

Abstract
Objective
Angelman syndrome (AS) is a severe neurodevelopmental disorder caused by loss of function of the maternally inherited UBE3A gene in neurons. Promising disease‐modifying treatments to reinstate UBE3A expression are under development and an early measure of treatment response is critical to their deployment in clinical trials. Increased delta power in EEG recordings, reflecting abnormal neuronal synchrony, occurs in AS across species and correlates with genotype. Whether delta power provides a reliable biomarker for clinical symptoms remains unknown.
Methods
We analyzed combined EEG recordings and developmental assessments in a large cohort of individuals with AS (N = 82 subjects, 133 combined EEG and cognitive assessments, 1.08–28.16 years; 32F) and evaluated delta power as a biomarker for cognitive function, as measured by the Bayley Cognitive Score. We examined the robustness of this biomarker to varying states of consciousness, recording techniques and analysis procedures.
Results
Delta power predicted the Bayley Scale cognitive score (P < 10−5, R 2 = 0.9374) after controlling for age (P < 10−24), genotype:age (P < 10−11), and repeat assessments (P < 10−8), with the excellent fit on cross validation (R 2 = 0.95). There were no differences in model performance across states of consciousness or bipolar versus average montages (ΔAIC < 2). Models using raw data excluding frontal channels outperformed other models (ΔAIC > 4) and predicted performance in expressive (P = 0.0209) and receptive communication (P < 10−3) and fine motor skills (P < 10−4).
Interpretation
Delta power is a simple, direct measure of neuronal activity that reliably correlates with cognitive function in AS. This electrophysiological biomarker offers an objective, clinically relevant endpoint for treatment response in emerging clinical trials.
Introduction
Novel disease‐modifying treatments have profound potential to transform the treatment landscape for neurodevelopmental diseases with strong genetic underpinnings. 1 , 2 Detection of a treatment response currently relies on clinical measurements of neurodevelopmental outcome, which can be slow, unreliable and challenging in young and neurologically impaired populations. The electroencephalogram (EEG) provides a direct, objective measure of neuronal activity and offers a promising non‐invasive tool to identify early and clinically meaningful biomarkers of neurological function for use in clinical trials.
Angelman syndrome (AS) is a genetic neurodevelopmental disorder occurring in 1 in 10,000 to 25,000 live births 3 , 4 , 5 characterized by severe cognitive deficits, motor impairments and a high comorbidity with epilepsy. 6 , 7 , 8 AS is caused by loss of function of the maternally inherited and paternally imprinted ubiquitin‐protein ligase UBE3A gene in neurons. 9 , 10 Targeted disease‐modifying treatments to reinstate UBE3A expression in neurons are under development, 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 and a robust, easily obtained and quantifiable biomarker that reliably assays disease severity is critical to an assessment of efficacy in clinical trials.
The baseline EEG is strikingly abnormal in AS, even in individuals without clinical seizures. 19 , 20 Diffuse, frequent bursts of notched or polyphasic delta activity (2–4 Hz) are the most characteristic EEG feature of AS and are observed across all genotypic classes of disease. 20 , 21 , 22 , 23 Quantitative EEG studies have shown increased delta power in individuals with AS compared to neurotypical control subjects 24 and have also demonstrated that delta power varies with age 25 and genotype 26 in patients with AS. Increased delta power is observed in AS mouse models relative to wild‐type mice, 24 confirming preservation of this EEG phenotype across species. Highlighting the potential utility of delta power as a biomarker in clinical trials, decreased delta power was found to correlate with treatment in a recent open‐label clinical trial. 27 Although delta power is a promising biomarker for AS, whether this EEG measure correlates with neurological symptoms in this disease remains unknown.
We hypothesized that delta power would predict cognitive function in individuals with AS. Using data gathered in a large natural history study of individuals with AS including concurrent EEG recordings and neurodevelopmental testing, we found that increased delta power strongly correlated with decreased cognitive function in AS. We tested this relationship across states of consciousness, EEG recording modalities and data analysis approaches, and found that delta power provides a robust predictor of cognitive function in each circumstance. This work suggests that EEG delta power provides a powerful, objective, non‐invasive, and reliable endpoint for treatment response in emerging clinical trials for AS.
Subjects and Methods
Subject enrollment
Data were obtained through the large, multicenter Angelman Syndrome Natural History Study (ClinicalTrials.gov identifier: NCT00296764) conducted as part of the Rare Diseases Clinical Research Network, Angelman, Rett and Prader‐Willi syndrome consortium. Consent was obtained according to the Declaration of Helsinki and was approved by the institutional review boards of the participating sites. Subjects were recruited at six sites between the years of 2006–2017, and EEGs from two sites (Rady Children’s Hospital/University of Califoria San Diego and Boston Children’s Hospital) comprised the cohort used in this study. We excluded data where more than 3 months had elapsed between EEG and neurodevelopmental data collection.
Neurodevelopmental testing
The Bayley Scales of Infant and Toddler Development, Third Edition, administered by psychologists with extensive testing experience in the AS population, was used to assess developmental functioning. 28 The Bayley Scales are normed for typically developing children between 1 and 42 months of age, but have been used to assess patients with developmental disabilities of all ages, including AS. 29 Five domains from the Bayley Scales (Cognitive, Expressive Communication, Receptive Communication, Fine Motor and Gross Motor) were assessed for each subject, where maximal scores possible for each test were: Cognitive (91), Receptive Communication (49), Expressive Communication (48), Fine Motor (66) and Gross Motor (72). Raw scores were used for analysis.
EEG data collection and processing
EEG recordings were collected using the international 10–20 EEG system on either BioLogic or Xltek systems. 30 At each session, 30 min of awake recording and 30 min of asleep recording were attempted.
The EEG data processing pipeline is shown in Figure 1. Data were collected at 200–512 Hz sampling rates. All data were bandpass filtered from 1 to 80 Hz and notch filtered at 60 Hz (using Hamming‐windowed finite impulse response filters) for visual inspection. On visual review, sections of data containing at least one channel contaminated by muscle, ocular, movement, or electrical artifacts were manually identified. We choose to perform a manual review, rather than apply an automated method for artifact removal, to avoid the potential subtle impacts of data processing on the results, and carefully distinguish the delta frequency rhythm of interest from artifacts with overlapping spectral content (e.g., due to eye blinks).
FIGURE 1.
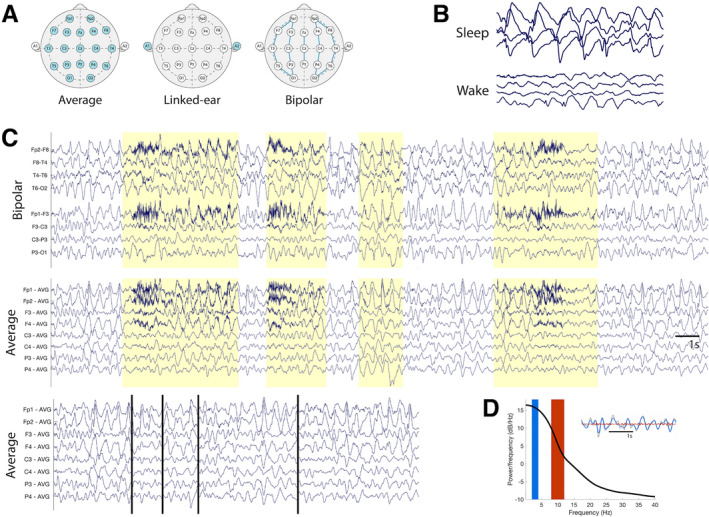
EEG data processing procedure. (A) Data were referenced to three standard montages for review and analysis: the linked‐ear, bipolar, and average references. Electrodes used in computing the reference are highlighted in blue. (B) EEG data were manually reviewed and staged for periods of sleep and wakefulness. (C) Data were visually inspected and manually cleaned to remove non‐cerebral artifacts (highlighted here in yellow). An example “clean” dataset is shown (bottom row) with breakpoints identified by vertical black lines. (D) Power spectral densities (PSDs) were calculated for each EEG recording. The delta [alpha] frequency range is indicated with a blue [red] rectangle on the plotted PSD. An example EEG trace (grey) showing filtered delta [alpha] activity in blue [red] in a sample EEG trace is shown in the inset.
Data were analyzed referenced to the common average reference, longitudinal bipolar reference and the linked‐ear reference. In addition, regional clusters of electrodes were group‐average referenced to assess the impact of subsampling channels on model quality.
EEG data were first manually staged for wake and sleep states by an experienced clinical neurophysiologist (CJC). As children with AS lack normal wake and sleep EEG architecture, here, all data with a blink, movement, or eyes open documented were categorized as awake. All data without a blink or movement artifacts in addition to either documentation of closed eyes or the presence of increased slowing at the vertex and frontocentral regions or well‐characterized sleep architecture (e.g., sleep spindles and K‐complexes) were classified as sleep. Data without sufficient evidence to categorize them as awake or asleep were excluded from the analysis.
Power spectral density estimates were derived using custom MATLAB scripts. Sliding, non‐overlapping 1‐sec windows were used to calculate delta power (2–4 Hz), theta power (5–7 Hz), alpha power (8–12 Hz), and beta power (13–30 Hz) using the fast Fourier Transform with a Hamming window taper. For relative power estimates, within each 1‐sec sample, power spectral densities were divided by the summed broad‐spectrum power (1–50 Hz) in the same 1‐sec sample, resulting in a unitless estimate of delta power.
Statistical modeling
To evaluate the relationship between cognitive function and delta power, we modeled cognitive score as a function of delta power and a subject‐specific intercept to account for multiple observations from some subjects. We included age and genotype:age as fixed effects in each model. To determine the partial R 2 for each predictor, we computed the full model sum of squared error (SSE) and then the reduced model SSE excluding the predictor of interest using the formula:
In our primary analysis, relative power in the delta band was computed from wake‐state EEG data from all 19 channels in the common average reference, excluding all epochs manually marked as containing artifact.
We subsequently evaluated the relationship between delta power and Bayley Cognitive Score in which delta power was estimated from varying states of consciousness (wake, sleep, or mixed states); montages (bipolar longitudinal, common average, or linked‐ear reference); pre‐processing steps (different subsamplings of electrodes, and cleaned or raw data); and spectral measures (absolute or relative delta power, and relative theta, alpha, or beta band power). To compare across nested models, we used the Akaike Information Criterion (AIC). Using the AIC, the degree of penalty can be approximately understood in terms of the number of parameters, where if comparing two models, the model with the lower AIC would still be preferable even if it had an additional ΔAIC/2 parameters. 31 We interpret a change in AIC (ΔAIC) of less than 2 as model equivalency, and ΔAIC ≥ 4 as model superiority. 32 To allow direct comparison of AIC scores between models, datasets of equal sizes were compared.
In additional post hoc analyses, we used spectral power in frequency bands outside the 2–4 Hz delta range, as well as raw delta power, to predict cognitive function. All relative power values were normalized to global power in the 1–50 Hz frequency band. To allow direct comparison of coefficient estimates amongst spectral bands with different power value ranges, spectral power values were z‐scored, and the standardized coefficient estimates (β ± SE) based on these z‐scores are reported. To assess the relationship of delta power to other developmental domains assessed by the Bayley, we evaluated the ability of delta power to predict dependent variables: Fine Motor (N = 131), Gross Motor (N = 133), Receptive Communication (N = 131), and Expressive Communication raw scores (N = 131).
Data availability
Derived data supporting the findings of this study are available from the corresponding author on request.
Results
Subject and EEG data characteristics
Eighty‐two unique participants with AS (6.52 ± 4.41 years, range 1.08–28.16 years; 32F) with 133 coordinated EEG recordings (including 82 first visits, 27 sec visits, 15 third visits, 6 fourth visits, 2 fifth visits, and 1 sixth visit) and Bayley Scale assessments were analyzed. All EEGs captured the awake state and 54 captured non‐rapid eye movement sleep. The average recording length was 27.08 ± 12.36 min (mean ± SD) for awake data, and 17.85 ± 10.19 min for sleep data. After manual removal of artifacts, an average of 7.32 ± 5.80 min of clean awake data and 17.53 ± 10.20 min of clean sleep data were available for analysis.
Fifty‐eight subjects (70.73%) had a deletion genotype, of which 20 subjects (24.39%) had a class 1 deletion, 33 (40.24%) had a class 2 deletion, and a further five subjects (6.10%) had an atypical or unspecified deletion. Twenty‐four subjects (29.27%) had a non‐deletion genotype, of which 10 (12.20%) had a UBE3A mutation, 5 (6.10%) had an imprinting defect, 6 (7.32%) had uniparental paternal disomy, and 3 (3.66%) had abnormal DNA methylation with a negative fluorescent in situ hybridization test, ruling out deletion. Consistent with prior literature, 24 , 26 on our visual inspection, Bayley Cognitive Scores reliably clustered between deletion and non‐deletion AS groups, but visual inspection did not suggest any differences in Bayley Cognitive Scores across subclassifications within deletion and non‐deletion groups. However, our sample sizes for some subgroups were small, limiting the power to detect a difference amongst deletion and non‐deletion genotype subclassifications.
The mean performances on the Bayley Scales domains were Cognitive 50.6 (range 18–83), Fine Motor 31.37 (range 8–52), Gross Motor 44.52 (range 13–65), Receptive Communication 17.07 (range 5–38), and Expressive Communication 11.08 (range 4–29). The median duration of time between assessment for the Bayley Scales and EEG recording was 0 days (range 0–72).
Delta power is a reliable biomarker of cognitive function in Angelman syndrome
To test for a relationship between delta power and cognitive performance, we first assessed for variation based on age and genotype. Consistent with prior studies, age and genotype both predicted cognitive function (Fig. 2A), 29 , 33 , 34 and model testing showed that an interaction term including both genotype and age outperformed genotype alone (ΔAIC > 5). We, therefore, included this interaction term (genotype:age) as a predictor. Among subjects with longitudinal data available, the change in delta predicted the change in the Bayley Cognitive Score (Fig. 2B), however, subjects were found to have different baseline values (Fig. 2C). We, therefore, included a subject‐specific intercept in the model to account for repeat visits by several subjects. Our final mixed effects model was:
where the dependent variable BCS represents the Bayley Cognitive Score. Fixed effects include: (1) log10(age), representing the log10‐transformed age in years; (2) I genotype, an indicator variable representing deletion or non‐deletion genotype; and, (3) spectral power, here, the relative delta power during wakefulness. A random effect on intercept is included for subject ID to account for repeat subject visits.
FIGURE 2.
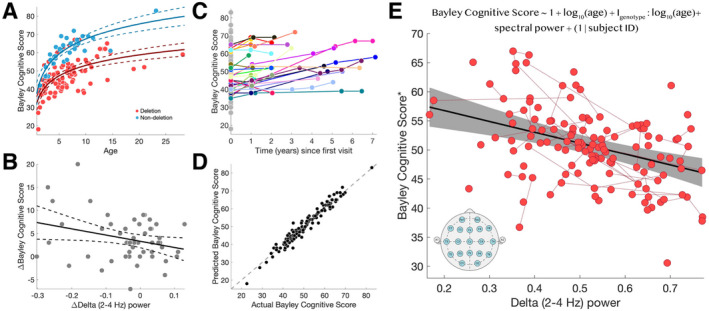
Delta power predicts cognitive function. (A) Both log10(age) (P < 10−31) and genotype:log10(age) interaction (P < 10−12) predict cognitive function. Each circle indicates a subject visit. The solid [dashed] curves indicate model fit [95% confidence intervals]. Subjects with deletion [non‐deletion] genotype are indicated in red [blue]. (B) Amongst longitudinal subjects (N = 27 subjects, N = 51 pairs), change in delta power predicts change in the Bayley Cognitive Score (R 2 = 0.0814, P = 0.0386, β = −13.40 [SE 6.30]). (C) Subjects had similar cognitive scores across longitudinal assessments, with different baseline values (e.g., y‐intercepts). Connected points in color indicate longitudinal data from the same subject. Points in gray indicate subjects with only one visit. (D) The observed Bayley Cognitive Scores and the Bayley Cognitive Scores predicted by the model were highly correlated (R 2 = 0.9374), indicating a good model fit. Each circle indicates a real and predicted subject score. The dashed line indicates identical values between the two scores. (E) There is a linear relationship between delta power and the Bayley Cognitive Score after controlling for age, age: genotype interaction, and repeat visits using the mixed‐effects model shown at the top of the panel (R 2 = 0.9374, delta power P < 10−5, log10(age) P < 10−24, genotype:log10(age) P < 10−11, repeat subjects P < 10−8). The solid black line shows the model fit and the gray shaded region indicates the 95% confidence intervals. Red circles indicate combined EEG and cognitive score visits. Red lines indicate repeat visits by the same subject. The insert shows the standard 10–20 EEG channels used to estimate delta power. *The Bayley Cognitive Score has been adjusted for a fixed age and genotype for visualization.
Fitting this model to the data, we find a strong negative relationship between delta power and cognitive function, wherein the model explains 93.74% of the variance in the Bayley Cognitive Score (Fig. 2D–E; R 2 = 0.9374, delta power P < 10−5, log10(age) P < 10−24, genotype:log10(age) P < 10−11, repeat subjects P < 10−8). For each 0.1 increase in relative delta power, the Bayley Cognitive Score decreased by 2.00 points (95% CI [−2.81, −1.19]). Within this model, we find that age explains most of the variance (partial R 2 = 0.7169), followed by delta power (partial R 2 = 0.2075), and then age:genotypic interaction (partial R 2 = 0.1027). Full model criteria and output statistics are shown in Table 1. We note that the majority of individuals in our sample were above the upper age limit for the norm‐referenced Bayley Scales standard scores. Age‐equivalent scores have been adapted for the AS population 35 but our model performed best using raw scores and including age as a continuous variable (using age‐equivalent Bayley Cognitive Scores in our model, R 2 = 0.7001).
Table 1.
Delta power correlates with cognitive function in AS.
| Predictor | Coefficient estimate | P | Partial R 2 |
|---|---|---|---|
| Delta power | −20.01 ± 4.08 | 2.76e‐06 | 0.2075 |
| log10(age) | 29.73 ± 2.28 | 3.07e‐25 | 0.7169 |
| Genotype:log10(age) | −13.57 ± 1.71 | 1.00e‐12 | 0.1027 |
Coefficient estimates (±SE), P‐values, and partial R 2 values for the fixed effects model predictors.
We assessed model accuracy and goodness‐of‐fit using a leave‐one‐out cross‐validation procedure, adapted for mixed‐effects models, where the number of iterations is equal to the number of subjects. Here, all data points from a single subject were excluded and model criteria recomputed and used to predict the Bayley Cognitive score for the excluded subject. This process was repeated for all subjects. We found that the residuals from this procedure are randomly distributed (Fig. 3A) and the model performs accurately on test data (Fig. 3B; R 2 = 0.9455), indicating good model generalizability. We conclude that delta power is a powerful predictor of cognitive function in AS and that the model generalizes across this large and varied AS dataset.
FIGURE 3.
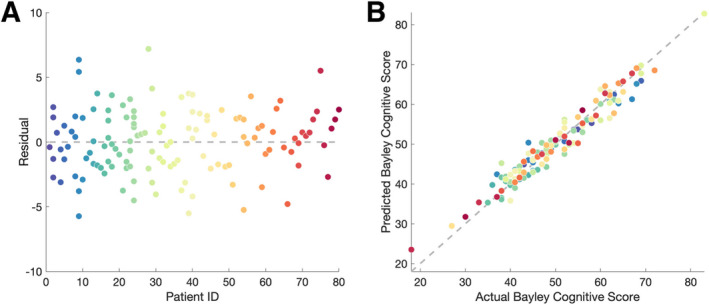
Model validation. (A) The residuals are normally distributed, with a low root mean squared prediction error (RMSPE = 2.50). (B) there is a strong correlation (R 2 = 0.9455) between the actual and predicted Bayley Cognitive Score values for each excluded subject, indicating a good model fit. Each circle indicates a subject visit and colors represent the same subject in (A) and (B).
Delta power measured from wake or sleep predicts cognitive function
To test whether state of consciousness impacted the predictive performance of EEG delta power as a biomarker for cognitive function in AS, we compared delta power derived from a subset of wake, sleep, and mixed state EEG segments available from the same cohort of subjects (N = 54). We found similar relationships between cognitive score and delta power using delta power in sleep (R 2 = 0.9613, P = 0.0048, coefficient estimate β = −22.01 [SE 7.45], AIC = 340.52), wake (R 2 = 0.9691, P = 0.0021, β = −18.97 [SE 5.84], AIC = 339.41), and mixed states of consciousness (50% wake and 50% sleep; R 2 = 0.9690, P = 0.0013, β = −22.57 [SE 6.60], AIC = 338.39; Fig. S1A–C). Delta power predicts cognitive function in each case (P < 0.008, R 2 values 0.94–0.95, and ΔAIC < 2 between models). To supplement this finding, we modeled (by linear regression) delta power in the awake state as a function of delta power in the sleep state in the 54 EEGs with both states available and found a strong correlation (R 2 = 0.5450; Fig. S1D). We conclude that the state of consciousness during the EEG recording does not impact the performance of delta power in predicting cognitive function in AS.
Delta power as a biomarker is robust to different referencing procedures
To determine the generalizability of delta power as a biomarker across recording configurations, we compared the performance of delta power estimated from EEG data referenced to the bipolar reference and linked‐ear reference to the results using the common average reference. We found that models with delta power computed from the common average reference (R 2 = 0.9372, AIC = 797.39) and the bipolar reference (R 2 = 0.9353, AIC = 798.06) outperformed the model with delta power computed from the linked‐ear reference (R 2 = 0.9354, AIC = 803.68, 5 < ΔAIC < 7). We found no evidence of a difference (ΔAIC < 1) in model performance between the common average reference and bipolar reference. The coefficient estimate for delta power was significant and similar for all montages (average reference: P < 10−5, β = −19.27 [SE 4.12]; bipolar reference: P < 10−4, β = −19.06 [SE 4.16]; linked‐ear reference: P < 10−3, β = −16.44 [SE 4.28]). We conclude that delta power estimated from the common average and bipolar references work equivalently well to predict cognitive function, and both slightly outperform the linked‐ear reference (Table S1).
Models of cognitive function improve with delta power estimated from posterior EEG channels
Delta power abnormalities have been observed to occur across the entire scalp recording in people with AS. 24 To test the relationship between delta power and cognitive score at different scalp locations, we evaluated the performance of delta power estimates obtained from posterior subsamples of electrodes. Six clusters of interest were defined referenced to a local common average reference. Most clusters excluded frontal channels, as they are most impacted by ocular artifacts and muscle artifacts, and focused on posterior channels least impacted by these artifacts (Fig. 4, Row 1–2).
FIGURE 4.
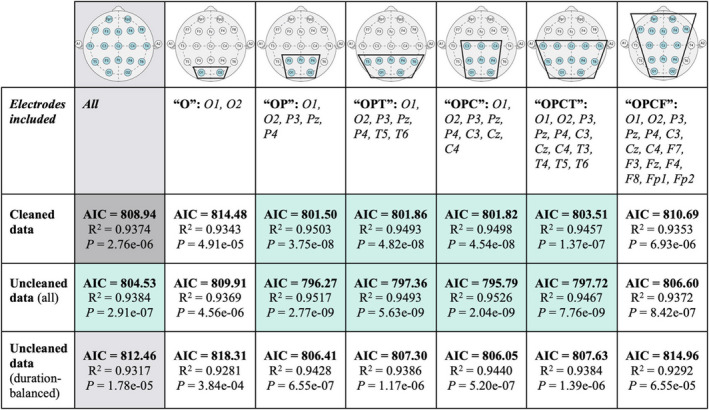
Sub‐sampled electrode clusters and raw EEG data predict cognitive function in AS. Sub‐sampled clusters of electrodes are shown in the first row (blue = channel included in montage), with the groupwise abbreviation and the names of channels included listed in the second row. The full average reference is shown in the second column in gray for comparison, and cleaned data in the full average reference, to which all other cells are compared, is shown in dark gray. For each of the sub‐sampled channel montages, and the full average reference, relative delta power was computed using cleaned, raw, and raw data with balanced duration to cleaned data, and used to predict the Bayley Cognitive Score. Cells with AIC scores superior (ΔAIC > 4) to those from the model using cleaned data from all electrodes in the full average reference are highlighted in green.
We found that the exclusion of the frontal electrodes improved model performance (ΔAIC > 4) when compared to using data from all channels (Fig. 4, Row 3). In particular, electrode subsets that only included occipital and parietal channels (OP, R 2 = 0.9503, delta power P < 10−7), occipital, parietal and temporal channels (OPT, R 2 = 0.9493, P < 10−7), occipital, parietal, and central channels (OPC, R 2 = 0.9498, P < 10−7), and occipital, parietal, central, and temporal channels (OPCT, R 2 = 0.9457, P < 10−6) yielded models with superior performance compared to the model utilizing the full EEG recording montage (5 < ΔAIC < 8). See Figure 4, Row 1–2 for graphic depictions of these electrode subsets tested.
Models of cognitive function using raw EEG data perform well
Manual removal of ocular, motor, and muscle artifacts required approximately 1–2 h per 30 min of wake EEG data. The greatest sources of noise were muscle and blink artifact, which contaminated at least one channel in greater than 50% of the awake EEG recordings. To determine the necessity of this time‐consuming manual step, we evaluated model performance with delta power estimated from the raw EEG data, without removing artifacts. We found that delta estimates from raw data using the OP (R 2 = 0.9517, delta power P < 10−8), OPT (R 2 = 0.9493, P < 10−8), OPC (R 2 = 0.9526, P < 10−8), and OPCT (R 2 = 0.9467, P < 10−8) montages outperformed the original model with delta power estimated from cleaned data using the full average reference montage (ΔAIC > 4, Fig. 4, Row 3–4).
Improved model performance with raw data could be explained by the longer total duration of EEG data included in the analyses. To avoid this confounder, we compared model performance using delta power estimated from raw EEG data of the same duration as cleaned data segments for each subject (mean ± STD, 7.32 ± 5.80 min). Even with equal data quantities, the raw data analyzed using the channel subsets excluding frontal electrodes showed slightly improved model fits compared to the cleaned data from the full EEG montage (1 < ΔAIC < 3; Fig. 4, Row 5). We conclude that with frontal electrodes removed, delta power estimates from raw data outperform a similar measure computed from manually cleaned data in predicting cognitive function in AS (Fig. 5; full model output statistics are shown in Table 2).
FIGURE 5.
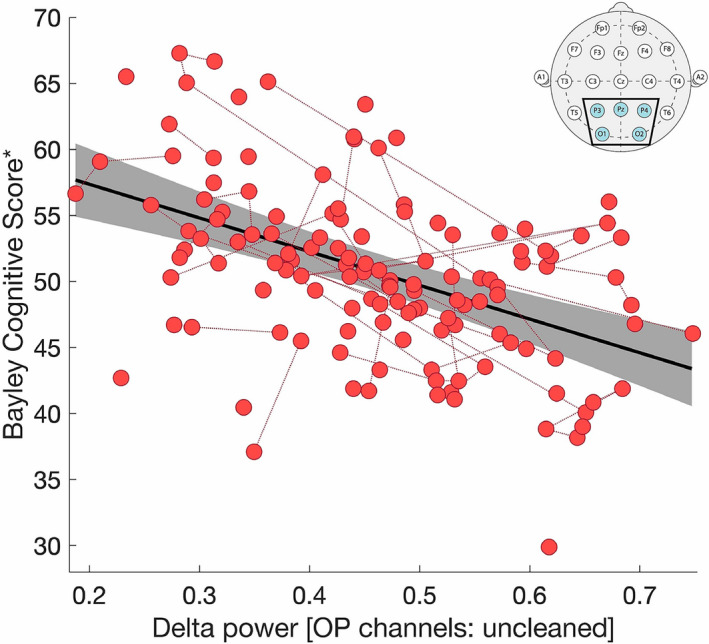
Delta power calculated from raw data in the OP montage predicts cognitive function. There is a linear relationship between delta power, estimated from raw data in the OP montage (inset), and the Bayley Cognitive Score after controlling for age, age:genotype interaction, and random effects for repeat subjects (R 2 = 0.9517, delta power P < 10−8, log10(age) P < 10−22, genotype:log10(age) P < 10−12, repeat subjects P < 10−8). The solid black line indicates the linear model fit, and the gray shaded region indicates the 95% confidence interval. Longitudinal same‐subject data are connected by red dotted lines. *The Bayley Cognitive Score has been adjusted for the impacts of age and genotype and the recast values plotted.
Table 2.
Delta power, estimated from raw EEG data in the OP montage, correlates with cognitive function in AS.
| Predictor | Coefficient estimate | P | Partial R 2 |
|---|---|---|---|
| Delta power | −26.94 ± 4.21 | 2.77e‐09 | 0.3806 |
| log10(age) | 27.87 ± 2.25 | 1.59e‐23 | 0.5902 |
| Genotype : log10(age) | −13.89 ± 1.67 | 1.12e‐13 | 0.0516 |
Coefficient estimates (±SE), P‐values, and partial R 2 values for the fixed effects model predictors.
Models of cognitive function with relative delta power outperform other canonical frequency bands
Relative measures of delta power can be impacted by prominent activities in other frequencies bands, such as the posterior dominant alpha rhythm. However, absolute power measures can be impacted by non‐cortical features, such as skull thickness. 36 Given these competing limitations, we evaluated whether absolute or relative delta power provided a better biomarker for cognitive function in AS. We found a significant negative relationship between absolute delta power and Bayley Cognitive Score (P < 10−3). However, the model using relative delta power outperformed the model with absolute delta power (ΔAIC = 11.22).
To evaluate the specificity of the model to the delta band, we evaluated the relationship between each of the canonical frequency bands (alpha, theta, beta) and cognitive function. We found a positive relationship between Bayley Cognitive Score and relative theta power (P = 0.0011), the log10‐transform of relative alpha power (P < 10−4), and the log10‐transform of relative beta power (P < 10−5; Table S2). Including both delta power and either relative theta power, relative log10 alpha power, or relative log10 beta power in the model, only relative delta power remained a significant predictor of cognitive function (Table S3), suggesting that relative power in frequency bands other than delta may simply provide a poor surrogate measure of relative delta power. We conclude that relative delta power is the optimal spectral band to predict cognitive function in AS.
Delta power predicts motor and language function
In addition to cognitive performance, we tested delta power as a predictor of performance in other developmental domains using the Fine and Gross Motor and Expressive and Receptive Communication Scores of the Bayley Scales. We note that although these measures are independently tested and scored in each subject, the Bayley Scales were highly collinear across developmental domains (Fig. S2). The strongest correlations were observed between the Cognitive, Fine Motor, and Receptive Communication Scores (R 2 > 0.7).
We found a negative correlation between delta power estimated from the OP montage on raw data and receptive communication (Fig. 6A; R 2 = 0.8718, P < 10−3, β = −10.77 [SE 3.12]), expressive communication (Fig. 6B; R 2 = 0.6801, P = 0.0209, β = −6.16 [SE 2.63]), and fine motor skills (Fig. 6C; R 2 = 0.9074, P < 10−4, β = −14.84 [SE 3.42]). We did not find evidence of a relationship between delta power estimated from the OP montage and gross motor skills (Fig. 6D; R 2 = 0.9382, P = 0.36, β = −4.02 [SE 4.34]). Using the cleaned data in the full‐channel montage, we found a negative relationship between delta power and fine motor skills (R 2 = 0.9026, P < 10−3, β = −11.91 [SE 3.15]) and receptive communication (R 2 = 0.8559, P = 0.0302, β = −6.36 [SE 2.90]), and a negative trend between delta power and expressive communication (R 2 = 0.6823, P = 0.0585, β = −4.58 [SE 2.40]). We did not find evidence of a relationship between delta power estimated from the full‐channel montage and gross motor skills (P > 0.2), though gross motor scores were noted to have a limited range of scores across AS patients, limiting the power to detect a relationship. We conclude that delta power predicts not only cognitive function, but additional domains of neurodevelopment—motor and language function—in AS, findings that are most evident using the OP montage.
FIGURE 6.
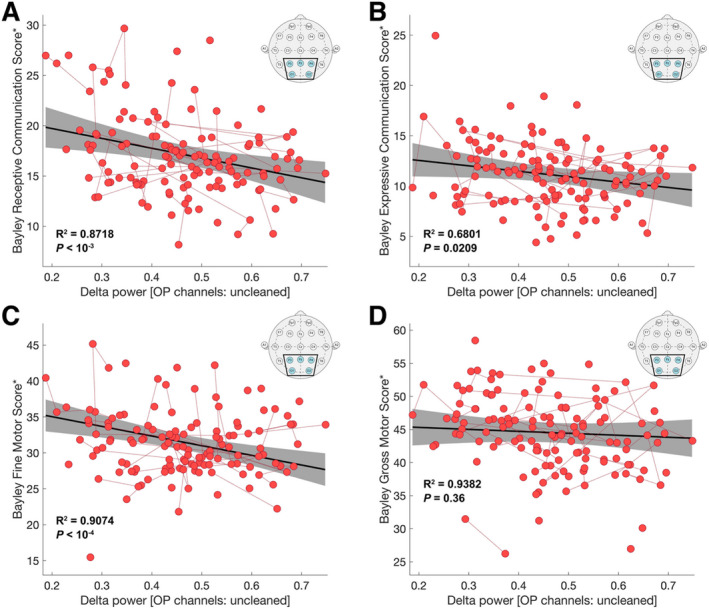
Motor and language function are predicted by delta power with reduced data collection criteria. Delta power estimated using the OP regional reference montage (insets), and raw EEG data was used to predict the (A) Bayley Receptive Communication Score, (B) Bayley Expressive Communication Score, (C) Bayley Fine Motor Score, and (D) Bayley Gross Motor Score. For all plots, the solid black line shows the linear fit, and the gray shaded region indicates the 95% confidence interval. Longitudinal same‐subject data are connected by red dotted lines. The R 2 value given by the model and the P value for delta power are shown in bolded text. *Scores have been adjusted for a fixed age and genotype for visualization.
Discussion
Using a large natural history database of individuals with combined short EEG recordings and cognitive assessments, we found that delta power provides a robust biomarker for cognitive function in Angelman syndrome. This relationship was evident after controlling for age and genotype and was generalizable across a large cohort of subjects with AS. These results support the viability of delta power as a promising surrogate biomarker for clinical trials to test emerging disease‐modifying treatments in this population.
EEG recordings provide a direct measure of neuronal activity, and delta activity has been proposed to reflect the dynamics of intrinsic membrane and synaptic conductances, with local generators in the thalamus 37 , 38 and the cortex. 39 , 40 The relationship between increased delta power with decreased cognitive function has been observed across many disease states. 41 , 42 , 43 , 44 The acquisition of cognitive skills also coincides with decreased delta power and increased prevalence of faster oscillations across normal development. 45 Hypersynchronization in the delta frequency band and decreased signal complexity observed in patients with AS have been inferred as a state of diminished consciousness. 46 Excessive delta activity in AS at least in part replaces or disrupts typically present rhythms, such as alpha band activity (8–12 Hz), which has known correlations with cognitive function, particularly in relation to the ability to orient and sustain attention, process information, and store and retrieve semantic memory. 47 , 48 , 49 , 50 , 51 Here, we demonstrated that increased delta power corresponds to decreased cognitive, motor, and language performance in AS, validating the utility of this biomarker as a surrogate for clinical disease severity. Given this strong relationship and the impact that these abnormal rhythms have on normal cognitive function, treatments that improve these abnormal brain rhythms would be expected to correlate with improvement in AS.
We found that the strong relationship between delta power and cognitive function in AS persists across different states of consciousness. This finding is of practical importance because disrupted sleep and the lack of well‐characterized sleep architecture in most patients with AS makes confident estimates of sleep states challenging in this population. Furthermore, we found that manual cleaning of the EEG data is unnecessary to reliably measure this high amplitude and diffuse signal. Finally, we found that the use of just five electrodes, placed in the posterior regions of the head to avoid the most common recording artifacts, provides optimal estimates of delta power to reliably predict cognitive function. Because the application of EEG electrodes is both time‐consuming and can be poorly tolerated in young children or those with severe developmental delay, fewer electrodes may improve the tolerance of an EEG recording session. The robustness of this biomarker to different brain regions, states of consciousness, and high‐artifact recordings support its practical deployment in clinical populations.
Although delta power abnormalities have historically been targeted as the canonical EEG feature in AS, abnormalities in other frequency bands have also been reported in AS. Theta power has been shown to be increased in approximately half of the AS population. 21 , 23 , 25 , 52 Decreased beta power has been found in patients with deletion‐positive AS, 26 in which the genes GABRB3, GABRA5, and GABRG3, encoding subunits of the GABAA receptor (involved in the production of healthy beta oscillations 53 ), are deleted in addition to UBE3A. Consistent with prior reports, we found relationships between cognitive function and relative theta and beta band activity, but these relationships did not persist after controlling for delta band activity. Thus, this work finds that delta power is the optimal frequency band to predict cognitive function in AS.
At present, there are no specific disease‐modifying therapies available for AS, but transformative treatments are on the horizon. Current supportive therapies include physical and occupational therapies, augmentative communication strategies, and medication for comorbid seizures, attention issues, sleep disturbances, and gastrointestinal issues, 54 as well as dietary adjustments to help reduce seizures. 55 , 56 , 57 Emerging therapies which restore neural UBE3A expression have the potential to directly modify the pathogenic disease process in AS. 12 In rodent models of AS, 11 successes in restoring neural UBE3A expression using antisense oligonucleotides 13 and topoisomerase inhibitors 14 , 15 have been achieved, and have fast‐approaching potential for transition to clinical therapies. Direct measures of UBE3A expression in these models were demonstrated through direct assays of neuronal tissue; behavioral measures provided a less robust response. A rapid, non‐invasive biomarker for target engagement and treatment response will be useful as these potential therapies are translated to human patients. Delta power in short EEG recordings may provide a non‐invasive measure to detect treatment response and a clinically meaningful reduction in disease severity.
AS results in severe functional impairments and, currently, supportive therapies are the only means of treatment. As new disease‐modifying treatments emerge and are transitioned to clinical trials, quantitative biomarkers are required for rapid and accurate assessment of intervention efficacy. Here we show that delta power correlates with cognitive function in AS, and that this correlation persists with changes in the state of consciousness, variable electrode configuration, and non‐neuronal artifacts in the data. Furthermore, the simplicity of the model is an attractive feature for clinical application; in an era of ever‐expanding machine learning models and complex approaches to computational analysis, the identification of a biomarker with a simple, direct relationship to outcome, without specific parameter requirements, increases usability and model interpretation. 58 Here, a simple and interpretable mixed‐effect linear model achieved near‐perfect prediction of cognitive function. We, therefore, propose that delta power provides a robust, easily obtained, and practical biomarker of clinical disease severity in AS. This work also demonstrates the utility of EEG to identify reliable biomarkers for neurological function in neurodevelopmental disease.
Conflict of Interest
C.J.C. and M.A.K. have provided consulting services for Biogen Inc. R.K. is employed at Biogen Inc. R.T. receives clinical support from the Angelman Foundation and consults for Ovid Pharmaceuticals and Roche Pharma.
Supporting information
Figure S1. Data from wake, sleep, and mixed states of consciousness correlate with cognitive function. Delta power was estimated (A) during wakefulness (R 2 = 0.9691, P = 0.0021, β = −18.97 [SE 5.84], AIC = 339.41), (B) during sleep (R 2 = 0.9613, P = 0.0048, β = −22.01 [SE 7.45], AIC = 340.52), and (C) from mixed states of consciousness, created from 50% to 50% sleep data (R 2 = 0.9690, P = 0.0013, β = −22.57 [SE 6.60], AIC = 338.39). (D) Delta power estimated from the same subjects during wake and sleep were highly correlated (R 2 = 0.5450). For all plots, the solid black line indicates the linear fit and the gray shaded region indicates the 95% confidence interval. Longitudinal same‐subject data are connected by red dotted lines. The insert shows the standard 10–20 EEG channels that were used to generate an estimate of delta power. *Scores have been adjusted for a fixed age and genotype for visualization.
Figure S2. Collinearity among the Bayley Scales in AS. Comparison of the Bayley Scores in four developmental domains. For each subject, the Bayley Scores are plotted against one another, per the vertical and horizontal labels. The R 2 values are shown in blue text. Particularly strong collinearities exist between the Bayley Cognitive, Fine Motor, and Receptive Communication Scores (R 2 > 0.7), whereas relatively weaker correlations exist between the Bayley Gross Motor and Expressive Communication Scores and all other Bayley Scores (R 2 < 0.7).
Table S1. Delta power was estimated from three montages to model cognitive function in Angelman syndrome. Detailed outputs for all three models are shown, including R 2, AIC, and coefficient estimates (β ± SE) and P‐values for each predictor.
Table S2. Spectral power in the delta (2–4 Hz), theta (5–7 Hz), alpha (8–12 Hz), and beta (13–30 Hz) frequency bands was used to model cognitive function. The standardized coefficient estimates (β ± SE) and P‐values for the spectral predictors in each model are shown, with P‐values that pass Bonferroni correction (P < 0.00714 for 7 tests) indicated in bold.
Table S3. Detailed model outputs for three models with two spectral predictors are shown. In all models, only delta power remains a significant predictor (P < 0.038).
Acknowledgements
M.A.K. acknowledges support by the National Science Foundation DMS Award #1451384. E.R.S. acknowledges support by the National Science Foundation GRFP. C.J.C. acknowledges support by the National Institutes of Health K23 Award NS092923. L.B. received support from the NIH to obtain the data used in this project.
Funding Information
This work was supported by an investigator‐initiated sponsored research award by Biogen Inc. L.M.O. and E.R.S. received financial support from the same investigator‐initiated sponsored research award. E.R.S. was also supported by a NSF Graduate Research Training Fellowship. C.J.C. is supported by NIH NINDS K23NS092923.
Funding Statement
This work was funded by Biogen Inc ; NSF Graduate Research Training Fellowship; NIH NINDS grant K23NS092923.
References
- 1. Deverman BE, Ravina BM, Bankiewicz KS, et al. Gene therapy for neurological disorders: progress and prospects. Nat Rev Drug Discov 2018;17:641–659. [DOI] [PubMed] [Google Scholar]
- 2. Bennett CF, Krainer AR, Cleveland DW. Antisense oligonucleotide therapies for neurodegenerative diseases. Annu Rev Neurosci 2019;42:385–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Petersen MB, Brøndum‐Nielsen K, Hansen LK, Wulff K. Clinical, cytogenetic, and molecular diagnosis of Angelman syndrome: estimated prevalence rate in a Danish county. Am J Med Genet 1995;60:261–262. [DOI] [PubMed] [Google Scholar]
- 4. Kyllerman M. On the prevalence of Angelman syndrome. Am J Med Genet 1995;59:405. [DOI] [PubMed] [Google Scholar]
- 5. Mertz LGB, Christensen R, Vogel I, et al. Angelman syndrome in Denmark. Birth incidence, genetic findings, and age at diagnosis. Am J Med Genet A 2013;161:2197–2203. [DOI] [PubMed] [Google Scholar]
- 6. Thibert RL, Larson AM, Hsieh DT, et al. Neurologic manifestations of Angelman syndrome. Pediatr Neurol 2013;48:271–279. [DOI] [PubMed] [Google Scholar]
- 7. Williams CA, Beaudet AL, Clayton‐Smith J, et al. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A 2006;140:413–418. [DOI] [PubMed] [Google Scholar]
- 8. Bird LM. Angelman syndrome: review of clinical and molecular aspects. Appl Clin Genet 2014;7:93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kishino T, Lalande M, Wagstaff J. UBE3A/E6‐AP mutations cause Angelman syndrome. Nat Genet 1997;15:70–73. [DOI] [PubMed] [Google Scholar]
- 10. Matsuura T, Sutcliffe JS, Fang P, et al. De novo truncating mutations in E6‐AP ubiquitin‐protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet 1997;15:74–77. [DOI] [PubMed] [Google Scholar]
- 11. Yang X. Towards an understanding of Angelman syndrome in mice studies. J Neurosci Res 2020;98:1162–1173. [DOI] [PubMed] [Google Scholar]
- 12. Bi X, Sun J, Ji AX, Baudry M. Potential therapeutic approaches for Angelman syndrome. Expert Opin Ther Targets 2016;20:601–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meng L, Ward AJ, Chun S, et al. Towards a therapy for Angelman syndrome by targeting a long non‐coding RNA. Nature 2015;518:409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang H‐S, Allen JA, Mabb AM, et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 2011;481:185–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee H‐M, Clark EP, Kuijer MB, et al. Characterization and structure‐activity relationships of indenoisoquinoline‐derived topoisomerase I inhibitors in unsilencing the dormant Ube3a gene associated with Angelman syndrome. Mol Autism 2018;9:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Daily JL, Nash K, Jinwal U, et al. Adeno‐associated virus‐mediated rescue of the cognitive defects in a mouse model for Angelman syndrome. PLoS One 2011;6:e27221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Woerden GM, Harris KD, Hojjati MR, et al. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci 2007;10:280–282. [DOI] [PubMed] [Google Scholar]
- 18. Egawa K, Kitagawa K, Inoue K, et al. Decreased tonic inhibition in cerebellar granule cells causes motor dysfunction in a mouse model of Angelman syndrome. Sci Transl Med 2012;4:163ra157. [DOI] [PubMed] [Google Scholar]
- 19. Tan W‐H, Bacino CA, Skinner SA, et al. Angelman syndrome: mutations influence features in early childhood. Am J Med Genet A 2011;155A:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boyd SG, Harden A, Patton MA. The EEG in early diagnosis of the Angelman (happy puppet) syndrome. Eur J Pediatr 1988;147:508–513. [DOI] [PubMed] [Google Scholar]
- 21. Valente KD, Andrade JQ, Grossmann RM, et al. Angelman syndrome: difficulties in EEG pattern recognition and possible misinterpretations. Epilepsia 2003;44:1051–1063. [DOI] [PubMed] [Google Scholar]
- 22. Laan LAEM, Brouwer OF, Begeer CH, et al. The diagnostic value of the EEG in Angelman and Rett syndrome at a young age. Electroencephalogr Clin Neurophysiol 1998;106:404–408. [DOI] [PubMed] [Google Scholar]
- 23. Wang PJ, Hou JW, Sue WC, Lee WT. Electroclinical characteristics of seizures – comparing Prader‐Willi syndrome with Angelman syndrome. Brain Dev 2005;27:101–107. [DOI] [PubMed] [Google Scholar]
- 24. Sidorov MS, Deck GM, Dolatshahi M, et al. Delta rhythmicity is a reliable EEG biomarker in Angelman syndrome: a parallel mouse and human analysis. J Neurodev Disord 2017;9:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vendrame M, Loddenkemper T, Zarowski M, et al. Analysis of EEG patterns and genotypes in patients with Angelman syndrome. Epilepsy Behav 2012;23:261–265. [DOI] [PubMed] [Google Scholar]
- 26. Frohlich J, Miller MT, Bird LM, et al. Electrophysiological phenotype in Angelman syndrome differs between genotypes. Biol Psychiatry 2019;85:752–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martinez LA, Born HA, Harris S, et al. Quantitative EEG Analysis in Angelman syndrome: candidate method for assessing therapeutics. Clin EEG Neurosci 2020. [DOI] [PubMed] [Google Scholar]
- 28. Piñon M. Theoretical background and structure of the Bayley scales of infant and toddler development, 3rd ed. Cambridge: Weiss & Oakland, 2010. [Google Scholar]
- 29. Gentile JK, Tan W‐H, Horowitz LT, et al. A neurodevelopmental survey of Angelman syndrome with genotype‐phenotype correlations. J Dev Behav Pediatr 2010;31:592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klem GH, Lüders HO, Jasper HH, Elger C. The ten‐twenty electrode system of the International Federation. The International Federation of Clinical Neurophysiology. Electroencephalogr Clin Neurophysiol Suppl 1999;52:3–6. [PubMed] [Google Scholar]
- 31. Kramer MA, Eden UT. Case studies in neural data analysis: a guide for the practicing neuroscientist. Cambridge: MIT Press, 2016. [Google Scholar]
- 32. Burnham KP, Anderson DR. Multimodel inference: understanding AIC and BIC in model selection. Sociol Methods Res 2004;33:261–304. [Google Scholar]
- 33. Lossie AC, Whitney MM, Amidon D, et al. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet 2001;38:834–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moncla A, Malzac P, Voelckel M‐A, et al. Phenotype‐genotype correlation in 20 deletion and 20 non‐deletion Angelman syndrome patients. Eur J Hum Genet 1999;7:131–139. [DOI] [PubMed] [Google Scholar]
- 35. Sadhwani A, Wheeler A, Gwaltney A, et al. Developmental skills of individuals with Angelman syndrome assessed using the Bayley‐III. J Autism Dev Disord 2021. Epub ahead of print. 10.1007/s10803-020-04861-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chauveau N, Franceries X, Doyon B, et al. Effects of skull thickness, anisotropy, and inhomogeneity on forward EEG/ERP computations using a spherical three‐dimensional resistor mesh model. Hum Brain Mapp 2004;21:86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pirchio M, Turner JP, Williams SR, et al. Postnatal development of membrane properties and δ oscillations in thalamocortical neurons of the cat dorsal lateral geniculate nucleus. J Neurosci 1997;17:5428–5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hughes SW, Cope DW, Blethyn KL, Crunelli V. Cellular mechanisms of the slow (<1 Hz) oscillation in thalamocortical neurons in vitro. Neuron 2002;33:947–958. [DOI] [PubMed] [Google Scholar]
- 39. Amzica F, Steriade M. Electrophysiological correlates of sleep delta waves. Electroencephalogr Clin Neurophysiol 1998;107:69–83. [DOI] [PubMed] [Google Scholar]
- 40. Carracedo LM, Kjeldsen H, Cunnington L, et al. A neocortical delta rhythm facilitates reciprocal interlaminar interactions via nested theta rhythms. J Neurosci 2013;33:10750–10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Holmes GL, McKeever M, Adamson M. Absence seizures in children: clinical and electroencephalographic features. Ann Neurol 1987;21:268–273. [DOI] [PubMed] [Google Scholar]
- 42. Englot DJ, Yang LI, Hamid H, et al. Impaired consciousness in temporal lobe seizures: role of cortical slow activity. Brain 2010;133:3764–3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kaplan PW. The EEG in metabolic encephalopathy and coma. J Clin Neurophysiol 2004;21:307–318. [PubMed] [Google Scholar]
- 44. Sutter R, Kaplan PW. Electroencephalographic patterns in coma: when things slow down. Epileptologie 2012;29:201–209. [Google Scholar]
- 45. Chu CJ, Leahy J, Pathmanathan J, et al. The maturation of cortical sleep rhythms and networks over early development. Clin Neurophysiol 2014;125:1360–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Frohlich J, Bird LM, Dell’Italia J, et al. Erratum: high‐voltage, diffuse delta rhythms coincide with wakeful consciousness and complexity in Angelman syndrome. Neurosci Conscious 2020;2020:niaa021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mahjoory K, Cesnaite E, Hohlefeld FU, et al. Power and temporal dynamics of alpha oscillations at rest differentiate cognitive performance involving sustained and phasic cognitive control. NeuroImage 2019;188:135–144. [DOI] [PubMed] [Google Scholar]
- 48. Limbach K, Corballis PM. Alpha‐power modulation reflects the balancing of task requirements in a selective attention task. Psychophysiology 2017;54:224–234. [DOI] [PubMed] [Google Scholar]
- 49. Jausovec N. Differences in EEG alpha activity related to giftedness. Intelligence 1996;23:159–173. [Google Scholar]
- 50. Fink A, Benedek M. EEG alpha power and creative ideation. Neurosci Biobehav Rev 2014;44:111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Klimesch W. EEG alpha and theta oscillations reflect cognitive and memory performance: a review and analysis. Brain Res Rev 1999;29:169–195. [DOI] [PubMed] [Google Scholar]
- 52. Laan LAEM, Renier WO, Arts WFM, et al. Evolution of epilepsy and EEG findings in Angelman syndrome. Epilepsia 1997;38:195–199. [DOI] [PubMed] [Google Scholar]
- 53. Porjesz B, Almasy L, Edenberg HJ, et al. Linkage disequilibrium between the beta frequency of the human EEG and a GABAA receptor gene locus. Proc Natl Acad Sci USA 2002;99:3729–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Margolis SS, Sell GL, Zbinden MA, Bird LM. Angelman syndrome. Neurotherapeutics 2015;12:641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Evangeliou A, Doulioglou V, Haidopoulou K, et al. Ketogenic diet in a patient with Angelman syndrome. Pediatr Int 2010;52:831–834. [DOI] [PubMed] [Google Scholar]
- 56. Ciarlone SL, Grieco JC, D'Agostino DP, Weeber EJ. Ketone ester supplementation attenuates seizure activity, and improves behavior and hippocampal synaptic plasticity in an Angelman syndrome mouse model. Neurobiol Dis 2016;96:38–46. [DOI] [PubMed] [Google Scholar]
- 57. Thibert RL, Pfeifer HH, Larson AM, et al. Low glycemic index treatment for seizures in Angelman syndrome. Epilepsia 2012;53:1498–1502. [DOI] [PubMed] [Google Scholar]
- 58. Mignan A, Broccardo M. One neuron versus deep learning in aftershock prediction. Nature 2019;574:E1–E3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Data from wake, sleep, and mixed states of consciousness correlate with cognitive function. Delta power was estimated (A) during wakefulness (R 2 = 0.9691, P = 0.0021, β = −18.97 [SE 5.84], AIC = 339.41), (B) during sleep (R 2 = 0.9613, P = 0.0048, β = −22.01 [SE 7.45], AIC = 340.52), and (C) from mixed states of consciousness, created from 50% to 50% sleep data (R 2 = 0.9690, P = 0.0013, β = −22.57 [SE 6.60], AIC = 338.39). (D) Delta power estimated from the same subjects during wake and sleep were highly correlated (R 2 = 0.5450). For all plots, the solid black line indicates the linear fit and the gray shaded region indicates the 95% confidence interval. Longitudinal same‐subject data are connected by red dotted lines. The insert shows the standard 10–20 EEG channels that were used to generate an estimate of delta power. *Scores have been adjusted for a fixed age and genotype for visualization.
Figure S2. Collinearity among the Bayley Scales in AS. Comparison of the Bayley Scores in four developmental domains. For each subject, the Bayley Scores are plotted against one another, per the vertical and horizontal labels. The R 2 values are shown in blue text. Particularly strong collinearities exist between the Bayley Cognitive, Fine Motor, and Receptive Communication Scores (R 2 > 0.7), whereas relatively weaker correlations exist between the Bayley Gross Motor and Expressive Communication Scores and all other Bayley Scores (R 2 < 0.7).
Table S1. Delta power was estimated from three montages to model cognitive function in Angelman syndrome. Detailed outputs for all three models are shown, including R 2, AIC, and coefficient estimates (β ± SE) and P‐values for each predictor.
Table S2. Spectral power in the delta (2–4 Hz), theta (5–7 Hz), alpha (8–12 Hz), and beta (13–30 Hz) frequency bands was used to model cognitive function. The standardized coefficient estimates (β ± SE) and P‐values for the spectral predictors in each model are shown, with P‐values that pass Bonferroni correction (P < 0.00714 for 7 tests) indicated in bold.
Table S3. Detailed model outputs for three models with two spectral predictors are shown. In all models, only delta power remains a significant predictor (P < 0.038).
Data Availability Statement
Derived data supporting the findings of this study are available from the corresponding author on request.