonederivatives against SARS-CoV-2. J. King Saud Univ. Sci. mars 2021;33(2):101352. doi: 10.1016/j.jksus.2021.101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kalantzi L., Goumas K., Kalioras V., Abrahamsson B., Dressman J.B., Reppas C. Characterization of the human upper gastrointestinal contents under conditions simulating bioavailability/bioequivalence studies. Pharm. Res. 2006;23(1):165–176. doi: 10.1007/s11095-005-8476-1. [DOI] [PubMed] [Google Scholar]
- 75.Pires D.E., Blundell T.L., Ascher D.B. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015;58(9):4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Speciale A., Muscarà C., Molonia M.S., Cimino F., Saija A., Giofrè S.V. Silibinin as potential tool against SARS-Cov-2: in silico spike receptor-binding domain and main protease molecular docking analysis, and in vitro endothelial protective effects. Phytother Res. 2021;n/a(n/a) doi: 10.1002/ptr.7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Han Y., Zhang J., Hu C.Q., Zhang X., Ma B., Zhang P. In silico ADME and toxicity prediction of ceftazidime and its impurities. Front. Pharmacol. 2019;10 doi: 10.3389/fphar.2019.00434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Domínguez-Villa F.X., Durán-Iturbide N.A., Ávila-Zárraga J.G. Synthesis, molecular docking, and in silico ADME/Tox profiling studies of new 1-aryl-5-(3-azidopropyl) indol-4-ones: potential inhibitors of SARS CoV-2 main protease. Bioorg. Chem. 2021;106:104497. doi: 10.1016/j.bioorg.2020.104497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zanger U.M., Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013;138(1):103–141. doi: 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 80.Rodrigues-Junior V.S. Nonclinical evaluation of IQG-607, an anti-tuberculosis candidate with potential use in combination drug therapy. Regul. Toxicol. Pharmacol. 2020;111:104553. doi: 10.1016/j.yrtph.2019.104553. [DOI] [PubMed] [Google Scholar]
- 81.Puccini A. Safety and tolerability of c-MET inhibitors in cancer. Drug Saf. 2019;42(2):211–233. doi: 10.1007/s40264-018-0780-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ferraz E.R.A. Differential toxicity of Disperse Red 1 and Disperse Red 13 in the Ames test, HepG2 cytotoxicity assay, and Daphnia acute toxicity test. Environ. Toxicol. 2011;26(5):489–497. doi: 10.1002/tox.20576. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on request.