

Summary
Cell type-specific pathway-based polygenic risk scores (PRSs) may better inform disease biology and improve the precision of PRS-based clinical prediction. For microRNA-137 (MIR137), a leading neuropsychiatric risk gene and a post-transcriptional master regulator, we conducted a cell type-specific gene set PRS analysis in both European and Han Chinese schizophrenia (SZ) samples. We found that the PRS of neuronal MIR137-target genes better explains SZ risk than PRS derived from MIR137-target genes in iPSC or from the reported gene sets showing MIR137-altered expression. Compared with the PRS derived from the whole genome or the target genes of TCF4, the PRS of neuronal MIR137-target genes explained a disproportionally larger (relative to SNP number) SZ risk in the European sample, but with a more modest advantage in the Han Chinese sample. Our study demonstrated a cell type-specific polygenic contribution of MIR137-target genes to SZ risk, highlighting the value of cell type-specific pathway-based PRS analysis for uncovering disease-relevant biological features.
Subject areas: biological sciences, genomics, Neuroscience
Graphical abstract
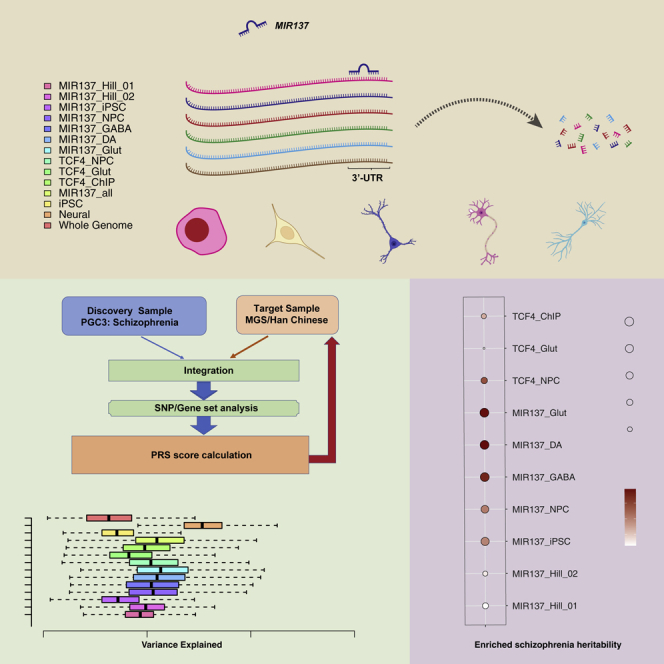
Highlights
-
•
PRS of neural MIR137 target genes better explains schizophrenia (SZ) risk variance
-
•
SZ risk and SNP heritability explained by MIR137 target genes is cell type-specific
-
•
MIR137 target genes explain a disproportionally larger SZ risk than genomic control
-
•
PRS of MIR137 target genes better explains SZ risk in Europeans than in Han Chinese
Biological sciences; Genomics; Neuroscience
Introduction
Genome-wide association studies (GWASs) of complex disorders are identifying an increasing number of common risk variants. The most recent schizophrenia (SZ) GWAS meta-analysis of Psychiatric Genomic Consortium (PGC3) with 69,369 patients with SZ and 236,642 controls identified 270 distinct risk loci (The Schizophrenia Working Group of the Psychiatric Genomics Consortium et al., 2020). Like other complex disorders, SZ is polygenic, i.e., there are possibly hundreds or even thousands of risk-associated genes, each contributing small effect. Beyond the polygenic model, an omnigenic model has also been hypothesized for complex disorders such as SZ, where most if not all genes, expressed in a relevant cell type may contribute to risk through interacting with a core set of genes (Boyle et al., 2017). The polygenic or omnigenic nature of SZ poses challenges to identifying causal variants/genes and better understanding pathogenesis. Polygenic risk scores (PRSs) derived from GWAS data of common variants in a large cohort have been commonly used to improve estimates of risk variance in a target sample and estimate individual risk. However, PRSs derived from genome-wide data can be noisy because they include many SNPs with no risk effect and many risk genes may not be expressed in a cell/tissue type relevant to the disorder. A more effective approach may be deriving PRS from more disorder relevant gene pathways and cell types that are more likely to harbor a higher proportion of causal SNPs (Baker et al., 2018; Zeng et al., 2017), an approach that has been understudied for SZ.
It has been increasingly recognized that many risk genes likely act as part of a gene network/pathway. It is thus imperative to identify core gene network(s) contributing to risk. As opposed to maximizing the predictive power of case-control status in PRS analyses by using genome-wide data, gene set PRS analyses can more specifically detect associations to some biologically informative features (Baker et al., 2018). For instance, a recent PRS study using SZ GWAS SNPs of gene sets highly expressed and dynamically modulated in placenta showed that considering environmental exposure (early-life complications) can increase the predicting accuracy of SZ risk (Ursini et al., 2018), although a later study did not replicate this finding (Vassos et al., 2021). Similarly, by incorporating cell type-specific or developmental stage-specific gene expression information, gene set PRS analysis may inform cell types or developmental stages that are more biologically relevant to a disorder. Furthermore, although genome-wide PRS of one specific ancestry has been commonly considered inappropriate for predicting risk variance of a different ancestry (Amariuta et al., 2020), a cell type-specific pathway-based PRS may be more portable between different populations. For example, PRS derived by prioritizing GWAS variants in the predicted cell type-specific regulatory elements has substantially increased trans-ancestry portability (Amariuta et al., 2020). Once a gene set is implicated by PRS analysis, the gene set risk scores for each individual can be used to stratify individuals for functional follow-up and to formulate a more tractable and testable hypothesis, e.g., by generating patient-specific cell models in those individuals with high PRS and perform pathway-specific genetic perturbation.
MicroRNAs (miRNAs) and their targets are ideal candidates for gene set-based PRS analyses. This is because each miRNA often has many possible target genes, i.e., be a “master regulator.” In addition, miRNAs are central to brain development and play an important role in the cause of neurodevelopmental disorders such as SZ (Guarnieri and DiLeone, 2008; Im and Kenny, 2012). Among GWAS-implicated SZ risk loci (Schizophrenia Psychiatric Genome-Wide Association Study (GWAS)Consortium, 2011; Lee et al., 2012; Purcell et al., 2009; Shi et al., 2009; Stefansson et al., 2009; Ripke and Consortium, 2014), one of the most strongly associated (p = 6.6 × 10−16) (Schizophrenia Psychiatric Genome-Wide Association Study (GWAS)Consortium, 2011; Ripke and Consortium, 2014) spans MIR137. The SZ risk alleles of the MIR137 locus also predict cognitive deficits (Green et al., 2012), and the deletion of the MIR137 locus has been associated with intellectual disability (Willemsen et al., 2011). MIR137 is highly expressed in brain and enriched at neuronal synapses (Willemsen et al., 2011). It regulates neuronal differentiation, migration, and dendritogenesis (Silber et al., 2008; Smrt et al., 2010; Sun et al., 2011; Szulwach et al., 2010; Volvert et al., 2012). Of interest, several SZ GWAS loci encompass MIR137 targets; for instance, MIR137 has been known to regulate other genes that harbor SZ-associated variants such as ZNF804A, TCF4, CACNA1C, CSMD1, and C10orf26 (Collins et al., 2014; Kwon et al., 2013; Remmers et al., 2014; Wright et al., 2013), suggesting a central hub role for MIR137 in an SZ gene network. A recent PRS analysis of genes whose expression was altered by artificial manipulation of MIR137 expression in a neural progenitor cell (NPC) line uncovered an interesting association of the MIR137 gene set PRS with cognitive function (Cosgrove et al., 2017). However, another study did not find a significant association between MIR137 PRS and cortical thickness, surface area, or hippocampal volume measures linked to memory function (Cosgrove et al., 2018). Although interesting, these MIR137 PRS analyses have several limitations: (1) the relative contribution of MIR137 gene set PRS to the overall SZ risk variance has not been estimated; (2) because of the notorious weak expression correlation of MIR137 and its targeting genes (Forrest et al., 2017; Topol et al., 2016), the gene set defined by artificially manipulating MIR137 expression in a single cell line may not be representative of an actual specific MIR137 gene network; and (3) NPCs used to define the gene sets for MIR137 PRS analyses may not represent the most relevant cell type for SZ (Skene et al., 2018).
Here, by using PGC3 (The Schizophrenia Working Group of the Psychiatric Genomics Consortium et al., 2020) SZ GWAS summary statistics of European ancestry samples, we have compared the PRS performance of different gene sets of MIR137 targets in explaining SZ risk variances in the Molecular Genetics of Schizophrenia (MGS) European ancestry cohort (Shi et al., 2009) and in a Han Chinese SZ case-control sample (Li et al., 2017; Yu et al., 2017) (Figure 1). We also compared with performances of PRSs derived from sets of target genes of TCF4, a transcriptional master regulator of SZ (Doostparast Torshizi et al., 2019). We discovered that the PRSs of neuronal MIR137-target gene sets explain disproportionally larger SZ risk variance, compared with the TCF4-target gene sets and genome-wide SNP sets. Furthermore, with human induced pluripotent stem cells (hiPSCs) as a model, we found that the PRS predictive efficacy of MIR137-target gene sets is neuron specific. Finally, we evaluated the PRS portability of MIR137 gene sets from European Ancestry to Han Chinese samples. Our study underscores the value of cell type-specific gene set PRS analysis in understanding the biology of a polygenic or omnigenic disorder.
Figure 1.

Workflow for PRS analysis
Results
To evaluate the contribution of gene pathway(s) pertinent to MIR137, a leading noncoding SZ risk locus, to genetic risk of SZ, we addressed three specific questions in a set of PRS analyses: (1) Between genes perturbed by altering MIR137 expression in a specific cell type (Cosgrove et al., 2017) and those predicted MIR137 target genes specifically expressed in a disease-relevant cell type, which constitutes an optimal gene set of the MIR137 pathway for PRS analysis? (2) What is the proportion of SZ risk variance that can be explained by disease risk variants in the MIR137 gene pathway? (3) Is the MIR137 gene pathway-based PRS portable across ancestry? To address these questions, we have compiled 10 gene sets (Tables 1 and S1), including the lists of genes perturbed by MIR137 alteration and previously used for MIR137 gene pathway PRS analysis by Hill et al. (Cosgrove et al., 2017), genes predicted to be MIR137 targets and expressed in hiPSC or in various hiPSC-derived neuronal cell types (Forrest et al., 2017; Zhang et al., 2018, 2020). For a comparison, we also included sets of genes that were perturbed by knocking down a transcriptional master regulator, TCF4 (also an SZ GWAS-implicated risk gene), in hiPSC-derived neuronal cells (Doostparast Torshizi et al., 2019), as well as empirical binding targets of TCF4 in chromatin immunoprecipitation (ChIP-seq) (Forrest et al., 2017). For each gene set, the “significant” SNPs were selected based on seven predetermined significance thresholds (Pd < 0.01, 0.05, 0.1, 0.2, 0.3, 0.4, and 0.5) in the discovery sample (the European ancestry PGC-SZ3 minus MGS) to derived PRS for each individual in two target samples, the European ancestry MGS (2,681 cases, 2,653 controls) (Shi et al., 2009) and a Han Chinese sample (4,288 cases, 5,512 controls) (Li et al., 2017; Yu et al., 2017). We estimated the proportion of variance explained by the aggregate risk score by each SNP/gene set in each sample (Figure 1).
Table 1.
Gene/SNP sets used in the PRS analysis
| List no. | List name | Genes | Description | Reference |
|---|---|---|---|---|
| 1 | MIR137_Hill_01 | 1,017 | Genes altered by over-expressing MIR137 in an NPC cell line (previous study) | (Cosgrove et al., 2017; Hill et al., 2014) |
| 2 | MIR137_Hill_02 | 934 | Genes altered by knocking down MIR137 in an NPC cell line (previous study) | (Cosgrove et al., 2017; Hill et al., 2014) |
| 3 | MIR137_iPSC | 909 | TargetScan MIR137 target genes expressed in hiPSC | (Zhang et al., 2020) |
| 4 | MIR137_NPC | 925 | TargetScan MIR137 target genes expressed in hiPSC-derived NPCs | (Zhang et al., 2020) |
| 5 | MIR137_GABA | 986 | TargetScan MIR137 target genes expressed in hiPSC-derived GABA neurons | (Zhang et al., 2018) |
| 6 | MIR137_DA | 1,034 | TargetScan MIR137 target genes expressed in hiPSC-derived DA neurons | (Zhang et al., 2018) |
| 7 | MIR137_Glut | 991 | TargetScan MIR137 target genes expressed in hiPSC-derived Glut neuron | (Forrest et al., 2017; Zhang et al., 2020) |
| 8 | TCF4_NPC | 1,000 | Top ranking genes altered by knocking down TCF4 in hiPSC-derived NPCs | (Doostparast Torshizi et al., 2019) |
| 9 | TCF4_Glut | 1,000 | Top ranking genes altered by knocking down TCF4 in hiPSC-derived Glut neurons | (Doostparast Torshizi et al., 2019) |
| 10 | TCF4_ChIP | 1,000 | Top ranking genes with promoter/enhancer bind by TCF4 (previous study) | (Forrest et al., 2017) |
The lists of MIR137_Hill_01 and MIR137_Hill_02 have been used by Hill et al. in previous PRS analysis (Cosgrove et al., 2017; Hill et al., 2014).
Neuronal MIR137 target genes better explain SZ risk variance than genes with expression altered in vitro by MIR137
Genes with expression altered by either over-expression or knocking-down of MIR137 in NPCs have been recently used to correlate PRS to SZ-related cognitive phenotypes (Cosgrove et al., 2017, 2018). Given the known weak expression correlation of MIR137 and its targeted genes (Forrest et al., 2017; Topol et al., 2016), we hypothesized that predicted MIR137 gene targets specifically expressed in a disease-relevant cell type may constitute a better gene set for PRS analysis. As shown in Figure 2A (Table S3), although with similar number of genes (Table S2), PRS derived from the predicted MIR137 target genes that are expressed in hiPSC-derived NPCs (Ngene = 925 in MIR137_NPC), GABA (Ngene = 986 in MIR137_GABA), DA neurons (Ngene = 1,034 of MIR137_DA), or Glut (Ngene = 991 of MIR137_Glut) neurons explains greater SZ risk variance in the MGS sample than PRS derived from genes expressed in hiPSCs (Ngene = 909 in MIR137_iPSC) or those with expression altered by MIR137 in NPC (Ngene = 1,017 and 934 in MIR137_Hill_01 and MIR137_Hill_02, respectively). The PRS of gene set in Glut showed the best performance (Figure 2A and Table S3), consistent with the notion that it is among the most disease-relevant cell types for SZ (The Schizophrenia Working Group of the Psychiatric Genomics Consortium et al., 2020). We observed similar patterns across different GWAS p-value thresholds (Pd < 0.01, 0.05, 0.1, 0.2, 0.3, 0.4, and 0.5) and at different gene intervals, i.e., 20 (Figure 2A) or 100 kb (Figure S1A) extended genic regions.
Figure 2.

PRS analysis of different lists of target genes of MIR137 and TCF4 annotated in a 20-kb window for the MGS sample
Gene sets in (A) and (B) were described in Table 1.
(A) PRS result using LD-pruned SNPs (r2 < 0.2) for each gene list. The variance explained in the target sample is based on risk scores derived from an aggregated sum of weighted SNP risk allele effect sizes estimated from the discovery samples at seven significance thresholds (p < 0.01, 0.05, 0.1, 0.2, 0.3, 0.4, and 0.5). The y axis indicates the percentage of phenotypic variance explained by the PRS (Nagelkerke's pseudo R2).
(B) The permutation (N = 1,000) PRS results based on randomly selected 1,500 SNPs for each gene set. Data are presented as mean ± SEM.
(C) Stratified LDSC analysis showing stronger enrichment of SZ GWAS SNP heritability in neural MIR137-target gene sets. Shown are the folds of enrichment of SNP heritability (proportion of explained heritability h2 normalized by the proportion of SNPs in each gene set) and the statistical significance (Bonferroni-corrected p values; in -log10 scale).
Although different gene sets in our above-described PRS analysis (Figure 2A) contain similar number of genes, the results may be cofounded by variable gene length (e.g., neuronal genes are often larger) (Zylka et al., 2015) and SNP numbers in different gene sets. To account for these confounding factors, we first estimated the relative PRS enrichment for each gene set in Figure 2A by correcting for the number of tested SNPs in each gene set (Figure S2). We found that MIR137 target gene sets in iPSC-derived neuronal cells explained disproportionally more SZ risk variance than SNPs from MIR137 target genes expressed in iPSC across all GWAS p-value thresholds (Figure S2). However, MIR137 target gene sets in iPSC-derived neuronal cells did not seem to outperform MIR137_Hill_01 and MIR137_Hill_02 in this analysis, especially at Pd < 0.01, 0.05, 0.1, 0.2, or 0.3.
We then performed permutation (N = 1,000) to randomly select the same number of SNPs in each gene set for calculating PRS (Table S3). As shown in Figures 2B and S1B, we found that MIR137 target gene sets expressed in hiPSC-derived NPCs or neurons explain more SZ risk variance in the MGS sample than the genes expressed in non-neuronal hiPSCs. Overall, these neural MIR137 target gene sets also explained disproportionally more SZ risk variance than SNPs of the two gene sets with expression altered by artificially changing MIR137 expression in NPCs (MIR137_Hill_01 and MIR137_Hill_02), especially with SZ GWAS p-value cut-offs of <0.3, <0.4, or <0.5 (Figure 2B). Although this assessment may still be confounded by the number of SNPs in each permutation at some SZ GWAS p value cut-offs, we noted that the SNP numbers in the permutation tests were overall similar between gene sets, especially with exactly the same number of SNPs (n = 1,500) at SZ GWAS p-value cut-off <0.5 (Figure S3), suggesting a greater relative enrichment of variance explained by MIR137 target gene sets from hiPSC-derived neural cells (versus those from iPSC, MIR137_Hill_01 or MIR137_Hill_02). We also compared the SZ PRS of these SNP sets with an equal number of SNPs from all the predicted MIR137 target genes independent of whether they were expressed in any particular cell type (MIR137_all) and found that SNPs from MIR137_all explained similar SZ risk variance as SNPs from neuronal gene sets, but significantly more variance than genes expressed in iPSC (Figure 2B and Table S4), suggesting a major contribution of neuronal MIR137 target genes to SZ risk (also see below). It is noteworthy that MIR137_Hill_01 and MIR137_Hill_02 gene sets, although explaining proportionally less variance than the neuronal MIR137 target gene sets, still explain proportionately more variance than the control genomic SNP set (Figure 2B and Table S4). The statistical significance of a Student's t-tests was shown in Figure 3 and Table S4 on 1,000 permuted Nagelkerke's pseudo R2 values between any two gene sets. Altogether, these results suggest the importance of PRS analysis using cell type-specific gene sets.
Figure 3.

-log(p-values) of t-tests for comparing the variance explained by the equal number of randomly selected SNPs (1,000 permutations) of two gene sets
The upper triangle indicates the results in the MGS sample, and the lower triangle indicates the results in the Han Chinese sample, both with SNPs within 20-kb flanking regions of a gene. The number of genes of each gene set is shown in the diagonal grid. The cells with ∗ indicate non-significant p-value > 0.0009 (from two-sample Student's t-test; Bonferroni corrected).
To independently validate the results from gene set-specific PRS analysis, we further conducted a partitioned SZ heritability analysis by a stratified linkage disequilibrium score regression (sLDSC) (Bulik-Sullivan et al., 2015; Finucane et al., 2015) for different gene sets. With PGC SZ wave_3 GWAS summary statistics, we estimated the proportion of explained SZ SNP heritability (h2) and the proportion of SNPs in each gene set (Figure 2C and Table S5). We found that all the tested MIR137 target gene sets are enriched (3- to 5-fold) for SZ SNP heritability, with neuronal gene sets, especially those expressed in Glut neurons, showing strongest enrichment, whereas MIR137_Hill_01 and MIR137_Hill_02 gene sets showed the least enrichment (Figure 2C and Table S5). On comparison with the enrichment score of each gene set, the coefficient from the sLDSC analysis gave even stronger support for proportionally more SZ SNP heritability explained by neuronal MIR137 target gene sets than those expressed in iPSC or MIR137_Hill_01 and MIR137_Hill_02 gene sets (Table S5). The result from the sLDSC analysis is thus largely consistent with our gene set-specific SZ PRS analysis using randomly selected SNPs from each gene set, supporting that neuronal MIR137 target genes better explain SZ risk variance/heritability than non-neuronal genes or those with expression altered in vitro by MIR137.
SZ PRS explained by MIR137 target genes is mainly attributed to a small number of neuron-specific genes
Given that the MIR137 target gene sets expressed in hiPSC-derived neurons explain 2- to 3-fold more SZ variance than those in hiPSCs (Figures 2A and 2B), we speculated that those MIR137 target genes specifically expressed in neurons contributed to the difference. As shown in Figure 4A, most genes (N = 784; Table S1) were expressed in both hiPSC and hiPSC-derived NPCs, DA, GABA, and Glut neurons. Therefore, we hypothesize that the PRS of a very small subset of MIR137 target genes expressed only in NPCs and/or neurons explained a larger proportion of the SZ risk variance. These genes include those at genome-wide significant SZ risk loci, such as CACNA1C uniquely expressed in Glut/DA/GABA neurons, CSMD1 and DPYD expressed only in Glut neurons, and GRIA1 expressed in all neuronal cells. It is noteworthy that the dysregulated synaptic expression of GRIA1 in dendritic protrusions of Glut neurons was found associated with the SZ risk allele of MIR137 (Forrest et al., 2017). Some other contributing neuronal genes are not among those top-ranking SZ GWAS risk genes, e.g., PDE10A (Table S1), a gene that was recently shown to be a key target that mediates the MIR137 function in heterozygous MIR137 knockout mice (Cheng et al., 2018).
Figure 4.

SZ PRS explained by MIR137 target genes (in Figure 2) is mainly attributed to a small number of neuron-specific genes
(A) Venn diagram of the five lists of MIR137 target genes expressed in different cell types derived from hiPSC. NPC, neuron progenitor cells; GABA, GABAergic neurons; DA, dopaminergic neurons; Glut, glutamatergic neurons.
(B) The PRS results from permutation (N = 1,000) for the small subset of MIR137 target genes expressed only in neuronal cell type (N = 182, denoted as “Neural”) or shared with hiPSC (N = 784, denoted as “iPSC”), using randomly selected 1,500 SNPs for each gene set at GWAS p-value threshold of <0.05. Data are represented as mean ± SEM.
(C) H-MAGMA analysis showing enrichment of SZ risk (PGC_wave3) in MIR137 target genes specific to each neuronal cell type but not in genes shared with iPSC (n = 784 from A). Shown in bubble plot are corresponding effect size (BETA) and enriched p-value (Bonferroni corrected; in -log10 scale) of each gene set.
To explore the hypothesis that a small subset of neuronal MIR137 target genes show a higher enrichment of SZ risk variance, we restricted the PRS analysis to the neural MIR137 target genes (N = 182; denoted as “Neural”) or those genes also expressed in iPSC (N = 784; denoted as “iPSC”) (Figure 4B). Similar to the SNP permutation test in Figure 2B, we compared the SZ variance explained by each subset of genes by using the same number of 1,500 SNPs in each permutation for each gene set at GWAS p-value threshold of <0.5 (Figure 4B). We found that the subset of neural MIR137 target genes (N = 182) disproportionally explained much greater SZ risk variance than those also expressed in iPSC (N = 784) or any other gene set tested in Figure 2B (Figures 3 and 4B).
We further conducted Hi-C-coupled MAGMA (H-MAGMA) analysis (de Leeuw et al., 2015; Sey et al., 2020) to orthogonally validate the enrichment of SZ GWAS risk in the small subset of neuronal MIR137 target genes (N = 75 in NPC to N = 161 in Glut) as opposed to genes expressed in both iPSC and neural cells (N = 784; Figure 4A). H-MAGMA improved the prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles (Sey et al., 2020), specifically the Hi-C data from iPSC-derived neurons (Rajarajan et al., 2018) for the current study (Figure 4C). Consistent with the result from our PRS analysis (Figure 4B), we found that MIR137 target genes specifically expressed in each subtype of neural cells, but not those also expressed in iPSC, showed robust enrichment of SZ GWAS risk (enrichment p-values ranging from 3.2 × 10−4 in GABA to 9.6 × 10−7 in Glut) (Figure 4C). Taken together, these analyses suggest that SZ PRS explained by MIR137 target genes is mainly attributed to a small number of neuron-specific genes.
SZ PRS of MIR137 target genes accounts for a disproportionally larger SZ risk variance of MGS sample
The maximum SZ risk variance explained by PRS of neuronal MIR137 target gene sets in the MGS sample was ~2%, whereas the whole genome SNP set explains up to ~10% SZ risk variance (Figure 2A). Considering the small number of MIR137 target genes (N = ~1,000) in each gene set in comparison with the number of all the RefSeq genes (N = ~20,000), the variance explained by neuronal MIR137 target gene sets suggests a disproportionally larger (4-fold) contribution of neuronal MIR137 target gene sets to genetic etiology of SZ. This observation is consistent with the “master regulator” role of MIR137 in post-transcriptional regulation. The larger proportion of SZ risk variance explained by neuronal MIR137 target gene sets was further supported by a comparative PRS analysis of gene targets of TCF4, a transcriptional “master regulator” (and also a prominent SZ GWAS risk locus): PRS of MIR137 target gene sets explained higher SZ risk variance in the MGS sample (Figures 2A and S1, and Table S3).
Permutation tests of gene set-specific PRS analysis accounting for SNP number in each gene set also demonstrated an overall better performance of the PRS of neuronal MIR137 target genes in explaining SZ risk variance (Table S4), compared with the PRS of TCF4 target gene sets or the genome-wide SNP set (Figures 2B and S1B). Our independent partitioned heritability analysis further showed neuronal MIR137 target gene sets explained disproportionally more SZ heritability (with PGC3 SZ GWAS summary statistics) than TCF4 target gene sets (Figure 2C and Table S5).
The European ancestry-based PRS of MIR137 target gene sets explains smaller SZ risk variance of Han Chinese in cell type-specific manner
Although genome-wide PRS is usually considered to not be portable between samples of different ancestry, PRS of a disease-relevant gene pathway/set may perform better in explaining disease risk variance across populations (Baker et al., 2018). Given the strong genetic association of the MIR137 locus with SZ in both European and Han Chinese samples, as well as our observed large contribution of neuronal MIR137 target genes to SZ risk variance in the MGS sample (Figure 2), we conducted a PRS analysis in a Han Chinese SZ sample for the same gene lists and by using the same European ancestry-based PGC-SZ3-minus-MGS summary GWAS statistics. As expected, we observed that the PGC-SZ3 genome-wide PRS explained much smaller (~2%) SZ risk variance in the tested Han Chinese sample (Figure 5A, Figure S4A, and Table S3) (versus ~10% in MGS). Similarly, PRS of MIR137 target genes also explained much less variance of the Han Chinese sample (up to ~0.3% with extended 20 kb; Figure 5A) than of the MGS sample (Figure 2A).
Figure 5.

PRS analysis of different SNP lists of MIR137 and TCF4 target genes annotated in a 20-kb window for the Han Chinese sample
(A) PRS result using LD-pruned SNPs (r2 < 0.2) for each gene list. The variance explained in the target sample is based on risk scores derived from an aggregated sum of weighted SNP risk allele effect sizes estimated from the discovery samples at seven significance thresholds (p < 0.01, 0.05, 0.1, 0.2, 0.3, 0.4, and 0.5). The y axis indicates the percentage of phenotypic variance explained by the PRS (Nagelkerke's pseudo R2).
(B) The permutation (N = 1,000) PRS results based on randomly selected 1,500 SNPs for each gene set. Data are presented as mean ± SEM. Gene sets in (A) and (B) were described in Table 1.
We further examined whether the PRS of neuronal MIR137 target genes also explain larger SZ risk variance of Han Chinese sample than that of genes with expression altered in vitro by MIR137 or the TCF4 target gene sets as we observed for MGS sample. For gene sets with the 20-kb extended genic region, although we observed a better predictive performance of PRS of the entire SNP sets of neuronal MIR137 genes at p < 0.01 (Figures 5A and 3), permutation testing accounting for SNP number in each gene list did not provide support (Figures 5B and 3). However, for gene sets with a 100-kb extended genic region at some SZ GWAS p-value cut-offs (p < 0.01, <0.05, and <0.5) both the PRS analysis of the entire SNP sets and the permutation test supported that the PRS of neuronal MIR137 target genes overall explain larger SZ risk variance of the Han Chinese sample than that of non-neuronal MIR137 target genes or genes simply altered by changing MIR137 expression (Figures S4 and 3). As observed for MGS, the PRS of neuronal MIR137 target genes with the extended 100-kb genic region overall explained larger SZ risk variance of the Han Chinese sample compared with the PRS of TCF4 target genes or over genome-wide random SNP sets (Figures S4B and 3). However, the same pattern was not observed for the gene sets with the 20-kb extended genic region, even with the same number of randomly selected SNPs in each gene set (Figure 5B), which may be due to the different LD structure in the Han Chinese sample that makes the PRS analysis very sensitive to the SNP numbers in each gene set. Altogether, these results suggest that, compared with the MGS sample of European ancestry, there was only a more modest proportional advantage of neuronal MIR137 gene sets over non-neuronal gene sets or genomic controls in explaining SZ risk variance in Han Chinese.
Discussion
Gene pathway- or set-based PRS analysis in a large cohort can help inform disease biology and identify core gene networks that contribute most risk to a polygenic or omnigenic disorder (Baker et al., 2018). Furthermore, because a disorder-relevant gene pathway may contain SNPs that show more convergent cross-ancestry genetic associations, the pathway-based PRS in a sample of one ancestry may be somewhat more portable for predicting individual risks in a population of another ancestry. Here, for MIR137, a leading SZ GWAS risk gene and a post-transcriptional master regulator, our gene pathway-based PRS analysis suggested a cell type-specific effect of PRS in explaining SZ risk variance and a disproportionally large contribution of MIR137 target gene set to SZ risk in a European ancestry sample and to a lesser degree in a Han Chinese sample.
MIR137 is a leading SZ risk gene that is predicted by TargetScan to post-transcriptionally regulate more than 1,000 genes. However, the relative genetic contribution of the MIR137 gene pathway to SZ etiology has not been examined. Through cell type-specific PRS analyses, we have found that neuronal MIR137 target genes can explain ~2% of SZ risk variance in the MGS European ancestry sample (Figure 2), which accounts for one-fifth of the total risk variance explained by the genome-wide SNP set. Despite an overall much smaller proportion (as expected) of SZ risk variance of Han Chinese explained by the European ancestry-based SZ PRS, a comparable proportion of PRS contribution from neuronal MIR137 target gene pathway was identified for Han Chinese. The cross-population convergent PRS contribution of the neuronal MIR137 target gene sets is not unexpected, because MIR137 is also a leading SZ GWAS locus in the Han Chinese sample (Li et al., 2017). The same GWAS SNP at the MIR137 locus has also been associated with brain imaging phenotypes in the Han Chinese population (Liu et al., 2014). The relatively large contribution of the MIR137 gene pathway to SZ risk in both populations (Figures 2B, 2C, 5B, and S4B) presents a clear contrast to the target gene sets of TCF4, another leading GWAS risk gene and a transcriptional master regulator (Doostparast Torshizi et al., 2019). These results suggest a possibly shared disease pathway involving MIR137 and its target genes across populations.
Our cell type-specific MIR137 target gene set PRS contribution to SZ risk also implies the importance of considering the most disease-relevant cell types in PRS analysis. Gene sets with expression altered by either over-expressing or knocking down MIR137 expression have been used in previous PRS analysis of SZ-relevant cognitive performance, but with mixed outcomes (Cosgrove et al., 2017, 2018). By comparing with the exact set of MIR137 gene sets used by these studies (Cosgrove et al., 2017, 2018), we have shown here that PRSs of neuronal MIR137 target gene sets rather than those genes simply altered by manipulating MIR137 expression better explain SZ risk variance in a target sample (Figure 2). The improved performance of neuronal MIR137 target gene sets was not unexpected, because many genes showing gene expression changes upon artificially changing MIR137 expression may just reflect the noise of transcriptomic data, i.e., not specific to MIR137 function. The importance of cell type specificity in PRS analysis is echoed by a recent study that shows PRSs derived from cell type-specific regulatory elements can substantially increase its trans-ancestry portability (Amariuta et al., 2020). Beyond disorder-relevant cell types, other considerations that reflect the convergent disease biology, such as developmental stage or variant functionality, may further improve the applicability of PRS.
Identifying the core gene networks and the functionally relevant cell types for a polygenic disorder has been a challenge. Leveraging the transcriptomic profiles of hiPSC-derived relatively pure (>80%) neuronal cultures (Forrest et al., 2017; Zhang et al., 2018, 2020), both our cell type-specific PRS analyses of MIR137 target gene sets with the same number of randomly selected SNPs (Figure 2B) and our independent partitioned heritability analysis for each gene set (Figure 2C) indicated that Glut, DA, and GABA neurons are all relevant cell types for MIR137 gene pathway to function and contribute SZ risk, which has not been previously demonstrated. Indeed, the implicated specific cell types where the PRS of MIR137 target gene sets better explain SZ risk variance are consistent with our previous studies that showed high-level expression of MIR137 in DA and Glut neurons, but not as much in NPCs or iPSCs (Forrest et al., 2017; Shi et al., 2014). Moreover, our observation that the PRS of the Glut neuronal MIR137 target gene set explained the highest SZ risk variance in MGS (Figure 2) is consistent with our recent report of a negative correlation of MIR137 expression and its target gene GRIA1 in dendritic protrusions (or spines) of hiPSC-derived Glut neurons (Forrest et al., 2017). Identifying disease relevant cell type(s) is important for understanding the biology of SZ (Skene et al., 2018). The significance of our current analysis is the identification of SZ-relevant cell types for a specific gene pathway through cell type-specific and cross-population PRS analyses (Figures 2 and 3), providing a more tractable mechanistic insight on understanding the biology of SZ.
Limitations of the study
We acknowledge the limitations in our analyses. First, although the number of genes in each list are comparable (N~1,000), the number of SNPs (and gene lengths) in each cell type or population may vary, which may confound the SZ risk variance explained by a specific gene/SNP set. Nevertheless, our permutation tests using the same number SNPs randomly selected from each gene list and our independent partitioned heritability analysis (Figures 2B, 2C, S1B, and 3B), which was expected to account for the varying SNP number and gene length in each gene list, revealed consistent results with the observation directly from all SNPs in each gene set. Second, some other cell types such as astrocytes and microglia might also be relevant to MIR137 function, which was not investigated in our study; however, compared with glutamatergic and GABAergic neurons, these cell types are less genetically relevant to SZ (Skene et al., 2018). Third, iPSC-derived neurons, although relatively pure, are commonly known to represent immature neurons (during the prenatal stages), and our observations may thus not reflect the disorder biology at the adult stage of SZ. Nevertheless, SZ is known to have a strong neurodevelopmental pathogenic aspect and hiPSC models have been effectively used for understanding SZ-relevant functional genomics and disease biology (Amiri et al., 2018; Forrest et al., 2017; Rajarajan et al., 2018). Finally, although MIR137 post-transcriptionally functions through targeting 3′-UTRs of its target genes, our gene set PRS analysis was conducted with genetic risk variants not only at 3′-UTRs of these MIR137 target genes but also in other parts of the genes such as enhancers. More comprehensive PRS analysis or partitioned heritability analysis of the MIR137 gene pathway in different cell types at various developmental stages, and by variant location in different genomic/epigenomic features, may shed further mechanistic insight onto how MIR137 confers individual susceptibility to SZ.
In summary, by combining pathway-based PRS analysis with cell type-specific transcriptomic profiles in hiPSC-derived neurons, we discovered that MIR137 target genes expressed in three major SZ-relevant neuronal cell types explain a larger proportion of SZ risk variance in European and, to a lesser degree, Han Chinese samples. Despite the limitations, our analysis informed the SZ-relevant cell types for MIR137 to function, highlighting the value of pathway-based PRS analysis in uncovering disorder-relevant biological features. Knowledge on cross-population high-risk gene pathways may not only help improve the specificity of PRS-based individual risk prediction but also have the potential to facilitate the translation of genetic findings into pathway-based therapeutic interventions.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jubao Duan (jduan@uchicago.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The PGC3 SZ GWAS summary statistics can be accessed through “schizophrenia data access portal” (https://www.med.unc.edu/pgc/pgc-workgroups/schizophrenia/). The Chinese Han SZ GWAS summary statistics have been deposited in the Peking University Open Research Data repository with accession number 10638 (https://doi.org/10.18170/DVN/IHROUE). The gene expression data used to generate the list of MIR137-target genes expressed in different neural cell types can be accessed through GEO: GSE129017 and GSE132757. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Method details
Genetic data sets
PGC-SZ3
The PGC-SZ3 (The Schizophrenia Working Group of the Psychiatric Genomics Consortium et al., 2020) dataset was downloaded through approved access from PGC SZ workgroup (www.med.unc.edu/pgc/pgc-workgroups/schizophrenia). This specific dataset is the summary statistics of PGC-SZ samples of European ancestry leaving out MGS. This PGC-SZ3 dataset contains the odds ratios (ORs) and p values that were calculated on 7,651,682 SNPs in the meta-analysis of all 75 European cohorts (except the MGS data), and was used as a discovery sample to identify SZ risk variants.
MGS
MGS was used as a target sample of European ancestry. This MGS dataset contains GWAS data for 2,681 SZ cases and 2,653 controls (3,134 males, 2,200 females; mean age = 46.8 with 14.7 SD) (Shi et al., 2009). Quality control (QC) performed on the genotype data was previously reported (Shi et al., 2009). Genotype imputation was conducted using the IMPUTE2 software program (version 2.1.2) (Howie et al., 2009, 2011), using haplotypes from all 2,504 individuals in the 1,000 Genomes Phase 3 panel (October 2014 Data Release) as a reference panel (Auton et al., 2015). The haplotypes were phased using SHAPEIT2 (version v2.r644) (Delaneau et al., 2011), which can perform the pre-phasing step for the study genotypes to produce ‘best-guess’ haplotypes. The imputed SNPs with low quality were excluded (IMPUTE2 info <0.6, IMPUTE2 certainty <0.8, or minor allele frequency (MAF) < 0.05). A total of 5,132,475 imputed SNPs was retained in the follow-up PRS analyses. The NorthShore University HealthSystem IRB approved the study.
Han Chinese
The Han Chinese dataset contains 4,288 SZ cases and 5,512 controls (4,395 males, 5,405 females; mean age = 31 with 10.03 SD) (Li et al., 2017; Yu et al., 2017). Genotyping of these samples was conducted using different types of Illumina Genome-Wide Arrays: GWAS1 dataset using HumanHap610-Quad BeadChips, GWAS2 dataset using Human660W-Quad BeadChip, and GWAS3 and GWAS4 datasets using Illumina Human OmniZhongHua BeadChips specifically designed for the Chinese population. The genotyping QC filters included: genotype call rate >98%, MAF >0.01, difference in SNP missingness between cases and controls <0.02, Hardy-Weinberg disequilibrium p > 1 × 10−6. Samples were excluded for gender discordance and genetic ancestry outliers. Genotypes were first phased using SHAPEIT (v2.r727) (Delaneau et al., 2011), and imputation was then performed over each 3 Mb-interval centered on all index SNPs using IMPUTE (v2.3.0) software (Howie et al., 2009, 2011). Haplotypes derived from Phase 1 of the 1000 Genomes Project (release v3) were used as reference data. After imputation, we excluded SNPs with an imputation quality score below a set threshold (info<0.6), call rate <0.98 in either cases or controls, MAF<0.01, Hardy-Weinberg disequilibrium p < 1 × 10−6.
PRS analysis
To explore the genetic relationship between the discovery sample (PGC-SZ3 without MGS) and target samples (MGS, Han Chinese) datasets, we used PRSice to conduct a standard PRS analysis (Euesden et al., 2015) as in the PRS workflow (Figure 1). Let Pd be the p values of the GWAS study in the discovery sample. Specifically, ‘significant’ SNPs with Pd<0.5 were pruned based on linkage disequilibrium (LD) (r2 < 0.2) in MGS data, and then selected based on seven predetermined significance thresholds (Pd<0.01, 0.05, 0.1, 0.2, 0.3, 0.4, and 0.5) in PGC-SCZ3. Within each pruned SNP set under each significance threshold, a quantitative aggregate risk score was calculated for each individual in the target samples, defined as a sum across SNPs of the number of reference alleles (0, 1, or 2) at that SNP multiplied by the effect size measures (log of OR) for that SNP estimated from the discovery sample. Association of aggregate risk score and actual SZ in the target samples were performed with logistic regression adjusted for gender, age, and 10 principal components to control for population stratification. We then calculated the percentage of phenotypic variance explained by the aggregate risk score (Nagelkerke's pseudo R2) of different SNP/gene-sets (see below).
Gene-sets
We have used the same lists of 900–1,000 genes previously used for MIR137 PRS analysis (Cosgrove et al., 2017; Hill et al., 2014) for comparison to our own gene sets. These lists of genes are those with expression altered by overexpressing MIR137 or knocking down MIR137 in a single NPC cell line, selected by the fold-change of gene expression and their statistical significance (Cosgrove et al., 2017; Hill et al., 2014).
For testing lists of cell type-specific MIR137 target genes (900–1,000 genes per list), we assembled them by cross-referencing the predicted MIR137 target genes (target prediction score >0.1 by TargetScan 6.2) with their expression value FPKM (fragments per kilobase per million reads) in hiPSC and hiPSC-derived NPCs, dopaminergic neurons (DA), GABAergic neurons (GABA), and glutamatergic neurons (Glut) (Forrest et al., 2017; Zhang et al., 2018, 2020). Briefly, RNA-seq data (GENCODE v28) from cell lines of 8 donors (genetic backgrounds) per cell type (iPSC, NPC, DA, and GABA) (Zhang et al., 2020) were used to estimate the average gene expression level. For Glut neurons, we used NGN2-induced excitatory neurons at day-27 post neuronal induction, and expression values were calculated as the average of all available samples (Forrest et al., 2017; Zhang et al., 2018, 2020). An expressed gene in each cell type was defined using a cutoff value of log2 FPKM >0.5. Subsequently, the lists of expressed genes were cross-referenced with predicted target gene list of MIR137 in each cell type.
For targets of TCF4, we have compiled 3 lists of ~1,000 genes each. These include genes with expression either up- or down-regulated by knockdown TCF4 expression in NPCs and in early-stage Glut neurons (Doostparast Torshizi et al., 2019). For genes in both cell types, we only selected the top 1,000 genes showing differential expression upon TCF4 knockdown. The direct binding targets of TCF4 were extracted from an empirical chromatin immune-precipitation (ChIP) study, and the top ranking 1,000 gene were selected according to their promoter and intergenic ChIP peak significance (Forrest et al., 2017).
Permutation to account for variable SNP number
The number of genes varies from 909 to 1,017 among 10 gene sets from both MIR137 and TCF4 (Table 1 and Table S1). Furthermore, the number of SNPs for each gene set varies in both MGS data and Chinese data before/after LD pruning (Table S2). Therefore, to adjust for different SNP number in each gene set, PRS were evaluated based on the same number of SNPs that were randomly selected within each gene set in 1,000 permutations. A total of 3,000 and 1,500 SNPs (Pd<0.5) was randomly selected in the permutation for both the MGS and the Han Chinese datasets annotated within 20kb and 100kb windows (i.e., extended genic regions), respectively. The detailed procedures were:
-
1)
Obtain GWAS summary statistics (Pd values and ORs) in the discovery sample (PGC-SZ3 without MGS).
-
2)
Cross-reference with target sample data with both genetic and phenotype data and identify SNPs in common between the discovery and target samples.
-
3)
Account for association redundancy due to LD by SNP pruning (LD-r2 = 0.2 as a cut-off) among all overlapped SNPs with Pd<0.5.
-
4)
Extract SNPs located within a specific gene-set, and randomly select Nrandom SNPs, where Nrandom was selected based on the number of SNPs after LD pruning (Table S2), and Nrandom = 1500/3000 for data within 20kb and 100kb window, respectively.
-
5)
Restrict to SNPs with various thresholds (Pd<0.01, 0.05, 0.1, 0.2, 0.3, 0.4, and 0.5).
-
6)
Construct PRS = sum of risk alleles weighted by log (OR) from regression.
-
7)
Regress trait in target sample onto PRS. Evaluate strength of this association adjusted by age, sex, and 10 principal components of ancestry (Nagelkerke's pseudo R2).
-
8)
Repeat steps 2-8 for 1,000 permutations.
These procedures were repeated for all the gene-sets including the whole-genome SNP set for both MGS and Han Chinese samples as target samples. Two-sample Student's t-tests were further performed on 1,000 permutated Nagelkerke's pseudo R2 values to test for the mean difference between any two gene-sets.
Partitioned heritability analysis by stratified LDSC
To independently validate the gene-set specific PRS analysis and account for potential confounding factors (e.g., numbers of SNPs) in PRS analysis, we performed a sLDSC analysis (Bulik-Sullivan et al., 2015; Finucane et al., 2015) for different gene sets using PGC SCZ wave_3 GWAS summary statistics. For calculating partitioned LD scores, a 20 kb window size (similar to what was used in Figures 2A and 2B) was applied to all genes within the ten gene sets. For partitioned heritability (h2), we used baseline model LD scores from the 1,000 Genomes project European cohort (1000G_EUR_Phase3_baseline v2.2). Finally, we used the cell-type group analysis algorithm as described in the LDSC documentation (Bulik-Sullivan et al., 2015; Finucane et al., 2015) to estimate the proportion of heritability and identify the enrichment of h2 in each gene set.
Gene-set GWAS risk enrichment analysis by H-MAGMA
To account for gene sizes and overlapping genetic variants in different gene sets as well as other potential cofounding factors, we performed H-MAGMA analysis using MAGMA version 1.08 (Sey et al., 2020) to evaluate the enrichment for SZ GWAS risk. Specifically, we first started by generating the MAGMA-required gene analysis data files using pre-compiled gene-SNP annotation files. Compared to the original MAGMA analysis, H-MAGMA improved the prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles (de Leeuw et al., 2015; Sey et al., 2020). Since we defined neural gene sets using iPSC-derived neurons, we used the gene-SNP annotation file pre-compiled with Hi-C data from iPSC-derived neurons (Rajarajan et al., 2018). With the gene-SNP annotation file, we then performed gene-level analysis on SNP p-values using the reference SNP data of 1,000 Genomes European panel (g1000_eur, --bfile) and the pre-computed SNP p-values from the PGC SZ GWAS wave 3 dataset (daner_PGC_SCZ_w3, --pval). The sample size (ncol = ) was directly taken from the column of sample sizes per SNP column of the PGC3 SZ dataset. Subsequently, the result files (--gene-results) from the gene-level analysis were read in for competitive gene-set analysis (--set-annot), where we used default setting (‘correct = all’) to control for gene sizes in number of SNPs and the gene density (a measure of within-gene LD). The gene-set analysis produced output file (gsa.out) with competitive gene-set analysis results that contained the effect size (BETA) and the statistical significance of the enrichment of each gene set for SZ GWAS risk.
Quantification and statistical analysis
To quantify the SZ risk variance explained, we used PRSice to conduct a standard PRS analysis (Euesden et al., 2015). A quantitative aggregate risk score was calculated for each individual in the target samples. The percentage of phenotypic variance explained by the PRS (Nagelkerke's pseudo R2) was presented in Figures 2, 3, 4, and 5. To compare the SZ risk variance explained by different gene sets, we accounted for SNP number used in each gene set by permutation test (N = 1,000) using the randomly selected 1,500 SNPs for each gene-set. Data are presented as mean ± SEM in Figures 2 and 5. To further determine whether the difference of the SZ risk variance explained by the equal number of randomly selected SNPs (1,000 permutations) of two gene-sets are statistically significant, we used two-sample Student's t-test and corrected p-values for multiple testing by Bonferroni method (Figure 3). To quantify the enrichment of SZ GWAS SNP heritability (h2) for each gene set, we used sLDSC analysis (Bulik-Sullivan et al., 2015; Finucane et al., 2015) to determine the folds of enrichment of SNP heritability (proportion of explained heritability h2 normalized by the proportion of SNPs in each gene set) and derive the statistical significance (Bonferroni-corrected p-values) in Figure 2.
Acknowledgments
MGS includes P. V. Gejman, A. R. Sanders, J. Duan (NorthShore University HealthSystem and University of Chicago, IL, USA); D. F. Levinson (Stanford University, CA, USA); J. Shi (National Cancer Institute, MD, USA); N. G. Buccola (Louisiana State University Health Sciences Center, LA, USA); B. J. Mowry (Queensland Center for Mental Health Research, Brisbane and Queensland Brain Institute, The University of Queensland, Australia); R. Freedman, A. Olincy (University of Colorado Denver, CO, USA); F. Amin (Atlanta Veterans Affairs Medical Center and Emory University, GA, USA); D. W. Black (University of Iowa Carver College of Medicine, IA, USA); J. M. Silverman (Mount Sinai School of Medicine, NY, USA); W. F. Byerley (University of California at San Francisco, CA, USA); and C. R. Cloninger, D. M. Svrakic (Washington University, MO, USA). We thank the study participants of MGS. MGS was mainly supported by R01MH059571, R01MH081800, and U01MH079469 (to P.V. Gejman.); and other NIH grants for other MGS sites (R01MH067257 to N. G. Buccola, R01MH059588 to B. J. Mowry, R01MH059565 to R. Freedman, R01MH059587 to F. Amin, R01MH060870 to W. F. Byerley, R01MH059566 to D. W. Black, R01MH059586 to J. M. Silverman., R01MH061675 to D. F. Levinson, R01MH060879 to C. R. Cloninger, U01MH046276 to C. R. Cloninger, and U01MH079470 to D. F. Levinson). We thank Dr. P. V. Gejman for helping with the interpretation of the MGS dataset. We also thank the PGC-SZ group for providing early access of wave-3 (PGC-SZ3) summary statistics. This study was supported by Shanghai Municipal Science and Technology Major Project (Grant No. 2017SHZDZX01) and by Department of Computational Biology and Institute of Life Sciences and Human Phenomics Institute (to Y.Y.), and NIH grants R21MH102685, R01MH106575, R01MH116281, and R01AG063175 (to J.D.).
Author contributions
W.G. performed the PRS analysis and wrote the manuscript; S.Z. prepared most genomics datasets and performed partitioned heritability and gene set enrichment analyses; H. Yu and H. Yan analyzed the Han Chinese GWAS dataset; H.Z. produced the iPSC-derived neuronal expression data; A.R.S. helped with the interpretation of MGS dataset and wrote the manuscript; W.Y. supervised the analysis of the Han Chinese GWAS and wrote the manuscript; Y.Y. supervised the overall PRS analyses and wrote the manuscript; J.D. conceived the study, coordinated and supervised the analyses, and wrote the manuscript.
Declaration of interests
The authors declare no conflicts of interest.
Published: July 23, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102785.
Contributor Information
Yin Yao, Email: yin_yao@fudan.edu.cn.
Jubao Duan, Email: jduan@uchicago.edu.
Supplemental information
References
- Amariuta T., Ishigaki K., Sugishita H., Ohta T., Koido M., Dey K.K., Matsuda K., Murakami Y., Price A.L., Kawakami E. Improving the trans-ancestry portability of polygenic risk scores by prioritizing variants in predicted cell-type-specific regulatory elements. Nat. Genet. 2020;52:1346–1354. doi: 10.1038/s41588-020-00740-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiri A., Coppola G., Scuderi S., Wu F., Roychowdhury T., Liu F., Pochareddy S., Shin Y., Safi A., Song L. Transcriptome and epigenome landscape of human cortical development modeled in organoids. Science. 2018;362:eaat6720. doi: 10.1126/science.aat6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A., Abecasis G.R., Altshuler D.M., Durbin R.M., Abecasis G.R., Bentley D.R., Chakravarti A., Clark A.G., Donnelly P., Eichler E.E. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker E., Schmidt K.M., Sims R., O'Donovan M.C., Williams J., Holmans P., Escott-Price V., Consortium W.T.G. POLARIS: polygenic LD-adjusted risk score approach for set-based analysis of GWAS data. Genet. Epidemiol. 2018;42:366–377. doi: 10.1002/gepi.22117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle E.A., Li Y.I., Pritchard J.K. An Expanded View of complex traits: from polygenic to omnigenic. Cell. 2017;169:1177–1186. doi: 10.1016/j.cell.2017.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik-Sullivan B.K., Loh P.-R., Finucane H.K., Ripke S., Yang J., Patterson N., Daly M.J., Price A.L., Neale B.M., Schizophrenia Working Group of the Psychiatric Genomics Consortium LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015;47:291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y., Wang Z.-M., Tan W., Wang X., Li Y., Bai B., Li Y., Zhang S.-F., Yan H.-L., Chen Z.-L. Partial loss of psychiatric risk gene Mir137 in mice causes repetitive behavior and impairs sociability and learning via increased Pde10a. Nat. Neurosci. 2018;21:1689–1703. doi: 10.1038/s41593-018-0261-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins A.L., Kim Y., Bloom R.J., Kelada S.N., Sethupathy P., Sullivan P.F. Transcriptional targets of the schizophrenia risk gene MIR137. Transl. Psychiatry. 2014;4:e404. doi: 10.1038/tp.2014.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Psychiatric Genome-Wide Association Study (GWAS)Consortium Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 2011;43:969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Schizophrenia Working Group of the Psychiatric Genomics Consortium, Ripke S., Walters J.T.R., O'Donovan M.C. Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. MedRxiv. 2020 doi: 10.1101/2020.09.12.20192922. [DOI] [Google Scholar]
- Cosgrove D., Harold D., Mothersill O., Anney R., Hill M.J., Bray N.J., Blokland G., Petryshen T., Richards A., Mantripragada K. MiR-137-derived polygenic risk: effects on cognitive performance in patients with schizophrenia and controls. Transl. Psychiatry. 2017;7:e1012. doi: 10.1038/tp.2016.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove D., Mothersill D.O., Whitton L., Harold D., Kelly S., Holleran L., Holland J., Anney R., Richards A., Mantripragada K. Effects of MiR-137 genetic risk score on brain volume and cortical measures in patients with schizophrenia and controls. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018;177:369–376. doi: 10.1002/ajmg.b.32620. [DOI] [PubMed] [Google Scholar]
- Delaneau O., Marchini J., Zagury J.F. A linear complexity phasing method for thousands of genomes. Nat. Methods. 2011;9:179–181. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- Doostparast Torshizi A., Armoskus C., Zhang H., Forrest M.P., Zhang S., Souaiaia T., Evgrafov O.V., Knowles J.A., Duan J., Wang K. Deconvolution of transcriptional networks identifies TCF4 as a master regulator in schizophrenia. Sci. Adv. 2019;5:eaau4139. doi: 10.1126/sciadv.aau4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euesden J., Lewis C.M., O'Reilly P.F. PRSice: polygenic risk score software. Bioinformatics. 2015;31:1466–1468. doi: 10.1093/bioinformatics/btu848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane H.K., Bulik-Sullivan B., Gusev A., Trynka G., Reshef Y., Loh P.-R., Anttila V., Xu H., Zang C., Farh K. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015;47:1228–1235. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest M.P., Zhang H., Moy W., McGowan H., Leites C., Dionisio L.E., Xu Z., Shi J., Sanders A.R., Greenleaf W.J. Open chromatin profiling in hiPSC-derived neurons prioritizes functional noncoding psychiatric risk variants and highlights neurodevelopmental loci. Cell Stem Cell. 2017;21:305–318.e8. doi: 10.1016/j.stem.2017.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green M.J., Cairns M.J., Wu J., Dragovic M., Jablensky A., Tooney P.A., Scott R.J., Carr V.J. Genome-wide supported variant MIR137 and severe negative symptoms predict membership of an impaired cognitive subtype of schizophrenia. Mol. Psychiatry. 2012;18:774–780. doi: 10.1038/mp.2012.84. [DOI] [PubMed] [Google Scholar]
- Guarnieri D.J., DiLeone R.J. MicroRNAs: a new class of gene regulators. Ann. Med. 2008;40:197–208. doi: 10.1080/07853890701771823. [DOI] [PubMed] [Google Scholar]
- Hill M.J., Donocik J.G., Nuamah R.A., Mein C.A., Sainz-Fuertes R., Bray N.J. Transcriptional consequences of schizophrenia candidate miR-137 manipulation in human neural progenitor cells. Schizophr. Res. 2014;153:225–230. doi: 10.1016/j.schres.2014.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie B.N., Donnelly P., Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie B., Marchini J., Stephens M. Genotype imputation with thousands of genomes. G3 (Bethesda) 2011;1:457–470. doi: 10.1534/g3.111.001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im H.I., Kenny P.J. MicroRNAs in neuronal function and dysfunction. Trends Neurosci. 2012;35:325–334. doi: 10.1016/j.tins.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon E., Wang W., Tsai L.H. Validation of schizophrenia-associated genes CSMD1, C10orf26, CACNA1C and TCF4 as miR-137 targets. Mol. Psychiatry. 2013;18:11–12. doi: 10.1038/mp.2011.170. [DOI] [PubMed] [Google Scholar]
- Lee S.H., DeCandia T.R., Ripke S., Yang J., Sullivan P.F., Goddard M.E., Keller M.C., Visscher P.M., Wray N.R. Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nat. Genet. 2012;44:247–250. doi: 10.1038/ng.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw C.A., Mooij J.M., Heskes T., Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015;11:e1004219. doi: 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Chen J., Yu H., He L., Xu Y., Zhang D., Yi Q., Li C., Li X., Shen J. Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nat. Genet. 2017;49:1576–1583. doi: 10.1038/ng.3973. [DOI] [PubMed] [Google Scholar]
- Liu B., Zhang X., Hou B., Li J., Qiu C., Qin W., Yu C., Jiang T. The impact of MIR137 on dorsolateral prefrontal-hippocampal functional connectivity in healthy subjects. Neuropsychopharmacology. 2014;39:2153–2160. doi: 10.1038/npp.2014.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S.M., Wray N.R., Stone J.L., Visscher P.M., O'Donovan M.C., Sullivan P.F., Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajarajan P., Borrman T., Liao W., Schrode N., Flaherty E., Casiño C., Powell S., Yashaswini C., LaMarca E.A., Kassim B. Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science. 2018;362:eaat4311. doi: 10.1126/science.aat4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmers C., Sweet R.A., Penzes P. Abnormal kalirin signaling in neuropsychiatric disorders. Brain Res. Bull. 2014;103C:29–38. doi: 10.1016/j.brainresbull.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke and Consortium Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sey N.Y.A., Hu B., Mah W., Fauni H., McAfee J.C., Rajarajan P., Brennand K.J., Akbarian S., Won H. A computational tool (H-MAGMA) for improved prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles. Nat. Neurosci. 2020;23:583–593. doi: 10.1038/s41593-020-0603-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Levinson D.F., Duan J., Sanders A.R., Zheng Y., Pe'er I., Dudbridge F., Holmans P.A., Whittemore A.S., Mowry B.J. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S., Leites C., He D., Schwartz D., Moy W., Shi J., Duan J. MicroRNA-9 and microRNA-326 regulate human dopamine D2 receptor expression and the microRNA-mediated expression regulation is altered by a genetic variant. J. Biol. Chem. 2014;289:13434–13444. doi: 10.1074/jbc.M113.535203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silber J., Lim D.A., Petritsch C., Persson A.I., Maunakea A.K., Yu M., Vandenberg S.R., Ginzinger D.G., James C.D., Costello J.F. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 2008;6:14. doi: 10.1186/1741-7015-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene N.G., Bryois J., Bakken T.E., Breen G., Crowley J.J., Gaspar H.A., Giusti-Rodriguez P., Hodge R.D., Miller J.A., Munoz-Manchado A.B. Genetic identification of brain cell types underlying schizophrenia. Nat. Genet. 2018;50:825–833. doi: 10.1038/s41588-018-0129-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smrt R.D., Szulwach K.E., Pfeiffer R.L., Li X., Guo W., Pathania M., Teng Z.Q., Luo Y., Peng J., Bordey A. MicroRNA miR-137 regulates neuronal maturation by targeting ubiquitin ligase mind bomb-1. Stem Cells. 2010;28:1060–1070. doi: 10.1002/stem.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H., Ophoff R.A., Steinberg S., Andreassen O.A., Cichon S., Rujescu D., Werge T., Pietilainen O.P., Mors O., Mortensen P.B. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun G., Ye P., Murai K., Lang M.F., Li S., Zhang H., Li W., Fu C., Yin J., Wang A. miR-137 forms a regulatory loop with nuclear receptor TLX and LSD1 in neural stem cells. Nat. Commun. 2011;2:529. doi: 10.1038/ncomms1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulwach K.E., Li X., Smrt R.D., Li Y., Luo Y., Lin L., Santistevan N.J., Li W., Zhao X., Jin P. Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J. Cell Biol. 2010;189:127–141. doi: 10.1083/jcb.200908151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topol A., Zhu S., Hartley B.J., English J., Hauberg M.E., Tran N., Rittenhouse C.A., Simone A., Ruderfer D.M., Johnson J. Dysregulation of miRNA-9 in a subset of schizophrenia patient-derived neural progenitor cells. Cell Rep. 2016;15:1024–1036. doi: 10.1016/j.celrep.2016.03.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursini G., Punzi G., Chen Q., Marenco S., Robinson J.F., Porcelli A., Hamilton E.G., Mitjans M., Maddalena G., Begemann M. Convergence of placenta biology and genetic risk for schizophrenia. Nat. Med. 2018;24:792–801. doi: 10.1038/s41591-018-0021-y. [DOI] [PubMed] [Google Scholar]
- Vassos E., Kou J., Tosato S., Maxwell J., Dennison C.A., Legge S.E., Walters J.T.R., Owen M.J., O’Donovan M.C., Breen G. Lack of support for the genes by early environment interaction hypothesis in the pathogenesis of schizophrenia. Schizophr. Bull. 2021:sbab052. doi: 10.1093/schbul/sbab052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volvert M.L., Rogister F., Moonen G., Malgrange B., Nguyen L. MicroRNAs tune cerebral cortical neurogenesis. Cell Death Differ. 2012;19:1573–1581. doi: 10.1038/cdd.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen M.H., Valles A., Kirkels L.A., Mastebroek M., Olde Loohuis N., Kos A., Wissink-Lindhout W.M., de Brouwer A.P., Nillesen W.M., Pfundt R. Chromosome 1p21.3 microdeletions comprising DPYD and MIR137 are associated with intellectual disability. J. Med. Genet. 2011;48:810–818. doi: 10.1136/jmedgenet-2011-100294. [DOI] [PubMed] [Google Scholar]
- Wright C., Turner J.A., Calhoun V.D., Perrone-Bizzozero N. Potential impact of miR-137 and its targets in schizophrenia. Front. Genet. 2013;4:58. doi: 10.3389/fgene.2013.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., Yan H., Li J., Li Z., Zhang X., Ma Y., Mei L., Liu C., Cai L., Wang Q. Common variants on 2p16.1, 6p22.1 and 10q24.32 are associated with schizophrenia in Han Chinese population. Mol. Psychiatry. 2017;22:954–960. doi: 10.1038/mp.2016.212. [DOI] [PubMed] [Google Scholar]
- Zeng Y., Navarro P., Fernandez-Pujals A.M., Hall L.S., Clarke T.K., Thomson P.A., Smith B.H., Hocking L.J., Padmanabhan S., Hayward C. A combined pathway and regional heritability analysis indicates NETRIN1 pathway is associated with major depressive disorder. Biol. Psychiatry. 2017;81:336–346. doi: 10.1016/j.biopsych.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Moy W., Zhang H., Leites C., McGowan H., Shi J., Sanders A.R., Pang Z.P., Gejman P.V., Duan J. Open chromatin dynamics reveals stage-specific transcriptional networks in hiPSC-based neurodevelopmental model. Stem Cell Res. 2018;29:88–98. doi: 10.1016/j.scr.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Zhang H., Zhou Y., Qiao M., Zhao S., Kozlova A., Shi J., Sanders A.R., Wang G., Luo K. Allele-specific open chromatin in human iPSC neurons elucidates functional disease variants. Science. 2020;369:561–565. doi: 10.1126/science.aay3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylka M.J., Simon J.M., Philpot B.D. Gene length matters in neurons. Neuron. 2015;86:353–355. doi: 10.1016/j.neuron.2015.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The PGC3 SZ GWAS summary statistics can be accessed through “schizophrenia data access portal” (https://www.med.unc.edu/pgc/pgc-workgroups/schizophrenia/). The Chinese Han SZ GWAS summary statistics have been deposited in the Peking University Open Research Data repository with accession number 10638 (https://doi.org/10.18170/DVN/IHROUE). The gene expression data used to generate the list of MIR137-target genes expressed in different neural cell types can be accessed through GEO: GSE129017 and GSE132757. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.