

Abstract
Objective
To compare the frequency and impact on the channel function of KCNH2 variants in SUDEP patients with epilepsy controls comprising patients older than 50 years, a group with low SUDEP risk, and establish loss‐of‐function KCNH2 variants as predictive biomarkers of SUDEP risk.
Methods
We searched for KCNH2 variants with a minor allele frequency of <5%. Functional analysis in Xenopus laevis oocytes was performed for all KCNH2 variants identified.
Results
KCNH2 variants were found in 11.1% (10/90) of SUDEP individuals compared to 6.0% (20/332) of epilepsy controls (p = 0.11). Loss‐of‐function KCNH2 variants, defined as causing >20% reduction in maximal amplitude, were observed in 8.9% (8/90) SUDEP patients compared to 3.3% (11/332) epilepsy controls suggesting about threefold enrichment (nominal p = 0.04). KCNH2 variants that did not change channel function occurred at a similar frequency in SUDEP (2.2%; 2/90) and epilepsy control (2.7%; 9/332) cohorts (p > 0.99). Rare KCNH2 variants (<1% allele frequency) associated with greater loss of function and an ~11‐fold enrichment in the SUDEP cohort (nominal p = 0.03). In silico tools were unable to predict the impact of a variant on function highlighting the need for electrophysiological analysis.
Interpretation
These data show that loss‐of‐function KCNH2 variants are enriched in SUDEP patients when compared to an epilepsy population older than 50 years, suggesting that cardiac mechanisms contribute to SUDEP risk. We propose that genetic screening in combination with functional analysis can identify loss‐of‐function KCNH2 variants that could act as biomarkers of an individual’s SUDEP risk.
Introduction
People with epilepsy have a two to threefold increased risk of premature mortality, with Sudden Unexpected Death in Epilepsy (SUDEP) the most common cause of death. 1 SUDEP occurs without warning, most frequently in young adults. Frequent tonic‐clonic seizures are the biggest risk factor for SUDEP. 2 , 3 , 4 Other risk factors are the markers of seizure severity including epilepsy duration and poly‐therapy. 3 , 4 , 5 However, SUDEP also occurs in patients with mild, well‐controlled epilepsy, suggesting that other risk factors exist. 6
The pathophysiological mechanism(s) responsible for SUDEP are unclear. A systematic retrospective analysis of 10 SUDEP deaths in the Incidence and Mechanisms of Cardiorespiratory Arrests in Epilepsy Monitoring Units (MORTEMUS) revealed that seizure‐mediated terminal apnea always preceded terminal asystole. 7 The patient cohort in this study was small and involved individuals who were undergoing long‐term video‐EEG monitoring, implying refractory epilepsy. These cases may, therefore, not be representative of all individuals with SUDEP. Aside from seizure severity, the presence of abnormal cardiac rhythms has been implicated in SUDEP risk. 8 , 9 Both human and animal studies show that seizure‐mediated changes in cardiac electrophysiology occur, including seizure‐driven cortical autonomic dysfunction and longer‐term altered cardiac ion channel expression. 10 , 11 Patients with epilepsy also have an increased risk of sudden cardiac death. 12 , 13 , 14 Furthermore, genetic studies have found variants in genes associated with cardiac arrhythmia syndromes in SUDEP cases, including genes that cause long QT syndrome (LQTS). 9 , 15 , 16 , 17 , 18 , 19
LQTS results from delayed myocardial repolarization that manifests as a prolonged QT interval on the electrocardiography increasing the risk of “torsades de pointes” that can trigger sudden cardiac death. 20 Here we focus on KCNH2, which was among the top 30 genes identified in a gene‐based rare variant collapsing analysis of SUDEP patients and matched ancestry controls. 16 KCNH2 encodes the pore‐forming α subunit of the voltage‐gated potassium channel Kv11.1. In vitro screening assays have established that the loss of Kv11.1 function leads to familial LQTS type 2 (LQTS2). 21 , 22 , 23 These same assays routinely assess the modulation of Kv11.1 by drug candidates, which block a strong predictor of cardiac toxicity. 24 KCNH2 variants have been identified in SUDEP patients, including both rare pathogenic and common variants although statistically significant enrichment has not been demonstrated. 16 , 17 , 18 We have proposed that when combined, seizures and a risk variant in an arrhythmogenic gene could interact to significantly increase SUDEP risk. 25 This hypothesis predicts that SUDEP individuals will have an enrichment of KCNH2 variants that impact channel function compared to epilepsy patients at low risk of SUDEP. To test this hypothesis, we measured the Kv11.1‐generated current of KCNH2 variants found in SUDEP patients. The results were compared to KCNH2 variants identified in a control sample comprising epilepsy patients who were over 50 years of age. Death rate is reported to be six times higher between the ages of 16 and 24 in epilepsy patients, with SUDEP accounting for ~40% of deaths under the age of 45. 26 , 27 Our epilepsy control cohort is, therefore, considered to have “escaped” SUDEP. An enrichment of loss‐of‐function KCNH2 variants in SUDEP compared to epilepsy controls argue that cardiac mechanisms may contribute to risk.
Methods
Identification of SUDEP and control variants
Our combined cohorts of 90 unrelated SUDEP patients were recruited from the epilepsy genetics research program in Melbourne, Australia, during life, or from coronial cases investigated at the Departments of Forensic Medicine in New South Wales, Victoria, Queensland, and South Australia, as previously described. 16 , 18 All exons of KCNH2 were Sanger‐sequenced in 29 SUDEP cases, and the remaining 61 SUDEP cases underwent exome sequencing as previously described. 16 We looked for variants in the KCNH2 protein‐coding regions and essential splice site dinucleotides with an allele frequency <5% in the gnomAD reference population database. 28 Variants identified by exome sequencing were Sanger‐verified. Eighty‐eight out of 90 of the SUDEP events occurred without witnesses. Hence there is limited information on the circumstances surrounding death, but they fulfilled the operational definition of SUDEP (definite or probable SUDEP) and most had post‐mortem anti‐seizure medication levels performed (Table S1).
KCNH2 variants were identified in our epilepsy control cohort of 332 well‐characterized Australian individuals, drawn from a broad spectrum of epilepsy patients, who were over the age of 50 years (born before 1970) and underwent whole‐exome sequencing as part of our contribution to the Epi25 Consortium 29 (Table S2). Diagnoses were non‐lesional focal epilepsies (n = 137), lesional focal epilepsies (n = 84), genetic generalized epilepsy (n = 76), GEFS+ or febrile seizures (n = 22), and developmental and epileptic encephalopathies (n = 13). Given that SUDEP is far more likely to occur between the ages of 20 and 40 years of age, 26 with a death rate reported to be six times higher between the ages of 16 and 24 in epilepsy patients and accounting for ~40% of deaths under the age of 45, 27 these patients were considered to have “escaped” SUDEP, providing a suitable comparison population. Given the lack of available clinical details on the SUDEP cases, and IRB constraints, we were unable to match cases and controls for epilepsy duration, seizure burden, frequency of generalized tonic–clonic seizures, and AED use. Variant calling was performed as previously described. 29 Variants in the 332 individuals with epilepsy over the age of 50 were annotated using ANNOVAR 29 and then filtered to obtain a list of single‐nucleotide variants in KCNH2 with an allele frequency <5% in gnomAD. 28
Ancestry predictions for the exome‐sequenced SUDEP and epilepsy controls inferred 95% of samples in each cohort to be European. Exome sequencing achieved mean coverage greater than 20× across 78% f the KCNH2 coding region in the SUDEP cohort and 94% in the epilepsy controls.
Standard protocol approvals, registrations, and patient consents
The study was approved by the Human Research Ethics Committees of Austin Health and Royal Prince Alfred Hospital. Signed consent was provided by patients themselves, their parents, next‐of‐kin, or legal guardian in the case of children or patients with intellectual disability. Some of the SUDEP cases were analyzed in a de‐identified manner. Some samples from the control cohort had been collected over a 20‐year period in some centers, so the consent forms reflected standards at the time of collection. Samples were only accepted if the consent did not exclude data sharing.
KCNH2 site‐directed mutagenesis and in vitro cRNA preparation
cDNA encoding a full‐length transcript (NM_000238.4 Ensembl database) of human KCNH2 was subcloned into the Xenopus oocyte expression vector pGEMHE‐MCS. Site‐directed mutagenesis to create human KCNH2 variants was completed by GenScript Biotech (Piscataway, NJ, USA). All clones were verified by Sanger sequencing. In vitro synthesis of cRNA was performed using linearized cDNA template and the mMessage mMachine® T7 transcription kit (Ambion, Thermo Fisher Scientific, Waltham, MA). RNA integrity was assessed both spectrophotometrically (NanoDrop) and by gel electrophoresis. All cRNAs were stored at −80°C.
Oocyte extraction
Adult female Xenopus laevis frogs were housed at the Florey Institute of Neuroscience and Mental Health. Animal procedures and oocyte preparation followed standard procedures in accordance with the conditions approved by the Florey’s Ethics Committee. Briefly, frogs were anesthetized with 1.3 mg/mL of tricaine methanesulfonate and oocytes were surgically removed via a small incision to the abdomen. Oocytes were defolliculated with 1.5 mg/mL of collagenase for 2 h and rinsed with OR‐2 solution (in mmol/L: 82.5 NaCl, 2 KCl, 1 MgCl2.6H2O, 5 HEPES, pH 7.4). Healthy mature oocytes stage V or VI were isolated.
Channel expression
The NM_000238.4 KCNH2 transcript was set as our control sequence and designated as the wild‐type (WT) channel. cRNAs of 0.5 ng (in 50 nL) coding for KCNH2 were manually injected into the oocytes. Injected oocytes were maintained in ND96 storage solution (in mmol/L: 96 NaCl, 2 KCl, 1 MgCl2·6H2O, 1.8 CaCl2·2H2O, 5 HEPES, 50 mg/L of gentamicin, pH 7.4) at 17°C for 2 days to allow translation and trafficking of channels prior to recording.
Two‐electrode voltage clamp electrophysiology
Standard two‐electrode voltage clamp hardware was used (TEC‐05X or TEC10X, NPI, Tamm, Germany). Oocytes were impaled with microelectrodes with an input resistance of between 0.2 and 2.0 MΩ, containing 3 mol/L KCl. During experiments, oocytes were continually perfused with high K+ solution (in mmol/L: 100 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES, pH 7.4) and clamped at −90 mV. Incremental 10 mV voltage steps from −90 to +50 mV were applied for 0.5 s, followed by a test pulse at −100 mV for 3.2 s. Data were sampled at 5000 Hz and low‐pass‐filtered at 1 kHz. Voltage clamp control and data acquisition were obtained using pCLAMP v8.10 software (Molecular Devices, USA). All recordings were performed at 18–20°C. Currents from WT channels were always measured contemporaneously using the same batch of oocytes.
Electrophysiology analysis
Raw currents were baseline‐adjusted and leak‐subtracted, and peak current amplitudes were analyzed offline using AxoGraph v1.7.4 (AxoGraph Scientific, Sydney, AU). Half‐maximal activation voltage and slope values were obtained from current–voltage curves that were fitted with the Boltzmann equation after normalizing the peak test pulse current after each voltage step to the maximum peak test pulse current. Wild‐type normalized current–voltage curves were obtained by normalizing peak test pulse current after each voltage step to the average maximum wild‐type peak test pulse current and fitted with the Boltzmann equation.
Statistical analysis
Functional analysis
An F‐test was used to check for variance equality. Standard one‐way ANOVA with Dunnett’s post hoc correction was used for statistical comparison to wild‐type values if variances were approximately equal (p > 0.05). Welch’s ANOVA with Dunnett’s post hoc correction was used instead, for unequal variances (p < 0.05). Statistical analysis for association used the Fisher’s Exact Test. Significance was set at p < 0.05. Graphs and statistical tests were generated and performed using Prism v8.1.0 (GraphPad, CA, USA). All data points are shown as mean ± SEM.
Results
KCNH2 variants in SUDEP patients and a control epilepsy population
KCNH2 variants previously described in our SUDEP cohorts are listed in Table 1. Ten out of 90 individuals suffering SUDEP carried a missense or truncation KCNH2 variant with <5% minor allele frequency. 16 , 18 These variants have a range of pathogenicity classifications according to the ClinVar database. 30 R744X is reported as pathogenic; Y54H, G924A, R176W, and R1047L are variants of uncertain clinical significance; while G749A is not reported in the ClinVar database. Clinical metadata available for the SUDEP cohort are presented in Table S2.
Table 1.
KCNH2 variants found in SUDEP and epilepsy control populations.
| Cohort | KCNH2 variant (number of cases) | Combined Annotation‐Dependent Depletion (CADD) score | gnomAD allele count | gnomAD allele frequency | Functional analysis |
|---|---|---|---|---|---|
| SUDEP (n = 90) | R1047L (5) | 25.9 | 3117 | 0.0180 | Loss‐of‐function |
| G924A (1) | 22.8 | 8 | 0.0000505 | Loss‐of‐function | |
| G749A (1) | 26.5 | 0 | 0 | Loss‐of‐function | |
| R744X (1) | 39.0 | 0 | 0 | Loss‐of‐function | |
| R176W (1) | 23.4 | 44 | 0.000406 | No change | |
| Y54H (1) | 26.6 | 0 | 0 | No change | |
| Epilepsy control (n = 332) | R1047L (10) | 25.9 | 3117 | 0.0180 | Loss‐of‐function |
| A913V (1) | 20.3 | 73 | 0.000482 | No change | |
| G903R (1) | 22.4 | 19 | 0.000120 | No change | |
| K897R (1) | 13.2 | 3 | 0.0000126 | No change | |
| S871C (1) | 28.0 | 0 | 0 | No change | |
| T436M (1) | 13.9 | 9 | 0.0000318 | No change | |
| R397H (1) | 27.0 | 4 | 0.0000159 | No change | |
| P347S (1) | 19.9 | 281 | 0.000998 | No change | |
| D259N (1) | 22.5 | 1 | 0.0000323 | No change | |
| A193V (1) | 19.9 | 2 | 0.0000286 | No change | |
| S140F (1) | 23.7 | 0 | 0 | Loss‐of‐function |
In our epilepsy control population of 332 living patients with epilepsy over 50 years of age with whole‐exome sequencing data, variants in KCNH2 that satisfied the filtering criteria were found in 20 out of 332 subjects (Table 1). These included the common R1047L variant also found in SUDEP cases (Table 1). Variants A913V, K897R, S871C, T436M, R397H, P347S, and D259N were classified as variants of uncertain significance in ClinVar, while A193V and S140F are not reported in ClinVar. Clinical metadata available for the control cohort are presented in Table S2.
KCNH2 variants were found in 11.1% of SUDEP cases (10/90) compared to 6.0% of epilepsy controls (20/332; p = 0.11).
In silico predictions of Kv11.1 channel dysfunction and SUDEP risk
In silico prediction tools provide a method of estimating the detrimental impact of a given variant on protein function. We determined the Combined Annotation Dependent Depletion (CADD) score for each KCNH2 variant (Table 1). Only the KCNH2 R744X variant had a CADD score above 30 indicating, as in ClinVar, that it was pathogenic.
Functional characterization of KCNH2 variants
Manual two‐electrode voltage clamp was used to record currents from wild‐type (WT; NM_000238.4 KCNH2 transcript) and mutated KV11.1 channels expressed in Xenopus oocytes (Fig. 1). Channels were activated by a series of depolarizing voltage excursions from a holding potential of −90 mV. Maximal tail currents were used to measure channel activity. In our SUDEP cohort, functional analysis revealed a significant reduction in current amplitude (> 20%) for R1047L, G924A, G749A, and R744X mutated channels relative to wild‐type channels (Fig. 1A–E and H). R176W and Y54H variants had no effect (Fig. 1F–H). In the epilepsy control group, the R1047L (also in the SUDEP cohort) and the S140F variant reduced current amplitude (Fig. 2). The other nine epilepsy control variants were without effect on KV11.1 channel current amplitude (Fig. 2). Other biophysical parameters measured, including the half‐maximal activation voltage and Boltzmann slope, are reported in Figure 3. In the SUDEP group, only G924A and G749A showed altered biophysical properties (Fig. 3A and C), whereas R744X could not be measured. No variants in the epilepsy control group showed any differences in other biophysical parameters (Fig. 3B and D). Changes in biophysical properties for SUDEP KCNH2 variants may contribute to overall channel dysfunction and consequently increase the risk of sudden death.
FIGURE 1.
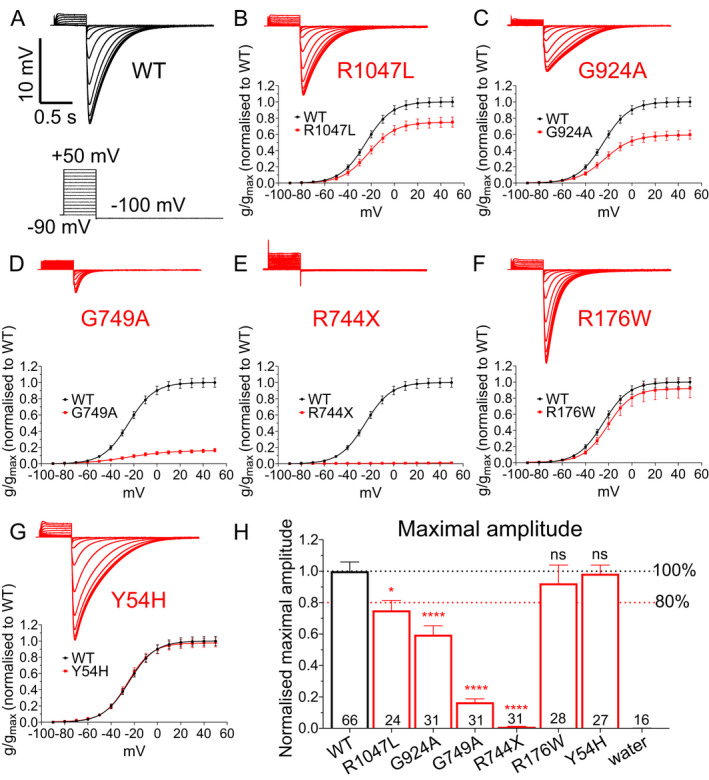
Functional analysis of KCNH2 variants from SUDEP patients. (A) Sample recording traces of Kv11.1 wild‐type (WT) channels. Insert: cartoon of the voltage protocol applied. (B–G) Sample recording traces of Kv11.1 variant channels (top) and average normalized conductance–voltage relationships (below) comparing Kv11.1 WT and variant channels for (B) R1047L, (C) G924A, (D) G749A, (E) R744X, (F) R176W, and (G) Y54H variants. (H) Average maximal amplitude for each variant. Number in each bar represents the number of independent oocytes recorded for each variant. Black and red dashed lines indicate 100% and 80%, respectively, of maximal current amplitude of Kv11.1WT channel. *p < 0.05, ****p < 0.0001.
FIGURE 2.
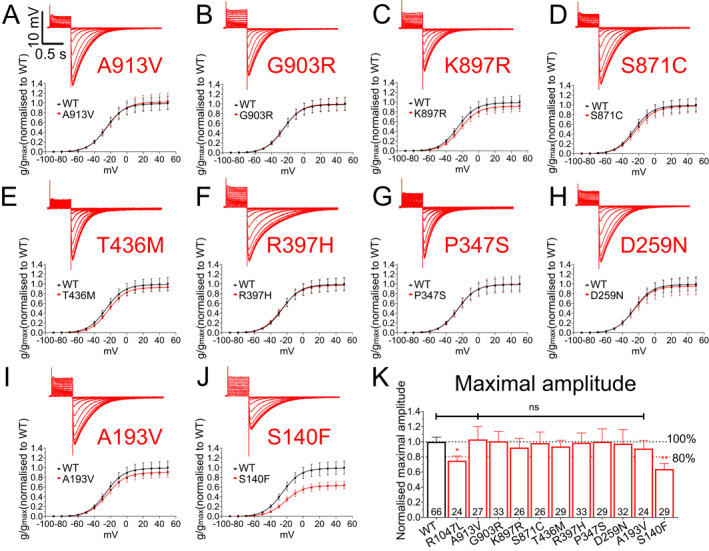
Functional analysis of KCNH2 variants from epilepsy control cohort. Sample recording traces of Kv11.1 variant channels (top) and average normalized conductance–voltage relationships (below) comparing Kv11.1 WT and variant channels for (A) A913V (B) G903R, (C) K897R, (D) S871C, (E) T436M, (F) R397H, (G) P347S, (H) D259N (I), A193V, and (J) S140F variants. (K) Average maximal amplitude for each variant in the epilepsy control cohort. Number in each bar represents the number of independent oocytes recorded for each variant. Black and red dashed lines indicate 100% and 80%, respectively, of maximal current amplitude of the Kv11.1 WT channel. *p < 0.05, **p < 0.01.
FIGURE 3.
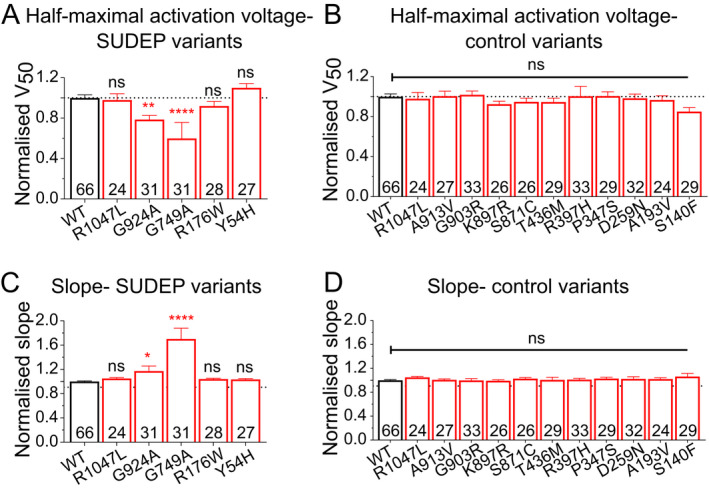
Biophysical properties of KCNH2 variants from SUDEP cases and epilepsy control population. (A) Average half‐maximal voltage of activation for each variant in SUDEP cohort. (B) Average half‐maximal voltage of activation for each variant in the epilepsy control cohort. (C) Average slope from the Boltzmann fit for each variant in the SUDEP cohort. (D) Average slope from the Boltzmann fit for each variant in the epilepsy control cohort. Number in each bar represents the number of independent oocytes recorded for each variant. *p < 0.05, **p < 0.01, ****p < 0.0001.
Enrichment of loss‐of‐function KCNH2 variants in SUDEP cases
Our functional data allow the classification of each variant as either loss‐of‐function, defined as a statistically significant reduction in a current amplitude of >20% (p < 0.05), or no change in function (Table 1). Based on these criteria, 8 out of 90 (8.9%) SUDEP cases carried a KCNH2 loss‐of‐function variant, while 2 out of 90 (2.2%) carried a variant in which function was not changed. In contrast, our epilepsy patient control population had 11 out of 332 (3.3%) patients with a loss‐of‐function variant, and 9 out of 332 (2.7%) carried a variant that did not alter function. The SUDEP cohort has approximately threefold enrichment for loss‐of‐function KCNH2 variants (OR = 2.7, 95% confidence interval (1.1, 7.4), Fisher’s exact test nominal p = 0.04) compared with the epilepsy control population (Fig. 4A). There was no enrichment of KCNH2 variants that did not change channel function in the SUDEP cohort compared to the epilepsy control cohort (OR = 0.8, 95% confidence interval 0.17 to 3.8, Fisher’s exact p > 0.99).
FIGURE 4.
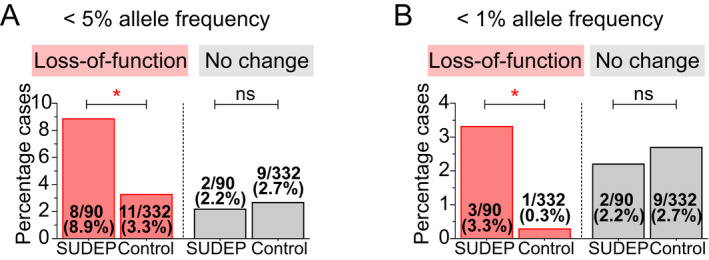
Enrichment of KCNH2 variants in SUDEP and epilepsy control cohorts. (A) KCNH2 variants with less than 5% allele frequency are enriched approximately three times in SUDEP compared to the epilepsy control cohort. (B) Enrichment of rare KCNH2 variants with less than 1% allele frequency is further increased to about 10 times in the SUDEP cohort. *p < 0.05.
In the SUDEP cohort, rare KCNH2 variants with a minor allele frequency of <1% associate with greater loss of function implicating increased risk (Fig. 1). Consistent with this, rare KCNH2 variants are enriched with a greater odds ratio (OR = 11.4, 95% confidence interval 1.2–111.1, Fisher’s exact p = 0.03) (Fig. 4B). R1047L is the most common variant that we tested. It is observed in approximately 3.5% of the general population 28 and was found in 5 out of 90 (5.6%) SUDEP cases and 10 out of 332 (3.0%) epilepsy control patients. The reduction in current amplitude for the R1047L variant shows that loss‐of‐function is not confined to rare KCNH2 variants.
Discussion
Previous genetic screening efforts in SUDEP have identified rare variants in genes that are associated with severe developmental and epileptic encephalopathies. 3 This is to be expected, as these variants cause severe epilepsies which carry a high SUDEP risk. 2 , 3 , 4 Whether other genetic risk factors contribute to SUDEP risk is less clear. Screening efforts in SUDEP cases have identified variants in cardiac genes that cause arrhythmia syndromes. 9 , 15 , 16 , 17 , 18 , 19 We have proposed that when combined, seizures and a risk variant in an arrhythmogenic gene could interact to significantly increase SUDEP risk. 25 Here we focused on KCNH2, in which loss‐of‐function variants are an established cause of LQTS leading to sudden death. We show an approximate threefold enrichment in loss‐of‐function KCNH2 variants in our SUDEP cohort relative to an older epilepsy control cohort that has “escaped” SUDEP. These data support the premise that KCNH2 loss‐of‐function variants act as genetic biomarkers of SUDEP risk and motivates the need to examine this hypothesis in additional, independent SUDEP cohorts.
A systematic retrospective analysis of 10 SUDEP deaths in the Incidence and Mechanisms of Cardiorespiratory Arrests in Epilepsy Monitoring Units (MORTEMUS) revealed that seizure‐mediated terminal apnea always preceded terminal asystole. 7 This is strong evidence implying respiratory factors as a cause of death. However, the patient cohort in this study was small and involved individuals who were undergoing long‐term video‐EEG monitoring, implying refractory epilepsy. These cases may, therefore, not be representative of all individuals with SUDEP and imply that the pathophysiological mechanism(s) responsible for SUDEP are likely to be multifactorial. Our data suggest that cardiac factors are important, at least in a subset of SUDEP cases.
Characterization of KCNH2 variants based on in silico predictions of protein dysfunction using the CADD method was uninformative. Only the truncation variant, R744X, had a CADD score greater than 30 and is therefore predicted to be deleterious. CADD scores for the G924A and G749A variants were relatively low yet functional analysis in Xenopus oocytes revealed significant reductions in current amplitude. As noted below, functional analysis methods are not without limitation. However, our findings highlight that functional analysis should remain the gold standard by which to judge potential pathogenicity.
In this study, we have categorized KCNH2 variants into either loss‐of‐function or no change in function. However, individual variants do vary in the degree of functional impairment shown in vitro and thus are unlikely to contribute equally to the risk of sudden death. Other factors are also likely to contribute to SUDEP. In the case of rare pathogenic variants, such as R744X, G749A, and G924A there is a large loss‐of‐function observed in the expression assay and we infer a stronger likelihood that cardiac arrhythmia contributed to death. Increased enrichment of rare KCNH2 variants (OR = 11.4) in the SUDEP cohort is consistent with a correlation between the extent of loss of function and increased risk. More common variants with lesser in vitro functional impairment may contribute less to individual risk. For example, R1047L has an allele frequency of 1.8% in the gnomAD database of population controls and has a smaller impact on channel function. The R1047L variant is likely to increase individual risk less when compared to variants that cause a large impact on channel function. However, at the population level, the R1047L variant impact is likely to be significant given its common nature, increasing risk to a small degree in many people. Additional studies looking at the functional impact of KCNH2 variants in a greater number of SUDEP patients will be required to fully understand attributable risk. It is also important to note that heterologous expression systems cannot report on more complex cell functions. More sophisticated model systems such as cardiac myocytes derived from stem cells will help further define the relationship between KCNH2 variants and arrhythmia risk.
There is surprisingly little evidence that directly links acute seizures to genetically caused cardiac arrhythmia and sudden death. The Kcnq1 T311I mouse model of LQTS provides some evidence with over half of the recorded abnormalities in cardiac rhythm associated with epileptiform discharges. 31 This has implications for more common variants, such as R1047L, that are unlikely to be pathogenic alone but may increase the risk of death in the context of seizures. Furthermore, both human and animal studies show that seizure‐mediated changes in cardiac electrophysiology occur. 10 This includes seizure‐driven dysfunction that can alter acute cardiac rhythm. 11 , 32 Studies have also observed altered cardiac ion channel expression with ongoing seizure activity. 33 Either acute and/or chronic changes in cardiac function due to seizures may increase susceptibility to cardiac arrhythmias in people with a “loss‐of‐function” KCNH2 variant to significantly increase SUDEP risk.
We acknowledge the small sample size of the SUDEP cohort and the retrospective nature of our study. Adequate seizure and medication metadata are also frequently missing. As such we cannot match our control and SUPDEP cohort precisely. We propose a framework on which to design prospective studies. Whole‐genome and exome sequencing (that include KCNH2) are becoming routine, providing an opportunity to identify variants. These could be functionally characterized or matched to the growing datasets of characterized KCNH2 variants that are now available. 34 This would allow the stratification of epilepsy patients and enable prospective cohorts to be captured that could be matched for seizure load, number, and types of antiepileptics and other variables. It would also be informative to identify a cohort with KCNH2 variants of similar functional deficits but without epilepsy to directly assess the impact of the combination of seizures plus arrhythmic predisposition on the risk of death.
The ability to identify patients at risk of SUDEP has important clinical implications. In patients with epilepsy carrying loss‐of‐function KCNH2 variants, prolonged electroencephalogram with cardiac monitoring might be informative to explore ictal and interictal changes in cardiac rhythm, while prolonged cardiac monitoring with cardiac loop recorders could be considered for the continuous interrogation of cardiac rhythm. This would allow the detection of additional biomarkers of risk, including arrhythmogenic markers such as prolonged QT intervals, especially during seizures. Patients identified to be at risk of cardiac arrhythmia could start prophylactic treatment with β‐blockers which are used effectively in LQTS. Drugs known to impact QT intervals should also be avoided in such epilepsy patients.
KCNH2 variants will only ever be one biomarker that will be part of a risk assessment. Seizure severity and frequency remain significant predictors of SUDEP risk. 2 , 3 Further investigation into other potential genetic biomarkers, including arrhythmogenic genes such as KCNQ1 and SCN5A is required, as are studies into how acute seizures or long‐term seizure‐related changes in cardiac function interact with genetic causes of arrhythmia. An ultimate goal is to develop a SUDEP risk matrix integrating the various clinical, genetic, and environmental factors, and to prevent SUDEP by targeting all modifiable risk factors.
In conclusion, our data provide evidence that both rare and more common KCNH2 variants that cause loss‐of‐function may act as biomarkers of SUDEP in epilepsy patients. These data need to be replicated in larger independent study cohorts. Our data motivate more focused clinical studies investigating the impact of loss‐of‐function KCNH2 variants on cardiac rhythm. Our study also motivates the development of more complex in vitro models, as well as animal models, that will allow the interaction between seizures and genetic cardiac abnormalities to be investigated. These models will also provide an opportunity to test novel therapeutic strategies for the prevention of SUDEP.
Conflict of Interest
SFB declares unrestricted educational grants from UCB Pharma, SCIgen, and Eisai and consultancy fees from Praxis Precision Medicines. IES has served on scientific advisory boards for UCB, Eisai, GlaxoSmithKline, BioMarin, Nutricia, Rogcon, and Xenon Pharmaceuticals; has received speaker honoraria from GlaxoSmithKline, UCB, BioMarin, Biocodex, and Eisai; has received funding for travel from UCB, Biocodex, GlaxoSmithKline, BioMarin, and Eisai; has served as an investigator for Zogenix, Zynerba, Ultragenyx, GW Pharma, UCB, Eisai, Anavex Life Sciences, Ovid Therapeutics, Epygenix, Encoded Therapeutics, and Marinus; and has consulted for Zynerba Pharmaceuticals, Atheneum Partners, Ovid Therapeutics, Epilepsy Consortium, and UCB. IES may accrue future revenue on pending patent WO61/010176 (filed: 2008): Therapeutic Compound; has a patent for SCN1A testing held by Bionomics Inc and licensed to various diagnostic companies; has a patent molecular diagnostic/theranostic target for benign familial infantile epilepsy (BFIE) [PRRT2] 2011904493 & 2012900190 and PCT/AU2012/001321 (TECH ID:2012‐009) with royalties paid. The remaining authors have no conflicts of interest.
Author Contributions
MSS, RDB, MFB, LEB, AMP, MH, DC, MB, CS, IES, SFB, and CAR developed the concept and designed the study. MSS and ESMS performed the experiments and analyzed the data. CAR drafted a significant portion of the manuscript. MSS prepared the figures and drafted the methods section. MSS, AMP, MDFC, and CEM performed the molecular biology. All authors contributed to revising and editing the manuscript and approved the submitted version.
Supporting information
Table S1. Characteristics of SUDEP patients without KCNH2 variant (orange‐shaded patients carried loss‐of‐function KCNH2 variants).
Table S2. Characteristics of control epilepsy patients with KCNH2 variant (orange‐shaded patients carried loss‐of‐function KCNH2 variants).
Table S3. Types of epilepsies in the control cohort.
Acknowledgments
We would like to thank Kate Esnault for her help with the epilepsy classification. This work was supported by the National Health and Medical Research Council (NHMRC) Program Grant (10915693) to SFB, IES, and CAR and by an anonymous philanthropic gift for SUDEP research to SFB and IES. CAR would like to acknowledge the CURE foundation for support. CS and IES are recipients of a National Health and Medical Research Council Practitioner Fellowships (#1154992 to CS and #1104831 to IES) and Senior Investigator Fellowship to IES (#1172897). MB was supported by an NHMRC Senior Research Fellowship (#1102971). LEB acknowledges the support of an Australian Government Research Training Program Scholarship. This work was also made possible through the Victorian State Government Operational Infrastructure Support and Australian Government National Health and Medical Research Council (NHMRC) independent research Institute Infrastructure Support Scheme (IRIISS).
*This manuscript has been uploaded to the preprint server bioRxiv (https://doi.org/10.1101/2021.03.19.436102).
Funding Information
This work was supported by the National Health and Medical Research Council (NHMRC) Program Grant (10915693) to SFB, IES, and CAR and by an anonymous philanthropic gift for SUDEP research to SFB and IES. CAR would like to acknowledge the CURE foundation for support. CS and IES are recipients of a National Health and Medical Research Council Practitioner Fellowships (#1154992 to CS and #1104831 to IES) and Senior Investigator Fellowship to IES (#1172897). MB was supported by an NHMRC Senior Research Fellowship (#1102971). LEB acknowledges the support of an Australian Government Research Training Program Scholarship. This work was also made possible through the Victorian State Government Operational Infrastructure Support and Australian Government National Health and Medical Research Council (NHMRC) independent research Institute Infrastructure Support Scheme (IRIISS).
Funding Statement
This work was funded by Victorian State Government ; National Health and Medical Research Council grants #1104831, #1154992, 10915693, #1172897, and #1102971; Australian Government National Health and Medical Research Council (NHMRC); Australian Government.
References
- 1. Thurman DJ, Logroscino G, Beghi E, et al. The burden of premature mortality of epilepsy in high‐income countries: a systematic review from the Mortality Task Force of the International League Against Epilepsy. Epilepsia 2017;58:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. DeGiorgio CM, Markovic D, Mazumder R, et al. Ranking the leading risk factors for sudden unexpected death in epilepsy. Front Neurol 2017;8:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Devinsky O, Hesdorffer DC, Thurman DJ, et al. Sudden unexpected death in epilepsy: epidemiology, mechanisms, and prevention. Lancet Neurol 2016;15:1075–1088. [DOI] [PubMed] [Google Scholar]
- 4. Hesdorffer DC, Tomson T, Benn E, et al. Combined analysis of risk factors for SUDEP. Epilepsia 2011;52:1150–1159. [DOI] [PubMed] [Google Scholar]
- 5. Nilsson L, Farahmand BY, Persson P‐G, et al. Risk factors for sudden unexpected death in epilepsy: a case control study. Lancet 1999;353:888–893. [DOI] [PubMed] [Google Scholar]
- 6. Verducci C, Hussain F, Donner E, et al. SUDEP in the North American SUDEP Registry: the full spectrum of epilepsies. Neurology 2019;93:e227–e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ryvlin P, Nashef L, Lhatoo SD, et al. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol 2013;12:966–977. [DOI] [PubMed] [Google Scholar]
- 8. Tomson T, Nashef L, Ryvlin P. Sudden unexpected death in epilepsy: current knowledge and future directions. Lancet Neurol 2008;7:1021–1031. [DOI] [PubMed] [Google Scholar]
- 9. Bagnall RD, Crompton DE, Semsarian C. Genetic basis of sudden unexpected death in epilepsy. Front Neurol 2017;8:348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ravindran K, Powell KL, Todaro M, et al. The pathophysiology of cardiac dysfunction in epilepsy. Epilepsy Res 2016;127:19–29. [DOI] [PubMed] [Google Scholar]
- 11. Nei M. Cardiac effects of seizures. Epilepsy Curr 2009;9:91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bardai A, Lamberts RJ, Blom MT, et al. Epilepsy is a risk factor for sudden cardiac arrest in the general population. PLoS One 2012;7:e42749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lamberts RJ, Blom MT, Wassenaar M, et al. Sudden cardiac arrest in people with epilepsy in the community: circumstances and risk factors. Neurology 2015;85:212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stecker EC, Reinier K, Uy‐Evanado A, et al. Relationship between seizure episode and sudden cardiac arrest in patients with epilepsy. Circ Arrhythm Electrophysiol 2013;6:912–916. [DOI] [PubMed] [Google Scholar]
- 15. Aurlien D, Leren TP, Taubøll E, et al. New SCN5A mutation in a SUDEP victim with idiopathic epilepsy. Seizure 2009;18:158–160. [DOI] [PubMed] [Google Scholar]
- 16. Bagnall RD, Crompton DE, Petrovski S, et al. Exome‐based analysis of cardiac arrhythmia, respiratory control, and epilepsy genes in sudden unexpected death in epilepsy. Ann Neurol 2016;79:522–534. [DOI] [PubMed] [Google Scholar]
- 17. Partemi S, Cestèle S, Pezzella M, et al. Loss‐of‐function KCNH2 mutation in a family with long QT syndrome, epilepsy, and sudden death. Epilepsia 2013;54:e112–e116. [DOI] [PubMed] [Google Scholar]
- 18. Tu E, Bagnall RD, Duflou J, et al. Post‐mortem review and genetic analysis of sudden unexpected death in epilepsy (SUDEP) cases. Brain Pathol 2011;21:201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tu E, Waterhouse L, Duflou J, et al. Genetic analysis of hyperpolarization‐activated cyclic nucleotide‐gated cation channels in sudden unexpected death in epilepsy cases. Brain Pathol 2011;21:692–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tester DJ, Ackerman MJ. Genetics of long QT syndrome. Methodist Debakey Cardiovasc J 2014;10:29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kozek KA, Glazer AM, Ng C‐A, et al. High‐throughput discovery of trafficking‐deficient variants in the cardiac potassium channel KV11.1. Heart Rhythm 2020;17:2180–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bellin M, Casini S, Davis RP, et al. Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long‐QT syndrome. EMBO J 2013;32:3161–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Smith JL, Anderson CL, Burgess DE, et al. Molecular pathogenesis of long QT syndrome type 2. J Arrhythm 2016;32:373–380. 10.1016/j.joa.2015.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Witchel H. Drug‐induced hERG block and long‐QT syndrome. Cardiovasc Ther 2011;29:251–259. [DOI] [PubMed] [Google Scholar]
- 25. Bleakley LE, Soh MS, Bagnall RD, et al. Are variants causing cardiac arrhythmia risk factors in sudden unexpected death in epilepsy? Front Neurol 2020;11:925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Langan Y. Sudden unexpected death in epilepsy (SUDEP): risk factors and case control studies. Seizure 2000;9:179–183. [DOI] [PubMed] [Google Scholar]
- 27. Morrish P, Duncan S, Cock H. Epilepsy deaths: learning from health service delivery and trying to reduce risk. Epilepsy Behav 2020;103:106473. [DOI] [PubMed] [Google Scholar]
- 28. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Epi25 Collaborative ; Feng YC, Howrigan DP, Abbott LE, et al. Ultra‐rare genetic variation in the epilepsies: a whole‐exome sequencing study of 17,606 individuals. Am J Hum Genet 2019;105:267–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014;42(D1):D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Goldman AM, Glasscock E, Yoo J, et al. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med 2009;1:2ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Seyal M, Pascual F, Lee C‐Y, et al. Seizure‐related cardiac repolarization abnormalities are associated with ictal hypoxemia. Epilepsia 2011;52:2105–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li MCH, O'Brien TJ, Todaro M, et al. Acquired cardiac channelopathies in epilepsy: evidence, mechanisms, and clinical significance. Epilepsia 2019;60:1753–1767. [DOI] [PubMed] [Google Scholar]
- 34. Ng C‐A, Perry MD, Liang W, et al. High‐throughput phenotyping of heteromeric human ether‐à‐go‐go‐related gene potassium channel variants can discriminate pathogenic from rare benign variants. Heart Rhythm 2020;17:492–500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristics of SUDEP patients without KCNH2 variant (orange‐shaded patients carried loss‐of‐function KCNH2 variants).
Table S2. Characteristics of control epilepsy patients with KCNH2 variant (orange‐shaded patients carried loss‐of‐function KCNH2 variants).
Table S3. Types of epilepsies in the control cohort.