

Abstract
The spinocerebellar ataxias (SCAs) are a group of dominantly inherited diseases that share the defining feature of progressive cerebellar ataxia. The disease process, however, is not confined to the cerebellum; other areas of the brain, in particular, the brainstem, are also affected, resulting in a high burden of morbidity and mortality. Currently, there are no disease‐modifying treatments for the SCAs, but preclinical research has led to the development of therapeutic agents ripe for testing in patients. Unfortunately, due to the rarity of these diseases and their slow and variable progression, there are substantial hurdles to overcome in conducting clinical trials. While the epidemiological features of the SCAs are immutable, the feasibility of conducting clinical trials is being addressed through a combination of strategies. These include improvements in clinical outcome measures, the identification of imaging and fluid biomarkers, and innovations in clinical trial design. In this review, we highlight current challenges in initiating clinical trials for the SCAs and also discuss pathways for researchers and clinicians to mitigate these challenges and harness opportunities for clinical trial development.
Introduction
The spinocerebellar ataxias (SCAs) comprise a group of dominantly inherited cerebellar disorders that typically begin in mid‐life. 1 The early ataxic symptoms of gait, speech, and eye movement incoordination are often followed by non‐ataxic symptoms, which include cognitive deficits, cranial nerve dysfunction, spasticity, rigidity, and dystonia. For many patients, neurodegeneration occurs in multiple brain regions including the brainstem, resulting in significantly reduced lifespan. 1 , 2 , 3 Though relatively rare compared to other human neurodegenerative syndromes, the SCAs produce significant long‐term disability and therefore place a significant burden on not just patients, but also the overall health care infrastructure.
Forty‐eight SCAs have been identified to date. 3 , 4 , 5 The SCAs are designated by the prefix SCA followed by a number, which reflects the order in which that genetic mutation was identified. The most common SCAs have all likely been identified. 6 While the genetic underpinning of the SCAs are diverse, the most prevalent SCAs are caused by expansion of CAG trinucleotide repeats, which encode a polyglutamine tract in the protein product. These include SCAs 1, 2, 3, 6, 7, 17, and a related ataxic syndrome, dentatorubral‐pallidoluyesian atrophy/DRPLA. 7 The polyglutamine SCAs account for more than half of the known SCAs and are the best characterized.
Six other ataxias—SCA8, 10, 12, 31, 36, and 37—are also caused by nucleotide repeats, but in these diseases, the repeats do not occur in the coding region of the gene. 7 , 8 , 9 Pathology here is thought to result from toxic RNA species or a peptide product that results from a non‐canonical form of translation, termed repeat‐associated non‐ATG initiated translation (RAN translation). 10 , 11 These pathogenic mechanisms could also contribute to the pathogenesis of the polyglutamine ataxias which is an active area of study. The remaining SCAs are caused by conventional mutations. These include point mutations or deletions in the coding regions of the genes, resulting in abnormal protein products. 3
As one moves toward finding therapies, advances in our understanding of these diseases at a mechanistic level are proving to be crucial. For the polyglutamine repeat SCAs in particular, research has benefited from knowledge gained from other polyglutamine repeat disorders, most notably Huntington’s disease (HD), another autosomal dominant disease caused by polyglutamine repeat expansion. There appear to be clear commonalities among the polyglutamine repeat disorders in pathogenic pathways at a subcellular level. Polyglutamine expansion causes the protein to misfold and accumulate in cells due to impaired clearance by quality control mechanisms in the cell, including proteasomal and autophagic clearance. 12 The accumulated polyglutamine protein is thought to exert its pathogenic effect mainly through a toxic gain of function mechanism. 12 A greater number of CAG repeats is associated with earlier age of onset, faster progression, and a broader spectrum of clinical symptoms. 2 , 13 The number of CAG repeats can increase over subsequent generations due to meiotic instability, which influences the disease severity in different family members. More recently, it has been found that the CAG repeat length can vary in different cells from somatic instability and this could also modify the disease process. 14 , 15
There are currently no disease‐modifying treatments for any of the SCAs and there are many challenges to be overcome in clinical trial development (Fig. 1). Research suggests that a promising strategy for at least the polyglutamine SCAs lies in reducing the level of the toxic mutant protein itself. This strategy is appealing since it does not require a complete understanding of downstream pathogenic pathways. 16 , 17 , 18 Several high‐throughput small‐molecule screens have revealed promising candidates that may lower levels of the mutant protein. 3 , 19 , 20 But perhaps more exciting are recently developed approaches based on newer RNA depleting or DNA editing tools. 21 These include small‐interfering RNA (siRNA), micro‐RNA (miRNA), or antisense oligonucleotide (ASO)‐based approaches to target RNA species, as well as CRISPR‐based approaches to target RNA or DNA. 3 , 22 , 23 Many of these strategies have been validated in animal models of SCAs. 3 , 24 , 25 Similar approaches are further along in Huntington disease (HD), a more common polyglutamine disease, where ASOs targeting the mutated huntingtin protein are already being tested in clinical trials. 26 , 27 The hope is that the results of the HD trials will inform work in the SCAs.
FIGURE 1.
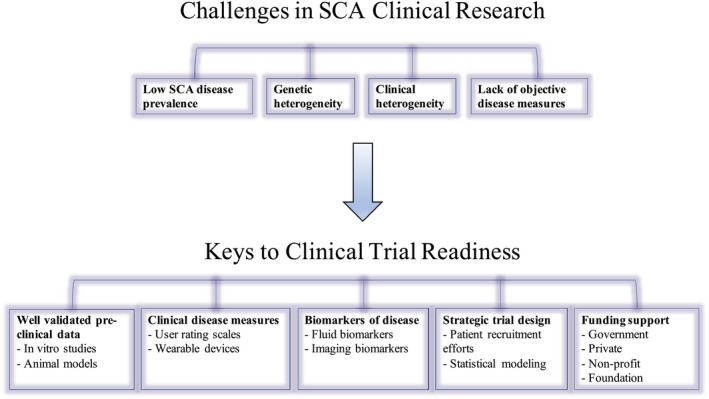
Schematic summary of challenges facing clinical research in the spinocerebellar ataxias and proposed elements that are critical to support clinical trial readiness.
In addition to targeting the mutant protein itself, other strategies are aimed at addressing down‐stream targets that contribute to disease pathogenesis. Pharmacologically addressable deficits include repleting neurotrophic factors such as brain‐derived neurotrophic factor (BDNF) and vascular endothelial growth factor (VEGF), ameliorating ion channel dysfunction, or addressing consequences of mitochondrial damage including the generation of reactive oxygen species. 28 , 29 , 30 , 31 , 32 , 33 Lastly, a less targeted approach to therapeutic development is to repurpose or modify drugs used for other neurodegenerative disorders. For instance, an ongoing trial is testing whether an analogue of riluzole provides symptomatic benefit for patients with polyglutamine SCAs. 34 , 35 Drugs used to treat non‐neurologic diseases can be repurposed as well. For example, the tyrosine kinase inhibitor nilotinib, which is used to treat chronic myelogenous leukemia, is under active clinical investigation for the SCAs. 36 , 37 , 38
Many of the potential treatment strategies outlined above could potentially be neuroprotective, meaning that these strategies could halt or slow disease progression. This also raises the exciting possibility that new treatments could be tested in pre‐manifest individuals to potentially prevent development of manifest disease. Given the autosomal dominant inheritance pattern of the SCAs, the children of affected individuals carry a 50% risk of inheriting an SCA mutation. It is important to recruit these at‐risk individuals to identify the earliest and most sensitive clinical signs of disease. For example, mutation carriers have been shown to have mild coordination deficits and brain abnormalities that gradually increase before the onset of clinically evident ataxia. 39 Furthermore, with recent advancements in imaging techniques like MRS and diffusion MRI, it is feasible to detect brain abnormalities in the pre‐symptomatic period. 40 , 41
Despite the lack of currently available treatments for the SCAs, promising treatment agents are entering the pipeline (Fig. 2). This review will provide an overview of the current challenges facing clinical trial development in the SCAs and the approaches being taken to overcome these challenges.
FIGURE 2.
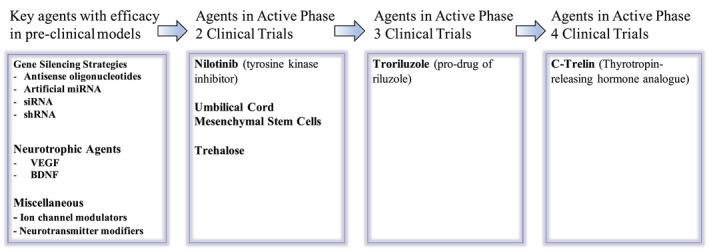
Schematic summary of major therapeutic agents being tested for the SCAs. Major agents that have been successful in preclinical disease models are summarized, including gene silencing strategies, neurotrophic agents, and ion channel modifiers. Current agents being tested in active clinical trials in the United States were identified from clinicaltrials.gov.
Challenges To Clinical Trials
Low disease prevalence and phenotypic variability
The first challenge to clinical trials for the SCAs is the rarity of these disorders. The global prevalence of the known SCAs is estimated to be between 1 and 6 per 100,000. 1 , 3 , 12 The low prevalence of the SCAs makes it difficult to recruit sufficient patients for any given trial. 1 , 42
Another constraint to clinical trial development is the significant clinical heterogeneity of the SCAs. Because of the genetic heterogeneity of this group of disorders, patient recruitment must take into account the specific SCA subtype. Even for the polyglutamine SCAs, which share a common mutational mechanism, clinical features differ. This clinical heterogeneity is true not just for disease severity but also for the range of symptoms and the rate of disease progression. SCA6, for instance, tends to have a relatively pure cerebellar syndrome, is milder in severity, and more protracted in course, while SCAs 1, 2, 3, and 7 are associated with brainstem dysfunction with variable involvement of the basal ganglia, cortex, and spinal cord. 1 , 3 , 43 There are other peculiarities of the SCAs: SCAs 1, 2, and 3 are often accompanied by peripheral neuropathy, while SCA7 is more prone to result in retinal degeneration and blindness. 39 , 44 , 45 , 46
Even more problematic is the variability within a particular SCA subtype. There is substantial individual variability in terms of disease penetrance, age at presentation, and rate of symptom progression, which complicates attempts to monitor disease progression and response to potential therapies. For example, the age of onset of the SCAs is highly variable, with one study reporting the onset of symptoms in SCA1, SCA2, and SCA3 ranging from the first to the seventh decade of life in all three disorders. 47 A major source of the variability in the polyglutamine SCAs arises from the length of the CAG expansion—the longer the repeat, the earlier the symptom onset and the worse the severity. 13 , 39 , 45 Longer expansions are also accompanied by more extracerebellar involvement. 7 , 48 Therefore, at least for the polyglutamine SCAs, CAG repeat length is one method by which patients can be stratified in terms of suspected disease severity.
Additional causes of variability likely arise from genetic and environmental variables. There are hints that the size of repeats in other genes could contribute to disease, while other studies suggest that ethnicity might play a role. 49 , 50 , 51 , 52 It is also likely that genes that play a role in DNA repair also modulate disease phenotype through effects on somatic instability, as has been described for HD. 53 From a clinical trial standpoint, all these factors influencing the disease phenotype require a sufficient number of patients of each SCA subtype to be recruited to prevent confounding variables that might interfere with the interpretation of interventional studies.
Efforts Toward Clinical Trial Readiness
Pre‐clinical data have identified several exciting potential treatments for the SCAs. This includes both symptomatic agents as well as neuroprotective treatment strategies. Several strategies are being pursued to translate the information learned from pre‐clinical models into fruitful and productive clinical trials. These include: (1) improvements in clinical rating scales to provide robust and precise outcome measures with less variability; (2) development of biomarkers to provide additional objective measures to monitor neurodegeneration; and (3) clinical trial design and statistical innovations to allow for optimization of the small number of patients recruited for these trials.
Improvements in clinical measures: rating scales
Ataxia, like most movement disorders, is difficult to characterize in an objective and accurate manner. For this reason, several rating scales have been developed to serve as instruments to capture ataxia severity.
The most widely used ataxia rating scales are the International Cooperative Rating Scale (ICARS) 54 and the Scale for the Assessment and Rating of Ataxia (SARA) scale. 55 The ICARS, the first of the two to be developed, has 19 items with four subdomains assessing posture and gait, kinetic function, speech, and oculomotor function. 56 The SARA scale is shorter, with only 8 items, and includes common clinical exam maneuvers that are frequently performed as part of routine assessments for evidence of cerebellar impairment. 57 It is similar to the ICARS, with several items removed in order to reduce redundancy. It also differs from the ICARS in that it does not test for eye movement abnormalities. Other scales include the Modified ICARS (MICARS), the Brief Ataxia Rating Scale (BARS), and the Neurological Examination Score for Spinocerebellar Ataxia (NESSCA). 58 None of these scales are meant to address non‐ataxic symptoms, for which additional scales have been developed. These additional scales include the Inventory of Non‐Ataxia Signs, which rates several neurological signs including spasticity, tremor, and dystonia, and the Cerebellar Cognitive Affective Syndrome Scale (CCAS), which is a dedicated cognitive scale to identify cognitive deficits in the context of cerebellar decline. 59
The ataxic rating scales are being combined with other quantifiable performance tests. These include the SCA Functional Index, which combines measures on an 8‐Minute Walk Test, a Nine‐Hole Peg Test, and a measure of the number of syllables that a patient can repeat in 10 sec; and the Composite Cerebellar Functional Severity Score (CCFS), which combines a click test of alternate tapping on two keys at a fixed distance with the Nine‐Hole Peg Test. 60 , 61
Some of these rating scales and performance measure are already being used in natural history studies. The rating scales are relatively reliable in their ability to detect disease, and can detect conversion from pre‐manifest to manifest disease over a period of 4–8 years. 13 , 47 , 62 They do, however, have their limitations. They are non‐linear scales, with different weights given to different ataxic symptoms. They have floor and ceiling effects, making them not very effective when the symptoms are either very mild or very severe. Furthermore, there is still some unavoidable subjectivity in scoring. 57 And finally, patient performance may fluctuate for a variety of reasons, including energy levels and the time of the day when patients are evaluated.
In a clinical trial, disease progression will likely be influenced by age of onset, repeat length, and age at trial inclusion. 47 , 62 For the most rapidly progressive polyglutamine SCA, namely SCA1, estimates suggest that approximately 142 patients would need to be recruited to power two arms of a clinical study to detect a 50% reduction in progression of the SARA score in a 1‐year trial. 3 The necessary recruitment numbers for other SCA subtypes for which there is natural history data are 172 patient for SCA2, 202 for SCA3, and 602 for SCA6. 47 Clearly, such long duration studies with large cohorts are difficult to conduct because of limitations in patient recruitment, patient attrition, comorbid conditions, and expense. Efforts are, therefore, ongoing to use new technological innovations to capture objective longitudinal data on patient symptoms in order to remove any bias from a human rater, and to provide additional objective data points assessing patients’ symptoms over time. These include the use of wearable sensors or smartphones to capture movements, allowing for remote long‐term tracking of patients’ movements so as to more accurately capture the full range and variability of symptoms on a day‐to‐day basis. Given that ataxia symptoms are known to fluctuate based on a variety of environmental factors, longitudinal wearable sensors may provide a more accurate representation of symptom burden, as opposed to intermittent clinical assessments. Similar technologies are being studied for other neurological diseases such as PD. 63 , 64 It is likely that advances in this arena will provide more precise and accurate rating scales for the SCAs that will improve statistical power in clinical trials. Potential downsides to wearable sensor technologies include patient inconvenience or discomfort as well as technical difficulties with device use or with data storage and interpretation.
While clinical rating scales and wearable devices allow us to assess ataxia and other neurologic symptoms, it is also important to keep in mind that the clinical changes measured must be clinically relevant. Therefore, from both a regulatory standpoint as well as from a patient advocacy standpoint, it will be important to incorporate quality of life metrics as well as other patient‐reported outcome (PRO) measures as a critical outcome in clinical trial design. This will ensure that any potential treatment interventions produce clinically relevant improvements for patients.
Identification of imaging and fluid biomarkers
Biomarkers are defined as “objectively‐measured characteristics that serve as indicators of normal biological processes, pathogenic processes or pharmacologic responses to therapeutic interventions.” 65 Strictly speaking, the motor performance measures described above could also be considered biomarkers. However, for the purposes of this review article, we focus our discussion of biomarkers on neuroimaging findings associated with disease state (Table 1) and biochemical markers obtained from body fluids (Table 2). The expectation is that, when validated, biomarkers for the SCAs could be used singly or in combination to provide effective ways to monitor disease onset, progression, or response to treatment, with less variability and more objectivity than clinical rating scales.
Table 1.
Current state of imaging biomarkers in the SCAs.
| Brain imaging |
|---|
| MRI: brain volume changes |
| ↓Cerebellum and brainstem volume in SCA1, SCA2, SCA3, SCA6, and SCA17 66 , 67 , 68 |
| ↓White matter and gray matter volume in cerebellum and brain stem in SCA1, SCA2, and SCA3 69 , 70 , 71 , 72 |
| ↓Spinal cord, basal ganglia, and cerebellar vermis size in SCA3 and SCA6 69 , 73 |
| ↓ Caudate size in SCA6 and SCA17 69 , 74 |
| MRS: chemical changes |
| ↓[Glu], [NAA], [NAAG], [tNAA], [Cho/Cr] ratio, and [Glu/Gln] ratio in SCA1, SCA2, SCA3, and SCA6 75 , 76 , 77 , 78 |
| ↑ [Glc], [Gln], [mI], [Tau], [tCr], and [Glc+Tau] in SCA1, SCA2, and SCA3 75 , 76 , 77 , 78 |
| ↓[GABA] in SCA6 41 |
| ↓ [NAA/Cho], [NAA/Cr] in SCA1, SCA2, SCA3, SCA6, and SCA17 75 , 76 , 77 , 78 |
| PET: metabolic changes |
| [18F]FDG, [11C]dMP, and [11C]MP4P: ↓metabolism in different brain regions in SCA1, SCA2, SCA3, SCA6, and SCA17 81 , 82 , 83 , 84 , 85 , 86 |
| SPECT: functional and metabolic changes |
| SCA2 |
| [99mTc]TRODAT‐1 SPECT:↓striatal DAT binding 87 |
| [123I]β‐CIT SPECT:↓striato‐cerebellar ratio 88 |
| [123I]IBZM SPECT:↓striato‐frontal IBZM binding ratio 88 |
| [123I]FP‐CIT SPECT:↓uptake in caudate and putamen 89 |
| SCA3 |
| [99mTc]TRODAT‐1 SPECT:↓nigrostriatal ratio 90 |
| [99mTc]HMPAO SPECT:↓perfusion in cerebellar hemispheres, 91 inferior, 91 and superior 91 frontal lobe, 91 lateral temporal lobe, 91 parietal lobe, and vermis 91 |
| [99mTc]ECD SPECT:↓perfusion in bilateral cerebellum 92 and vermis 92 |
| [123I]iomazenil SPECT:↓binding in cerebellum, 93 cerebral cortex, 93 thalamus, 93 and striatum 93 |
| SCA6 |
| [99mTc]ECD SPECT:↓perfusion in cerebellar hemisphere 94 and cerebral vermis 94 |
| SCA17 |
| [99mTc]TRODAT‐1 SPECT:↓striatal DAT binding 87 |
Table is modified from Meng‐LingChen et al. 95
Table 2.
Current state of fluid biomarkers in the SCAs.
| Fluid biomarkers |
|---|
| Poly‐Q expanded ataxin‐3 |
| ↑ in plasma and CSF of SCA3 104 , 105 |
| Oxidative stress markers |
| ↑ in serum of SCA3 106 |
| Carboxyl terminus of Hsp70‐interacting protein |
| ↑ in serum and CSF of SCA3 107 |
| Oxidation of 2′,7′‐dichlorofluorescein diacetate (DCFH‐DA) |
| ↑ in serum of SCA3 108 |
| Glutathione peroxidase activity |
| ↓ in serum of symptomatic SCA3 108 |
| IGFBP‐1 and IGFBP‐3 |
| ↑ in serum of SCA3 109 |
| Insulin |
| ↓ in serum of SCA3 109 |
| Neuron‐specific enolase |
| ↑ in serum of SCA3 110 , 111 |
| Neurofilament light chain |
| ↑ in serum and CSF of SCA1 112 and SCA3 112 , 113 , 114 |
| Phosphorylated neurofilament heavy chain |
| ↑ in serum of SCA3 112 |
| miRNA |
| ↑ miR‐34b, 114 miR‐7014 115 in serum, and CSF of SCA3 |
| Differential expression of seventy one miRs in plasma of SCA7 12 |
| ↓miR‐25, 114 miR‐29a, 114 miR‐125b, 114 and miR‐7014 115 in serum/plasma of SCA3 |
| Different expression of various exosomal miRs in plasma and CSF of SCA3 115 |
| S100B |
| ↑ in serum of SCA3 111 |
| Tau |
| ↑ in CSF of SCA2 117 |
| Valine, leucine, and tyrosine |
| ↓ in plasma of SCA7 118 |
Imaging biomarkers
Candidate imaging biomarkers have arisen from our understanding of findings garnered from neuroimaging performed on patients in the clinic (Table 1). 41 , 66 , 67 , 68 , 69 , 70 , 71 , 72 , 73 , 74 , 75 , 76 , 77 , 78 , 79 , 80 , 81 , 82 , 83 , 84 , 85 , 86 , 87 , 88 , 89 , 90 , 91 , 92 , 93 , 94 , 95 Focal degeneration can be quantified structurally using region of interest (ROI)‐based analysis. Structural abnormalities are accompanied by disturbances in white matter tracts as demonstrated by diffusion tensor imaging (DTI), with abnormalities discernible not just in the cerebellum and cerebellar peduncles but also in the brainstem and cerebral white matter. 3 , 96 , 97 These imaging abnormalities align closely with clinical rating scales, and indeed can help identify pre‐manifest abnormalities and predict phenotypic conversion. 25 , 98 , 99
Structural changes visible on MRI are accompanied by functional changes as well. While resting state functional MRI shows a few abnormalities, a more promising strategy is magnetic resonance spectroscopy (MRS). In MRS‐specific neurochemical, abnormalities are identified by comparing spectroscopic peaks corresponding to different metabolites. Up to 15 metabolites are measured with current MRS technologies. The most useful so far are N‐acetylaspartate (NAA, a marker of neuronal density and function) and glutamate that are altered with neuronal degeneration. 41 , 98 , 100 Furthermore, the glial marker myoinositol has been shown to increase with gliosis. 100 Other metabolites that can be monitored include the metabolic marker creatine/phosphocreatine and the membrane marker choline. MRS abnormalities are influenced by the specific SCA subtype, and they are even more sensitive to progression and phenotypic conversion than structural MRI. 3 , 101 , 102 , 103
Other imaging modalities are also being tested. These include positron emission tomography (PET) and single‐photon emission computed tomography (SPECT). In both modalities, tracers are used to interrogate specific circuit dysfunction, such as dopamine transmission. 103 The current data on imaging biomarkers in the SCAs are summarized in Table 1. There are many pros and cons to the different modalities available including variable sensitivity and specificity, and the use of certain technologies may be challenging in resource‐limited settings. While these efforts are still in their infancy, the hope is that several imaging modalities used together could produce a multimodal imaging signature to track disease onset and progression.
Fluid biomarkers
Fluid biomarkers are biomolecules or metabolites in the blood or cerebrospinal fluid (CSF) that correlate with an underlying pathological process. For the SCAs, the purpose is not to identify a biomarker for diagnosis, since diagnosis is achievable by genetic testing alone. Rather, the goal is to identify markers that predict phenoconversion (from the asymptomatic to symptomatic condition), or provide objective evidence of disease progression.
While no fluid biomarkers have yet been validated for the SCAs, there are some promising candidates (Table 2). 104 , 105 , 106 , 107 , 108 , 109 , 110 , 111 , 112 , 113 , 114 , 115 , 116 , 117 , 118 The first candidate is the mutant protein itself in the case of the polyglutamine SCAs. In all these ataxias, the protein tends to accumulate and aggregate. Following the levels of the mutant polyglutamine protein would be analogous to following the levels of other proteins in the more common neurodegenerative proteinopathies: Aβ 42 and tau (including phosphorylated tau) in AD, alpha synuclein in PD, and huntingtin protein in HD. 119 , 120 , 121 , 122
The comparison with HD is particularly apt since, as a polyglutamine disorder, it serves as an exemplar that can provide important insight into what we might observe in the polyglutamine SCAs. Levels of mutant huntingtin protein have been found to be elevated in the CSF of patients with HD, and importantly, the level correlates with diminished cognitive and motor function in HD patients. 123 , 124 Huntingtin levels are also being followed in clinical trials attempting huntingtin reduction by ASOs to show target engagement of the ASOs. 26 Techniques to detect mutant huntingtin in the CSF have required technical advances in antibody‐mediated detection, and similar approaches are being developed for the polyglutamine SCAs. 125 For instance, a recent strategy for monitoring ataxin‐3 levels in blood has been identified, although it is still unclear the extent to which blood levels of ataxin‐3 reflect the levels in the central nervous system. 105
In addition to biomarkers based on the biology of the specific disease, there are candidate fluid biomarkers that are more broadly applicable to diverse neurodegenerative disorders. The most promising of these are the neurofilament proteins, which are constituents of the neuronal cytoskeleton that leak out of damaged neurons. These proteins are showing considerable promise as a general marker of neuroaxonal damage in a wide range of neurological disorders, including HD, and can be detected not just in CSF but also in blood. 126 , 127 , 128 Neurofilament light chain (NFL) and a phosphorylated form of neurofilament heavy chain (pNFH) have been the most thoroughly studied to date. 128 In HD, for instance, the level of NFL in blood correlates with disease severity as assessed by standardized functional capacity scoring, brain atrophy on imaging, and CAG‐repeat length. 121
While not all SCAs have been evaluated for NFL levels, in SCA3, serum levels of NFL are significantly elevated in patients compared to controls. Importantly NFL levels correlate with disease severity as measured via clinical SARA scores. 104 , 112 , 113 NFL is also elevated in genetic carriers who have not yet developed ataxia. 104 Pilot studies suggest that NFL proteins are likely to be elevated in other SCA subtypes as well. 112 , 117
Biomarkers are also being identified by additional candidate‐based approaches focusing on neurotrophic factors, cytokines, or markers of oxidative stress that are known to be affected in preclinical models of the disease. 108 , 129 There is also the possibility of using unbiased biomarker discovery approaches such as mass‐spectroscopy techniques, which have the advantage of accelerating the identification of biomarkers since multiple potential candidates can be screened in parallel. These large‐scale screens need not be confined to proteins. For instance, metabolites could be identified using dedicated metabolomic or lipidomic approaches as is being done for other neurodegenerative diseases. 130 , 131 RNA molecules could be detected by RNA isolation from leukocytes or exosomes (including microRNA and mRNA). 132 , 133 Clearly, the identification and validation of a biomarker signature in the blood or CSF could be an invaluable tool in evaluating the response to any investigational drug, especially when used in conjunction with clinical rating scales and imaging biomarkers.
Innovations in clinical trial design
Unlike the more prevalent neurodegenerative disorders, clinical trial implementation for rare diseases such as the SCAs can pose unique challenges. However, in many respects, ataxia researchers are going over well‐ploughed terrain based on the lessons learned from other neurodegenerative diseases, most notably HD. In the HD field, there have been large‐scale biomarker studies such as TRACK‐HD that have helped establish the optimal outcome measures for tracking clinical trial results. 134 Clinical trials of antisense oligonucleotide therapies to inhibit HTT messenger RNA are now underway, along with biomarker monitoring of mutant HTT to monitor target engagement and neurofilament light chain to measure neurodegeneration. 22 , 26 , 135 Successful strategies used in HD can hopefully translate to the SCA field.
To recruit sufficient patients for clinical trials, setting up collaborations and patient registries is critical. Fortunately, SCA registries are already in place in the United States and Europe and efforts are ongoing for global initiatives. Multiple successful international research cohorts have been developed (Table 3). 2 , 47 , 62 , 97 , 136 , 137 For instance, In the United States, there is a large consortium for the SCAs called the Clinical Research Consortium for Spinocerebellar Ataxias (CRC‐SCA), while in Europe, the Ataxia Study Group has organized multiple international collaborative research projects including the European Integrated Project on Spinocerebellar Ataxia (EuroSCA) and the European Spinocerebellar Ataxia Type 3/Machado‐Joseph Disease Initiative (ESMI). Looking forward, there are on‐going global initiatives like SCA Global, which aims to accelerate biomarker discovery and treatment development via standardization of research procedures and expansion of research efforts around the world. 138
Table 3.
Summary of international SCA research cohorts.
| Research consortium | Genetic ataxia | Biomarker sample type | Imaging studies | Target sample size | Participating countries and funding sources | Time period |
|---|---|---|---|---|---|---|
| Clinical Research Consortium for Spinocerebellar Ataxias (CRC‐SCA) 97 | SCA1 | Blood | MRI/MRS at 4 sites | 800 | Country: USA | 2010‐present |
| SCA2 | Funding: National Ataxia Foundation; National Institute of Neurological Disorders and Stroke (NINDS) of the National Institute of Health (NIH) | |||||
| SCA3 | ||||||
| SCA6 | ||||||
| SCA7 | ||||||
| SCA8 | ||||||
| SCA10 | ||||||
| European Integrated Project on Spinocerebellar Ataxias (EUROSCA) 2 , 47 | SCA1 | Blood | MRI | 1255 | Countries: Germany, Spain, France, Italy, UK, Netherlands, Belgium, Poland, Hungary | 2005–2016 |
| SCA2 | Urine | Funding: European Union Sixth Framework Programme FP‐6 | ||||
| SCA3 | ||||||
| SCA6 | ||||||
| European Spinocerebellar Ataxia Type 3/Machado‐Joseph Disease Initiative (ESMI) 136 | SCA3 | Blood | MRI | 800 | Countries: Germany, UK, Portugal, Netherlands | 2016‐present |
| CSF | Funding: EU Joint Programme – Neurodegenerative Disease Research (JPND); German Ministry of Education and Research; Medical Research Council (MRC, United Kingdom); Fundação para a Ciência e a Tecnologia (FCT; Portugal); Netherlands Organisation for Health Research and Development | |||||
| Individuals at Risk for SCA (RISCA) 62 | SCA1 | Blood | MRI at 8 sites | 480 | Countries: France, Austria, Germany, Hungary, Italy, Poland, Spain | 2009‐present |
| SCA2 | Urine | Funding: European Research Area Network for Research Programmes on Rare Diseases, Polish Ministry of Science and Higher Education, Italian Ministry of Health, European Community's Seventh Framework Programme | ||||
| SCA3 | ||||||
| SCA6 | ||||||
| Clinical trial Readiness for SCA1 & SCA3 (READISCA) 97 | Early‐stage SCA1 and SCA3 | Blood | MRI/MRS at 6 sites in 3 countries | 200 | Countries: USA, Germany, France, UK, Poland, Hungary, Italy, Austria, Belgium, Portugal, Spain, Netherlands, Norway, Denmark, Serbia, Egypt, Morocco | 2018‐present |
| Funding: National Institute of Neurological Disorders and Stroke (NINDS) of the National Institute of Health (NIH); National Ataxia Foundation | ||||||
| SCA3 Biomarkers and Genetic modifiers in a Study pre‐ and post‐symptomatic carriers (BIGPRO) 137 | SCA3 | Blood | Not applicable | 95 | Countries: Brazil, Germany | 2017–2020 |
| Funding: CAPES Foundation |
For the SCAs, it is important that manifest patients as well as asymptomatic individuals at risk are recruited for both biomarker development and clinical trials. Recruited patients should have a confirmed diagnosis of a genetic ataxia based on standardized genetic testing. It is important that in global patient registries both genetic and phenotypic information be catalogued closely since trials of treatments aimed largely at symptomatic improvement would require that patient symptomatology be a major inclusion criteria and that those with minimal symptoms be excluded. On the other hand, trials aimed at assessing neuroprotective strategies could recruit patients with manifest disease but could also potentially assess pre‐manifest individuals with known SCA mutations to determine if progression to manifest disease can be halted.
Due to the constraints in patient recruitment, innovations in clinical design are likely to play a role especially for the rarer SCAs. One recourse is for experimental drugs to be compared to historical controls rather than a placebo arm, but this is not ideal given the role of placebo effects. Therefore, an alternative approach would be to test multiple drugs against a single placebo arm. 139 , 140 This arm would serve as a control for multiple interventions, thus reducing the number of patients necessary for recruitment. Such an approach may also be appealing to patients who may prefer the increased likelihood of being randomized to an experimental arm. Another approach that eliminates the need for a dedicated placebo cohort is a cross‐over design in which individuals could be placed on an experimental drug for a certain duration of time, then given time for the drug to wash out. The disease process can be observed in both conditions, with the process of cross‐over repeated (if necessary) to document whether the patient did in fact improve on the drug. 140 , 141
Trials could also be run to demonstrate non‐effectiveness rather than effectiveness. Such studies are similar to establishing non‐inferiority of experimental interventions in other diseases and require fewer patients. By paring down candidate therapies through an early rejection of candidates, fewer patients will be tied down in non‐productive trials and more patients will be available to participate in trials testing drugs that are found to be more likely to show efficacy after this initial screening process. Other statistical techniques include Bayesian methods where information from natural history studies could be borrowed to make prior distributions about expected endpoints and clinical outcomes. 142
Another challenge in clinical trial development that needs to be addressed is financial. Clinical research to be sure is expensive, and funds are especially hard to come by for rare disorders. Creative and collaborative approaches, therefore, need to be employed to raise support for these disorders from philanthropic sources, government agencies, and the pharmaceutical industry. Fortunately government regulatory agencies in the United States and Europe have set up mechanisms to help accelerate the development of drugs for rare disorders. In particular, in 1983, the Orphan Drug Act was passed in the United States with the help of advocacy groups such as the National Organization for Rare Disorders (NORD). This legislation helped promote and accelerate drug development for rare disorders by providing financial incentives such as tax credits and a period of market exclusivity, as well as the establishment of a grant support program. Such support is critical for helping to develop effective clinical trials for these rare disorders.
Conclusion
This review summarizes the major challenges to clinical trials in the SCAs. But we would like to end on an optimistic note. As outlined above, these challenges are being addressed by substantial technological advancements along several fronts, and new therapies are on the horizon.
Some of the biggest operational hurdles for clinical trials have already been accomplished. Collaborative consortia have been established and patients are being recruited in registries throughout North America and Europe. Natural history studies, rating scale optimization, and biomarker development are ongoing at an active pace. We are also gaining important information from clinical trials in other neurodegenerative diseases, most notably Alzheimer’s disease, Parkinson’s disease, and perhaps most relevant, Huntington’s disease. For each of these diseases, researchers have had to work through similar steps of infrastructure development and clinical trial readiness, and we can therefore learn from the steps taken in these disorders. But perhaps most importantly, the anticipation of truly groundbreaking protein lowering strategies is creating opportunities for pharmaceutical companies to attempt to test novel treatments for the SCAs. This is providing the incentives to provide further funding for clinical trials in the years ahead. These are exciting times for SCA research with emerging optimism that treatments will soon be brought to bear on these otherwise incurable diseases.
Conflict of Interest
P. O. receives support from the NIH (1R01NS062051, 1R01NS082351, and R56NS108639). He has received funding from the following sources for clinical trials: Biohaven Pharmaceuticals, U01NS104326 (site PI), and the National Ataxia Foundation (CRC‐SCA natural history study). C.R.E received support from National Ataxia Foundation (YISCA award) and NUCATS & NMF (Dixon translational Research Grants Initiative award). S.H.K receives support from the NIH (1R01NS104434, 1R01NS118179, 1R03NS114871, and 1R13NS117005), and also receives funding support from Biohaven Pharmaceuticals and Sage Therapeutics for clinical trial research. S.M.B., S.M.A, and C.R.E. have no competing interests.
Funding Information
P. O. receives support from the NIH (1R01NS062051, 1R01NS082351, and R56NS108639). He has received funding from the following sources for clinical trials: Biohaven Pharmaceuticals, U01NS104326 (site PI), and the National Ataxia Foundation (CRC‐SCA natural history study). C.R.E received support from National Ataxia Foundation (YISCA award) and NUCATS & NMF (Dixon translational Research Grants Initiative award). S.H.K receives support from the NIH (1R01NS104434, 1R01NS118179, 1R03NS114871, and 1R13NS117005), and also receives funding support from Biohaven Pharmaceuticals and Sage Therapeutics for clinical trial research.
Funding Statement
This work was funded by NIH grants 1R01NS062051, 1R01NS082351, R56NS108639, 1R01NS104434, 1R01NS118179, 1R03NS114871, and 1R13NS117005; Biohaven Pharmaceuticals grant U01NS104326; National Ataxia Foundation ; NUCATS; NMF.
Contributor Information
Sarah M. Brooker, Email: sarah-brooker@northwestern.edu.
Chandrakanth Reddy Edamakanti, Email: chandu@northwestern.edu.
Puneet Opal, Email: p-opal@northwestern.edu.
References
- 1. Klockgether T, Mariotti C, Paulson HL. Spinocerebellar ataxia. Nat Rev Dis Primers 2019;5:24. [DOI] [PubMed] [Google Scholar]
- 2. Diallo A, Jacobi H, Cook A, et al. Survival in patients with spinocerebellar ataxia types 1, 2, 3, and 6 (EUROSCA): a longitudinal cohort study. Lancet Neurol 2018;17:327–334. [DOI] [PubMed] [Google Scholar]
- 3. Ashizawa T, Öz G, Paulson HL. Spinocerebellar ataxias: prospects and challenges for therapy development. Nat Rev Neurol 2018;14:590–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sullivan R, Yau WY, O’Connor E, Houlden H. Spinocerebellar ataxia: an update. J Neurol 2019;266:533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lieto M, Riso V, Galatolo D, et al. The complex phenotype of spinocerebellar ataxia type 48 in eight unrelated Italian families. Eur J Neurol 2020;27:498–505. [DOI] [PubMed] [Google Scholar]
- 6. Didonna A, Opal P. Advances in sequencing technologies for understanding hereditary ataxias: a review. JAMA Neurol 2016;73:1485–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 2010;9:885–894. [DOI] [PubMed] [Google Scholar]
- 8. Swinnen B, Robberecht W, Van Den Bosch L. RNA toxicity in non‐coding repeat expansion disorders. EMBO J 2020;39:e101112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lieberman AP, Shakkottai VG, Albin RL. Polyglutamine repeats in neurodegenerative diseases. Annu Rev Pathol 2019;14:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Banez‐Coronel M, Ranum LPW. Repeat‐associated non‐AUG (RAN) translation: insights from pathology. Lab Invest 2019;99:929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nguyen L, Cleary JD, Ranum LPW. Repeat‐associated non‐ATG translation: molecular mechanisms and contribution to neurological disease. Annu Rev Neurosci 2019;42:227–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Paulson HL, Shakkottai VG, Clark HB, Orr HT. Polyglutamine spinocerebellar ataxias—from genes to potential treatments. Nat Rev Neurosci 2017;18:613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jacobi H, Bauer P, Giunti P. The natural history of spinocerebellar ataxia type 1, 2, 3, and 6: a 2‐year follow‐up study. Neurology 2011;77:1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kraus‐Perrotta C, Lagalwar S. Expansion, mosaicism and interruption: mechanisms of the CAG repeat mutation in spinocerebellar ataxia type 1. Cerebellum Ataxias 2016;3:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mouro Pinto R, Arning L, Giordano JV, et al. Patterns of CAG repeat instability in the central nervous system and periphery in Huntington's disease and in spinocerebellar ataxia type 1. Hum Mol Genet 2020;29:2551–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Friedrich J, Kordasiewicz HB, O’Callaghan B, et al. Antisense oligonucleotide‐mediated ataxin‐1 reduction prolongs survival in SCA1 mice and reveals disease‐associated transcriptome profiles. JCI insight 2018;3:e123193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scoles DR, Meera P, Schneider MD, et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 2017;544:362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Scoles D, Schneider M, Pratap M, et al. Antisense oligonucleotides for the treatment of spinocerebellar ataxia type 2 (SCA2) (S32.002). Neurology 2015;84:S32.002. [Google Scholar]
- 19. Yang WY, Gao R, Southern M, et al. Design of a bioactive small molecule that targets r(AUUCU) repeats in spinocerebellar ataxia 10. Nat Commun 2016;7:11647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Khan E, Mishra SK, Mishra R, et al. Discovery of a potent small molecule inhibiting Huntington's disease (HD) pathogenesis via targeting CAG repeats RNA and Poly Q protein. Sci Rep 2019;9:16872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gustincich S, Zucchelli S, Mallamaci A. The Yin and Yang of nucleic acid‐based therapy in the brain. Prog Neurogibol 2017;155:194–211. [DOI] [PubMed] [Google Scholar]
- 22. Scoles DR, Pulst SM. Oligonucleotide therapeutics in neurodegenerative diseases. RNA Biol 2018;15:707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov 2020;19:673–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zesiewicz TA, Wilmot G, Kuo S‐H, et al. Comprehensive systematic review summary: treatment of cerebellar motor dysfunction and ataxia: report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology 2018;90:464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Coarelli G, Brice A, Durr A. Recent advances in understanding dominant spinocerebellar ataxias from clinical and genetic points of view. F1000Research 2018;7:1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tabrizi SJ, Ghosh R, Leavitt BR. Huntingtin lowering strategies for disease modification in Huntington's disease. Neuron 2019;101:801–819. [DOI] [PubMed] [Google Scholar]
- 27. Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, et al. Targeting Huntingtin expression in patients with Huntington’s disease. N Engl J Med 2019;380:2307–2316. [DOI] [PubMed] [Google Scholar]
- 28. Bushart DD, Chopra R, Singh V, et al. Targeting potassium channels to treat cerebellar ataxia. Ann Clin Transl Neurol 2018;5:297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hu Y‐S, Do J, Edamakanti CR, et al. Self‐assembling vascular endothelial growth factor nanoparticles improve function in spinocerebellar ataxia type 1. Brain 2019;142:312–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cvetanovic M, Patel JM, Marti HH, et al. Vascular endothelial growth factor ameliorates the ataxic phenotype in a mouse model of spinocerebellar ataxia type 1. Nat Med 2011;17:1445–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Palmer EE, Hong S, Al Zahrani F, et al. De novo variants disrupting the HX repeat motif of ATN1 cause a recognizable non‐progressive neurocognitive syndrome. Am J Hum Genet 2019;104:542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carroll LS, Massey TH, Wardle M, Peall KJ. Dentatorubral‐pallidoluysian atrophy: an update. Tremor Other Hyperkinet Mov 2018;8:577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mellesmoen A, Sheeler C, Ferro A, et al. Brain derived neurotrophic factor (BDNF) delays onset of pathogenesis in transgenic mouse model of spinocerebellar ataxia type 1 (SCA1). Front Cell Neurosci 2018;12:509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Romano S, Coarelli G, Marcotulli C, et al. Riluzole in patients with hereditary cerebellar ataxia: a randomised, double‐blind, placebo‐controlled trial. Lancet Neurol 2015;14:985–991. [DOI] [PubMed] [Google Scholar]
- 35. Ristori G, Romano S, Visconti A, et al. Riluzole in cerebellar ataxia: a randomized, double‐blind, placebo‐controlled pilot trial. Neurology 2010;74:839–845. [DOI] [PubMed] [Google Scholar]
- 36. Sarva H, Shanker VL. Treatment options in degenerative cerebellar ataxia: a systematic review. Movement Disord Clin Pract (Hoboken, NJ) 2014;1:291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ogawa M. Pharmacological treatments of cerebellar ataxia. Cerebellum 2004;3:107–111. [DOI] [PubMed] [Google Scholar]
- 38. Chen Y‐S, Hong Z‐X, Lin S‐Z, Harn H‐J. Identifying therapeutic targets for spinocerebellar ataxia type 3/Machado‐Joseph disease through integration of pathological biomarkers and therapeutic strategies. Int J Mol Sci 2020;21:3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jacobi H, Reetz K, du Montcel ST, et al. Biological and clinical characteristics of individuals at risk for spinocerebellar ataxia types 1, 2, 3, and 6 in the longitudinal RISCA study: analysis of baseline data. Lancet Neurol 2013;12:650–658. [DOI] [PubMed] [Google Scholar]
- 40. Falcon MI, Gomez CM, Chen EE, et al. Early cerebellar network shifting in spinocerebellar ataxia type 6. Cereb Cortex 2016;26:3205–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Joers JM, Deelchand DK, Lyu T, et al. Neurochemical abnormalities in premanifest and early spinocerebellar ataxias. Ann Neurol 2018;83:816–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ruano L, Melo C, Silva MC, Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 2014;42:174–183. [DOI] [PubMed] [Google Scholar]
- 43. Paulson HL. The spinocerebellar ataxias. J Neuroophthalmol 2009;29:227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jayadev S, Bird TD. Hereditary ataxias: overview. Genet Med 2013;15:673–683. [DOI] [PubMed] [Google Scholar]
- 45. Tezenas du Montcel S, Durr A, Rakowicz M, et al. Prediction of the age at onset in spinocerebellar ataxia type 1, 2, 3 and 6. J Med Genet 2014;51:479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Campos‐Romo A, Graue‐Hernandez EO, Pedro‐Aguilar L, et al. Ophthalmic features of spinocerebellar ataxia type 7. Eye 2018;32:120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jacobi H, du Montcel ST, Bauer P, et al. Long‐term disease progression in spinocerebellar ataxia types 1, 2, 3, and 6: a longitudinal cohort study. Lancet Neurol 2015;14:1101–1108. [DOI] [PubMed] [Google Scholar]
- 48. Driessen TM, Lee PJ, Lim J. Molecular pathway analysis towards understanding tissue vulnerability in spinocerebellar ataxia type 1. eLife 2018;7:e39981. 10.7554/eLife.39981.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tezenas du Montcel S, Durr A, Bauer P, et al. Modulation of the age at onset in spinocerebellar ataxia by CAG tracts in various genes. Brain 2014;137:2444–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gan S‐R, Figueroa KP, Xu H‐L, et al. The impact of ethnicity on the clinical presentations of spinocerebellar ataxia type 3. Parkinsonism Relat Disord 2020;72:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Suh J, Romano DM, Nitschke L, et al. Loss of ataxin‐1 potentiates Alzheimer's pathogenesis by elevating cerebral BACE1 transcription. Cell 2019;178:1159–1175.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Becker LA, Huang B, Bieri G, et al. Therapeutic reduction of ataxin‐2 extends lifespan and reduces pathology in TDP‐43 mice. Nature 2017;544:367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Genetic Modifiers of Huntington’s Disease C . Identification of genetic factors that modify clinical onset of Huntington's disease. Cell 2015;162:516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Burk K, Schulz SR, Schulz JB. Monitoring progression in Friedreich ataxia (FRDA): the use of clinical scales. J Neurochem 2013;126(Suppl 1):118–124. [DOI] [PubMed] [Google Scholar]
- 55. Subramony SH. SARA–a new clinical scale for the assessment and rating of ataxia. Nat Clin Pract Neurol 2007;3:136–137. [DOI] [PubMed] [Google Scholar]
- 56. Dworak EM, Revelle W, Doebler P, Condon DM. Using the International Cognitive Ability Resource as an open source tool to explore individual differences in cognitive ability. Personality Individ Differ 2021;169:109906. [Google Scholar]
- 57. Schmitz‐Hubsch T, du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 2006;66:1717–1720. [DOI] [PubMed] [Google Scholar]
- 58. Perez‐Lloret S, Warrenburg B, Rossi M, et al. Assessment of ataxia rating scales and cerebellar functional tests: critique and recommendations. Mov Disord 2021;36:283–297. [DOI] [PubMed] [Google Scholar]
- 59. Hoche F, Guell X, Vangel MG, et al. The cerebellar cognitive affective/Schmahmann syndrome scale. Brain 2018;141:248–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. du Montcel ST, Charles P, Ribai P, et al. Composite cerebellar functional severity score: validation of a quantitative score of cerebellar impairment. Brain 2008;131:1352–1361. [DOI] [PubMed] [Google Scholar]
- 61. Barbuto S, Mackenzie S, Kuo S‐H, et al. Measurements of Hand function in degenerative cerebellar disease: a case‐control pilot study. Am J Phys Med Rehabil 2020;99:795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jacobi H, du Montcel ST, Romanzetti S, et al. Conversion of individuals at risk for spinocerebellar ataxia types 1, 2, 3, and 6 to manifest ataxia (RISCA): a longitudinal cohort study. Lancet Neurol 2020;19:738–747. [DOI] [PubMed] [Google Scholar]
- 63. Johansson D, Malmgren K, Alt MM. Wearable sensors for clinical applications in epilepsy, Parkinson's disease, and stroke: a mixed‐methods systematic review. J Neurol 2018;265:1740–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lonini L, Dai A, Shawen N, et al. Wearable sensors for Parkinson’s disease: which data are worth collecting for training symptom detection models. npj Digital Med 2018;1:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Biomarkers Definitions Working G . Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther 2001;69:89–95. [DOI] [PubMed] [Google Scholar]
- 66. Guerrini L, Lolli F, Ginestroni A, et al. Brainstem neurodegeneration correlates with clinical dysfunction in SCA1 but not in SCA2. A quantitative volumetric, diffusion and proton spectroscopy MR study. Brain 2004;127:1785–1795. [DOI] [PubMed] [Google Scholar]
- 67. Schulz JB, Borkert J, Wolf S, et al. Visualization, quantification and correlation of brain atrophy with clinical symptoms in spinocerebellar ataxia types 1, 3 and 6. NeuroImage 2010;49:158–168. [DOI] [PubMed] [Google Scholar]
- 68. Goel G, Pal PK, Ravishankar S, et al. Gray matter volume deficits in spinocerebellar ataxia: an optimized voxel based morphometric study. Parkinsonism Relat Disord 2011;17:521–527. [DOI] [PubMed] [Google Scholar]
- 69. Reetz K, Costa AS, Mirzazade S, et al. Genotype‐specific patterns of atrophy progression are more sensitive than clinical decline in SCA1, SCA3 and SCA6. Brain 2013;136:905–917. [DOI] [PubMed] [Google Scholar]
- 70. Guimarães RP, D'Abreu A, Yasuda CL, et al. A multimodal evaluation of microstructural white matter damage in spinocerebellar ataxia type 3. Mov Disord 2013;28:1125–1132. [DOI] [PubMed] [Google Scholar]
- 71. Kang JS, Klein JC, Baudrexel S, et al. White matter damage is related to ataxia severity in SCA3. J Neurol 2014;261:291–299. [DOI] [PubMed] [Google Scholar]
- 72. D'Abreu A, Franca MC Jr, Yasuda CL, et al. Neocortical atrophy in Machado‐Joseph disease: a longitudinal neuroimaging study. J Neuroimaging 2012;22:285–291. [DOI] [PubMed] [Google Scholar]
- 73. Fahl CN, Branco LM, Bergo FP, et al. Spinal cord damage in Machado‐Joseph disease. Cerebellum 2015;14:128–132. [DOI] [PubMed] [Google Scholar]
- 74. Brockmann K, Reimold M, Globas C, et al. PET and MRI reveal early evidence of neurodegeneration in spinocerebellar ataxia type 17. J Nucl Med 2012;53:1074–1080. [DOI] [PubMed] [Google Scholar]
- 75. Öz G, Hutter D, Tkáč I, et al. Neurochemical alterations in spinocerebellar ataxia type 1 and their correlations with clinical status. Mov Disord 2010;25:1253–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Oz G, Iltis I, Hutter D, et al. Distinct neurochemical profiles of spinocerebellar ataxias 1, 2, 6, and cerebellar multiple system atrophy. Cerebellum 2011;10:208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Doss S, Brandt AU, Oberwahrenbrock T, et al. Metabolic evidence for cerebral neurodegeneration in spinocerebellar ataxia type 1. Cerebellum 2014;13:199–206. [DOI] [PubMed] [Google Scholar]
- 78. Wang PS, Chen HC, Wu HM, et al. Association between proton magnetic resonance spectroscopy measurements and CAG repeat number in patients with spinocerebellar ataxias 2, 3, or 6. PLoS One 2012;7:e47479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Viau M, Marchand L, Bard C, Boulanger Y. (1)H magnetic resonance spectroscopy of autosomal ataxias. Brain Res 2005;1049:191–202. [DOI] [PubMed] [Google Scholar]
- 80. Lirng J‐F, Wang P‐S, Chen H‐C, et al. Differences between spinocerebellar ataxias and multiple system atrophy‐cerebellar type on proton magnetic resonance spectroscopy. PLoS One 2012;7:e47925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wüllner U, Reimold M, Abele M, et al. Dopamine transporter positron emission tomography in spinocerebellar ataxias type 1, 2, 3, and 6. Arch Neurol 2005;62:1280–1285. [DOI] [PubMed] [Google Scholar]
- 82. Wang PS, Liu RS, Yang BH, Soong BW. Regional patterns of cerebral glucose metabolism in spinocerebellar ataxia type 2, 3 and 6: a voxel‐based FDG‐positron emission tomography analysis. J Neurol 2007;254:838–845. [DOI] [PubMed] [Google Scholar]
- 83. Oh M, Kim JS, Oh JS, et al. Different subregional metabolism patterns in patients with cerebellar ataxia by 18F‐fluorodeoxyglucose positron emission tomography. PLoS One 2017;12:e0173275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gilman S, Sima AAF, Junck L, et al. Spinocerebellar ataxia type 1 with multiple system degeneration and glial cytoplasmic inclusions. Ann Neurol 1996;39:241–255. [DOI] [PubMed] [Google Scholar]
- 85. Soong B, Cheng C, Liu R, Shan D. Machado‐Joseph disease: clinical, molecular, and metabolic characterization in Chinese kindreds. Ann Neurol 1997;41:446–452. [DOI] [PubMed] [Google Scholar]
- 86. Soong BW, Liu RS. Positron emission tomography in asymptomatic gene carriers of Machado‐Joseph disease. J Neurol Neurosurg Psychiatry 1998;64:499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yun JY, Lee W‐W, Kim HJ, et al. Relative contribution of SCA2, SCA3 and SCA17 in Korean patients with parkinsonism and ataxia. Parkinsonism Relat Disord 2011;17:338–342. [DOI] [PubMed] [Google Scholar]
- 88. Boesch SM, Donnemiller E, Muller J, et al. Abnormalities of dopaminergic neurotransmission in SCA2: a combined 123I‐betaCIT and 123I‐IBZM SPECT study. Mov Disord 2004;19:1320–1325. [DOI] [PubMed] [Google Scholar]
- 89. Varrone A, Salvatore E, De Michele G, et al. Reduced striatal [123 I]FP‐CIT binding in SCA2 patients without parkinsonism. Ann Neurol 2004;55:426–430. [DOI] [PubMed] [Google Scholar]
- 90. Yen TC, Lu CS, Tzen KY, et al. Decreased dopamine transporter binding in Machado‐Joseph disease. J Nucl Med 2000;41:994–998. [PubMed] [Google Scholar]
- 91. Etchebehere ECSC, Cendes F, Lopes‐Cendes I, et al. Brain single‐photon emission computed tomography and magnetic resonance imaging in Machado‐Joseph disease. Arch Neurol 2001;58:1257–1263. [DOI] [PubMed] [Google Scholar]
- 92. Braga‐Neto P, Pedroso JL, Gadelha A, et al. Psychosis in Machado‐Joseph disease: clinical correlates, pathophysiological discussion, and functional brain imaging. Expanding the cerebellar cognitive affective syndrome. Cerebellum 2016;15:483–490. [DOI] [PubMed] [Google Scholar]
- 93. Ishibashi M, Sakai T, Matsuishi T, et al. Decreased benzodiazepine receptor binding in Machado‐Joseph disease. J Nucl Med 1998;39:1518–1520. [PubMed] [Google Scholar]
- 94. Honjo K, Ohshita T, Kawakami H, et al. Quantitative assessment of cerebral blood flow in genetically confirmed spinocerebellar ataxia type 6. Arch Neurol 2004;61:933–937. [DOI] [PubMed] [Google Scholar]
- 95. Chen M‐L, Lin C‐C, Rosenthal LS, et al. Rating scales and biomarkers for CAG‐repeat spinocerebellar ataxias: implications for therapy development. J Neurol Sci 2021;424:117417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Meira AT, Arruda WO, Ono SE, et al. Analysis of diffusion tensor parameters in spinocerebellar ataxia type 3 and type 10 patients. Parkinsonism Relat Disord 2020;78:73–78. [DOI] [PubMed] [Google Scholar]
- 97. Lin C‐C, Ashizawa T, Kuo S‐H. Collaborative efforts for spinocerebellar ataxia research in the United States: CRC‐SCA and READISCA. Front Neurol 2020;11:902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Oz G, Nelson CD, Koski DM, et al. Noninvasive detection of presymptomatic and progressive neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci 2010;30:3831–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Meira AT, Arruda WO, Ono SE, et al. Neuroradiological findings in the spinocerebellar ataxias. Tremor Other Hyperkinet Mov 2020;9. 10.7916/tohm.v7910.7682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Adanyeguh IM, Henry P‐G, Nguyen TM, et al. In vivo neurometabolic profiling in patients with spinocerebellar ataxia types 1, 2, 3, and 7. Mov Disord 2015;30:662–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Mandelli ML, De Simone T, Minati L, et al. Diffusion tensor imaging of spinocerebellar ataxias types 1 and 2. Am J Neuroradiol 2007;28:1996–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Adanyeguh IM, Perlbarg V, Henry P‐G, et al. Autosomal dominant cerebellar ataxias: imaging biomarkers with high effect sizes. Neuroimage Clin 2018;19:858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Miletich RS. Positron emission tomography and single‐photon emission computed tomography in neurology. Continuum (Minneap Minn) 2016;22:1636–1654. [DOI] [PubMed] [Google Scholar]
- 104. Wilke C, Haas E, Reetz K, et al. Neurofilaments in spinocerebellar ataxia type 3: blood biomarkers at the preataxic and ataxic stage in humans and mice. EMBO Mol Med 2020;12:e11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Prudencio M, Garcia‐Moreno H, Jansen‐West KR, et al. Toward allele‐specific targeting therapy and pharmacodynamic marker for spinocerebellar ataxia type 3. Sci Transl Med 2020;12:eabb7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pacheco LS, da Silveira AF, Trott A, et al. Association between Machado‐Joseph disease and oxidative stress biomarkers. Mutat Res 2013;757:99–103. [DOI] [PubMed] [Google Scholar]
- 107. Hu Z‐W, Yang Z‐H, Zhang S, et al. Carboxyl terminus of Hsp70‐interacting protein is increased in serum and cerebrospinal fluid of patients with spinocerebellar ataxia type 3. Front Neurol 2019;10:1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. de Assis AM, Saute JAM, Longoni A, et al. Peripheral oxidative stress biomarkers in spinocerebellar ataxia type 3/Machado‐Joseph disease. Front Neurol 2017;8:485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Saute JAM, da Silva ACF, Muller AP, et al. Serum insulin‐like system alterations in patients with spinocerebellar ataxia type 3. Mov Disord 2011;26:731–735. [DOI] [PubMed] [Google Scholar]
- 110. Tort ABL, Portela LVC, Rockenbach IC, et al. S100B and NSE serum concentrations in Machado Joseph disease. Clin Chim Acta 2005;351:143–148. [DOI] [PubMed] [Google Scholar]
- 111. Zhou J, Lei L, Shi Y, et al. Serum concentrations of NSE and S100B in spinocerebellar ataxia type 3/Machado‐Joseph disease. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2011;36:504–510. [DOI] [PubMed] [Google Scholar]
- 112. Wilke C, Bender F, Hayer SN, et al. Serum neurofilament light is increased in multiple system atrophy of cerebellar type and in repeat‐expansion spinocerebellar ataxias: a pilot study. J Neurol 2018;265:1618–1624. [DOI] [PubMed] [Google Scholar]
- 113. Li Q‐F, Dong YI, Yang LU, et al. Neurofilament light chain is a promising serum biomarker in spinocerebellar ataxia type 3. Mol Neurodegener 2019;14:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Shi Y, Huang F, Tang B, et al. MicroRNA profiling in the serums of SCA3/MJD patients. Int J Neurosci 2014;124:97–101. [DOI] [PubMed] [Google Scholar]
- 115. Hou X, Gong X, Zhang L, et al. Identification of a potential exosomal biomarker in spinocerebellar ataxia Type 3/Machado‐Joseph disease. Epigenomics 2019;11:1037–1056. [DOI] [PubMed] [Google Scholar]
- 116. Borgonio‐Cuadra VM, Valdez‐Vargas C, Romero‐Córdoba S, et al. Wide profiling of circulating microRNAs in spinocerebellar ataxia type 7. Mol Neurobiol 2019;56:6106–6120. [DOI] [PubMed] [Google Scholar]
- 117. Brouillette AM, Öz G, Gomez CM. Cerebrospinal fluid biomarkers in spinocerebellar ataxia: a pilot study. Dis Markers 2015;2015:413098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Nambo‐Venegas R, Valdez‐Vargas C, Cisneros B, et al. Altered plasma acylcarnitines and amino acids profile in spinocerebellar ataxia type 7. Biomolecules 2020;10:390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Thijssen EH, La Joie R, Wolf A, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer's disease and frontotemporal lobar degeneration. Nat Med 2020;26:387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Borsche M, König IR, Delcambre S, et al. Mitochondrial damage‐associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain 2020;143:3041–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Byrne LM, Rodrigues FB, Blennow K, et al. Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington's disease: a retrospective cohort analysis. Lancet Neurol 2017;16:601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Rodrigues FB, Byrne LM, Tortelli R, et al. Longitudinal dynamics of mutant huntingtin and neurofilament light in Huntington’s disease: the prospective HD‐CSF study. medRxiv 2020:2020.2003.2031.20045260. 10.1101/2020.03.31.20045260 [DOI] [Google Scholar]
- 123. Wild EJ, Boggio R, Langbehn D, et al. Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington's disease patients. J Clin Invest 2015;125:1979–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Byrne LM, Wild EJ. Cerebrospinal fluid biomarkers for Huntington's disease. J Huntington's Dis 2016;5:1–13. [DOI] [PubMed] [Google Scholar]
- 125. Fodale V, Boggio R, Daldin M, et al. Validation of ultrasensitive mutant huntingtin detection in human cerebrospinal fluid by single molecule counting immunoassay. J Huntingtons Dis 2017;6:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Norgren N, Rosengren L, Stigbrand T. Elevated neurofilament levels in neurological diseases. Brain Res 2003;987:25–31. [DOI] [PubMed] [Google Scholar]
- 127. Gaiottino J, Norgren N, Dobson R, et al. Increased neurofilament light chain blood levels in neurodegenerative neurological diseases. PLoS One 2013;8:e75091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Khalil M, Teunissen CE, Otto M, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol 2018;14:577–589. [DOI] [PubMed] [Google Scholar]
- 129. da Silva Carvalho G, Saute JAM, Haas CB, et al. Cytokines in Machado Joseph disease/spinocerebellar ataxia 3. Cerebellum 2016;15:518–525. [DOI] [PubMed] [Google Scholar]
- 130. Schwarzschild MA, Schwid SR, Marek K, et al. Serum urate as a predictor of clinical and radiographic progression in Parkinson disease. Arch Neurol 2008;65:716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Mapstone M, Cheema AK, Fiandaca MS, et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat Med 2014;20:415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Hosaka T, Yamashita T, Tamaoka A, Kwak S. Extracellular RNAs as biomarkers of sporadic amyotrophic lateral sclerosis and other neurodegenerative diseases. Int J Mol Sci 2019;20:3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Wang L, Zhang L. Circulating exosomal miRNA as diagnostic biomarkers of neurodegenerative diseases. Front Mol Neurosci 2020;13:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Tabrizi SJ, Scahill RI, Owen G, et al. Predictors of phenotypic progression and disease onset in premanifest and early‐stage Huntington's disease in the TRACK‐HD study: analysis of 36‐month observational data. Lancet Neurol 2013;12:637–649. [DOI] [PubMed] [Google Scholar]
- 135. Southwell AL, Kordasiewicz HB, Langbehn D, et al. Huntingtin suppression restores cognitive function in a mouse model of Huntington's disease. Sci Transl Med 2018;10:eaar3959. [DOI] [PubMed] [Google Scholar]
- 136. ESMI Ataxia Study Group . http://www.ataxia‐study‐group.net/html/studies/esmi.
- 137. SCA3 BIGPRO BIGPRO Study . https://bigpro.webnode.com/.
- 138. SCA Global ‐ National Ataxia Foundation . https://ataxiaorg/sca‐global/.
- 139. Chow SC, Huang Z. Innovative design and analysis for rare disease drug development. J Biopharm Stat 2020;30:537–549. [DOI] [PubMed] [Google Scholar]
- 140. Mulberg AE, Bucci‐Rechtweg C, Giuliano J, et al. Regulatory strategies for rare diseases under current global regulatory statutes: a discussion with stakeholders. Orphanet J Rare Dis 2019;14:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Friede T, Posch M, Zohar S, et al. Recent advances in methodology for clinical trials in small populations: the InSPiRe project. Orphanet J Rare Dis 2018;13:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Diallo A, Jacobi H, Tezenas du Montcel S, Klockgether T. Natural history of most common spinocerebellar ataxia: a systematic review and meta‐analysis. J Neurol 2020. 10.1007/s00415-020-09815-2 [DOI] [PubMed] [Google Scholar]